

































-
铬是一种易致癌、致突变和高毒性的环境污染物[1],环境中铬含量主要来自于电镀、冶金和制革行业等工业活动废水的排放[2]。铬主要为Cr(Ⅲ)和Cr(Ⅵ),Cr(Ⅲ)可以通过简单加碱沉淀的方法予以去除。相较而言,Cr(Ⅵ)毒性是Cr(Ⅲ)的100倍,且高迁移性使得Cr(Ⅵ)更容易在生物体和人体中积累,对生态环境及人体健康造成了严重的威胁[3-4]。当前含铬废水的处理方法主要有离子交换法[5]、膜分离法[6]和化学还原法[7]等,然而大量实践表明,这些处理方法仍然存在处理费用高和较严重的二次污染问题。目前,高毒性的Cr(Ⅵ)向低毒性的Cr(Ⅲ)转化的高效还原方法,由于降低了其对环境的危害,已成为是含铬废水处理的研究热点之一。高级还原技术(ARPs)是在高级氧化技术(AOPs)基础上发展起来的,区别在于前者可产生还原性自由基,包括水合电子(
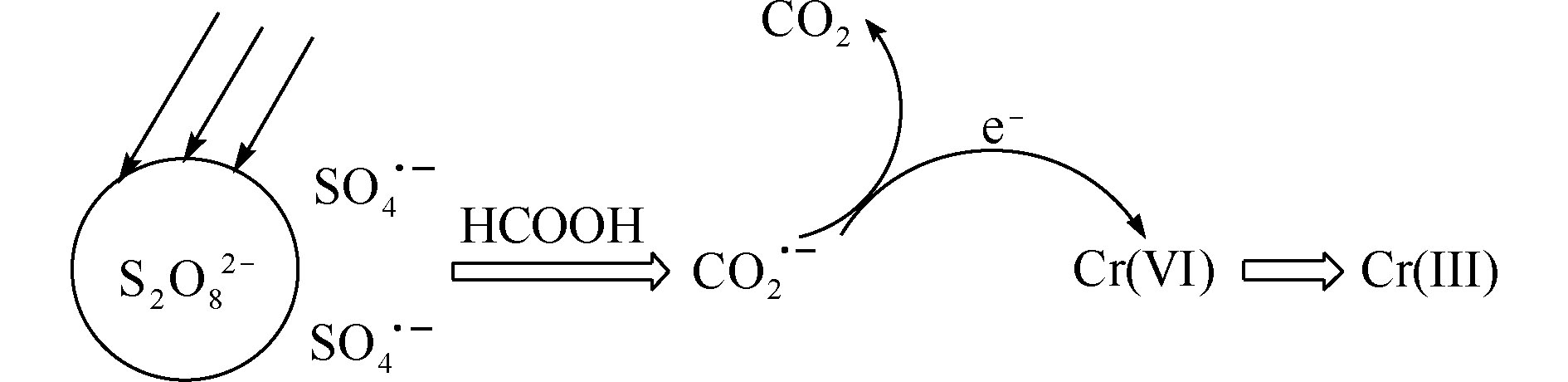
e−aq )、原生态氢(H · )和二氧化碳阴离子自由基(CO⋅−2 )等。近年来,对于CO⋅−2 的研究屡有报道[8-9],CO⋅−2 被证实是一种强还原性自由基,氧化还原电位为−1.9 V[10],目前有研究[11-12]表明,CO⋅−2 可以与Hg(Ⅱ)、取代苯化合物[13]、四氯化碳[10]、Cr(Ⅵ)[14]等反应,且均具有较好的还原去除效果。有研究[13]表明,CO⋅−2 产生的主要方法为向AOPs体系中加入甲酸或者甲酸盐,使含有SO⋅−4 、HO · 的氧化性体系转化为还原性体系(如式(1)和式(2)所示),进而对高氧化态的污染物表现出明显的去除效果。JIANG等[15]向过碳酸钠/Fe(Ⅱ)和过氧化钙/Fe(Ⅱ)类芬顿体系中加入HCOOH之后,可以明显降解四氯化碳。REN等[14]在Cr(Ⅵ)、Na2S2O8和HCOO−初始浓度分别为50、30和30 mmol·L−1,反应温度为70 ℃条件下,热活化Na2S2O8/HCOO−体系,反应240 min之后,Cr(Ⅵ)去除率可以达到99%。本研究采用紫外(UV)活化过硫酸盐(PS)/甲酸(FA)体系还原高浓度Cr(Ⅵ),利用UV、UV/PS和UV/PS/FA体系对Cr(Ⅵ)还原效果的对比,结合电子自旋共振(ESR)对体系中产生的主要自由基进行了检测分析,研究了UV/PS/FA体系的活化机制及其对Cr(Ⅵ)的还原机制,并考察体系主要反应条件以及复杂水体环境中天然有机物(NOM)、无机阴离子对体系还原Cr(Ⅵ)的影响,以期为高浓度Cr(Ⅵ)还原提供新型高效的、实用性较强的均相光催化处理方法。
-
丙酮(C3H6O)、硝酸钠(NaNO3)、氯化钠(NaCl)、碳酸氢钠(NaHCO3)、硫酸(H2SO4)、磷酸(H3PO4)、氢氧化钠(NaOH)、甲酸(HCOOH)、二苯碳酰二肼(C13H14N4O)、过硫酸钠(Na2S2O8)均为分析纯;重铬酸钾(K2Cr2O7)为优级纯;腐植酸(黄腐酸FA≥90%)。
紫外分光光度计(TU-1901,北京普析通用仪器有限责任公司)、光催化反应仪(PL-02,北京普林塞斯科技有限公司)、pH计(PHS-3C,上海天达仪器有限公司)、ESR检测仪(A300,布鲁克)。
-
用重铬酸钾和去离子水配置模拟不同浓度的Cr(Ⅵ)废水。采用石英玻璃圆底试管作为反应瓶(V=50 mL),先将一定浓度的PS、FA和模拟废水取至反应瓶混合均匀,置于光催化反应仪里,开启磁力搅拌(400 r·min−1)和紫外光源(P=500 W)进行反应(预开启紫外灯管30 min保证光强稳定),按照既定时间取样分析,每组实验另外设置2个平行实验组。
Cr(Ⅵ)含量采用二苯碳酰二肼分光光度法(GB 7467-1987)测定;采用ESR进行检测分析主要自由基种类,ESR检测条件:中心磁场为350.00 mT;扫描宽度为15.00 mT;扫描时间为30.00 s;微波功率为3.99 mW;调制幅度为0.10 mT;转化时间为40.00 s。
-
在Cr(Ⅵ)初始浓度为200 mg·L−1条件下,分别对比了单独UV、UV/PS和UV/PS/FA体系对Cr(Ⅵ)的还原效率,实验结果如图1所示。由图1可知,当PS和FA分别为20 mmol·L−1和40 mmol·L−1,初始pH为2.4(未调节),单独UV和UV/PS体系在50 min内对Cr(Ⅵ)没有还原去除效果;当向UV/PS体系中加入FA之后,在50 min内Cr(Ⅵ)的去除率高达100%。由此可见,UV/PS/FA体系对高浓度Cr(Ⅵ)具有很好的去除效果。
有研究表明,水分子在真空紫外(VUV,λ=185 nm)照射的条件下能够产生具有还原性的原生态氢(H · )和水合电子(
e−aq )[16-19],见式(3)~式(5)。为避免H · 和e−aq 对本体系带来的干扰,实验所用紫外光波长范围为200~350 nm,故单独UV对Cr(Ⅵ)不存在还原效果。UV活化PS过程中会产生SO⋅−4 和HO · ,加入FA之后,Cr(Ⅵ)去除效果明显增强,这是由于FA可以与SO⋅−4 反应产生CO⋅−2 ,通过CO⋅−2 与Cr(Ⅵ)的相互作用,进而高效还原Cr(Ⅵ)为Cr(Ⅲ)。 -
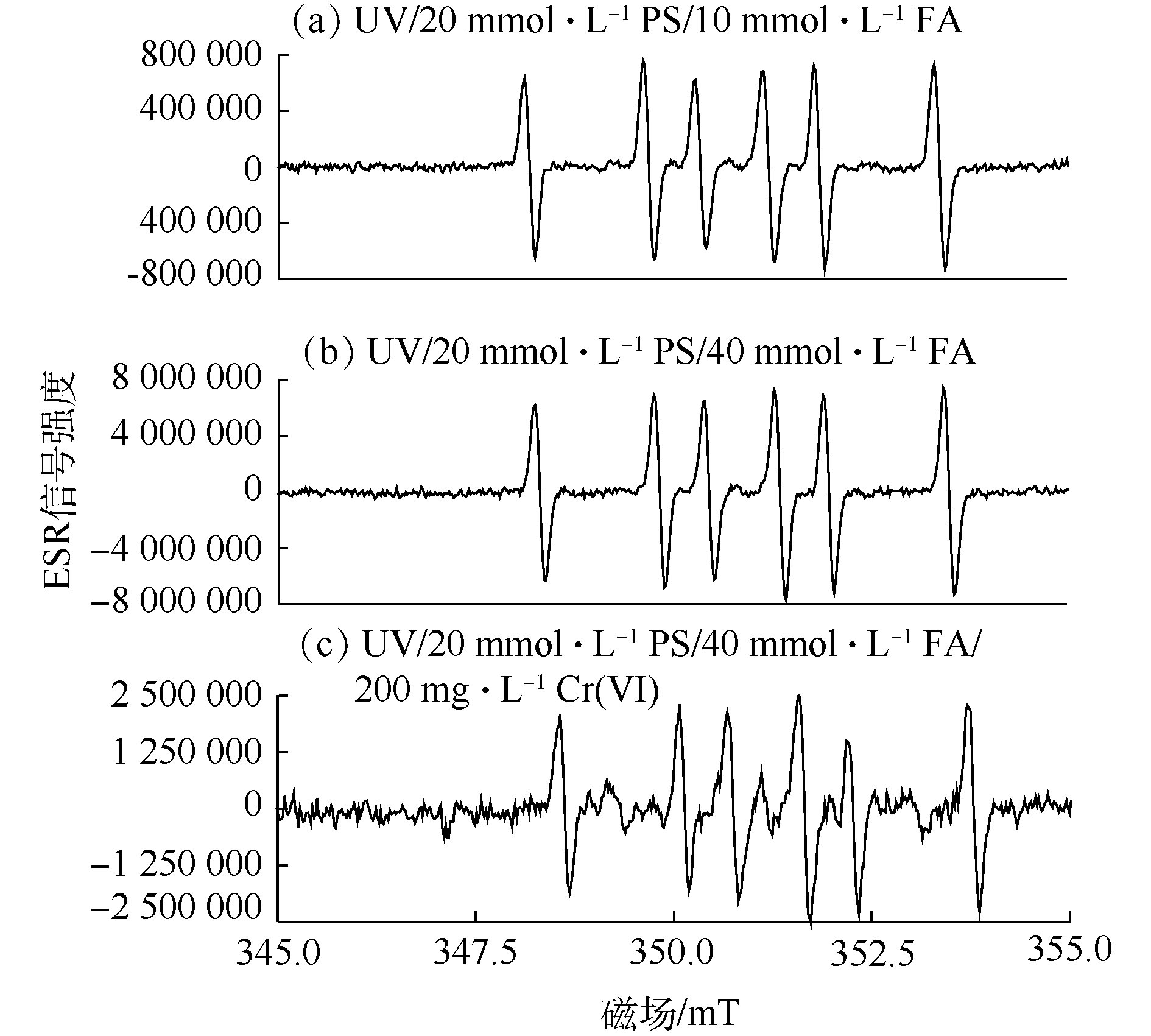
为了进一步探明UV/PS/FA的活化机制及对Cr(Ⅵ)的还原机理,以DMPO为自由基捕获剂,采用ESR对体系中的自由基进行了检测分析。在PS浓度为20 mmol·L−1条件下,通过改变FA和Cr(Ⅵ)的浓度,以UV照射15 min所捕获的自由基信号峰强度为依据,考察了自由基信号强度与FA浓度、Cr(Ⅵ)浓度之间的变化关系,结果如图2所示。由ESR表征结果可知,图2(a)~图2(c)均出现了明显且强度相同的DMPO-
CO⋅−2 加合物信号峰,与LIU等[20]和VILLAMENA等[21]报道的DMPO-CO⋅−2 加合物峰值相一致,从而证实了UV/PS/FA体系确实可以产生CO⋅−2 。通过比较图2(a)和图2(b)可知,在没有Cr(Ⅵ)干扰的条件下,FA浓度由10 mmol·L−1上升至40 mmol·L−1时,UV/PS/FA体系中出现的DMPO-CO⋅−2 加合物信号峰强度得到明显提高,这表明升高FA浓度可以提高CO⋅−2 产率,证明FA直接参与了CO⋅−2 的生成反应。由图2(b)和图2(c)可以看出,UV/PS/FA体系中有Cr(Ⅵ)存在时,DMPO-CO⋅−2 加合物信号峰强度明显减弱,这说明体系中产生的CO⋅−2 可以与Cr(Ⅵ)发生反应,并将Cr(Ⅵ)还原去除,初步推断反应机理如图3所示。 -
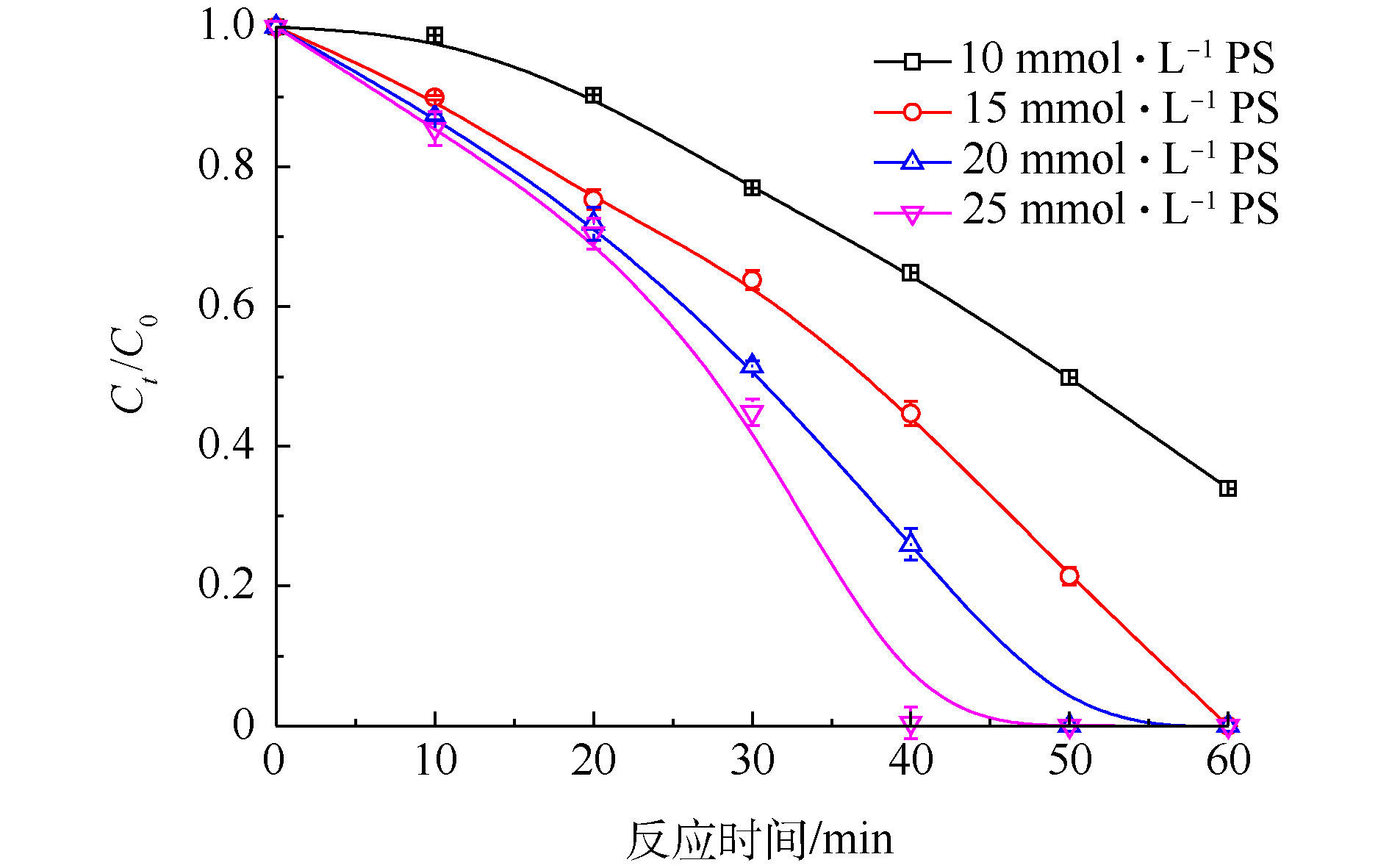
当初始Cr(Ⅵ)浓度为200 mg·L−1,FA浓度为40 mmol·L−1,PS浓度为10~25 mmol·L−1时,反应在60 min内,Cr(Ⅵ)去除率的变化情况如图4所示。从图4可以看出,当PS浓度为10 mmol·L−1时,在40 min内Cr(Ⅵ)去除率仅为35.2%;当PS增加为25 mmol·L−1时,Cr(Ⅵ)去除率提高至100%,PS对Cr(Ⅵ)的还原具有明显的促进效果。导致上述结果的主要原因是:随着PS浓度的增加,体系可以产生更多的
SO⋅−4 [22],而随着SO⋅−4 的增加,提高了FA的利用率,加快了体系产生CO⋅−2 的速率,进而增强了Cr(Ⅵ)还原的效率。 -
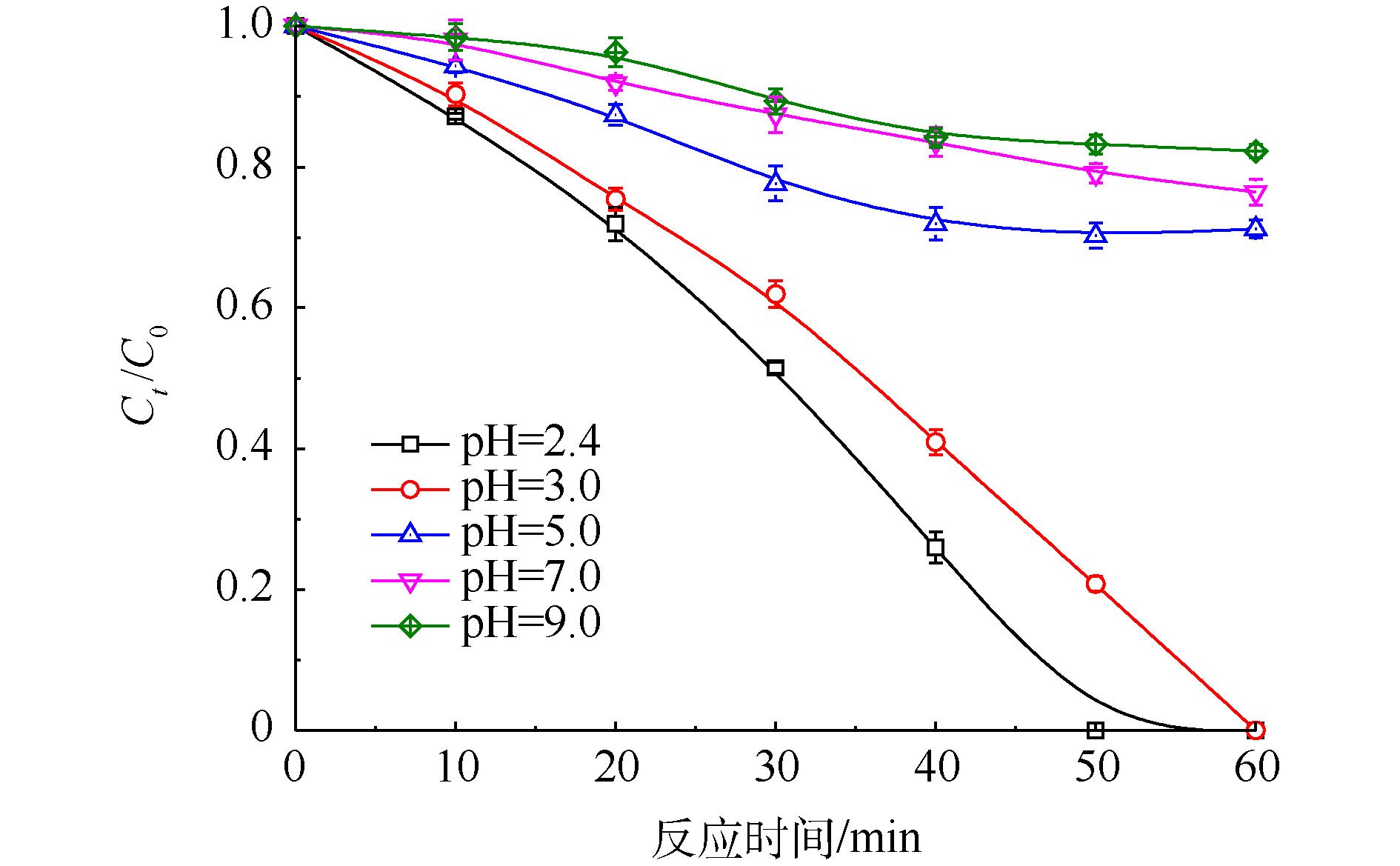
初始Cr(Ⅵ)浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,考察在60 min内,随着初始pH改变,Cr(Ⅵ)去除率的变化趋势,实验结果见图5。反应前后体系pH存在微弱改变,由图5可知,随着pH由2.4升高到9.0时,Cr(Ⅵ)还原效率呈现明显的下降趋势。当pH=2.4时,在50 min内Cr(Ⅵ)可以完全被去除;当pH=9.0时,Cr(Ⅵ)在50 min内的去除率仅为16.8%。这说明UV/PS/FA在酸性条件下具有较好的还原效果,而在中性尤其是碱性条件下,还原性能会受到明显抑制。这是因为FA的pKa=3.75,改变体系pH会影响FA在溶液中的存在形态,即分子态(HCOOH)或离子态(HCOO−),两者与
SO⋅−4 的反应速率常数存在差异性,FA分子态更容易与SO⋅−4 反应产生CO⋅−2 [20],因此,随着pH的升高,Cr(Ⅵ)还原效率逐渐降低。 -
图6为初始Cr(Ⅵ)浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1,水中常见阴离子Cl−、
HCO−3 和NO−3 对UV/PS/FA体系降解Cr(Ⅵ)的影响,每种离子浓度均为100 mmol·L−1。由图6可知,Cl−、HCO−3 和NO−3 3种离子均会抑制Cr(Ⅵ)的还原去除,在相同浓度条件下,其抑制程度强弱的顺序为HCO−3 >NO−3 >Cl−。当Cl−浓度为100 mmol·L−1时,50 min内Cr(Ⅵ)的去除率由100%降低为74.3%,这表明高浓度的Cl−对Cr(Ⅵ)降解存在严重的抑制效果。其主要原因是:Cl−可以与SO−4 反应生成低活性的氯自由基(Cl · )和超氯自由基(Cl⋅−2 ),从而减少了体系SO−4 的量[23],见式(6)和式(7);同时,生成的Cl⋅−2 会进一步与HCOOH发生反应[24],故Cl−抑制了体系中CO⋅−2 的产生,从而进一步抑制了Cr(Ⅵ)的降解。XU等[25]在四氯化碳降解研究中也发现,低浓度Cl−存在微弱抑制效果,高浓度Cl−的抑制效果更加明显。当
NO−3 投加量是100 mmol·L−1时,50 min内Cr(Ⅵ)去除率降低为41.6%。推断原因是:溶液中NO−3 的增加会消耗体系中氧化性自由基SO⋅−4 [26](式(8)),虽然反应产物为硝酸根自由基(NO⋅3 ),可以替代SO4·−与HCOOH反应产生主要还原性自由CO⋅−2 [27](式(9)),但增加的反应步骤,大大降低了整体的反应速率,因此,NO−3 对Cr(Ⅵ)还原亦表现出抑制作用。当体系中
HCO−3 浓度为100 mmol·L−1时,对体系还原Cr(Ⅵ)的抑制效果最为明显,50 min去除率降低至26.7%。这是由于当溶液中加入HCO−3 ,会使溶液pH升高,由2.3.2节可知,提高pH不利于Cr(Ⅵ)降解。此外,随着HCO−3 浓度的增加,其会与体系产生的SO⋅−4 发生反应并产生具有氧化性的碳酸根自由基(CO⋅−3 )(见式(10)),从而降低SO⋅−4 与FA反应产生CO⋅−2 的概率[28],且过量的HCO−3 还可能与CO⋅−2 相互反应产生CO⋅−3 [29](见式(11)),故高浓度HCO−3 也会抑制Cr(Ⅵ)的还原。 -
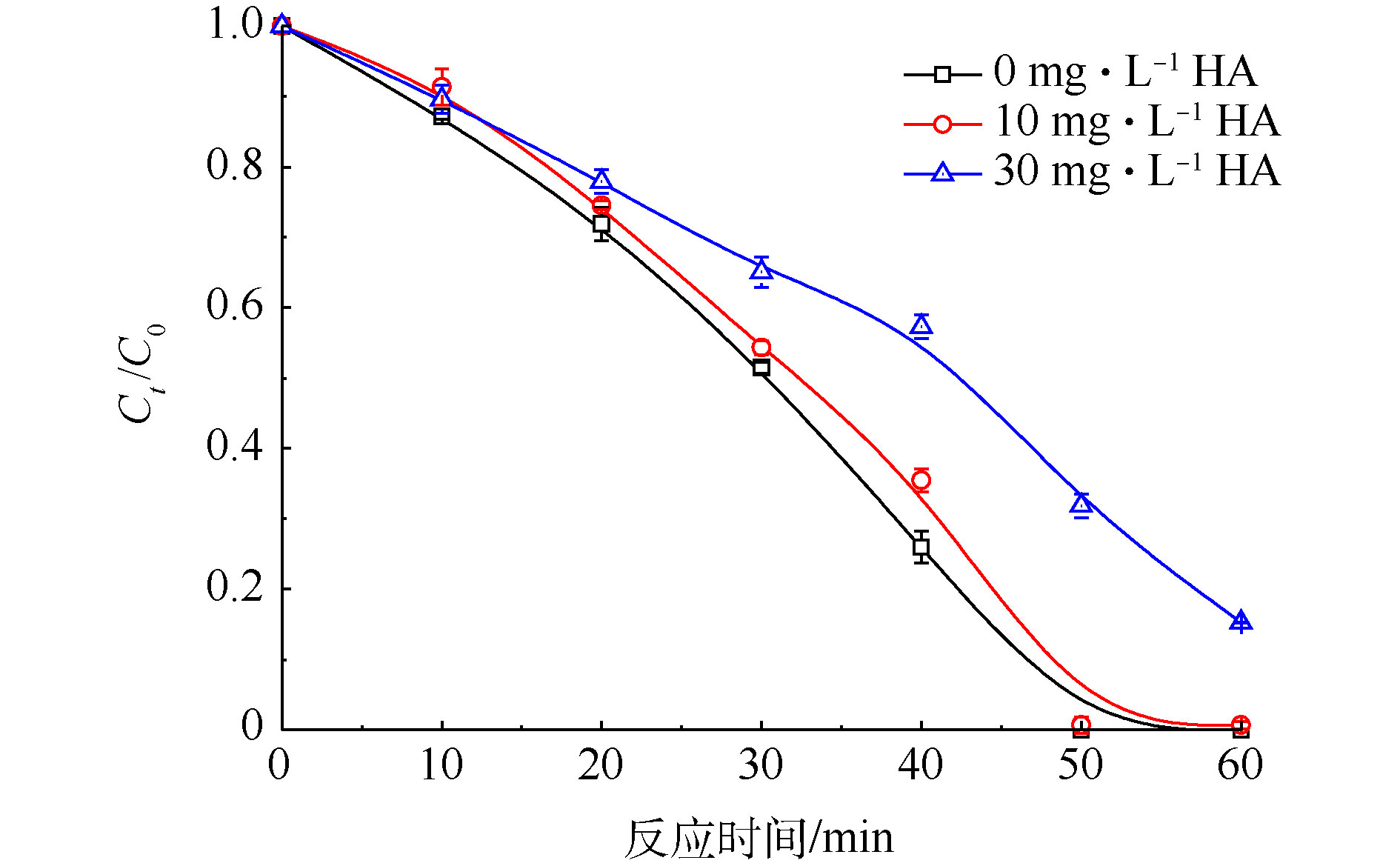
腐殖酸(HA)是天然水体中的主要有机物[30]。实验中以HA模拟自然水体中的有机物,当Cr(Ⅵ)初始浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,考察不同浓度HA对Cr(Ⅵ)降解效果的影响,实验结果见图7。由图7可知,当HA浓度为0 mg·L−1时,50 min内Cr(Ⅵ)的去除率为100%;当HA浓度提高到30 mg·L−1时,Cr(Ⅵ)的去除率降低为68.1%。因此,HA会抑制Cr(Ⅵ)的还原。这可能是由于HA是一种天然有机物,会与强氧化性的自由基反应得到矿化[31],故HA的加入会减少
SO⋅−4 的量,从而抑制CO⋅−2 的产生,最终降低了Cr(Ⅵ)还原去除速率。 -
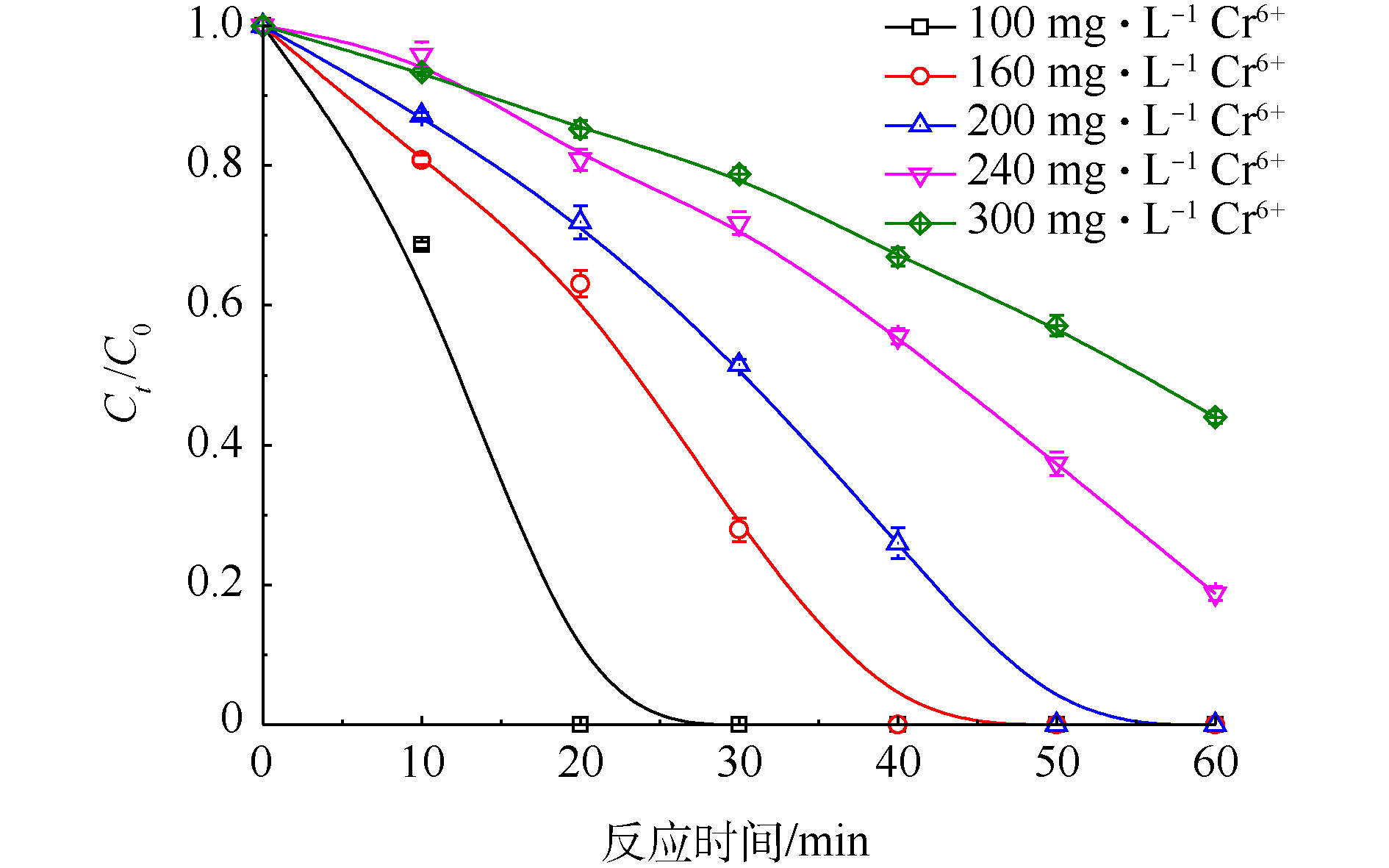
当PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,改变Cr(Ⅵ)初始浓度,考察Cr(Ⅵ)初始浓度对Cr(Ⅵ)去除的影响,实验结果见图8。由图8可知,Cr(Ⅵ)去除率随着Cr(Ⅵ)初始浓度的增加而降低,但UV/PS/FA体系对不同浓度的Cr(Ⅵ)都有很好的去除效果。当Cr(Ⅵ)初始浓度为100 mg·L−1时,20 min内Cr(Ⅵ)去除率就可以到达100%,将Cr(Ⅵ)完全去除,当Cr(Ⅵ)初始浓度提高为300 mg·L−1时,在60 min内的去除率仅为56%。这是因为当PS和FA浓度一定时,单位时间内产生的
SO⋅−4 和CO⋅−2 的量基本维持不变,CO⋅−2 还原Cr(Ⅵ)的能力也是一定的,随着Cr(Ⅵ)浓度的增加,被还原Cr(Ⅵ)的量与起始Cr(Ⅵ)的量的比值下降,表观去除率变小。当初始Cr(Ⅵ)浓度为200、240和300 mg·L−1时,在一定的反应时间内,对Cr(Ⅵ)的还原过程进行反应动力学拟合,拟合结果如图9所示。由图9可知,dC与dt有很好的线性关系,这说明UV/PS/FA降解Cr(Ⅵ)的反应符合零级反应动力学。通过拟合得到Cr(Ⅵ)不同初始浓度下还原速率常数(见表1)。由表1可知,当Cr(Ⅵ)初始浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,Cr(Ⅵ)还原反应速率常数为78.467 μmol·(L·min)−1。WANG等[32]发现Fe(III)浓度为179.1 μmol·L−1,pH为1.0时,均相光催化还原Cr(Ⅵ)也遵循零级动力学反应方程式,且反应动力学常数仅为0.934 μmol·(L·min)−1,相对而言,UV/PS/FA体系对Cr(Ⅵ)具有更加高效的还原效率。
-
1)采用UV/PS/FA体系,实现了对高浓度Cr(Ⅵ)的还原处理。当Cr(Ⅵ)初始浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,在反应50 min后Cr(Ⅵ)基本完全被还原去除。对反应过程中的活性自由基采用ESR技术进行表征,证实了
CO⋅−2 的存在。2)UV/PS/FA体系在酸性条件下具有较强的还原能力,随着体系pH的升高还原作用呈现明显下降趋势。随着体系中PS浓度的升高,Cr(Ⅵ)去除率逐渐提高。水溶液中存在的HA、Cl−、
HCO−3 和NO−3 都会在不同程度对Cr(Ⅵ)的还原产生抑制作用。3)对不同初始浓度下Cr(Ⅵ)降解过程进行动力学分析推导,可知其符合零级反应动力学,且初始Cr(Ⅵ)浓度为200、240和300 mg·L−1时对应的反应速率常数分为78.467、70.156和56.577 μmol·(L·min)−1。本研究为Cr(Ⅵ)废水的处理提供了高效的还原方法。
紫外活化过硫酸盐/甲酸体系还原水中Cr(Ⅵ)机理及影响因素
Mechanism and influencing factors of aqueous Cr(Ⅵ) reduction by carbon dioxide anion radical based on the UV-activated sodium persulfate/formic acid system
-
摘要: 采用紫外活化过硫酸盐/甲酸体系所产生的还原性二氧化碳阴离子自由基(
CO⋅−2 ),研究了水溶液中高浓度Cr(Ⅵ)的去除效果;使用电子自旋共振(ESR)技术,鉴定识别了体系中产生的活性自由基;分析了体系的活化机理及其对Cr(Ⅵ)的还原机制;考察了过硫酸盐投加量、初始pH、腐殖酸、无机阴离子(Cl−、HCO−3 和NO−3 )及初始Cr(Ⅵ)浓度等对Cr(Ⅵ)去除的影响。结果表明:紫外活化过硫酸盐/甲酸体系可以有效还原Cr(Ⅵ),当过硫酸钠与甲酸浓度分别为20 mmol·L−1和40 mmol·L−1,未调初始pH为2.4时,初始浓度为200 mg·L−1 Cr(VI)在50 min内基本完全可被还原;此外,Cr(Ⅵ)还原去除率随过硫酸盐浓度升高而增强,在酸性条件下(pH=2.4),体系对Cr(Ⅵ)的还原效率最高,随着pH的增大,还原效率明显降低。进一步研究表明,Cl−、HCO−3 和NO−3 对Cr(Ⅵ)的还原都存在抑制作用,在相同浓度下,其抑制程度分别为HCO−3 >NO−3 >Cl−,腐殖酸也对Cr(Ⅵ)的去除存在抑制作用。紫外活化过硫酸盐/甲酸体系还原Cr(Ⅵ)过程符合零级反应动力学方程,其动力学常数为78.467 μmol·(L·min)−1。本研究结果为Cr(Ⅵ)废水的处理提供了一种高效的还原新技术。-
关键词:
- 紫外活化过硫酸盐/甲酸体系 /
- 二氧化碳阴离子自由基 /
- 六价铬还原
Abstract: The removal of high concentration of Cr(Ⅵ) in aqueous solution was studied by using ultraviolet activated persulfate/formic acid system to produce carbon dioxide anion radicals (CO⋅−2 ). The active free radicals produced in the system were identified by Electron paramagnetic resonance (EPR), and the mechanisms of radical activation and Cr(Ⅵ) reduction in this system were also discussed. The effects of persulfate dosages, initial pH, humic acid, inorganic anions (Cl−,HCO−3 ,NO−3 ) and the initial concentration of Cr(Ⅵ) on Cr(Ⅵ) removal were systematically investigated. The results showed that an effective Cr(Ⅵ) reduction occurred in ultraviolet activated persulfate/formic acid system, and Cr(Ⅵ) with an initial concentration of 200 mg·L−1 could be almost completely reduced within 50 min at the sodium persulfate concentration of 20 mmol·L−1, formic acid concentration of 40 mmol·L−1, and the initial pH of 2.4 (without the adjustment). In addition, the reduction efficiency of Cr(Ⅵ) increased with the increase of persulfate concentration. The highest reduction efficiency of Cr(Ⅵ) appeared at acidic conditions of pH=2.4. The reduction efficiency decreased significantly with the increase of pH. Further studies revealed that Cl−,HCO−3 andNO−3 inhibited the reduction of Cr(Ⅵ), the order of their inhibition degree at the same concentration wasHCO−3 >NO−3 >Cl−, and humic acid was also an inhibition factor for the removal of Cr(Ⅵ) at the same concentration. Ultraviolet activated persulfate/formic acid system was well fitted with the zero-order kinetic equation with the kinetic constant of 78.467 μmol·(L·min)−1, which provides an efficient new reduction technology for Cr(Ⅵ) wastewater treatment. -
铬是一种易致癌、致突变和高毒性的环境污染物[1],环境中铬含量主要来自于电镀、冶金和制革行业等工业活动废水的排放[2]。铬主要为Cr(Ⅲ)和Cr(Ⅵ),Cr(Ⅲ)可以通过简单加碱沉淀的方法予以去除。相较而言,Cr(Ⅵ)毒性是Cr(Ⅲ)的100倍,且高迁移性使得Cr(Ⅵ)更容易在生物体和人体中积累,对生态环境及人体健康造成了严重的威胁[3-4]。当前含铬废水的处理方法主要有离子交换法[5]、膜分离法[6]和化学还原法[7]等,然而大量实践表明,这些处理方法仍然存在处理费用高和较严重的二次污染问题。目前,高毒性的Cr(Ⅵ)向低毒性的Cr(Ⅲ)转化的高效还原方法,由于降低了其对环境的危害,已成为是含铬废水处理的研究热点之一。高级还原技术(ARPs)是在高级氧化技术(AOPs)基础上发展起来的,区别在于前者可产生还原性自由基,包括水合电子(
e−aq )、原生态氢(H · )和二氧化碳阴离子自由基(CO⋅−2 )等。近年来,对于CO⋅−2 的研究屡有报道[8-9],CO⋅−2 被证实是一种强还原性自由基,氧化还原电位为−1.9 V[10],目前有研究[11-12]表明,CO⋅−2 可以与Hg(Ⅱ)、取代苯化合物[13]、四氯化碳[10]、Cr(Ⅵ)[14]等反应,且均具有较好的还原去除效果。有研究[13]表明,CO⋅−2 产生的主要方法为向AOPs体系中加入甲酸或者甲酸盐,使含有SO⋅−4 、HO · 的氧化性体系转化为还原性体系(如式(1)和式(2)所示),进而对高氧化态的污染物表现出明显的去除效果。JIANG等[15]向过碳酸钠/Fe(Ⅱ)和过氧化钙/Fe(Ⅱ)类芬顿体系中加入HCOOH之后,可以明显降解四氯化碳。REN等[14]在Cr(Ⅵ)、Na2S2O8和HCOO−初始浓度分别为50、30和30 mmol·L−1,反应温度为70 ℃条件下,热活化Na2S2O8/HCOO−体系,反应240 min之后,Cr(Ⅵ)去除率可以达到99%。HO⋅+HCOOH⟶CO⋅−2+H++H2O (1) SO⋅−4+HCOOH→CO⋅−2+2H++SO2−4 (2) 本研究采用紫外(UV)活化过硫酸盐(PS)/甲酸(FA)体系还原高浓度Cr(Ⅵ),利用UV、UV/PS和UV/PS/FA体系对Cr(Ⅵ)还原效果的对比,结合电子自旋共振(ESR)对体系中产生的主要自由基进行了检测分析,研究了UV/PS/FA体系的活化机制及其对Cr(Ⅵ)的还原机制,并考察体系主要反应条件以及复杂水体环境中天然有机物(NOM)、无机阴离子对体系还原Cr(Ⅵ)的影响,以期为高浓度Cr(Ⅵ)还原提供新型高效的、实用性较强的均相光催化处理方法。
1. 材料与方法
1.1 试剂与仪器
丙酮(C3H6O)、硝酸钠(NaNO3)、氯化钠(NaCl)、碳酸氢钠(NaHCO3)、硫酸(H2SO4)、磷酸(H3PO4)、氢氧化钠(NaOH)、甲酸(HCOOH)、二苯碳酰二肼(C13H14N4O)、过硫酸钠(Na2S2O8)均为分析纯;重铬酸钾(K2Cr2O7)为优级纯;腐植酸(黄腐酸FA≥90%)。
紫外分光光度计(TU-1901,北京普析通用仪器有限责任公司)、光催化反应仪(PL-02,北京普林塞斯科技有限公司)、pH计(PHS-3C,上海天达仪器有限公司)、ESR检测仪(A300,布鲁克)。
1.2 实验与分析方法
用重铬酸钾和去离子水配置模拟不同浓度的Cr(Ⅵ)废水。采用石英玻璃圆底试管作为反应瓶(V=50 mL),先将一定浓度的PS、FA和模拟废水取至反应瓶混合均匀,置于光催化反应仪里,开启磁力搅拌(400 r·min−1)和紫外光源(P=500 W)进行反应(预开启紫外灯管30 min保证光强稳定),按照既定时间取样分析,每组实验另外设置2个平行实验组。
Cr(Ⅵ)含量采用二苯碳酰二肼分光光度法(GB 7467-1987)测定;采用ESR进行检测分析主要自由基种类,ESR检测条件:中心磁场为350.00 mT;扫描宽度为15.00 mT;扫描时间为30.00 s;微波功率为3.99 mW;调制幅度为0.10 mT;转化时间为40.00 s。
2. 结果与讨论
2.1 UV活化PS耦合FA体系降解Cr(Ⅵ)对比及还原机制分析
在Cr(Ⅵ)初始浓度为200 mg·L−1条件下,分别对比了单独UV、UV/PS和UV/PS/FA体系对Cr(Ⅵ)的还原效率,实验结果如图1所示。由图1可知,当PS和FA分别为20 mmol·L−1和40 mmol·L−1,初始pH为2.4(未调节),单独UV和UV/PS体系在50 min内对Cr(Ⅵ)没有还原去除效果;当向UV/PS体系中加入FA之后,在50 min内Cr(Ⅵ)的去除率高达100%。由此可见,UV/PS/FA体系对高浓度Cr(Ⅵ)具有很好的去除效果。
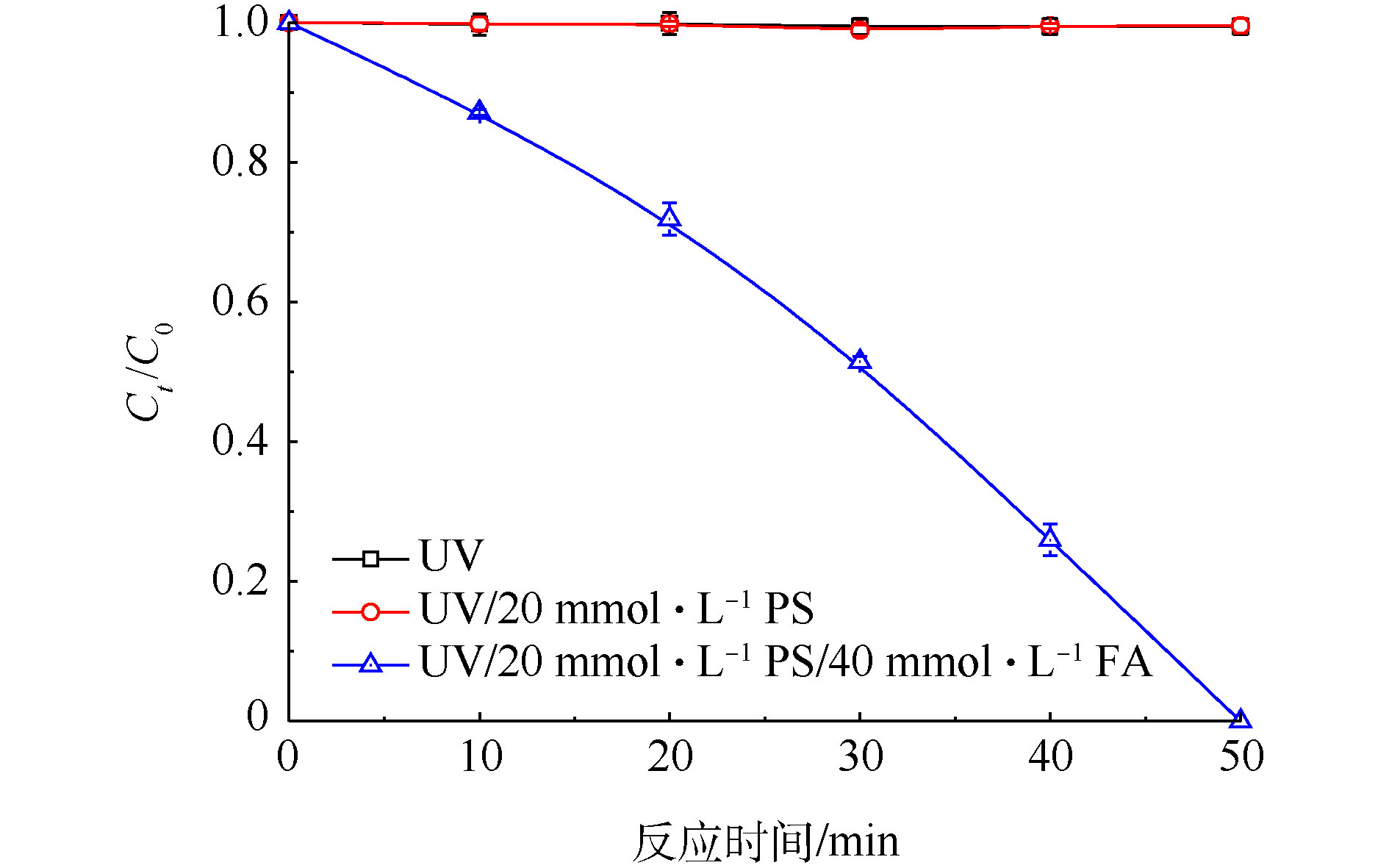 图 1 UV、UV/PS、UV/PS/HCOOH还原Cr(Ⅵ)Figure 1. Reduction of Cr(Ⅵ) by UV, UV/PS and UV/PS/HCOOH
图 1 UV、UV/PS、UV/PS/HCOOH还原Cr(Ⅵ)Figure 1. Reduction of Cr(Ⅵ) by UV, UV/PS and UV/PS/HCOOH有研究表明,水分子在真空紫外(VUV,λ=185 nm)照射的条件下能够产生具有还原性的原生态氢(H · )和水合电子(
e−aq )[16-19],见式(3)~式(5)。为避免H · 和e−aq 对本体系带来的干扰,实验所用紫外光波长范围为200~350 nm,故单独UV对Cr(Ⅵ)不存在还原效果。UV活化PS过程中会产生SO⋅−4 和HO · ,加入FA之后,Cr(Ⅵ)去除效果明显增强,这是由于FA可以与SO⋅−4 反应产生CO⋅−2 ,通过CO⋅−2 与Cr(Ⅵ)的相互作用,进而高效还原Cr(Ⅵ)为Cr(Ⅲ)。H2O+hv185nm⟶e−aq+H++HO⋅ (3) H2O+hv185mm⟶H⋅+HO⋅ (4) H⋅+H2O⟶H3O++e−aq,k=3.6×108L⋅(mol⋅s)−1 (5) 2.2 反应自由基的ESR表征
为了进一步探明UV/PS/FA的活化机制及对Cr(Ⅵ)的还原机理,以DMPO为自由基捕获剂,采用ESR对体系中的自由基进行了检测分析。在PS浓度为20 mmol·L−1条件下,通过改变FA和Cr(Ⅵ)的浓度,以UV照射15 min所捕获的自由基信号峰强度为依据,考察了自由基信号强度与FA浓度、Cr(Ⅵ)浓度之间的变化关系,结果如图2所示。由ESR表征结果可知,图2(a)~图2(c)均出现了明显且强度相同的DMPO-
CO⋅−2 加合物信号峰,与LIU等[20]和VILLAMENA等[21]报道的DMPO-CO⋅−2 加合物峰值相一致,从而证实了UV/PS/FA体系确实可以产生CO⋅−2 。通过比较图2(a)和图2(b)可知,在没有Cr(Ⅵ)干扰的条件下,FA浓度由10 mmol·L−1上升至40 mmol·L−1时,UV/PS/FA体系中出现的DMPO-CO⋅−2 加合物信号峰强度得到明显提高,这表明升高FA浓度可以提高CO⋅−2 产率,证明FA直接参与了CO⋅−2 的生成反应。由图2(b)和图2(c)可以看出,UV/PS/FA体系中有Cr(Ⅵ)存在时,DMPO-CO⋅−2 加合物信号峰强度明显减弱,这说明体系中产生的CO⋅−2 可以与Cr(Ⅵ)发生反应,并将Cr(Ⅵ)还原去除,初步推断反应机理如图3所示。2.3 单因素影响实验
2.3.1 PS浓度对Cr(Ⅵ)还原效果的影响
当初始Cr(Ⅵ)浓度为200 mg·L−1,FA浓度为40 mmol·L−1,PS浓度为10~25 mmol·L−1时,反应在60 min内,Cr(Ⅵ)去除率的变化情况如图4所示。从图4可以看出,当PS浓度为10 mmol·L−1时,在40 min内Cr(Ⅵ)去除率仅为35.2%;当PS增加为25 mmol·L−1时,Cr(Ⅵ)去除率提高至100%,PS对Cr(Ⅵ)的还原具有明显的促进效果。导致上述结果的主要原因是:随着PS浓度的增加,体系可以产生更多的
SO⋅−4 [22],而随着SO⋅−4 的增加,提高了FA的利用率,加快了体系产生CO⋅−2 的速率,进而增强了Cr(Ⅵ)还原的效率。2.3.2 初始pH的影响
初始Cr(Ⅵ)浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,考察在60 min内,随着初始pH改变,Cr(Ⅵ)去除率的变化趋势,实验结果见图5。反应前后体系pH存在微弱改变,由图5可知,随着pH由2.4升高到9.0时,Cr(Ⅵ)还原效率呈现明显的下降趋势。当pH=2.4时,在50 min内Cr(Ⅵ)可以完全被去除;当pH=9.0时,Cr(Ⅵ)在50 min内的去除率仅为16.8%。这说明UV/PS/FA在酸性条件下具有较好的还原效果,而在中性尤其是碱性条件下,还原性能会受到明显抑制。这是因为FA的pKa=3.75,改变体系pH会影响FA在溶液中的存在形态,即分子态(HCOOH)或离子态(HCOO−),两者与
SO⋅−4 的反应速率常数存在差异性,FA分子态更容易与SO⋅−4 反应产生CO⋅−2 [20],因此,随着pH的升高,Cr(Ⅵ)还原效率逐渐降低。2.3.3 无机离子的影响
图6为初始Cr(Ⅵ)浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1,水中常见阴离子Cl−、
HCO−3 和NO−3 对UV/PS/FA体系降解Cr(Ⅵ)的影响,每种离子浓度均为100 mmol·L−1。由图6可知,Cl−、HCO−3 和NO−3 3种离子均会抑制Cr(Ⅵ)的还原去除,在相同浓度条件下,其抑制程度强弱的顺序为HCO−3 >NO−3 >Cl−。当Cl−浓度为100 mmol·L−1时,50 min内Cr(Ⅵ)的去除率由100%降低为74.3%,这表明高浓度的Cl−对Cr(Ⅵ)降解存在严重的抑制效果。其主要原因是:Cl−可以与SO−4 反应生成低活性的氯自由基(Cl · )和超氯自由基(Cl⋅−2 ),从而减少了体系SO−4 的量[23],见式(6)和式(7);同时,生成的Cl⋅−2 会进一步与HCOOH发生反应[24],故Cl−抑制了体系中CO⋅−2 的产生,从而进一步抑制了Cr(Ⅵ)的降解。XU等[25]在四氯化碳降解研究中也发现,低浓度Cl−存在微弱抑制效果,高浓度Cl−的抑制效果更加明显。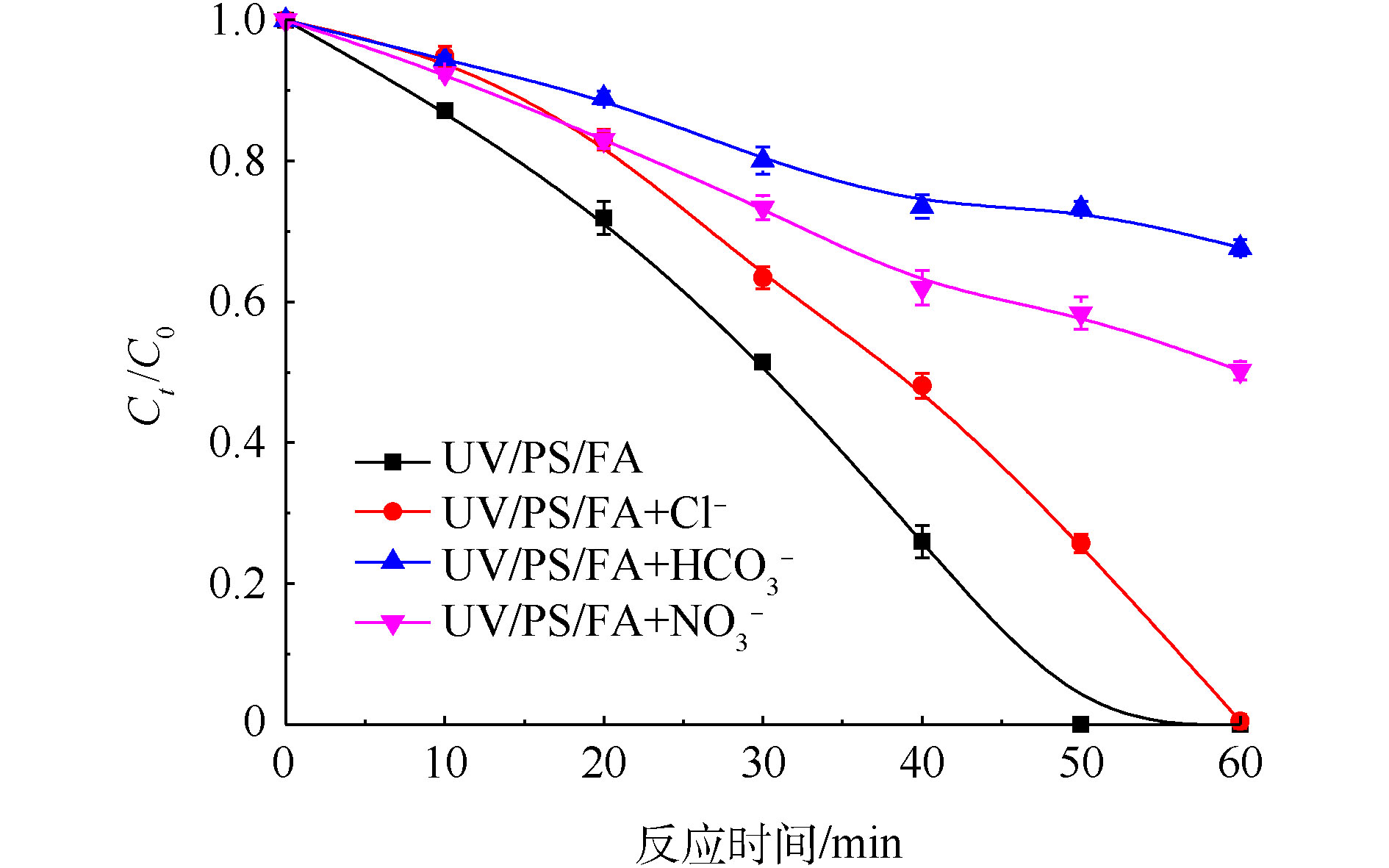 图 6 Cl−、
图 6 Cl−、HCO−3 和NO−3 浓度对Cr(Ⅵ)还原去除效果的影响Figure 6. Effects of Cl−,HCO−3 andNO−3 on Cr(Ⅵ) removal efficiency当
NO−3 投加量是100 mmol·L−1时,50 min内Cr(Ⅵ)去除率降低为41.6%。推断原因是:溶液中NO−3 的增加会消耗体系中氧化性自由基SO⋅−4 [26](式(8)),虽然反应产物为硝酸根自由基(NO⋅3 ),可以替代SO4·−与HCOOH反应产生主要还原性自由CO⋅−2 [27](式(9)),但增加的反应步骤,大大降低了整体的反应速率,因此,NO−3 对Cr(Ⅵ)还原亦表现出抑制作用。当体系中
HCO−3 浓度为100 mmol·L−1时,对体系还原Cr(Ⅵ)的抑制效果最为明显,50 min去除率降低至26.7%。这是由于当溶液中加入HCO−3 ,会使溶液pH升高,由2.3.2节可知,提高pH不利于Cr(Ⅵ)降解。此外,随着HCO−3 浓度的增加,其会与体系产生的SO⋅−4 发生反应并产生具有氧化性的碳酸根自由基(CO⋅−3 )(见式(10)),从而降低SO⋅−4 与FA反应产生CO⋅−2 的概率[28],且过量的HCO−3 还可能与CO⋅−2 相互反应产生CO⋅−3 [29](见式(11)),故高浓度HCO−3 也会抑制Cr(Ⅵ)的还原。SO⋅−4+Cl−⟶SO2−4+Cl⋅ (6) Cl−+Cl⋅⟶Cl⋅−2 (7) NO−3+SO⋅−4⟶SO2−4+NO⋅−3 (8) HCOOH+NO⋅3⟶NO−3+CO⋅−2+2H+ (9) SO⋅−4+HCO−3⟶CO⋅−3+SO2−4+H+ (10) HCO−3+CO⋅−2⟶HCO−2+CO⋅−3 (11) 2.3.4 天然有机物(NOM)的影响
腐殖酸(HA)是天然水体中的主要有机物[30]。实验中以HA模拟自然水体中的有机物,当Cr(Ⅵ)初始浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,考察不同浓度HA对Cr(Ⅵ)降解效果的影响,实验结果见图7。由图7可知,当HA浓度为0 mg·L−1时,50 min内Cr(Ⅵ)的去除率为100%;当HA浓度提高到30 mg·L−1时,Cr(Ⅵ)的去除率降低为68.1%。因此,HA会抑制Cr(Ⅵ)的还原。这可能是由于HA是一种天然有机物,会与强氧化性的自由基反应得到矿化[31],故HA的加入会减少
SO⋅−4 的量,从而抑制CO⋅−2 的产生,最终降低了Cr(Ⅵ)还原去除速率。2.3.5 Cr(Ⅵ)初始浓度的影响及动力学拟合
当PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,改变Cr(Ⅵ)初始浓度,考察Cr(Ⅵ)初始浓度对Cr(Ⅵ)去除的影响,实验结果见图8。由图8可知,Cr(Ⅵ)去除率随着Cr(Ⅵ)初始浓度的增加而降低,但UV/PS/FA体系对不同浓度的Cr(Ⅵ)都有很好的去除效果。当Cr(Ⅵ)初始浓度为100 mg·L−1时,20 min内Cr(Ⅵ)去除率就可以到达100%,将Cr(Ⅵ)完全去除,当Cr(Ⅵ)初始浓度提高为300 mg·L−1时,在60 min内的去除率仅为56%。这是因为当PS和FA浓度一定时,单位时间内产生的
SO⋅−4 和CO⋅−2 的量基本维持不变,CO⋅−2 还原Cr(Ⅵ)的能力也是一定的,随着Cr(Ⅵ)浓度的增加,被还原Cr(Ⅵ)的量与起始Cr(Ⅵ)的量的比值下降,表观去除率变小。当初始Cr(Ⅵ)浓度为200、240和300 mg·L−1时,在一定的反应时间内,对Cr(Ⅵ)的还原过程进行反应动力学拟合,拟合结果如图9所示。由图9可知,dC与dt有很好的线性关系,这说明UV/PS/FA降解Cr(Ⅵ)的反应符合零级反应动力学。通过拟合得到Cr(Ⅵ)不同初始浓度下还原速率常数(见表1)。由表1可知,当Cr(Ⅵ)初始浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,Cr(Ⅵ)还原反应速率常数为78.467 μmol·(L·min)−1。WANG等[32]发现Fe(III)浓度为179.1 μmol·L−1,pH为1.0时,均相光催化还原Cr(Ⅵ)也遵循零级动力学反应方程式,且反应动力学常数仅为0.934 μmol·(L·min)−1,相对而言,UV/PS/FA体系对Cr(Ⅵ)具有更加高效的还原效率。
表 1 不同初始浓度下Cr (VI) 的还原反应速率常数Table 1. Zero-order rate constant of Cr (VI) reduction with different initial concentrationsCr(VI)初始浓度/(mg·L−1) k/(μmol·(L·min)−1) R2 200 78.467 0.981 240 70.156 0.986 300 56.577 0.985 | Show Table DownLoad:
CSV
DownLoad:
CSV
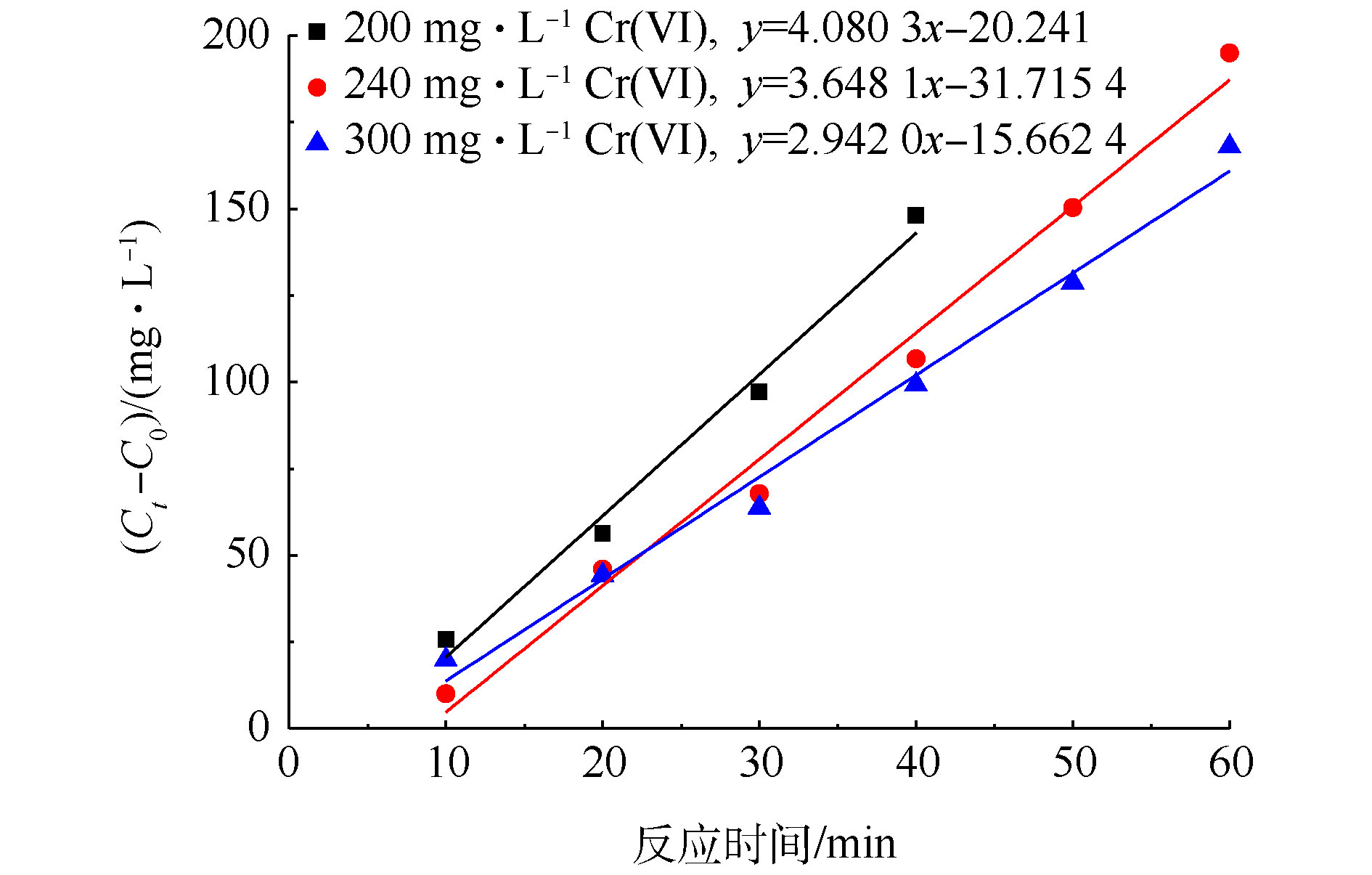 图 9 不同初始浓度下Cr(Ⅵ)还原过程的零级动力学分析Figure 9. Zero-order kinetics of Cr(Ⅵ) reduction with different initial concentrations
图 9 不同初始浓度下Cr(Ⅵ)还原过程的零级动力学分析Figure 9. Zero-order kinetics of Cr(Ⅵ) reduction with different initial concentrations3. 结论
1)采用UV/PS/FA体系,实现了对高浓度Cr(Ⅵ)的还原处理。当Cr(Ⅵ)初始浓度为200 mg·L−1、PS浓度为20 mmol·L−1、FA浓度为40 mmol·L−1时,在反应50 min后Cr(Ⅵ)基本完全被还原去除。对反应过程中的活性自由基采用ESR技术进行表征,证实了
CO⋅−2 的存在。2)UV/PS/FA体系在酸性条件下具有较强的还原能力,随着体系pH的升高还原作用呈现明显下降趋势。随着体系中PS浓度的升高,Cr(Ⅵ)去除率逐渐提高。水溶液中存在的HA、Cl−、
HCO−3 和NO−3 都会在不同程度对Cr(Ⅵ)的还原产生抑制作用。3)对不同初始浓度下Cr(Ⅵ)降解过程进行动力学分析推导,可知其符合零级反应动力学,且初始Cr(Ⅵ)浓度为200、240和300 mg·L−1时对应的反应速率常数分为78.467、70.156和56.577 μmol·(L·min)−1。本研究为Cr(Ⅵ)废水的处理提供了高效的还原方法。
-
图 1 UV、UV/PS、UV/PS/HCOOH还原Cr(Ⅵ)
Figure 1. Reduction of Cr(Ⅵ) by UV, UV/PS and UV/PS/HCOOH
图 6 Cl−、
HCO−3 和NO−3 浓度对Cr(Ⅵ)还原去除效果的影响Figure 6. Effects of Cl−,
HCO−3 andNO−3 on Cr(Ⅵ) removal efficiency
图 9 不同初始浓度下Cr(Ⅵ)还原过程的零级动力学分析
Figure 9. Zero-order kinetics of Cr(Ⅵ) reduction with different initial concentrations
表 1 不同初始浓度下Cr (VI) 的还原反应速率常数
Table 1. Zero-order rate constant of Cr (VI) reduction with different initial concentrations
Cr(VI)初始浓度/(mg·L−1) k/(μmol·(L·min)−1) R2 200 78.467 0.981 240 70.156 0.986 300 56.577 0.985
下载: 导出CSV
-
[1] CHAKRABARTI S, CHAUDHURI B, BHATTACHARJEE S, et al. Photo-reduction of hexavalent chromium in aqueous solution in the presence of zinc oxide as semiconductor catalyst[J]. Chemical Engineering Journal, 2009, 153(1): 86-93. [2] BRAHIMI R, BESSEKHOUAD Y, NASRALLAH N, et al. Visible light CrO2−4 reduction using the new CuAlO2/CdS hetero-system[J]. Journal of Hazardous Materials, 2012, 219-220(6): 19-25.[3] MITRA P, SARKAR D, CHAKRABARI S, et al. Reduction of hexa-valent chromium with zero-valent iron: Batch kinetic studies and rate model[J]. Chemical Engineering Journal, 2011, 171(1): 54-60. doi: 10.1016/j.cej.2011.03.037 [4] WU P X, LI S Z, JU L T, et al. Mechanism of the reduction of hexavalent chromium by organo-montmorillonite supported iron nanoparticles[J]. Journal of Hazardous Materials, 2012, 219-220(6): 283-288. [5] BALAN C, VOLF I, BILBA D. Chromium (VI) removal from aqueous solutions by purolite base anion-exchange resins with gel structure[J]. Chemical Industry and Chemical Engineering Quarterly, 2013, 19(4): 615-628. doi: 10.2298/CICEQ120531095B [6] CHEN S S, LI C W, HSU H D, et al. Concentration and purification of chromate from electroplating wastewater by two-stage electrodialysis processes[J]. Journal of Hazardous Materials, 2009, 161(2): 1075-1080. [7] SU C, LUDWING R D. Treatment of hexavalent chromium in chromite ore processing solid waste using a mixed reductant solution of ferrous sulfate and sodium dithionite[J]. Environmental Science and Technology, 2005, 39(16): 6208-6216. doi: 10.1021/es050185f [8] TACHIKAWA T, TOJO S, FUJITSUKA M, et al. Direct observation of the one-electron reduction of methyl viologen mediated by the CO2 radical anion during TiO2 photocatalytic reactions[J]. Langmuir, 2004, 20(22): 9441-9444. doi: 10.1021/la048100w [9] XU M H, GU X G, LU S G, et al. Degradation of carbon tetrachloride in thermally activated persulfate system in the presence of formic acid[J]. Frontiers of Environmental Science and Engineering, 2016, 286(3): 1-9. [10] JIANG W C, TANG P, LU S G, et al. Enhanced reductive degradation of carbon tetrachloride by carbon dioxide radical anion-based sodium percarbonate/Fe(II)/formic acid system in aqueous solution[J]. Frontiers of Environmental Science and Engineering, 2018, 12(2): 6-15. doi: 10.1007/s11783-017-0987-6 [11] BERKOVIC A M, GONZALEZ M C, RUSSO N, et al. Reduction of mercury(II) by the carbon dioxide radical anion: A theoretical and experimental investigation[J]. Journal of Physical Chemistry A, 2010, 114(49): 12845-12850. doi: 10.1021/jp106035m [12] FAN L, ZHOU A L, ZHONG L R, et al. Photoinduced reduction of high concentration Hg(II) to Hg2Cl2 from acid wastewater with the presence of fulvic acid under anaerobic conditions[J]. Chemosphere, 2018, 198: 13-20. doi: 10.1016/j.chemosphere.2018.01.123 [13] ROSSO J A, BERTOLOTTI S G, BRAUN A M, et al. Reactions of carbon dioxide radical anion with substituted benzenes[J]. Journal of Physical Organic Chemistry, 2010, 14(5): 300-309. [14] REN H J, HOU Z M, HAN X, et al. Highly reductive radical CO⋅−2 deriving from a system withSO⋅−4 and formate anion: Implication for reduction of Cr(VI) from wastewater[J]. Chemical Engineering Journal, 2017, 309: 638-645. doi: 10.1016/j.cej.2016.10.071[15] JIANG W C, TANG P, LU S G, et al. Comparative studies of H2O2/Fe(II)/formic acid, sodium percarbonate/Fe(II)/formic acid and calcium peroxide/Fe(II)/formic acid processes for degradation performance of carbon tetrachloride[J]. Chemical Engineering Journal, 2018, 344: 453-461. doi: 10.1016/j.cej.2018.03.092 [16] GONZALEZ M C, BRAUN A M. VUV photolysis of aqueous solutions of nitrate and nitrite[J]. Research on Chemical Intermediates, 1995, 21(8/9): 837-859. [17] IMOBERDORF G, MOHSENI M. Degradation of natural organic matter in surface water using vacuum-UV irradiation[J]. Journal of Hazardous Materials, 2011, 186(1): 240-246. doi: 10.1016/j.jhazmat.2010.10.118 [18] BUXTON G V, GREENSTOCK C L, HELMAN W P, et al. Critical view of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O−) in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 1988, 17(2): 513-886. doi: 10.1063/1.555805 [19] MOUSSAVI G, HOSSAINI H, JAFARI S J, et al. Comparing the efficacy of UVC, UVC/ZnO and VUV processes for oxidation of organophosphate pesticides in water[J]. Journal of Photochemistry and Photobiology A: Chemistry, 2014, 290(1): 86-93. [20] LIU X W, ZHONG J E, FANG L, et al. Trichloroacetic acid reduction by an advanced reduction process based on carboxyl anion radical[J]. Chemical Engineering Journal, 2016, 303: 56-63. doi: 10.1016/j.cej.2016.05.130 [21] VILLAMENA F A, LOCIGNO E J, ROCKENBAUER A, et al. Theoretical and experimental studies of the spin trapping of inorganic radicals by 5, 5-dimethyl-1-pyrroline N-oxide (DMPO). 2. Carbon dioxide radical anion[J]. Journal of Physical Chemistry A, 2006, 110(49): 13253-13258. doi: 10.1021/jp064892m [22] XIE P C, MA J, LIU W, et al. Impact of UV/persulfate pretreatment on the formation of disinfection byproducts during subsequent chlorination of natural organic matter[J]. Chemical Engineering Journal, 2015, 269: 203-211. doi: 10.1016/j.cej.2015.01.043 [23] DENG J, SHAO Y S, GAO N Y, et al. Degradation of the antiepileptic drug carbamazepine upon different UV-based advanced oxidation processes in water[J]. Chemical Engineering Journal, 2013, 222(2): 150-158. [24] HASEGAWA K, NETA P. Rate constants and mechanisms of reaction of Cl2− radicals[J]. Journal of Physical Chemistry, 1978, 82(8): 854-857. doi: 10.1021/j100497a003 [25] XU M H, GU X G, LU S G, et al. Degradation of carbon tetrachloride in thermally activated persulfate system in the presence of formic acid[J]. Frontiers of Environmental Science and Engineering, 2016, 286(3): 438-446. [26] NETA P, HUIE R E. Rate constants for reactions of nitrogen oxide (NO3) radicals in aqueous solutions[J]. Meat Technology, 1986, 90(19): 4644-4648. [27] EXNER M, HERRMANN H, ZELLNER R. Rate constants for the reactions of the NO3 radical with HCOOH/HCOO– and CH3COOH/CH3COO– in aqueous solution between 278 and 328 K[J]. Journal of Atmospheric Chemistry, 1994, 18(4): 359-378. doi: 10.1007/BF00712451 [28] DRAGANIC Z D, NEGRON-MENDOZA A, SEHESTED K, et al. Radiolysis of aqueous solutions of ammonium bicarbonate over a large dose range[J]. International Journal of Radiation Applications and Instrumentation Part C: Radiation Physics and Chemistry, 1991, 38(3): 317-321. doi: 10.1016/1359-0197(91)90100-G [29] PADMAJA S, NETA P, HUIE R E. Rate constants for some reactions of inorganic radicals with inorganic ions. Temperature and solvent dependence[J]. International Journal of Chemical Kinetics, 2010, 25(6): 445-455. [30] HALL E S, PACKHAM R F. Coagulation of organic color with hydrolyzing coagulants[J]. Journal American Water Works Association, 1965, 57(9): 1149-1166. doi: 10.1002/j.1551-8833.1965.tb01506.x [31] LIU S, LIM M, FABRIS R, et al. Removal of humic acid using TiO2 photocatalytic process: Fractionation and molecular weight characterisation studies[J]. Chemosphere, 2008, 72(2): 263-271. doi: 10.1016/j.chemosphere.2008.01.061 [32] WANG S L, CHEN C C, TZOU Y M, et al. A mechanism study of light-induced Cr(VI) reduction in an acidic solution[J]. Journal of Hazardous Materials, 2009, 164(1): 223-228. doi: 10.1016/j.jhazmat.2008.07.145 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5274
- HTML全文浏览数: 5274
- PDF下载数: 73
- 施引文献: 0
