

-
近年来,与传统的化学计量发动机相比,柴油和其他贫燃汽油车辆因其更好的燃油效率和低CO2排放量而拥有广阔的发展前景[1],但三效催化剂对NOx的催化还原效果较差[2]。随着氮氧化物排放造成的环境污染问题的日趋严重,在未来几年,交通运输部门对氮氧化物排放的监管也将更为严格。因此,有必要开发能够在柴油和其他贫燃汽油车辆的发动机处理系统之后还原NOx的催化系统[3]。为了解决这个问题,许多学者研究了NSR(NOx储存还原)技术及以NH3或烃为还原剂的NH3-SCR和HC-SCR催化体系[1]。HC-SCR因其不必通过外部加入还原剂而引起人们的广泛关注[2],并且已有研究证明HC-SCR是一种有效且经济的技术[4]。研究表明,TiO2不仅在光催化领域效果显著,在SCR应用中也是一种优良的催化剂载体[5]。TiO2具有强抗硫中毒能力、高比表面积[6]、低毒性和价格低廉的优势[7]。同时Ce基材料可以用作三效催化剂的重要组分,它们能够储存和释放氧气,进而起到促进CO和NO转化的作用[8]。王淑勤等[9]以TiO2为载体,负载Ce和Co元素,研究其脱硝性能,发现与纯TiO2相比,其效率提高了近50%。JIN等[10]将Mn-Ce活性组分负载在TiO2和Al2O3载体上,发现在80~150 ℃,Mn-Ce/TiO2的脱氮活性高于Mn-Ce/Al2O3。此外,杂多酸(HPAs)因具有假液相特性、强氧化还原能力、活泼的晶格氧、强质子酸性、无毒和非挥发性的特点而引起关注[11]。研究[12]表明,极性分子(如NO、NH3、吡啶)等可以进入HPAs内部,从而引发HPAs表面和内部反应。实验发现,NOx的去除率与杂多酸的酸度密切相关,杂多酸的酸性越强,去除率越高。其中HPW酸性最强,具有最高的NOx去除率。在HPW的各种结构中,已有研究探讨了Keggin型H3PW12O40的物理化学和催化性质,并且已经证明它是一种可以用于均相或非均相的有效超级酸[12]。但是在实际应用中,纯杂多酸也存在很多缺点,如比表面积小(<10 m2·g−1)、热稳定性低、机械强度差、在极性溶剂中溶解且难以回收,因此,应将其负载到载体上来克服这些缺点[13]。目前,负载磷钨酸的主要载体有活性炭、SiO2、TiO2、MCM-41、分子筛等中性载体[14]。
本研究采用浸渍法制备了TiO2、Ce-TiO2和Ce与H3PW12O40(HPW)共掺杂TiO2 3种催化剂,模拟烟气进行脱硝活性测试,使用X射线衍射(XRD)、傅里叶变换红外(FTIR)和扫描电镜(SEM)对制备的催化剂进行表征测试,同时进行了原位傅里叶变换红外光谱实验,阐明HPW和Ce在SCR反应中的作用,推测其可能的反应机理,为进一步深入研究烃类选择性催化还原NO反应机理提供参考。
-
通过浸渍法合成负载Ce和HPW的催化剂。首先,在剧烈搅拌下,将2份0.3 g HPW溶解在去离子水中;然后将2份HPW溶液(15%)逐滴分别加入到2 g TiO2中,剧烈搅拌2 h,将沉淀物静置12 h,100 ℃下干燥过夜,得到HPW-TiO2粉末;将其中1份HPW-TiO2样品在空气中400 ℃下煅烧4 h,得到HPW-TiO2(CM),另一个未煅烧的样品标记为HPW-TiO2(UCM)。
将3份0.186 g Ce(NO3)·6H2O(3%)溶解在去离子水中,完全溶解后,将硝酸铈溶液滴加到TiO2、HPW-TiO2(CM)和HPW-TiO2(UCM)中,剧烈搅拌2 h。然后将3个样品静置12 h,100 ℃下干燥过夜。最后,将2 g TiO2与其他3个样品一起在空气气氛中400 ℃煅烧4 h,以获得TiO2、3%Ce-TiO2、3%Ce-15%HPW-TiO2(CM)和3%Ce-15%HPW-TiO2(UCM)催化剂,简称为TiO2、Ce-TiO2、Ce-HPW-TiO2(CM)和Ce-HPW-TiO2(UCM)。
-
采用具有Cu Kα(λ=0.154 06 nm)的BRUKER D8 ADVANCE衍射仪进行粉末X射线衍射(XRD)测试表征,管电压为40 kV,管电流为40 mA,扫描速率为11.8 (°)·min−1,扫描角度为5°~80°;采用S-4800场发射扫描电子显微镜(SEM)观察分析催化剂的微观形貌;通过BRUKER VERTEX 70光谱仪获得傅里叶变换红外光谱(FTIR)的数据,扫描波数为400~4 000 cm−1。
-
以固定床装置评价各催化剂的C3H6-SCR反应活性。U型石英反应管内径为5 mm,外径为6 mm。催化剂用量为0.2 g,粒径为20~40目。标准C3H6-SCR反应的初始气体条件为:[NO]=[C3H6]=0.1%,[O2]=10%,以He作为平衡气,气体流量为100 mL·min−1,气体空速约为30 000 h−1。混合气体中NO浓度由化学传感器多组分气体分析仪进行监测,以NO脱除率
ηNO 表示催化剂脱硝效率,计算方法见式(1)。式中:
CNO,in 为入口NO浓度,μg·L−1;CNO,out 为出口NO浓度,μg·L−1。 -
原位FT-IR测试在FT-IR光谱仪(BRUKER VERTEX 70)上进行,仪器分辨率为4 cm−1。在每次实验运行前,样品在400 ℃下,Ar气氛中以10 mL·min−1的总流速预处理20 min,除去吸附的杂质,然后冷却至目标反应温度。在C3H6(或NO+O2)吸附实验中,将样品在200 ℃下暴露于0.1% C3H6(或0.1% NO+10% O2)气流中60 min,然后Ar吹扫10 min。在瞬态研究中,首先将样品预先暴露于0.1% C3H6(或0.1% NO+5% O2)中,在200 ℃下吸附60 min,然后用Ar吹扫10 min,随后将气体转换为0.1% NO+5% O2(或0.1% C3H6),以获得FT-IR光谱的动态变化。
-
TiO2、Ce-TiO2、Ce-HPW-TiO2(UCM)和Ce-HPW-TiO2(CM)催化剂的XRD结果如图1所示。据研究[7],在25.28°、36.84°、37.78°、38.42°、47.97°、53.79°、55.08°、62.70°、68.83°、70.32°和75.03°处出现的衍射峰对应于锐钛矿型TiO2的特征峰。在27.42°、36.06°、41.24°和56.64°处出现的衍射峰与金红石型TiO2的特征峰完全吻合。图1中未显示出Ce和H3PW12O40特征峰,仅出现了TiO2的特征峰。而且随着Ce和HPW的掺杂,TiO2衍射峰的强度逐渐减弱。这证明Ce和HPW的掺杂影响了TiO2的晶体结构,并且在载体表面上以非晶态高度分散。
图2是TiO2、Ce-TiO2、Ce-HPW-TiO2(UCM)和Ce-HPW-TiO2(CM)的SEM图。图2(a)为纯TiO2的SEM图像,其表面表现为具有不光滑表面和不均匀颗粒的聚集体。掺杂Ce后,Ce-TiO2的形貌发生了明显的变化,催化剂表面变得更加光滑。HPW修饰的催化剂表现出与Ce-TiO2相似的形态和微观结构,如图2(c)和图2(d)所示。显然,Ce-HPW-TiO2(UCM)和Ce-HPW-TiO2(CM)的颗粒形态更规则,分散性更好。这可能是由于各组分之间的强相互作用导致的,这种相互作用有利于催化活性的增强,可抑制TiO2颗粒的团聚过程、干扰晶体颗粒的生长以及增加催化剂表面积。
TiO2、Ce-TiO2、Ce-HPW-TiO2(UCM)和Ce-HPW-TiO2(CM)的FT-IR谱图如图3所示。据研究[15-16],Ti―O、Ce―O、Ti―O―Ce键的伸缩振动峰值出现在800~400 cm−1。与TiO2的谱图相比,Ce-TiO2相应的透射带呈蓝移状态,这证明Ce成功掺入了TiO2骨架[17]。曲线Ce-HPW-TiO2(UCM)和Ce-HPW-TiO2(CM)的趋势类似于曲线TiO2,峰值位置也存在蓝移现象,表明HPW、Ce和TiO2在催化剂表面存在相互作用。
与HPW相关的FT-IR研究[18]指出,P―Oa是指连接杂原子P和金属氧化物簇W3O13的一种氧原子,其伸缩振动峰出现在大约1 083 cm−1处,并且在802 cm−1处左右的峰可以归属于边缘共享W3O13八面体与W―Oc―W之间的拉伸。这几个特征峰可被用作证明HPW的Keggin结构存在的常用方法[19]。在图3中的800~1 100 cm−1处,HPW修饰的催化剂谱图存在Keggin结构典型振动带特征峰,表明Keggin型聚阴离子成功负载在催化剂表面。与Ce-HPW-TiO2(CM)相比,Ce-HPW-TiO2(UCM)的HPW特征峰不明显,这可能是由于在负载HPW后没有进行煅烧导致的,使其没有很好地固定在催化剂表面上。
-
图4显示了纯TiO2以及掺杂Ce、HPW后的催化剂的C3H6-SCR活性随温度的变化情况。由图4可知,在中低温范围内(150~350 ℃),随着温度的升高,各催化剂对NO的转化率均呈先增大后减小的趋势。NO转化能力顺序为Ce-HPW-TiO2(CM)>Ce-HPW-TiO2(UCM)>Ce-TiO2>TiO2。Ce-HPW-TiO2(CM)催化剂显示最高的NO转化率,约为73%,这说明Ce和HPW的共掺杂显著促进了催化剂在中低温区的C3H6-SCR脱硝活性。由此还可看出,Ce-HPW-TiO2(CM)和Ce-HPW-TiO2(UCM)虽然脱硝活性差距一直很小,但中间煅烧过的催化剂即Ce-HPW-TiO2(CM)的活性还是优于Ce-HPW-TiO2(UCM)的活性。
-
1) NO+O2共吸附。采用原位FT-IR技术测定了TiO2、Ce-TiO2、Ce-HPW-TiO2(UCM)和Ce-HPW-TiO2(CM)上的NOx吸附物种,如图5所示。可以看出,图5中出现了归属于螯合双齿硝酸盐(1 035 cm−1)、单齿硝酸盐(1 076、1 294、1 354、1 367、1 383、1 536和1 679 cm−1)、双齿硝酸盐(1 163、1 305、1 543、1 566和1 580 cm−1)、桥联硝酸盐(1 646 cm−1)、单齿亚硝酸盐(1 408 cm−1)、气态吸附的NO(1 849 cm−1和1 907 cm−1)、亚硝鎓离子(NO+)(2 208 cm−1和2 237 cm−1)、亚硝酰阳离子(NO+)(2 334 cm−1)和[NO2]+物种(2 361 cm−1)[8, 20-27]的特征峰,这些特征峰的强度均随时间逐渐增强。TiO2(图5(a))催化剂在Ar吹扫10 min后,1 566 cm−1处吸收峰逐渐增强,而1 536 cm−1处的吸收峰强度逐渐降低。推测单齿硝酸盐不稳定,转化为双齿硝酸盐。Ce-TiO2的谱图与TiO2相似,但峰强度更高(图5(b)),这表明Ce的负载促进了TiO2对NOx的吸附能力。由图5(c)可以明显地看出,在将NO+O2引入反应池1 min后,立即出现了具有不同构型的硝酸盐物质。在Ar吹扫10 min后,1 850 cm−1和1 901 cm−1处的特征峰消失,这种现象归因于NO的气态吸附。图5(c)与图5(b)得到的硝酸盐的特征峰不同,表明HPW的添加改变了由Ce-TiO2吸附形成的硝酸盐物质。如图5(d)所示,Ce-HPW-TiO2(CM)与Ce-HPW-TiO2(UCM)催化剂上NO+O2共吸附的原位FT-IR谱图基本相同。可以观察到,位于2 334 cm−1和2 361 cm−1处的峰随时间增强,2 208 cm−1和2 237 cm−1处的特征峰在2 min后消失,这种现象可能是由于吸附的NO+和O2之间的反应。此外,图5(d)中的特征峰强度比图5(c)中的特征峰强度增强,表明在二次浸渍之前,煅烧样品有利于增加结晶度并由此增强吸附NOx的能力。由此可知,Ce和HPW的掺杂可以增加吸附在催化剂表面上的硝酸盐物质的数量。
2) C3H6吸附。图6为通入C3H6的过程中TiO2、Ce-TiO2、Ce-HPW-TiO2(UCM)和Ce-HPW-TiO2(CM)上吸附物种的动态变化情况。图6中的特征峰可归属于甲酸盐(1 368、1 382、1 598和2 885 cm−1)、乙酸盐(1 458、1 465、1 535、1 565、1 582和2 981 cm−1)、C―C振动(1 049、1 079、1 094、1 179、1 211和1 245 cm−1)、甲酸盐(COO−)振动特征峰(1 352 cm−1)、H2O(1 641 cm−1)、C3H6物种(1 658 cm−1)、碳酸盐(1 338、1 408、1 440、1 470和1 552 cm−1)、丙酮(1 670、1 682和1 723 cm−1)、CO2(2 357 cm−1)、烷烃的C―H伸缩振动峰(2 862、2 947和3 104 cm−1)、甲酸盐的COO−不对称伸缩振动和CH变形振动峰(2 954 cm−1)、碳氢化合物的C―C振动(1 299 cm−1)和未反应的丙烯(1 648 cm−1)[2, 8, 22, 28-32]。由图6可知,所有特征峰强度都随时间的增加而增强。如图6(a)所示,对于TiO2催化剂,Ar吹扫10 min后,1 368 cm−1和1 598 cm−1处的特征峰消失,同时出现1 582 cm−1处的新特征峰。这是由于甲酸盐稳定性较低,并转化成了乙酸盐。与TiO2相比,Ce-TiO2出现了更多的丙烯吸附物种(图6(a)),表明Ce的负载可促进TiO2对丙烯的吸附,从而产生更为丰富的C3H6吸附物种。Ce-HPW-TiO2(UCM)和Ce-HPW-TiO2(CM)催化剂对于C3H6的吸附具有相同的趋势。由图6(c)可以看出,以1 440 cm−1和1 470 cm−1为中心的特征峰强度先增加,然后逐渐减小,直到完全消失。同时出现了位于1 458 cm−1的特征峰,这可能是由于碳酸盐不稳定,转化为乙酸盐。与Ce-TiO2相比,添加HPW的催化剂可增加丙烯的吸附种类,且主要吸附物种由乙酸盐转变为丙酮。
3)预吸附的NO+O2物种与C3H6之间的反应。预吸附的NOx物质与C3H6在200 ℃下的反应的原位FT-IR谱图如图7(a)~图7(d)所示。由图7(a)可知,在TiO2上1 567 cm−1处,峰强逐渐减弱,证明在反应过程中消耗了双齿硝酸盐,而双齿硝酸盐和C3H6吸附物种之间进行反应得到的COO−,与预吸附的单齿硝酸盐共吸附,使得1 536 cm−1处的峰强增加。将气体切换为C3H6后,Ce-TiO2催化剂未检测到表面物质的明显变化。由此可知,负载Ce对于优化C3H6与预吸附的NO+O2物质之间的反应过程是无效的。
Ce-HPW-TiO2(UCM)在预吸附NO+O2后,催化剂表面覆盖着各种NOx吸附物种,气体切换至C3H6后,归属于C―H弯曲振动的位于1 099 cm−1处的特征峰强度在前10 min内逐渐增加,随后减小。同时单齿硝酸盐的强度逐渐下降甚至消失,2 886 cm−1和2 962 cm−1处的O―H特征峰也消失,表明吸附的NOx物种在这一反应过程中被消耗了。另外部分出现了由于C3H6氧化产生的H2O(1 639 cm−1)、双齿碳酸盐(1 663 cm−1)、丙酮的C=O伸缩振动(1 679 cm−1)、甲酸盐的COO−不对称伸缩振动+CH变形振动(2 951 cm−1)和CH伸缩振动(2 980和3 095 cm−1)[8, 22, 32-33]等新峰,而且各特征峰的强度随时间的增加逐渐增强。但双齿硝酸盐物种相对稳定,因此,推断双齿硝酸盐在Ce-HPW-TiO2(UCM)催化剂上的反应是惰性的。Ce-HPW-TiO2(CM)与Ce-HPW-TiO2(UCM)的红外谱图基本相似,通入C3H6后,归属于C―H弯曲振动(1 100 cm−1)的特征峰在前10 min逐渐增强,随后减弱。同时归属于C―C振动(1 031 cm−1)的特征峰在10 min后增强。表明C―H弯曲振动是在10 min内通入丙烯产生的,之后,该物质与吸附的NOx物种进行反应而被消耗,同时这一反应过程也伴随着C―C振动的形成和积累。此外,单齿硝酸盐和双齿硝酸盐强度逐渐减弱甚至消失,位于2 889 cm−1和2 962 cm−1处的O―H特征峰也逐渐消失,说明NOx吸附物种被消耗了。在这一过程中,出现了归属于乙酸盐、在C3H6氧化过程中产生的H2O(1 638 cm−1)、亚硝酸乙酯(1 673 cm−1)、甲酸盐的COO−不对称伸缩振动+CH的变形振动和C―H伸缩振动(2 978和3 099 cm−1)[8, 28, 29, 33]的新特征峰,而且它们的强度随时间的延长而增强。随后1 673 cm−1处的峰位移动到归属于丙酮C=O伸缩振动[32]的1 681 cm−1处,表明亚硝酸乙酯与吸附的C3H6物种进行了反应。观察到位于2 361 cm−1处的峰强略微增加,这要归因于[NO2]+和CO2的共吸附。可以看出,负载HPW后,催化剂吸附形成的硝酸盐物种基本都可以与丙烯物种反应,在催化剂上形成了丰富的活性物质,参与到C3H6-SCR反应中。
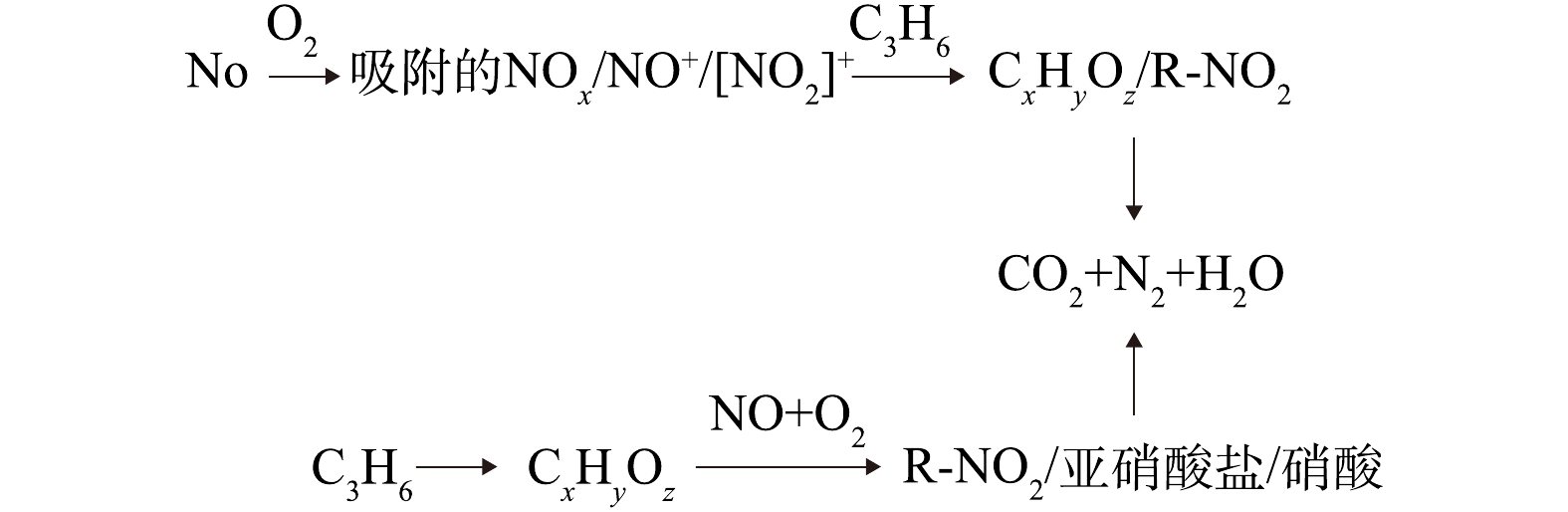
4)预吸附的C3H6物种与NO+O2之间的反应。NO+O2与预吸附的C3H6吸附物种在4种催化剂上反应的原位FTIR谱图如图8(a)~图8(d)所示。TiO2上发生的瞬态反应如图8(a)所示,归属于C―C振动的峰强度降低,同时归属于单齿硝酸盐(1 355和1 385 cm−1)、亚硝酸盐(1 406 cm−1)和双齿硝酸盐(1 534、1 563和1 583 cm−1)[8, 22, 30]的峰强在原峰的基础上增加,表明吸附的C3H6物种和相应的NOx吸附物种的共吸附。在1 299、2 860和2 949 cm−1处的峰保持相对稳定。Ce-TiO2与TiO2反应趋势相同,主要特征峰位置也相同。通入NO+O2后,归属于C―C振动和表面碳酸盐的峰强大大降低。同时单齿硝酸盐(1 353和1 384 cm−1)、亚硝酸盐(1 408 cm−1)、双齿硝酸盐(1 536、1 549、1 564和1 580 cm−1)和CO2(2 353 cm−1)[8, 22, 28, 30]的峰在原C3H6吸附物种峰的基础上逐渐增强,表明C3H6吸附物种和相应的NOx吸附物种存在共吸附现象。在反应过程中,1 299、1 408~1 536、1 641~1 723、2 862和2 954 cm−1处的其他特征峰保持相对稳定,这表明Ce-TiO2催化剂上吸附的大多数C3H6物种在SCR反应中是惰性的。CO2特征峰增强可能是由于在引入NO+O2后,气体吸附的C3H6被氧化成CO2。所以Ce的负载对催化剂上C3H6吸附物种的反应活性有轻微的促进作用。
通入NO+O2后,Ce-HPW-TiO2(UCM)上几乎所有C3H6吸附物种,如甲酸盐、乙酸盐、未反应的丙烯、烷烃的C―H伸缩(vCH)和C―H振动都减少,甚至消失,表明它们在与NOx吸附物种反应的过程中被消耗了。同时观察到在1 845和1 904 cm−1处,出现了归属于表面NO[8]的新特征峰。位于1 569 cm−1处的乙酸盐特征峰在4 min后向低波数移动,最终与逐渐增强的1 564 cm−1处的双齿硝酸盐重叠[8]。1 641 cm−1(未反应的丙烯)和1 689 cm−1(丙酮羧基C=O振动和单齿硝酸盐的共吸附[34])处的特征峰强度分别逐渐减小和增大,4 min后,这2个特征峰均与逐渐增强的1 668 cm−1处的
NO−3 [35]物种重叠。在反应过程中,没有出现亚硝酸盐,但出现了NO−3 。与Ce-TiO2相比,HPW的负载丰富了参与C3H6-SCR反应的活性物质。Ce-HPW-TiO2(CM)趋势与Ce-HPW-TiO2(UCM)基本相同,甲酸盐、乙酸盐、未反应的丙烯、烷烃的C―H不对称伸缩振动和C―H振动都减少甚至消失,且位于1 458 cm−1和1 565 cm−1处的乙酸盐分别在通入NO+O2 2 min和6 min后,逐渐向较低的波数移动,并逐渐与位于1 421 cm−1(亚硝酸盐)和1 556 cm−1(双齿硝酸盐)[8, 30]的特征峰重叠。归属于丙酮中羧基C=O振动的1 676 cm−1处特征峰一直在增强,推断这是由于通入NO+O2后丙酮和亚硝酸乙酯[29]共吸附造成的。上述结果表明Ce-HPW-TiO2(CM)上几乎所有的C3H6吸附物种都参与了C3H6-SCR反应。基于上述实验结果,我们提出了可能存在的反应机理(如图9所示)。NO被O2氧化为NO2,并在催化剂活性位上以硝酸盐的形式储存,随后在C3H6-SCR反应中,与丙烯(通过产生甲酸盐,乙酸盐,亚硝酸乙酯和丙酮)产生相互作用,最终得到氧化产物(CO2,H2O)。同时还可能存在一条平行反应路径,即C3H6通过吸附在催化剂活性位上产生甲酸盐、丙酮和未反应的丙烯,这些吸附物种会进一步与NO和O2进行反应,最终得到CO2、H2O和N2。
-
1)用浸渍法制备了掺杂Ce、HPW的TiO2催化剂,在模拟烟气的情况下,考察了各催化剂在中低温区的脱硝活性,可以看出,Ce-HPW-TiO2(CM)具有最佳的NO转化率,大约为73%。
2)催化剂吸附NO和C3H6的原位FT-IR分析表明,Ce和Keggin结构HPW的掺杂可以促进催化剂表面硝酸盐物质和丙烯吸附物种的形成。
3)在瞬态反应过程中发现,无论是在预吸附NO还是在预吸附C3H6的情况下,Ce-HPW-TiO2(CM)催化剂表面发生的反应活性最高,基本上所有的NO或C3H6吸附物种都参与到了C3H6-SCR反应中。
4)基于瞬态反应,推出Ce-HPW-TiO2(CM)催化剂的反应机理,发现该反应的中间体主要为无机硝酸盐、甲酸盐、乙酸盐和有机氮化合物。
5) Ce-HPW-TiO2(UCM)和Ce-HPW-TiO2(CM)的活性测试、表征结果和FT-IR的研究表明,在浸渍负载HPW后、负载Ce之前对样品进行1次煅烧,有利于第2种活性物质的成功负载且有利于优化催化剂活性。
Ce-HPW-TiO2催化剂利用C3H6选择性催化还原NO反应的机理
Mechanism of C3H6 selective catalytic reduction reaction of NO by Ce-HPW-TiO2 catalyst
-
摘要: 为提高C3H6-SCR脱硝催化剂的低温脱硝性能,采用浸渍法合成了几种由铈和Keggin型磷钨酸改性的TiO2催化剂。在模拟烟气的实验条件下,考察了不同催化剂在150~350 ℃的脱硝活性,通过XRD、FT-IR和SEM对催化剂的理化性质进行了分析,并且通过原位FT-IR探究并对比了不同催化剂在吸附NO和C3H6时产生的吸附物种。结果表明:铈和磷钨酸的共掺杂大大提高了TiO2催化剂在中低温区的脱硝效率;Ce和H3PW12O40(HPW)成功负载于TiO2上,负载的HPW也保留了其Keggin结构,而且负载后的催化剂表面更加光滑,形态更加规则,分散性更好;原位FT-IR结果显示:Ce和HPW的掺杂可以促进催化剂表面硝酸盐物质和丙烯吸附物种的形成;同时发现无论是在预吸附NO还是在预吸附C3H6的情况下,Ce-HPW-TiO2(CM)催化剂表面发生的反应活性最高。由此提出了Ce-HPW-TiO2(CM)催化剂的反应机理,发现其反应中间体主要为无机硝酸盐、甲酸盐、乙酸盐和有机氮化合物。Abstract: In order to improve the low-temperature denitration performance of C3H6-SCR denitration catalyst, several modified TiO2 catalysts with ceria and Keggin-type tungstophosphoric acid were synthesized by impregnation method. Under the experimental conditions of simulated flue gas, the denitrification activities of different catalysts at 150~350 ℃ was investigated. The physicochemical properties of these catalysts were analyzed by XRD, FT-IR and SEM. In situ FT-IR was used to investigate and compare the adsorbed species produced on different catalysts when they absorbed NO and C3H6. The results showed that the co-doping of cerium and phosphotungstic acid greatly improved the denitration efficiency of TiO2 catalyst in the middle and low temperature regions. Ce and H3PW12O40(HPW) were successfully supported on TiO2, and the supported HPW also retained its Keggin structure, and the catalysts after loading had more smooth surface, more regular shape, and better dispersion. The in situ FTIR spectra showed that the doping of Ce and HPW could promote the formation of nitrate and propylene adsorbed species on the surface of catalysts. At the same time, the surface of Ce-HPW-TiO2(CM) catalyst had the highest reactivity whether it was pre-adsorbed with NO or C3H6. Therefore, the reaction mechanism of Ce-HPW-TiO2(CM) catalyst was proposed, and the reaction intermediates were mainly inorganic nitrate, formate, acetate and organic nitrogen compounds.
-
Key words:
- phosphotungstic acid /
- NO reduction /
- C3H6-SCR /
- in situ FT-IR /
- mechanism
-
挥发性有机化合物(VOCs)作为PM2.5和臭氧的重要前驱体,已经对大气环境质量和人体健康造成直接和间接的危害[1-2],VOCs废气治理成为了近年来的环境热点话题之一。在VOCs废气的众多处理技术中,催化燃烧技术因处理效率高、能耗低、二次污染小而在工业上应用广泛[3]。为达到VOCs催化燃烧所需温度,工业上多采用电加热使VOCs废气达到起燃温度。由于电加热是采用热传导的加热方式,因而对大气量的VOCs废气加热时能耗巨大。另外,VOCs催化燃烧时,持续的高温环境会使催化剂活性组分烧结而影响VOCs降解效果[4]。微波加热应用于VOCs催化燃烧是一种新技术,它利用电磁波的选择性仅对催化剂进行加热,因而能耗低且催化剂受热均匀、快速[5]。而且,微波对催化剂活性组分的热点效应有利于引发VOCs的催化燃烧,同时微波的偶极极化作用还可降低VOCs的反应阈能而促进其氧化降解[6]。卜龙利等[7]、姚泽等[8]利用吸波型催化剂证实了微波催化燃烧VOCs效果优于电加热催化燃烧。
微波催化燃烧技术的关键是高效而稳定的催化剂。过渡金属氧化物催化剂种类丰富、经济性好、不易中毒,但其对于某些难降解有机物的催化活性差、低温活性以及热稳定性有待提高[9]。贵金属催化剂具有催化活性高、高温下稳定性好和适用范围广的优点,但也存在资源稀少和价格高昂的问题[10]。目前,市面上较为常见的是铜锰铈三元金属氧化物催化剂。胡旭睿[11]研究证实,铜锰铈氧化物催化剂对芳香烃类、醇类、酮类等有机物具有良好的氧化性能,但其矿化效果不佳且对芳香烃的降解效果低于醇、酮类。贵金属Pt、Pd等在低温时对芳烃类和3个碳以上的直链烷烃活化能力强[12]。已有研究证实,将贵金属与过渡金属复合可以增强催化剂活性。SHI等[13]通过还原法和离子交换法合成了Pt /Ce-USY催化剂,其对1,2-二氯乙烷的催化活性高于Pt/USY催化剂,Pt与CeO2之间的相互作用抑制了碳物质的沉积,从而增强了催化剂的耐久性。LEE等[14]将贵金属金、钯沉积在CeO2表面,证实适量Au的加入使得Pd/CeO2催化剂活性增强。CHEN等[15]合成了Pd/Fe3O4催化剂用于去除CO,当沉积适量Fe3O4时,CO氧化的起燃温度明显降低。因此,本研究在铜锰铈三元催化剂基础上复合微量贵金属Pt,以期提高催化剂对芳烃类VOCs的活化能力,进而提高总VOCs的去除效果。
本研究选取油墨印刷VOCs废气中含量最多的2种物质甲苯和乙酸乙酯作为目标污染物,以蜂窝状堇青石为载体制备Pt复合铜锰铈(CMC)负载型催化剂(Pt-CMC/堇青石),重点考察Pt复合前后催化剂对模拟VOCs废气催化燃烧效果的差异,以探究低含量贵金属添加对铜锰铈氧化物催化剂催化活性的影响程度。
1. 材料与方法
1.1 实验原料与仪器
蜂窝状正方体堇青石(7目,边长150 mm);硝酸铜、硝酸锰(50 wt%)、硝酸铈、氯铂酸,分析纯;硅溶胶,分析纯。超声波清洗器(KQ3200,昆山市超声仪器有限公司);电热鼓风干燥箱(101-3AB,天津泰斯特仪器有限公司); 箱式电阻炉(SX-4-10,天津泰斯特仪器有限公司);单模腔微波装置(ZDM-2,南京汇研微波系统工程有限公司);气相色谱仪(GC/FID,6890 N,美国安捷伦科技公司);泵吸式VOC检测仪(XS-2000-VOC,希思智能科技);扫描电子显微镜(JSM-6510LV型,日本电子);比表面积及孔径分析仪(V-sorb2800P型,北京金埃谱公司);X射线衍射仪(X’Pert型,荷兰帕纳科)。
1.2 催化剂制备与表征
本研究采用等体积浸渍法进行负载型催化剂的制备,即载体吸水率测试基础上配置适量体积的浸渍液、然后浸渍液被载体完全吸收的一种催化剂制备方法。首先,将正方体堇青石切割成圆柱型载体(D×L=27×150 mm),称重并测其吸水率,根据吸水率确定助剂硅溶胶的用量;其次,按3∶3∶1的质量负载比例分别称取铜(2.5%(质量分数))、锰、铈的金属盐,称取一定量的硅溶胶以及少量氯铂酸(Pt的质量分数0.01%),加入去离子水超声震荡30 min配制完全溶解的浸渍液;最后,将堇青石载体置于浸渍液中将浸渍液完全吸收,样品静置风干后放入烘箱中80 ℃过夜烘干,电阻炉内500 ℃煅烧4 h,自然冷却后即可制得Pt-CMC/堇青石催化剂。不加入氯铂酸、仅加入氯铂酸和其余步骤均相同情况下,可分别制得CMC/堇青石、Pt /堇青石催化剂。
利用扫描电子显微镜观察催化剂表面形貌及活性组分分布状况,借助BET测试仪分析催化剂的比表面积及孔径孔体积,使用X射线衍射仪测试活性组分的晶体结构及晶粒大小。
1.3 实验装置与方法
实验装置如图1所示,整个装置分为配气、催化燃烧和尾气净化3部分。在配气系统中,空气依次通过变色硅胶柱和活性炭柱去除水分和有机物,其流量通过气体流量计控制;实验时采用微量注射泵将液态甲苯和乙酸乙酯(按一定比例混合)以恒定速度连续均匀地注入三口烧瓶中,三口烧瓶被电加热套加热而将有机物气化,气化的甲苯和乙酸乙酯被空气带出并在混合瓶内混合均匀。实验开始前催化剂置于石英管内,石英管填装催化剂的长度为360 mm,锥形收口的长度是20 mm,上下两端开口外径分别为32 mm和9 mm,壁厚2 mm;石英管固定在单模腔微波装置上形成固定床反应器,实验开始后微波通过单模腔持续辐照在固定床上,催化剂吸波升温达到VOCs催化燃烧温度;石英管下端连接混合瓶,通入的VOCs气体在固定床上发生催化燃烧反应,床中插入的热电偶探针与温度显示仪连接以实时监测床层温度变化,石英管上下分别设进、出气采样口。催化燃烧后的VOCs尾气分别通过缓冲瓶和装有乙醇和碱液的尾气吸收瓶,净化后的废气经通风橱排空。
 图 1 微波催化燃烧VOCs实验装置示意图Figure 1. Schematic diagram of the experimental device for microwave catalytic combustion of VOCs
图 1 微波催化燃烧VOCs实验装置示意图Figure 1. Schematic diagram of the experimental device for microwave catalytic combustion of VOCs本研究利用CMC/堇青石、Pt-CMC/堇青石和Pt /堇青石催化剂在处理气量0.12 m3·h−1、气时空速(gas hourly space velocity,GHSV)1 500 h−1条件下,考察了不同微波功率(30、40、50、60、70 W)和不同VOCs浓度(1 000、1 500、2 000 mg·m−3)下甲苯和乙酸乙酯的催化燃烧效率,分析微波催化燃烧甲苯反应动力学及Pt复合影响,比较3种催化剂的催化活性及Pt复合对总VOCs去除效率的提升作用,探究Pt-CMC/堇青石催化剂对甲苯、乙酸乙酯和总VOCs催化活性的稳定性。
1.4 分析方法
本研究主要利用气相色谱仪对甲苯和乙酸乙酯的进气和出气质量浓度进行定量分析,气相色谱检测条件为[16]:柱箱初始温度100 ℃,以20 ℃·min−1的速率升温至180 ℃,保持3 min,加热器温度为190 ℃,检测器温度300 ℃;设定分流比为50∶1;尾气吹脱用氮气,流量30 mL·min−1。
使用手持泵吸式VOC检测仪对反应器进气和出气的总VOCs浓度进行检测,从而分析反应过程中总VOCs的催化燃烧效率。文中数据为2次平行实验的平均值,以此消除实验偶然性误差的影响。
2. 结果与讨论
2.1 催化剂表征
1) BET。根据表1数据可知,与堇青石载体相比,CMC/堇青石与Pt-CMC/堇青石2种催化剂的比表面积有不同程度的增加,这表明催化剂的吸附能力也相应增强。与堇青石载体相比,CMC/堇青石催化剂总孔容相对减小,平均孔径有所增大。推测其原因是,活性组分负载后,载体表面的部分孔隙被活性组分填充和覆盖,载体结构的原有孔道被拓宽。Pt-CMC/堇青石催化剂活性晶粒间相互积聚生成了包括微孔和介孔在内的新孔道,催化剂的比表面积及孔体积都明显增大,这有利于污染物分子在催化剂表面的吸附与活化。因此,Pt的复合在一定程度上增强了催化剂的吸附活化性能。
表 1 堇青石载体和2种催化剂的比表面积、孔体积及孔径数据表Table 1. Data table of specific surface area, pore volume and pore diameter of Cordierite and two catalysts供试样品 比表面积/(m2·g−1) 微孔面积/(m2·g−1) 孔体积/(cm3·g−1) 总孔容/(cm3·g−1) 平均孔径/nm 堇青石 0.11 ND ND 0.30 36.79 CMC/堇青石 0.66 ND ND 0.09 41.23 Pt-CMC/堇青石 2.95 1.11 0.000 47 0.38 36.85 备注:ND为未检出。 | Show Table DownLoad:
CSV
DownLoad:
CSV
2) XRD。如图2所示,2 θ在10°~40°之间存在着较为密集的堇青石特征衍射峰。对比XRD谱图可以发现,活性组分的负载和制备时的高温煅烧不会改变堇青石自身的晶体结构;与堇青石载体相比,CMC/堇青石和Pt-CMC/堇青石催化剂的特征衍射峰强度减弱,活性晶粒的平均尺寸有不同程度的减小。活性组分负载后堇青石特征峰峰强减弱的原因可能是活性组分一定程度上覆盖和屏蔽了堇青石表面的特征峰[17];贵金属Pt复合后,金属氧化物等活性组分晶体特征峰的峰高有微弱增大,这可能是不同尖晶石活性组分共存所致。6个处于17.5°、21.9°、28.7°、34.1°、36.7°、38.3°位置较为明显的特征峰,均归属于铜锰铈及其金属氧化物,这说明在贵金属Pt复合前后,铜锰铈金属氧化物都是微波催化燃烧VOCs的主要活性组分。在堇青石载体、CMC/堇青石催化剂和Pt-CMC/堇青石催化剂上,3种催化剂的晶粒平均尺寸分别为50.7、13.9和14.5 nm。Pt复合后,催化剂晶粒尺寸变化较小,而比表面积却从0.66 m2·g−1增大到2.95 m2·g−1,由此可以说明贵金属Pt的复合使活性颗粒的分散性增大[18]。
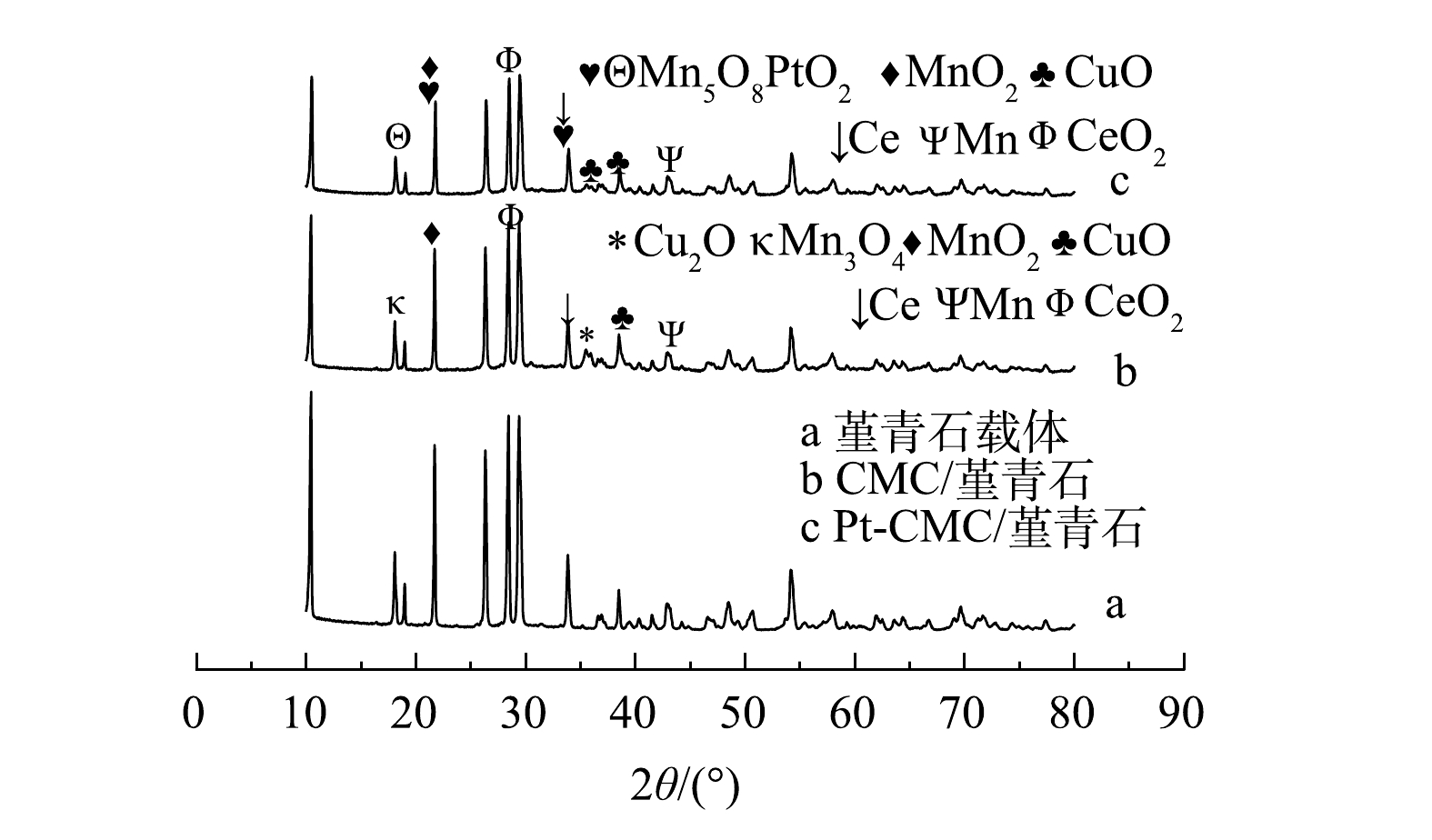 图 2 堇青石载体及CMC/堇青石、Pt-CMC/堇青石催化剂的XRD谱图Figure 2. XRD spectra of Cordierite support and CMC/Cordierite and Pt-CMC/Cordierite catalysts
图 2 堇青石载体及CMC/堇青石、Pt-CMC/堇青石催化剂的XRD谱图Figure 2. XRD spectra of Cordierite support and CMC/Cordierite and Pt-CMC/Cordierite catalysts结合特征峰组分分析可知,少量贵金属Pt的添加改变了催化剂上铜和锰的晶相。贵金属Pt复合前,锰以+4价的MnO2和中间价态的Mn3O4形式存在;贵金属Pt复合后,部分金属锰的价态升高,以Mn5O8形式存在,且Cu2O的晶体在掺杂后未检测到,检测出以+4价存在的PtO2。有文献报道,Cu、Mn金属离子价态升高,可以增强催化剂的活性[19]。
3) SEM。图3给出堇青石载体和CMC/堇青石、Pt-CMC/堇青石催化剂的表面形貌。可以看出,堇青石载体呈明显的层状结构,表面孔隙较多且分布均匀,有利于活性组分的负载。CMC/堇青石催化剂表面存在着较多微米尺寸的活性颗粒,不均匀地分布在载体表面;部分活性组分经高温煅烧后团聚成大块与堇青石结构连结在一起,填充载体原有孔隙的同时也产生了新的孔隙。Pt-CMC/堇青石催化剂表面的活性颗粒在载体表面上分布更均匀、分散度更高。对比Pt复合前后催化剂的微观形貌,可以看出,Pt的复合改变了活性颗粒的分散性,活性物质的再分散可能导致催化剂的吸附性改变。
 图 3 堇青石载体、CMC/堇青石与Pt-CMC/堇青石催化剂的微观表面形貌Figure 3. Microscopic surface morphology of Cordierite, CMC/Cordierite and Pt-CMC/Cordierite catalyst
图 3 堇青石载体、CMC/堇青石与Pt-CMC/堇青石催化剂的微观表面形貌Figure 3. Microscopic surface morphology of Cordierite, CMC/Cordierite and Pt-CMC/Cordierite catalyst2.2 催化剂性能实验
1)吸波升温曲线。图4为堇青石载体、CMC/堇青石、Pt-CMC/堇青石和Pt/堇青石在200 W微波功率辐照下的吸波升温曲线。由图4可见,堇青石载体温度几乎不变,这表明其吸波性能很差;而Pt/堇青石催化剂在15 min内温度也仅升高了3 ℃,其原因是贵金属具有高热稳定性,且吸波性能差。CMC/堇青石和Pt-CMC/堇青石催化剂升温效果明显好于前2种,微波辐照15 min时分别升温至300和355 ℃,说明铜锰铈及其金属氧化物具有良好的吸波性能,是2种催化剂吸波升温的主要原因;Pt复合催化剂升温速率更快,这可能是Pt的复合提高了催化剂上活性晶粒的分散性,降低了金属对微波的反射,以及颗粒尺寸变大增强了微波辐照时的局部热点效应,从而使得催化剂的吸波升温性能提高[20-21]。催化剂的快速升温有助于微波热点效应和高温活性区的出现,从而有助于甲苯和乙酸乙酯的快速氧化与彻底降解。
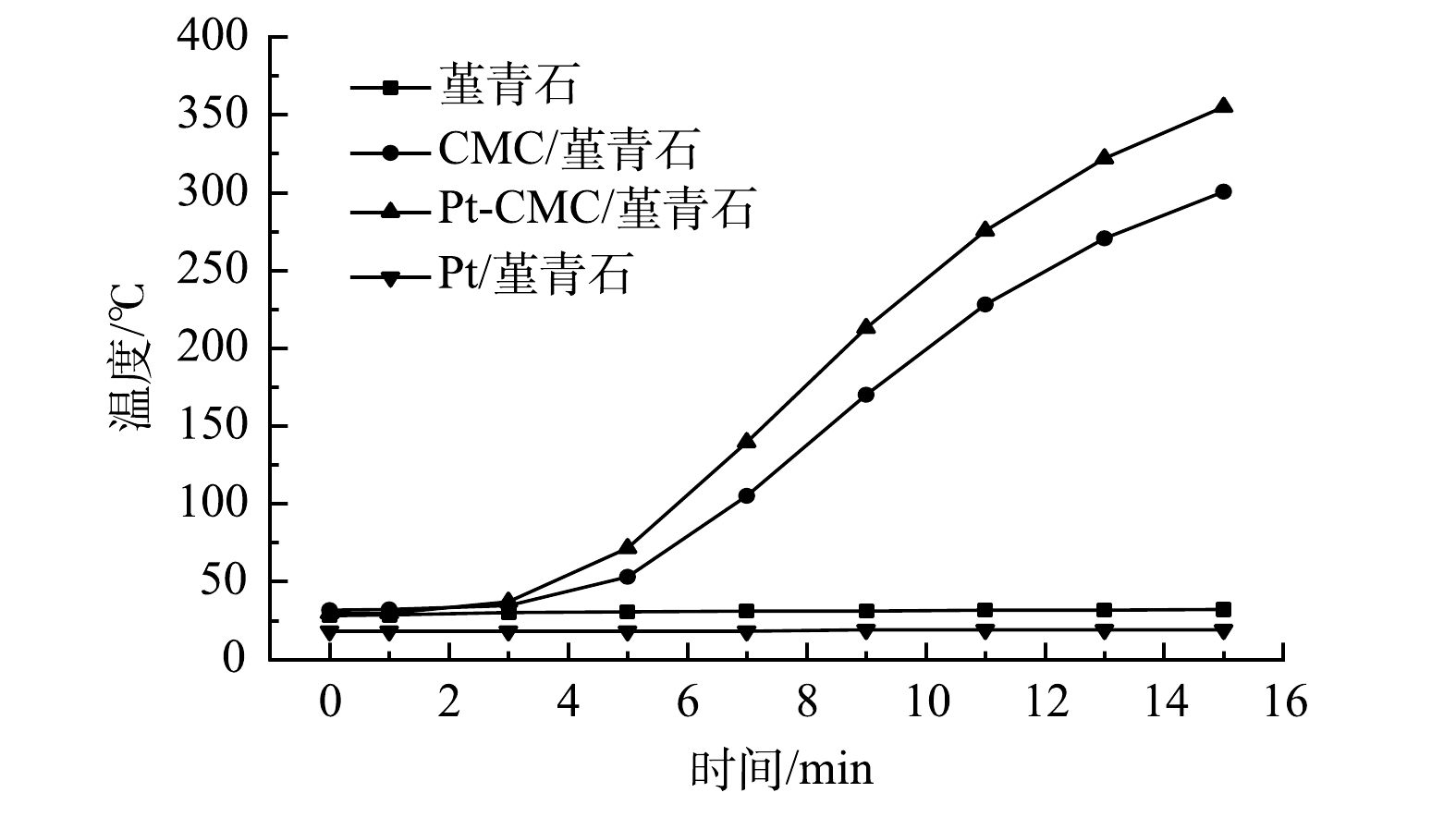 图 4 堇青石载体及3种催化剂的吸波升温曲线Figure 4. Wave absorption heating curve of Cordierite and three catalysts
图 4 堇青石载体及3种催化剂的吸波升温曲线Figure 4. Wave absorption heating curve of Cordierite and three catalysts2)催化剂活性。在甲苯和乙酸乙酯的进气浓度分别为1 000和500 mg·m−3,进气量为0.12 m3·h−1,微波功率分别选取30、40、50、60、70 W的条件下,开展CMC/堇青石和Pt-CMC/堇青石催化剂对甲苯和乙酸乙酯双组分VOCs废气的催化燃烧性能试验,结果如图5所示。图5中纵坐标C/C0为反应实时浓度与进气浓度比值(图6同),可直观表示降解效率并使数据间具有可比性。
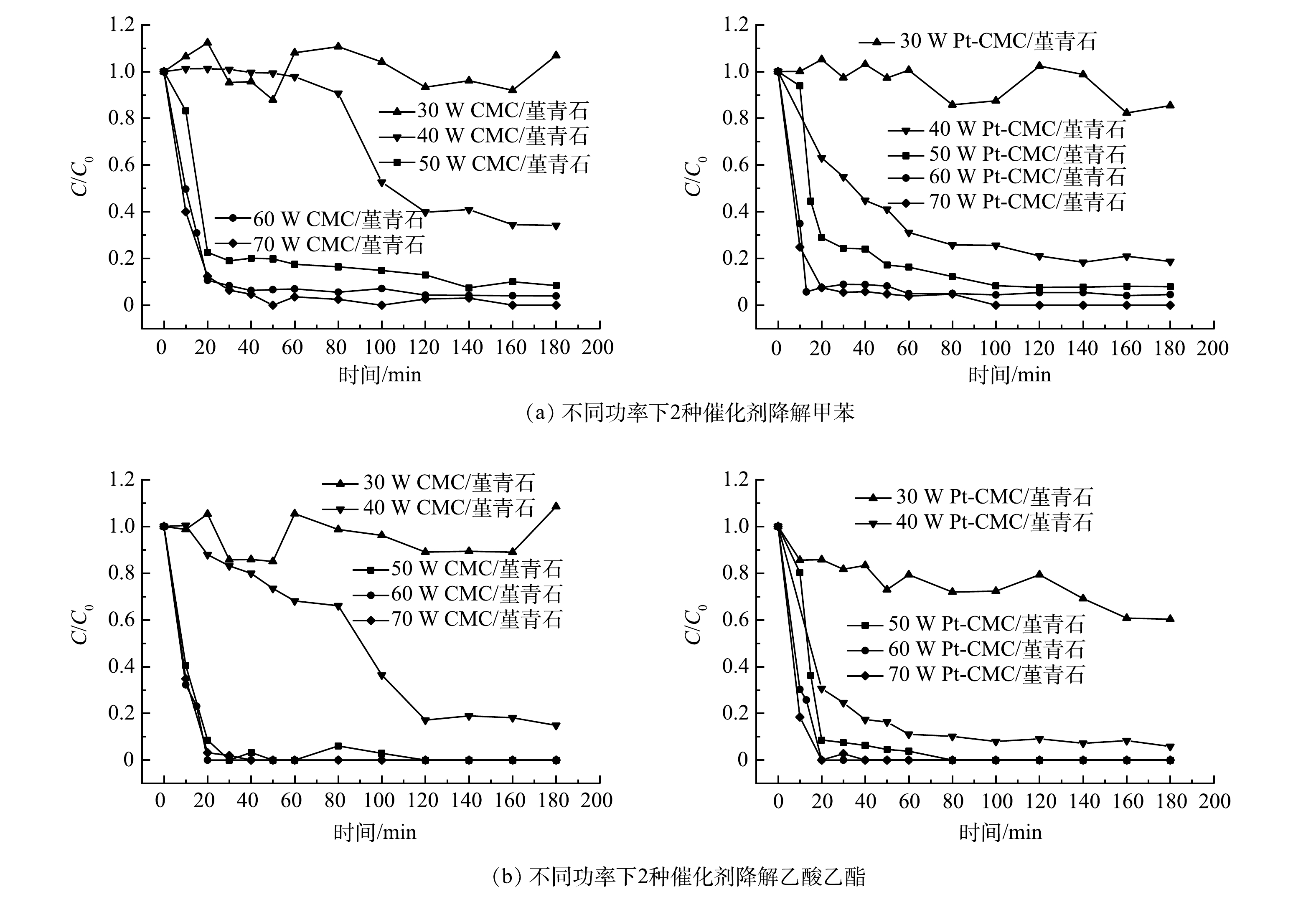 图 5 不同微波功率下CMC/堇青石和Pt-CMC/堇青石催化燃烧甲苯和乙酸乙酯效率曲线Figure 5. CMC/Cordierite and Pt-CMC/Cordierite catalytic combustion efficiency curves of toluene and ethyl acetate under different microwave power
图 5 不同微波功率下CMC/堇青石和Pt-CMC/堇青石催化燃烧甲苯和乙酸乙酯效率曲线Figure 5. CMC/Cordierite and Pt-CMC/Cordierite catalytic combustion efficiency curves of toluene and ethyl acetate under different microwave power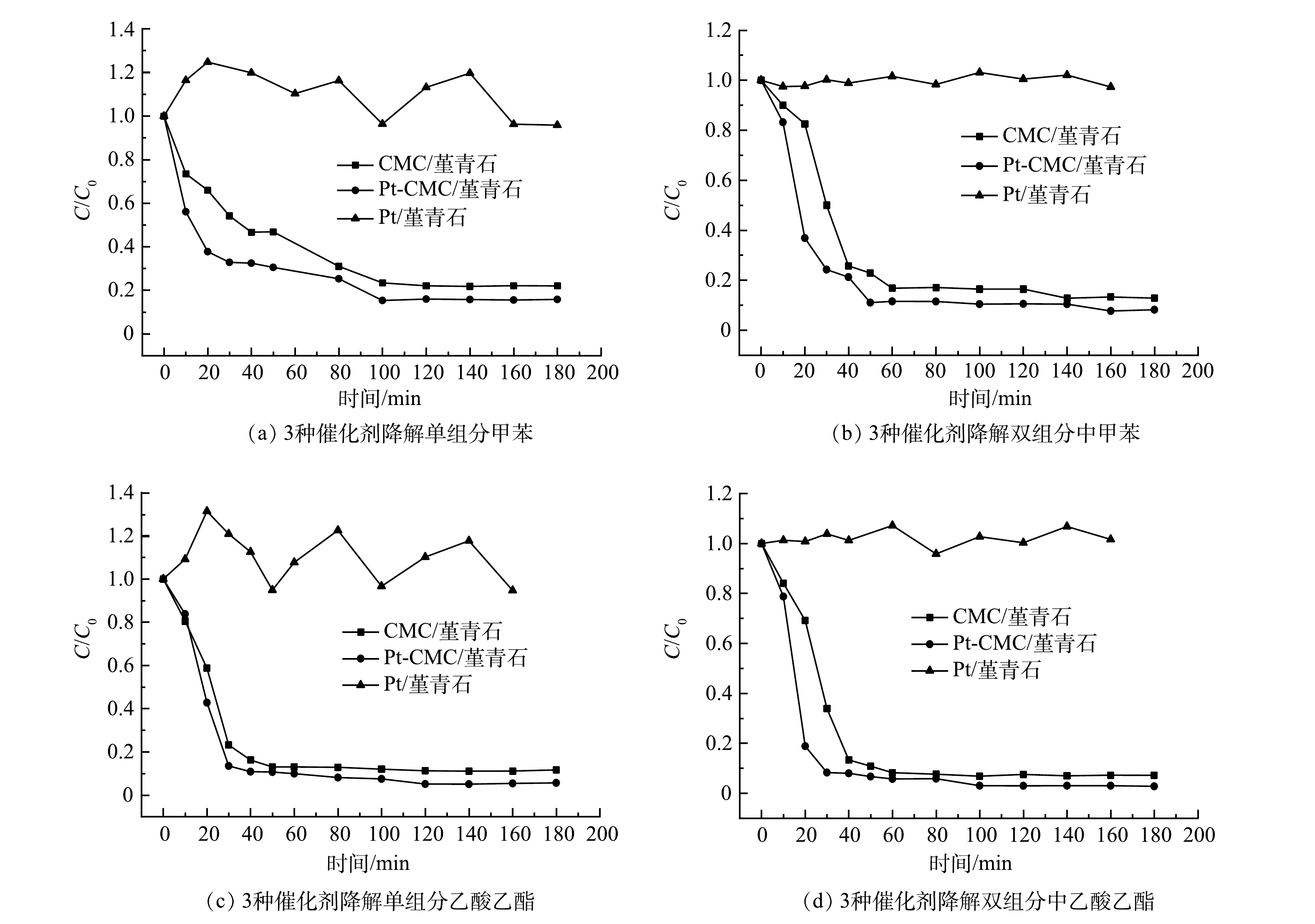 图 6 不同催化剂对甲苯和乙酸乙酯的催化活性比较Figure 6. Comparison of catalytic activity of three catalysts to toluene and ethyl acetate
图 6 不同催化剂对甲苯和乙酸乙酯的催化活性比较Figure 6. Comparison of catalytic activity of three catalysts to toluene and ethyl acetate由图5可见,微波功率30 w时,2种催化剂对甲苯和乙酸乙酯的降解呈不稳定态势,其原因是床层温度偏低(CMC/堇青石床层温度低于66 ℃),催化剂对污染物的去除以吸附-脱附为主。随着微波功率增大,甲苯和乙酸乙酯的降解效率也随之升高,但2种催化剂对甲苯和乙酸乙酯降解效率的差异却由大变小:40 W时,催化剂对甲苯和乙酸乙酯降解效率的差异最大;70 W时,这种差异最小,此时固定床温度高,污染物在催化剂表面完全燃烧,温度的作用掩盖了Pt的复合作用。
同一微波功率下,降解甲苯与乙酸乙酯时, Pt-CMC/堇青石催化剂表现出更高的VOCs催化活性。40 W微波功率下,Pt-CMC/堇青石对甲苯和乙酸乙酯的降解效率分别为82%和93%,而CMC/堇青石降解甲苯和乙酸乙酯的效率分别为66%和82%。推测其原因是,Pt-CMC/堇青石的比表面积相比CMC/堇青石大幅增加,从而有利于甲苯与乙酸乙酯的吸附与活化;同时,活性组分更好的分散性使得Pt-CMC/堇青石升温更快,而高温则有利于VOCs的催化燃烧。如果要使CMC/堇青石催化剂对甲苯和乙酸乙酯的降解效率与Pt-CMC/堇青石相同,则须提高微波功率至47 W。由此可见,Pt的复合提高了催化剂的低温催化活性,有效降低了VOCs的催化燃烧成本。
贺丽娜等[16]证明了甲苯的微波催化燃烧为假一级反应。因此,依据一级反应动力学方程ln(C0/C)=kt,对CMC/堇青石和Pt-CMC/堇青石3种催化剂在微波功率分别为50、60、70 W时降解双组分VOCs废气中甲苯的反应动力学进行线性拟合,相应方程与参数如表2所示。结果表明,CMC/堇青石和Pt-CMC/堇青石微波催化燃烧甲苯的反应符合假一级反应动力学,同时R2值随微波功率的增加而增大,此时一级动力学拟合程度越高,证明高温下的催化燃烧反应为一级反应。反应速度常数k值对比发现,微波功率越大,k值越大,催化反应速度越快;微波功率60 W时,Pt-CMC/堇青石降解甲苯的k值为0.129,明显高于CMC/堇青石的0.111 9。可见,Pt的复合增强了甲苯的吸附活化作用而加快了反应速率,进而提高CMC活性组分对甲苯的微波催化燃烧效率。
表 2 微波催化燃烧甲苯反应动力学Table 2. Reaction kinetics of microwave catalytic combustion of toluene功率条件 催化剂 ln(C0/C)=kt k R2 50 W Pt-CMC/堇青石 y=0.0594x−0.0204 0.0594 0.9828 60 W CMC/堇青石 y=0.1119x−0.1398 0.1119 0.9553 60 W Pt-CMC/堇青石 y=0.129x−0.0797 0.129 0.9887 70 W CMC/堇青石 y=0.1045x−0.0428 0.1045 0.995 70 W CMC/堇青石 y=0.1299x+0.031 0.1299 0.9983 | Show TableDownLoad:
CSV
3)催化剂比较。图6为微波功率40 W、进气量0.12 m3·h−1,甲苯和乙酸乙酯浓度各为1 000 mg·m−3条件下,CMC/堇青石、Pt-CMC/堇青石和Pt/堇青石催化剂降解单组分甲苯、乙酸乙酯及二者混合的效率曲线。无论是单、双组分甲苯和乙酸乙酯的催化燃烧降解,均是Pt-CMC/堇青石的活性最高,CMC/堇青石次之,而Pt/堇青石则未表现出催化活性。对于Pt/堇青石而言,其在微波辐照下几乎不升温,从而无法为VOCs的催化燃烧提供所需的温度条件,因此甲苯和乙酸乙酯几乎不降解。无论是甲苯或乙酸乙酯的单组分降解,还是双组分废气降解,乙酸乙酯的降解效率均高于甲苯。以Pt-CMC/堇青石为例,单组分甲苯和乙酸乙酯的降解效率分别为84%和95%(此时床层温度相近)。乙酸乙酯比甲苯更易于降解的原因可能为:一是乙酸乙酯等含氧有机物比甲苯更易被催化剂吸附活化;二是乙酸乙酯上的C-O键比甲苯的C-H键键能低而更易断裂[22-23]。
比较分析图6中甲苯和乙酸乙酯的降解效率发现:对于单组分甲苯和乙酸乙酯的降解,Pt-CMC/堇青石较CMC/堇青石有约7%和6%的提升;对于甲苯与乙酸乙酯的双组分降解,Pt-CMC/堇青石较CMC/堇青石有约5%和4%的提升。由此可见,Pt复合对甲苯降解效率的提升大于乙酸乙酯。分析认为,CMC/堇青石对于一些C-H键较弱、吸附性好的含氧有机物如乙酸乙酯、丙酮等具有良好的活化性能,但催化氧化C-H键较强的芳香类物质的能力则稍弱,而贵金属Pt活化C-H键的能力很强,弥补了铜锰铈金属氧化物活化苯环能力的不足[24]。此外,CMC/堇青石和Pt-CMC/堇青石对甲苯和乙酸乙酯双组分废气的降解效率明显高于各自单组分,其原因是双组分VOCs废气的初始浓度更高,催化燃烧时放出更多热量使得床层温度更高,从而有利于VOCs的氧化降解。
4)总VOCs去除。微波功率40 W时,对催化剂催化燃烧1 000 mg·m−3甲苯的总VOCs去除率进行测试,其结果和床层温度变化如图7所示。由图7(a)可见,Pt/堇青石的总VOCs去除率为负值且波动幅度大,CMC/堇青石和Pt-CMC/堇青石的总VOCs去除率分别为25%和40%,Pt的复合将总VOCs的去除效率提高了15%。如图7(b)所示,Pt/堇青石的不吸波使其床层温度为常温,催化燃烧反应未发生,甲苯在催化剂表面进行吸附-脱附动态平衡过程,因而其总VOCs去除率波动大且多为负值;Pt-CMC/堇青石床层升温速率快于CMC/堇青石,稳定后的床层温度高出CMC/堇青石床层30 ℃,这是总VOCs去除效率提高的主要原因。分析认为,Pt的复合在提升催化剂吸波升温能力的同时,其对甲苯C-H键的活化能力也有助于提高总VOCs的去除效率。随着微波功率的增加,床层温度急剧升高,总VOCs去除率也随之升高,催化剂对总VOCs去除效率的差别也随之减小,Pt复合的影响被床层温度影响掩盖而体现不出。因此,Pt的复合提高了催化剂的低温催化活性,有助于低温下VOCs的催化氧化。
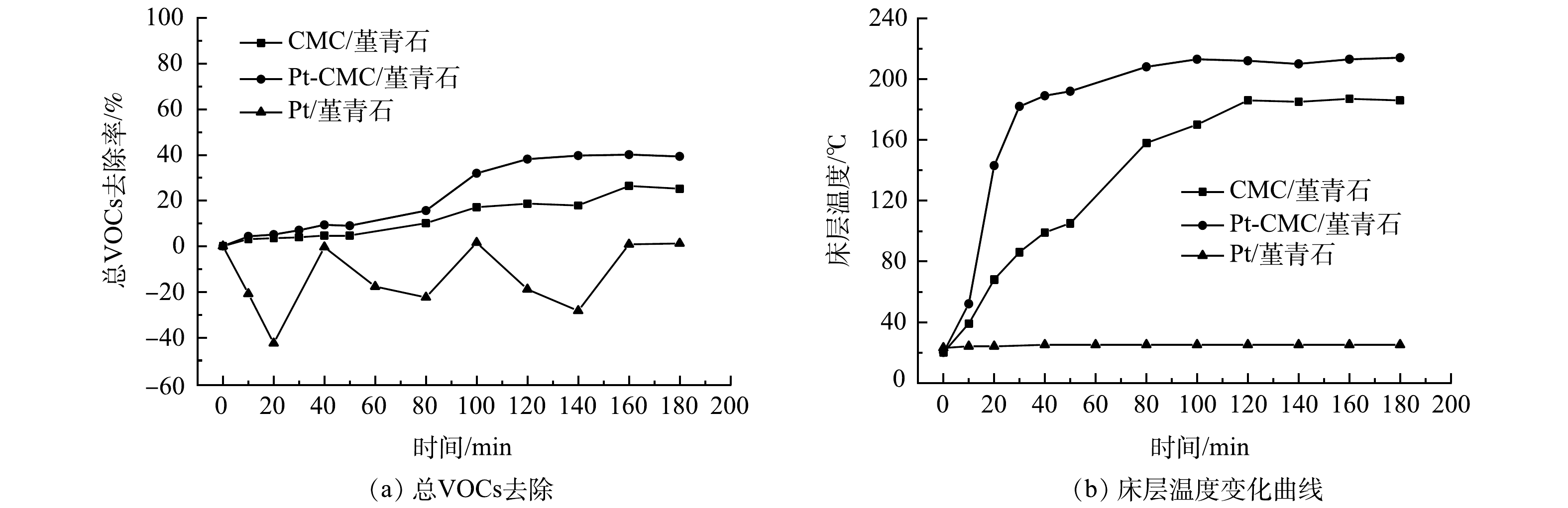 图 7 不同催化剂的总VOCs去除效率及床层温度曲线Figure 7. The total VOCs removal efficiency and bed temperature curves of the three catalysts
图 7 不同催化剂的总VOCs去除效率及床层温度曲线Figure 7. The total VOCs removal efficiency and bed temperature curves of the three catalysts5)稳定性实验。根据图5的实验结果可知,微波功率70 W时Pt-CMC/堇青石催化剂对甲苯和乙酸乙酯的催化效果最好,同时总VOCs的去除率也最高。因此,在微波功率70 W、进气量0.12 m3·h−1、双组分气体中甲苯和乙酸乙酯初始浓度各为1 000 mg·m−3和500 mg·m−3条件下对Pt-CMC/堇青石催化剂进行了连续7次(每次实验3 h)的稳定性测试,结果如图8所示。
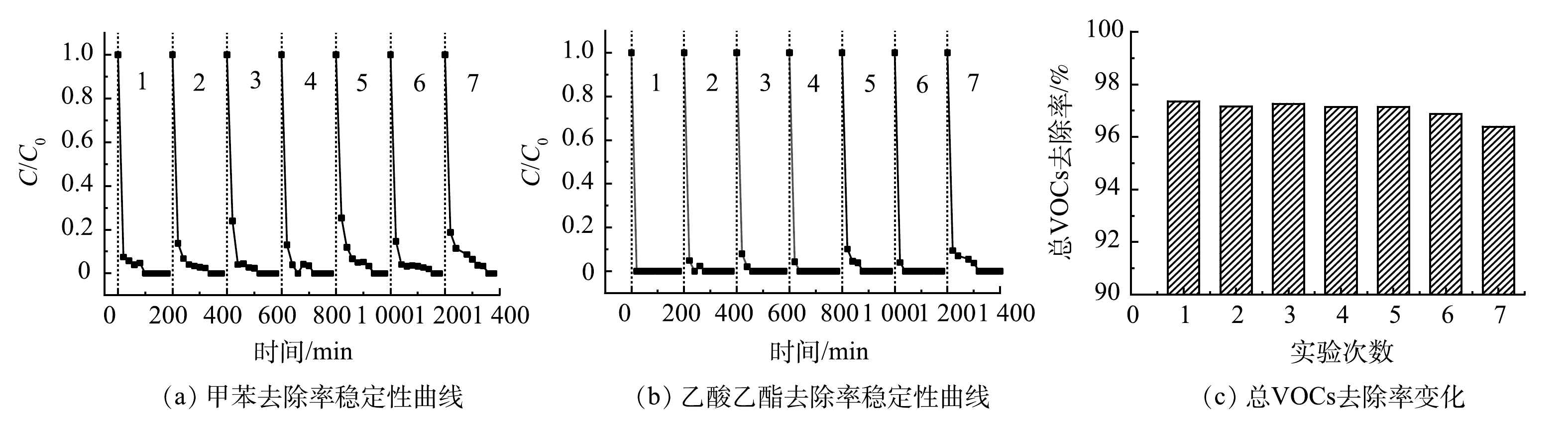 图 8 Pt-CMC/堇青石催化燃烧甲苯和乙酸乙酯稳定性测试结果Figure 8. Pt-CMC/Cordierite catalytic combustion toluene and ethyl acetate stability test results
图 8 Pt-CMC/堇青石催化燃烧甲苯和乙酸乙酯稳定性测试结果Figure 8. Pt-CMC/Cordierite catalytic combustion toluene and ethyl acetate stability test results如图8(a)、8(b)所示,连续稳定性试验中,甲苯和乙酸乙酯被完全去除,乙酸乙酯的去除效率较甲苯更稳定,催化剂展示出良好的催化活性和稳定性。图8(c)的总VOCs去除率从97.5%略微降至96.5%,表明Pt-CMC/堇青石对双组分VOCs废气具有高的矿化效率与稳定性。微波功率70 W的稳定性实验中,催化剂床层温度320 ℃,高于40 W时的210 ℃,可见,床层温度的升高是提升总VOCs去除率的有效手段之一。连续的高温稳定性实验,会引起催化剂孔隙结构的微变和活性颗粒的团聚,进而引起双组分VOCs(特别是甲苯)去除效率的波动;然而,Pt复合增强了催化剂表面活性颗粒的分散性,增大了催化剂的比表面积与孔容,从而有效提高了催化剂活性,保证了催化剂性能的稳定。
3. 结论
1)等体积浸渍法制备的Pt-CMC/堇青石催化剂比CMC/堇青石催化剂具有更大的活性颗粒尺寸,Pt的再分散作用使得催化剂表面活性组分分布更加均匀;Pt的复合提高了催化剂的孔隙率,进而有利于VOCs分子在催化剂表面的吸附与活化;Pt复合后的尖晶石活性组分峰有微弱增大,Cu、Mn价态升高而使催化剂活性增强。
2)复合Pt后,催化剂的吸波升温能力有一定的增强,这与吸波活性组分分布均匀、散热能力增强有关。反应动力学分析表明,Pt的复合加快了催化燃烧反应速率,高温下甲苯的催化燃烧反应为一级反应;复合Pt后的催化剂其低温催化活性明显提高,Pt-CMC/堇青石催化剂表面总VOCs去除效率比CMC/堇青石提高15%。
3)对于Pt-CMC/堇青石和CMC/堇青石催化剂而言,乙酸乙酯的降解效率高于甲苯,可认为含氧的乙酸乙酯比甲苯更易被催化剂吸附活化,C-O键比甲苯的C-H键键能低而更易断裂;Pt复合对甲苯降解效率的提高大于乙酸乙酯,其原因是Pt对苯环中C-H键的活化能力强而有助于苯环的氧化与开环。
-
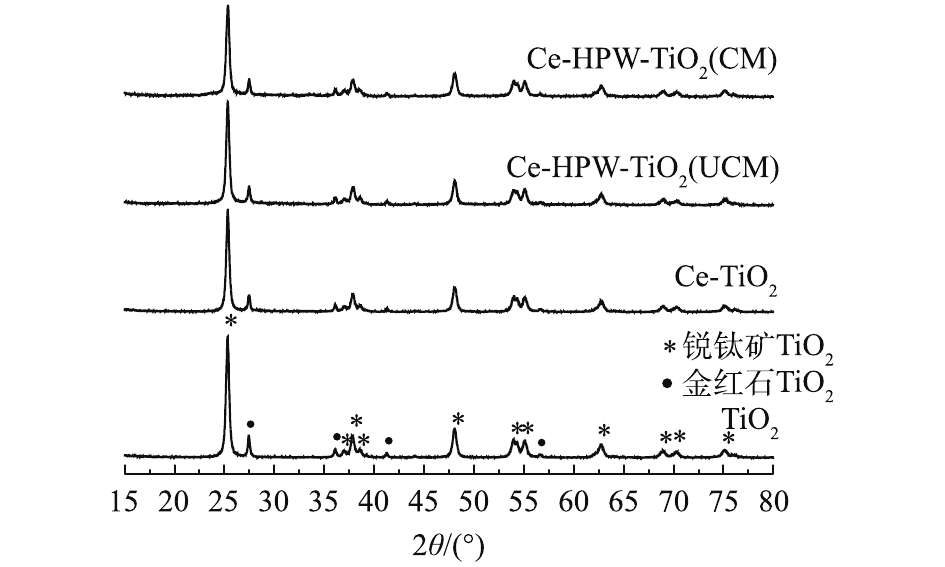
图 1 TiO2、Ce-TiO2、Ce-HPW-TiO2 (UCM)和Ce-HPW-TiO2 (CM)催化剂的XRD图
Figure 1. XRD patterns of the catalysts of TiO2, Ce-TiO2, Ce-HPW-TiO2 (UCM) and Ce-HPW-TiO2 (CM)

图 2 TiO2、Ce-TiO2、Ce-HPW-TiO2 (UCM)和Ce-HPW-TiO2 (CM)催化剂的SEM图
Figure 2. SEM images of the catalysts of TiO2, Ce-TiO2, Ce-HPW-TiO2 (UCM) and Ce-HPW-TiO2 (CM)
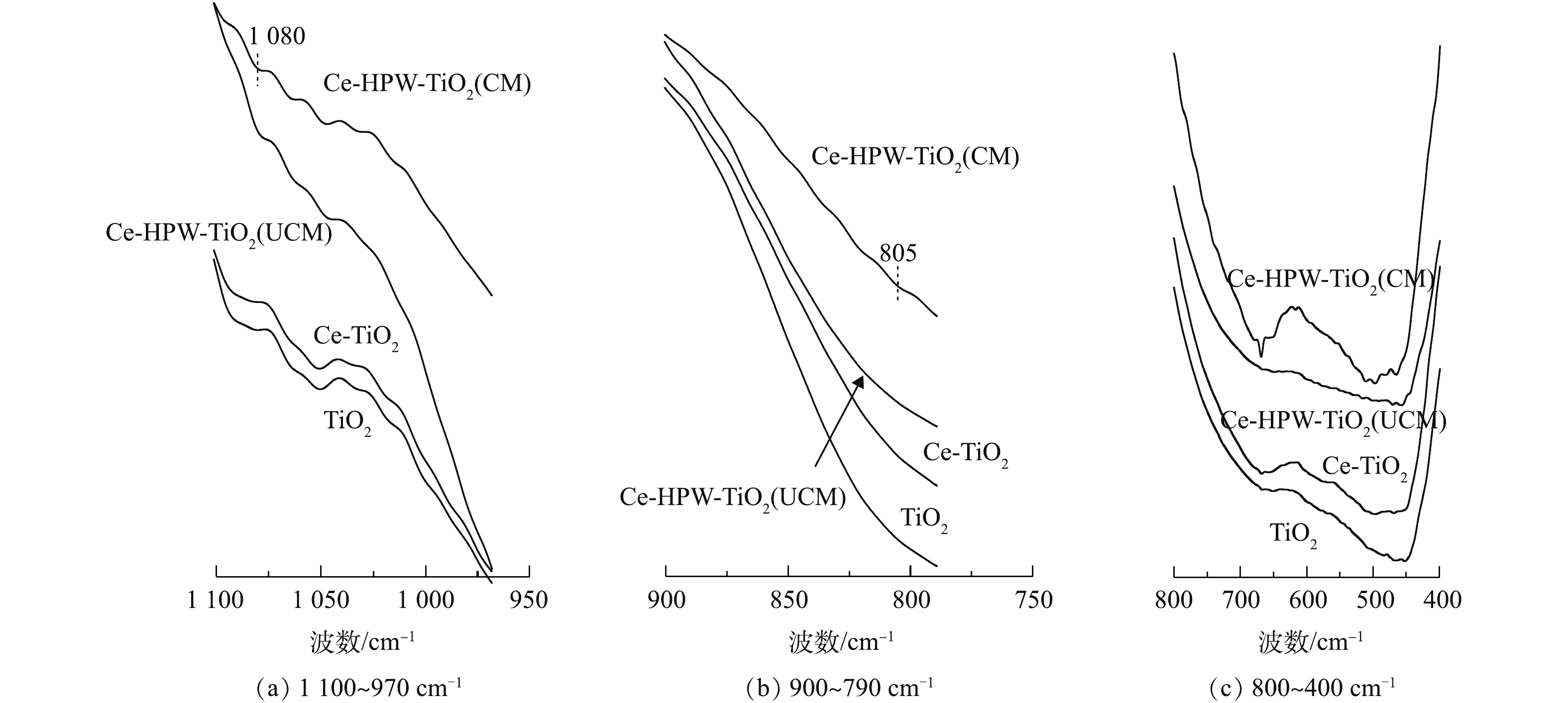
图 3 TiO2、Ce-TiO2、Ce-HPW-TiO2 (UCM)和Ce-HPW-TiO2 (CM)催化剂的FT-IR图
Figure 3. FT-IR spectra of the catalysts of TiO2, Ce-TiO2, Ce-HPW-TiO2 (UCM) and Ce-HPW-TiO2 (CM)
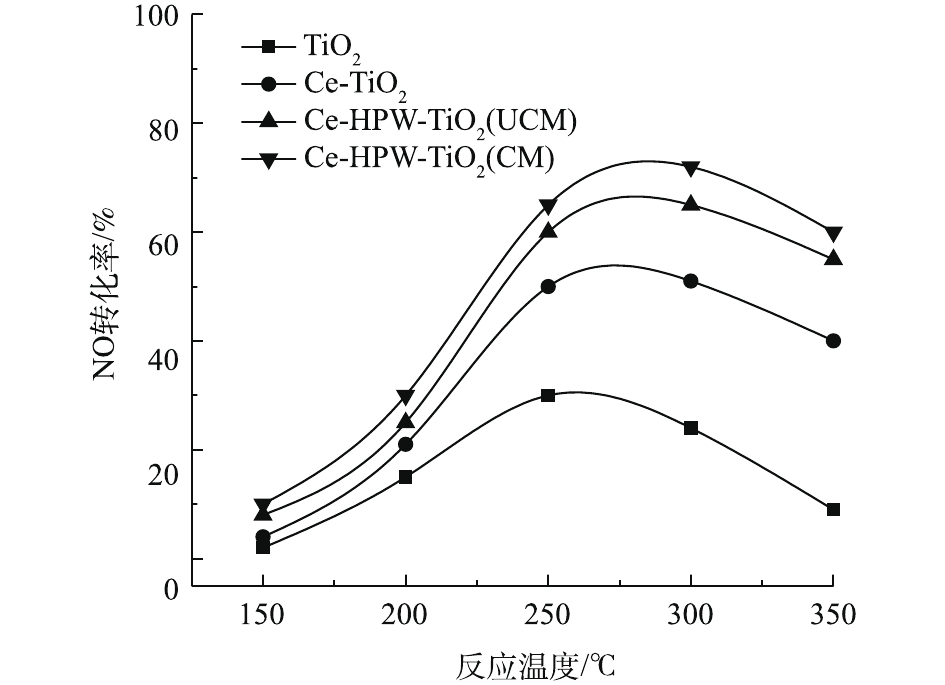
图 4 TiO2、Ce-TiO2、Ce-HPW-TiO2 (UCM)和Ce-HPW-TiO2 (CM)催化剂在不同反应温度下的NO转化率
Figure 4. NO conversion of the catalysts of TiO2, Ce-TiO2, Ce-HPW-TiO2 (UCM) and Ce-HPW-TiO2 (CM) at the different reaction temperatures
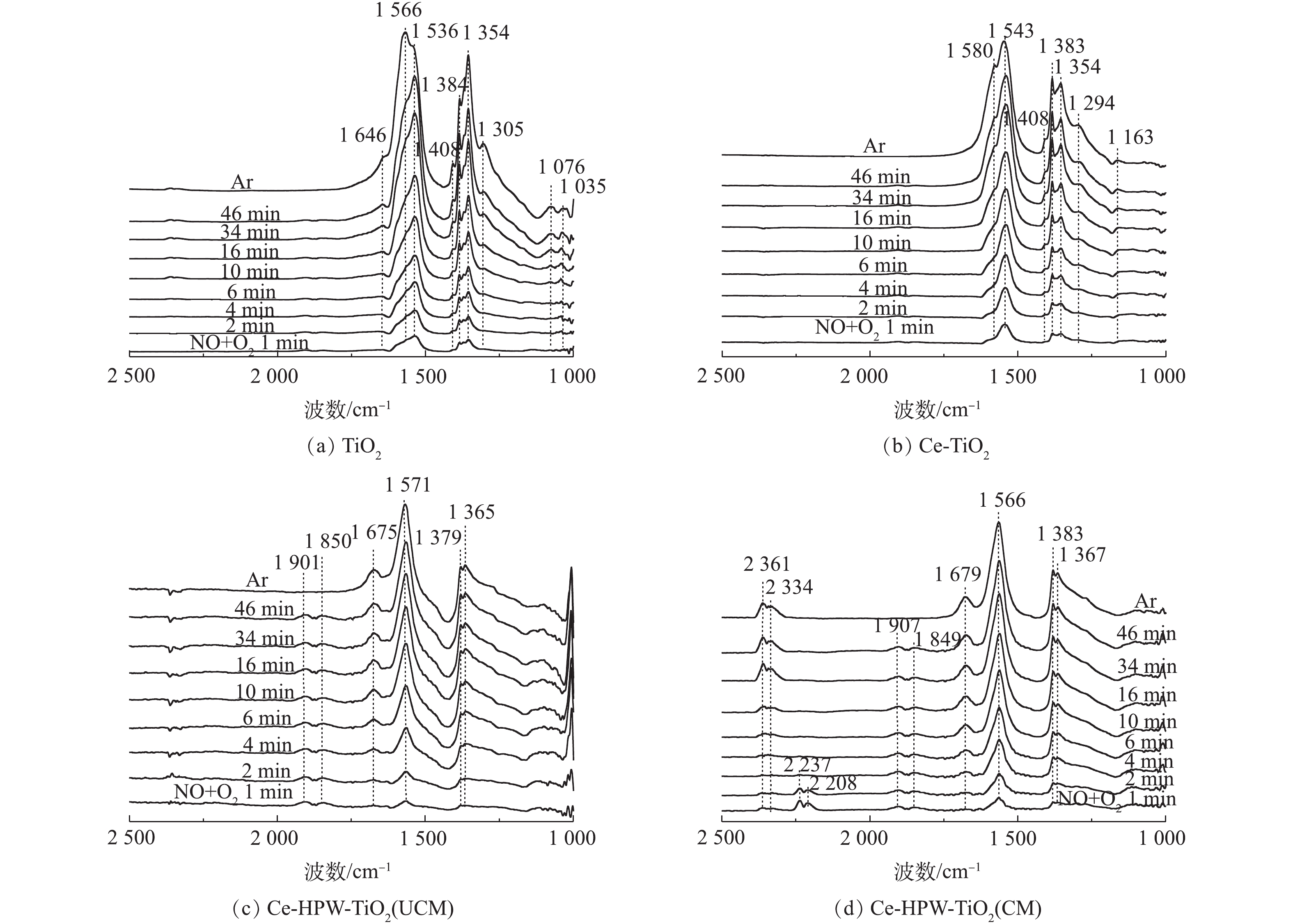
图 5 200 ℃下TiO2、Ce-TiO2、Ce-HPW-TiO2 (UCM)和Ce-HPW-TiO2 (CM)催化剂吸附NO+O2的原位红外谱图
Figure 5. In situ FT-IR spectra of NO+O2 adsorption on TiO2, Ce-TiO2, Ce-HPW-TiO2 (UCM) and Ce-HPW-TiO2 (CM) samples at 200 ℃

图 6 200 ℃下TiO2、Ce-TiO2、Ce-HPW-TiO2 (UCM)和Ce-HPW-TiO2 (CM)催化剂吸附C3H6的原位红外谱图
Figure 6. In situ FT-IR spectra of C3H6 adsorption on TiO2, Ce-TiO2, Ce-HPW-TiO2 (UCM) and Ce-HPW-TiO2 (CM) samples at 200 ℃
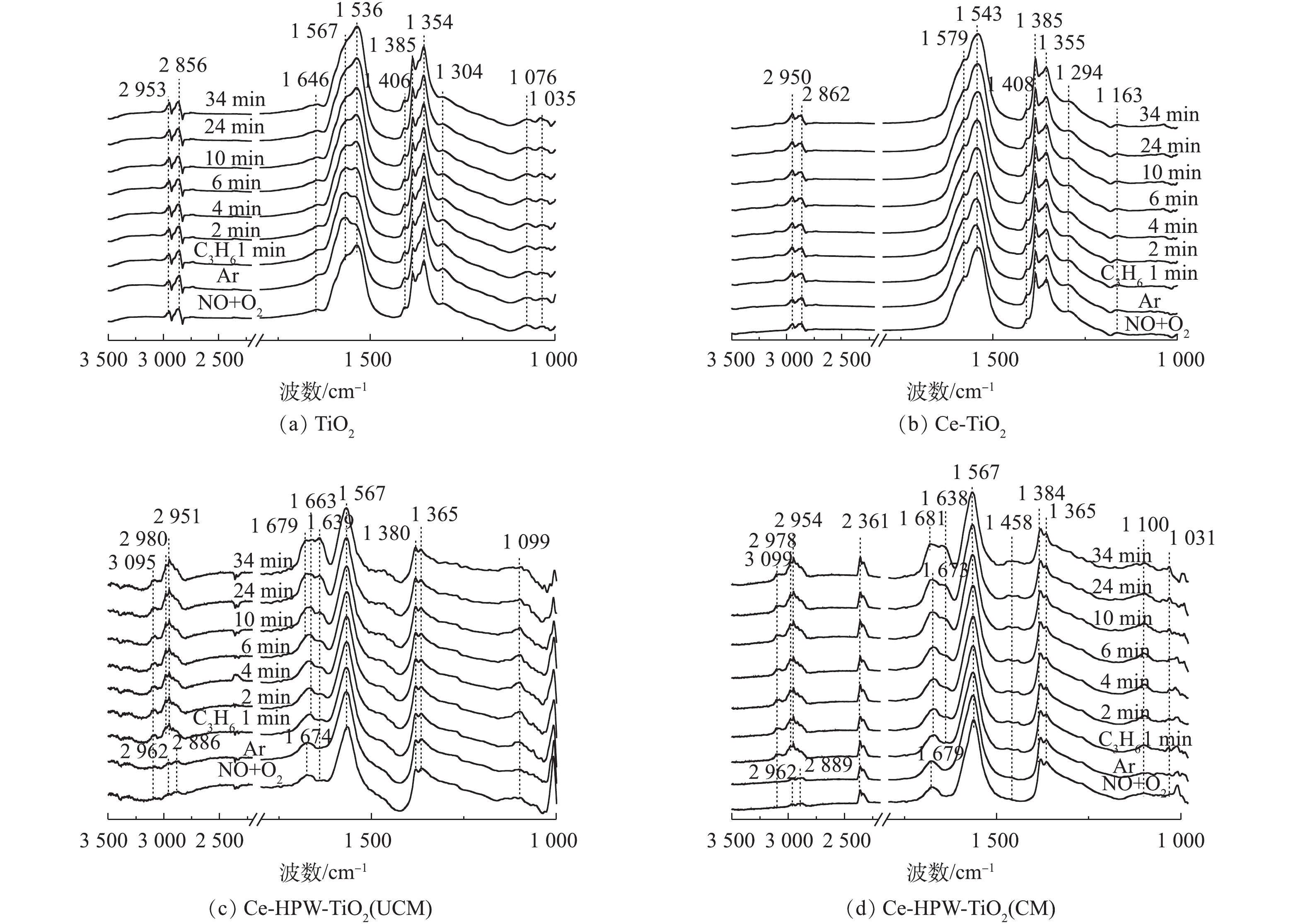
图 7 200 ℃下TiO2、Ce-TiO2、Ce-HPW-TiO2 (UCM)和Ce-HPW-TiO2 (CM)催化剂C3H6与预吸附的NOx物种反应的原位红外谱图
Figure 7. In situ FT-IR spectra of C3H6 reacted with pre-adsorbed NOx species on TiO2, Ce-TiO2, Ce-HPW-TiO2 (UCM) and Ce-HPW-TiO2 (CM) samples at 200 ℃
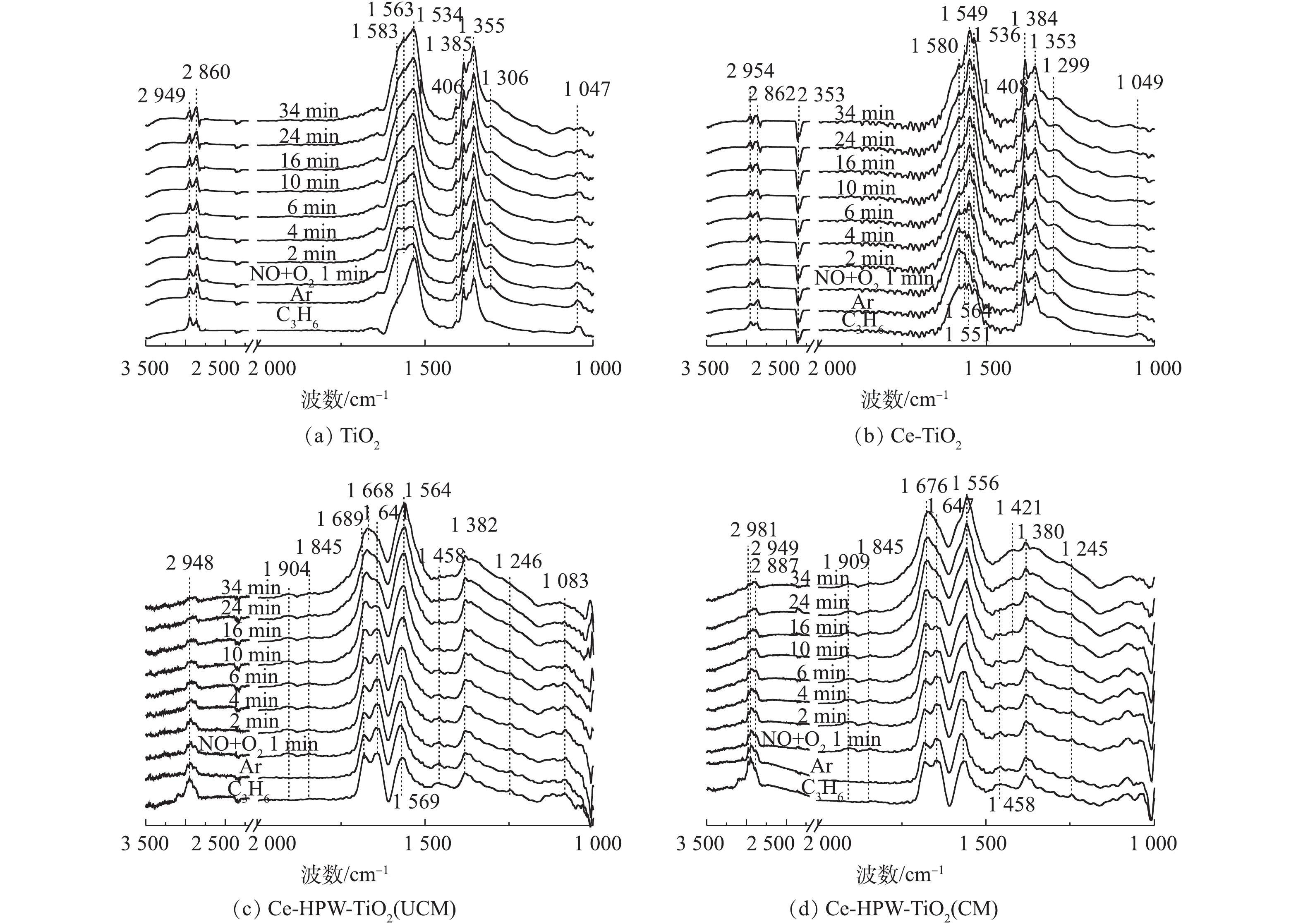
图 8 200 ℃下TiO2、Ce-TiO2、Ce-HPW-TiO2 (UCM)和Ce-HPW-TiO2 (CM)催化剂NO+O2与预吸附的C3H6物种反应的原位红外谱图
Figure 8. In situ FT-IR spectra of NO+O2 reacted with pre-adsorbed C3H6 species on TiO2, Ce-TiO2, Ce-HPW-TiO2 (UCM) and Ce-HPW-TiO2 (CM) samples at 200 ℃
-
[1] MRAD R, COUSIN R, POUPIN C, et al. Propene oxidation and NO reduction over MgCu-Al(Fe) mixed oxides derived from hydrotalcite-like compounds[J]. Catalysis Today, 2015, 257: 98-103. doi: 10.1016/j.cattod.2015.02.020 [2] YUAN D L, LI X Y, ZHAO Q D, et al. A novel CuTi-containing catalyst derived from hydrotalcite-like compounds for selective catalytic reduction of NO with C3H6 under lean-burn conditions[J]. Journal of Catalysis, 2014, 309: 268-279. doi: 10.1016/j.jcat.2013.09.010 [3] AZIS M M, HÄRELIND H, CREASER D. On the role of H2 to modify surface NOx species over Ag-Al2O3 as lean NOx reduction catalyst: TPD and DRIFTS studies[J]. Catalysis Science & Technology, 2014, 5: 296-309. [4] 刘欣, 苏亚欣, 董士林, 等. Co/Fe/Al2O3/cordierite催化C3H6选择性还原NO的实验研究[J]. 燃料化学学报, 2018, 46(6): 743-753. doi: 10.3969/j.issn.0253-2409.2018.06.013 [5] KIM Y J, KWON H J, NAM I S, et al. High deNOx performance of Mn/TiO2 catalyst by NH3[J]. Catalysis Today, 2010, 151(3/4): 244-250. [6] PUTLURU S S R, MOSSIN S, RIISAGER A, et al. Heteropoly acid promoted Cu and Fe catalysts for the selective catalytic reduction of NO with ammonia[J]. Catalysis Today, 2011, 176(1): 292-297. doi: 10.1016/j.cattod.2010.11.087 [7] YAO S H, CHEN S, SHI Z L. Preparation and photocatalytic activity of Ce, H3PW12O40 co-doped TiO2 hollow fibers[J]. Chinese Journal of Chemical Physics, 2014, 27: 343-349. doi: 10.1063/1674-0068/27/03/343-349 [8] LIU J, LI X Y, ZHAO Q D, et al. Combined spectroscopic and theoretical approach to sulfur-poisoning on Cu-supported Ti-Zr mixed oxide catalyst in the selective catalytic reduction of NOx[J]. ACS Catalysis, 2014, 4(8): 2426-2436. doi: 10.1021/cs5005739 [9] 王淑勤, 武金锦, 杜志辉. Co-Ce共掺杂对TiO2催化剂室温可见光催化脱硝性能的影响[J]. 燃料化学学报, 2019, 47(3): 361-369. [10] JIN R B, LIU Y, WU Z B, et al. Low-temperature selective catalytic reduction of NO with NH3 over Mn-Ce oxides supported on TiO2 and Al2O3: A comparative study[J]. Chemosphere, 2010, 78(9): 1160-1166. doi: 10.1016/j.chemosphere.2009.11.049 [11] 宋忠贤. 固体酸改性CeO2催化剂的制备及其NH3-SCR机理研究[D]. 昆明: 昆明理工大学, 2017. [12] WENG X L, DAI X X, ZENG Q S, et al. DRIFT studies on promotion mechanism of H3PW12O40 in selective catalytic reduction of NO with NH3[J]. Journal of Colloid and Interface Science, 2016, 461: 9-14. doi: 10.1016/j.jcis.2015.09.004 [13] 宋淑美, 王睿. 具有低温活性的高效脱硝催化体系研究进展[J]. 现代化工, 2007, 27(s1): 108-112. [14] GÓMEZ-GARCÍA M A, PITCHON V, KIENNEMANN A, et al. Sorption-desorption of NOx from a lean gas mixture on H3PW12O40·6H2O supported on carbon nanotubes[J]. Topics in Catalysis, 2004, 30-31(1/2/3/4): 229-233. [15] PALACIO M, VILLABRILLE P I, ROMANELLI G P, et al. Ecofriendly liquid phase oxidation with hydrogen peroxide of 2,6-dimethylphenol to 2,6-dimethyl-1,4-benzoquinone catalyzed by TiO2-CeO2 mixed xerogels[J]. Applied Catalysis A: General, 2009, 359(1/2): 62-68. [16] XUE W L, ZHANG G W, XU X F, et al. Preparation of titania nanotubes doped with cerium and their photocatalytic activity for glyphosate[J]. Chemical Engineering Journal, 2011, 167(1): 397-402. doi: 10.1016/j.cej.2011.01.007 [17] WANG Y J, CUI Y X, SUO Y H, et al. Influences of cerium on structure and catalytic performance of n-heptane hydroisomerization of Ni-HPW/MCM-48[J]. Journal of Rare Earths, 2015, 33(1): 46-55. doi: 10.1016/S1002-0721(14)60382-3 [18] MICEK-ILNICKA A, BIELAŃSKA E, LITYŃSKA-DOBRZYŃSKA L, et al. Carbon nanotubes, silica and titania supported heteropolyacid H3PW12O40 as the catalyst for ethanol conversion[J]. Applied Catalysis A: General, 2012, 421-422: 91-98. doi: 10.1016/j.apcata.2012.02.001 [19] REN Z Y, TENG Y F, ZHAO L Y, et al. Keggin-tungstophosphoric acid decorated Fe2O3 nanoring as a new catalyst for selective catalytic reduction of NOx with ammonia[J]. Catalysis Today, 2017, 297: 36-45. doi: 10.1016/j.cattod.2017.06.036 [20] CHANSAI S, BURCH R, HARDACRE C, et al. The use of short time-on-stream in situ spectroscopic transient kinetic isotope techniques to investigate the mechanism of hydrocarbon selective catalytic reduction (HC-SCR) of NOx at low temperatures[J]. Journal of Catalysis, 2016, 281(1): 98-105. [21] GENG Y, XIONG S C, LI B, et al. H3PW12O40 grafted on CeO2: A high-performance catalyst for the selective catalytic reduction of NOx with NH3[J]. Industrial & Engineering Chemistry Research, 2018, 57(3): 856-866. [22] GUNNARSSON F, PIHL J A, TOOPS T J, et al. Lean NOx reduction over Ag/alumina catalysts via ethanol-SCR using ethanol/gasoline blends[J]. Applied Catalysis B: Environmental, 2017, 202: 42-50. doi: 10.1016/j.apcatb.2016.09.009 [23] GUO R T, LI M Y, SUN P, et al. Mechanistic investigation of the promotion effect of Bi modification on the NH3-SCR performance of Ce/TiO2 catalyst[J]. Journal of Physical Chemistry C, 2017, 121(49): 27535-27545. doi: 10.1021/acs.jpcc.7b10342 [24] JIANG H X, WANG Q Y, WANG H Q, et al. MOF-74 as an efficient catalyst for the low-temperature selective catalytic reduction of NOx with NH3[J]. ACS Applied Materials & Interfaces, 2016, 8(40): 26817-26826. [25] ZHA K W, CAI S X, HU H, et al. In situ DRIFTs investigation of promotional effects of tungsten on MnOx-CeO2/meso-TiO2 catalysts for NOx reduction[J]. Journal of Physical Chemistry C, 2017, 121(45): 25243-25254. doi: 10.1021/acs.jpcc.7b08600 [26] ZHANG Q L, FAN J, NING P, et al. In situ DRIFTS investigation of NH3-SCR reaction over CeO2/zirconium phosphate catalyst[J]. Applied Surface Science, 2018, 435: 1037-1045. doi: 10.1016/j.apsusc.2017.11.180 [27] HADJIIVANOV K. Identification of neutral and charged NxOy surface species by IR spectroscopy[J]. Catalysis Reviews: Science and Engineering, 2000, 42(1/2): 71-144. [28] KRISTIANSEN T, MATHISEN K. On the promoting effect of water during NOx removal over single-site copper in hydrophobic silica APD-aerogels[J]. Journal of Physical Chemistry C, 2014, 118(5): 2439-2453. doi: 10.1021/jp406610v [29] SHIMIZU K, SATSUMA A. Selective catalytic reduction of NO over supported silver catalysts-practical and mechanistic aspects[J]. Physical Chemistry Chemical Physics, 2006, 8(23): 2677-2695. doi: 10.1039/B601794K [30] SOBCZAK I, MUSIALSKA K, PAWLOWSKI H, et al. NO and C3H6 adsorption and coadsorption in oxygen excess: A comparative study of different type zeolites modified with gold[J]. Catalysis Today, 2011, 176(1): 393-398. doi: 10.1016/j.cattod.2010.11.028 [31] XU G Y, YU Y B, HE H. Silver valence state determines the water tolerance of Ag/Al2O3 for the H2-C3H6-SCR of NOx[J]. Journal of Physical Chemistry C, 2018, 122(1): 670-680. doi: 10.1021/acs.jpcc.7b10860 [32] YU Y B, HE H, ZHANG X L, et al. A common feature of H2-assisted HC-SCR over Ag/Al2O3[J]. Catalysis Science & Technology, 2014, 4(5): 1239-1245. [33] KAMEOKA S, KITA K, TANAKA S I, et al. Enhancement of C2H6 oxidation by O2 in the presence of N2O over Fe ion-exchanged BEA zeolite catalyst[J]. Catalysis Letters, 2002, 79(1/2/3/4): 63-67. [34] HAMADA S, HIBARINO S, IKEUE K, et al. Preparation of supported Pt-M catalysts (M=Mo and W) from anion-exchanged hydrotalcites and their catalytic activity for low temperature NO-H2-O2 reaction[J]. Applied Catalysis B: Environmental, 2007, 74(3/4): 197-202. [35] LI Y H, DENG J L, SONG W Y, et al. Nature of Cu species in Cu-SAPO-18 catalyst for NH3-SCR: combination of experiments and DFT calculations[J]. Journal of Physical Chemistry C, 2016, 120(27): 14669-14680. doi: 10.1021/acs.jpcc.6b03464 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4517
- HTML全文浏览数: 4517
- PDF下载数: 47
- 施引文献: 0
