













 下载:
下载:

</td><td class="table_top_border" align="center" valign="middle">Cl<sup>−</sup>/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle"><inline-formula>${\rm{CO}}_3^ - $<img class="inline-formula" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">pH</td><td class="table_top_border" align="center" valign="middle">TOC/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">Cl<sup>−</sup>/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle"><inline-formula>${\rm{CO}}_3^ - $<alternatives><img class="graphic" src="201905182_M42.jpg"><img class="graphic" src="201905182_M42.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">7.82</td><td class="table_top_border2" align="center" valign="middle">13.2</td><td class="table_top_border2" align="center" valign="middle">38.6</td><td class="table_top_border2" align="center" valign="middle">95</td></tr><tr><td colspan="4" class="table_top_border2" style="line-height:2pt;height:2pt;"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"><inline-formula>${\rm{NO}}_3^ - $<alternatives><img class="graphic" src="201905182_M43.jpg"><img class="graphic" src="201905182_M43.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><inline-formula>${\rm{SO}}_4^{2 - }$<alternatives><img class="graphic" src="201905182_M44.jpg"><img class="graphic" src="201905182_M44.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle">Fe<sup>2+</sup>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle">总铁离子/(mg·L<sup>−1</sup>)</td></tr><tr><td class="table_bottom_border table_top_border2" align="center" valign="middle">&lt; 0.1</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">46.4</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">1.2</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">2.1</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula>/(mg·L<sup>−1</sup>)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">7.82</td><td class="table_top_border2" align="center" valign="middle">13.2</td><td class="table_top_border2" align="center" valign="middle">38.6</td><td class="table_top_border2" align="center" valign="middle">95</td></tr><tr><td colspan="4" class="table_top_border2" style="line-height:2pt;height:2pt;"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"><inline-formula>${\rm{NO}}_3^ - $<img class="inline-formula" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">pH</td><td class="table_top_border" align="center" valign="middle">TOC/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">Cl<sup>−</sup>/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle"><inline-formula>${\rm{CO}}_3^ - $<alternatives><img class="graphic" src="201905182_M42.jpg"><img class="graphic" src="201905182_M42.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">7.82</td><td class="table_top_border2" align="center" valign="middle">13.2</td><td class="table_top_border2" align="center" valign="middle">38.6</td><td class="table_top_border2" align="center" valign="middle">95</td></tr><tr><td colspan="4" class="table_top_border2" style="line-height:2pt;height:2pt;"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"><inline-formula>${\rm{NO}}_3^ - $<alternatives><img class="graphic" src="201905182_M43.jpg"><img class="graphic" src="201905182_M43.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><inline-formula>${\rm{SO}}_4^{2 - }$<alternatives><img class="graphic" src="201905182_M44.jpg"><img class="graphic" src="201905182_M44.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle">Fe<sup>2+</sup>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle">总铁离子/(mg·L<sup>−1</sup>)</td></tr><tr><td class="table_bottom_border table_top_border2" align="center" valign="middle">&lt; 0.1</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">46.4</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">1.2</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">2.1</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><inline-formula>${\rm{SO}}_4^{2 - }$<img class="inline-formula" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">pH</td><td class="table_top_border" align="center" valign="middle">TOC/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">Cl<sup>−</sup>/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle"><inline-formula>${\rm{CO}}_3^ - $<alternatives><img class="graphic" src="201905182_M42.jpg"><img class="graphic" src="201905182_M42.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">7.82</td><td class="table_top_border2" align="center" valign="middle">13.2</td><td class="table_top_border2" align="center" valign="middle">38.6</td><td class="table_top_border2" align="center" valign="middle">95</td></tr><tr><td colspan="4" class="table_top_border2" style="line-height:2pt;height:2pt;"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"><inline-formula>${\rm{NO}}_3^ - $<alternatives><img class="graphic" src="201905182_M43.jpg"><img class="graphic" src="201905182_M43.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle"><inline-formula>${\rm{SO}}_4^{2 - }$<alternatives><img class="graphic" src="201905182_M44.jpg"><img class="graphic" src="201905182_M44.png"></alternatives></inline-formula>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle">Fe<sup>2+</sup>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle">总铁离子/(mg·L<sup>−1</sup>)</td></tr><tr><td class="table_bottom_border table_top_border2" align="center" valign="middle">&lt; 0.1</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">46.4</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">1.2</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">2.1</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle">Fe<sup>2+</sup>/(mg·L<sup>−1</sup>)</td><td class="table_top_border2" align="center" valign="middle">总铁离子/(mg·L<sup>−1</sup>)</td></tr><tr><td class="table_bottom_border table_top_border2" align="center" valign="middle">< 0.1</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">46.4</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">1.2</td><td class="table_bottom_border table_top_border2" align="center" valign="middle">2.1</td></tr></tbody>
</table></div></foreignObject></svg>)













-
三氯乙烯(TCE)是一种优良的溶剂,常作为清洗剂和脱脂剂大量应用于金属脱脂和干洗等行业。由于使用过程中的不当处置等原因,导致其成为土壤和地下水中最常见的有机污染物之一[1]。TCE具有“三致”效应,密度大于水,属重质非水相液体(DNAPLs),进入环境后可长期滞留于含水层底部,持续污染地下水并危害人类健康[2]。
近年来,基于活化过硫酸盐的原位化学氧化(ISCO)技术被广泛应用于修复受有机物污染的土壤和地下水[3]。在热、光、碱和过渡金属离子等活化条件下,过硫酸盐可以产生强氧化性的硫酸根自由基(
SO−4⋅ )[4],能够氧化降解包括氯代烃、多环芳烃、苯系物等在内的多种有机污染物[5-6],过渡金属铁活化是其中主要的一种方式。铁是一种可以大规模应用于污染治理的环境友好型材料,相比普通铁粉,纳米零价铁(nZVI)由于其粒径小,比表面积大等特点,具有优越的吸附性能、更高的还原能力和更好的迁移性能,因而被广泛应用于土壤和地下水原位修复[7]。有研究[8]表明,在有氧和厌氧条件下,纳米零价铁表面均可以发生腐蚀反应且可释放Fe(II),进而活化过硫酸盐,产生SO−4⋅ ,以降解目标污染物,如式(1)~式(3)所示。然而,在酸性介质中,纳米零价铁的腐蚀过快,短时间内释放出大量的Fe(Ⅱ),高浓度的Fe(Ⅱ)反而会清除反应体系中的
SO−4⋅ ,降低污染物的降解率[9];在反应过程中,Fe(Ⅱ)被迅速氧化为Fe(Ⅲ),如式(4)所示,致使催化剂的活化能力降低,也不利于氧化反应的持续进行[10-11];此外,当溶液pH大于4时,Fe(Ⅱ)和Fe(Ⅲ)易形成沉淀,导致溶解性铁离子浓度降低[12]。有研究[13-15]表明,加入螯合剂(如柠檬酸、乙二胺四乙酸等)可以消除过量Fe(Ⅱ)对活性氧自由基的清除,减缓Fe(Ⅱ)向Fe(Ⅲ)的转化,阻碍铁离子的沉淀,提高氧化剂和催化剂的利用效率,从而提高污染物的降解率。ZHANG等[16]发现,在纳米零价铁活化过硫酸钠体系中,加入螯合剂柠檬酸,可以促进2,4,6-三氯苯甲醚的降解。DANISH等[17]发现,螯合剂草酸、柠檬酸、谷氨酸的加入,可以提高纳米零价铁活化过碳酸钠体系中游离Fe(Ⅱ)的浓度,强化活性氧自由基的产生,进而提高三氯乙烷的降解率。在众多螯合剂中,广泛存在于自然界中的天然有机酸柠檬酸(CA),是一种鳌合能力优良、可生物降解的绿色螯合剂,可大规模应用于环境修复领域。到目前为止,CA对nZVI活化过硫酸盐降解有机污染物的影响尚鲜有报道。本研究选用CA为螯合剂,过硫酸钠(PS)为氧化剂,TCE为目标污染物,主要研究了CA对PS/nZVI体系降解TCE的强化作用,考察了PS、CA和nZVI投加量、溶液初始pH和无机阴离子对PS/nZVI/CA体系中TCE降解的影响,探究了反应体系中活性氧自由基的产生及其降解TCE的作用机制,最终在实际地下水中验证PS/nZVI/CA体系降解TCE的效果,为该技术应用于实际污染场地地下水修复提供技术支持。
全文HTML
-
试剂包括PS、TCE、CA、正己烷、异丙醇(IPA)、叔丁醇(TBA)、三氯甲烷(CF)、硫酸(H2SO4)、氢氧化钠(NaOH)、5,5-二甲基-1-氧化吡咯啉(DMPO)、氯化钠、碳酸氢钠,以上试剂均为分析纯,购于上海晶纯试剂有限公司。纳米零价铁购于上海超威纳米科技有限公司(型号:CW-Fe-001,平均粒径50 nm,纯度>99%)。实验中用水均为自制超纯水(18.2 MΩ·cm)。
仪器包括电子分析天平(AL204,瑞士Mettler-Toledo集团)、pH计(DELTA320,瑞士Mettler-Toledo集团)、超纯水机(Classic DI,英国ELGA公司)、电子顺磁共振仪(EMX-8/2.7C,德国Bruker公司)、恒温磁力搅拌器(85-2,上海闵行虹浦仪器厂)、涡旋振荡器(XW-80A,上海虹浦仪器厂)、低温恒温槽(SDC-6,宁波新芝生物科技股份有限公司)、气相色谱分析仪(7890 A,安捷伦科技有限公司)。
-
所有实验均在250 mL密闭的定制玻璃反应器中进行(内径6 cm,内高9 cm),反应器设有夹层,采用低温恒温槽控制反应温度为20 ℃,用超纯水配置TCE储备液。在实验时,移取一定量的TCE储备液至反应器中,稀释至实验所需的浓度(0.15 mmol·L−1),除在研究溶液初始pH对TCE降解效果的影响时,采用0.1 mol·L−1 H2SO4或0.1 mol·L−1 NaOH预先调节溶液的pH,其他反应条件下pH不做调整。加入磁力搅拌子,搅拌器转速设置为600 r·min−1,然后依次向反应器中加入所需的CA、nZVI和PS并开始计时,在此搅拌强度下,nZVI加入反应器后,可以形成分散均一的悬浊液,之后在既定时间点取样分析,每组实验设2个平行样,结果取平均值。
-
反应溶液中TCE的浓度采用正己烷萃取法进行测定:取1 mL含有TCE的反应溶液,加入到预先含有1 mL正己烷的萃取瓶中,旋涡振荡萃取3 min后,静置5 min,取上层有机相到进样瓶中密封保存,使用气相色谱仪分析。气相色谱条件:色谱柱型号DB-VRX(长60 m,内径250 μm,膜厚1.4 μm),电子俘获检测器(ECD),进样量为1.0 μL,进样口温度为220 °C,柱温100 °C,检测器温度240 °C,载气为氮气,分流比为20∶1。
1.1. 试剂与仪器
1.2. 实验方法
1.3. 分析方法
-
图1反映了不同反应体系对TCE的降解效果,PS、nZVI、CA、TCE初始浓度分别为0.6、0.3、0.15、0.15 mmol·L−1。实验结果表明,在不投加氧化剂和催化剂时,TCE在60 min内的挥发率小于6%;只加nZVI或PS时,60 min内,TCE的降解率也仅有8.3%或11.8%。nZVI可以有效还原TCE,但通常需要较高的nZVI投加量和较长的反应时间[18],而在本实验中,少量的nZVI在短时间内无法有效降解TCE,且在单独PS体系中的降解效果也不佳;但是同时加入PS和nZVI后,60 min内,TCE降解率提升至79.7%,表明nZVI可以活化PS,产生活性氧自由基,氧化降解溶液中的TCE。值得注意的是,在PS/nZVI体系中加入螯合剂CA后,TCE的降解率可达到94.7%,明显高于PS/nZVI体系中TCE的降解率,这说明CA的加入可以有效促进nZVI催化PS体系中TCE的降解。在nZVI催化PS体系中投加CA可以从多方面影响TCE的降解过程。首先,柠檬酸作为一种有机酸,降低了溶液的pH,投加0.15 mmol·L−1CA后,溶液初始pH由6.88降低至3.95。溶液pH的降低可以加快nZVI的腐蚀,促进Fe(Ⅱ)的释放,有利于PS/nZVI体系中活性氧自由基的产生和TCE的降解[19]。其次,CA可以鳌合Fe(Ⅱ)和Fe(Ⅲ),形成稳定的配合物,抑制铁离子沉淀,增强体系中可溶性Fe(Ⅱ)的含量,并减缓Fe(Ⅱ)向Fe(Ⅲ)的转化,强化自由基的持续不断产生,从而提高氧化剂和催化剂的利用效率,促进TCE的降解[20]。综上可知,CA可以降低溶液的初始pH,稳定溶液中的Fe(Ⅱ)/Fe(Ⅲ),并阻止铁离子沉淀的产生,进而显著提高TCE的降解率。
-
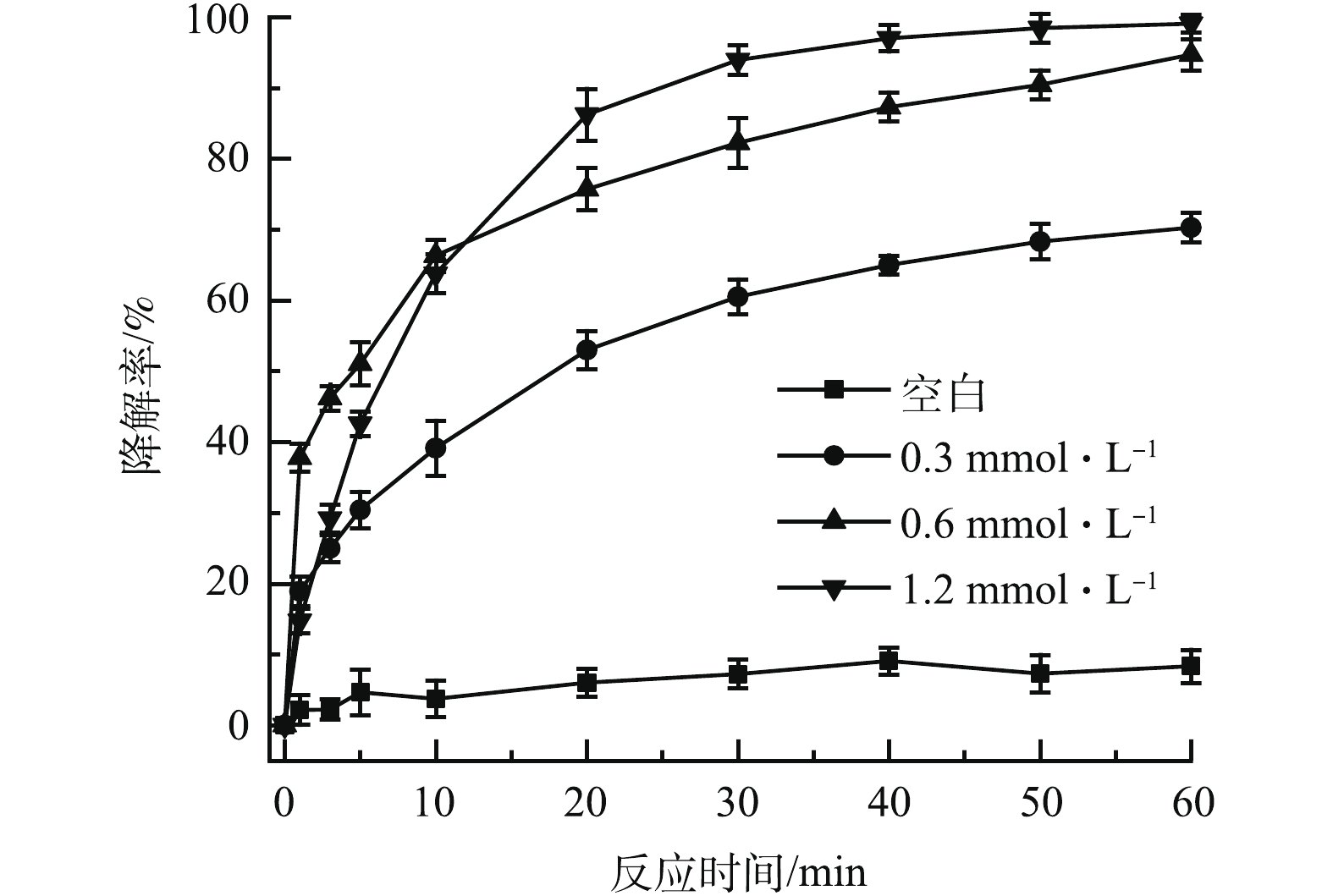
控制nZVI、CA、TCE初始浓度分别为0.3、0.15、0.15 mmol·L−1,通过改变PS的初始浓度,考察氧化剂投加量对PS/nZVI/CA体系降解TCE的影响,结果如图2所示。当PS浓度为0.3 mmol·L−1和0.6 mmol·L−1时,60 min内,TCE的降解率分别达到70.3%和94.7%;进一步增加氧化剂浓度至1.2 mmol·L−1,TCE的降解率并无明显变化,说明适当增加氧化剂浓度,有助于PS/nZVI/CA体系中产生更多的活性氧自由基,提高TCE的降解率。但是氧化剂浓度过高时,产生的高浓度的
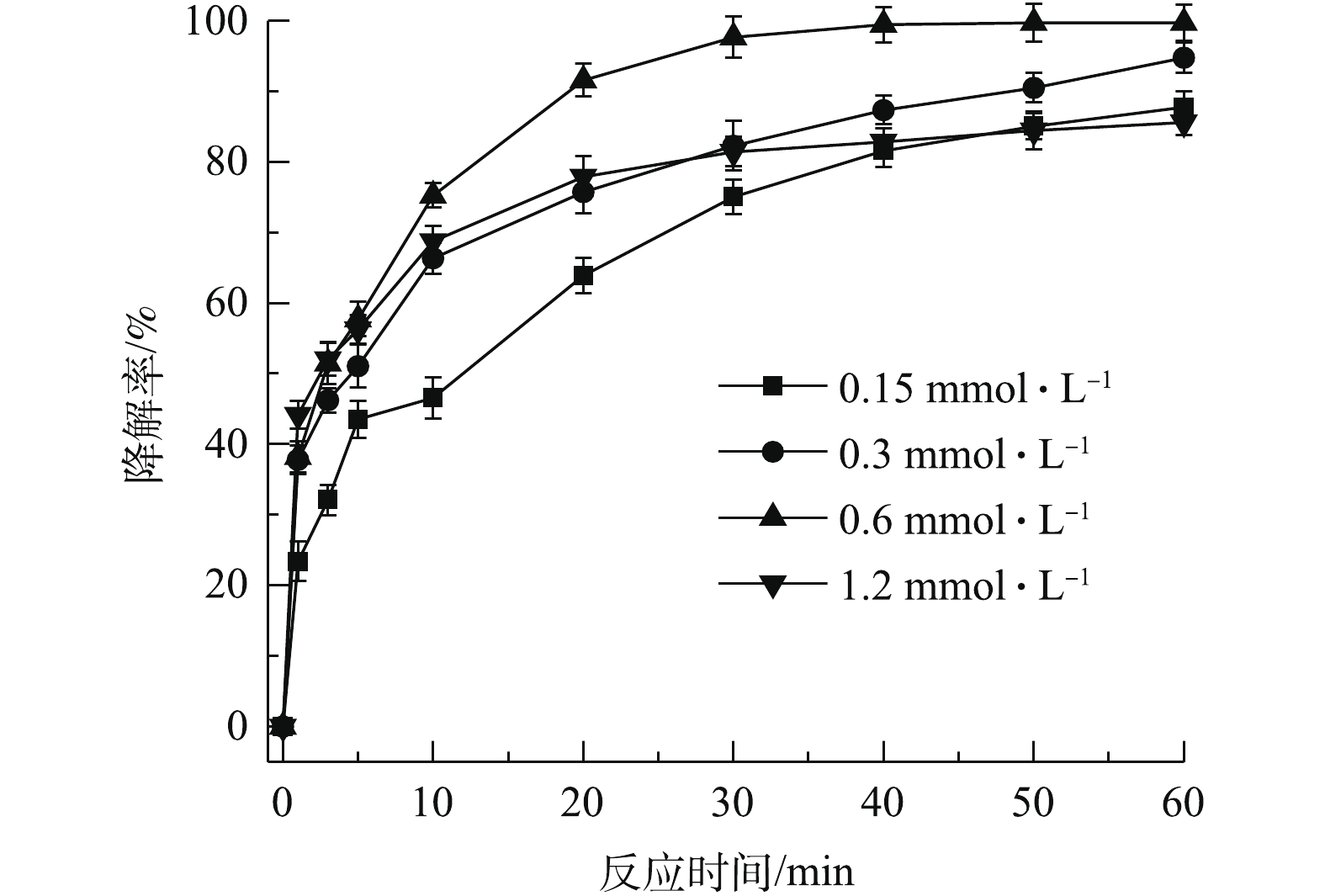
SO−4⋅ 自由基会相互消耗,如式(5)所示,降低自由基的有效反应,从而不能达到提高TCE的降解目的[21-22]。控制PS、nZVI、TCE初始浓度分别为0.6、0.3、0.15 mmol·L−1,通过改变柠檬酸的投加量,考察其对PS/nZVI/CA体系降解TCE的影响,结果如图3所示。反应体系中没有加CA时,60 min内,TCE的降解率为79.7%;当加入0.15 mmol·L−1的CA后,TCE降解率提高至94.7%;但继续增加CA至0.3 mmol·L−1和0.6 mmol·L−1时,TCE降解受到抑制,降解率分别降低至91.3%和84.9%。这表明在PS/nZVI/CA体系中,投加适量的CA可以有效促进TCE的降解,但当CA浓度过高时,TCE的降解反而受到抑制。这主要是因为过量的CA将会与污染物竞争活性氧自由基[23-24],不利于TCE的降解;此外,CA具有较强的金属鳌合能力,过量的CA会与溶液中游离的Fe(Ⅱ)形成过于稳定的螯合物,削弱了Fe(Ⅱ)的活化能力,使反应体系中产生的活性氧自由基减少[25],最终导致TCE的降解率降低。因此,在实际应用中须确定适当的CA浓度,提高目标污染物降解率并降低成本。
控制PS、CA、TCE初始浓度分别为0.6、0.15、0.15 mmol·L−1,通过改变nZVI的投加量,考察nZVI对PS/nZVI/CA体系降解TCE的影响,结果如图4所示。随着nZVI投加量的提高,TCE降解率先升后降。当nZVI初始浓度由0.15 mmol·L−1提高至0.3 mmol·L−1和0.6 mmol·L−1时,TCE降解率由87.7%上升至94.7%和99.7%,但继续提高nZVI浓度至1.2 mmol·L−1,TCE降解率降低至85.5%。这是因为过量的nZVI会释放大量的Fe(II),清除体系中的
SO−4⋅ ,如式(4)所示,导致TCE降解率下降[26]。 -
控制PS、nZVI、CA、TCE初始浓度分别为0.6、0.3、0.15、0.15 mmol·L−1,考察在PS/nZVI/CA体系中溶液初始pH对TCE降解率及终点pH的影响,溶液初始pH为3.0~11.0,结果如表1所示。可以看出,当溶液初始pH由3升至9时,TCE的降解率无明显变化,60 min内,TCE降解率均可以达到90%以上,并且反应终点的pH都可以维持在3.33以下,这说明在PS和CA的共同作用下,该反应体系有较宽的pH适用范围,最终溶液pH均回归酸性,这与PS和CA的性质密切相关[27]。有研究[28]指出,酸性环境下活化过硫酸盐体系中可以产生更多的
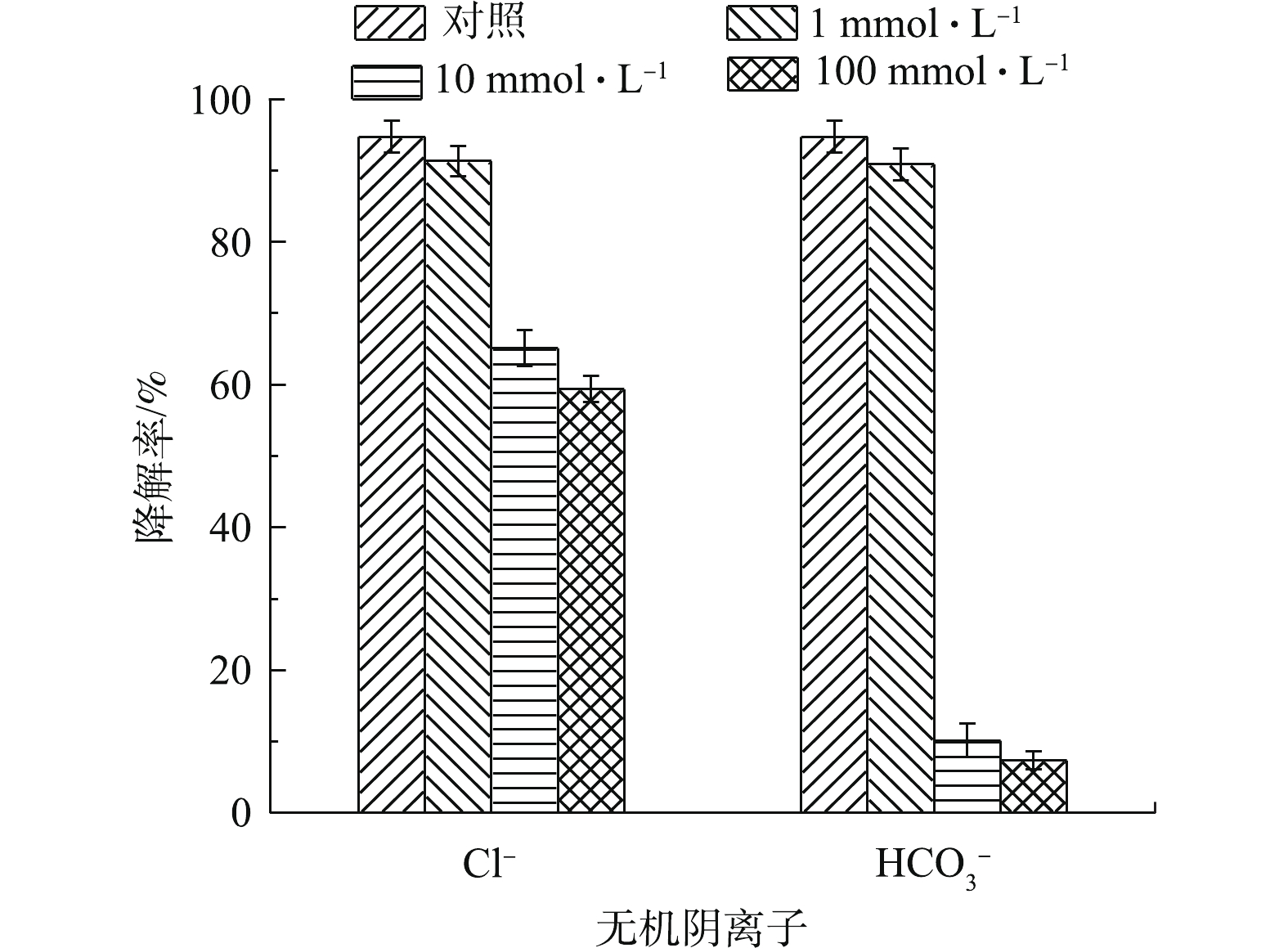
SO−4⋅ ,从而提高污染物的降解率;而CA的加入可以降低溶液的pH,有利于纳米零价铁的腐蚀,维持溶液中游离态Fe(Ⅱ)的浓度,持续活化PS产生活性氧自由基降解TCE。然而,当溶液初始pH由9升至11时,TCE的降解明显受到抑制,降解率由90.35%降低到11.57%,相应的终点pH为10.81,表明在强碱性的溶液中,TCE的降解明显受到抑制。首先,在强碱性溶液中,纳米零价铁的腐蚀速度过慢,短时间内无法释放足量的游离态Fe(Ⅱ)催化PS,最终的pH为碱性,导致释放的Fe(Ⅱ)被沉淀,不利于TCE的降解[29];其次,碱性环境下过硫酸盐氧化体系倾向于生成更多HO·,如式(6)所示[30],而HO·在碱性溶液中的氧化能力较弱[31],故导致TCE的降解率下降。综上可知,PS/nZVI/CA体系具有较宽的pH适用范围,且酸性条件更有利于该体系中TCE的降解。实验选取地下水中普遍存在的Cl−、
HCO−3 作为阴离子的代表,考察上述阴离子对PS/nZVI/CA体系降解TCE的影响。体系中PS、nZVI、CA、TCE初始浓度分别设为0.6、0.3、0.15、0.15 mmol·L−1,阴离子浓度为0~100 mmol·L−1,结果如图5所示。可以看出,在实验所选的浓度范围内,2种阴离子对TCE的降解均有不同程度的抑制作用,低浓度的阴离子(1 mmol·L−1)对TCE的降解影响不大,但随着阴离子浓度的增加,抑制作用逐渐增强。当Cl−浓度由0 mmol·L−1升高至10 mmol·L−1和100 mmol·L−1时,60 min内,TCE的降解率由94.7 %分别降低至65.1%和59.4%。有研究[32]表明,Cl−是SO−4⋅ 和HO·的清除剂,在反应过程中,会与TCE竞争活性氧自由基,从而生成低反应活性的氯自由基,如式(7)和式(8)所示,进而影响TCE降解。相比于Cl−,HCO−3 对TCE的抑制作用更为显著。当HCO−3 浓度由0升高至10 mmol·L−1和100 mmol·L−1时,60 min内,TCE的降解率由94.7%分别降低至10.2%和7.4%。一方面,HCO−3 的加入提高了溶液的pH,加入10 mmol·L−1和100 mmol·L−1的HCO−3 后,溶液初始pH分别升高至7.03和7.86,并且HCO−3 具有一定的缓冲能力,因而不利于纳米零价铁的腐蚀和铁的溶解,不利于体系中活性氧自由基的生成;另一方面,HCO−3 会清除体系中的自由基,导致体系的氧化能力下降[33]。因此,当体系中存在高浓度的HCO−3 时,TCE的降解可能会被完全抑制。 -
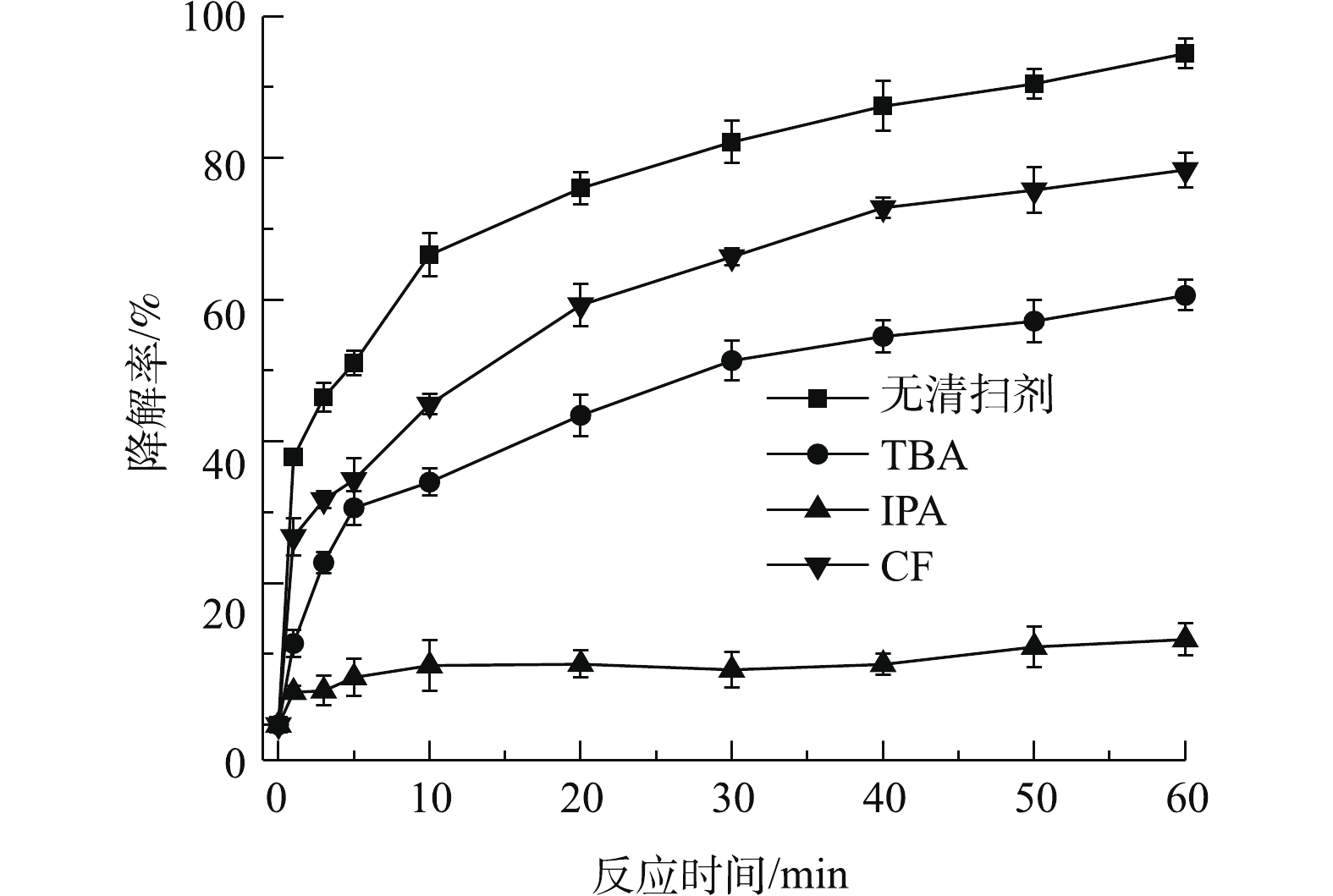
通过自由基清除实验确定PS/nZVI/CA体系降解TCE过程中的主导自由基。实验中选取TBA为HO·清除剂,IPA为HO·和
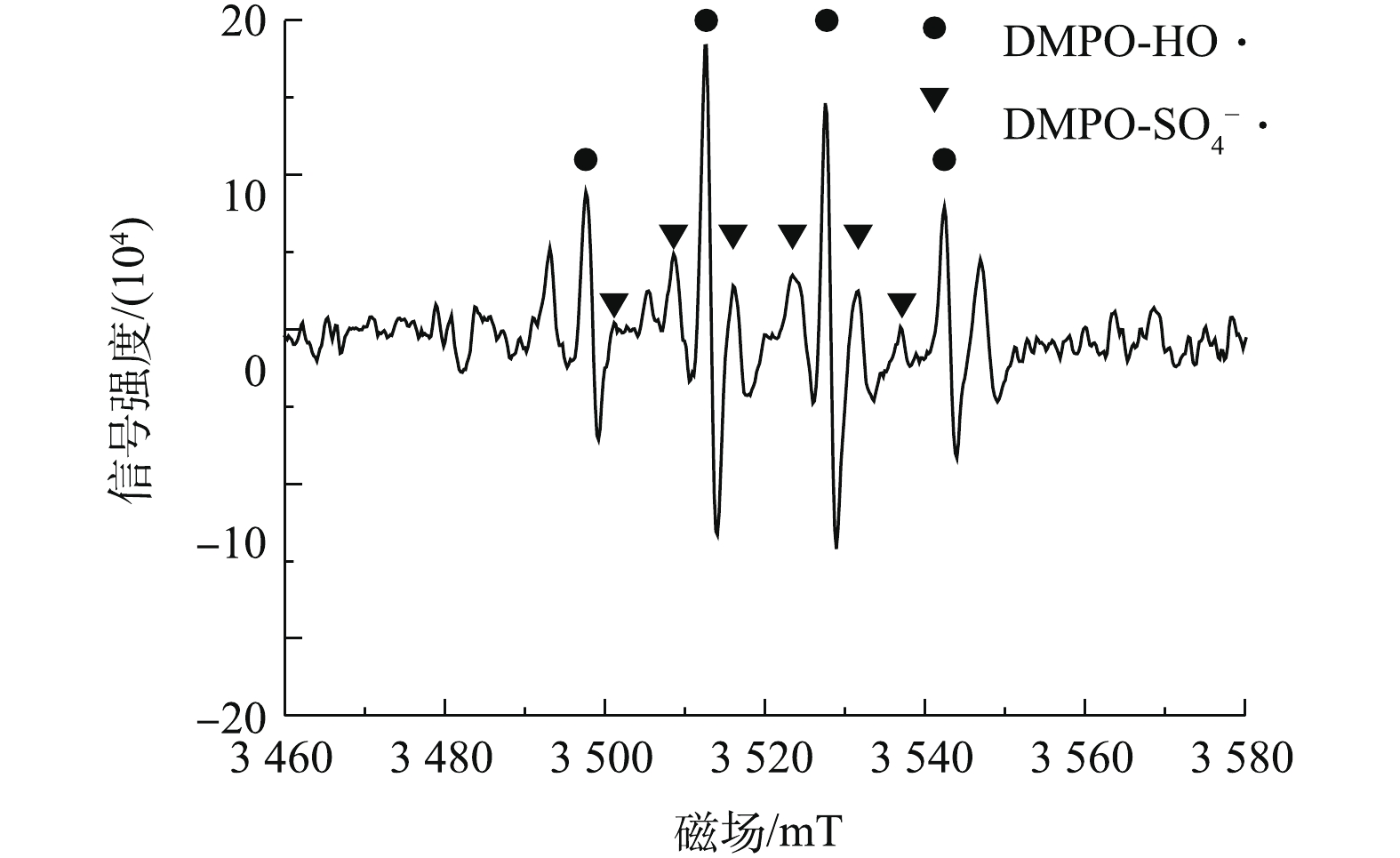
SO−4⋅ 共同的清除剂,CF为O−2⋅ 清除剂[34]。实验控制PS、nZVI、CA、TCE初始浓度分别为0.6、0.3、0.15、0.15 mmol·L−1,TBA、IPA、CF浓度分别为100、100、50 mmol·L−1,反应时间为60 min。如图6所示,不加清除剂时,PS/nZVI/CA体系中TCE的降解率为94.7%,加入100 mmol·L−1 TBA后,TCE降解率下降至60.2%,这说明HO·是PS/nZVI/CA体系中的主导自由基之一;加入100 mmol·L−1 IPA后,TCE降解率仅有12.1%,表明SO−4⋅ 在TCE降解中也起到了重要作用;加入50 mmol·L−1的CF后,TCE降解率降低至78.3%,这表明O−2⋅ 同样参与了PS/nZVI/CA体系中TCE的降解过程,O−2⋅ 主要表现在通过自由基链式反应参与其中,并影响HO·和SO−4⋅ 自由基的生成。综上可知,在PS/nZVI/CA体系中产生了HO·、SO−4⋅ 和O−2⋅ ,其中,HO·和SO−4⋅ 是降解TCE的主要自由基,O−2⋅ 则起次要作用。为了进一步直观地检测PS/nZVI/CA体系中产生的活性氧自由基,选择DMPO作为自由基的捕捉剂[35],通过EPR检测DMPO与活性氧自由基生成的加合物来确定自由基的类型。PS/nZVI/CA体系中PS、nZVI、CA、TCE初始浓度分别为0.6、0.3、0.15、0.15 mmol·L−1时,反应进行3 min时EPR谱图如图7所示。在EPR谱图中发现了DMPO-HO·(峰高比为1∶2∶2∶1)和DMPO-
SO−4⋅ 加合物的典型特征峰,证明在PS/nZVI/CA体系中产生了HO·和SO−4⋅ 。但是在EPR谱图中未发现DMPO与O−2⋅ 的加合物DMPO-OOH的特征峰,这可能是由于该体系中O−2⋅ 产生量较少,导致加合物DMPO-OOH的强度较低,而被HO·和SO−4⋅ 的特征峰所遮蔽,而且DMPO-OOH在酸性条件下极不稳定,易转化为DMPO-OH。 -
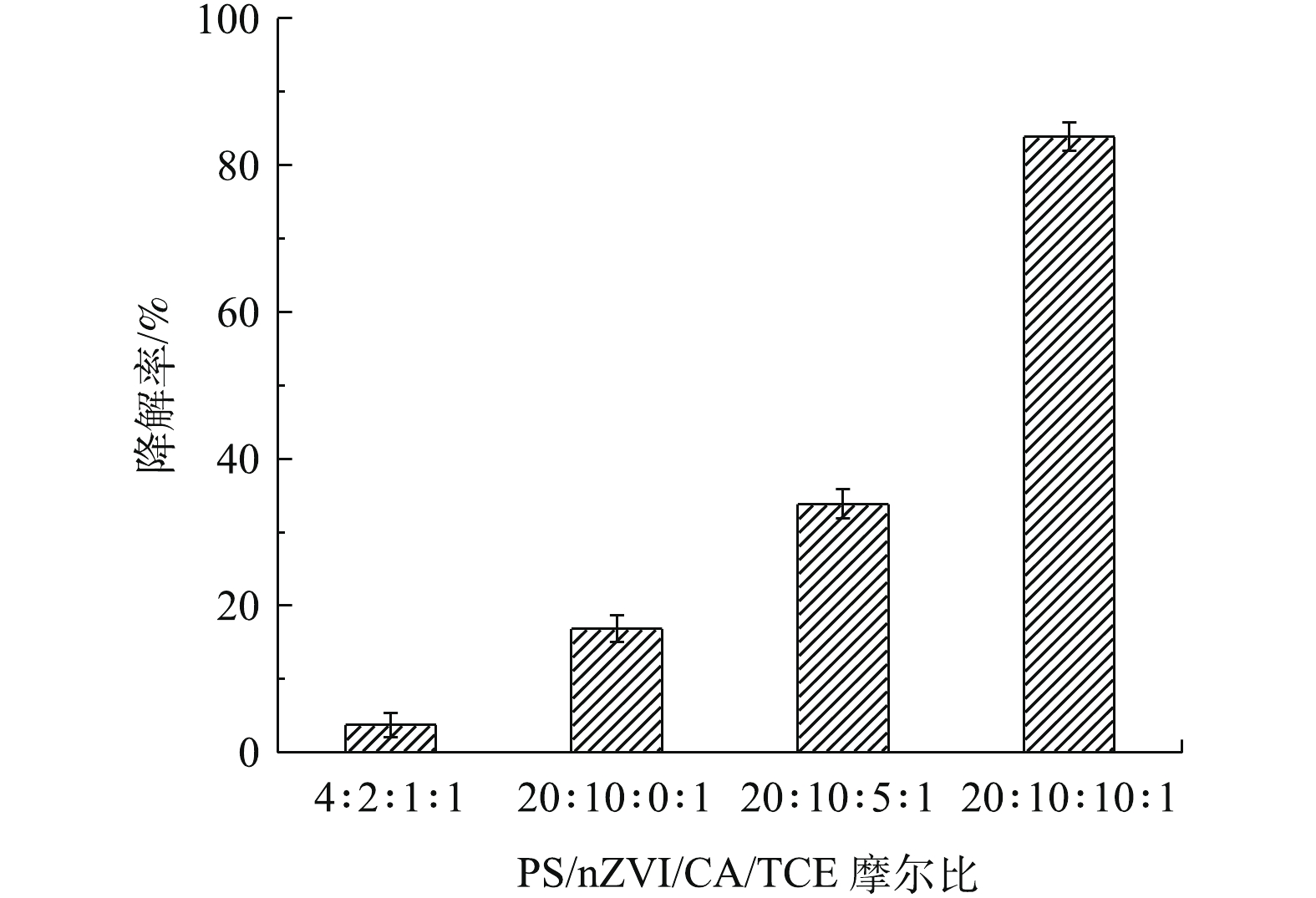
为了验证PS/nZVI/CA体系降解实际地下水中TCE的效果,采用实际地下水代替超纯水进行实验,所用实际地下水的水质参数如表2所示。图8显示了实际地下水中PS/nZVI/CA体系降解TCE的效果,TCE初始浓度为0.15 mmol·L−1。可以看出,与超纯水相比,相同药剂投加量下PS/nZVI/CA体系在实际地下水中TCE的降解率显著降低。当PS/nZVI/CA/TCE摩尔比为4∶2∶1∶1和20∶10∶5∶1时,60 min内,TCE的降解率只有3.7%和33.8%。这主要是因为实际地下水的pH较高,并且具有一定的缓冲能力。如前所述,在较高的pH条件下,nZVI的腐蚀速率过慢,溶液中游离态Fe(Ⅱ)的浓度较低,PS无法被有效活化;此外,由表2可知,实际地下水中成分复杂,含有的天然有机物和阴离子也会影响体系中TCE的降解。为了提高实际地下水中TCE的降解率,保持PS和nZVI投加量不变,进一步提高CA的投加量。当PS/nZVI/CA/TCE摩尔比增加到20∶10∶10∶1时,60 min内,TCE的降解率可以提高至83.8%,而当PS/nZVI/CA/TCE摩尔比为20∶10∶0∶1时,即在反应体系中不投加CA时,TCE的降解率仅有16.8%。这表明提高药剂投加量可以消除实际地下水水质条件对氧化反应的不利影响,提高TCE的降解率。提高CA投加量,有助于降低溶液的pH,加快nZVI的腐蚀,促进Fe(II)的释放,保持溶液中Fe(II)的浓度,并减缓铁沉淀的形成,强化TCE的降解。因此,PS/nZVI/CA体系相比PS/nZVI体系更适应实际地下水中各种水质条件的冲击影响,具有应用于实际地下水修复的潜势。
2.1. 不同反应体系对TCE的降解效果比较
2.2. PS、CA、nZVI投加量对TCE降解效果的影响
2.3. 溶液水质条件对TCE降解的影响
2.4. PS/nZVI/CA体系中活性氧自由基鉴定
2.5. 实际地下水中柠檬酸强化nZVI活化PS降解TCE的效果验证
-
1)投加适量的CA,可以明显提高PS/nZVI体系中TCE的降解效果,PS/nZVI/CA/TCE摩尔比为4∶2∶1∶1时,60 min内,TCE降解率为94.7%;PS、nZVI不足或过量均不利于TCE的降解。
2)PS/nZVI/CA体系具有较宽的pH适用范围,溶液初始pH为3~9时,TCE的降解效果良好;溶液中Cl−和
HCO−3 的存在会影响TCE的降解,其中HCO−3 的影响程度较大。3) PS/nZVI/CA体系中有HO·、
SO−4⋅ 和O−2⋅ 活性氧自由基产生,其中HO·和SO−4⋅ 对TCE的降解起主导作用。4)与超纯水体系相比,实际地下水中PS/nZVI/CA体系降解TCE的效果不佳,但是适当提高药剂投加量,改变药剂投加比即可提高TCE的降解率;PS/nZVI/CA体系相比PS/nZVI体系更适应实际地下水中各种水质条件冲击的影响,具有良好的实际应用前景。
