

-
化学氧化修复是有机污染场地常见的修复技术之一[1],其修复机理主要是通过强氧化剂与有机污染物发生氧化还原反应,使得污染物转化为稳定、低毒或无毒性物质[2]。常见的化学氧化剂有活化过硫酸盐、Fenton、类Fenton、高锰酸钾、臭氧等,其中活化过硫酸盐以其易活化、适应性广、效果好等优点而成为研究热点[3-4]。化学氧化能快速而高效地去除土壤中的各类有机污染物,具有修复效率高、修复速率快、普适性强等优点[5]。然而,加入大量化学氧化剂往往会导致土壤理化性质改变、微生物生态系统破坏、带来二次污染等隐患[6]。
微生物修复技术通过微生物的生长代谢作用,将有机污染物转化成简单无机物,从而达到去除环境中有机污染物的目的[7],包括生物刺激(添加营养物质)、生物强化(添加高效降解菌或生物催化剂(基因和酶))和曝气系统(曝气增氧)等[8]。与化学氧化等其他技术相比,微生物修复技术能够有效避免二次污染问题,其成本更低,更易于维护,但也存在修复时间长、对环境要求比较严格等缺点[9],故在实际污染场地的应用中,仍具有一定的局限性。
为了解决单项修复技术的局限性,实现更高的有机污染物去除率的目标,可以使用多种方法联合修复[10]。近年来的研究表明,化学氧化和微生物联合修复是一种可行的联合修复方法,具有广泛的应用前景[11-12]。然而,在此前化学氧化-微生物联用技术的研究中,微生物降解主要依赖土著微生物,重点关注的是化学氧化剂对土壤微生物生态系统的影响,关于化学预氧化联合微生物强化或微生物刺激技术(即预氧化后强化微生物降解作用)的研究较少[13-14]。
菲作为土壤中常见的多环芳烃污染物之一,对人类健康具有严重威胁[15]。针对菲污染土壤修复的研究主要集中于化学氧化、微生物降解等单一方法[16],本研究将化学氧化和微生物修复技术相结合,旨在探究化学预氧化后强化微生物降解对土壤中菲的降解效应,重点关注低浓度过硫酸盐预氧化耦合生物强化和生物刺激技术对菲降解的促进效应,以及修复期间土壤各项理化性质的变化情况,为化学氧化-微生物联用修复技术的应用提供参考。
-
实验采用人工模拟的污染土壤,其制备方法如下:在5 kg洁净土壤中加入50 mL 10 g·L−1菲的丙酮溶液,充分混匀后,老化1个月。土壤各项理化指标:菲浓度(98.70±3.23) mg·kg−1,pH 7.42±0.06,有机质含量(34.79±0.89) g·kg−1,总磷含量(0.617±0.02) g·kg−1,总氮含量(0.732±0.02) g·kg−1,微生物数量(5.73±0.70)×107 CFU·g−1。土壤机械组成:(2.71±0.02)%黏粒,(36.28±1.21)%粉粒,(61.01±2.11)%砂粒。
-
1)化学试剂。PHE(纯度>98%)购自Sigma-Aldrich(中国上海);色谱纯试剂:丙酮,正己烷,二氯甲烷;分析纯试剂:Na2S2O8,KH2PO4,NaNO3等。这些试剂均购自国药集团化学试剂有限公司(中国北京)。
2)菌剂。使用实验室筛选、保存的高效菲降解菌Acidovorax sp.JG5制备菌剂,该菌在1 d内对浓度100 mg·L−1菲的降解率为90%以上。菌剂制备方法如下:从斜管培养基中挑取1环Acidovorax sp.JG5菌株,接种至富集培养基中,在30 ℃、180 r·min−1条件下,恒温振荡培养至对数生长期,于4 ℃、8 000g下,离心分离10 min,收集菌体,用无菌生理盐水洗涤2次后重悬,并调节OD600值为0.3。
-
实验共设10个处理组(如表1所示),每个处理组设3个重复。取菲污染土壤150 g于250 mL锥形瓶内,根据表1中设定的浓度,加入过硫酸钠和适量去离子水,调节水土比为3∶5。搅拌均匀后,盖上无菌透气膜,放置于50 ℃水浴锅中,反应72 h后取出,转移至30 ℃恒温培养箱中静置。在化学氧化降解期间,每隔24 h取样测定体系中过硫酸盐浓度;第1、3、5、7 天分别取样测定土壤pH、微生物数量;化学氧化前后,测定土壤中菲浓度。化学预氧化后,通过添加不同药剂,将化学预氧化后的实验组分为表1所示的9组实验组。其中:C-CK实验组加入抑菌剂NaN3,以抑制后续微生物活性,作为单独化学氧化对照组;仅加入等量去离子水的实验组命名为CK组,作为化学氧化-土著微生物降解对照组;B-CK组仍然仅添加等量去离子水,以作为单独微生物降解对照组。所有实验组均根据表1中设定的浓度,加入菌剂、各类营养物质或去离子水。放置于30 ℃、80%湿度恒温培养箱中,静置培养,每隔7 d取样测定土壤pH、微生物数量和菲浓度。
-
使用紫外分光光度法[17]测定体系中过硫酸盐浓度。使用pH计(PB-10,Sartorius)测定体系中pH。微生物计数(土壤中活细菌数)的测定参考标准ISO 6222(1999)。按照赵丹等[18]的研究方法,使用气相色谱-质谱联用仪(Agilent 7890A-5975C)测定土壤中菲浓度。
-
使用Origin 2016软件(OriginLab Corporation,USA)制作图表,SPSS软件和R语言包进行相关性分析和显著性分析,并绘制heatmap图。
-
使用热活化过硫酸盐对菲污染土壤进行预氧化,土壤中过硫酸盐浓度随时间的变化情况及化学氧化前后土壤菲降解率如图1所示。加入氧化剂过硫酸钠反应1 d后,土壤中过硫酸盐浓度由0.1 mmol·g−1下降至0.052 mmol·g−1,降幅达近50%。随后的几天内,过硫酸盐持续消耗,到第7天,土壤中过硫酸盐基本消耗完毕。化学预氧化阶段到此结束,土壤菲的降解率达22.7%。随后加入降解菌剂或营养物质,开始进行菲的微生物降解。
-
化学预氧化后强化微生物降解对土壤中菲的降解率如图2所示。前7 d内(化学氧化阶段),单独土著微生物降解对照组(B-CK)仅去除了0.73%的菲,而活化过硫酸钠降解了22.7%的菲(对照组CK)。化学预氧化后,进一步培养B-CK、C-CK和CK等3组对照组,到第28天,B-CK最终降解率为18.43%,加入了抑菌剂NaN3的单独化学氧化对照组C-CK最终降解率为23.85%,而进行了化学预氧化-土著微生物降解的对照组CK的最终降解率为28.39%,分别较C-CK和B-CK提高4.54%和9.96%。上述结果表明,化学预氧化较单独微生物降解更能快速降解土壤中的菲,且化学预氧化后,残余的土壤微生物仍能对土壤中的菲进一步降解,进而取得更高的菲降解率,这为化学预氧化后强化微生物降解土壤中菲的研究提供了理论基础。
为进一步提高化学预氧化后土壤中多环芳烃菲的微生物降解率,通过生物强化(添加外源降解菌)和生物刺激(添加营养物质)2种手段,强化预氧化后土壤中微生物对菲的降解,结果如图2(微生物降解阶段)所示。可以看出:化学预氧化后,同时添加营养物质N和高效降解菌的实验组CBA+N,对土壤中菲的降解率最高,达41.29%;其次为添加营养物质N的实验组C+N(38.65%)。营养物质N和高效降解菌的加入,有效促进了土壤中菲的微生物降解,各实验组较对照组C-CK的降解率提高13%~17.44%,较未经生物强化的对照组CK提高8.46%~12.9%。
相比于营养物质N,添加营养物质P及NP复合营养液,对土壤中菲的微生物降解的促进有一定的迟滞性,营养物质添加1周后,菲的降解率增幅最高,仅为1.02%。自第2周起,才表现出明显的降解,最终实验组C+P、C+NP、CBA+P和CBA+NP中降解率分别达到33.86%、34.54%、35.06%和36.03%,较对照组C-CK提高10.01%~12.18%,较未经生物强化的对照组CK提高5.47%~7.64%。菲的降解结果表明,化学预氧化后耦合生物强化和生物刺激技术能够有效强化污染土壤中菲的降解。
-
如图3所示,化学预氧化会导致土壤中微生物数量急剧下降,但随着营养物质和高效降解菌的加入及培养时间的延长,微生物数量逐渐恢复。化学预氧化后同时加入菌剂和N盐,对微生物生长促进作用最强,P盐的加入对微生物生长的影响为先抑制后促进。在化学预氧化处理的实验组中,过硫酸钠的加入导致土壤中微生物数量急剧下降,从7.76lgN(N表示微生物菌落数,单位为CFU·g−1,下同)下降至2.33lgN。培养7 d后,微生物数量缓慢恢复至3.84lgN。营养物质P(实验组C+P、C+NP、CBA+P和CBA+NP)的加入,在短期内(第7~14天),抑制了土壤微生物数量的增长,各处理中微生物数量下降了3~63 CFU·g−1;而在后续培养中(第14~28天),微生物数量迅速增长。添加营养物质N及外源降解菌(实验组C+N、CBA、CBA+N),可有效促进土壤中微生物数量的增长。在第14~28天,除添加抑菌剂NaN3的对照组C-CK外,所有化学氧化-微生物联用实验组中土壤微生物数量均逐渐回升,增长率达4.10%~71.74%。这是生态系统稳定性的体现[19],也是化学氧化-微生物联用修复技术得以实现的基础。
-
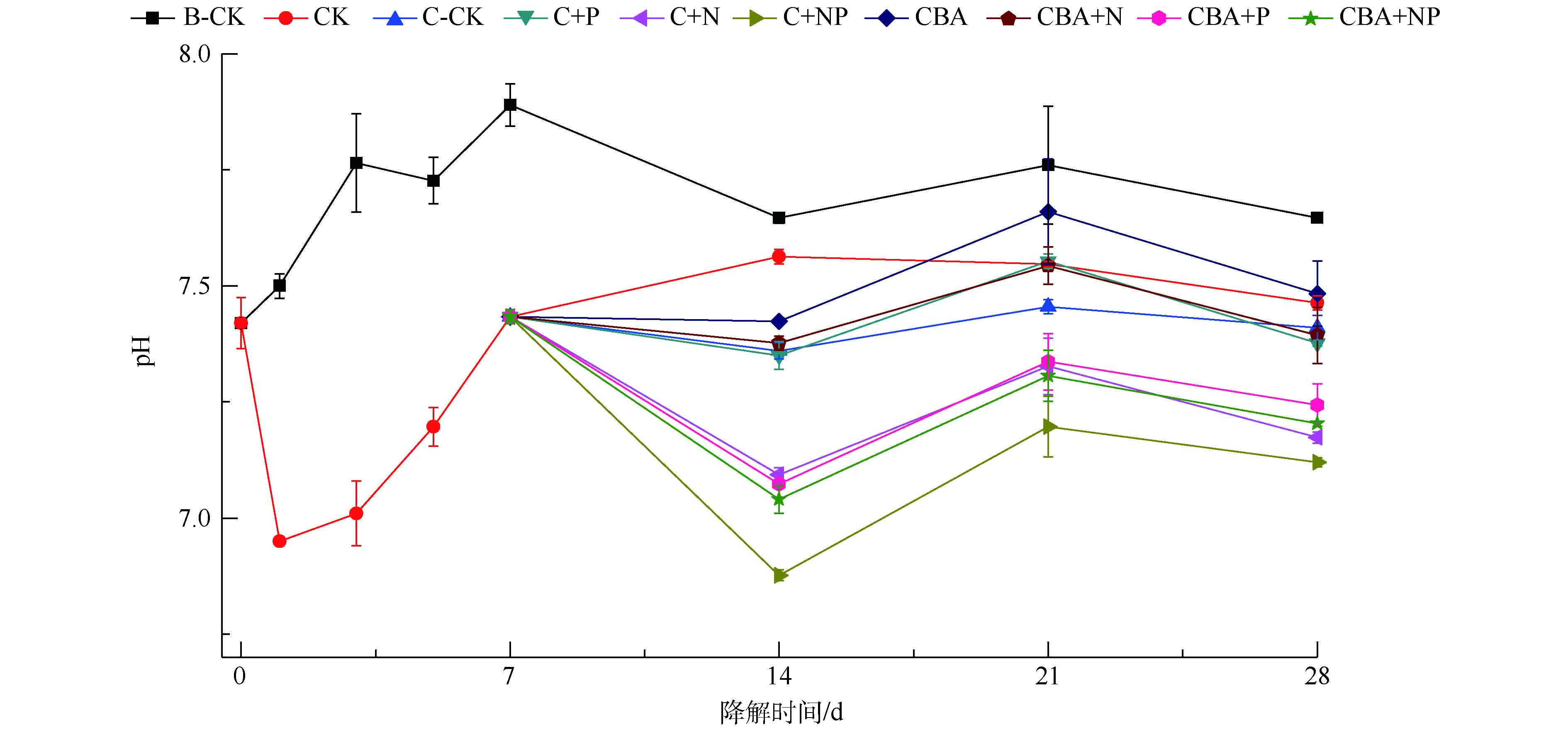
在化学预氧化作用下,强化微生物降解土壤中多环芳烃菲期间,土壤pH的变化情况见图4。化学氧化-微生物联用修复实验组中pH变化规律为先下降,后上升,最后维持稳定。在单独微生物降解对照组B-CK中,土壤pH从7.42上升至7.89,最后相对稳定地维持在7.70左右。这可能是土壤中土著微生物生长代谢期间,产生了某种碱性中间产物而导致的。过硫酸盐和P盐会导致土壤pH急剧下降,加入菌剂和N盐对pH影响较小。在化学预氧化实验组中,过硫酸盐的加入使得土壤pH由7.42迅速下降至6.95,随着过硫酸盐的消耗,pH逐渐恢复至氧化前水平。加入营养物质或菌剂,短期内会不同程度抑制土壤pH的持续升高。添加营养物质N和菌剂,对土壤pH抑制的程度较小,土壤pH仅轻微降低0.01~0.08,最终维持在7.37~7.48。而在添加了营养物质P的实验组C+P、C+NP、CBA+P和CBA+NP中,土壤pH下降幅度较大,分别下降了0.34、0.55、0.36和0.39。随着培养时间的增加,各实验组pH均逐渐回升,最终维持在7.12~7.24。
-
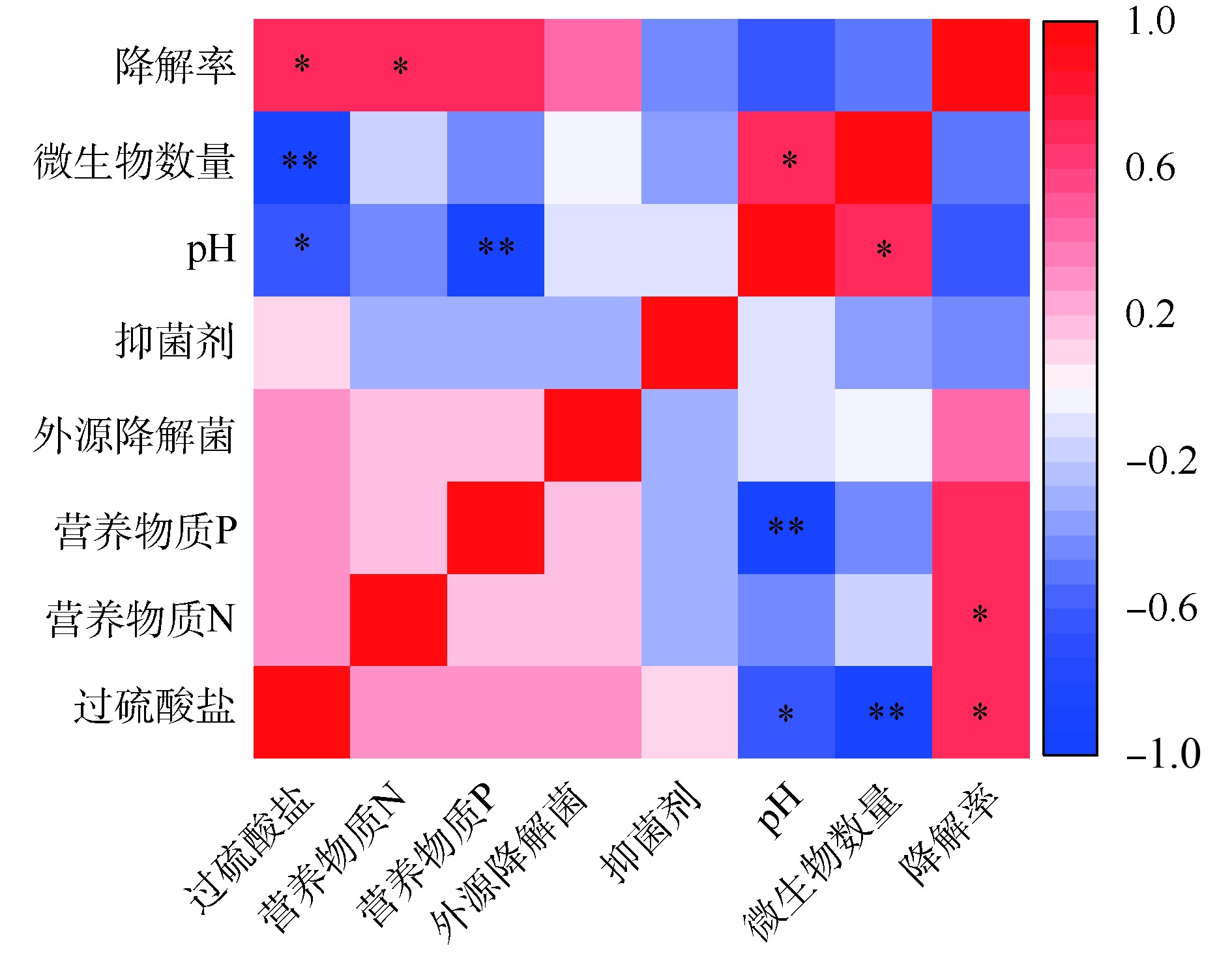
使用Pearson相关系数、T检验,对氧化剂、营养物质、菌剂和抑菌剂与土壤pH、微生物数量和菲降解率间的相关性进行分析,结果如图5所示。土壤中菲的降解率与过硫酸盐、营养物质和菌剂添加呈正相关,与抑菌剂、土壤pH呈负相关,其中过硫酸盐和营养物质N 2个因素与菲的降解率之间均具有显著的正相关性(P<0.05)。这也进一步证实了化学氧化结合生物强化和生物刺激,对土壤中菲的降解的促进作用。土壤pH与营养物质P呈显著负相关(r=−0.869,P<0.01),与过硫酸盐浓度呈显著负相关(r=−0.642,0.01<P<0.05)。微生物数量与过硫酸盐呈显著负相关(r=−0.898,P<0.01),与土壤pH呈显著正相关(r=0.724,0.01<P<0.05)。这说明过硫酸盐的加入对土壤pH和微生物数量具有严重负面影响。
-
氧化剂的加入会导致土壤pH和微生物数量急剧下降,但随着培养时间的增加,二者逐渐回升。土壤pH的下降可能是由于加入到污染土壤中的过硫酸钠活化分解,产生·
SO2−4 自由基的同时生成了大量SO2−4 和H+,使得土壤pH下降[20]。微生物数量的下降,一方面可能是由于pH骤然降低,土壤微生物难以适应,进而大量死亡。OGAWA等[21]指出,过高或过低的pH均会抑制土壤中微生物的生长。这也解释了在微生物降解阶段,加入酸式盐KH2PO4作为P源后,土壤微生物数量随着pH的降低而再一次下降的原因。另一方面,过硫酸盐分解产生的强氧化性自由基·SO2−4 也可能是导致土壤微生物数量迅速减少的原因之一。有研究表明,强氧化性的自由基及各种活性物质能够渗透进入细菌的细胞膜(壁),并破坏细胞内的成分,导致细胞死亡[22-23]。·SO2−4 可能会攻击细菌的细胞膜(壁),使得细胞膜(壁)破裂,导致细菌大量死亡。然而,化学氧化剂虽然在短期内会对微生物造成较大影响,但最终会随着时间的增加而逐渐恢复[19]。这使得化学氧化-微生物联用修复有机污染土壤成为可能。化学预氧化后添加营养物质和高效降解菌能够有效强化微生物对菲的降解,进而提高化学氧化-微生物联合修复对土壤中有机污染物的去除率。有研究表明,生物添加和生物刺激能够有效促进微生物对土壤中有机污染物的降解[24]。本实验使用热活化过硫酸钠进行化学预氧化后,添加外源高效降解菌和不同类型的营养物质,强化土壤微生物对残余多环芳烃的降解。各个处理最终降解率依次为CBA+N>C+N>CBA+NP>CBA+P>C+NP>C+P>CBA>CK>C-CK>B-CK。同时加入高效降解菌和营养物质N,能达到最优的降解效果。这与前人的研究结果稍有不同。ROY等[25]研究了加入降解菌和营养物质对土壤微生物降解总石油烃的促进作用,结果表明,同时加入降解菌和N、P 2种营养物质,对微生物的降解促进作用最强,其次为同时加入降解菌和营养物质N。造成这一差异的原因:一方面可能是在不同土壤中,微生物对不同类型营养物质的敏感性不同;另一方面则可能是由于作为P源添加的营养液为KH2PO4。KH2PO4属于酸式盐,导致降解体系中pH降低(如图3所示),在反应初期抑制了微生物活性,从而影响了微生物的降解效果。总的来说,在化学预氧化后,对土壤进行生物强化和生物刺激,可有效强化土壤微生物对残余有机污染物的降解,提高联用修复技术对有机污染物的修复效率。
将化学氧化与微生物修复技术相结合,对实际污染土壤的修复而言更具有参考价值。有研究表明,高浓度的过硫酸盐、高锰酸钾、Fenton等化学氧化剂对土壤微生物活性和理化性质均会带来不同程度的负面影响[26]。在使用化学氧化法修复各类有机污染土壤时,为保证修复效率,往往依赖于高浓度高剂量化学氧化剂的添加,这会带来严重的二次污染问题[27]。而微生物修复技术则存在修复时间长、难以修复高浓度污染场地等缺点,使其在实际污染场地修复的应用上仍存在较大的局限性[28]。本实验采用低浓度过硫酸盐对菲污染土壤进行预氧化,在降低了污染物浓度的同时有效减少了对土壤的二次污染,降低了土壤毒性;随后辅以生物强化和生物刺激2种典型的微生物修复技术对土壤中的菲进一步降解,最终获得了较高的降解率。这从绿色和可持续发展角度为有机污染土壤修复技术提供了新思路。
-
1)化学预氧化后耦合生物强化和生物刺激等手段能够有效强化微生物对土壤中菲的降解,是一种可行的化学氧化-微生物联合修复技术。活化过硫酸盐预氧化后,同时加入营养物质N和高效降解菌,对土壤中菲降解的促进作用最强;营养物质P(KH2PO4)的加入对菲降解的促进作用存在滞后现象;仅添加高效降解菌,对土壤中菲降解的促进作用最差。
2)氧化剂过硫酸钠和营养物质P(KH2PO4)的加入,在短期内会导致土壤pH和微生物数量急剧下降。随着培养时间的延长,pH和微生物数量会逐渐恢复。
3)土壤中菲的降解率与过硫酸盐和营养物质N的添加呈显著正相关。微生物数量与过硫酸盐的添加呈负相关,与土壤pH呈正相关。土壤pH受过硫酸盐和营养物质P的负面影响较大。
化学预氧化-生物强化-生物刺激对土壤中菲降解的联合效应
Joint degradation effects of phenanthrene in soil by chemical pre-oxidation-bioaugmentation-biostimulation
-
摘要: 通过室内实验,探究了低浓度过硫酸盐预氧化耦合生物强化或生物刺激技术处理下土壤中菲的降解率和修复效应。结果表明,浓度为0.1 mmol·g−1、温度为50 ℃热活化的过硫酸钠对土壤中菲7 d的降解率为22.7%。预氧化后,加入高效降解菌和营养物质,强化微生物对菲的降解,继续培育21 d,最终降解率较第7天可提高8.08%~18.59%。同时添加高效降解菌和营养物质N,对土壤中菲的降解促进作用最强,最终降解率可达41.29%,较仅进行化学氧化的对照组和仅进行微生物降解的对照组分别提高17.44%和22.86%,较预氧化后不进行微生物强化的对照组提高12.9%。降解期间,土壤微生物数量和pH呈先下降,后上升趋势,最终维持在相对稳定水平。相关性分析结果表明,土壤中菲的降解率与氧化剂和营养物质N的添加呈显著正相关,土壤微生物数量与pH呈正相关,与氧化剂呈负相关,土壤pH与氧化剂及营养物质P呈负相关。研究结果证实了化学预氧化耦合生物强化和生物刺激技术能有效促进微生物对菲污染土壤的修复。Abstract: The present study was conducted to investigate the degradation of phenanthrene in soil by low-concentration persulfate pre-oxidation combined bioaugmentation and biostimulation using high-efficiency degrading bacteria and different types of nutrients. The results showed that degradation efficiency of phenanthrene was 22.7% after 7 days treatment with 0.1 mmol·g−1 persulfate concentration and thermal activation at 50 ℃. Then the phenanthrene degradation bacteria and nutrients were added into the pre-oxidized matrix, and performed 21 days continuous cultivation, the final degradation efficiency of phenanthrene increased by 8.08%~18.59%. The simultaneous addition of degradation bacteria and nutrient N was the most effective way to promote the degradation of phenanthrene in soil, and the final degradation efficiency could reach 41.29%. This was 17.44% higher than that of the chemical oxidation control group, 22.86% higher than that of the microbial degradation control group and 12.9% higher than that of pre-oxidation without biofortification control group. During the degradation process, soil microbial biomass and pH decreased at first, then increased, and finally maintained at a relatively stable level. The results of correlation analysis showed that degradation efficiency of phenanthrene in soil was significantly positively correlated with the addition of oxidant and nutrient N, soil microbial biomass was positively correlated with pH, negatively correlated with the addition of oxidant. Soil pH was negatively correlated with the addition of oxidant and nutrient P. These results confirmed that chemical pre-oxidation combined with bioaugmentation and biostimulation can effectively promote the bioremediation of phenanthrene-contaminated soil.
-
Key words:
- chemical oxidation /
- biodegradation /
- bioaugmentation /
- biostimulation /
- phenanthrene
-
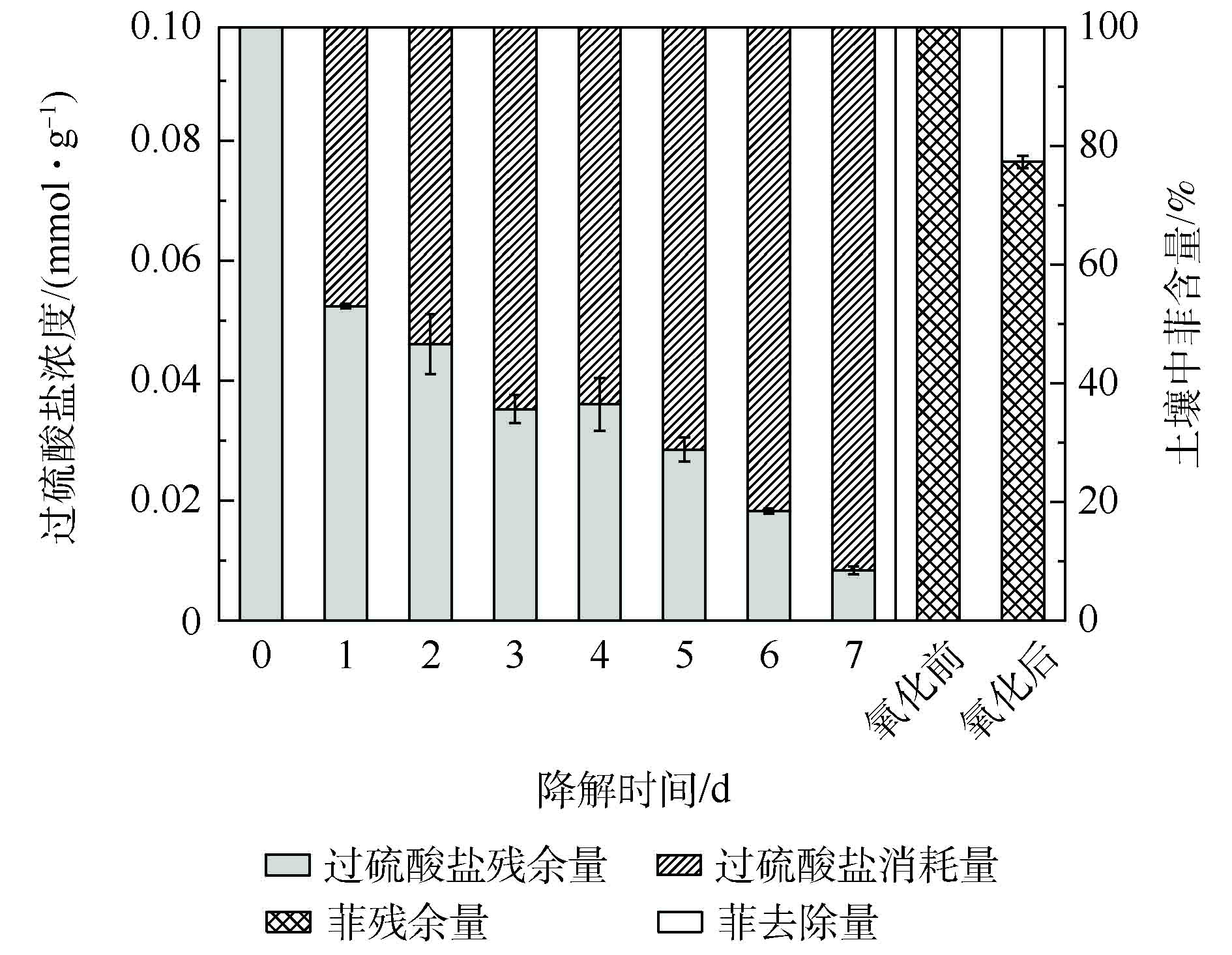
图 1 化学预氧化处理菲的降解率和过硫酸盐含量的变化
Figure 1. Changes of persulfate concentrations and PHE degradation efficiency in the chemical pre-oxidation treatments
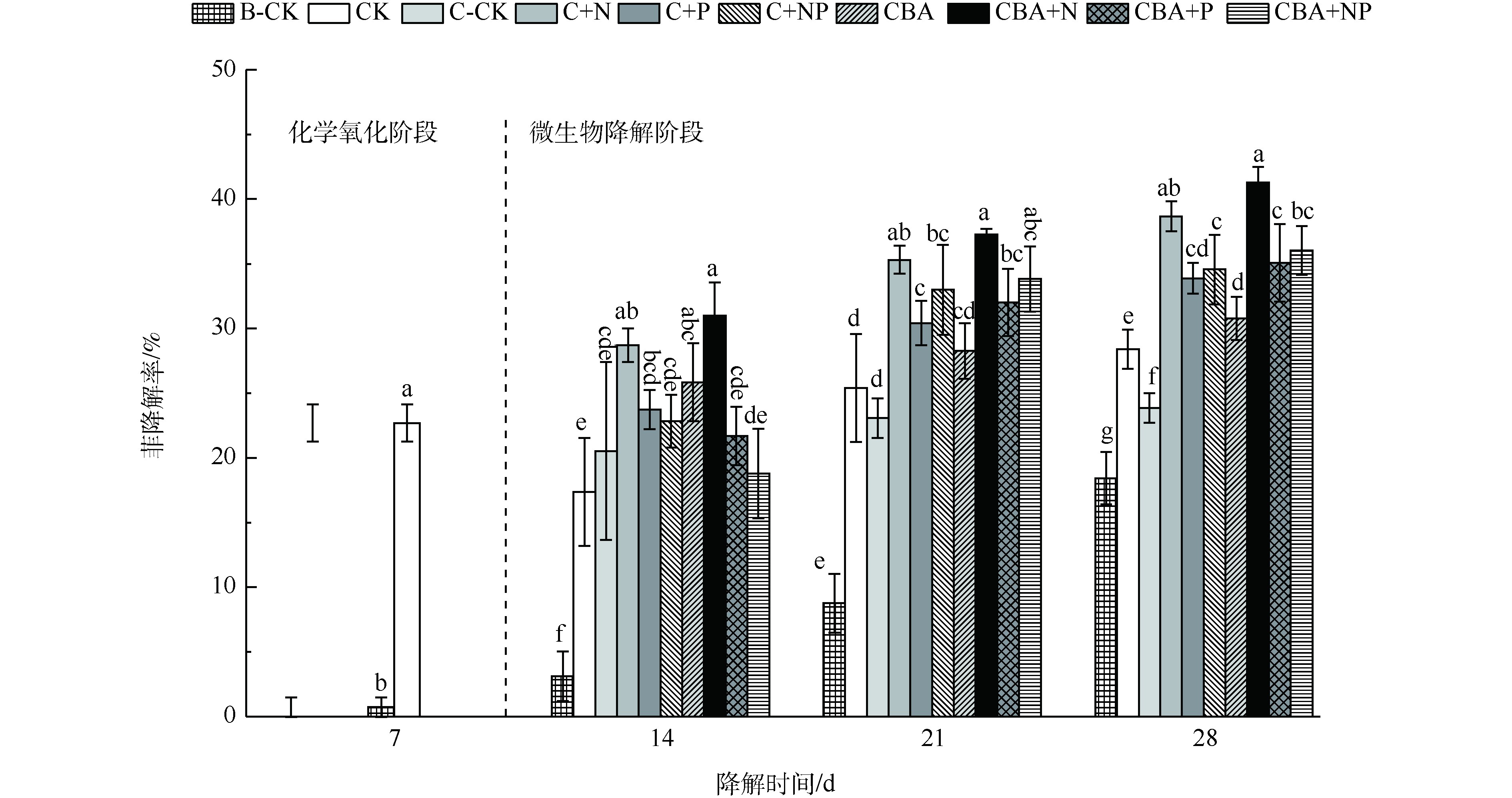
图 2 不同处理下土壤菲的降解率
Figure 2. Degradation efficiency of soil phenanthrene by different treatments
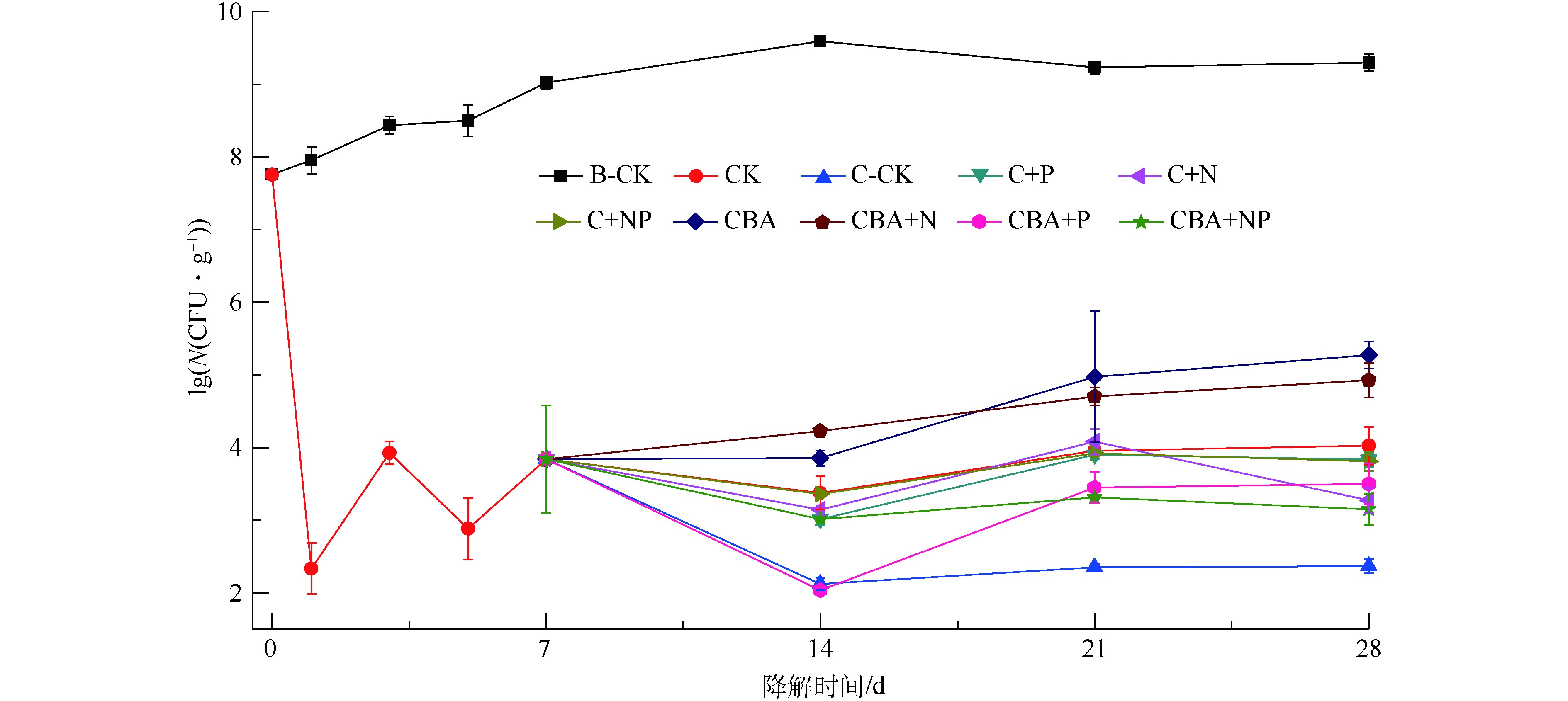
图 3 降解期间土壤微生物数量的变化
Figure 3. Variations of soil microbial biomass in the degradation process
表 1 实验处理组
Table 1. Experimental treatment groups
实验处理组 处理组简称 添加药剂 单独微生物降解对照 B-CK 化学氧化阶段:不进行化学氧化,仅加入等量去离子水 微生物降解阶段:等量去离子水 单独化学氧化对照 C-CK 化学氧化阶段:0.1 mmol·g−1 Na2S2O8 微生物降解阶段:0.1 mL·g−1 0.2 mol·L−1 NaN3 化学氧化-土著微生物降解对照 CK 化学氧化阶段:0.1 mmol·g−1 Na2S2O8 微生物降解阶段:等量去离子水 化学氧化+营养物质N刺激 C+N 化学氧化阶段:0.1 mmol·g−1 Na2S2O8 微生物降解阶段:0.187 mol·g−1 NaNO3 化学氧化+营养物质P刺激 C+P 化学氧化阶段:0.1 mmol·g−1 Na2S2O8 微生物降解阶段:0.018 7 mol·g−1 KH2PO4 化学氧化+营养物质NP刺激 C+NP 化学氧化阶段:0.1 mmol·g−1 Na2S2O8 微生物降解阶段:0.187 mol·g−1 NaNO3;0.018 7 mol·g−1 KH2PO4 化学氧化+生物强化 CBA 化学氧化阶段:0.1 mmol·g−1 Na2S2O8 微生物降解阶段:0.1 mL·g−1 菌液 化学氧化+生物强化+营养物质N刺激 CBA+N 化学氧化阶段:0.1 mmol·g−1 Na2S2O8 微生物降解阶段:0.1 mL·g−1 菌液;0.187 mol·g−1 NaNO3 化学氧化+生物强化+营养物质P刺激 CBA+P 化学氧化阶段:0.1 mmol·g−1 Na2S2O8 微生物降解阶段:0.1 mL·g−1 菌液;0.018 7 mol·g−1 KH2PO4 化学氧化+生物强化+营养物质NP刺激 CBA+NP 化学氧化阶段:0.1 mmol·g−1 Na2S2O8 微生物降解阶段:0.1 mL·g−1 菌液;0.187 mol·g−1 NaNO3;0.018 7 mol·g−1 KH2PO4  下载: 导出CSV
下载: 导出CSV
-
[1] 杨坤, 李尤, 刘琼枝, 等. 多环芳烃(PAHs)在不同腐殖酸组分中的赋存特征和氧化降解效果研究[J]. 环境科学学报, 2017, 37(11): 4277-4286. [2] ESPLUGAS S, BILA D M, KRAUSE L G T, et al. Ozonation and advanced oxidation technologies to remove endocrine disrupting chemicals (EDCs) and pharmaceuticals and personal care products (PPCPs) in water effluents[J]. Journal of hazardous materials, 2007, 149(3): 631-642. doi: 10.1016/j.jhazmat.2007.07.073 [3] WANG J, WANG S. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants[J]. Chemical Engineering Journal, 2018, 334: 1502-1517. doi: 10.1016/j.cej.2017.11.059 [4] 刘楚琛, 阎秀兰, 刘琼枝, 等. Fenton试剂和活化过硫酸钠氧化降解土壤中的二氯酚和三氯酚[J]. 环境工程学报, 2018, 12(6): 1749-1758. [5] GITIPOUR S, SORIAL G A, GHASEMI S, et al. Treatment technologies for PAH-contaminated sites: A critical review[J]. Environmental Monitoring and Assessment, 2018, 190(9): 546. doi: 10.1007/s10661-018-6936-4 [6] KAKOSOVA E, HRABAK P, CERNIK M, et al. Effect of various chemical oxidation agents on soil microbial communities[J]. Chemical Engineering Journal, 2016, 314: 257-265. [7] AYDIN S, KARACY H A, SHAHI A, et al. Aerobic and anaerobic fungal metabolism and omics insights for increasing polycyclic aromatic hydrocarbons biodegradation[J]. Fungal Biology Reviews, 2017, 31(2): 61-72. doi: 10.1016/j.fbr.2016.12.001 [8] GAUR N, NARASIMHULU K, PYDISETTY Y. Recent advances in the bio-remediation of persistent organic pollutants and its effect on environment[J]. Journal of Cleaner Production, 2018, 198: 1602-1631. doi: 10.1016/j.jclepro.2018.07.076 [9] 吴昊, 孙丽娜, 王辉, 等. 活化过硫酸钠原位修复石油类污染土壤研究进展[J]. 环境化学, 2015, 34(11): 2085-2095. doi: 10.7524/j.issn.0254-6108.2015.11.2015052601 [10] GAN S, LAU E V, NG H K. Remediation of soils contaminated with polycyclic aromatic hydrocarbons (PAHs)[J]. Journal of Hazardous Materials, 2009, 172(2): 532-549. [11] LIAO X Y, WU Z Y, LI Y, et al. Enhanced degradation of polycyclic aromatic hydrocarbons by indigenous microbes combined with chemical oxidation[J]. Chemosphere, 2018, 213: 551-558. doi: 10.1016/j.chemosphere.2018.09.092 [12] SUTTON N B, LANGENHOFF A A M, RIJNAARTS H H M. Efforts to improve coupled in situ chemical oxidation with bioremediation: A review of optimization strategies[J]. Journal of Soils & Sediments, 2011, 11(1): 129-140. [13] XU J, DENG X, CUI Y, et al. Impact of chemical oxidation on indigenous bacteria and mobilization of nutrients and subsequent bioremediation of crude oil-contaminated soil[J]. Journal of Hazardous Materials, 2016, 320: 160-168. doi: 10.1016/j.jhazmat.2016.08.028 [14] MORA V C, MADUENO L, PELUFFO M, et al. Remediation of phenanthrene-contaminated soil by simultaneous persulfate chemical oxidation and biodegradation processes[J]. Environmental Science & Pollution Research, 2014, 21(12): 7548-7556. [15] SANTANA M S, SANDRINI-NETO L, NETO F F, et al. Biomarker responses in fish exposed to polycyclic aromatic hydrocarbons (PAHs): Systematic review and meta-analysis[J]. Environmental Pollution, 2018, 242: 449-461. doi: 10.1016/j.envpol.2018.07.004 [16] KUPPUSAMY S, THAVAMANI P, VENKATESWARLU K, et al. Remediation approaches for polycyclic aromatic hydrocarbons (PAHs) contaminated soils: Technological constraints, emerging trends and future directions[J]. Chemosphere, 2017, 168: 944-968. doi: 10.1016/j.chemosphere.2016.10.115 [17] LIANG C, HUANG C F, MOHANTY N, et al. A rapid spectrophotometric determination of persulfate anion in ISCO[J]. Chemosphere, 2008, 73(9): 1540-1543. doi: 10.1016/j.chemosphere.2008.08.043 [18] 赵丹, 阎秀兰, 廖晓勇, 等. 不同化学氧化剂对焦化污染场地苯系物的修复效果[J]. 环境科学, 2011, 32(3): 849-856. [19] SAHL J, MUNAKATA-MARR J. The effects of in situ chemical oxidation on microbiological processes: A review[J]. Remediation Journal, 2006, 16(3): 57-70. doi: 10.1002/(ISSN)1520-6831 [20] WALDEMER R H, TRATNYEK P G, JOHNSON R L, et al. Oxidation of chlorinated ethenes by heat-activated persulfate: Kinetics and products[J]. Environmental Science & Technology, 2007, 41(3): 1010-1015. [21] OGAWA M, OKIMORI Y. Pioneering works in biochar research, Japan[J]. Soil Research, 2010, 48(7): 489-500. doi: 10.1071/SR10006 [22] CHANDANA L, SANGEETHA C J, SHASHIDHAR T, et al. Non-thermal atmospheric pressure plasma jet for the bacterial inactivation in an aqueous medium[J]. Science of the Total Environment, 2018, 640-641: 493-500. doi: 10.1016/j.scitotenv.2018.05.342 [23] GAUNT L F, BEGGS C B, GEORGHIOU G E. Bactericidal action of the reactive species produced by gas-discharge nonthermal plasma at atmospheric pressure: A review[J]. IEEE Transactions on Plasma Science, 2006, 34(4): 1257-1269. doi: 10.1109/TPS.2006.878381 [24] JASMINE J, MUKHERJI S. Characterization of oily sludge from a refinery and biodegradability assessment using various hydrocarbon degrading strains and reconstituted consortia[J]. Journal of Environmental Management, 2015, 149(7): 118-125. [25] ROY A, DUTTA A, PAL S, et al. Biostimulation and bioaugmentation of native microbial community accelerated bioremediation of oil refinery sludge[J]. Bioresource Technology, 2018, 253: 22-32. doi: 10.1016/j.biortech.2018.01.004 [26] CHEN K F, CHANG Y C, CHIOU W T. Remediation of diesel-contaminated soil using in situ chemical oxidation (ISCO) and the effects of common oxidants on the indigenous microbial community: A comparison study[J]. Journal of Chemical Technology & Biotechnology, 2016, 91(6): 1877-1888. [27] CHENG M, ZENG G, HUANG D, et al. Hydroxyl radicals based advanced oxidation processes (AOPs) for remediation of soils contaminated with organic compounds: A review[J]. Chemical Engineering Journal, 2016, 284: 582-598. doi: 10.1016/j.cej.2015.09.001 [28] YONG Y C, ZHONG J J. Recent advances in biodegradation in China: New microorganisms and pathways, biodegradation engineering, and bioenergy from pollutant biodegradation[J]. Process Biochemistry, 2010, 45(12): 1937-1943. doi: 10.1016/j.procbio.2010.04.009 -

 点击查看大图
点击查看大图
图( 5) 表( 1)
计量
- 文章访问数: 4186
- HTML全文浏览数: 4186
- PDF下载数: 58
- 施引文献: 0
