







-
铀矿冶理虽经过数十年的发展,对铀尾矿等中低放固废的处置方法仍以近地表处置为主[1-3]。长期露天堆放的铀尾矿,经过雨水淋滤后能析出铀等具有一定生物毒性和放射性的核素[4-6],他们进入土壤后对环境产生了极大的污染[7]。学者们对土壤剖面中铀的分布规律进行了大量研究,目前已知土壤pH、有机质、针铁矿、碳酸盐、磷酸盐、石英等众多因素均能影响铀在土壤剖面中的吸附和空间分布[5, 8-13]。近年来,针对土壤吸附铀的机理研究虽逐渐增加,但相关成果大多聚焦于单一组分对铀的影响[14-16]。
与许多研究结果不同,自然环境中土壤剖面各土层受地表生物活动、风化等因素的影响,土壤形成和发育过程中分化为多个土层,各土层间的理化性质存在较大区别,使得它们对铀吸附能力差异明显。目前,针对铀尾矿库中各土层对铀吸附研究,大多集中在土壤表层(腐殖质层)[17]。绝大部分铀尾矿库,由于年代久远,加上建设之初未设置防渗措施[18],铀等放射性核素不仅污染与其直接接触的腐殖质-淋溶层,也可能同有机胶体、无机胶体络合迁移至土壤剖面中更深的淀积层、母质层甚至地下水中,但目前针对这一情况的研究较少。
本研究采用静态吸附法考察了南方某铀尾矿库附近棕红壤剖面中的淋溶层、淀积层和母质层等土层对U(Ⅵ)吸附差异,从热力学和动力学方面分析吸附过程;并使用XRF、XRD、SEM、FT-IR等表征手段对吸附规律进行了分析。研究了土壤剖面中各土层对外源铀的吸附,有利于探讨铀尾矿库等核设施污染土壤中铀的空间分布及各土层对铀的吸附规律,本研究可为铀尾矿库土壤剖面中的外源铀污染综合治理及地下水防治等工作提供参考。
-
取南方某铀尾矿库附近洁净棕红壤剖面中1.5 m处淋溶层、3 m处淀积层和6 m处母质层土样各4个,每个样约重5 kg,选出石块、树根等杂物后破碎后过8目筛,室内阴干30 d,采用二分法混匀后,密封保存至干燥瓶中备用。按中国土壤颗粒分级(DCHN)进行筛分,结果见表1。
-
实验中所有的试剂均为化学纯。取1.215 7 g纯度为97%的U3O8,使用少量硝酸加热氧化直至完全溶解,冷却至常温后定容至1 L容量瓶中,配制成1 g·L−1的铀标准溶液,按需稀释而成含U(Ⅵ)溶液。参照南方某铀尾矿库滤液的实测pH(6.2)来调整模拟含U(Ⅵ)溶液的pH。
-
各土层理化性质测定结果见表2。其中自然密度、干密度、含水率和有机质含量根据GB 50123-1999进行测定;pH由FE20型pH计测得;比表面积采用V-Sorb 2800P型全自动比表面和孔径分布分析仪测定;使用Niton XL3t型X射线荧光光谱分析仪对各土层主要组成元素进行分析,结果见表3。
研究单因素条件对吸附U(Ⅵ)的影响时,使用BSA623S-CW型电子天平称取0.1 g待测土样,移入250 mL具塞锥形瓶中。加入100 mL浓度为30 mg·L−1的含U(Ⅵ)溶液,加塞密封,置于THZ-100 B型水浴恒温振荡器中振荡90 min(母质层土样为120 min),静置至上部液体澄清后,测量溶液中残余U(Ⅵ)浓度以确定各土层对U(Ⅵ)吸附量。调整吸附时间、U(Ⅵ)浓度、温度、pH和土壤粒径等因素,以确定不同条件下对U(Ⅵ)吸附效果的影响。每组做3次平行实验,结果取平均值。
对吸附前后各土层进行电子显微镜扫描(JSM-7500F型,日本JOEL株式会社),以分析各土层间的形貌差异;使用Bruker D8型X射线衍射仪和Nicolet-460型傅里叶变换红外光谱仪分别对各土层的矿物晶体组成与官能团进行表征分析。
-
吸附完成后,上清液过滤,移取5 mL至150 mL锥形瓶,加入8 mL纯H3PO4和1 mL纯HCl,不断振荡锥形瓶并逐滴加入15%三氯化钛,直至溶液呈稳定紫红色,并过量2滴。逐滴加入15%亚硝酸钠至溶液呈透明状,随后立即加入6 mL 20%尿素并不断震荡锥形瓶至气泡消失,静置5 min。滴入3滴0.2%二苯胺磺酸钠,使用0.05%的钒酸铵标准溶液滴定至溶液呈微紫红色且30 s不褪色即为终点。由钒酸铵溶液体积和浓度计算消耗钒酸铵总量,由此确定U(Ⅵ)溶液浓度,计算方法见《铀矿石中铀的测定-三氯化钛还原钒酸铵氧化滴定法》(EJ 267.1-267.5-84)。
-
除吸附时间、U(Ⅵ)溶液初始浓度、pH、温度和土壤粒径等影响因素外,有机质、微生物、
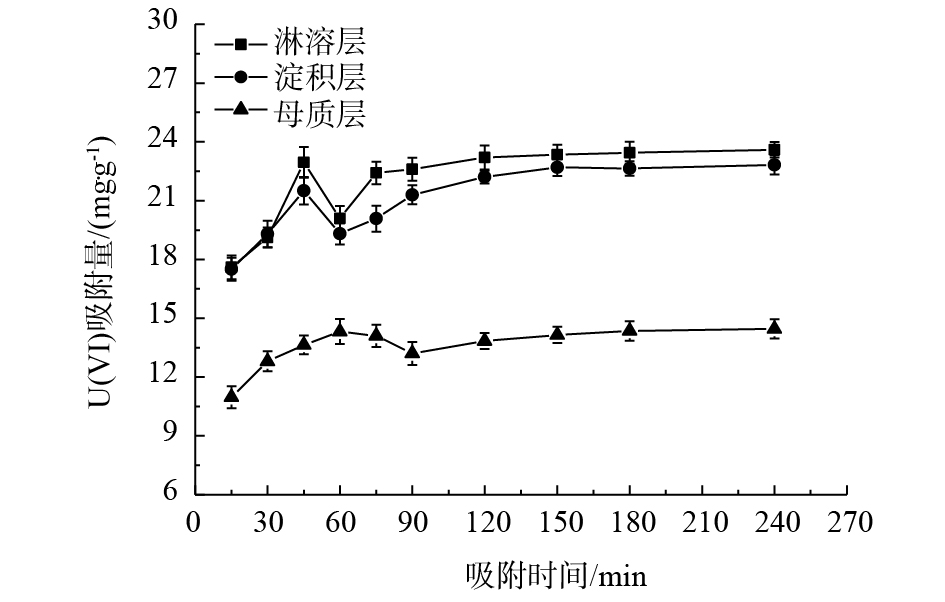
CO2−3 、PO3−4 等离子强度及土壤中其他矿物组分等的改变,同样能使各土层对U(Ⅵ)吸附能力产生变化,这些在其他学者研究中得到证实[19-21]。实验主要针对不同土层本身理化性质的调变对吸附U(Ⅵ)的影响开展讨论。1)吸附时间。吸附时间对各土样吸附U(Ⅵ)的影响结果如图1所示。淋溶层、淀积层和母质层分别在90、90和120 min时达到吸附平衡,平衡时各土层对U(Ⅵ)吸附量分别为23.60、22.82和13.05 mg·g−1。淋溶层、淀积层在45~60 min时、母质层在60~90 min时吸附量均开始降低,出现“解吸”现象。这是由于吸附初期U(Ⅵ)随溶液在土壤颗粒空隙间进行扩散,以孔隙吸附等物理吸附为主。随着时间递增,各层土样开始出现解吸[22],之后土壤有机质中—OH及C=O、C=C等基团和丰富的Fe—OH/Al—OH等无机胶体与
UO2+2 逐渐络合沉淀使得土壤对U(Ⅵ)吸附量重新增长[23]。土壤投加量一定的情况下,很难增加新的吸附位点,最终土壤颗粒活性位点与溶液中游离的U(Ⅵ)结合的空间位阻增大直至平衡。由此可见,各土壤对U(Ⅵ)吸附效果主要集中在0~120 min,随着时间的延长,吸附效果变化较小。2)U(Ⅵ)初始浓度。U(Ⅵ)初始浓度对各土层吸附U(Ⅵ)的影响结果如图2所示。本研究中,U(Ⅵ)溶液最高初始浓度为960 mg·L−1,此时,对应各土层对铀的吸附量达到最大值,分别为42.12、40.26和31.69 mg·g−1。在初始浓度为15~120 mg·L−1时,3类土样对U(Ⅵ)吸附量增长迅速;当U(Ⅵ)初始浓度上升至120~960 mg·L−1时,各土层对铀吸附量增长趋于缓慢。这是因为加入土样质量不变时,总的吸附位点基本不变,使得吸附量趋于饱和。同时淋溶层和淀积层对U(Ⅵ)吸附能力明显强于母质层。分析其原因,主要是由于不同土层中有机质、Fe、Mn等元素含量、土壤孔隙率与比表面积等理化性质不同所致。
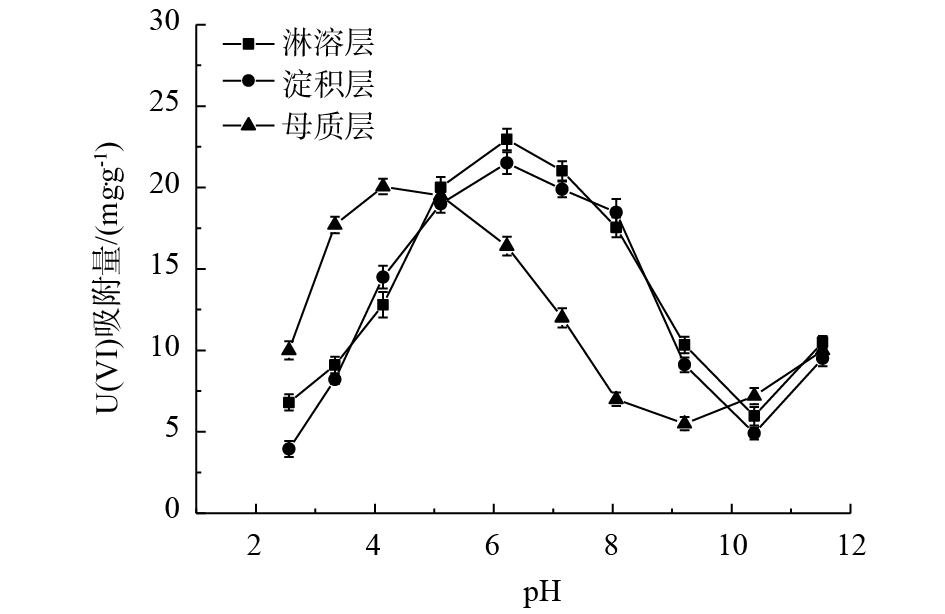
3) pH。溶液pH对各层土壤吸附铀的影响结果见图3。当pH为6.2时,淋溶层、淀积层土样对U(Ⅵ)吸附量达到最大值,其后随着pH的升高,吸附量开始下降;pH超过10.4时吸附量重新增加。pH对母质层吸附U(Ⅵ)的影响变化趋势类似,但各拐点的pH有所不同,在pH为4.1时,达到最大吸附值。已有的研究结果[24-25]很好地解释了这一现象,pH为3.7时,Fe(OH)3与U(Ⅵ)开始产生少量沉淀物;pH升至6.2时铀酰离子易水解成UO2OH+,同Fe(OH)3等胶体吸附沉淀。随着pH继续升高,UO2OH+开始转化为(UO2)2CO3(OH)3−、UO2(CO3)22−,这些阴离子同土样配位点及有、无机胶体表面产生静电斥力,阻碍了各类胶体对U(Ⅵ)的吸附,吸附量开始降低。pH到达10后,溶液中铀酰离子以UO2(CO3)34−沉淀为主,且棕红壤中丰富的Fe(OH)3胶体开始增大复溶,重新同铀酰离子进行配位络合吸附[26]。而母质层在pH为4.1时,吸附效果优于其他2层土样,这可能是母质层pH高于前两者,与溶液混合后相对较高的pH使溶液中UO22+更易水解成UO2OH+,同Fe(OH)3等胶体吸附沉淀。可见,pH对各土层吸附U(Ⅵ)的影响较大。
4)温度。温度对各土层中的U(Ⅵ)的吸附影响结果见图4。升温使母质层土样对U(Ⅵ)的吸附量不断增加,在55 ℃时可达到最大值。而淋溶层与淀积层土样在25 ℃时对U(Ⅵ)的吸附达到最大值,温度继续升高,2个土层的吸附量均开始缓慢降低。温度达到55 ℃时,3类土样对U(Ⅵ)的吸附量的排序为淋溶层>淀积层>母质层,但差距并不显著,这与冯明明[26]的研究结论相一致。结合FT-IR表征数据可知,淋溶层、淀积层中含有C=C、—COOH、—OH、—CH2、C=O、O=C—NH2等众多有机官能团,这些官能团均参与对U(Ⅵ)吸附,但升温会导致各官能团逐渐失活,由于各官能团对温度响应不完全相同,故削弱了升温对单一基团失活的影响[27]。另一方面,升温导致土壤中的矿物和Fe—OH、Al—OH等无机胶体的表面位点吸附活化能升高,提高了吸附位点的有效密度,吸附熵也随之变大,导致了母质层等土层吸附容量的增大[28],这一过程也缓解了由升温导致淋溶层和淀积层中的有机官能团失活带来的“解吸”现象。
5)土壤粒径。按1.3节实验方法,取不同粒径的土样探讨其对U(Ⅵ)吸附效果,结果如图5所示。淋溶层、淀积层和母质层土样对U(Ⅵ)去除率和吸附量均随粒径的减小而增加,各层土样在粒径≤200目对U(Ⅵ)吸附量时均达到最大值。一方面,粒径减小使得土样的单位比表面积更大;另一方面,颗粒越细的土样在水中的胶体聚合体往往越多。淋溶层土样同淀积层和母质层土样相比,其风化程度更好,粒径更小,土壤孔隙更多,有、无机胶体的含量更高,使得淋溶层土样对铀酰离子吸附能力强于淀积层和母质层。
-
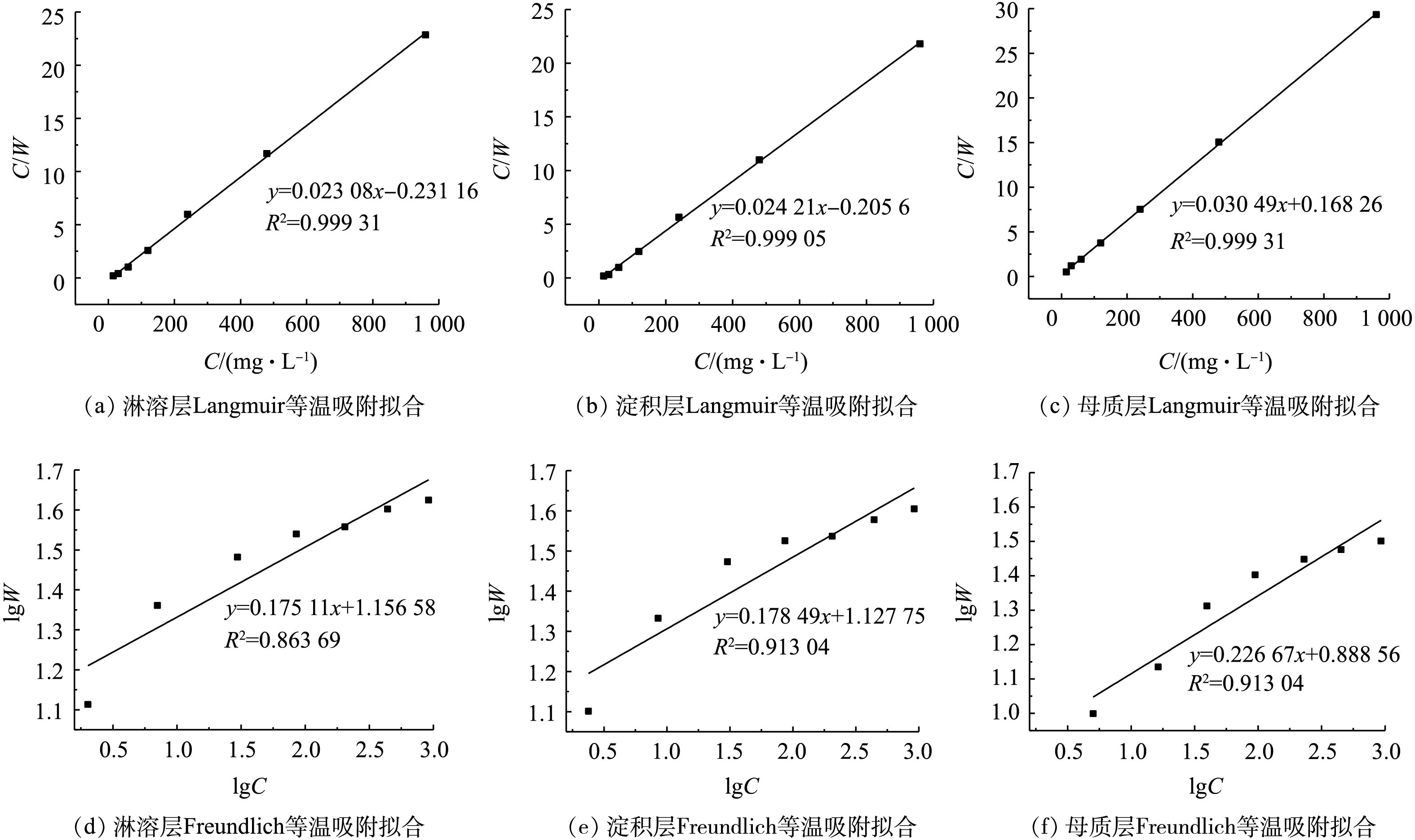
各土层对U(Ⅵ)吸附行为分别用Langmuir及Freundlich[29-30]模型进行描述,分别如式(1)和式(2)所示。
式中:C为吸附平衡时溶液浓度,mg·L−1;W为平衡时的吸附量,mg·g−1;WS为最大吸附量,mg·g−1;KL、KF和1/n为等温吸附常数。图6中的2种等温吸附拟合结果表明,3类土样均有较佳的拟合度,这说明物理和化学吸附过程并存。从可决系数R2可以看出,3类土样的拟合度均为:Langmuir方程>Freundlich方程,且Langmuir方程对3类土样的拟合程度均较高(R2>0.99),这说明3类土样对U(Ⅵ)实际吸附行为更贴合单层,均质吸附[31]。故土样单位比表面积越大,对U(Ⅵ)吸附能力越强,这一点也被粒径实验结果所证实。
-
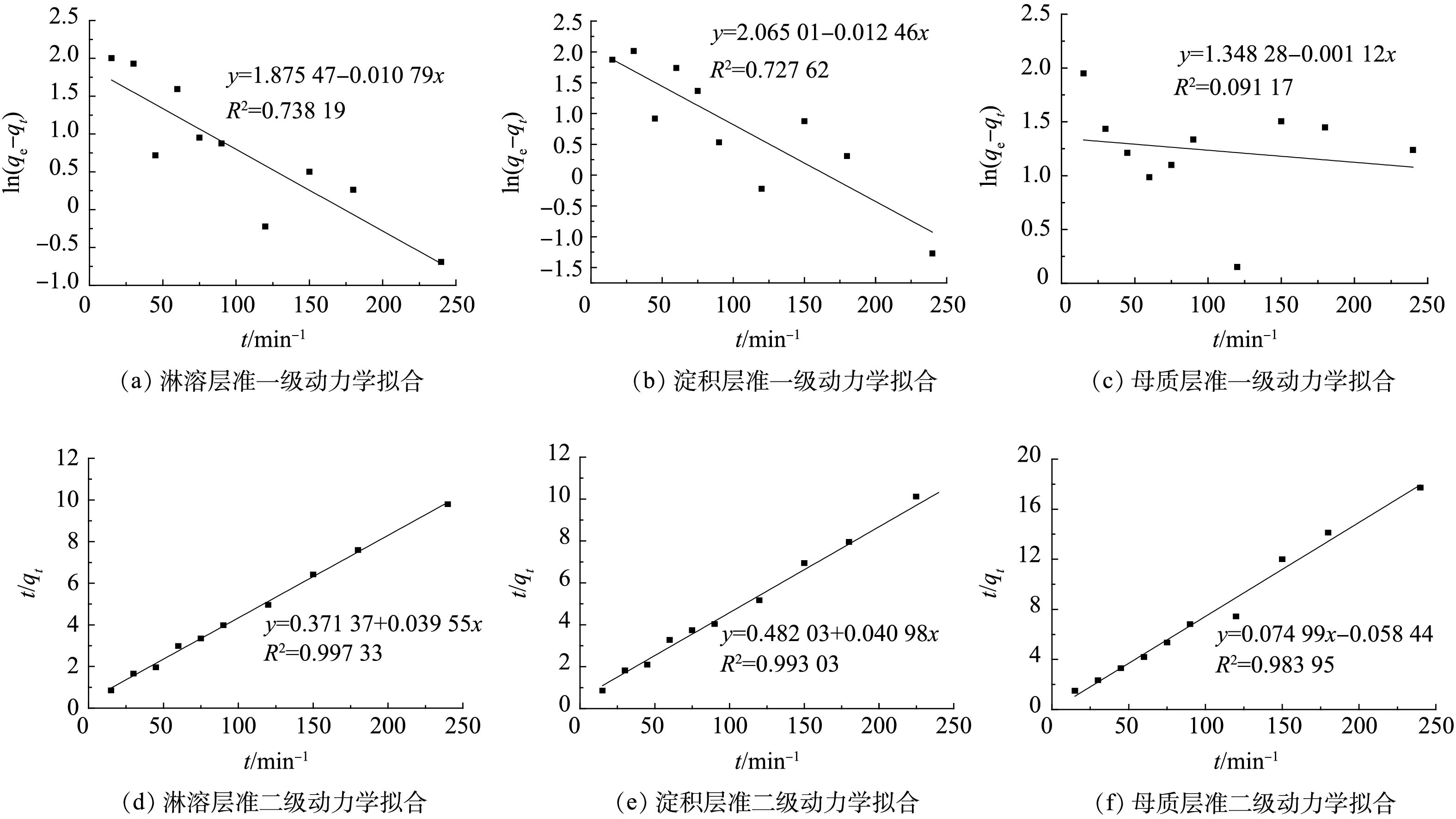
吸附动力学模型可以反映出吸附过程的快慢,因此,本研究使用准一级和准二级速率方程[29-30]来描述3类土样对铀的吸附速率,其计算公式如式(3)~式(4)所示。
式中:qt为单位土样t时刻对U(Ⅵ)的吸附量,mg·g−1;qe为平衡时单位土样对U(Ⅵ)吸附量,mg·g−1;t为吸附时间,min;k1为准一级速率方程吸附常量,h−1;k2为准二级速率方程吸附常量,g·(mg·h)−1,qe、k1和k2由拟合直线的斜率和截距计算得出。
从图7可以看出,准二级动力学方程拟合结果明显优于准一级动力学方程,这表明准二级动力学方程更贴合3类棕红壤土层对U(Ⅵ)的吸附过程。这是因为准二级动力学模型考虑了外部扩散、表面吸附和内部扩散作用,对U(Ⅵ)的吸附机理描述更加全面。同时,3类土样对准二级动力学方程的拟合度为淋溶层>淀积层>母质层,说明淋溶层对U(Ⅵ)吸附能力优于淀积层和母质层,且主要以化学吸附为主。
-
1)扫描电镜分析。图8为各层土样SEM表征结果。可以看出,样品的形貌大小不一,粒径分布差异较大。由图8(a)和图8(b)可知,U(Ⅵ)吸附前后的淋溶层土样并无大的结构变化,吸附后,土壤颗粒表面整体相对较平整,这说明表面已被吸附的UO22+填充。比较图8(a)、图8(b)、图8(c)可以明显看出,虽各土层基于同一岩层风化而成,但淋溶层土样颗粒更为松散且呈不规则状,孔隙更多,土样的单位比表面积相对更大。淀积层、母质层土样颗粒呈团聚体,与淋溶层相比粒径明显更大,且母质层颗粒更为致密,说明风化程度较低,BET测试数据也很好地解释了这一结果。
2)傅里叶红外光谱分析。各层土样吸附前的红外光谱如图9所示。淋溶层、淀积层土样在3 699 cm−1处出现吸收峰,此处多为石英等黏土矿物风化的Si—O单键,而母质层未见该峰,这证实淋溶层、淀积层中石英等土壤矿物的风化程度更高。3层土样在3 622 cm−1均存在伸缩振动峰,该峰通常表征为有机物上的羟基,但3层土样间明显能看出强度差异,这与表2中各土层的有机胶体含量的差异相符合。3 433 cm−1处淋溶层、淀积层吸收峰较宽,是C=C、C=N、N=O等螯合键的伸缩振动峰。而1 632 cm−1吸收峰多为C=O。1 025~1 035 cm−1以及797 cm−1处均为Si—O—Si的伸缩振动特征峰。913 cm−1和694 cm−1附近吸收峰分别表征为C—N键和羟基,而533 cm−1和471 cm−1的双峰多为C—Cl键。综上所述,各土样谱峰位置的差异主要为羟基和羰基(羧基羰基、酮羰基)等,这说明有机物上—OH和羰基(羧基羰基、酮羰基)等为吸附过程中的主要吸附位点。
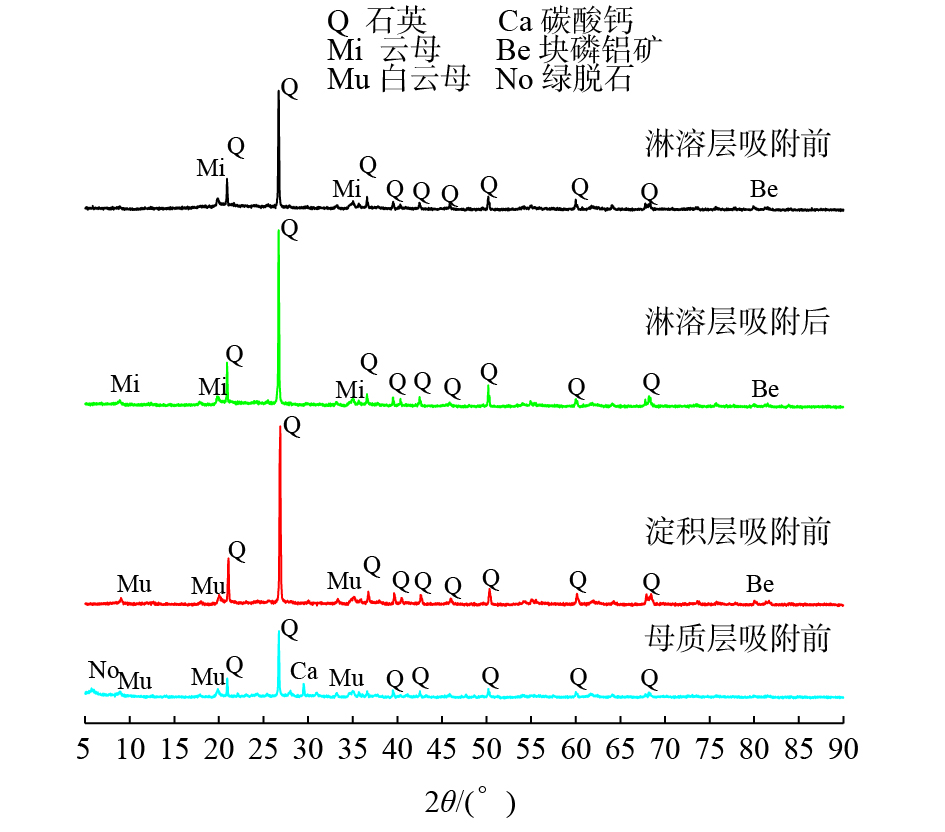
3) X射线晶体衍射分析。图10为各层土样XRD图谱,显示的矿物组成与XRF化学成分一致。淋溶层土样吸附前后衍射峰位置未发生偏移,这说明土壤吸附铀过程中各晶体形态未发生变化。由于3类土样风化程度不同,在物相组成上存在一定差异:淋溶层中含有云母,块磷铝矿(AlPO4);淀积层矿物组成中的白云母与云母组成元素基本相同,但云母结构为六方形片状晶形,成分更复杂,白云母结构为2层硅氧四面体夹着一层铝氧八面体构成的复式硅氧层(含K、Na、Fe、Al、Si、O、H等元素),风化程度比云母更低[32]。与淋溶层、淀积层土样相比,母质层土样中未发现块磷铝矿,但其含有绿脱石(Na0.33
Fe3+2 ((Al, Si)4O10)(OH)2·nH2O)和CaCO3,这与母质层土壤液pH远高于淋溶层、淀积层现状相吻合。XRD表征结果进一步证实了各土层在土壤矿物组成和风化程度上存在一定差异。 -
1)当淋溶层、淀积层pH为6.2、母质层pH为4.1且温度为25 ℃时,各土层样对U(Ⅵ)的单位吸附量可达到最大值,分别为23.60、22.82和13.05 mg·g−1,即各土层样对铀吸附能力为淋溶层>淀积层>母质层。吸附等温和动力学模型拟合结果分别表明,Langmuir方程和准二级动力学方程对各土层拟合度最佳,说明3类土样对U(Ⅵ)的吸附均以单层、均质吸附为主,且吸附过程主要为化学吸附。
2) BET、SEM、XRD和FT-IR分析结果表明,3类土样由于风化程度不同,在比表面积、吸附点位、土壤颗粒、矿物组成以及Si—O、—OH、C=C、C=N、N=O、C=O、C—Cl等官能团/化学键含量上存在显著差异。这也是各层土样对U(Ⅵ)吸附能力不同的主要原因。
3)外部U(Ⅵ)进入土壤剖面后大多聚集在地表腐殖质—淋溶层,随剖面深度增加铀浓度逐渐降低,少量进入淀积层、母质层。在铀尾矿库退役和治理期间,可在表层土中添加还原剂将淋溶层中的U(Ⅵ)还原成U(IV),同时调节土壤pH从而控制其进一步迁移扩散。
某尾矿库附近各土壤层对U(VI)的吸附特性
Adsorption characteristics of uranium in soil horizons near tailings impoundment area
-
摘要: 针对南方某铀尾矿库附近棕红壤剖面中的淋溶层、淀积层和母质层等土层对铀的吸附机理和空间分布规律进行了探讨。采用比表面测定、X射线荧光、扫描电镜、傅里叶变换和X射线衍射等方法对各土层样的理化性质、结构和形貌进行了表征分析;采用静态吸附法考查了时间、U(Ⅵ)初始浓度、pH、温度、粒径等因素对各土层吸附U(Ⅵ)的影响;并使用热力学和动力学方程对吸附过程进行了模拟分析。结果表明,25 ℃下淋溶层和淀积层pH均为6.2、母质层pH为4.1时,其对U(Ⅵ)最大吸附量分别可达23.60、22.82和13.05 mg·g−1。热力学和动力学拟合结果分别表明,各土层对U(Ⅵ)更符合Langmuir方程(R2>0.999)和准二级动力学模型(R2>0.98)。风化程度,Fe、Mn、Al、Ca等元素及有机质含量,pH和土壤粒径等因素是各土层对U(Ⅵ)吸附能力不同的主要原因;同时,外源铀进入土壤剖面后,大部分聚集在土壤表层,随着深度的下降的铀含量也逐渐降低。研究结果可为土壤剖面中的铀和其他重金属的污染防治提供参考。Abstract: In this study, the adsorption mechanism and spatial distribution of U(Ⅵ) in the eluvial, illuvial and parent material horizon of a brown-red soil profile near a Uranium tailing impoundment in South China were discussed. The physical and chemical properties of the soil horizons were characterized by using surface area measurement, X-ray fluorescence, scanning electron microscope, fourier transform infrared spectroscopy and X-ray diffraction. The effects of time, U(Ⅵ) initial concentration, pH, temperature, and particle sizes on U(Ⅵ) adsorption were investigated by static adsorption experiments. Thermodynamic and kinetic equations were used to simulate the adsorption process. The results showed that the maximum U(Ⅵ) adsorption capacities were 23.60, 22.82 and 13.05 mg·g−1 in the eluvial, illuvial and parent material horizon, respectively, at pH 6.2 for the former two horizons, pH 4.1 for the later one and 25 ℃. The isothermal adsorption models of the soil horizons were more consistent with the Langmuir equation (R2>0.999), and the kinetic adsorption process fitted better with the pseudo-second order kinetic model (R2>0.98). The different adsorption capacities towards U(Ⅵ) on each layer could be caused by the differences of their physico-chemical properties, such as weathering degree, Fe, Mn, Al, Ca, organic matter contents, pH and soil particle sizes. As the external uranium entered the soil profiles, it could accumulate in the surface layer of soil, and the content of uranium decreased with the decrease of depth. The results of this study can provide references for the prevention and control of external uranium or other heavy metal pollution in the soil profile.
-
Key words:
- U(Ⅵ) /
- near-surface disposal /
- uranium tailing impoundment /
- soil profile /
- static adsorption
-
硝化菌是活性污泥体系中起硝化作用的主要因素,但硝化菌生长缓慢,易受外界环境影响,pH、温度、溶解氧(dissolved oxygen, DO)和C/N等会使硝化菌活性受到抑制[1]。研究团队在对全国58座污水处理厂进行的全流程调研分析中发现,污水处理厂平均硝化速率仅为2.9 mg·(g·h)−1(以VSS计),远低于理论硝化速率4 mg·(g·h)−1 (GB 50014-2006)。这可能是因为硝化菌易受环境影响导致菌体流失[2],因此,将硝化性能良好的活性污泥进行固定化处理具有重要意义。
硝化菌富集包括纯菌扩大培养和活性污泥富集培养2种主要培养方式[3]。使用活性污泥富集法中的序批式间歇反应器(sequencing batch reactor, SBR)富集方法进行硝化菌富集时,具有成本低、菌种稳定、生物相容性好等优点,该富集方法通过调节水质中氨氮负荷,抑制异养型菌属的增长,促进硝化菌的繁殖,达到富集硝化菌的目的,即依靠硝化菌自我代谢提高硝化菌占比,但是富集效果相对较弱。
PVA水凝胶空间网络稳定,有丰富的孔隙结构,微生物亲和性好,可用作活性污泥包埋剂[4],但是PVA凝胶球总孔容相对较小且胶连过程中易发生曳尾现象。而聚氧化丙烯三醇是一种聚醚多元醇,常用作增韧剂[5],可用作凝胶球改性。
本研究通过基于降低C/N比的活性污泥富集法提高活性污泥硝化性能,并将驯化后的活性污泥制作成改性凝胶包埋颗粒,分析比较了改性材料性能和活性污泥在不同阶段的微生物群落变化及污泥活性指标,为研究活性污泥包埋颗粒在废水治理领域的可行性提供参考。
1. 材料与方法
1.1 活性污泥培养
以无锡市某市政污水处理厂好氧池活性污泥(activated sludge, AS)作为接种污泥,采用SBR反应装置进行培养驯化。驯化过程中,调节进水水质COD为300 mg·L−1;TP为4 mg·L−1;进水
NH+4 -N浓度以5 d为一周期,每周期进行一次调整,依次为25、35、45 mg·L−1,通过此方式改变进水碳氮比,实现硝化菌的富集,并添加一些稀有元素,促进活性污泥中微生物更好地生长繁殖。培养过程中不断添加适量Na2CO3溶液,调节进水pH为7.5~8.5,以满足硝化菌最佳生存条件。1.2 凝胶颗粒制备
实验选择PVA作为主要的包埋材料,添加少量的海藻酸钠(sodium alginate, SA),以缓解PVA的附聚性,并添加少量PPT(PPT是一种聚醚多元醇,常用作增韧剂[6]),改善凝胶球的机械强度和传质性能,同时通过延时包埋等方法来改善材料的水溶膨胀性。实验选择饱和硼酸CaCl2溶液作为交联剂,通过NaHCO3调节交联剂的pH,冷冻-解冻来增加固定化小球的强度,并采用Na2SO4溶液进一步交联,以减轻硼酸对微生物活性的影响。包埋颗粒的制备包括以下5个步骤。
1)将PVA(国药1799型)、去离子水分别以体积分数为10%和90%于烧杯中混合,采用电磁炉加热2~3 h,在加热过程中,须不断搅拌使溶液受热均匀,直至PVA颗粒完全溶解后,停止加热并冷却至室温。
2)驯化后的硝化污泥在3 000 r·min−1条件下离心15 min,再用0.9% NaCl溶液冲洗,重复该操作2次。
3)将浓缩后的硝化污泥、SA、PPT以及静置后的混合物以体积比10:1:0.2:88.8混合均匀后,滴入含2%氯化钙的饱和硼酸溶液形成球形颗粒,于4 ℃冰箱中放置30 min交联固化,然后将颗粒滤出,用0.9% NaCl溶液清洗表面残留的硼酸溶液。
4)为增加包埋颗粒的机械强度,将颗粒在-20 ℃的冰箱里反复冻融3次。
5)冻融后的颗粒用去离子水清洗,放置于模拟生活污水中,于4 ℃条件下保藏待用。
1.3 分析方法
取约10 mL泥水混合液,50 W功率超声1 min,后向混合液中加入60 μL甲酰胺及5 mL NaOH溶液(1 mol·L−1),并于4 ℃条件下振荡3 h后,在4 ℃、10 000 r·min−1条件下离心15 min,所得上清液用0.22 μm滤膜过滤,得到污泥的EPS。采用考马斯亮蓝法和蒽酮-硫酸比色法[7]对胞外聚合物(extracellular polymeric substances,EPS)中蛋白(PN)和多糖(PS)含量进行测定。
将待测包埋凝胶球在纯水中曝气30 min,以去除颗粒内部及表面的氨氮,烧杯中配置一定氨氮浓度的模拟废水,颗粒与废水混合的体积比为1:4。调节溶液pH小于5,以抑制包埋颗粒的硝化反应;采用恒温水浴锅将烧杯中混合液温度控制在(30.0±0.1) ℃,采用磁力搅拌器确保溶液充分混合,测试氨氮浓度的变化,计算凝胶颗粒氨传质系数。将包埋颗粒与去离子水以1:4的体积比混合,盛满250 mL的溶氧瓶,持续通入氮气2 h,使包埋颗粒内部的溶解氧完全消耗,采用恒温水浴锅控制温度为(30.0±0.1) ℃,采用磁力搅拌器使溶液充分混合,溶解氧仪记录混合液中溶解氧的变化,计算凝胶颗粒氧传质系数。
在测定氨氧化速率(ammonia uptake rate, AUR)时,将活性污泥或含有活性污泥的物体放入到碘量瓶中,碘量瓶中加入适量NH4Cl和NaHCO3,曝气并维持DO约为2 mg·L−1,同时不断搅拌,保证溶液与活性污泥混合均匀,通过测量碘量瓶中混合液在不同时间点的氨氮浓度,并结合混合液中活性污泥的浓度计算氨氧化速率。在测定比耗氧速率(specific oxygen uptake rate, SOUR)时,将活性污泥或含有活性污泥的物体放入到碘量瓶中,碘量瓶中加入模拟生活污水并充分曝气,然后将碘量瓶密封,同时不断搅拌,保证溶液与活性污泥混合均匀,通过测量碘量瓶中混合液在不同时间点的DO值,并结合混合液中活性污泥的浓度计算比呼吸速率。
污泥粒径采用BT-2003百特激光粒度分布仪分析系统测试;热重分析采用TGA5500型热重分析仪测试,温度为30~500 ℃,升温速率为10 ℃·min−1;SEM采用SU8010型微生物扫描电镜测试;BET采用Belsorp-Max型全自动比表面及孔隙分布测定仪测试;DO浓度使用WTW FDO 925-3便携式多参数水质分析仪测试;
NH+4 -N浓度采国标法[8]测定;样品DNA的提取采用土壤基因组提取试剂盒(柱形,艾比根),并送样到上海晶能生物有限公司完成16S rRNA基因的高通量测序和微生物群落分析。2. 结果与讨论
2.1 活性污泥污泥特性及胞外聚合物的变化
由图1和表1可以看出,在不断提高进水中氨氮占比的条件下,活性污泥对氨氮的去除效率略有降低,由100%降低至92%,再降低至85%左右;但单位时间内氨氮的去除量在提升,由25 mg·L−1提升至32 mg·L−1,再提升至38 mg·L−1,说明活性污泥的氨氮去除效果在驯化过程中得到了提高。经计算,经过15 d的驯化培养,活性污泥的AUR由2.044 mg·(g·h)−1(以VSS计)提升至2.645 mg·(g·h)−1,SOUR由19.317 mg·(g·h)−1(以VSS计)提升至21.868 mg·(g·h)−1。硝化性能较原活性污泥有了明显的提高,可以用作制备生物包埋凝胶球。
表 1 驯化前后活性污泥生物活性Table 1. Biological activity of activated sludge before and after domestication阶段 MLSS/(mg·L−1) MLVSS/(mg·L−1) AUR/(mg·(g·h)−1) SOUR/(mg·(g·h)−1) 驯化前 4 368 2 460 2.044 19.317 驯化后 4 008 2 387 2.645 21.868 | Show Table DownLoad:
CSV
DownLoad:
CSV
由图2和图3可以看出,EPS对活性污泥的理化性质有较大影响,蛋白和多糖是其主要成分。在驯化过程中,随着氨氮投加量的提高,水质C/N比不断降低,多糖和蛋白的浓度也均有不同程度降低,PN/PS不断提高。这是因为在低C/N条件下,由于碳源的缺乏,微生物将多糖用作营养物质分解使用。在驯化培养过程中,过量的曝气使活性污泥絮体破坏,导致活性污泥粒径相对减少,粒径分布向低粒径移动,中位径从最初的28.38 μm降低到27.09 μm。蛋白是微生物絮体骨架的重要成分,因此,蛋白含量也降低。C/N不断降低,微生物所需碳源增多,对多糖的消耗也增多。驯化过程中曝气程度保持稳定,在曝气过程中,活性污泥逐渐适应反应体系,蛋白降低幅度相对较小。因此,多糖降低幅度大于蛋白,PN/PS不断升高。
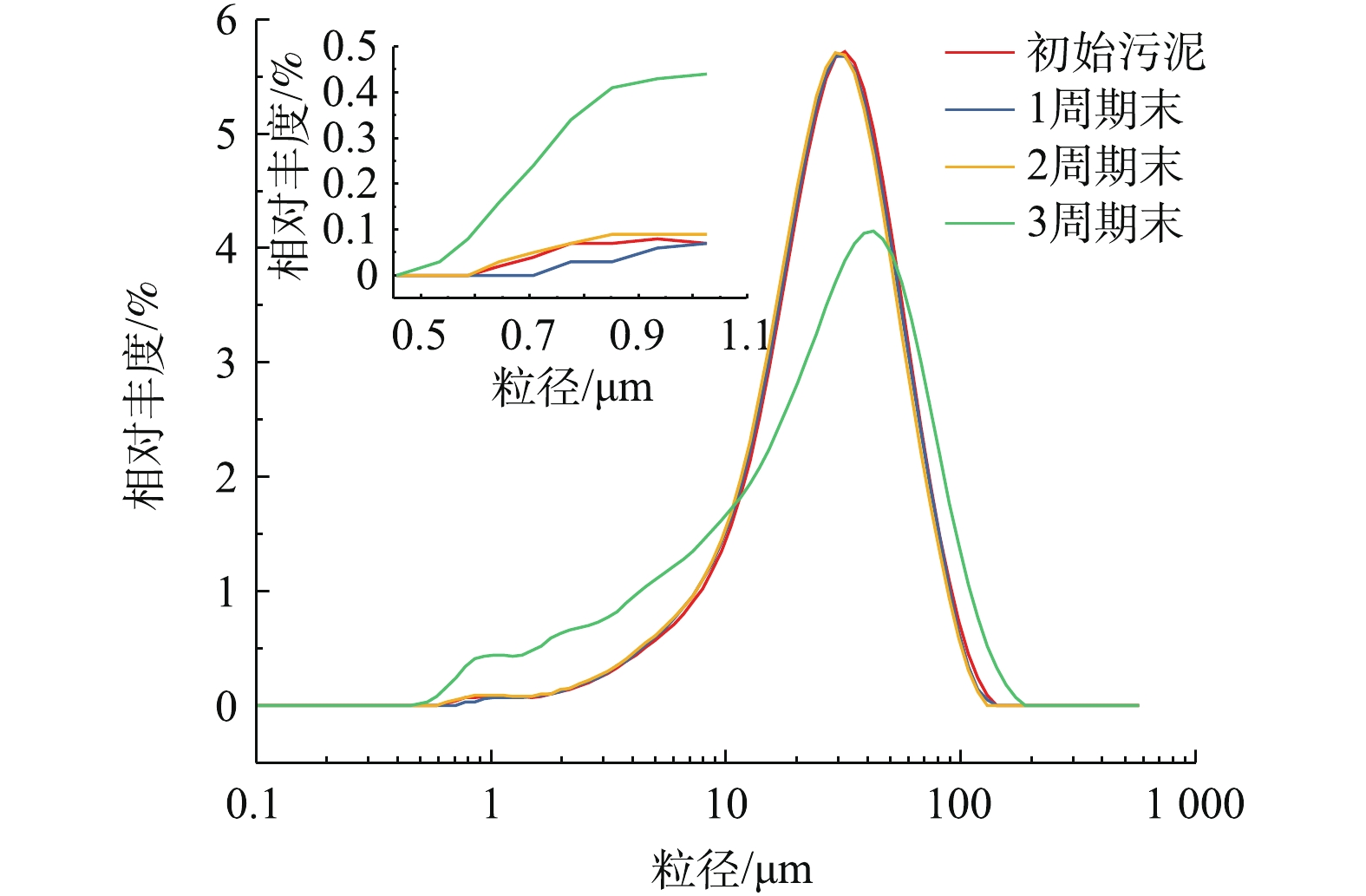 图 2 不同驯化阶段活性污泥粒径分布Figure 2. Particle size distribution of activated sludge at different domestication stages
图 2 不同驯化阶段活性污泥粒径分布Figure 2. Particle size distribution of activated sludge at different domestication stages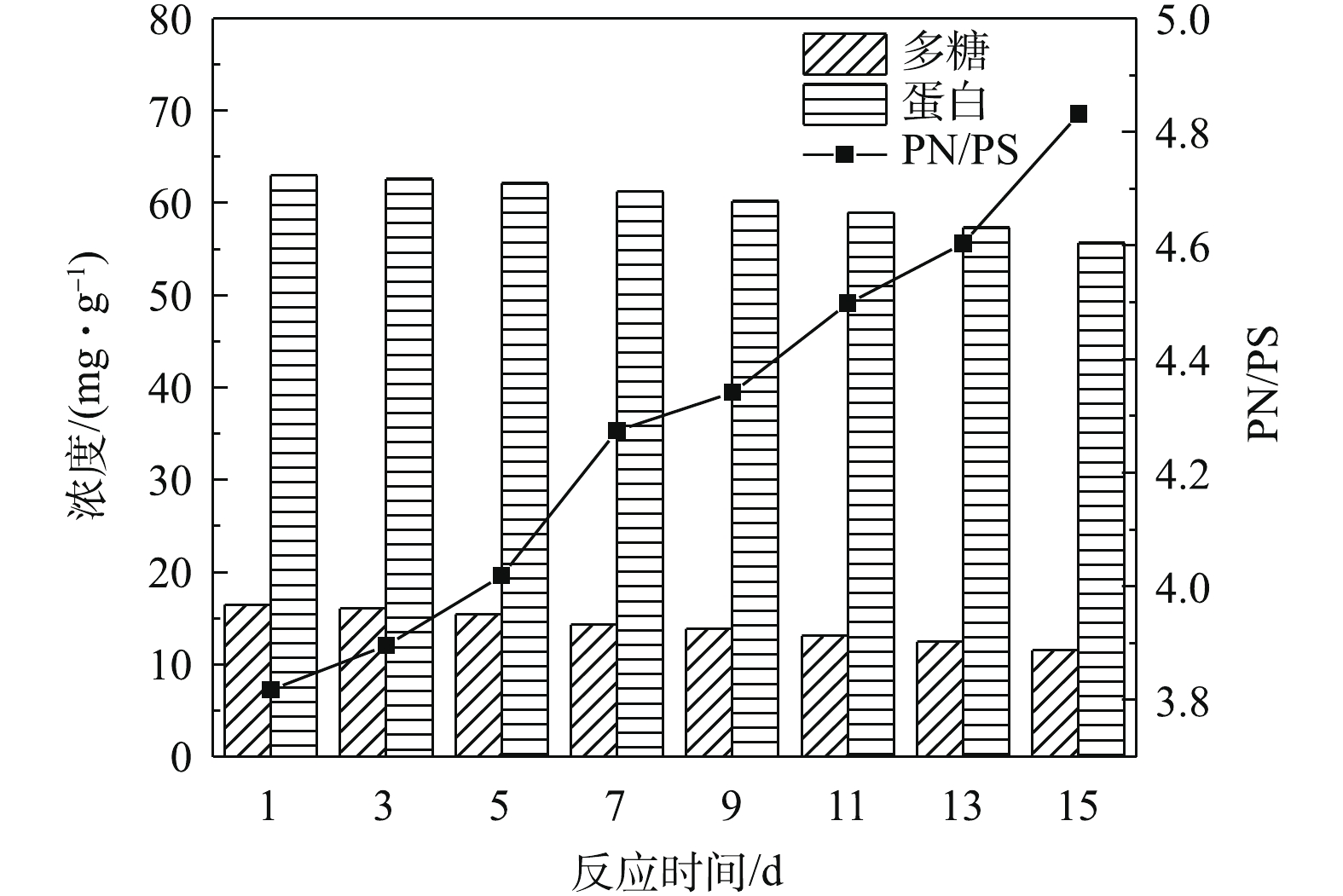 图 3 驯化过程中活性污泥PN、PS变化Figure 3. Changes of PN and PS in activated sludge during domestication
图 3 驯化过程中活性污泥PN、PS变化Figure 3. Changes of PN and PS in activated sludge during domestication2.2 改性凝胶颗粒理化性能
凝胶球半径约为3.4 mm,单颗凝胶球质量冻干前约为112.26 mg,冻干后约为33.85 mg,改性后粒径、质量与改性前大致相等。
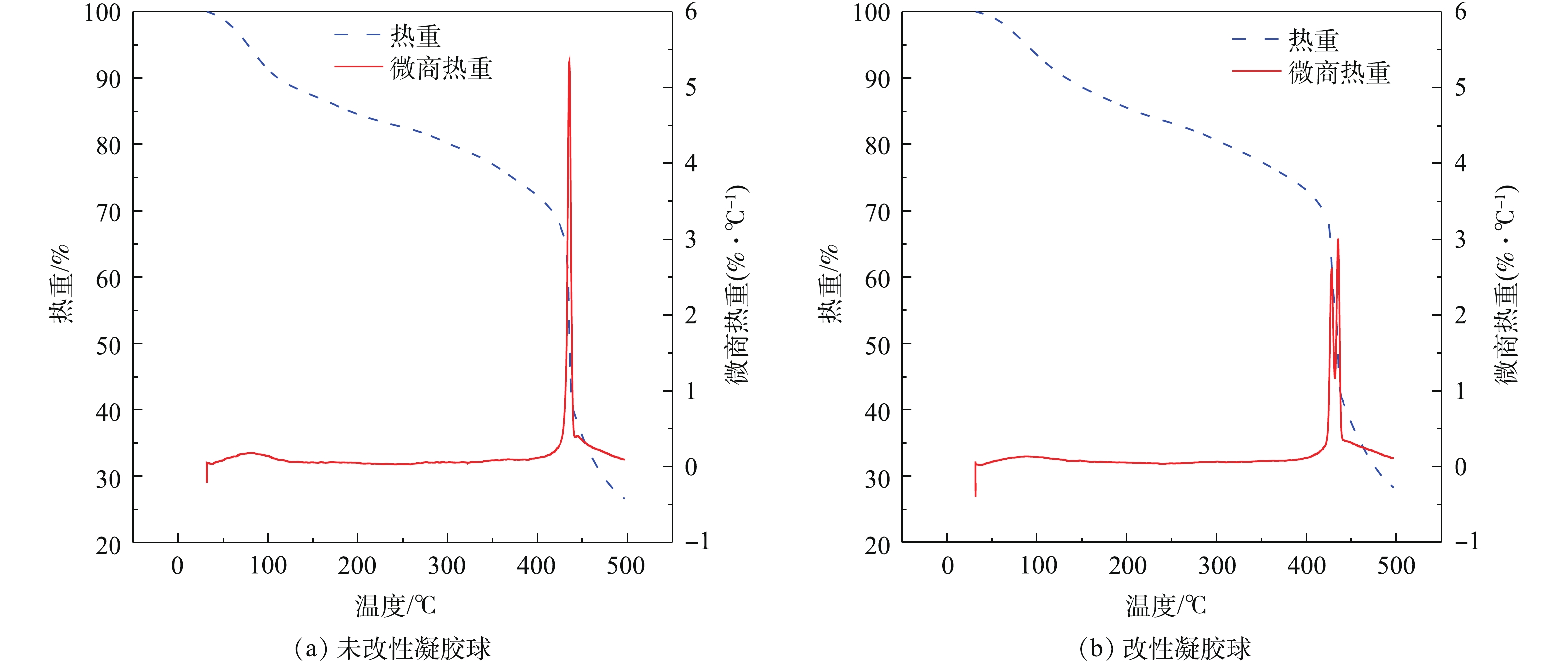
由图4可以看出,在常温条件(30~50 ℃)下,凝胶颗粒质量损失极小,改性前后分别为1.1166%和0.8015%,说明凝胶颗粒常温下热稳定性能好。凝胶颗粒在435 ℃左右失重速率最大。等温条件下,改性凝胶球失重量和失重速率都比改性前小,说明改性后,凝胶颗粒热稳定性能有所提高。
图5为改性前后凝胶球扫描电镜图,放大倍数分别为1 000、5 000和10 000。可以看出,改性前孔隙结构比较松散,且孔径较大;而改性后,孔隙结构相对规整,且孔径大小略有降低,这与之后测得传质速率略有降低相符合。
表2反映了未改性凝胶颗粒和PPT改性凝胶颗粒比表面积和孔径分布情况。可以看出,使用PPT改性后的凝胶球的比表面积和总孔容积增大,平均孔径减小,分别由0.315 m2·g−1、0.004 cm3·g−1和51.870 nm变为1.527 m2·g−1,0.015 cm3·g−1和39.362 nm。这说明PPT改性使凝胶包埋更加均匀,比表面积和总孔容也得到增加,能够包埋的微生物量也增多。周素红等[9]研究发现,适合微生物附着生长的孔径以大孔和中孔为主,PPT改性凝胶颗粒孔径大小符合微生物生长条件。与谭炳琰等[10]制备的PVA-SA水凝胶生物载体相比,PPT改性凝胶颗粒孔容与比表面积较小,孔径几乎相等,这说明PPT改性凝胶颗粒负载微生物量仍具有一定提升空间。许晓毅等[11]研究表明,凝胶包埋颗粒比表面积增加可以增强包埋活性污泥生物硝化和脱氮性能。因此,PPT改性凝胶颗粒可以有效用作一种活性污泥包埋材料并应用于废水治理中。
表 2 改性包埋颗粒孔结构参数Table 2. Pore structural parameters of modified embedded particles样品 比表面积/(m2·g−1) 总孔容积/(cm3·g−1) 平均孔径/nm 未改性凝胶球 0.315 0.004 51.870 改性凝胶球 1.528 0.015 39.362 | Show TableDownLoad:
CSV
包埋颗粒是一种多孔材料,凝胶颗粒内外物质会通过孔隙进行传输,直至达到平衡。氨氮和氧气是活性污泥新陈代谢必不可少的物质。氨、氧传质特性是凝胶颗粒能否用作活性污泥载体的重要评价指标。实验选取稳定运行的凝胶包埋颗粒,探究不同污泥投加方式对凝胶球氨、氧传质性能的影响。根据PU等[12]的研究,凝胶颗粒内基质有效扩散系数De的计算模型见式(1)。
De=ln(6Cb0a(1+a)(9+9a+q12a2)⋅(Cb(1+a)−Cb0a))⋅R2q12t (1) 式中:De为有效扩散系数,m2·s−1;Cb为主体溶液中基质的瞬时浓度,mg·L−1;Cb0为主体溶液中基质的初始浓度,取50 mg·L−1;a为液相体积与颗粒容积比,取值为4;q1为非零正根;R为颗粒半径,取值为3.4×10−3 m。
为简化计算过程,将式(1)进行整理变换,变换后得式(2)。
ln(Cb(1+a)Cb0a−1)=ln(6(1+a)9+9a+q12a2)−(Deq12R2)t (2) 图6反映了氨扩散实验结果,将拟合线代入式(2)得:空白凝胶球和PPT改性凝胶球的氨扩散系数De分别为4.867×10−10 m2·s−1和4.463×10−10 m2·s−1。
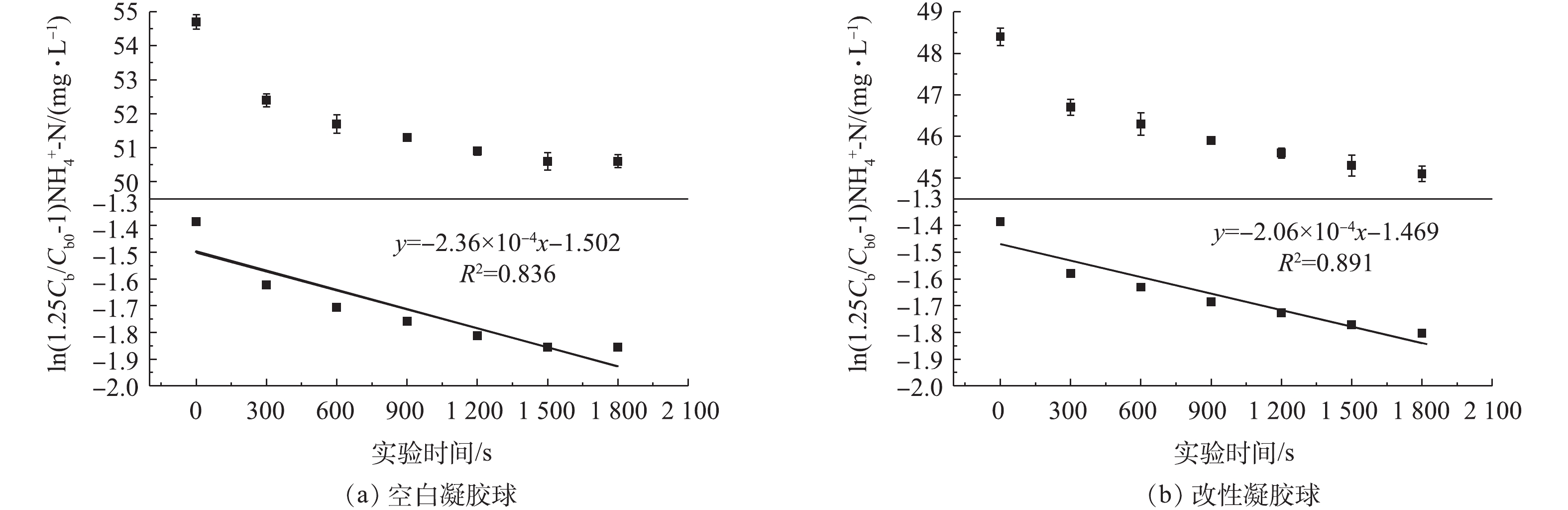 图 6 氨扩散实验结果及有效扩散系数的拟合Figure 6. Ammonia diffusion experiments and the fitting curve of effective diffusion coefficient in immobilized particles
图 6 氨扩散实验结果及有效扩散系数的拟合Figure 6. Ammonia diffusion experiments and the fitting curve of effective diffusion coefficient in immobilized particles图7反映了氧扩散实验结果,将拟合线代入式(2)得:空白凝胶球和PPT改性凝胶球的氧扩散系数De分别为2.108×10−10 m2·s−1和1.838×10−10 m2·s−1。
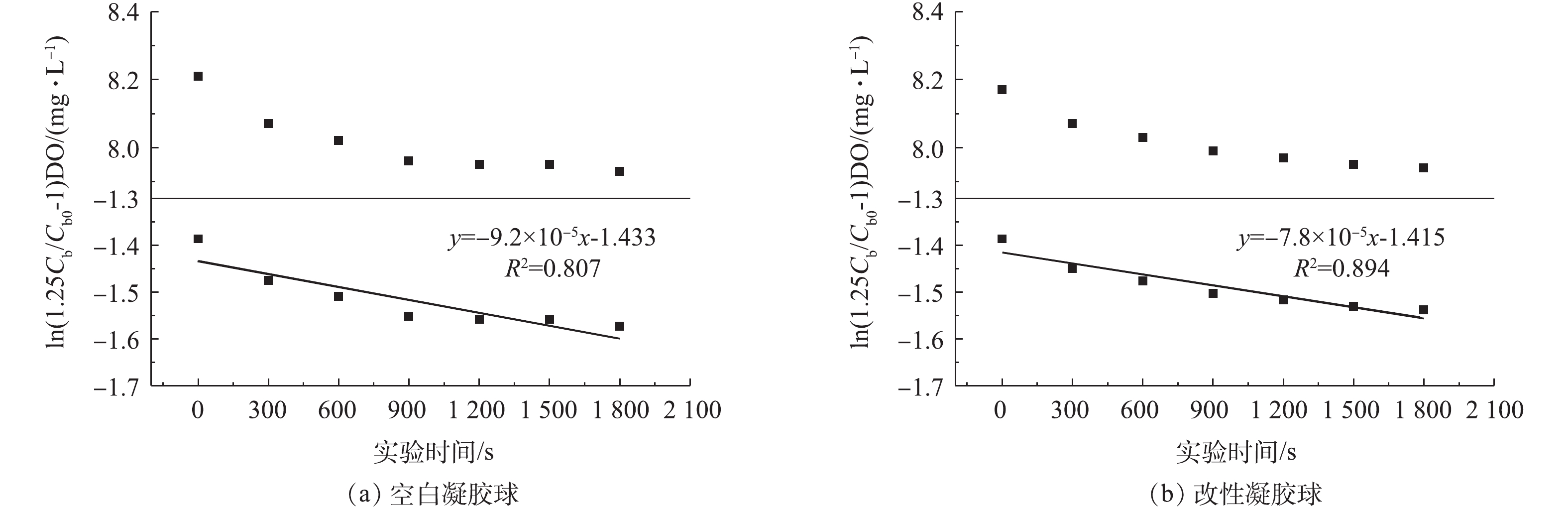 图 7 氧扩散实验结果及有效扩散系数的拟合Figure 7. Oxygen diffusion experiments and the fitting curve of effective diffusion coefficient in immobilized particles
图 7 氧扩散实验结果及有效扩散系数的拟合Figure 7. Oxygen diffusion experiments and the fitting curve of effective diffusion coefficient in immobilized particles由实验结果可知,添加改性剂后,凝胶球氨传质性能和氧传质性能均有不同程度的降低,这可能是由于凝胶球中添加改性材料时,凝胶球中总物质量增多,部分空隙被堵塞。曹国民等[13]测得15%PVA空白凝胶膜中氨氮扩散系数为6.9×10−10 m2·s−1。WIESMANN等[14]验证包埋微生物空心球的氧扩散系数为1×10−10~10×10−10 m2·s−1。本实验测得改性凝胶包埋颗粒的有效扩散系数均相对较低,原因可能是温度、搅拌条件以及载体成分等对凝胶包埋颗粒传质性能的影响。
2.3 凝胶包埋活性污泥微生物活性变化
如图8所示,包埋活性污泥后,凝胶颗粒颜色由纯白色变为黄灰色。半径约为3.4×10−3 m,质量约为114.027 mg。
由表3可知,OTU数量和Chao指数呈现递增趋势,说明驯化培养过程中,在持续曝气和营养底物充足的条件下,污泥中微生物物种丰度得到提高;包埋后由于凝胶颗粒形成较好的厌氧、缺氧和好氧环境,兼氧菌和厌氧菌有效繁殖,微生物物种丰度大幅提高。在驯化培养过程中,Shannon和Shannoneven生物多样性指数降低,包埋成活性污泥后升高,说明驯化培养过程中,水体环境更适合好氧菌和自养菌生长繁殖,因此,微生物菌群多样性及相对均匀程度均降低;包埋成凝胶球后,在较好的厌氧、缺氧和好氧环境以及模拟废水充足碳源条件下,各类微生物菌群都得以充分生长繁殖,因此,微生物菌群多样性及相对均匀程度升高,菌群相对均匀。
表 3 Alpha多样性指数Table 3. Alpha diversity indexes样品 OTU Chao Shannon shannoneven 初始污泥 1 322 1 518 5.14 0.71 活性污泥 1 392 1 636 4.90 0.67 凝胶包埋污泥 1 638 1 856 5.89 0.79 | Show TableDownLoad:
CSV
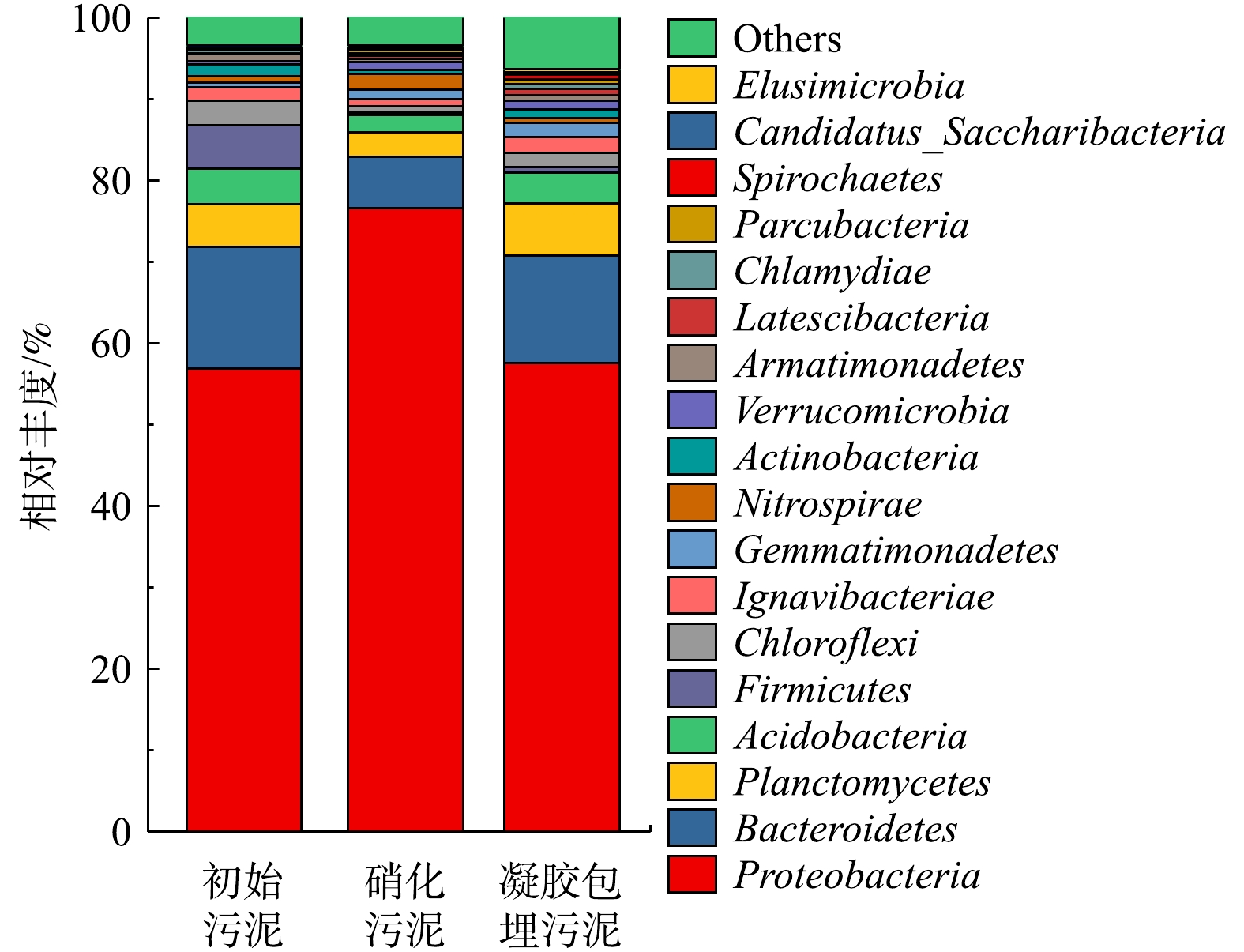
由图9可知,3个样品中的细菌门都比较丰富,以Proteobacteria(变形菌)、Bacterioidetes(拟杆菌)、Planctomycetes(浮霉状菌)和Acidobacteria(酸杆菌)为主。其中Proteobacteria和Bacterioidetes是活性污泥系统中的功能性菌群,主要参与有机物的降解和脱氮除磷过程[15]。Planctomycetes是一个独立的细菌门,其细胞壁缺少肽聚糖,通过细胞内膜(intracytoplasmic membrane, ICM)将细胞分为不同的部分[16]。大量的证据表明,浮霉状菌与赤潮存在一定联系,如能形成玫瑰环的浮霉状菌(如Planctomyces bekefii、Planctomyces guttaeformis和Planctomyces stramskae),在藻类或是蓝细菌的赤潮暴发后丰度上升[17],其中厌氧氨氧化菌(Anammox bacteria)是浮霉状菌中最有特点的一类菌,在环境氮循环中具有重要作用。凝胶包埋后,Acidobacteria占比略有提高,是因为凝胶包埋过程会加入一定量的硼酸试剂,而Acidobacteria是一种嗜酸菌。除此之外,Nitrospirae(硝化菌门)是活性污泥硝化反应的主要菌门,微生物群落门水平分析表明,3个阶段中Nitrospirae丰度分别为0.008、0.020、0.006,这是因为C/N低、DO高的环境适合硝化菌门的繁殖代谢,驯化培养后,硝化菌门大幅提高;但凝胶包埋后,功能性菌群在不断应对环境变化,引起微生物群落结构的改变,硝化菌门受到抑制,占比降低。
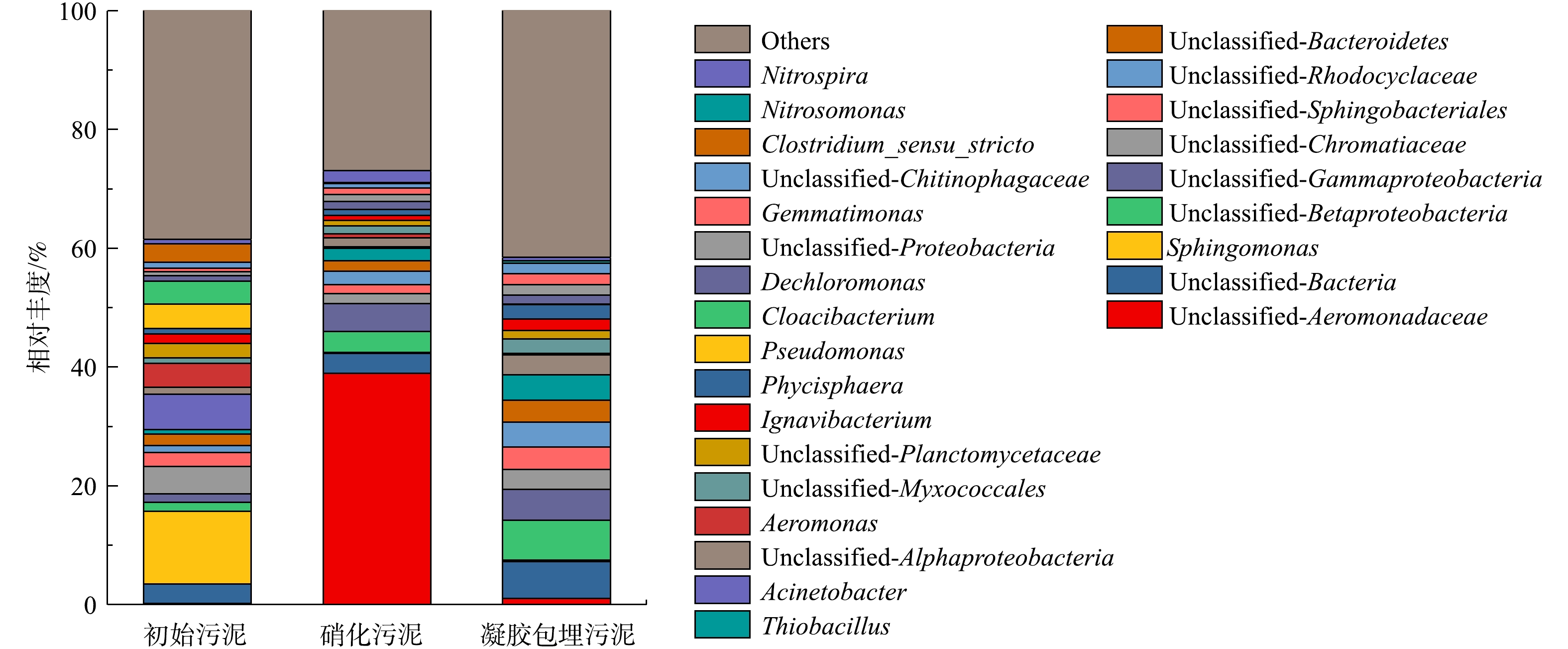
由图10可知,属水平在活性污泥培养3个阶段中变化较大,其中Nitrospira(硝化菌属)和Nitrosomonas(亚硝化菌属)在活性污泥脱氮过程中起着重要作用,3个阶段中Nitrospira丰度分别为0.008、0.020、0.006,Nitrosomonas丰度分别为0、0.002、0.004均先上升后下降,这是因为驯化培养过程成效明显,硝化类菌属丰度大幅提升;包埋后硝化类菌属丰度有所下降,这可能是因为硼酸等不利环境使硝化菌活性受到抑制。
图11反映了包埋后凝胶颗粒的AUR和SOUR的变化情况。AUR和SOUR分别为2.418 mg·(g·h)−1和18.929 mg·(g·h)−1,与初始污泥相比,AUR显著升高,有18.28%的升幅,有利于水体氨氮的去除;SOUR略有下降,降低幅度为2.01%,这与包埋过程环境的改变有关。这是因为包埋过程中,投加硼酸、反复冻融等条件对微生物活性有一定的抑制作用,部分微生物不适应环境被淘汰。
如图12和图13所示,刚制备好的凝胶球,由于制备过程中采用了硼酸等实验试剂,且为了增强凝胶球机械强度,在−20 ℃环境下反复冻融,均对活性污泥的微生物活性有一定程度的影响,因此,采用模拟废水对凝胶球进行微生物活性恢复实验,进行序批式实验,每12 h为一个实验周期,6个实验周期后,凝胶球颜色由刚制备好的石灰白转变为灰黑色。通过6个周期的活性恢复,凝胶球包埋活性污泥在1个运行周期内能够有效地去除15 mg·L−1氨氮,包埋凝胶球中活性污泥的微生物活性得到有效恢复。
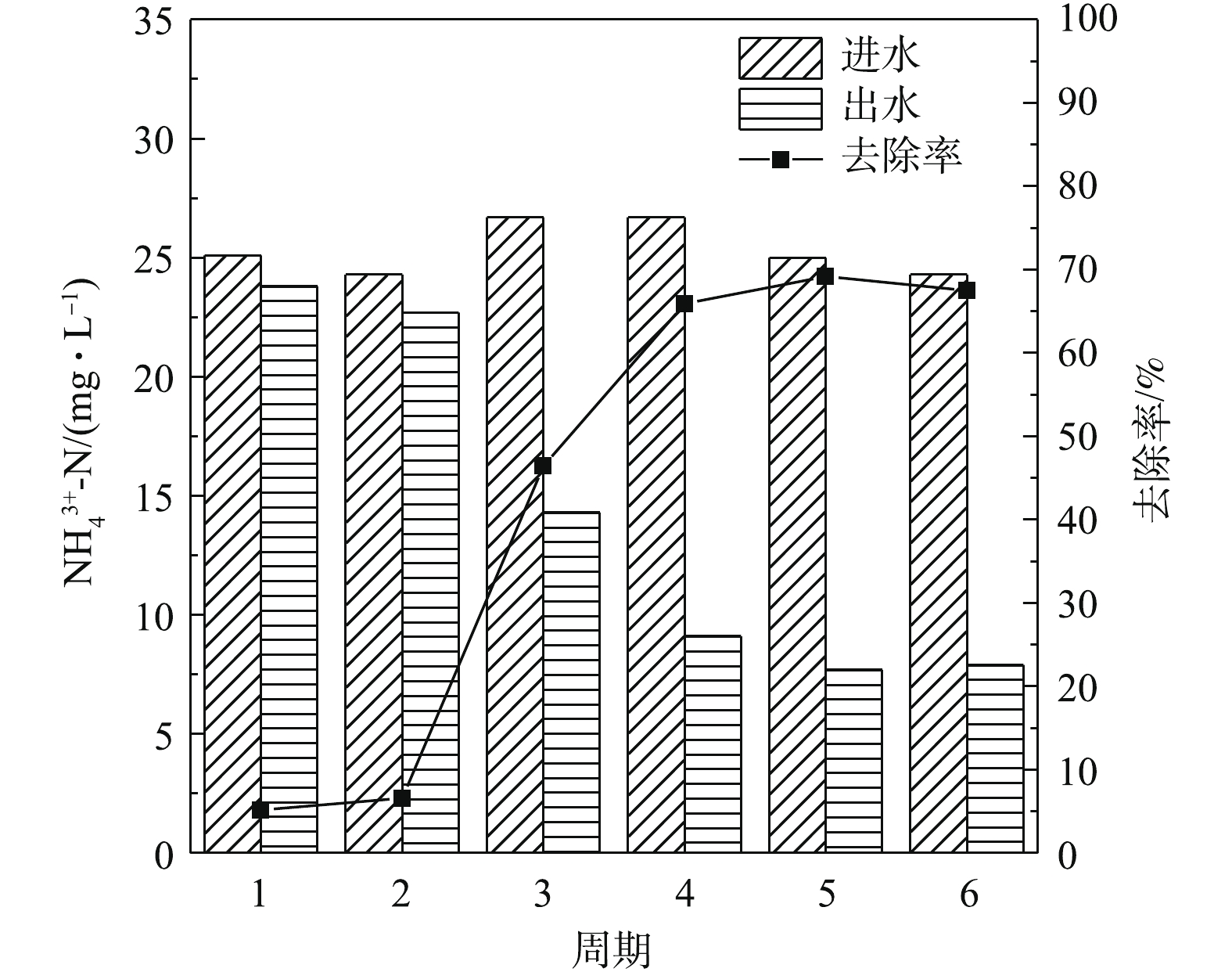 图 13 恢复活性凝胶包埋活性污泥氨氮去除性能Figure 13. Ammonia nitrogen removal performance of gel embedded activated sludge after activity recovery
图 13 恢复活性凝胶包埋活性污泥氨氮去除性能Figure 13. Ammonia nitrogen removal performance of gel embedded activated sludge after activity recovery3. 结论
1)提高进水氨氮浓度,降低C/N比,可以使活性污泥中硝化菌含量增加,硝化性能得到提升,培养后的活性污泥AUR和SOUR分别由2.044 mg·(g·h)−1和19.317 mg·(g·h)−1提升至2.645 mg·(g·h)−1和21.868 mg·(g·h)−1,菌群分析结果显示,Nitrospirae相对丰度提升150%。
2)改性凝胶颗粒具有热稳定性好、孔容大、比表面积大等优点,可有效用作活性污泥包埋材料。改性后凝胶颗粒在常温下质量损失仅为0.802%,可以在常温下稳定运行;总孔容增加至0.015 cm3·g−1,可以容纳更多的包埋物质;比表面积也增大至1.528 m2·g−1,可以有效增加水体中物质与包埋活性污泥的接触概率。
3)凝胶包埋会使部分微生物死亡,但经过活性恢复后,硝化性能得到恢复,AUR显著升高,升幅达到18.28%,有利于水体氨氮的去除;而SOUR略有下降,但降低幅度仅为2.01%,这与包埋过程环境的改变有关。用恢复活性的凝胶包埋颗粒处理25 mg·L−1的氨氮废水时,氨氮去除率达到70%左右,凝胶包埋颗粒污泥在氨氮废水治理中具有重要研究意义。-
-
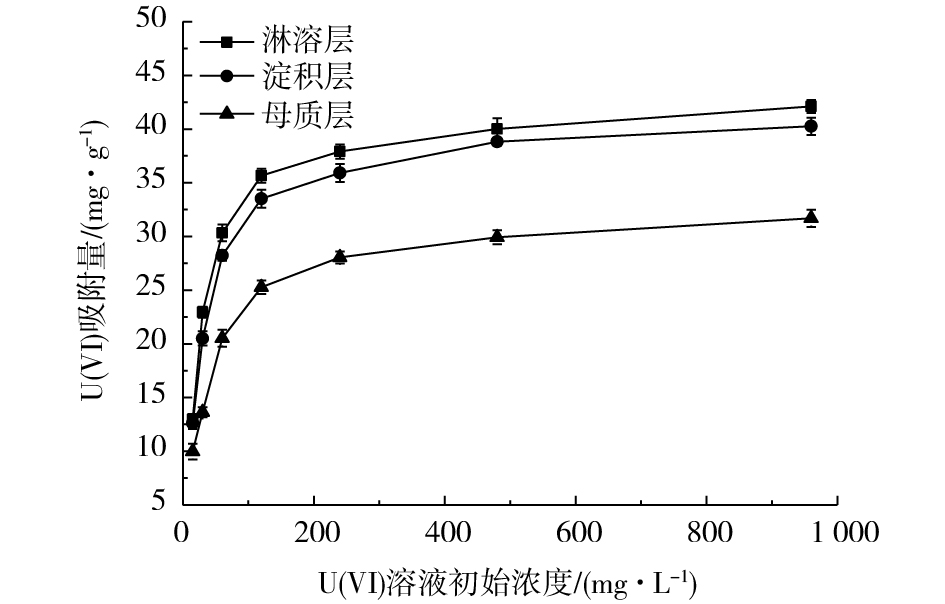
图 2 初始溶液中铀浓度对各土层吸附U(Ⅵ)的影响
Figure 2. Effect of U initial concentration on U(VI) adsorption in soil horizons
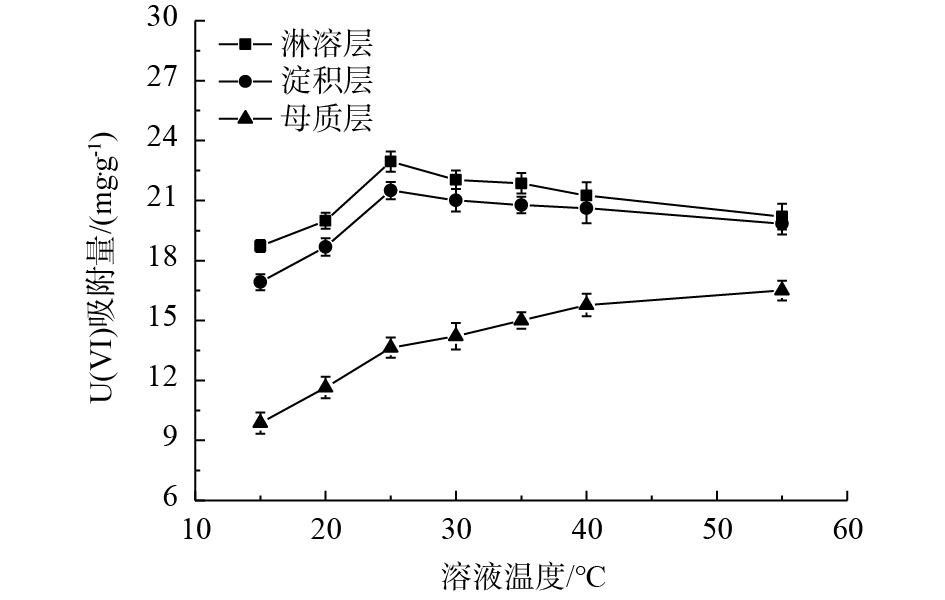
图 4 温度对各土层吸附U(Ⅵ)的影响
Figure 4. Effect of temperature on U(VI) adsorption in soil horizons

图 5 粒径对各土层吸附U(Ⅵ)的影响
Figure 5. Effect of particle size on U(VI) adsorption in soil horizons
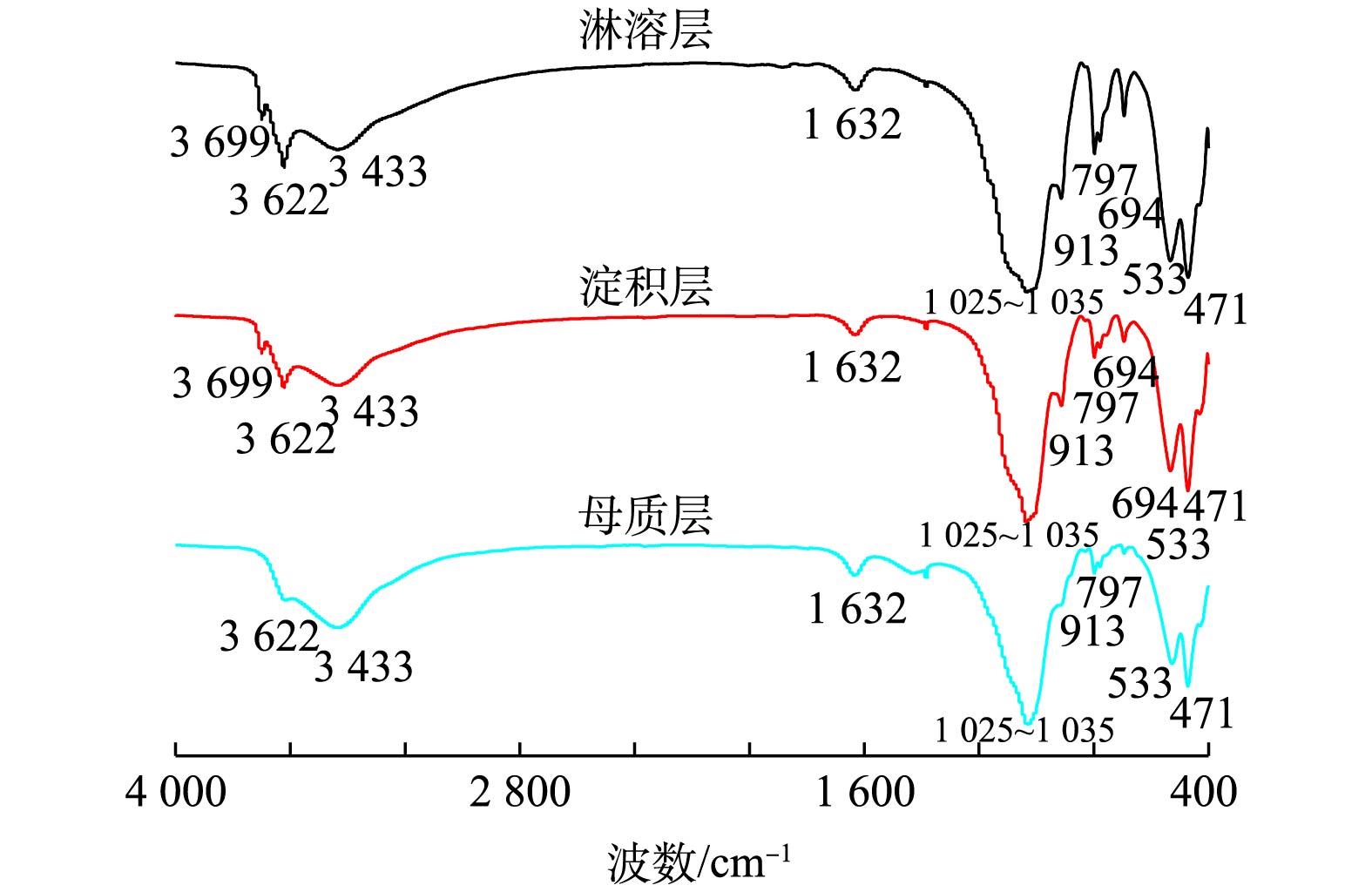
图 9 各层土样吸附前的傅里叶红外光谱
Figure 9. Fourier transform infrared spectroscopy spectra of soil horizons before adsorption
表 1 各土样粒径分级
Table 1. Particle size classification of soil horizons
土层类型 ≥8目 8~40目 40~60目 60~200目 ≤200目 淋溶层 2.83 50.96 9.29 17.04 19.88 淀积层 10.64 51.22 8.42 13.36 16.36 母质层 32.22 55.62 3.84 4.50 3.82
下载: 导出CSV
表 2 各土层理化性质
Table 2. Physico-chemical properties of soil horizons
土层类型 自然密度/(g·cm−3) 干密度/(g·cm−3) 含水率/% pH BET/(g·m−2) 有机质/(g·kg−1) 淋溶层 1.56 1.25 19.87 4.1 40.33 36.51 淀积层 1.66 1.35 18.67 4.6 33.99 25.74 母质层 1.71 1.50 12.28 7.2 17.56 2.10
下载: 导出CSV
表 3 各土层主要组成元素
Table 3. Main elements of soil horizons
土层类型 Fe Ca Mn K S Ti P 淋溶层 69.46 1.20 0.21 27.60 3.30 7.56 2.49 淀积层 65.35 1.26 0.15 27.71 4.56 5.68 3.31 母质层 48.51 4.81 1.01 32.74 2.86 4.04 1.85 注:各土层中U含量均低于XRF仪检测下限(十亿分之一)。
下载: 导出CSV
-
[1] 李春光, 谭凯旋, 刘振中, 等. 砂岩铀矿不同铀组分活度比特征及其对地浸采铀的指示[J]. 原子能科学技术, 2017, 51(8): 1358-1363. doi: 10.7538/yzk.2017.51.08.1358 [2] 周书葵, 刘迎九, 邓文静, 等. 木屑季铵盐型螯合吸附剂对U(Ⅵ)的吸附性能研究[J]. 原子能科学技术, 2016, 50(1): 24-30. doi: 10.7538/yzk.2016.50.01.0024 [3] ZUO R, LIU L, JIANG X, et al. Factor influencing U(VI) adsorption onto soil from a candidate very low level radioactive waste disposal site in China[J]. Nuclear Technology & Radiation Protection, 2016, 31(3): 268-276. [4] NEGREL P, DE V B, REIMANN C, et al. U-Th signatures of agricultural soil at the European continental scale (GEMAS): Distribution, weathering patterns and processes controlling their concentrations[J]. Science of the Total Environment, 2018, 622(1): 1277-1293. [5] LI X, WU J, LIAO J, et al. Adsorption and desorption of uranium(VI) in aerated zone soil[J]. Journal of Environmental Radioactivity, 2013, 115(1): 143-150. [6] SALBU B, KASHPAROV V, LIND O C, et al. Challenges associated with the behaviour of radioactive particles in the environment[J]. Journal of Environmental Radioactivity, 2018, 186(SI): 101-115. [7] 范婷, 张晓文, 吕俊文, 等. 放射性污染土壤生物修复的研究进展[J]. 安全与环境学报, 2011, 11(6): 65-68. doi: 10.3969/j.issn.1009-6094.2011.06.017 [8] ECHEVARRIA G, SHEPPARD M I, MOREL J. Effect of pH on the sorption of uranium in soils[J]. Journal of Environmental Radioactivity, 2001, 53(2): 257-264. doi: 10.1016/S0265-931X(00)00116-8 [9] SANTOS E A, LADEIRA A C Q. Recovery of uranium from mine waste by leaching with carbonate-based reagents[J]. Environmental Science & Technology, 2011, 45(8): 3591-3597. [10] VANDENHOVE H, HEES M V, WOUTERS K, et al. Can we predict uranium bioavailability based on soil parameters? Part 1: Effect of soilparameters on soil solution uranium concentration[J]. Environmental Pollution, 2007, 145(2): 587-595. doi: 10.1016/j.envpol.2006.04.011 [11] TAO C, MARK B O, ERIC R E, et al. Effects of phosphate on uranium(VI) adsorption to goethite-coated sand[J]. Environmental Science & Technology, 2004, 38(22): 6059-6065. [12] YU Y, JAMES S E, NA X, et al. Impact of natural organic matter on uranium transport through saturated geologic materials: From molecular to column scale[J]. Environmental Science & Technology, 2012, 46(11): 5931-5938. [13] MASSEY M S, LEZAMA-PACHECO J S, NELSON J M, et al. Uranium incorporation into amorphous silica[J]. Environmental Science & Technology, 2014, 48(15): 8636-8644. [14] MALKOVSKY V I, PETROV V A, DIKOV Y P, et al. Colloid-facilitated transport of uranium by groundwater at the U-Mo ore field in eastern Transbaikalia[J]. Environmental Earth Sciences, 2014, 73(10): 6145-6152. [15] FRANCES F S, PACHECO E G, GRANA A M, et al. Concentration of uranium in the soils of the west of Spain[J]. Environmental Pollution, 2018, 236(1): 1-11. [16] ROUT S, RAVI P M, KUMAR A, et al. Spectroscopic investigation of uranium sorption on soil surface using X-ray photoelectron spectroscopy[J]. Journal of Radioanalytical and Nuclear Chemistry, 2017, 313(3): 565-570. doi: 10.1007/s10967-017-5336-5 [17] LIU J, LUO X W, WANG J, et al. Provenance of uranium in a sediment core from a natural reservoir, South China: Application of Pb stable isotope analysis[J]. Chemosphere, 2018, 193(1): 1172-1180. [18] WANG J, LIU J, CHEN Y H, et al. Preliminary results of spatial distribution of uranium and thorium in soil profiles near a uranium industrial site, Guangdong province, China[J]. Nukleonika, 2016, 61(3): 367-371. doi: 10.1515/nuka-2016-0061 [19] 向明文, 王丹, 姚天月, 等. 8种植物对铀和镉的富集特性[J]. 环境工程学报, 2017, 11(1): 594-601. doi: 10.12030/j.cjee.201606213 [20] 蒋海燕, 张伟, 周书葵, 等. 腐殖酸修饰凹凸棒对U(VI)的吸附性能及机理[J]. 环境工程学报, 2015, 9(2): 705-710. doi: 10.12030/j.cjee.20150233 [21] 李合莲, 陈家军, 顾志杰. 铀尾矿对地下水的环境影响研究[J]. 环境污染治理技术与设备, 2000,1(3): 82-88. [22] LIU H, CHENG W, WANG M, et al. Investigation of U(VI) desorption behavior from natural sediment, Oak Ridge[J]. Journal of Environmental Radioanalytical and Nuclear Chemistry, 2017, 314(1): 167-175. doi: 10.1007/s10967-017-5384-x [23] 夏良树, 黄欣, 曹存存, 等. 红壤胶体对U(Ⅵ)的吸附性能及机理[J]. 原子能科学技术, 2013, 47(10): 1692-1699. doi: 10.7538/yzk.2013.47.10.1692 [24] CRAWFORD S E, LOFTS S, LIBER K. The role of sediment properties and solution pH in the adsorption of uranium(Ⅵ) to freshwater sediments[J]. Environmental Pollution, 2017, 220((Pt B): 873-881. [25] CUMBERLAND C, SUSAN A, DOUGLAS A, et al. Uranium mobility in organic matter-rich sediments : A review of geological and geochemical processes[J]. Earth Science Reviews, 2016, 159(1): 160-185. [26] 冯明明. 铀矿山尾矿库区土壤中有机质吸附铀效果与机制研究[D]. 抚州: 东华理工大学, 2016. [27] FIOL N, VILLAESCUSA I. Determination of sorbent point zero charge: usefulness in sorption studies[J]. Environmental Chemistry Letters, 2009, 7(1): 79-84. doi: 10.1007/s10311-008-0139-0 [28] PU Y, YANG X, ZHENG H, et al. Adsorption and desorption of thallium(I) on multiwalled carbon nanotubes[J]. Chemical Engineering Journal, 2013, 219(1): 403-410. [29] 李亚娟, 赵传起, 洪沛东, 等. 磁性还原石墨烯的制备及其对抗生素的吸附性能[J]. 环境工程学报, 2018, 12(1): 15-24. doi: 10.12030/j.cjee.201705157 [30] 张亚峰, 安路阳, 尚书, 等. 废玻璃/铝渣人工沸石对水中Ca2+的吸附[J]. 环境工程学报, 2019, 13(1): 49-61. doi: 10.12030/j.cjee.201806101 [31] TINNACHER R M, NICO P S, DAVIS J A, et al. Effects of fulvic acid on uranium(VI) sorption kinetics[J]. Environmental Science & Technology, 2013, 47(12): 6214-22. [32] DAINYAK L G, DRITS V A. A model for the interpretation of Mossbauer spectra of muscovite[J]. European Journal of Mineralogy, 2009, 21(1): 99-106. doi: 10.1127/0935-1221/2008/0020-1835 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3924
- HTML全文浏览数: 3924
- PDF下载数: 44
- 施引文献: 0
