
































-
砷(As)主要伴生在硫铁矿和有色金属矿中[1]。长期以来,我国以硫铁矿生产硫酸和有色冶炼烟气制酸为主,在制酸废水处理过程中产生了大量高As污泥,且As以溶解态As(Ⅲ)为主[2]。高As污泥的随意堆放对周边环境及人群健康危害性极大。近年来,对其进行固化后安全填埋,已成为重要处理途径之一[3]。一般的材料很难使高As污泥中As浸出浓度满足《危险废物填埋污染控制标准》(GB 18598-2001) [4]填埋入场要求,因此,固As材料的选择成为关键。
无机硫化物、含钙(石灰、石灰石等)材料、含铁锰或铝材料(Fe0、亚铁盐、铁盐、铁锰铝氧化物、铁的氢氧化物等)和水泥等[5-9]对As的固定机制不同,目前,用于高As污泥方面的研究较少。其中,无机硫化物主要通过与As形成螯合物,或与Fe、As形成共沉淀物质,如三硫化二砷(As2S3)或硫铁化砷(AsFeS) [10-11],但无机硫化物的实际固As效果须进一步验证。含钙材料(如CaO等)主要与As形成CaHAsO4和Ca3(AsO4)2) 沉淀[12],但有研究[13-14]认为,CaO固As效果不佳,As经CaO固定后,在高pH条件和酸性浸出条件下容易活化。含铁材料对As主要进行化学专性吸附并将其固定到氧化物晶格层间,可生成FeAsO4和FeAsO4·2H2O等[15]。但不同的含铁材料对As的固定效果具有明显的差异[16-18]。水泥是国内外处理危险废物最常用的也最廉价的固化材料,其通过水化过程的吸附、物理包裹、晶格化等作用抑制As和重金属的渗滤扩散[19-23]。有研究[19, 24-25]表明,在水泥固化前,添加其他固As材料,可通过吸附和共沉淀等作用进一步降低As的释放风险。然而,采用固定化技术处理高As污泥的可行性仍不确定,须根据其污染特性进行固As材料的筛选。
本研究以南方某硫酸厂产生的高浓度含As污泥为处理对象,选用硫化物、含钙、含铁或铝共10种固As材料,采用3种毒性浸出法评估了材料的固As效果,考察了各材料处理对污泥中As结合态和价态的影响,最终筛选出了固As效果最佳的材料,并与水泥进行了联合固化研究,为高As污泥的安全处理提供参考。
-
供试污泥取自南方某硫酸厂工业废水处理后的蓄泥池,为灰黑色。将污泥采集后,自然风干、除杂、混匀、磨碎,过2 mm尼龙筛后,用于固As实验,过0.16 mm筛后,测As含量和重金属总量。供试污泥As含量最高,达到27.33%,其次是Zn(3.63%)和Cu(3.08%),Pb、Cd、Fe、Mn含量依次为0.66%、0.46%、0.17%、0.01%,pH为2.50。
供试固As材料分3类共10种(表1),均为分析纯,包括硫化物(Na2S·9H2O)、含钙材料(CaO)、含铁或铝材料(Fe0、亚铁盐(FeSO4·7H2O)、铁盐(Fe(NO3)3·9H2O、FeCl3、Fe2(SO4)3、Fe(OH)3)、Fe2O3、Al2O3)。供试水泥为工业级PO 425#。
-
1)固As材料的筛选实验。称量20.00 g污泥置于200 mL三角瓶中,按表1的设计方法,投加各种材料,充分搅拌混匀后,加去离子水,保持含水率为25%,室温养护7 d后,进行样品pH、As毒性浸出、As结合态、As价态分析,设置空白对照(CK),每种处理设3个重复。硫化物、CaO、含铁或铝材料中有效元素与污泥总As的理论摩尔比分别按反应产物As2S3、Ca3(AsO4)2、FeAsO4·2H2O、AlAsO4计算,如S∶As=3∶2、Ca∶As=3∶2、Fe∶As=1∶1,因污泥总As含量很高(表1),材料投加量初按理论摩尔数的30%计算。
2) FeCl3与水泥配伍(复配)实验。将FeCl3按与污泥干重质量比为50∶100、100∶100、150∶100、200∶100、250∶100进行固定化处理,将水泥分别按25%、50%、75%、100%、125%投加比进行固化处理,并将不同比例的水泥与250% FeCl3进行配伍处理(见表2),处理和养护过程同上。
-
污泥pH测定采用《土壤pH的测定》(NY/T 1121.2-2006)中的方法[26],使用酸度计(pHs-3C型,上海仪电科学仪器股份有限公司)测定;As浸出采用TCLP法[27](L/S=1∶20)、H2SO4-HNO3法[28](L/S=1∶10)、H2O浸法[29](L/S=1∶10)、SBET法[30](L/S=1∶100);As结合态前处理采用WENZEL(2001)化学连续浸提法[13];污泥中As和重金属含量以及As结合态中的残渣态测试的前处理均采用HNO3-HF-HClO4消解法[31];消解液中重金属含量采用电感耦合等离子体发射光谱仪(ICP-MS 7500,美国Agilent公司)测定。浸出As含量、结合态As含量和消解液中的As含量均采用原子荧光分光光度计(AFS-9120,北京吉天仪器有限公司)测定;污泥As价态采用1.0 mol·L−1 H3PO4+0.1 mol·L−1抗坏血酸[32]提取,采用液相-原子荧光联用仪(LC-AFS(SA-20),北京吉天仪器有限公司)测定。
修复效果评估根据式(1)进行计算。
式中:η为As固定率;C0为废渣在处理前的As浸出浓度,mg·L−1;Ct为处理后As的浸出浓度,mg·L−1。
-
污泥中As浸出特性见表3。可以看出,污泥As浸出浓度均很高,依次为H2SO4-HNO3>H2O>TCLP>SBET。其中,TCLP和H2SO4-HNO3浸提As浓度分别可高达10 634.05 mg·L−1和14 961.25 mg·L−1,超出《危险废物鉴别标准浸出毒性鉴别》(GB 5085.3-2007)标准值(5 mg·L−1)的2 125.81倍和2 991.25倍,各种方法浸提出的As总量占污泥总As的比例均很高,为44.93%~98.01%,大小顺序依次为SBET(98.01%)>TCLP(77.83%)>H2SO4-HNO3(54.75%)>H2O(44.93%)。
污泥中As结合态分布见表4。可以看出,污泥中As主要以F1非专性吸附态和F2专性吸附态为主,占总As的比例达56.09%,与H2SO4-HNO3浸出As水平相当,残渣态占比不到20%,因此,As泥的毒性和危害性极大。
-
TCLP法模拟了污泥在生活垃圾填埋场中有机弱酸浸提条件下的环境风险。如图1所示,除CaO和Fe2O3外,各材料均可明显降低污泥As的浸出浓度,固As能力依次为FeCl3>FeSO4·7H2O>Fe2O12S3>Fe0>Fe(NO3)3·9H2O>Al2O3>Na2S·9H2O>Fe(OH)3。其中,FeCl3处理使As浸出由10 634.05 mg·L−1降至1 487.66 mg·L−1,固砷率η高达86.01%;CaO和Fe2O3效果最差,CaO甚至活化As,使As浸出增加了15.16%。在10种材料中,FeCl3在应对有机弱酸浸出方面表现出了最强的固As能力。
H2SO4-HNO3浸提法模拟了污泥在强酸降雨情境下As的淋滤风险。结果表明,材料固As能力依次为FeCl3>FeSO4·7H2O≈Fe0>Al2O3,其他材料均无效甚至活化As。其中,FeCl3效果最好,使As浸出从14 961.25 mg·L−1降至8 674.35 mg·L−1,η可达42.02%;其次是FeSO4·7H2O和Fe0。CaO、Na2S·9H2O、Fe2O3、Fe(NO3)3·9H2O、Fe2O12S3 等5种处理材料均对As产生了活化效果。其中,以CaO和Na2S·9H2O最为明显,相应的As浸出率分别增加了37.44%和17.42%。
H2O浸提法模拟了污泥在自然情景下中性H2O对As的浸沥风险。结果表明,FeCl3效果最好,使As浸出从12 276.50 mg·L−1降至5 049.40 mg·L−1,η为58.87%;其次是Fe0,η为35.52%。CaO、Fe(NO3)3·9H2O、Fe2O3、Na2S·9H2O、Fe2O12S3则明显增加了As的浸出,Fe(OH)3对As的浸出影响不明显。
FeCl3均明显降低了污泥的3种As浸出浓度,在各种材料中,FeCl3固As效果均最优,应对有机弱酸淋滤风险的能力最强,其次是H2O和强酸,这可能与FeCl3可促进污泥As向固定态转化有关。FeCl3表面的双配位基对As有很强的吸附能力[33],除提供Fe3+与砷酸根生成砷酸铁外,还可同时产生氢氧化铁胶体,与As发生吸附共沉淀作用。Fe(OH)3对As的浸出影响不大,这可能是因Fe主要以胶体形式存在,不易直接与As结合,其对As的吸附能力主要与pH有关,在中性和酸性条件下,As可能只以双配位表面络合的质子化的FeO2As(O)(OH)−和非质子化的≡FeO2As(O)2−形态存在于Fe(OH)3表面;在弱酸性和弱碱性条件下,吸附As的能力可能变强[34]。Fe2O3和Al2O3对As主要以专性吸附作用为主,与Fe0、亚铁盐和大多三价铁盐一样,与As(Ⅲ)不易形成固定化合物,可能更适用于弱酸性条件下含As(V)为主的污泥固定化处理[35-36]。
CaO在3种浸提条件下固As效果均最差,单一CaO并不适用于高As污泥处理。在还原条件下,污泥中As主要以还原态As(Ⅲ)或三元含氧酸H3AsO3形式存在,pH>12.13时,方可形成溶解度较低的As-Ca化合物[37];而在氧化条件下,pH为中性或为4.5~8.5,As-Ca才较易形成[10]。受污泥中pH和As(Ⅲ)含量的影响,可能须先对污泥中As(Ⅲ)进行预氧化,才利于形成砷酸钙盐及其水合化合物[38]。但也有研究[13-14]表明,As-Ca并不稳定,易被酸性浸提液和水提取出来,单独的CaO处理效果并不佳,须与其他固定化材料联合处理,才可能达到有效固As的目的[39]。
虽然Na2S·9H2O在一定程度上降低了污泥TCLP浸出As,但明显增加了H2SO4-HNO3和H2O浸出As浓度,这说明对于高As污泥,S2−与As形成的溶度积较小的硫化物沉淀或螯合物,在应对有机弱酸浸滤风险方面有一定效果,但应对强酸和H2O浸滤风险的能力并不强,这也可能与S2−在反应水解过程中会产生OH−、不利于As的固定有关[40]。
综上所述,处理以As(Ⅲ)为主的高As污泥时,FeCl3为首选固定化剂。采用TCLP法评估的固As率优于H2SO4-HNO3和H2O的原因还须进一步研究。
-
10种材料处理后,污泥As结合态分布变化情况见图2。结果表明,污泥F1~F5态As占总As的比例分别为0.84%~48.26%、10.04%~37.41%、7.03%~28.96%、2.58%~17.66%、3.16%~64.42%。F1和F2结合态As与介质结合弱,迁移能力较强,对环境风险较大。FeCl3、Fe0和Al2O3处理均可降低F1态和F2态占比,与CK相比,分别降低了80.60%、38.13%和13.15%。环境释放风险最大的F1态分别降低了96.51%、76.69%和27.07%;CaO、Na2S·9H2O、Fe(NO3)3·9H2O和Fe2O12S3的F1态和F2态占比均增加>28%。其中CaO和Na2S·9H2O的F1态和F2态占比分别增加38.71%和31.72%。
由图1和图2可知,FeCl3固As效果突出主要与促进污泥As从非专性/专性吸附态向结晶铁锰或铁铝水化氧化物结合态和残渣态转化有关。与CK相比,F4和F5占比分别增加了50.84%和239%;Fe0固As主要与F1向F3、F4转化有关,F3和F4分别增加119%和24.82%,F5仅增加14.40%;Al2O3促进部分F1和F2态转为固定态,F3、F4和F5态占比分别增加了20.29%、12.50%、17.02%。有研究[41]表明,Al2O3八面体的表层与As可形成双齿单核、单齿单核和双齿双核络合物,与Fe2O3一样,对As均有较好的吸附作用。但本研究中Al2O3固As效果有限,稍优于Fe2O3的原因可能与其和As可形成结构更为固定的双齿双核配合物有关[42]。综上所述,各材料可有效固As与促进As从非专性和专性吸附态向稳定态转化密切相关。
-
原污泥中的As主要以As(Ⅲ)形态存在,As(Ⅲ)占比高达77.14%(图3)。由图3可知,除Na2S·9H2O外,各材料处理后,污泥As(Ⅲ)比例均有所降低,降低幅度依次为FeCl3>Fe(NO3)3·9H2O>CaO>Fe2O12S3>Fe0≈FeSO4·7H2O。这些材料在高As污泥固定化过程中表现出一定的氧化特性,Fe(OH)3、Fe2O3和Al2O3处理对As(Ⅲ)比例分布的影响则不明显,Na2S·9H2O本身还原性强,与污泥反应后,产生H2S,处理后As(Ⅲ)占比升至85.84%,与CK相比,增加了45.85%,As(V)占比降低了2.7%。
经FeCl3、Fe(NO3)3·9H2O和CaO处理后,As(Ⅲ)占比分别降至19.72%、32.28%、47.38%。尤其经FeCl3处理后,与CK相比,污泥As(Ⅲ)占比降低了74.44%,As(V)则增加了2.51倍。这说明FeCl3处理不仅有效降低了As的总浸出浓度(图1),且能有效降低污泥As(Ⅲ)的相对毒性。铁基材料表现出的氧化性与铁的价态有关[43-44],Fe(NO3)3·9H2O与污泥反应产生的HNO3氧化性强,FeCl3的致酸性和腐蚀性比较强,两者和CaO处理高As污泥时发生放热反应,这可能会促进污泥As(Ⅲ)向As(Ⅴ)的转化。Fe0吸附污泥中的As(Ⅲ)后,向Fe(Ⅱ)氧化物缓慢转化[45],Fe0和Fe(Ⅱ)的耦合也可将部分高溶性As(Ⅲ)氧化为不溶性As(Ⅴ),最终也可能会形成As(Ⅲ)-Fe(Ⅲ)和As(Ⅴ)-Fe(Ⅲ)沉淀[44]。
-
FeCl3处理可使污泥TCLP、H2SO4-HNO3和H2O 浸出As浓度均明显降低(图4),随FeCl3投加量的增加,3种As浸出浓度均呈先降低,后稍升,后继续降低的趋势。在最低投加量为50% FeCl3处理后,As浸出浓度均急剧下降,分别降至2 746.18、6 805.90、7 656.85 mg·L−1,与CK相比,η分别达到74.18%、54.51%、37.63%。其中,TCLP浸出As降幅最大。3种As浸出浓度在100% FeCl3时,已降至较低水平,之后降幅变化趋于平缓。250% FeCl3处理后,污泥TCLP-As降至最低,为727.03 mg·L−1,η最高可达到93.16%,H2SO4-HNO3和H2O浸出As此时趋于接近,分别降至1 889.94 mg·L−1和1 840.63 mg·L−1,η均高于85%,但As浸出仍未达到危险废物填埋入场要求。
-
如图5所示,单独添加水泥可大大降低污泥As浸出毒性,随着水泥添加量的增加,污泥TCLP和H2SO4-HNO3浸出As浓度均呈下降的趋势,但25%的水泥投加量的固As效果并不明显,As浸出仅分别降至9 282.36、13 492.20 mg·L−1,η仅为12.71%、9.82%。污泥H2O浸出As呈先升高后降低的趋势,25%水泥投加处理,使污泥pH明显升高,增加了污泥颗粒表面负电荷,降低了污泥中带正电荷胶体对亚砷酸和砷酸根的吸附,导致H2O浸出As浓度升至13 521.95 mg·L−1,接近于H2SO4-HNO3浸出As值。与CK相比,H2O浸出As浓度被活化了10.14%,之后,随水泥投加量的增加,As浸出不断下降。3种方法浸提后,As浸出浓度均在水泥最高投加量125%时降至最低,且数值趋于2 200~2 600 mg·L−1,η均在80%左右。在同等投加量时,水泥固As效果明显弱于FeCl3。
250% FeCl3与水泥复配结果如图6所示,3种As浸出变化规律相似,As浸出浓度均先急剧下降,然后上升,后再下降,H2SO4-HNO3和H2O浸出As浓度和变化基本趋于一致,固As效果均弱于TCLP。水泥最低复配比为25%时,3种As浸出浓度可分别降至1 106.15、2 816.99、2 716.97 mg·L−1,η分别达到89.60%、81.17%、77.87%。与单一水泥相比,各配伍处理的固As效果均明显提高。与单一250% FeCl3处理相比,水泥复配比<100%时,配伍固As效果有所下降,这说明低量水泥的添加对FeCl3固As产生了拮抗作用,水泥易升高污泥pH,FeCl3则更易降低pH,在FeCl3与水泥的交互影响下,FeCl3虽使污泥正电荷增加,仍能维持固As效果较好的酸性条件[46],但配伍并未达到协同增效的目的。在水泥复配比≥100%后,配伍固As效果占优;在水泥复配比125%条件下,3种As浸出均降至最低,分别为113.81、399.28、347.27 mg·L−1,η均高于97%,但材料高投加量仍未使As浸出达到危险废物填埋控制标准。
铁盐和水泥虽是处理含As固废常用材料[47-48],但本研究结果表明,针对高As污泥,FeCl3和水泥复配不一定优于单一材料,这与材料种类、特性和投加量等有关。材料的高投加量虽可使固As率有所提高,但仍难达标,且存在增容比高、FeCl3强腐蚀性等问题。因此,须进一步研发更为高效的固As材料,可考虑联合其他技术或措施(如淋洗后再固定化固化、烧结固化等),以尽量降低As释放风险,满足危险废物填埋入场要求。
-
1)固As材料筛选结果表明,FeCl3固As效果最好。TCLP、H2SO4-HNO3、H2O浸提法评估的固砷率分别为86.01%、42.02%、58.87%,均强于其他9种材料,且明显促进了As的非专性/专性吸附态向结晶铁锰或铁铝水化氧化物结合态和残渣态转化,非专性和专性吸附态As占比降低了80.60%,非专性态降低96.51%。
2)在10种材料处理中,有6种材料对As(Ⅲ)具一定的氧化作用。其中,FeCl3处理对As(Ⅲ)的氧化作用最强,使As(Ⅲ)占比由77.14%降至19.72%。Fe(OH)3、Fe2O3和Al2O3处理对As(Ⅲ)的氧化性不明显,Na2S·9H2O处理使As(Ⅲ)占比升至85.84%。
3) FeCl3联合水泥固化结果表明,单一FeCl3和FeCl3与水泥配伍的固As效果均优于单一水泥。水泥复配比≥100%时,配伍处理的固As效果优于单一FeCl3;在250% FeCl3+125%水泥条件下,TCLP、H2SO4-HNO3和H2O浸出As浓度降至最低,分别为113.81、399.28、347.27 mg·L−1,η均高于97%。高投加量并未使As浸出达到危险废物填埋控制标准。
不同材料对高As污泥中As的固定效果
Immobilization effect of different materials on high As-bearing sludge
-
摘要: 针对制酸行业产生的高砷(As)污泥浸出毒性过高且难处理的问题,选择典型无机硫化物、含钙材料、含铁或铝材料(Fe0、铁盐、Fe2O3/Al2O3) 对高As污泥进行固定化,采用醋酸(TCLP)、H2SO4-HNO3和H2O 3种浸提法评估了各材料对As的固定效果;考察了固定化对污泥中As结合态和价态分布的影响,筛选最佳固定化材料,进行联合水泥固化。结果表明,FeCl3固化As的效果最好,3种浸提法评估的固砷率分别为86.01%、42.02%、58.87%。FeCl3和Fe0处理能够促进污泥As向固定态转化,非专性和专性吸附态占比分别降低了80.60%和38.13%。FeCl3促进非专性和专性吸附态向结晶铁锰或铁铝水化氧化物结合态和残渣态转化。CaO、Fe0、FeSO4·7H2O、Fe(NO3)3·9H2O、FeCl3和Fe2O12S3处理对As(Ⅲ)具有一定的氧化作用。FeCl3处理后,As(Ⅲ)占比由77.14%降为19.72%。Fe(OH)3、Fe2O3和Al2O3处理对As(Ⅲ)的氧化性不明显,Na2S·9H2O处理使As(Ⅲ)占比升至85.84%。随FeCl3、水泥和两者配伍材料投加量的增加,在3种方法中,污泥中As的浸出量均明显降低,单一FeCl3和配伍固砷效果均优于水泥;水泥配伍比≥100%时的固砷效果优于单一FeCl3;250% FeCl3+125%水泥可使3种方法中浸出砷浓度降至最低,分别为113.81、399.28、347.27 mg·L−1,固砷率均高于97%。本研究结果可为高As污泥的固定化提供参考。供参考。Abstract: The high arsenic (As)-bearing sludge produced from acid industry has too high leaching toxicity to dispose. In this study, three kinds of typical materials: inorganic sulfide, calcium-based material and several iron-aluminum-based materials (Fe0, ferric salt, Fe2O3/Al2O3), were used to immobilize the high As-bearing sludge. Their As immobilization effects were assessed by three leaching methods of TCLP(toxicity characteristic leaching procedure), H2SO4-HNO3 and H2O. The effect of immobilization on the valence and binding form distribution of arsenic in sludge was investigated. Then the optimal material for As immobilization was determined, which was used to conduct the subsequent joint solidification with cement. The screening result of materials showed that FeCl3 had the best As immobilization effect, and its As immobilization efficiencies assessed by above three leaching methods were 86.01%, 42.02%, 58.87%, respectively. The FeCl3 or Fe0 treatment could promote the As transformation to stable speciation, and the proportions of non-specific and specific bound As fractions decreased by 80.60% and 38.13%, respectively. In which FeCl3 treatment promoted the transformation from non-specific and specific bound As fractions to crystalline hydrous Fe(Mn, Al)oxide fraction and residual fraction. CaO, Fe0, FeSO4·7H2O, Fe(NO3)3·9H2O, FeCl3 and Fe2O12S3 could oxide As(Ⅲ) to As(V). Among them, the proportion of As(Ⅲ) in FeCl3 treated sludge decreased from 77.14% to 19.72%, and no obvious oxidation of As(Ⅲ) occurred in Fe(OH)3, Fe2O3 and Al2O3 treated sludge. However, due to the strong reducibility of Na2S·9H2O, the proportion of As(Ⅲ) in Na2S·9H2O treated sludge increased to 85.84%. With the increase of the dosage of FeCl3, cement or composite materials, the leaching amount of As in sludge decreased significantly. The As immobilization effect of FeCl3 alone or FeCl3+cement was better than that of cement alone. When the cement ratio was set to above 100%, FeCl3-cement As immobilization effect was better than that of FeCl3 alone. The As leaching concentrations of TCLP, H2SO4-HNO3 and H2O in 250% FeCl3+125% cement treatment were reduced to the lowest values of 113.81, 399.28 and 347.27 mg·L−1, respectively, and As immobilization efficiency reached above 97%. This study can provide reference for the immobilization of high As-bearing sludge.
-
随着工业化和城镇化的加速推进,对废水的集中处理备受关注[1]。1932年开始应用的Wuhrmann工艺是最早的脱氮工艺,称之为O/A工艺,遵循硝化、反硝化的流程顺序而设置[2]。然而,在硝化过程中需要供氧,反硝化过程中需要外加碳源,这造成了能耗和碳源的双重浪费。对此,将生物单元的顺序进行倒置,便产生了A/O工艺,A/O工艺成为最早使用的生物脱氮技术。这是工艺单元不同排列顺序构成组合工艺的开端,后续发展的废水生物处理工艺几乎均为厌氧、缺氧/水解、好氧单元的组装(图1)。典型的工艺有A/A/O和O/A/O,组合工艺中的不同单元反应器排序会影响碳源利用和脱氮效果,因此,需要根据废水组成与处理目标选择合适的工艺技术。
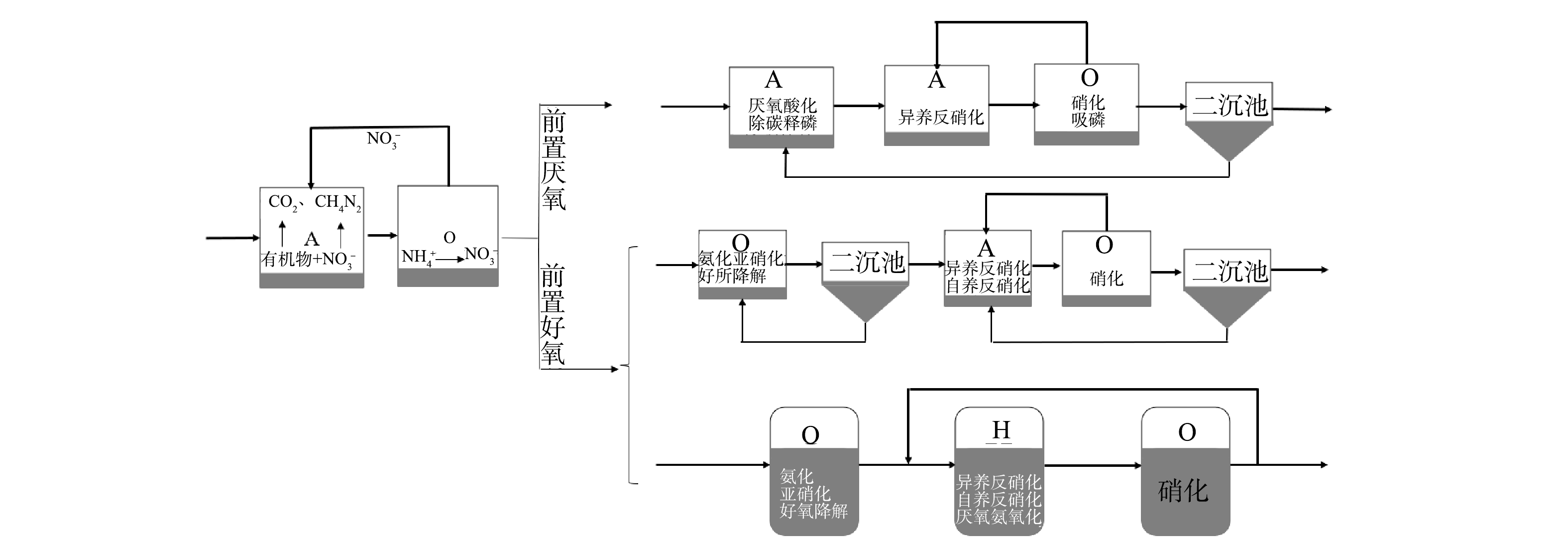 图 1 前置厌氧与前置好氧工艺的演化配置Figure 1. Evolutionary configuration of pre-anaerobic and pre-aerobic processes.
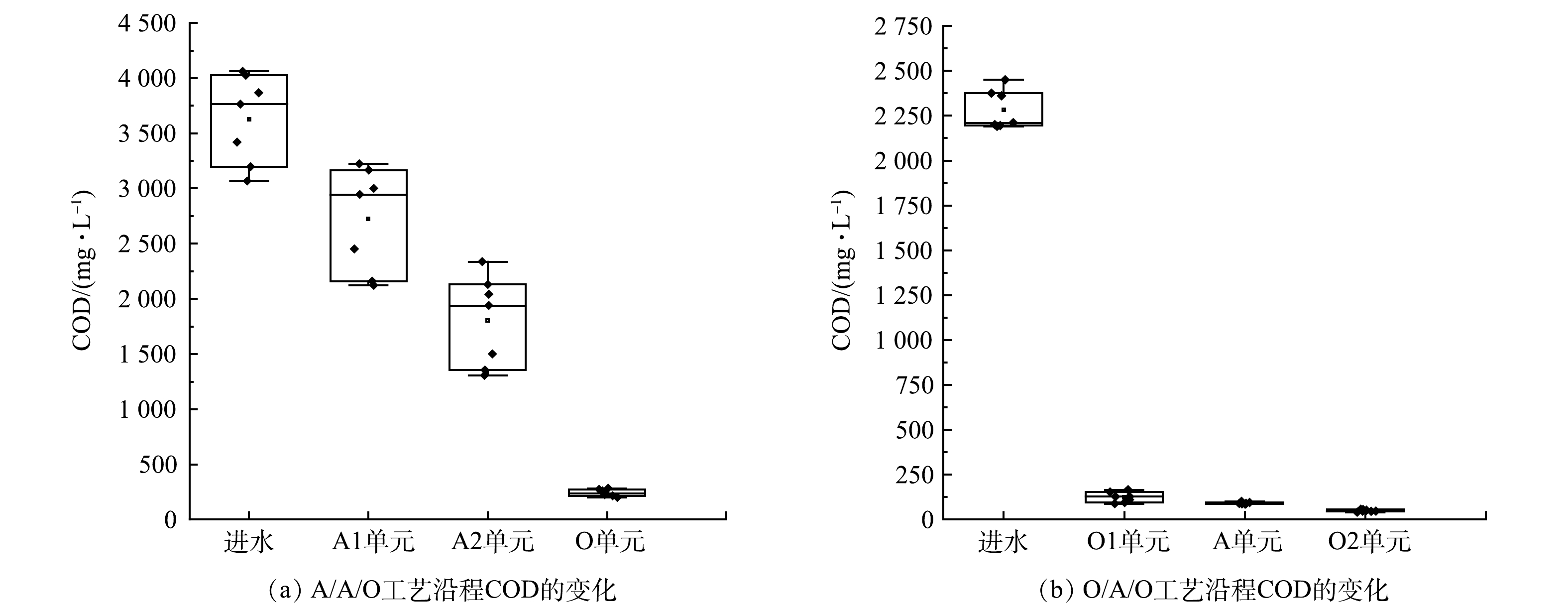
图 1 前置厌氧与前置好氧工艺的演化配置Figure 1. Evolutionary configuration of pre-anaerobic and pre-aerobic processes.厌氧置前的工艺可以控制碳源转化为小分子有机物或者甲烷,提高废水的可生化性,为后续反硝化反应提供碳源。HAO等[3]采用A/A/O工艺处理制革废水,考察了沿程溶解性有机物的浓度变化,发现A1的厌氧水解单元能优先去除小分子量的物质和蛋白质,后续的A/O工艺可更彻底地去除残余有机物。O/A/O工艺可在O1单元反应器中好氧降解部分有机物,实现含氮有机物的氨化,有助于硝化反应的实现。李国令等[4]对比了O/A/O和A/O工艺处理同一城镇污水的结果,在O1单元反应器中降解了大部分有机物,可为O2提供良好的硝化环境,因此,O/A/O脱氮效果优于A/O工艺。A/A/O工艺对高毒性工业废水的处理不具有优势,这是因为A1中的微生物增殖速度慢,难以消除毒性抑制作用。兼顾脱氮和除磷是A/A/O工艺的特征,脱氮效率受回流比的影响,无法实现完全脱除总氮,也存在着与除磷菌在碳源利用分配之间的矛盾。然而,前置好氧的O/A/O工艺因大幅度削减了毒性物质而有利于后续单元硝化菌的生长。与A/A/O工艺不同的是,该工艺不能利用废水中存在的易降解有机物作为碳源进行反硝化脱氮,造成一定程度的碳源浪费。由此可见,前置厌氧或者前置好氧对后续的脱氮工艺有着不同的影响机制,A/A/O工艺多用于生活污水[5-6],而O/A/O工艺可能更适合于工业废水[7]。
焦化废水是典型的高碳氮比工业废水,含有多种高浓度有毒物质。其中的有机污染物主要包括酚类[8]、苯系物、杂环芳烃和多环芳烃等物质[9];其无机物中,S2-、SCN−、CN−等均为典型的毒性物质,并且对废水的COD值有较大的贡献[10]。LI等[11]研究了在相同水力停留时间下A/A/O与A/O工艺分别对焦化废水中COD和NH4+-N的去除效果,发现两者的去除率几乎相同,但A/A/O比A/O工艺对总氮的去除效果更好。汤清泉等[12]比较了A/A/O与O/A/O工艺对焦化废水的处理效果,认为碳氮比是决定二者对总氮去除效果的关键因素。当碳氮比为15~20时可以选择A/A/O工艺,当碳氮比为20~35时则O/A/O工艺效果更好。其原因是:前置好氧单元可以去除高碳氮废水中的有机物而降低后续处理的负荷。本课题组在长期实践的基础上开发了针对焦化废水处理三污泥系统的好氧-水解-好氧流化床脱氮工艺(命名为O/H/O工艺,其中,O1为除碳氨化单元,H为水解脱氮单元,O2为完全硝化单元) [13-15],已有 5个实际工程应用案例,最长运行时间达到12年。O/H/O工艺具有独特的三相分离器,可以保证在不需要污泥回流的情况下实现各个单元反应器独立的污泥特征和生物量,节省了能耗,并促进了污泥生态与水质环境的相容性[16]。新型结构生物三相流化床作为O1反应器,在进水有机负荷达到2.4 kg ·(m3·d)−1 的运行情况下,其耗氧有机物的去除率可以达到93.0%以上,反应器中氧的利用率为50%~60%。面对高毒性、高浓度的焦化废水,A/A/O工艺需要1~2倍稀释后才能进入生物系统,而O/A/O或O/H/O工艺则不需要稀释。
厌氧、水解、好氧单元不同顺序的排列组合构成了不同的废水生物处理工艺技术。在废水性质转化方面,厌氧单元可提高B/C值[17],而好氧单元可降低B/C值,分别有利于异养反硝化与硝化反应;在脱氮模式中,要考虑硝化反硝化[18]、短程硝化反硝化[19]、厌氧氨氧化[20]、自养反硝化[21]、好氧反硝化[22]等原理的选用、协同及条件控制。A/A/O工艺和O/A/O工艺都需要回流才能保持反应器内的污泥浓度,A/A/O工艺的运行属于单污泥系统,O/A/O工艺中设置了2个二沉池,属于双污泥系统,而O/H/O工艺属于三污泥系统。根据废水的性质选择合适的工艺,可以在达标排放的基础上实现能耗与物耗的减量化。由于目前缺乏不同工艺特征的比较,为此,本文分析了不同工艺的碳源利用模式和脱氮模式,提出了一种代表性的焦化废水组成并通过研究A/A/O、O/A/O、O/H/O的组合工艺对焦化废水中核心污染物的去除及其能耗分配关系,阐明了工艺技术选择的原则,为复杂工业废水生物处理技术的工艺优选提供参考。
1. 研究方法
1.1 数据来源
本课题组对国内38个焦化厂进行了实地调查和数据采集,分析了焦化废水的水质特征与地域差异的关系,发现华北、华中、华东地区废水中的COD值略高,华中和西南地区废水的氨氮浓度略低[23]。焦化废水中的含氮物质主要由氨氮、有机氮、SCN−、CN−等组分构成,由于蒸氨工艺的差异,含氮物质的比例各有不同。综合国内外的焦化废水原水水质[24-26],结合我们的调查,为了消除差异性和增强可比性,本文定义代表性的焦化废水组成为: COD为4 000 mg·L−1,苯酚、
NH+4 A/A/O工艺借鉴宝武韶钢公司的运行数据,水量为60 m3·h−1,3个单元反应器的水力停留时间分别为34、22和52 h,COD负荷分别为1.22、1.46和0.47 kg·(m3·d)−1;O/H/O工艺参考实验室和焦化厂的运行数据[27-28],废水处理量为60 m3·h−1,3个单元反应器的水力停留时间分别为36、40和24 h,COD负荷分别为2.30、0.38和0.55 kg·(m3·d)−1;选取韩国某厂实验室数据作为O/A/O工艺的案例[29],实验规模为0.03 L·h−1,3个反应器的水力停留时间分别为28.8、12和19.2 h,进水中添加KH2PO4和Na2CO3以维持碱度,在缺氧池中加入3倍总氮浓度的甲醇作为碳源,工艺装置总水力停留时间为2.5 d。通过实际与假设相结合的方法进行分析,以3个焦化厂的实际废水数据(见表1)来剖析不同工艺的碳源利用和脱氮模式。O/A/O和O/H/O工艺的反应器排列顺序相同,反应器的性能和运行模式不同。因此,在分析碳源利用和脱氮模式时只考虑A/A/O与O/A/O的对比,而在能耗分析时,再考虑O/A/O与O/H/O的差异性。
表 1 3种工艺实际运行水质Table 1. Actual operating water quality in three processesmg·L−1 工艺 COD 挥发酚 NH+4 SCN− CN− A/A/O 1 727±60 742±69 173±12 175±18 26.2±4.5 O/A/O 2 300±100 635±15 235±15 375±25 - O/H/O 3 451±215 973±74 245±15 450±17 25±3 注:以集水调池的水质作为生物上水。 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 过程分析
根据污染物的降解途径计算了污染物的COD当量和TN当量,结果见表2,在生物系统里,SCN−和CN−中的氮转化为氨氮[30-31]。
表 2 不同污染物对COD和总氮的贡献Table 2. Contribution of various pollutants to COD and nitrogen mg·mg-1当量 挥发酚 SCN− CN− S2− NO−3 NO−2 COD当量 2.380 1.100 0.615 2.000 - 0.348 N当量 - 0.241 0.538 - 0.226 0.304 | Show TableDownLoad:
CSV
通过分析不同污染物对COD和总氮的贡献,检验废水组成的合理性。如式(1)所示,废水中的含氮量主要由
NO−3 NO−2 NH+4 CTN= 0.226CNO−3+0.538CCN−+0.241CSCN−+CNH+4−N+0.304CNO−2+C其他含氮物质 (1) CCOD= 2.380Cphenol+1.100CSCN−+0.615CCN−+2.000CS2−+0.348CNO−2+C其他有机物 (2) 式中:CTN、
CNO−3 CNO−2 CCN− CSCN− CNH+4 在每一个单元反应器的出水中,都通过以上的方法进行检验,以确定废水组成的合理关系。
1.3 数据处理
根据式(3)~式(7)计算A/A/O工艺中每个反应器对污染物i总体去除的贡献率,分别以
PiA1 PiA2 PiO PiO1 PiA PiO2 PiA1=(1+R1)×CiA1-I-CiA1-ECi0×100% (3) PiA2=(1+R1+R2)×CiA2-I-CiA2-ECi0×100% (4) PiO=(1+R1+R2)×CiO-I-CiO-ECi0×100% (5) CiA2-I=(1+R1)×CiA1-E+R1×CiO-E1+R1+R2 (6) CiO-I=CiA2-E (7) PiO1=(1+R3)CiO1-I-CiO1-ECi0×100% (8) PiA=(1+R4+R5)×CiA-I-CiA-ECi0×100% (9) PiO2=(1+R4+R5)×CiO2-I-CiO2-ECi0×100% (10) CiO1-I=Ci0+R3CiO1-E1+R3 (11) CiA-I=CiO1-E+(R4+R5)×CiO2-E1+R4+R5 (12) CiO2-I=CiA-E (13) 式中:i为各种污染物(COD、苯酚、硫氰化物、氰化物、氨氮、亚硝酸根、硝酸根和总氮)。R1和R2分别为A/A/O工艺中污泥回流比和硝化液回流比,污泥回流比取值1,硝化液回流比取值3;R3、R4、R5分别为O/A/O工艺中初沉池回流至O1的污泥回流比、二沉池回流至A的污泥回流比以及硝化液回流比,均取值为1。C0i为未处理废水中污染物i的质量浓度,mg·L−1;
CiA1−I CiA1−E CiA2−I CiA2−E CiO−I CiO−E CiO1−I CiO1−E CiA−I CiA−E CiO2−I CiO2−E 排除水力停留时间对工艺对比造成差异,假设A/A/O与O/A/O工艺具有相同的总水力停留时间,结合文献调研和实际考虑,每个工艺各个反应器的体积比为1:1:2,处理水量为60 m3·h−1。
污染物在反应器中会进行到氨化碳氧化、亚硝化氮氧化或硝化氮氧化3种不同的处理阶段,不同阶段的耗氧量分别根据式(14)~式(16)进行计算。
OS=[a⋅KCOD⋅CCOD+CDO]Q24 000 (14) OS=[a⋅KCOD⋅CCOD+b(1-Kd)×(CN+CCN1.86+CSCN4.14)+(1+RS+Rd)⋅CDO]Q24 000 (15) OS=[a⋅KCOD⋅CCOD+c(1-Kd)×(CN+CCN1.86+CSCN4.14)+(1+RS+Rd)⋅CDO]Q24 000 (16) Kd=(1-NoNi)×100% (17) 式中:Q为生物系统进水量,m3·d−1;a、b、c分别为氧化COD、氨氮到亚硝氮、氨氮到硝态氮的有关的耗氧系数,在本研究中为1.4、3.43、4.57;Os为好氧单元的理论需氧量,kg·h−1;CCOD为耗氧有机物(以COD计)的质量浓度,mg·L−1;CDO为好氧单元溶解氧的质量浓度,mg·L−1;KCOD为COD去除率,%;Rs、Rd分别为活性污泥和硝化液回流比;CN、CCN、CSCN分别为以脱氮为目标的好氧池中含氨氮、氰化物、硫氰化物的质量浓度,mg·L−1;Kd为反硝化率,%;Ni、No分别为脱氮系统进、出水总氮的质量浓度,mg·L−1。
A/A/O中的好氧单元主要发挥硝化作用,通过式(16)和式(17)计算可知其耗氧量;O/A/O工艺中,O1易氧化降解耗氧有机物(以COD计),不考虑硝化作用,耗氧量通过式(14)计算可知;在O2中进行硝化作用,耗氧量通过式(16)和式(17)计算可知;O/H/O工艺与O/A/O工艺相似,但不需要污泥回流,因此,在计算O/H/O工艺中O2的曝气能耗时,式(16)的污泥回流比Rs为0。
污泥回流的能耗是A/A/O与O/A/O工艺所必不可少的,只有通过污泥回流才能保证生物池活性污泥的浓度,回流泵的能耗通过式(18)进行计算。
WS=K⋅Q⋅H (18) K=k183.5 (19) 式中:Ws为污泥回流泵的能耗,kW·h;K是安全系数,由式(19)计算,当水泵功率和污泥回流泵功率超过5 kW时,式(19)中的k取值1.15[32];Q为回流的流量,m3·d−1;H为水泵总水头损失,m。
由于回流污泥含水率高达99.5%~99.9%,所以,污泥回流与废水回流的能耗以相同方法计算。A/A/O与O/A/O工艺的回流比已经明确,O/H/O工艺仅存在硝化液回流,回流比为1,能耗估算值可由泵能耗的公式给出。A/A/O工艺中污泥回流至厌氧池的水头损失为1.5~2 m,硝化液回流至缺氧池的水头损失为1~1.2 m。O/A/O工艺有2个污泥回流系统,二沉池至O1的水头损失为0.5~0.8 m,另一个二沉池至A的水头损失为1~1.5 m。O/H/O不存在污泥回流,硝化液回流的水头损失为1~1.6 m。
2. 结果与讨论
2.1 碳源利用
首先考察了2种工艺中COD的沿程变化,分析2种工艺的碳源利用模式差异。由图2可以看出,在A/A/O工艺中,O单元对耗氧有机物(以COD计)的去除效果最好,A1的水解作用使难降解有机物断链、开环,转化为小分子有机酸,为后续的反硝化脱氮所利用;而在O/A/O工艺中,O1对COD的去除率高达90.0%以上,使后续单元工艺主要为脱氮服务。两者不同的是,A/A/O工艺通过微生物反硝化作用去除了废水中的耗氧有机物,而O/A/O工艺则通过生物耗氧直接氧化废水中的耗氧有机物。
LI等[11]对比了A/A/O与A/O工艺的处理效果,指出2个工艺对于有机物和氨氮的去除效果几乎相同,但A/A/O工艺更有利于总氮脱除,这是因为A/A/O工艺设置了产酸阶段。CHAKRABORTY等[33]发现,在A1中COD的去除率为5%~11%,CN−降解率为35.0%,没有发现苯酚降解的中间产物和甲烷的生成。王子兴等[34]指出,在A/A/O-MBR工艺处理焦化废水的过程中,单个反应器COD去除率分别为9.2%、73.5%、14.7%;经过GC/MS检测分析,苯酚在A1中的降解率为26.7%,而含氮杂环化合物以及苯系物的去除率分别为49.5%和65.8%。此外,有研究[35]表明,在A/A/O工艺中,A1单元去除污染物效果不明显,COD去除率低于10%;A2单元的COD去除率最高,尤其是易降解有机物在此阶段几乎全部被利用;在O单元中,利用异养微生物好氧氧化残留的有机物,CN−和SCN−在O2中也被彻底去除。SHARMA[36]研究了厌氧、缺氧、好氧单个单元的处理效果时发现,好氧单元可去除83.3%的CN− 和62.0%的COD;当加入氰化物后,好氧单元中COD的去除率下降到52.0%。由此可见,废水组成的复杂性会影响单组分的去除效果。马昕等[37]采用O/A/O工艺处理焦化废水时发现,在O1停留时间为16 h时对COD的去除率达到75.0%,这与我们调查的工艺结果相似。由图2(b)可见,在O/A/O工艺中,O1对COD的去除率很高,浪费了部分有机碳源,而添加的外部有机碳源是造成A单元COD去除率降低的原因之一[38];另一方面,O1中的氨化过程可为O2提供良好的硝化环境。以上研究结果表明,2种工艺对废水中碳源的利用在原理上存在非常大的差异。
2.2 脱氮模式
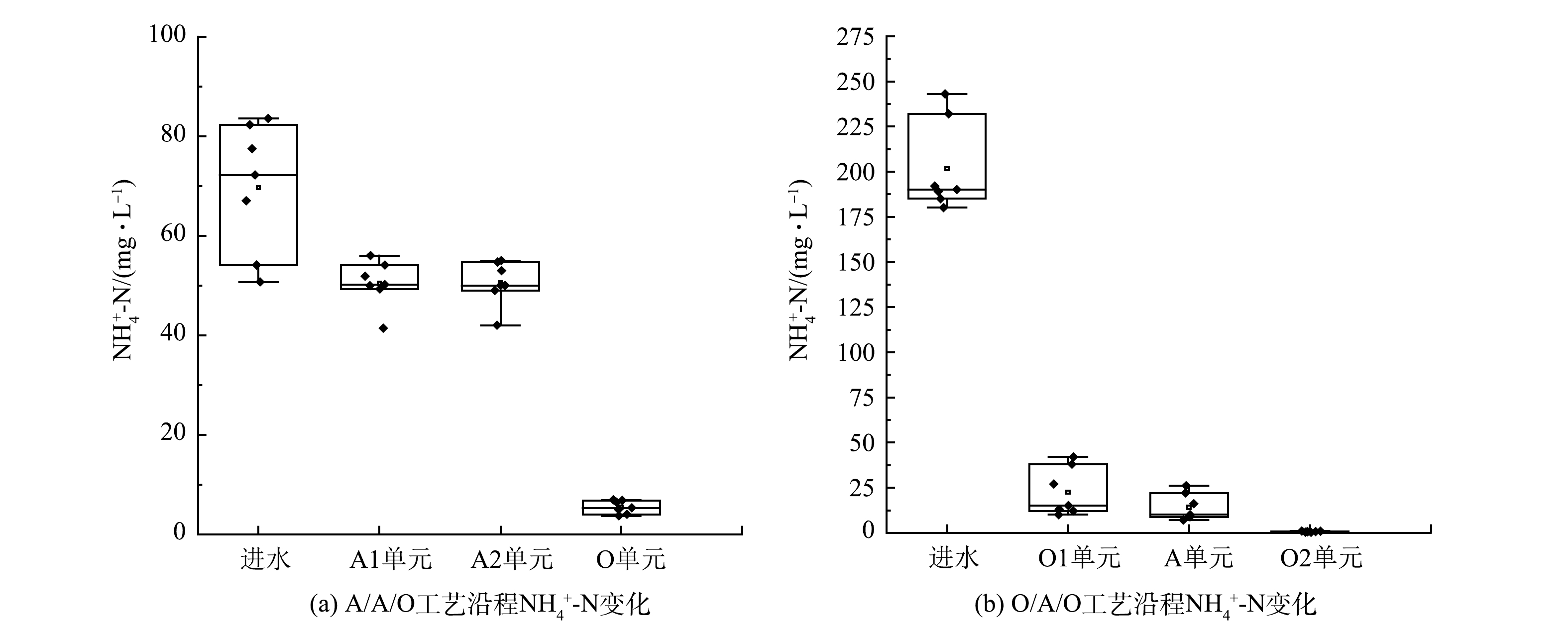
脱氮的效果可通过协调碳源、电子供受体以及DO等因素来实现,故根据2种工艺中氨氮浓度沿流程变化来分析不同脱氮模式的有效性。 由图3可见,虽然O/A/O工艺进水氨氮偏高,但出水氨氮却很低,在O2单元中已经彻底硝化。可见,前置好氧工艺可以为后续O2创造良好的硝化条件。A1去除了27.0%的氨氮,而O1去除了87.5%的氨氮,即在A1中仍然保留着较高浓度的氨氮,而在O1中氨氮几乎完全硝化,这与在进水中是否添加磷盐有关[39]。O1、A1中氨氮浓度的变化以及微生物同化、有机氮氨化、氰化物及硫氰化物氨化等可以同时发生。在工程研究中发现,O1中还存在亚硝化和硝化的可能性[17]。
焦化废水中的含氮物质除了铵离子/氨分子外,还有SCN−、CN−以及含氮有机物。ZHANG等[40]发现,A/A/O中各个单元对氨氮的去除率分别为-2.5%、3%、97%,A1出水中氨氮升高的原因是其他含氮物质氨化作用所致。吕鹏飞等[41]的研究表明,2种流化床工艺的前置厌氧单元对氨氮有少量的降解,氨氮去除率分别为18.1%和35.6%,体现出反应器对于处理效果的影响不同,流化床反应器面对复杂毒性废水比传统的沸腾床反应器表现出更好的耐毒性抑制作用。经过缺氧反应器A2后,氨氮浓度的变化主要有回流导致的直接稀释以及微生物降解的共同作用。GUI等研究了2个A/A/O系统,在硝化液回流比为200%的情况下,氨氮的质量浓度由250 mg·L−1降低至80 mg·L−1[42]。易欣怡等[28]考察了O/H/O工艺的焦化废水处理,发现O1单元能够把氰化物、硫氰化物氧化为氨氮,有机氮全部氨化,从而造成O1出水氨氮浓度的升高;而在H单元中,环状含氮化合物通过水解作用可实现分子开环转变为氨氮,回流液中的硝态氮实现反硝化转变为氮气;接下来的O2单元能够将残余低价状态的含氮化合物转变为硝态氮,所以对氨氮的去除非常彻底。由于多种含氮物质之间具有不同价态转化机制,工艺中合理安排碳源进行脱氮,以及通过回流/超越或微生物功能调控实现总氮的彻底去除将是工艺理论中具有挑战性的研究方向。
2.3 能耗分析
1)各单元反应器的去除效率。能耗分配受工艺的单元反应器组合的影响。单元反应器的不同组合顺序可构成多样的生物处理工艺,前置好氧与厌氧工艺对同一种废水会产生不同的污染物去除效率,较优的工艺应该是在达标排放(即核心污染物去除)的基础上实现时间和空间上的减量化,还要降低二次污染。图4反映了A/A/O和O/A/O工艺污染物浓度的沿程变化。沿流程图中的百分比数据代表反应单元出水污染物浓度占进水中污染物浓度的比例。除了内部降解外,还要考虑因回流引起的反应器内污染物浓度的稀释作用。结合文献调查,综合实际情况,总结出代表性焦化废水典型污染物在单元反应器中的去除效率,如图5所示。其中,假设SCN−和CN−在O/A/O工艺的O1中完全氨化。
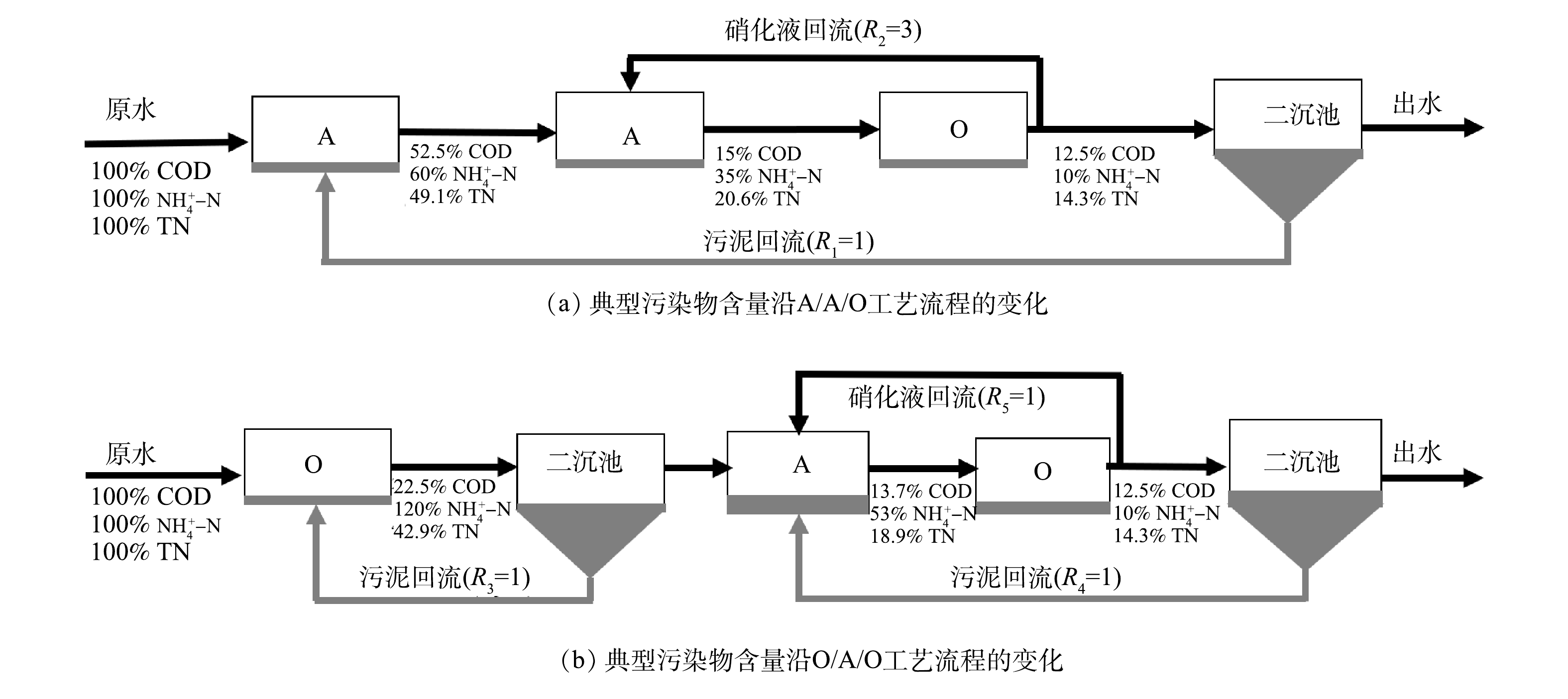 图 4 典型污染物含量沿各工艺流程的变化Figure 4. The variation of typical pollutant content along each process.
图 4 典型污染物含量沿各工艺流程的变化Figure 4. The variation of typical pollutant content along each process. 图 5 典型污染物在各工艺单元反应器中的降解率Figure 5. The degradation rate of typical pollutants in each process unit reactor.
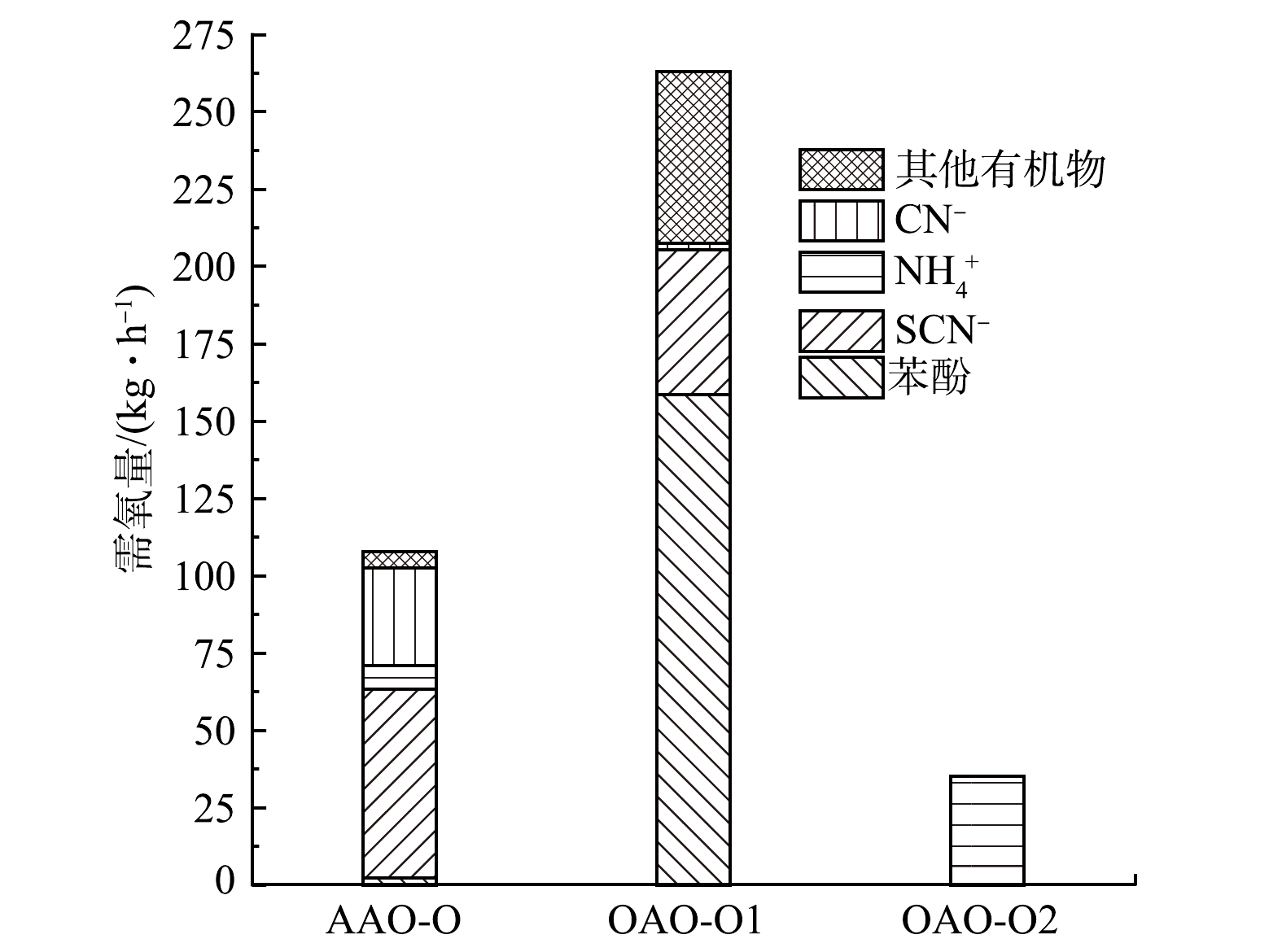
图 5 典型污染物在各工艺单元反应器中的降解率Figure 5. The degradation rate of typical pollutants in each process unit reactor.2)不同工艺的能耗分配。废水中的污染物在不同工艺各单元反应器中的总体去除率如图6所示。A/A/O工艺对污染物的降解主要集中在O单元中,O/A/O工艺的降解则集中在O1单元中。这两者的差异反映了前置好氧工艺与前置厌氧工艺在曝气能耗上的差别。通过式(14)~式(16)计算,各工艺需氧单元的曝气量如图7所示。A/A/O工艺中O单元的需氧量为102.7 kg·h−1,O/A/O中O1和O2的需氧量分别为260.8 kg·h−1和35.1 kg·h−1。由图7可看出,O/A/O工艺的O1大部分的曝气量是用来去除易降解有机物,因此,需氧量较高。但当废水中有机物的浓度很低时(当不考虑有机物耗氧时),A/A/O工艺氧化含氮物质需氧量为100.4 kg·h−1,O/A/O工艺氧化含氮类物质的需氧量为83.9 kg·h−1。因此,对于脱氮性能,O/A/O工艺比A/A/O工艺能耗更高。这归因于:在O1中解除了SCN−、CN−等有毒物质对A反应器微生物的抑制作用,使得在A中降解的含氮物质相对较多,可以实现O2单元的低能耗硝化反应。因此,当废水中的耗氧有机物的预处理较为彻底时,前置好氧工艺可以实现低耗能高效率脱氮。O/H/O工艺在保留了O/A/O工艺优点的基础上,实现了反应器内部流态化的颗粒污泥特征,氧传质系数是一般活性污泥的2倍左右[43],因此,与O/A/O工艺相比,O/H/O工艺在耗氧量的节能方面更能体现出优势。本课题组根据多年的O/H/O运行经验数据统计得出,在仅考虑脱氮目标时,O/H/O工艺的需氧量约为53.26 kg·h−1。
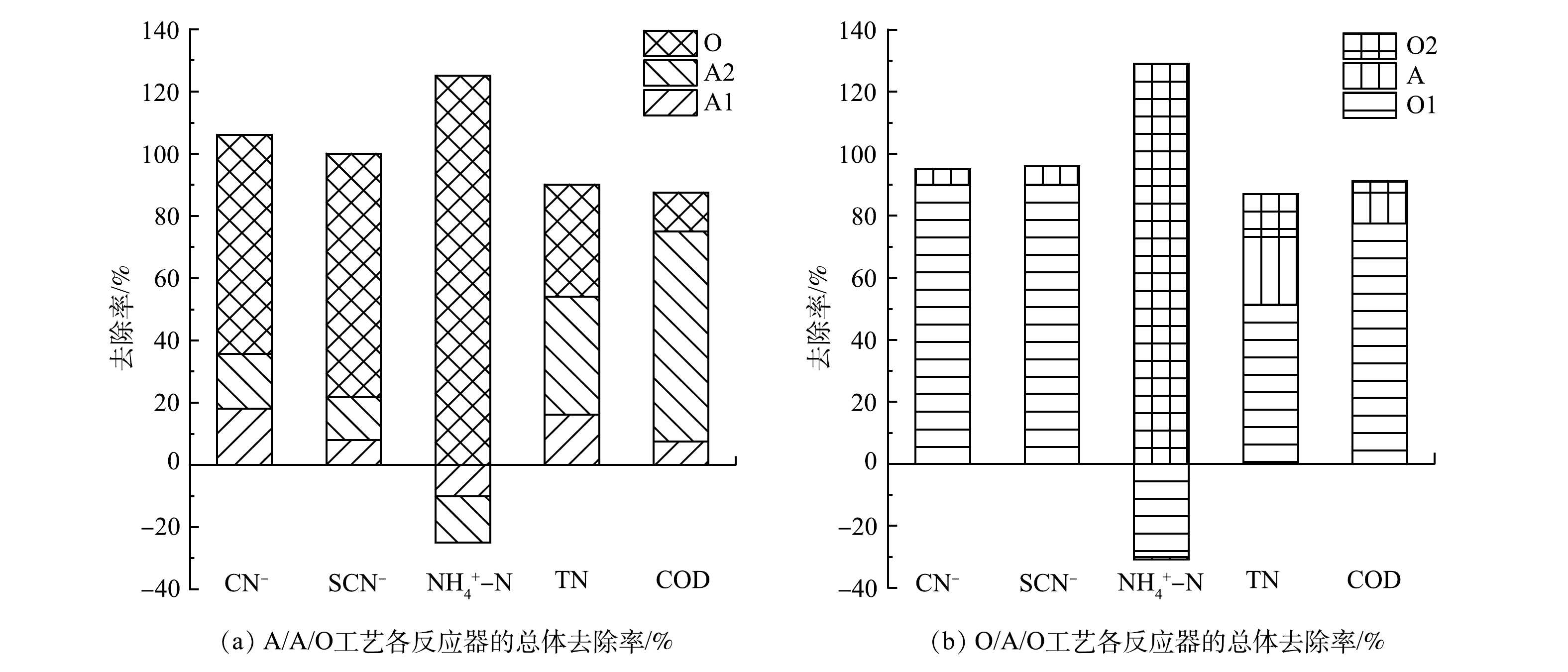 图 6 不同工艺单元反应器对各污染物的总体去除率Figure 6. Overall removal rate of various pollutants in the unit reactor of different process.
图 6 不同工艺单元反应器对各污染物的总体去除率Figure 6. Overall removal rate of various pollutants in the unit reactor of different process.由图4所示的计算可得出,在A/A/O工艺中,进入A2的废水COD为1 140.0 mg·L−1,硝化液回流的硝酸根为84.4 mg·L−1,在A2中主要去除总氮中的硝酸根,其余的氨氮、SCN−、CN−等含氮物质只是发生了少量的生物降解,经过A2可去除80.0 mg·L−1左右的硝态氮,满足微生物生长的碳源需求量为723.2 g·m−3 (缺氧条件下C∶N∶P = 200∶5∶1),因为废水中含有一定量的有机物,故实际可以供微生物利用的量约为540.0 g·m−3,需要外加碳源122.1 g·m−3 (以甲醇计)。在O/A/O工艺中,进入A单元的废水COD值为633.3 mg·L−1,其总氮类型为硝酸根和氨氮,浓度分别为93.9 mg·L−1和50.0 mg·L−1,在A中降解90.0 mg·L−1的硝态氮,满足微生物正常生长的碳源需求量约为813.6 g·m−3,进入A的废水中可降解有机物的含量约为83.3 g·m−3,不足的碳源需要从外部添加486.9 g·m−3(以甲醇计)。以上的讨论是在不考虑O/A/O工艺中有超越进水的情况,但在实际工程中,往往会使部分集水调节池中的出水以超越O1池的方式进入A池,这样既可以降低O1的曝气能耗,又可减少A单元的外部碳源的需求量。当超越1/3处理量的废水进入A单元时,O1的曝气量变为174.0 kg·h−1 ,超越之后A单元进水的有机物浓度达到977.8 mg·L−1,可供微生物利用的量约为427.8 mg·L−1,因此,折合计算1 m3废水仅需要257.2 g的外加碳源,节省了229.7 g的外部碳源(以甲醇计)。可以看出,O/A/O系统的模式多样性,可以实现总氮的低能耗高效率去除。在实际运行的O/H/O工艺中,由于不需要污泥回流,每个反应器可以灵活调控,因此,O/H/O工艺比O/A/O工艺更容易实现厌氧氨氧化反应,并且可以利用FeS进行自养反硝化脱氮而节省能耗,故实际的O/H/O工艺的外部碳源需求约0~220 g·m−3,具体的需求量取决于厌氧氨氧化与自养反硝化的耦合性能[44]。
污泥回流可以保证生物单元中的污泥浓度即生物量。通过式(18)和式(19)的计算,A/A/O工艺的污泥回流和硝化液回流的总能耗约为42.37 kW·h;O/A/O系统污泥回流与硝化液回流的总能耗约为23.55 kW·h;O/H/O系统只存在硝化液回流,回流能耗约为9.42 kW·h。除了曝气和回流的能耗外,考虑综合因素,3种工艺归纳为2大类:厌氧-缺氧-好氧以及好氧-水解/缺氧-好氧。由于反应器的设置不同,好氧-水解/缺氧-好氧工艺又可以分类为O/A/O和O/H/O,分化出二污泥法和三污泥法,反应器的类型决定了工艺的耗能。若只考虑生物阶段的处理,废水COD在3 000~4 000 mg·L−1、铵离子质量浓度在100~200 mg·L−1时,A/A/O的处理费用为6~8 元·t−1[45,46],O/A/O的处理费用为7~9 元·t−1 [47-49],而O/H/O流化床工艺的处理费用仅为4~5元·t−1,体现了不同技术的成本差异。
2.4 性能比较
单元工艺的摆放顺序不仅决定了整体工艺运行的能耗,还会对冲击负荷、系统中微生物菌落和处理效果产生很大的影响。李国令等[4]指出,热单胞菌属、脱氯单胞菌属是O/A/O工艺好氧池中的优势菌属;热单胞菌属、脱氯单胞菌属、球形红假单胞菌属是O/A/O工艺缺氧池中的优势菌属。WANG等也发现[50],热单胞菌属与硝酸盐还原酶基因呈正相关,对同时厌氧氨氧化-反硝化系统中的硝酸盐还原起重要作用。 WEI等[15]指出,丛毛单胞菌属在反应器O1中对COD去除起到了关键作用,有助于去除O1反应器中的NH4+-N;硫杆菌则在H反应器中起着主要的反硝化作用,AOB和NOB(亚硝化单胞菌和硝化螺菌)对反应器中硝化作用的贡献最大。三污泥法的O/H/O工艺各单元在污染物组成、去除、功能和微生物群落等方面存在显著进步,有望实现厌氧氨氧化脱氮与深度脱氮的结合,也表明废水水质和反应器的组合对微生物功能分布具有调控功能。
根据污泥回流的设置与否,A/A/O、O/A/O、O/H/O工艺可以分为单污泥系统、双污泥系统及三污泥系统,3个工艺的主要区别见表3。据报道,A/A/O工艺中A1单元对COD去除效率小于10%,检测不到甲烷的产生[51]。因此,A/A/O工艺仅仅在缺氧和有氧反应器中实现了对COD的去除。由于回流的存在,A/A/O工艺表现为单污泥特征,异养细菌具有较高的比生长速率,因污泥排放量高而导致其在处理高COD/TN废水时,大量自养硝化细菌被排洗。前置好氧工艺对高浓度毒性废水有很好的抗负荷冲击能力,并且O/H/O工艺中的新型结构流化床反应器的强化传质功能与污泥原位分离原理加强了各单元反应器中的微生物能力[22]。在H单元中,根据投加的电子供体不同而具有多种反硝化模式:如利用O1池的剩余COD作为碳源及其他电子供体进行异养反硝化脱氮;通过投加无机还原性电子供体以利用其作为营养源进行自养反硝化脱氮[21,52],还可以避免二次碳源的污染。另外,有研究表明,控制O1反应器在短程硝化水平,可使亚硝酸盐直接得到富集和积累,然后实现厌氧氨氧化模式脱氮,从而使工艺过程节能效果更好[17,19]。可见,复杂废水的脱氮模式多种多样,需要根据实际情况合理选择或耦合新原理,从而进一步实现低能耗、低物耗目标下的总氮去除。
表 3 不同工艺系统的特点Table 3. Characteristics of different process systems工艺 污泥系统 毒性物质的去除 COD/TN 脱氮途径 能耗影响因素 平均运行单价/(元·m-3) 优点 缺点 A/A/O 单污泥系统 A1对大分子有机物的去除 11.4 异养反硝化 一次回流、一次曝气 7 有利于含氮有机物的水解;反硝化可利用废水中有机物作为碳源 不耐冲击负荷,受毒性抑制,需要稀释进水 O/A/O 双污泥系统 O1对SCN−、CN−的去除及氨化 12.5 异养反硝化、自养反硝化 二次回流、二次曝气 8 耐冲击负荷,进水不需要稀释;硝化效果好 耗氧量大,污泥回流频繁,耗能多 O/H/O 三污泥系统 O1对SCN−、CN−的去除及氨化 13.8 异养反硝化、自养反硝化、厌氧氨氧化及其耦合脱氮 二次曝气 4.5 耐冲击负荷,颗粒污泥耐毒性抑制,硝化效果好,不需要沉淀池;不需要回流 耗氧量大 | Show TableDownLoad:
CSV
3. 结论与展望
1)每处理1 m3设定浓度的焦化废水(不考虑O/A/O的超越进水),A/A/O和O/A/O工艺分别需要122.1 g 与486.9 g的外部碳源(以甲醇计)。当废水中的易降解有机物较少且只考虑脱氮目标时,O/A/O工艺的曝气需氧量为83.9 kg·h−1,A/A/O工艺的曝气需氧量为100.4 kg·h−1;当O/A/O工艺中有1/3的进水流量超越至A单元时,其碳源需求量由486.9 g·m−3减至257.2 g·m−3(以甲醇计),曝气量也将显著降低。
2)由于废水组成的复杂性,污染物的降解效率除了受到彼此的相互制约外,工艺条件和反应器的设计也至关重要。具有高毒性、高碳氮含量的焦化废水,更适合于选择前置好氧的工艺。O/H/O工艺由于其独特的三相分离器的设置而节省了污泥回流部分的能耗,反应器中的颗粒污泥更加耐毒性抑制和抗冲击负荷,并且传氧速率高,工艺耗氧量仅为53.26 kg·h−1,外部碳源的消耗可以由486.9 g·m−3降至0~220 g·m−3。
3)反应器的高效性和可控性,使O/H/O工艺比O/A/O工艺更容易实现自养反硝化与异养反硝化协同脱氮、自养型短程反硝化与厌氧氨氧化的协同脱氮等其他脱氮途径,进而使O/H/O工艺成为一种更具潜力的低能耗、低物耗的生物脱氮技术工艺。针对不同的废水水质与物质组成特征,O/H/O工艺能够对不同功能的单元进行组合和编辑,从时间与空间、药剂与能耗、处理效率等方面追求更加丰富的优化模式,以满足各种不同的出水需求,特别是满足总氮浓度趋零的要求。
-
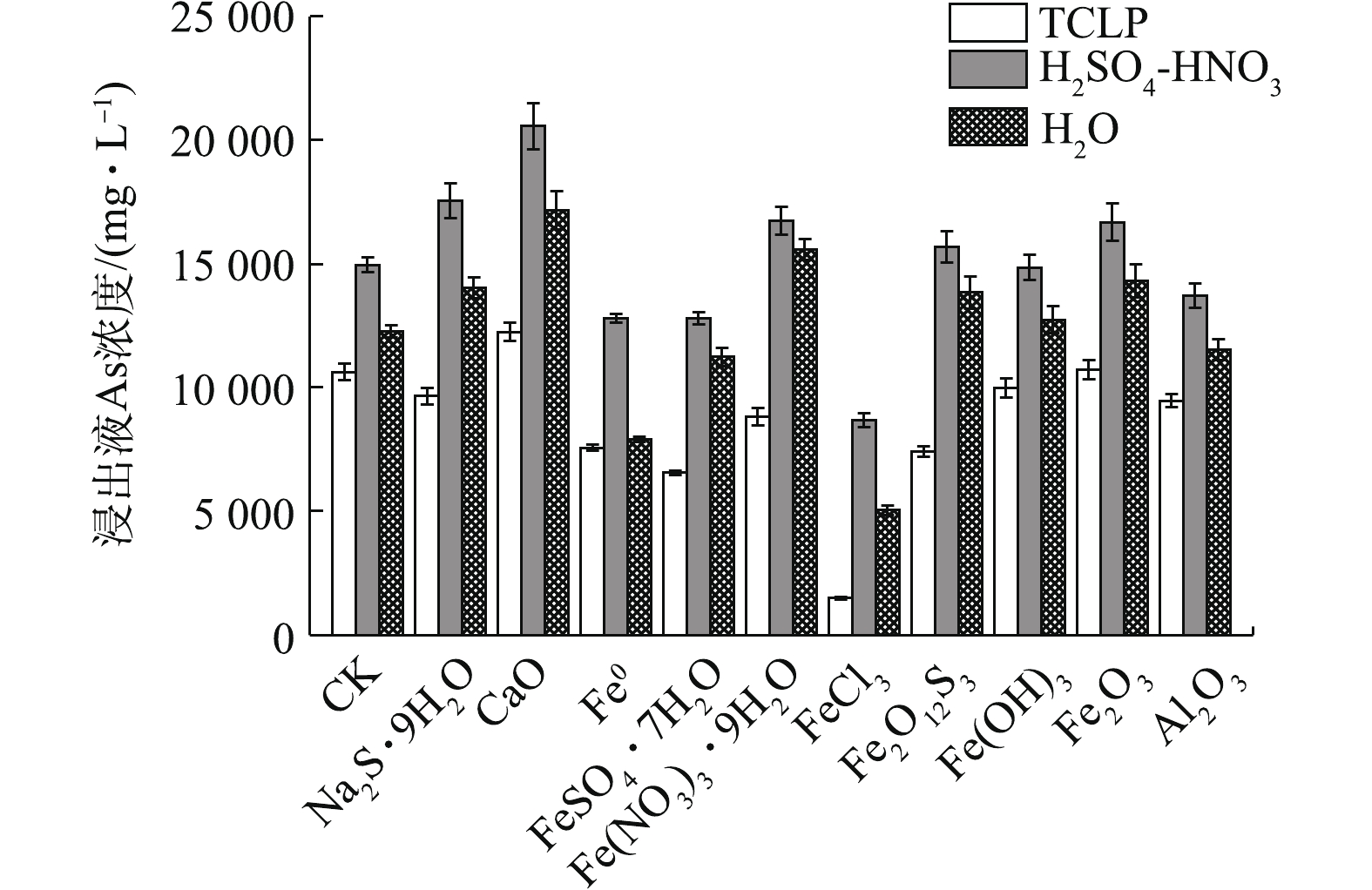
图 1 不同固定化处理对污泥中As浸出浓度的影响
Figure 1. Effects of different immobilization materials on As leaching concentration in sludge
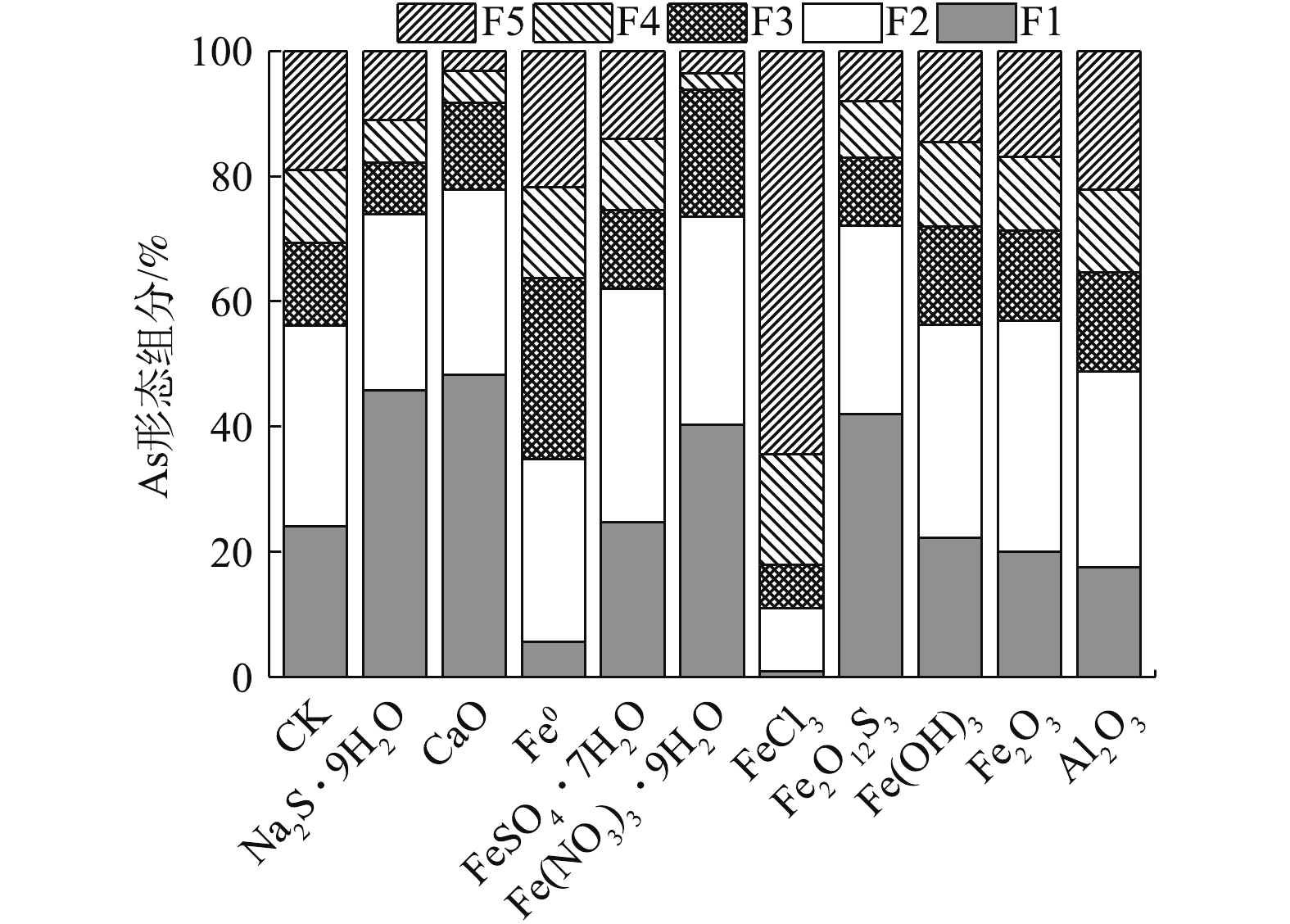
图 2 不同固定化处理对As结合态的影响
Figure 2. Effects of different immobilization treatments on the distribution of As binding form in sludge
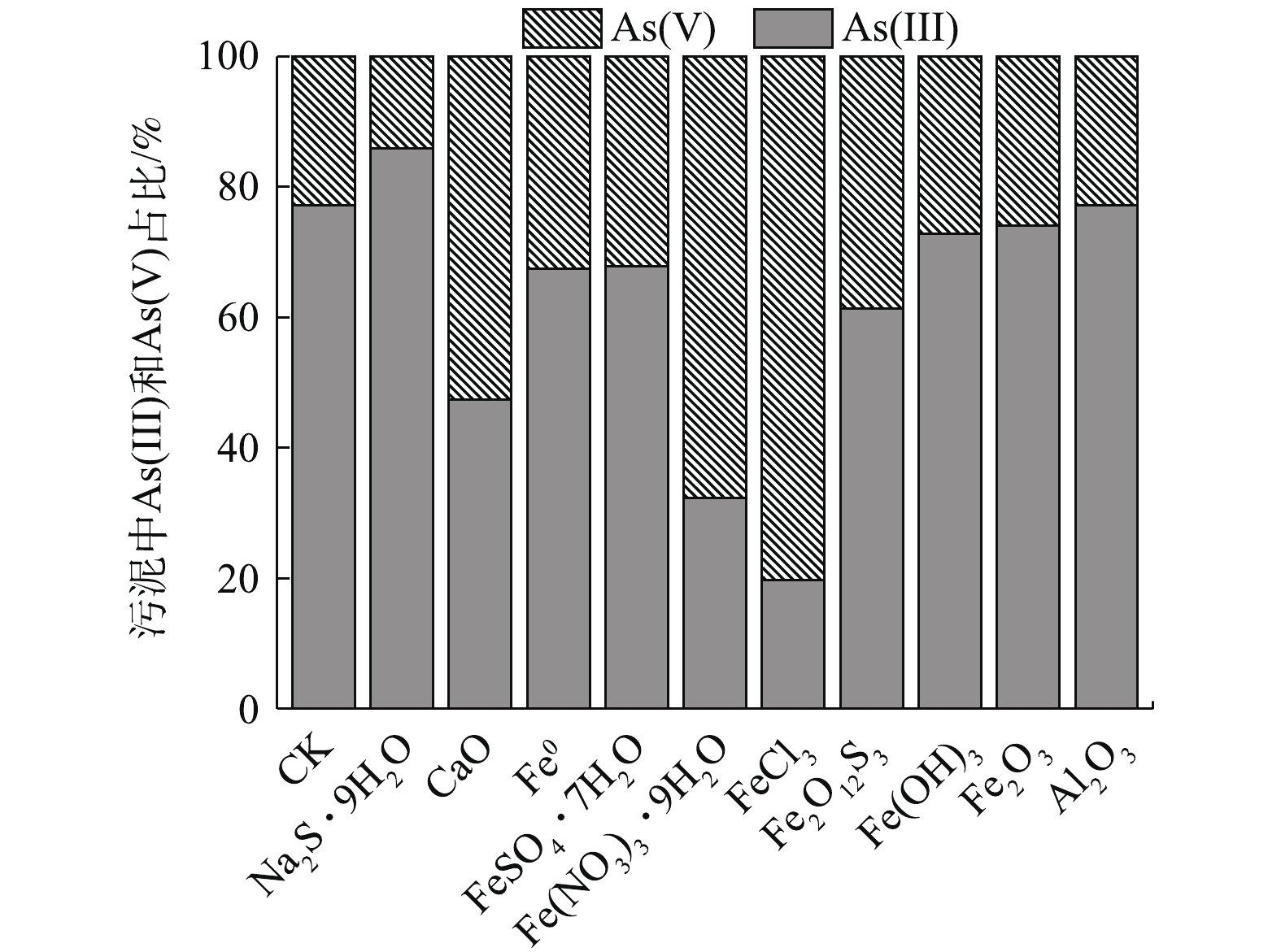
图 3 不同材料处理对污泥As价态分布的影响
Figure 3. Effects of different immobilization materials on As valence distribution in sludge
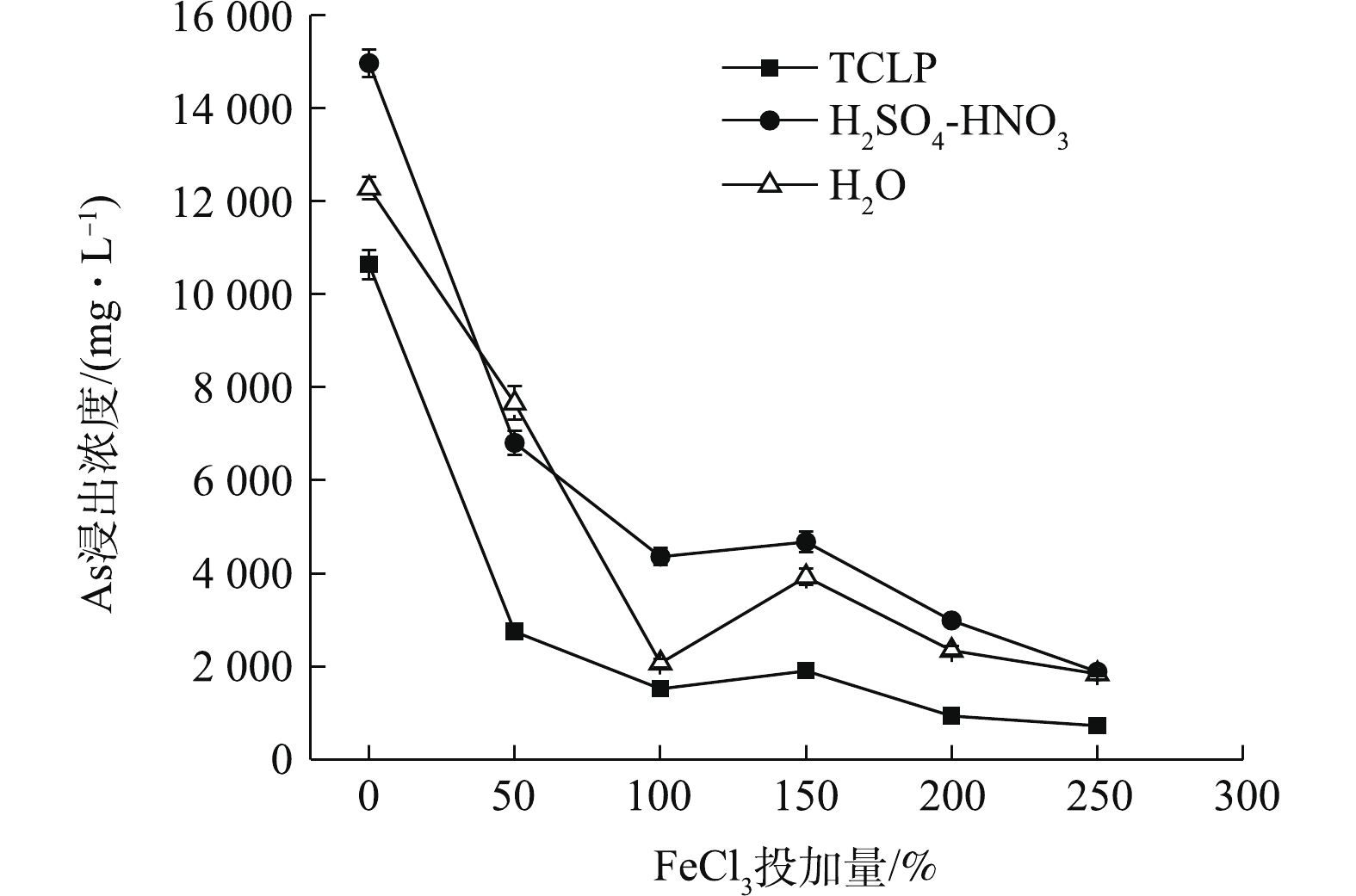
图 4 FeCl3处理对污泥As浸出浓度的影响
Figure 4. Effects of FeCl3 treatment on the leaching concentration of As in sludge
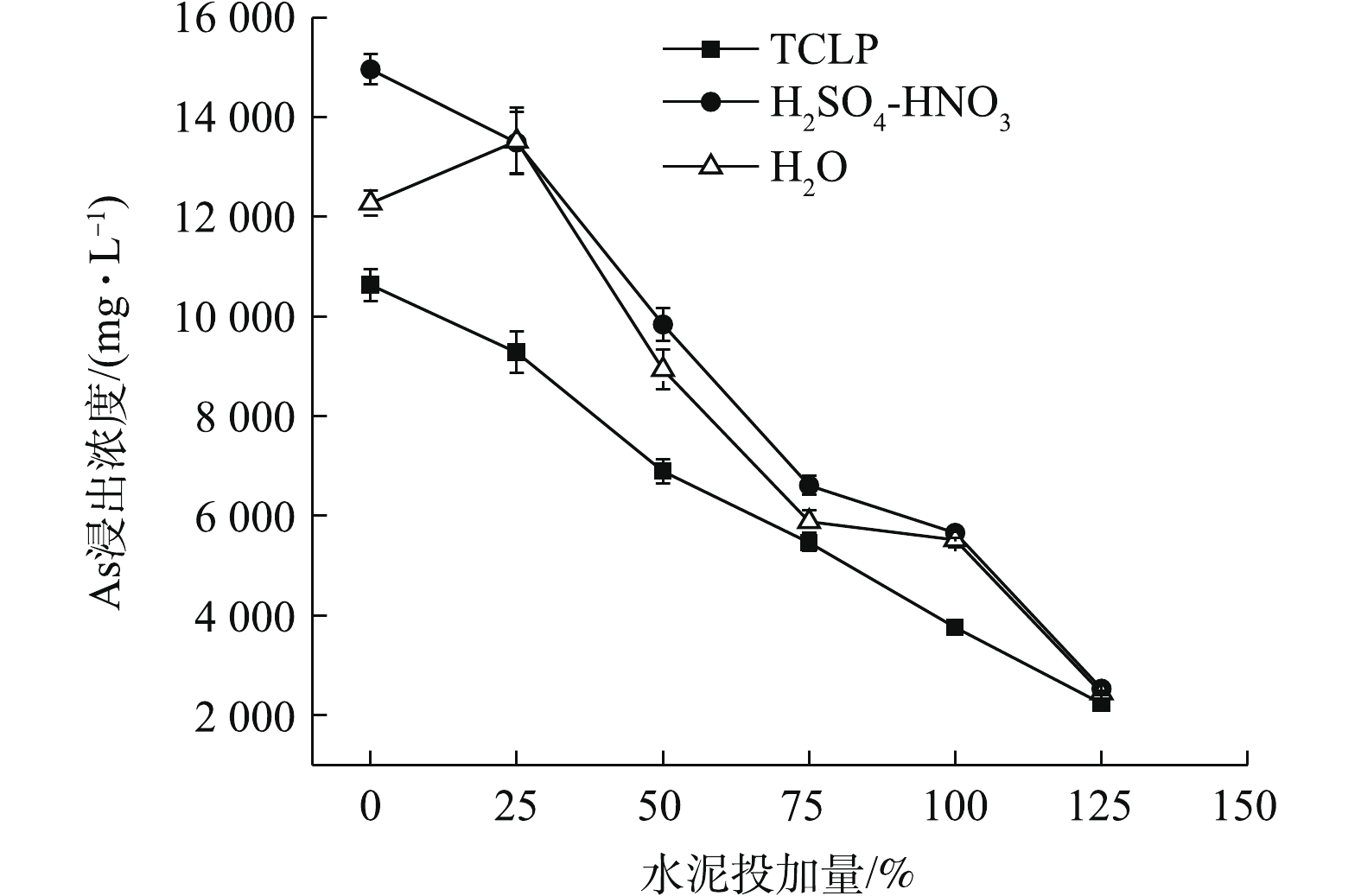
图 5 水泥处理对污泥As浸出浓度的影响
Figure 5. Effects of cement treatment on the leaching concentration of As in sludge
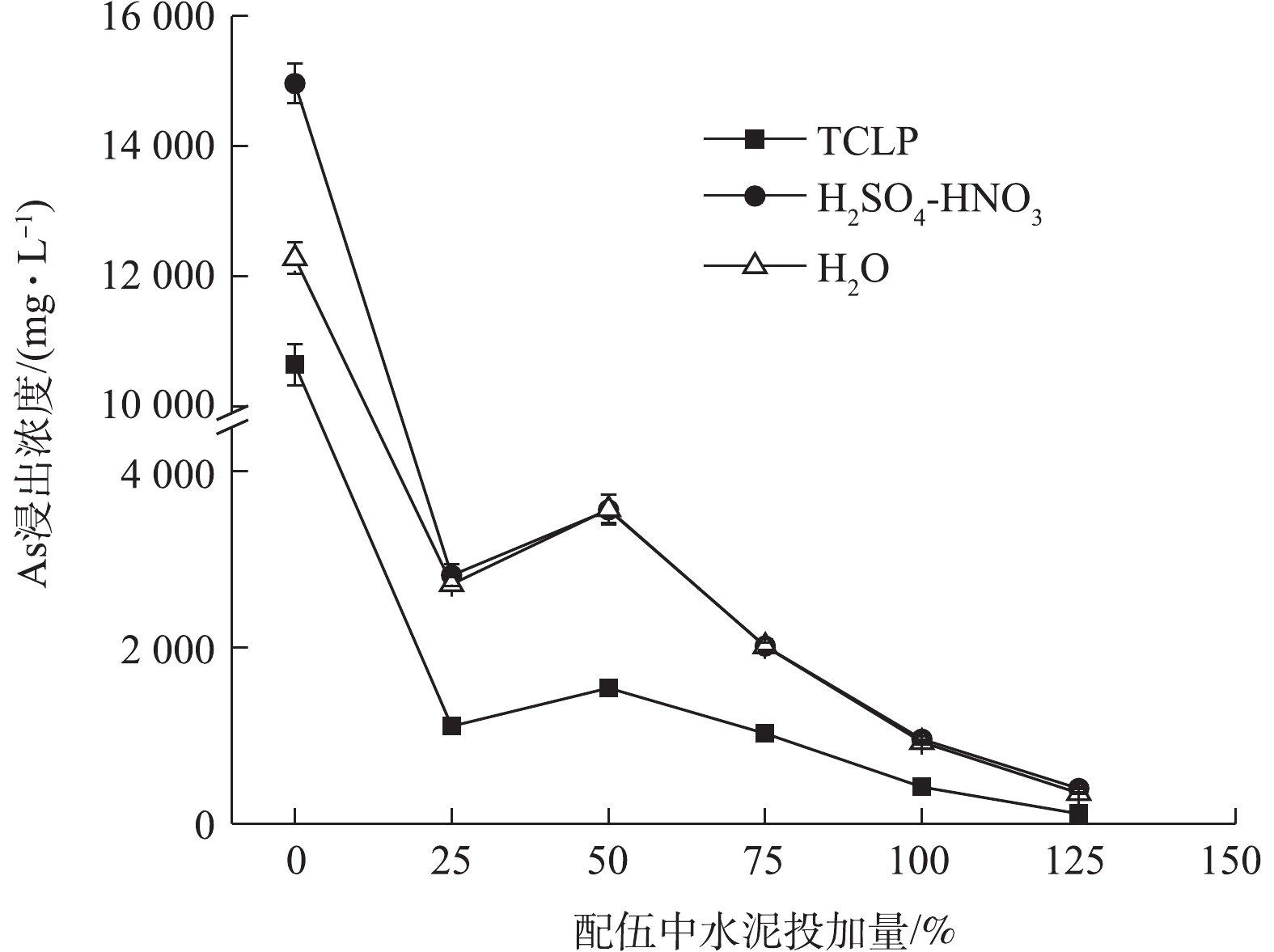
图 6 FeCl3复配水泥对污泥As浸出浓度的影响
Figure 6. Effects of FeCl3+cement treatment on the leaching concentration of As in sludge
表 1 固定化材料筛选实验设计
Table 1. Design of As immobilization materias screening experiment
实验处理 固定化材料名称 材料投加量/g 摩尔比 1 Na2S·9H2O 7.88 S/As=0.5∶1 2 CaO 1.84 Ca/As=0.5∶1 3 Fe0 1.22 Fe/As=0.3∶1 4 FeSO4·7H2O 6.08 Fe/As=0.3∶1 5 Fe(NO3)3·9H2O 8.84 Fe/As=0.3∶1 6 FeCl3 3.55 Fe/As=0.3∶1 7 Fe2O12S3 4.37 Fe/As=0.3∶1 8 Fe(OH)3 2.34 Fe/As=0.3∶1 9 Fe2O3 1.75 Fe/As=0.3∶1 10 Al2O3 1.11 Al/As=0.3∶1 空白对照 — — —
下载: 导出CSV
表 2 FeCl3与水泥配伍实验设计
Table 2. Design of the composite experiments of FeCl3 and cement
实验处理 砷泥/g FeCl3/g 水泥/g 1 20 10 — 2 20 20 — 3 20 30 — 4 20 40 — 5 20 50 — 6 20 — 5 7 20 — 10 8 20 — 15 9 20 — 20 10 20 — 25 11 20 50 5 12 20 50 10 13 20 50 15 14 20 50 20 15 20 50 25 空白对照 20 — — 注:—表示未添加,药剂添加顺序为先加FeCl3,再加水泥。
下载: 导出CSV
表 3 污泥中As浸出浓度和浸出量
Table 3. As leaching concentration and quantity of tested sludge
浸出方法 浸出浓度/(mg·L−1) 浸出量/(mg·kg−1) TCLP 10 634.05 21 2681.00 H2SO4-HNO3 14 961.25 149 612.50 H2O 12 276.50 122 765.00 SBET 2 678.14 26 7814.00
下载: 导出CSV
表 4 污泥中As结合态分布
Table 4. Distribution of As binding form in tested sludge
As结合态 含量/(mg·kg−1) 占总As百分比/% F1非专性吸附态 99 264.50±3 176.46 24.04 F2专性吸附态 132 332.50±5 425.63 32.05 F3无定形和弱结晶铁铝或铁锰水化氧化物结合态 54 535.50±2 399.56 13.21 F4结晶铁锰或铁铝水化氧化物结合态 48 348.25±1 257.05 11.71 F5残渣态 78 369.00±2 899.65 18.98
下载: 导出CSV
-
[1] RIVEROS P A, DUTRIZAC J E, SPENCER P, et al. Arsenic disposal practices in the metallurgical industry[J]. Canadian Metallurgical Quarterly, 2001, 40: 395-420. doi: 10.1179/cmq.2001.40.4.395 [2] CLANCY T M, HAYES K F, RASKIN L. Arsenic waste management: A critical review of testing and disposal of arsenic-bearing solid wastes generated during arsenic removal from drinking water[J]. Environmental Science & Technology, 2013, 47(19): 10799-10812. [3] KAMESWARI K S B, BHOLE A G, PARAMASIVAM R, et al. Evaluation of solidification/stabilization(S/S) process for the disposal of arsenic-bearing sludges in landfill sites[J]. Environmental Engineering Science, 2002, 18(3): 167-176. [4] 国家环境保护总局, 国家质量监督检验检疫总局. 危险废物填埋污染控制标准: GB 18598-2001[S]. 北京: 中国环境科学出版社, 2001. [5] MOORE T J, RIGHTMIRE C M, VEMPATI R K, et al. Ferrous iron treatment of soils contaminated with arsenic containing wood-preserving solution[J]. Soil and Sediment Contamination, 2000, 9(4): 375-405. doi: 10.1080/10588330091134310 [6] KUMPIENE J, LAGERKVIST A, MAURICE C. Stabilization of As, Cr, Cu, Pb and Zn in soil using amendments: A review[J]. Waste Management, 2008, 28: 215-225. doi: 10.1016/j.wasman.2006.12.012 [7] KOMÁREK M, VANEK A, ETTLER V. Chemical stabilization of metals and arsenic in contaminated soils using oxides: A review[J]. Environmental Pollution, 2013, 172(172C): 9-22. [8] 纪冬丽, 孟凡生, 薛浩, 等. 国内外土壤砷污染及其修复技术现状与展望[J]. 环境工程技术学报, 2016, 6(1): 90-99. doi: 10.3969/j.issn.1674-991X.2016.01.014 [9] NAZARI A M, RADZINSKI R, GHAHREMAN A. Review of arsenic metallurgy: Treatment of arsenical minerals and the immobilization of arsenic[J]. Hydrometallurgy, 2017, 174: 258-281. doi: 10.1016/j.hydromet.2016.10.011 [10] PORTER S K, SCHECKEL K G, IMPELLITTERI C A, et al. Toxic metals in the environment: Thermodynamic considerations for possible immobilisation strategies for Pb, Cd, As, and Hg[J]. Critical Reviews in Environmental Science & Technology, 2004, 34(6): 495-604. [11] 高峰, 贾永忠, 孙进贺, 等. 锌冶炼废渣浸出液硫化法除砷的研究[J]. 环境工程学报, 2011, 5(4): 812-814. [12] MOON D H, DERMANTAS D, MENOUNON N. Arsenic immobilization by calcium-arsenic precipitates in lime treated soil[J]. Science of the Total Environment, 2004, 330(1/2/3): 171-185. [13] WENZEL W W, KIRCHBAUMER N, PROHASKA T, et al. Arsenic fractionation in soils using an improved sequential extraction procedure[J]. Analytica Chimica Acta, 2001, 436(2): 309-323. doi: 10.1016/S0003-2670(01)00924-2 [14] SEAMAN J C, HUTCHISON J M, JACKSON B P, et al. In situ treatment of metals in contaminated soils with phytate[J]. Journal of Environmental Quality, 2003, 32: 153-161. doi: 10.2134/jeq2003.1530 [15] CARLSON L, BIGHAM J M, SCHWERTMANN U, et al. Scavenging of As from acid mine drainage by schwertmannite and ferrihydrite: A comparison with synthetic analogues[J]. Environmental Science & Technology, 2002, 36: 1712-1719. [16] HARTLEY W, EDWARDS R, LEPP N W. Arsenic and heavy mental mobilization in iron oxide-amended contaminated soils as evaluated by short and long-term leaching tests[J]. Environmental Pollution, 2004, 131(3): 495-504. doi: 10.1016/j.envpol.2004.02.017 [17] 胡立琼, 曾敏, 雷鸣, 等. 含铁材料对污染水稻土中砷的固定化效果[J]. 环境工程学报, 2014, 8(4): 13-18. [18] 黄玲, 雷鸣, 胡立琼, 等. 2种含铁物质对底泥中砷的固化效果[J]. 水土保持学报, 2014, 28(1): 253-256. doi: 10.3969/j.issn.1009-2242.2014.01.048 [19] DERMATAS D, MENG X. Utilization of fly ash for stabilization/solidification of heavy metal contaminated soils[J]. Engineering Geology, 2003, 70: 377-394. doi: 10.1016/S0013-7952(03)00105-4 [20] COUSSY S, PAKTUNC D, ROSE J, et al. Arsenic speciation in cemented paste backfills and synthetic calcium-silicate-hydrates[J]. Minerals Engineering, 2012, 39(10): 51-61. [21] VOGLARA G E, LESTAN D. Efficiency modeling of solidification/stabilization of multi-metal contaminated industrial soil using cement and additives[J]. Journal of Hazardous Materials, 2011, 192: 753-762. doi: 10.1016/j.jhazmat.2011.05.089 [22] VINTER S, MONTANES M T, BEDNARIK V, et al. Stabilization/solidification of hot dip galvanizing ash using different binders[J]. Journal of Hazardous Materials, 2016, 320: 105-113. doi: 10.1016/j.jhazmat.2016.08.023 [23] LIU D G, MIN X B, KE Y, et al. Cotreatment of flotation waste, neutralization sludge, and arsenic-containing gypsum sludge from copper smelting: Solidification/stabilization of arsenic and heavy metals with minimal cement clinker[J]. Environment Science Pollution, 2017, 25(8): 7600-7607. [24] KOGBARA R B. A review of the mechanical and leaching performance of stabilized/solidified contaminated soils[J]. Environment Reviews, 2013, 22: 66-86. [25] LI J S, WANG L, CUI J L, et al. Effects of low-alkalinity binders on stabilization/solidification of geogenic As-containing soils: Spectroscopic investigation and leaching tests[J]. Science of the Total Environment, 2018, 631-632: 1486-1494. doi: 10.1016/j.scitotenv.2018.02.247 [26] 中华人民共和国农业部. 土壤检测 第2部分: 土壤pH的测定: NY/T 1121.2-2006[S]. 北京: 中国农业出版社, 2006. [27] US EPA. Test methods for evaluating solid waste, physical/chemical methods[DB/OL]. [2019-05-01]. http://www.epa.gov/SW-846/main.htm. [28] 国家环境保护总局. 固体废物浸出毒性浸出方法 硫酸硝酸法: HJ/T 299-2007[S]. 北京: 中国环境科学出版社, 2007. [29] 中华人民共和国环境保护部. 固体废物浸出毒性浸出方法 水平振荡法: HJ 557-2010[S]. 北京: 中国环境科学出版社, 2010. [30] RAURET G, LOPEZ-SANCHEZ J F, SAHUQUILLO A, et al. Improvement of the BCR three step sequential extraction procedure prior to the certification of new sediment and soil reference materials[J]. Journal of Environmental Monitoring, 1999, 1(1): 57-61. doi: 10.1039/a807854h [31] BI C, ZHOU Y, CHEN Z, et al. Heavy metals and lead isotopes in soils, road dust and leafy vegetables and health risks via vegetable consumption in the industrial areas of Shanghai, China[J]. Science of the Total Environment, 2018, 619-620: 1349-1357. doi: 10.1016/j.scitotenv.2017.11.177 [32] GARCIA-MANYES S, JIMENEZ G, PADRO A, et al. Arsenic speciation in contaminated soils[J]. Talanta, 2002, 58(1): 97-109. doi: 10.1016/S0039-9140(02)00259-X [33] MARTA I, LITTER, MARIA E, et al. Possible treatments for arsenic removal in Latin American waters for human consumption[J]. Environmental Pollution, 2010, 158(5): 1105-1118. doi: 10.1016/j.envpol.2010.01.028 [34] 刘辉利, 梁美娜, 朱义年, 等. 氢氧化铁对砷的吸附与沉淀机理[J]. 环境科学学报, 2009, 29(5): 1011-1020. doi: 10.3321/j.issn:0253-2468.2009.05.019 [35] BASKAN M B, PALA A. Determination of arsenic removal efficiency by ferric ions using response surface methodology[J]. Journal of Hazardous Materials, 2009, 166(2/3): 796-801. [36] GUTIERREZ C, HANSEN H K, NUNEZ P, et al. Electrochemical peroxidation using iron nanoparticles to remove arsenic from copper smelter wastewater[J]. Electrochimica Acta, 2015, 181: 228-232. doi: 10.1016/j.electacta.2015.01.070 [37] YOKOYAMA Y, TANAKA K, TAKAHASHI Y. Differences in the immobilization of arsenite and arsenate by calcite[J]. Geochimica et Cosmochimica Acta, 2012, 91: 202-219. doi: 10.1016/j.gca.2012.05.022 [38] 张淑媛, 童宏祥, 徐诗琦, 等. 次氯酸钙/氧化钙对高砷污泥的氧化固定化处理[J]. 环境工程学报, 2018, 12(2): 625-629. doi: 10.12030/j.cjee.201706216 [39] WARREN G P, ALLOWAY B J. Reduction of arsenic uptake by lettuce with ferrous sulfate applied to contaminated soil[J]. Journal of Environmental Quality, 2003, 32(3): 767-772. doi: 10.2134/jeq2003.7670 [40] 曹宇, 陈明, 张佳文, 等. 硫化钠对土壤中铅镉的固定效果[J]. 环境工程学报, 2014, 8(2): 782-788. [41] LADEIRA A C Q, CIMINELLI V S T, DUARTE H A, et al. Mechanism of anion retention from EXAFS and density functional calculations: Arsenic(V) adsorbed on gibbsite[J]. Geochimica Et Cosmochimica Acta, 2001, 65: 1211-1217. doi: 10.1016/S0016-7037(00)00581-0 [42] WANG Y N, ZENG X B, LU Y H, et al. Effect of aging on the bioavailability and fractionation of arsenic in soils derived from five parent materials in a red soil region of Southern China[J]. Environmental Pollution, 2015, 207: 79-87. doi: 10.1016/j.envpol.2015.08.033 [43] 王强, 锦春, 魏世强. 赤铁矿对砷的吸附解吸及氧化特征[J]. 环境科学学报, 2008, 28(8): 1612-1617. doi: 10.3321/j.issn:0253-2468.2008.08.018 [44] RAMOS M A V, YAN W, LI X, et al. Simultaneous oxidation and reduction of arsenic by zero-valent iron nanoparticles: understanding the significance of the core-shell structure[J]. Journal of Physical Chemistry C, 2009, 113(33): 14591-14594. doi: 10.1021/jp9051837 [45] LEUPIN O X, HUG S J. Oxidation and removal of arsenic(III) from aerated groundwater by filtration through sand and zero-valent iron[J]. Water Research, 2005, 39(9): 1729-1740. doi: 10.1016/j.watres.2005.02.012 [46] 雷鸣, 曾敏, 胡立琼, 等. 3种含铁材料对重金属和砷复合污染底泥固定化处理[J]. 环境工程学报, 2014, 8(9): 3983-3988. [47] KIM J Y, DAVIS A P, KIM K W. Stabilization of available arsenic in highly contaminated mine tailings using iron[J]. Environmental Science & Technology, 2003, 37(1): 189-195. [48] 熊正为, 朱雷, 杨博豪, 等. 水泥回转窑共处置含砷污泥[J]. 环境工程学报, 2016, 10(1): 301-305. doi: 10.12030/j.cjee.20160149 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3659
- HTML全文浏览数: 3659
- PDF下载数: 41
- 施引文献: 0
