



 DownLoad:
DownLoad:










 百度学术
百度学术


-
间甲酚是酚类化合物中具有代表性的一种物质,广泛存在于煤化工、石油化工、制药废水等工业废水中,它的存在是造成工业废水难被生物降解的原因之一,具有腐蚀性和强烈刺激性,已被美国环保署列为11种难降解酚类化合物之一,同时也是我国水污染优先控制污染物黑名单中的一类。间甲酚的处理技术一般采用高级氧化法,可以在较短的时间内使间甲酚转化为其他小分子脂肪族化合物或完全矿化,为煤气化废水的治理奠定了基础[1-3]。
催化臭氧氧化法利用O3,在催化剂的作用下,产生了氧化能力极强的 · OH,以降解水中的有机物,具有反应速度快、不产生污泥和二次污染、显著提高废水可生化性等优点[4-6],但其O3转化率较低,工艺成本比较高。在此体系中加入H2O2后,H2O2和O3协同作用,可以显著加快O3分解产生 · OH的速率,既能提高处理效果,又能帮助降低经济成本。为了获得这一优势,可以将O3和H2O2进行联用。催化臭氧氧化技术常选用非均相氧化法,催化剂多以金属氧化物、金属负载于载体上及经金属改进的沸石、活性炭为主 [7]。与均相催化剂相比,非均相催化剂具有活性高、流失少、便于回收等优点,因此,选用非均相催化剂投入到O3/H2O2体系中考察其处理效果。
本研究采用等体积浸渍法制备了Fe-Mn/γ-Al2O3催化剂,考察O3氧化法、催化O3氧化法、催化H2O2氧化法以及催化O3-H2O2氧化法对间甲酚的降解效果,系统分析了pH、空速等对催化O3-H2O2氧化间甲酚的影响,同时表征了催化剂的性能,并对比了4种催化氧化条件下的氧化产物,为2种技术联用降解煤化工废水提供了参考。
全文HTML
-
间甲酚(99.8%)购自百灵威化学技术有限公司,H2O2 (30%) 购自天津市富宇精细化工有限公司,九水合硝酸铁、硝酸锰、硫代硫酸钠、甲醇、二氯甲烷购自天津科密欧化学试剂有限公司,草酸钛钾购自上海阿拉丁生化科技股份有限公司,碘化钾购自天津市大茂化学试剂厂,可溶性淀粉购自沈阳市新西试剂厂,pH计购自上海精密科学仪器有限公司,型号为PHSJ-3F。
-
使用的Fe-Mn/γ-Al2O3催化剂采用等体积浸渍法制备。具体的制备过程为:称取50 g Al2O3小球,按Fe、Mn质量分数为4%和0.5%,分别加入硝酸铁及硝酸锰,配成等体积的浸渍液,浸渍24 h,放入120 ℃的电热鼓风干燥箱中,干燥2 h,把得到的催化剂放入马弗炉中,在空气氛围下,以3 ℃·min−1的升温速度升至530 ℃,恒温焙烧3 h,冷却至室温,得到催化剂Fe-Mn/γ-Al2O3。
-
SEM表征采用Carl Zeiss Jena公司的FE-SEM SUPRA 55电镜,操作电压为20 kV。
X射线衍射(XRD)谱图在Panalytical X’pert PRO型的粉末衍射仪上测定,扫描的角度为10°~90°,管电压为40 kV,管电流为40 mA。
X射线荧光光谱(XRF)采用荷兰Philips公司Magix X型,使用X射线荧光光谱仪测定无机元素,测定条件:电压40 kV,电流40 mA。
X射线光电子波谱(XPS)在ESCALAB 250Xi仪器上测定,选用A1Kα射线,结合能(BE)通过C1s峰的位置((284.6 ± 0.2) eV)进行校正。
催化剂的比表面积及孔隙结构使用Quanta仪器公司的NOVAe型全自动比表面和孔隙度分析仪测定,将样品预先在真空条件下于90 ℃和300 ℃分别处理0.5 h和5 h,以氮气为吸附质于77 K恒温吸附。通过N2吸附等温线和BET方程计算比表面积,采用t-plot法计算微孔比表面积,采用BJH法计算总孔容及平均孔径。
-
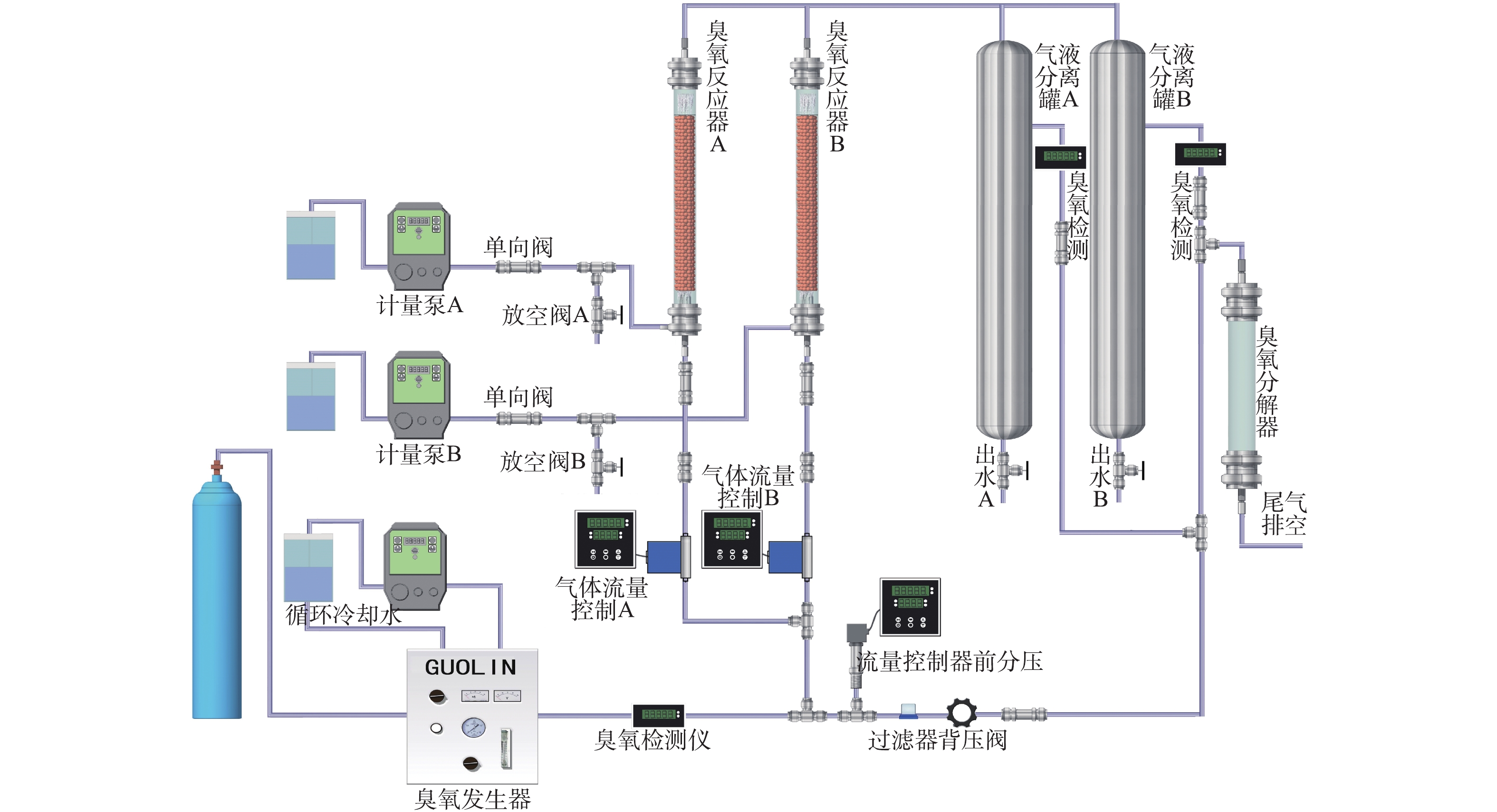
在反应器床层中加入100 mL Fe-Mn/γ-Al2O3催化剂,间甲酚溶液加入一定量的H2O2后,通过计量泵将混合溶液通入到玻璃反应器中,O3气体由氧气通过臭氧发生器制得,经反应器底部的曝气头通入反应器,形成微小O3气泡,与反应液进行充分接触,每隔固定时间,由反应装置尾部的气液分离罐中取样,通过0.45 μm滤膜后,测量出水TOC、间甲酚的浓度以及出水中剩余O3和H2O2的含量。水中残余O3及O3尾气均用碘量法测定。本实验装置流程见图1。
采用高效液相色谱法对间甲酚的浓度进行测定,选用大连依利特分析仪器有限公司生产的P1201型高效液相色谱仪进行分析,色谱柱采用依利特C18色谱柱(4.6 mm × 250 mm, 5 µm),流动相为甲醇∶水=80∶20(体积比),流速为1.0 mL·min−1,紫外检测波长为272 nm。TOC采用日本岛津公司生产的TOC -VCPH/CPN分析仪进行测定。使用GC-MS法对中间产物进行分析,通过Thermo DSQⅡ 型气相色谱/质谱联用仪,色谱柱:DB-5(30 m×0.25 mm)。分析前,将溶液进行处理,用H2SO4调节至pH= 2,用二氯甲烷萃取,无水硫酸钠脱水后浓缩保存。
出水的有机物相对分子质量采用LC-OCD法进行分析,使用德国DOC-Labor公司生产的LC-OCD液相色谱有机碳联用仪,测定出水中氧化降解生成的物质的相对分子质量。
依据我国建设部发布的《臭氧发生器臭氧浓度、产量、电耗的测量》(CJ/T 3028.2-1994)中的规定,采用碘量滴定法对O3尾气含量和出水中O3含量进行测定。在催化O3氧化反应的过程中,O3转化率的计算方法如式(1)所示。
式中:CU为O3转化率;CT为O3总投加量,mg;CR为水中剩余O3量,mg;CG 为尾气中O3含量,mg。
采用钛盐分光光度法测定H2O2浓度[8],测定仪器采用上海舜宇恒平科学仪器有限公司生产的722型可见分光光度计。H2O2转化率及出水中H2O2含量的计算方法如式(2)所示。
式中:CV为H2O2转化率;CI为进水H2O2浓度,mg·L−1;CO为出水H2O2浓度,mg·L−1。
可以看出,催化O3氧化和催化H2O2氧化这2种高级氧化技术都是在反应过程中首先生成具有强氧化性的 · OH,然后再由 · OH氧化降解有机污染物。因此,在实验中引入协同因子AF值来进行协同作用的评价[9]。在O3-H2O2联用体系中,其协同因子(AF)值(FAF)可通过式(3)计算得出。
式中:FAF为协同因子;
X1 为催化O3-H2O2法处理废水的TOC去除率;X2 为催化O3氧化法处理废水的TOC去除率;X3 为催化H2O2氧化法处理废水的TOC去除率。因此,当FAF大于1时,说明在此催化剂的作用下,O3和H2O2有协同效果。
1.1. 试剂与仪器
1.2. 催化剂的制备
1.3. 催化剂的表征
1.4. 实验方法及计算方法
-
图2(a)和图2(b)分别为催化剂载体γ-Al2O3的SEM图,图2(c) 和图2(d)分别为制备的催化剂Fe-Mn/γ-Al2O3的SEM图。可以看出,活性组分在载体表面负载良好,催化剂表面结构规整,粗糙程度较大,表明制备出的Fe-Mn/γ-Al2O3催化剂活性组分分散均匀,接触面积较大。
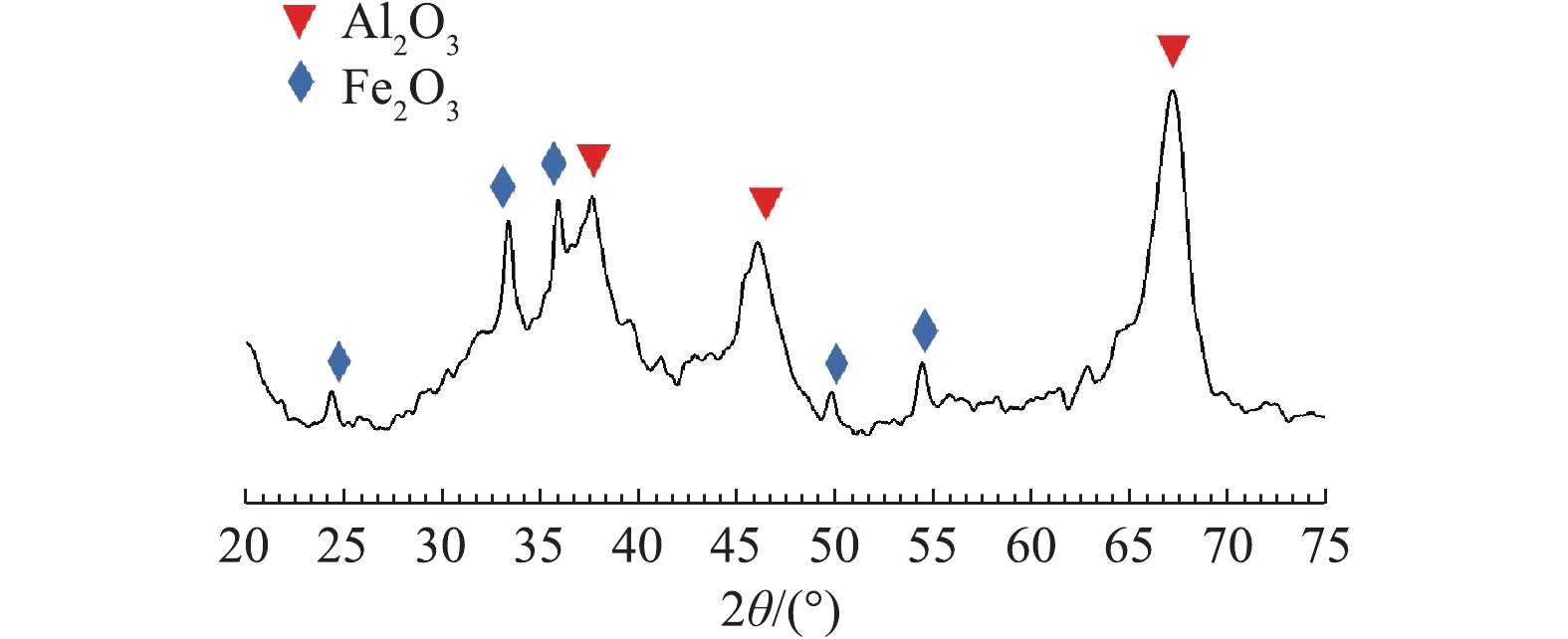
图3为Fe-Mn/Al2O3催化剂的XRD表征图谱。在2θ= 37.604°、45.79°、67.307°出现的衍射峰对应了 (311)、(400)、(440) 晶面,d值分别为2.39,1.98和1.39 nm,归属于γ-Al2O3[10];在2θ= 24.298°、33.389°、35.833°、49.784°和54.457°出现的衍射峰的d值分别为3.66、2.68、2.50、1.83和1.68 nm,对应了 (012)、(104)、(110)、(024) 和 (116) 晶面,归属于赤铁矿型α-Fe2O3[11]。催化剂的XRD谱图中未检测到负载Mn的衍射峰,这可能是由于Mn的负载量较少,在载体表面高度分散以及各金属颗粒过小等原因造成的。
载体γ-Al2O3和催化剂Fe-Mn/γ-Al2O3的表面结构性质如表1所示。由此可知:载体在浸渍了金属盐溶液制备成催化剂后,比表面积减小,原因可能为载体的孔内负载上了金属氧化物;总孔容和孔径减小是由于金属硝酸盐在焙烧时,硝酸根离子转化为NOx气体析出。制备出的催化剂Fe-Mn/γ-Al2O3的比表面积为181.08 m2·g−1。从平均孔径可以看出,该催化剂为中孔结构。总孔容为0.44 cm3·g−1,说明该催化剂有较为良好的多孔结构和比表面积,有利于催化反应的进行。
通过XRF对Fe-Mn/γ-Al2O3催化剂中重要化学元素组成及含量进行表征。经分析,催化剂中的活性组分Fe在Fe-Mn/γ-Al2O3催化剂的质量分数为4.013%,活性组分Mn在Fe-Mn/γ-Al2O3催化剂的质量分数为0.537%,另外还含有少量Na、Ca、Si、S等元素。
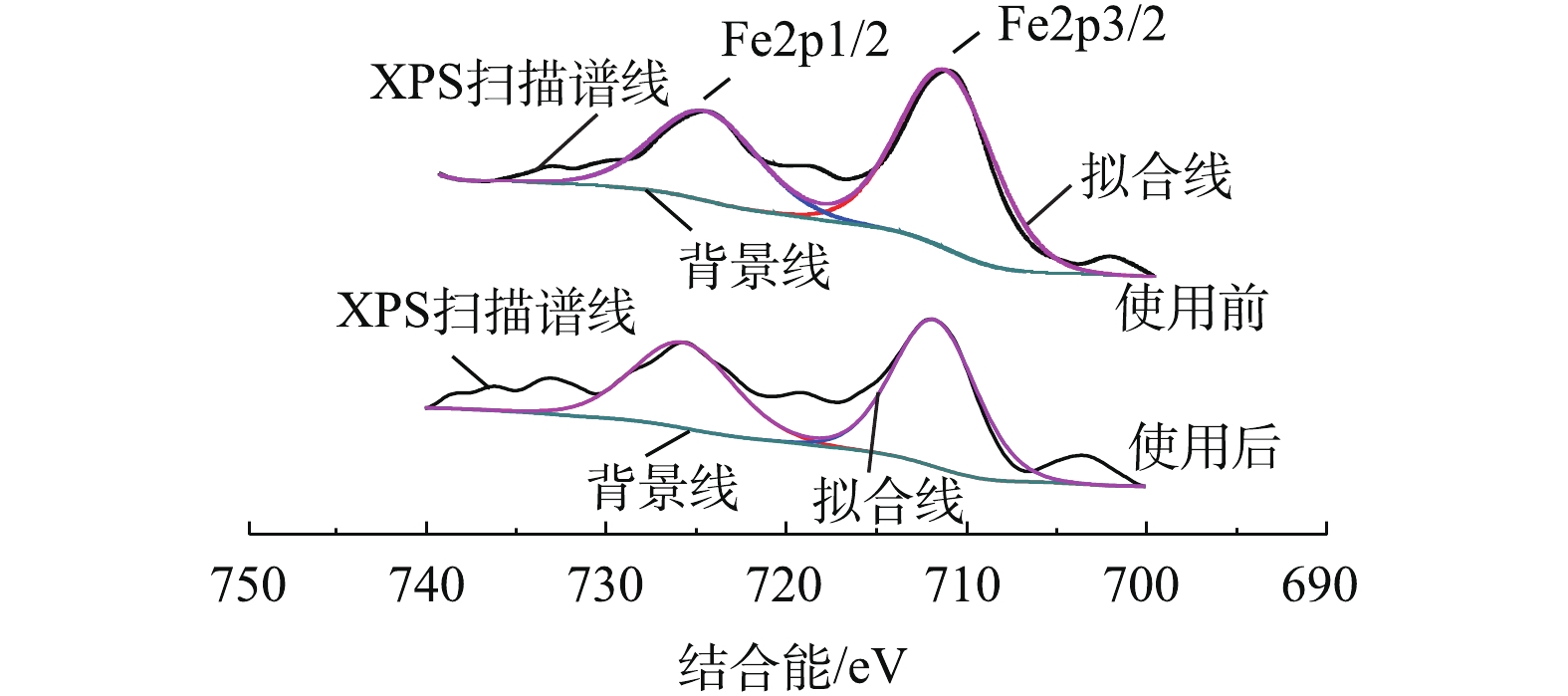
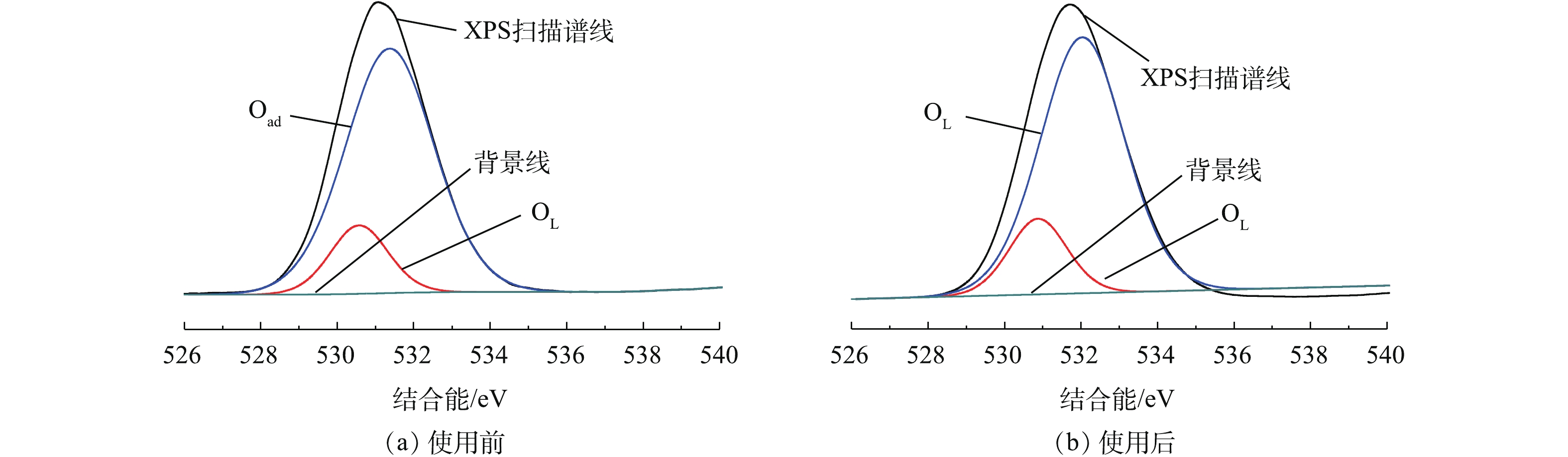
对反应前后的Fe-Mn/γ-Al2O3催化剂表面进行XPS测试。由图4可以看出,新鲜Fe-Mn/γ-Al2O3催化剂的Fe2p1/2及Fe2p3/2的电子结合能分别为724.98 eV和711.56 eV,参与反应后的催化剂的Fe2p1/2及Fe2p3/2的电子结合能分别为725.98 eV和711.95 eV。Fe的2p峰对应于Fe2O3[12-14],且Fe2p3/2存在关联卫星峰,卫星峰的位置比Fe2p3/2峰高8 eV,故反应前、后的催化剂中活性组分Fe都是以Fe2O3形式存在的,这与XRD的表征结果[14-16]一致,且氧化物在反应前、后无变化,较为稳定。反应前、后Mn的XPS表征结果如图5所示,由于其含量较少,未测出其对应的明显特征峰。
催化剂表面氧的存在形式对催化剂的活性也有着重要的影响。图6是反应前、后Fe-Mn/γ-Al2O3催化剂表面的O1s的XPS图。可以看出,O1s峰是对称的,说明催化剂表面的氧以化合物状态存在[17]。反应后的O1s峰结合能从531.07 eV升高到531.53 eV,峰面积有显著减小,O含量从87.95%下降到71.95%,这说明催化剂表面的O1s在反应前、后发生了变化。对反应前、后的O1s进行分峰拟合,结果如表2所示。可以看出,新鲜及使用后的催化剂表面O1s分峰拟合结果可以得到2个拟合峰。随结合能的增加,依次对应晶格氧和吸附氧[18]。吸附氧含有较高的流动性,能够参与到氧化还原反应过程中。根据催化剂参与反应后的表面O1s分峰拟合结果,可以看出,吸附氧含量略有降低,说明其催化活性在反应后有下降[19],但实际差别很小,催化剂性质较为稳定。
-
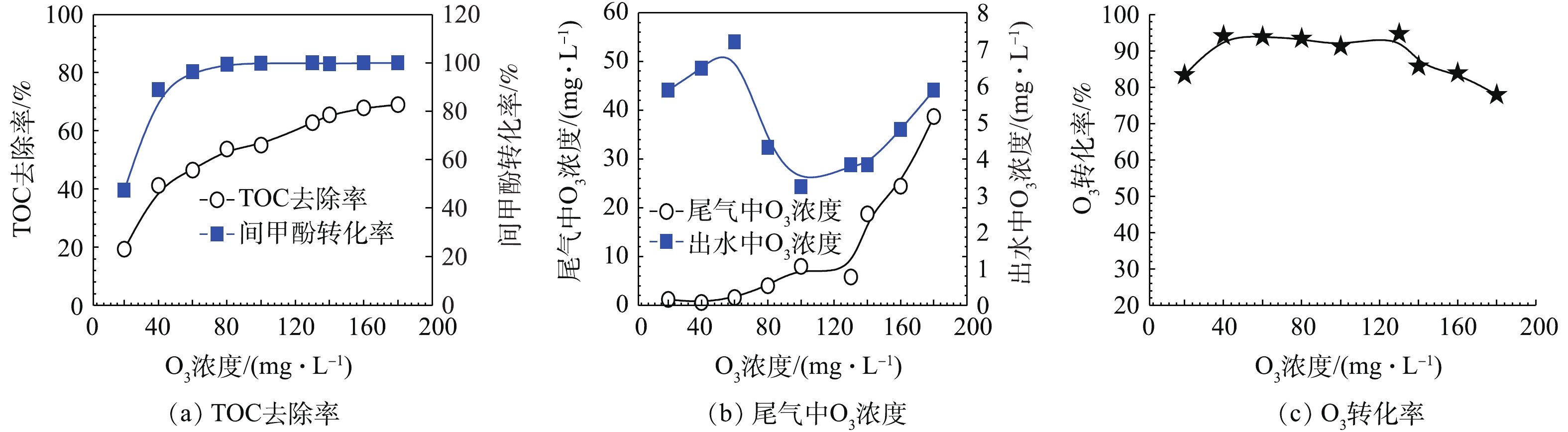
首先,在单独使用催化O3氧化技术时,考察不同O3投加量下对间甲酚的转化率、TOC去除率、O3残余量及O3转化率的影响,实验结果如图7所示。从图7(a)可以看出,在O3流量为37 mL·min−1,通入间甲酚溶液的流量为600 mL·h−1,反应时间为10 min的条件下,O3投加量越多,对间甲酚的氧化效果越好,当O3浓度达到60 mg·L−1时,间甲酚转化率接近100%,TOC去除率接近45%,在此过程中,间甲酚被转化成其他有机物,还可以进一步被降解;当O3浓度达到130 mg·L−1时,TOC去除率可达60%左右,此时继续增加O3投加量,无法使TOC去除率有更大幅度的提高,间甲酚转化率为100%。从图7(b)和图7(c)可以看出,尾气及出水中残余的O3量也随着O3投加量的增加而越来越高,故提高O3投加量虽然可使TOC的去除效率得到提高,但过量将会导致过多的O3没有被有效利用,引起O3转化率下降。当O3浓度为130 mg·L−1时,尾气及出水中残余O3量均较小,O3转化率也在此时达到最高。
与单独O3催化氧化相比,H2O2的量在一定合适范围内,可与O3反应生成
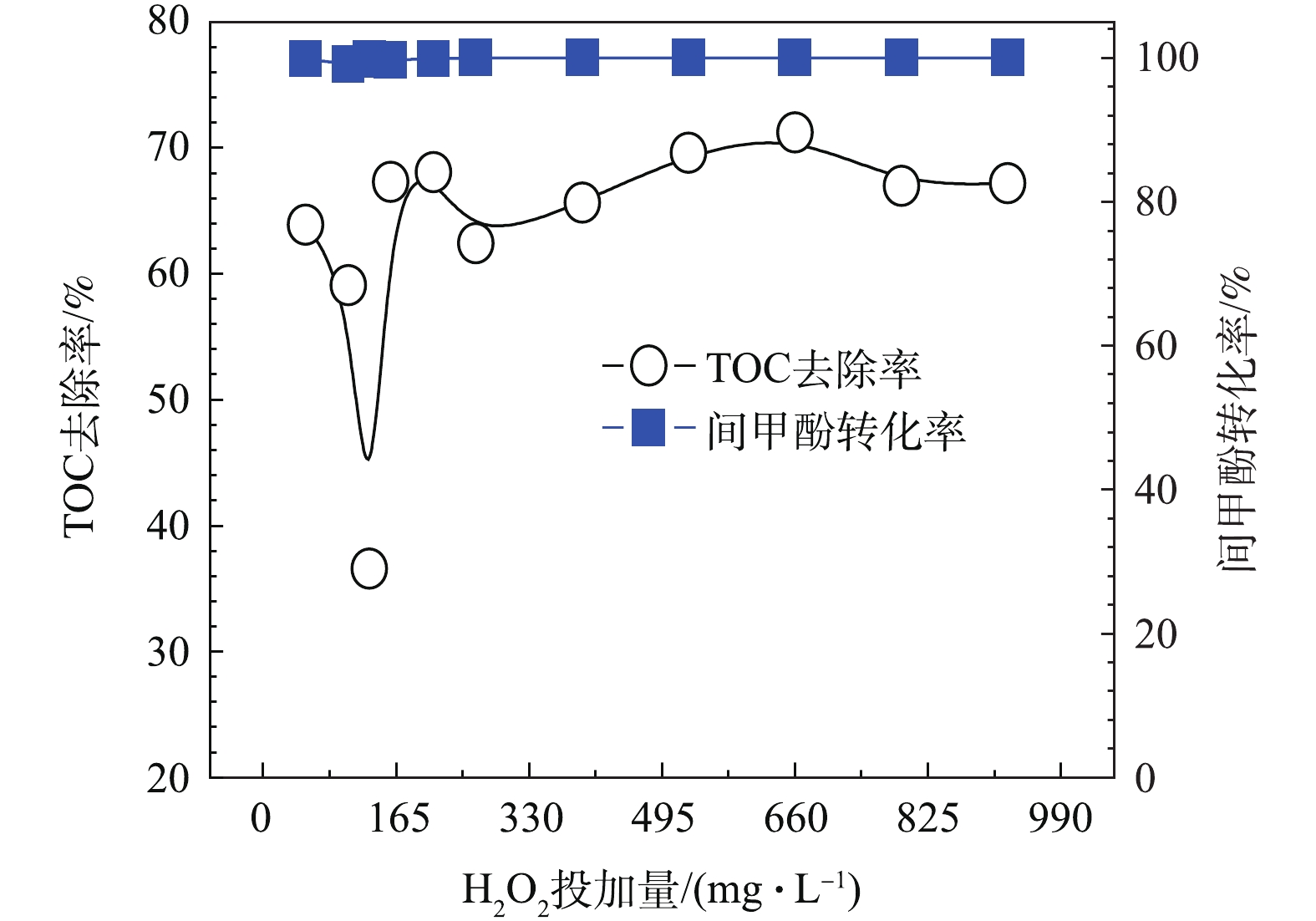
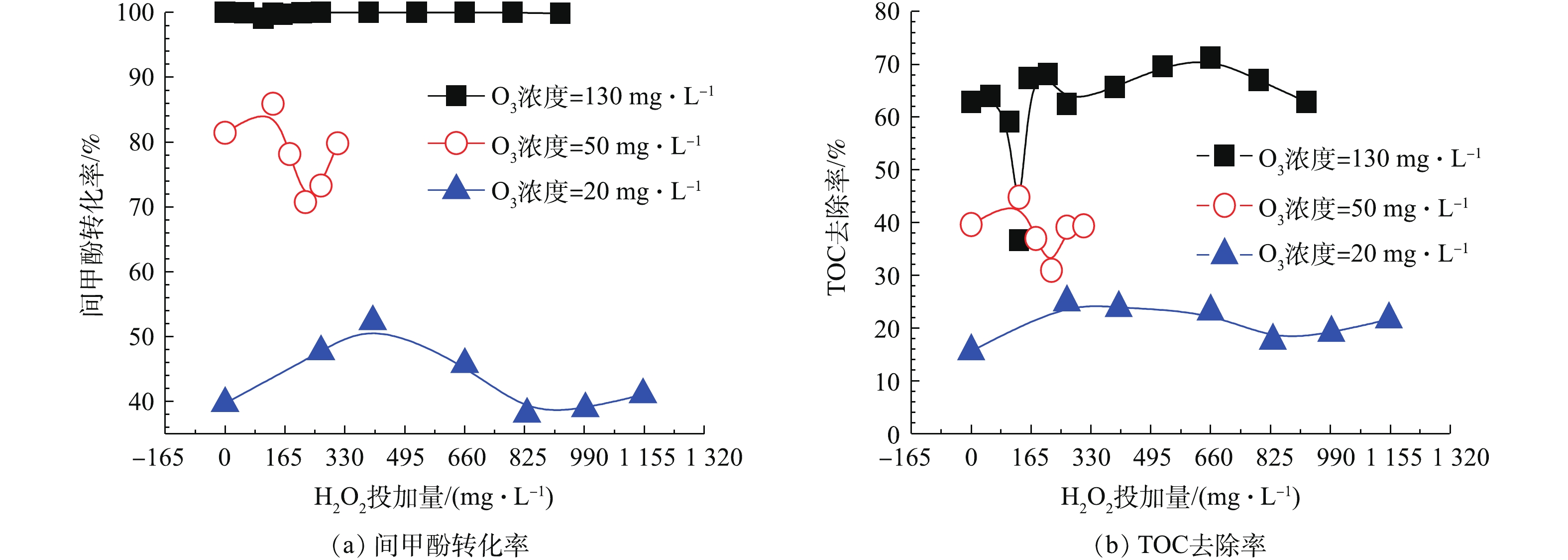
HO−2⋅/O−2⋅ 等物质,HO−2⋅ 继而能与O3反应产生O−3⋅ ,其迅速质子化产生 · OH,且H2O2解离出的HO−2⋅ 分解O3的能力要比OH− 高5个数量级,故H2O2是比传统OH−更有效的O3分解引发剂[20-22]。在O3投加量设定为481 mg·L−1的条件下,考察H2O2的投加量的影响。由图8可以看出,随着H2O2投加量的增加,TOC去除率呈先下降后上升的趋势,并在H2O2的投加量为211 mg·L−1时趋于平稳,TOC去除率接近70%且经济合理,间甲酚大部分已被转化为其他有机物,可能为小分子酸[23-24]。因此,O3与H2O2在一定投加配比的条件下,才能充分利用二者的氧化作用并产生协同效果,即最佳实验条件:在通入间甲酚溶液的流量为600 mL·h−1,反应时间为10 min,O3投加量481 mg·L−1时,H2O2的投加量为211 mg·L−1。由图9中可以看出,在3种O3投加浓度下,随着H2O2投加量的增多,TOC去除率均呈先上升再下降再上升的趋势,且均有最佳协同点。并呈现一定的规律:通入O3的浓度越高,对应此现象出现的H2O2投加量越小。
此外,由图9可知,在特定O3投加量的条件下,调节H2O2投加量并不能使其去除效果高于更高浓度的O3投加量的去除效果,即当O3投加量较低时,即使加入更多的H2O2,也不能使TOC及间甲酚有更好的去除效果,也可以得出O3投加量比H2O2投加量对于去除结果的影响更为显著。
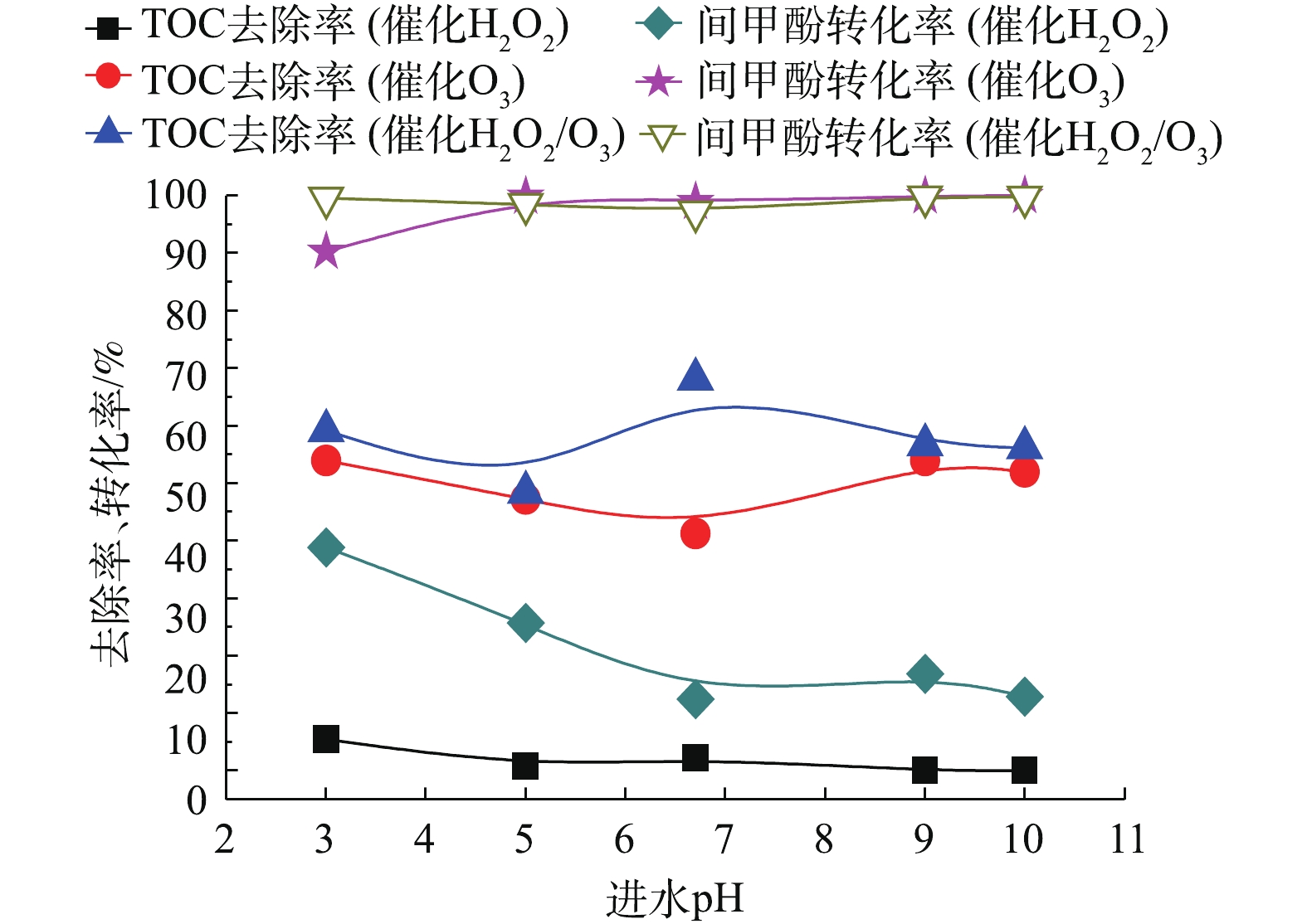
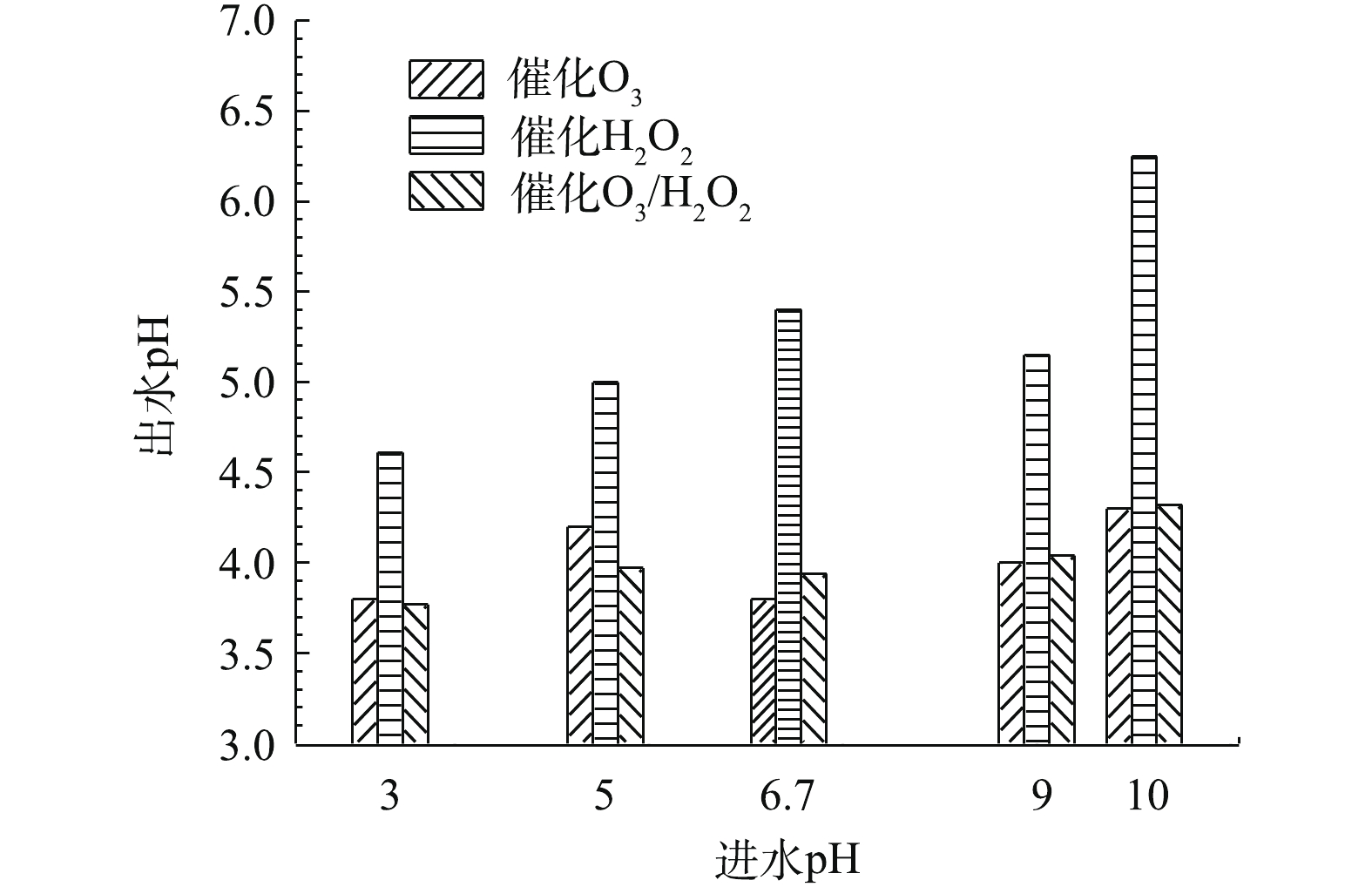
当确定实验最佳O3投加量为481 mg·L−1时,H2O2的投加量为211 mg·L−1,在通入间甲酚溶液的流量为600 mL·h−1,反应时间为10 min的条件下,进一步研究进水的初始pH对氧化效果的影响。由图10可知,pH对反应的影响并不显著。在不同pH下,催化O3-H2O2氧化技术对间甲酚的氧化效果均优于催化O3氧化和催化H2O2氧化技术的效果。由图11可以看出,催化O3-H2O2氧化和催化H2O2氧化的出水pH均为酸性,说明间甲酚被氧化降解后产生了小分子酸类物质。催化H2O2氧化的出水pH高于催化O3氧化和催化O3-H2O2氧化法的出水pH,可能原因为间甲酚被氧化分解为其他芳香族物质,没有进一步氧化成小分子酸,故氧化的效果不如催化O3氧化和催化O3-H2O2氧化。
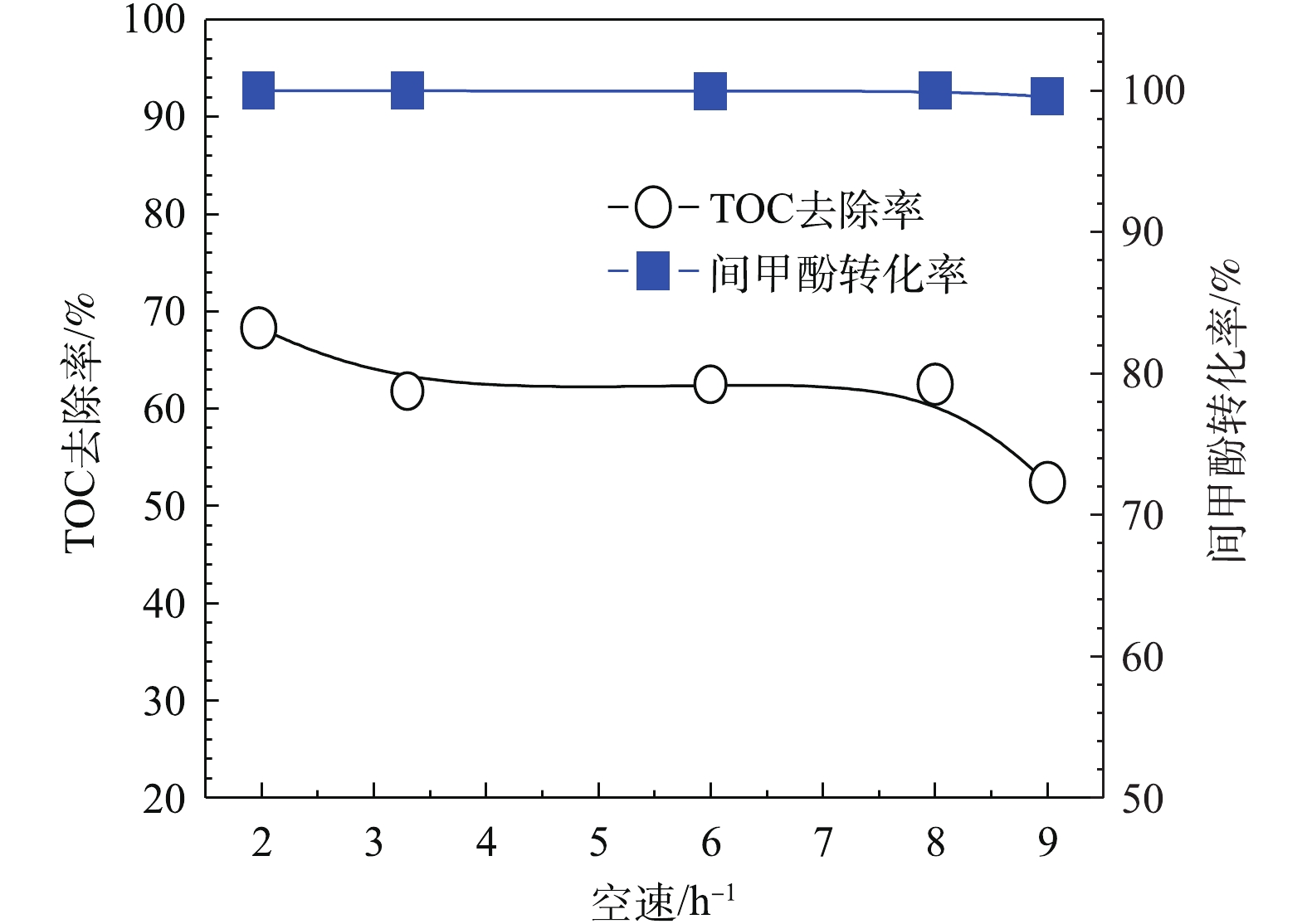
为进一步提高TOC去除率,考察进水的体积空速对模型废水的降解效果的影响。体积空速是指在一定的条件下,在单位时间内通过单位体积催化剂处理的废水体积。由图12可以看出,当O3投加量为481 mg·L−1时,H2O2的投加量为211 mg·L−1,在通入间甲酚溶液的流量为600 mL·h−1,溶液初始pH为6.7,反应时间为10 min的条件下,保证O3投加量不变,改变空速,间甲酚均可以完全转化,TOC去除率随着空速增大而降低,原因在于气水相与催化剂的接触时间短,气、液、固三相没有充分反应便脱离反应器。但总体来说,空速为1~8 h−1时,空速的影响不是主要因素。
-
在O3投加量为481 mg·L−1,O3浓度为130 mg·L−1,流量为37 mL·min−1,反应时间为10 min,空速为6 h−1,H2O2的投加量为211 mg·L−1,进水pH为6.7的最佳实验条件下,以不加催化剂只通O3氧化作为空白实验,分别做催化O3氧化、催化H2O2氧化和催化O3-H2O2氧化处理间甲酚废水,将出水中间产物分别做GC-MS和LC-OCD分析。
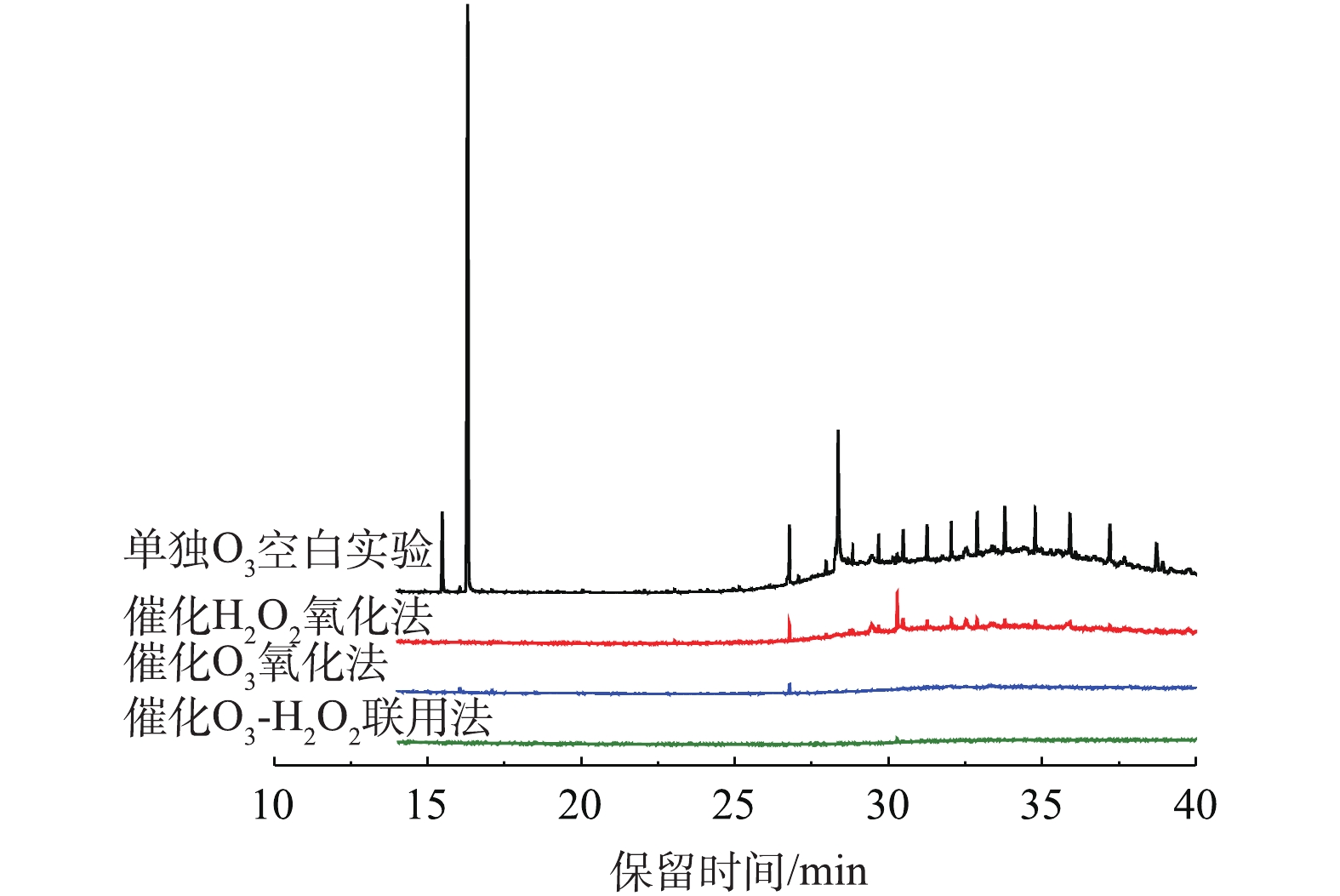
间甲酚降解产物的总离子色谱图如图13所示。从GC-MS结果来看,O3氧化法将间甲酚转化成二甲基对苯醌(t=16.053 min)、邻苯二甲酸二丁酯(t=26.733)及长链酸等物质;催化H2O2氧化及催化O3氧化均可将间甲酚完全转化,催化O3氧化使间甲酚转化为呋喃酮类(t=16.053 min)、2-烷基-5-甲基-1,4苯醌(t=17.097 min)、邻苯二甲酸二丁酯(t=26.773 min)及邻苯二甲酸酯类(t=28.302、31.593 min)等。单独O3氧化、催化O3氧化及催化H2O2氧化这3种氧化技术的中间产物均存在邻苯二甲酸二丁酯类,这种物质是持久性有机污染物POPs的一种,较难生物降解。在催化O3及H2O2的协同作用下,可将中间产物邻苯二甲酸酯类及对苯醌类完全降解,将间甲酚氧化成链状烷烃类物质。同时,单独O3氧化、催化H2O2氧化这2种氧化技术的出水仍存在大量酯类化学物质,催化O3氧化法的出水在t=28~32 min内的物质几乎被氧化,而在催化O3-H2O2氧化法的协同作用下,有机物几乎完全被矿化,出水结果明显优于上面3种方法。这也证明了协同作用下的氧化效果更好。
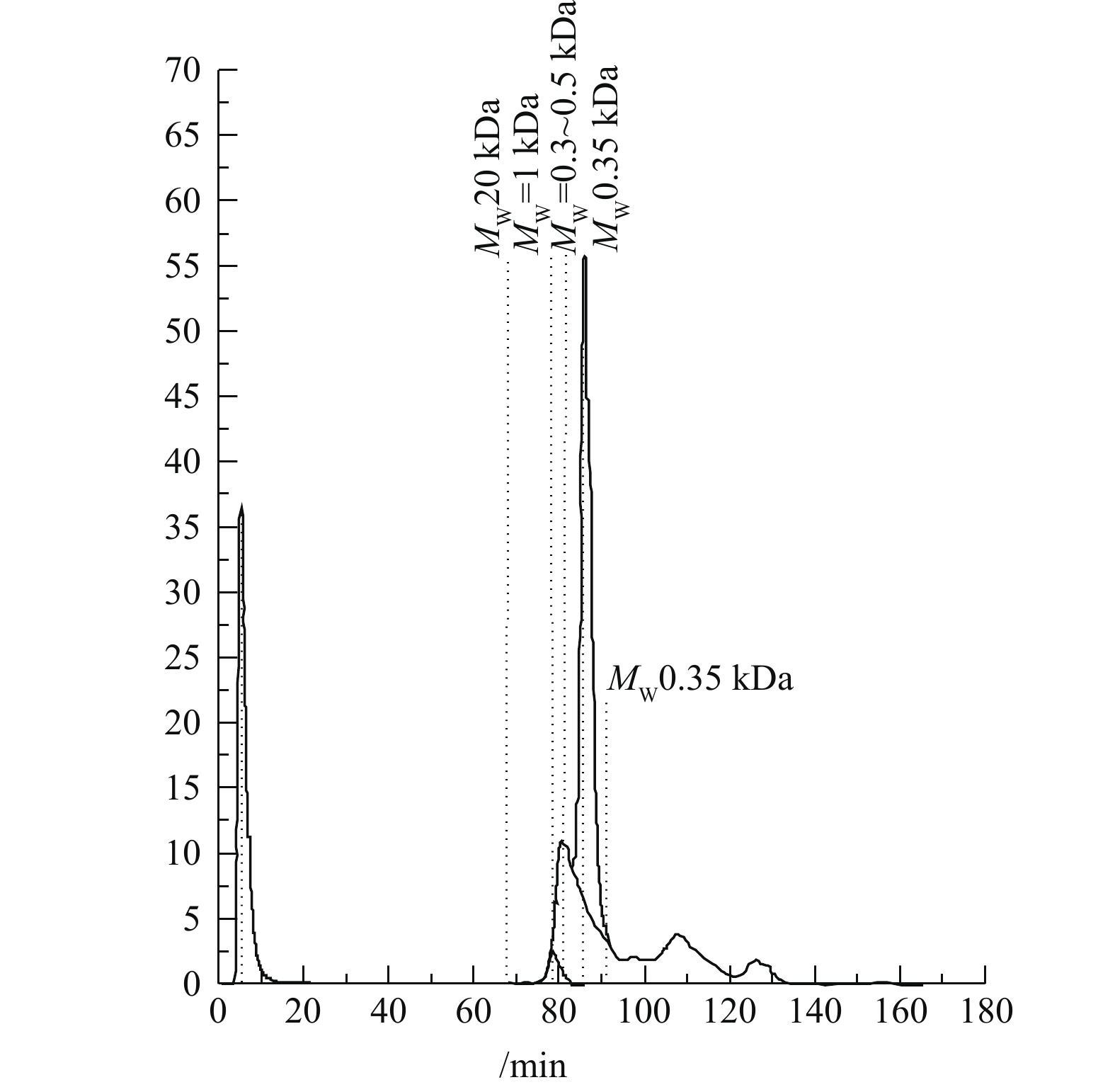
将催化O3-H2O2联用法处理后的出水进行LC-OCD分析,出水中有机物的分子的测定谱图如图14所示,测出该水样中有机物主要集中在相对分子质量小于0.35 kDa的范围内,说明较大分子的有机分子被氧化分解,生成小分子有机酸[25],其分析结果也与GC-MS的结果相吻合。
-
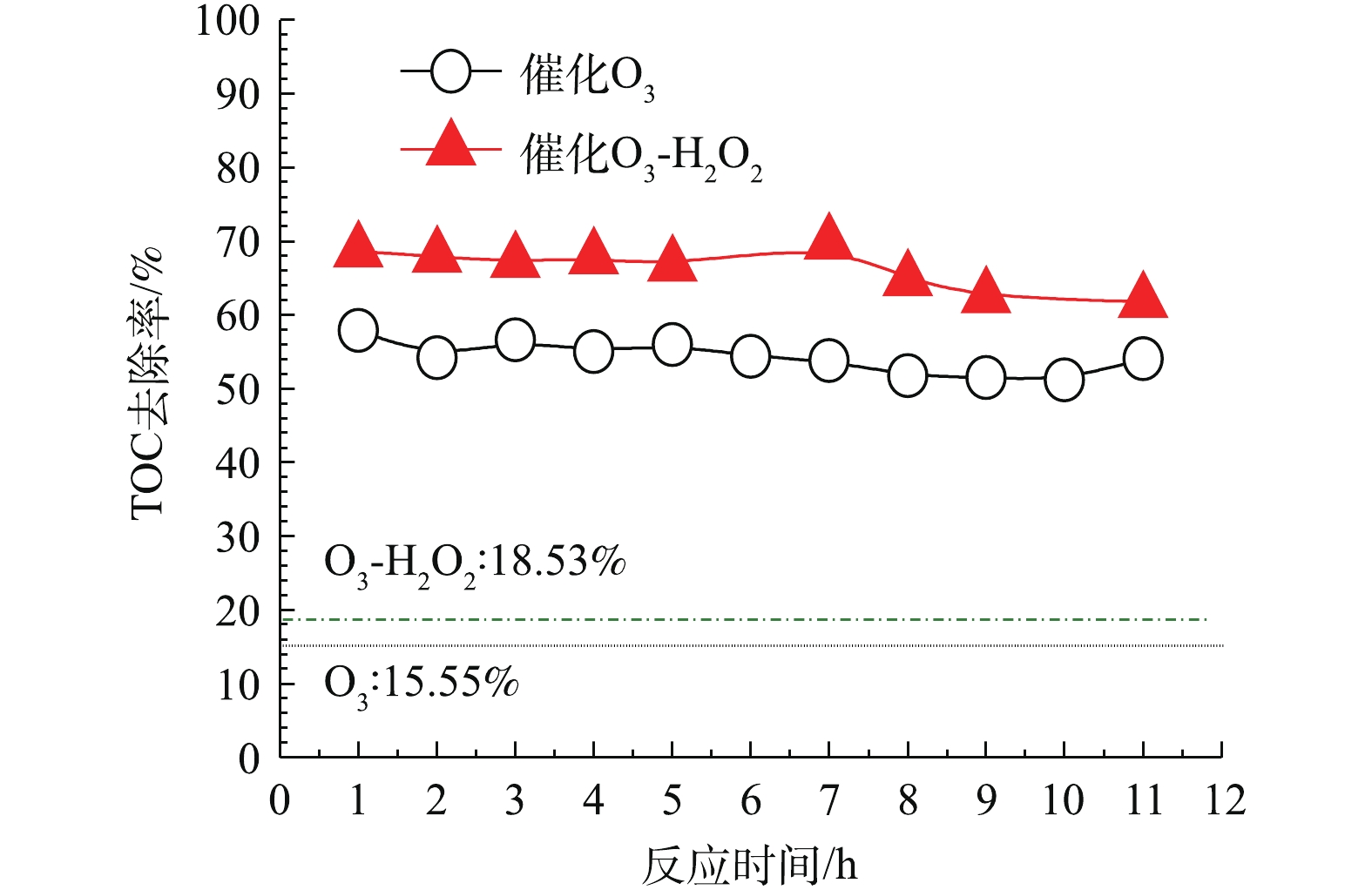
在最佳实验条件下,考察催化剂通过连续反应的催化活性的变化。图15中虚线为在同一条件下不使用催化剂的单独O3氧化和O3-H2O2反应的TOC去除率,分别为15.55%及18.53%,并进一步比较催化O3氧化和催化O3-H2O2联用氧化的氧化效果。可以看出,在使用催化剂的条件下,TOC去除率有很大的提高。催化剂在经过11 h的单独催化O3氧化连续反应后,TOC去除率比反应初始时略有下降;催化剂在经过11 h的催化O3-H2O2联用氧化连续反应期间,反应TOC去除率基本不变。说明该催化剂在使用一段时间后,催化性能基本不变,稳定性较好。
在O3投加量为481 mg·L−1,O3浓度为130 mg·L−1,流量为37 mL·min−1,反应时间为10 min,空速为6 h−1,H2O2的投加量为211 mg·L−1,进水pH为6.7的最佳实验条件下,计算出的协同因子AF为1.17 >1,因此,说明在催化剂的作用下,O3与H2O2起协同作用。
2.1. 催化剂的表征结果
2.2. 催化氧化处理间甲酚模型废水
2.3. 中间产物分析及出水有机物分子大小的测定
2.4. 催化剂稳定性评价
-
1) 实验研究了O3氧化、催化H2O2氧化、催化O3氧化和催化O3-H2O2联用氧化技术对间甲酚的降解处理效果,根据实验结果并通过GC-MS分析可知4种氧化作用按照以下顺序依次增强:O3氧化<催化H2O2氧化法<催化O3氧化<催化O3-H2O2氧化。
2) 在催化O3-H2O2复合氧化体系中,反应温度为室温,初始pH= 6.7,O3投加量为481 mg·L−1,O3浓度为130 mg·L−1,流量为37 mL·min−1,反应时间为10 min,空速为6 h−1,H2O2投加量为211 mg·L−1,初始间甲酚浓度为100 mg·L−1,在Fe-Mn/γ-Al2O3催化剂的催化作用下,间甲酚转化率及TOC去除率分别为100%、66.3%。TOC去除率在相同条件下比催化O3氧化高22.3%。协同因子AF=1.17 >1,说明催化O3-H2O2联用氧化技术具有协同效果。
3) 催化O3-H2O2协同氧化的最终产物可将其余3种氧化技术中间产生的持久性难降解有机物裂解开环,生成开环链状烷烃类物质,几乎被全部降解,催化效果最好。
