




-
印制电路板行业在生产印制电路板(PCB)过程中涉及大量有机溶剂、醚类有机物以及表面活性剂等,产生的废液中包含的有机污染物组成复杂、浓度高、毒性大且难以降解[1-2]。目前,企业针对高浓度难降解PCB有机废液的处理方法主要是Fenton氧化法,但该方法存在有机物降解不彻底、Fenton试剂不能循环利用且处理费用高等问题。因此,急需寻找高效、安全且低成本的处理技术来对该类废液进行治理。
高级氧化工艺(AOP)主要包括Fenton/photo-Fenton[3-4]、湿法氧化[5-6]、光催化氧化[7-8]以及臭氧氧化[9-10]等技术。其中,催化臭氧氧化技术克服了臭氧氧化法中存在的不足,能在常温常压下高效降解大部分有机污染物且无二次污染[11-14],从而备受关注。大部分研究[15-17]表明,催化剂的加入会促进活性自由基的产生。其中,羟基自由基( · OH)是催化臭氧氧化过程的主要活性自由基,对有机物的降解起关键性作用。目前,使用较为广泛的是非均相催化剂,主要包括金属和金属氧化物负载、金属氧化物,活性炭以及其他多孔材料等[18-19]。金属氧化物催化剂(如MnO2[20-21]、MgO[22-24]、ZnO[25-26]、TiO2[27-28]、Al2O3[29-30]和CeO2[31]等)、金属氧化物载体的材料(如Al2O3[32]、TiO2[33]等)、多孔材料载体(如石墨烯[34]、碳纳米管[35]等)都已经用于催化臭氧氧化过程,并且已经被证明具有良好的催化活性。然而,很多催化剂在制备及应用时存在一些缺点,如制备工艺复杂、成本较高、重复利用率低等,这些因素限制了催化臭氧氧化技术在实际高浓度难降解工业废液的应用。在催化臭氧化过程中,氧化钙(CaO)很少用于催化臭氧化过程;但初步实验表明,CaO结合臭氧氧化法,在处理实际工业废液中有机污染物时具有很大的优势,并且CaO具有活性高、成本低、毒性低、pH稳定性好和环境友好的特点,因此,将其应用于催化臭氧氧化过程有良好的发展前景[36]。
本研究探讨了PCB废液降解过程中的催化降解机理以及有机物降解途径,考察了催化剂的循环稳定性并分析催化剂失活的可能原因;通过单纯形优化实验考察了CaO在PCB废液的臭氧氧化过程中的催化性能,包括CaO质量、pH、臭氧浓度、降解时间和废液深度对废液中有机物降解率的影响;最后,将CaO催化臭氧过程应用于实际高浓度难降解废水并探讨其应用潜能,为实际工业废水的处理提供参考。
-
氧化钙(CaO)、氢氧化钠(NaOH)、硫酸(H2SO4)、异丙醇(C3H8O)、正己烷(C6H14)、乙二醇单丁醚(C6H14O2)、吐温-80、碘化钾(KI)、硫代硫酸钠(Na2S2O3)、叔丁醇(C4H10O)和水杨酸(C7H6O3)购于中国成都科隆化学试剂厂。二乙二醇单乙醚(C6H14O3),2,3-二羟基苯甲酸(C6H14O4)和2,5-二羟基苯甲酸(C6H14O4)购于梯希爱(上海)化成工业发展有限公司。所有试剂均为分析级,无须进一步处理,所有溶液均是由超纯水净化机(ATSro)获得的去离子水制备。
PCB废液来源于某工厂制造PCB过程,PCB废液为黄色,略带刺激性气味,pH为10.10,COD高达20 246.4 mg·L−1,属于碱性高浓度有机废液,主要成分为异丙醇、正己烷、二乙二醇单乙醚、乙二醇单丁醚和吐温-80等。
-
通过UV光谱(Shimadzu,Japan)检测水杨酸及其与 · OH的反应产物,波长扫描范围为260~400 nm。通过LC-MS(LCMS-8060)对水杨酸羟基化产物进行定量分析。PCB废液中的降解过程中的中间产物通过GC/MS(Agilent 7890A)检测。采用扫描电子显微镜(SEM)检测催化剂使用前后的形貌。通过X射线衍射分析仪(Empryean PANalytical B.V.)检测催化剂使用前后的组成,测定X射线为Cu靶Kα射线(λ=0.154 18 nm,加速电压为40 kV,发射电流为40 mA,扫描角度为10°~85°。用重铬酸钾法测量废液处理前后的COD。通过pH计(SevenEasyS20,Mettler Toledo)测定废液pH。
-
催化臭氧化过程在半连续反应器(内径可调,总高度25 cm)中进行,该反应器盛有250 mL PCB废液和一定量的CaO用作催化剂,在常温常压条件下进行反应。臭氧由臭氧发生器产生,并通过曝气石将O3分散到废液中,并且通过磁力搅拌使废液与O3接触更充分。在实验过程中,气体流速为4 L·min−1,臭氧浓度通过靛蓝法检测,残余臭氧用20%KI溶液吸收。
在催化臭氧化过程中,以异丙醇(IPA)、正己烷(nHA)、二乙二醇单乙醚(DGDE)、乙二醇单丁醚(EB)和吐温-80(Tween-80)为原料,模拟PCB实际废液。将3.0 g CaO加入到含有250 mL模拟废液的反应器中,然后通入一定浓度的臭氧以降解废液中的有机污染物。在此过程中,以一定的时间间隔从反应器中取出10 mL的降解液样品,加入Na2S2O3钠溶液淬灭样品中残余臭氧。将获得的样品通过0.22 μm微孔膜过滤,并将过滤的样品用于GC/MS检测分析。叔丁醇(TBA)用作 · OH淬灭剂,水杨酸(SA)用作 · OH捕获剂,添加到催化臭氧化过程中以研究催化臭氧氧化过程的主要活性自由基。使用过后的催化剂经过过滤、洗涤、50 ℃干燥后,用于催化剂的稳定性实验。
-
臭氧与有机物的反应主要有2种途径,即直接反应和间接反应。直接反应是指O3直接氧化有机物,间接反应主要是通过O3分解产生的活性自由基对有机物进行氧化[37-38]。通过研究 · OH淬灭剂的影响进行对比实验,研究 · OH对有机物的降解作用。TBA是一种常见的 · OH淬灭剂,它与臭氧分子基本不反应,反应速率仅3×10-3 L·(mol·s)−1,而其与 · OH的反应速率高达6×108 L·(mol·s)−1。因此,可以通过加入TBA到臭氧或催化臭氧过程,间接检测体系中是否有 · OH的产生[39]。图1为单独臭氧氧化过程以及催化臭氧氧化过程加入TBA前后废液的COD去除率对比图。由此可知,CaO催化臭氧氧化过程和单独臭氧氧化过程加入TBA后,处理180 min后,COD去除率分别降低13.04%和5.71%,表明TBA的加入对2个过程降解率均造成负面影响,从而间接证明单独臭氧氧化过程与催化臭氧氧化过程都有 · OH产生。此外,从TBA对2个过程的影响程度上可以看出,CaO可以促进O3产生更多的 · OH,表明CaO催化臭氧氧化过程遵循羟基自由基机理。
-
水杨酸(SA)羟基化实验是另外一种间接检测羟基自由基的方法[40]。羟基自由基具有存在时间短、不稳定的特点,但SA可以作为 · OH的捕捉剂,SA与 · OH反应后会生成较为稳定的2,3-二羟基苯甲酸(2,3-DHBA)和2,3-二羟基苯甲酸(2,5-DHBA)。本研究结合紫外-可见分光光度计跟踪SA与 · OH反应后产物,再结合液相色谱-质谱联用仪对2,3-DHBA和2,5-DHBA进行定量分析。
图2为SA、2,3-DHBA和2,5-BHBA的紫外-可见吸收光谱图以及CaO催化臭氧氧化处理后的紫外-可见吸收光谱。可以看出,SA在302 nm处有最大吸收峰,而2,3-DHBA和2,5-DHBA分别在315 nm和330 nm处出现最大吸收峰,SA经过催化臭氧氧化处理后最大吸收峰波长向右移动,在2,3-DHBA和2,5-DHBA的大吸收峰处有一定的吸收,证明在该过程中有 · OH产生。
从图2中可以观察到2,3-DHBA和2,5-DHBA的存在,接下来使用LC-MS联用仪对2,3-DHBA和2,5-DHBA的含量进行定量分析。图3(a)和图3(b)分别表示2,3-DHBA和2,5-DHBA的液相色谱标准曲线,根据该标准曲线求得CaO催化臭氧处理不同时间溶液中2,3-DHBA和2,5-DHBA的含量,结果如表1所示。从表1中可以看出,CaO催化臭氧处理4、8和12 min后,溶液中2,3-DHBA的含量分别为0.037 3、0.022 1和0.020 mg·L−1,2,5-DHBA的含量分别为0.015 5、0.014 4和0.013 7 mg·L−1。可以看出,随着时间的增加,2,3-DHBA和2,5-DHBA的含量都不断减少,表明催化臭氧过程中SA与羟基自由基结合的同时,羟基化产物2,3-DHBA和2,5-DHBA也被氧化降解。
-
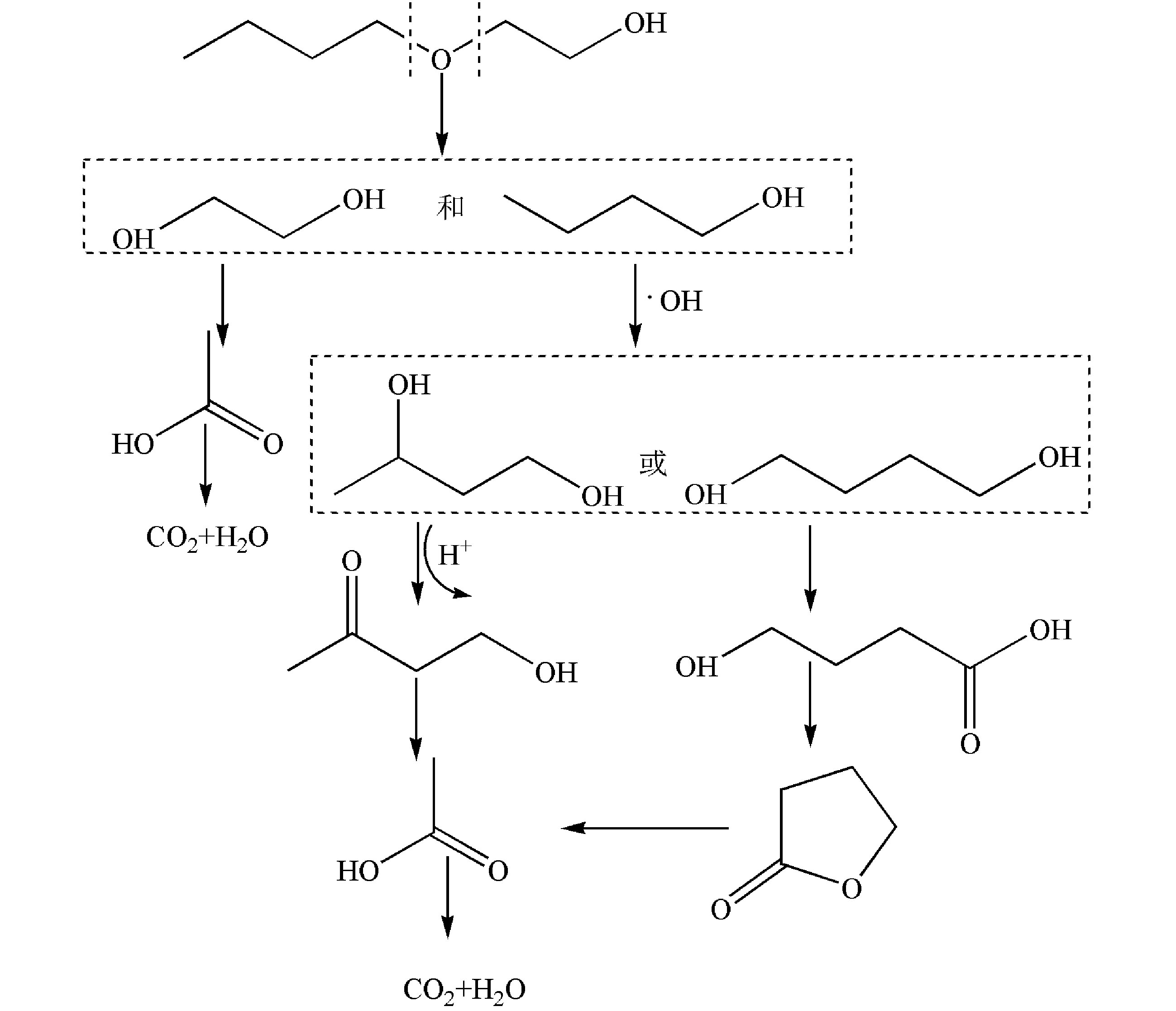
为了研究废液的降解路径,以废液主要成分异丙醇和乙二醇单丁醚为例进行探讨,对2种物质进行降解并对降解产物用GC/MS进行检测。
异丙醇降解30 min和60 min的GC/MS结果如图4所示,对应的中间产物信息如表2所示。异丙醇溶液经催化臭氧氧化降解30~60 min后,检测到的中间产物有羟基丙酮、1,2-羟基丙二醇以及乙酸,由此推测出异丙醇的可能降解路径为:异丙醇与 · OH结合形成了1,2-羟基丙二醇,1,2-羟基丙二醇再被氧化为羟基丙酮,然后再进一步被氧化为小分子酸乙酸,结果如图5所示。
乙二醇单丁醚降解30 min和60 min后的总离子流色谱图如图6所示,对应的中间产物信息如表3所示。乙二醇单丁醚的降解中间产物主要有1-丁醇、乙二醇、4-羟基-2-丁酮、丁内酯和乙酸。由此可以推测乙二醇单丁醚的可能降解路径如图7所示,乙二醇单丁醚的降解一部分是被氧化断链形成乙二醇,然后再接着被氧化为乙酸。另外有一部分乙二醇单丁醚被氧化断链形成1-丁醇,1-丁醇与 · OH结合,结合产物再被氧化形成酮或酸,最后再被氧化形成小分子酸乙酸,最后形成CO2和H2O。
-
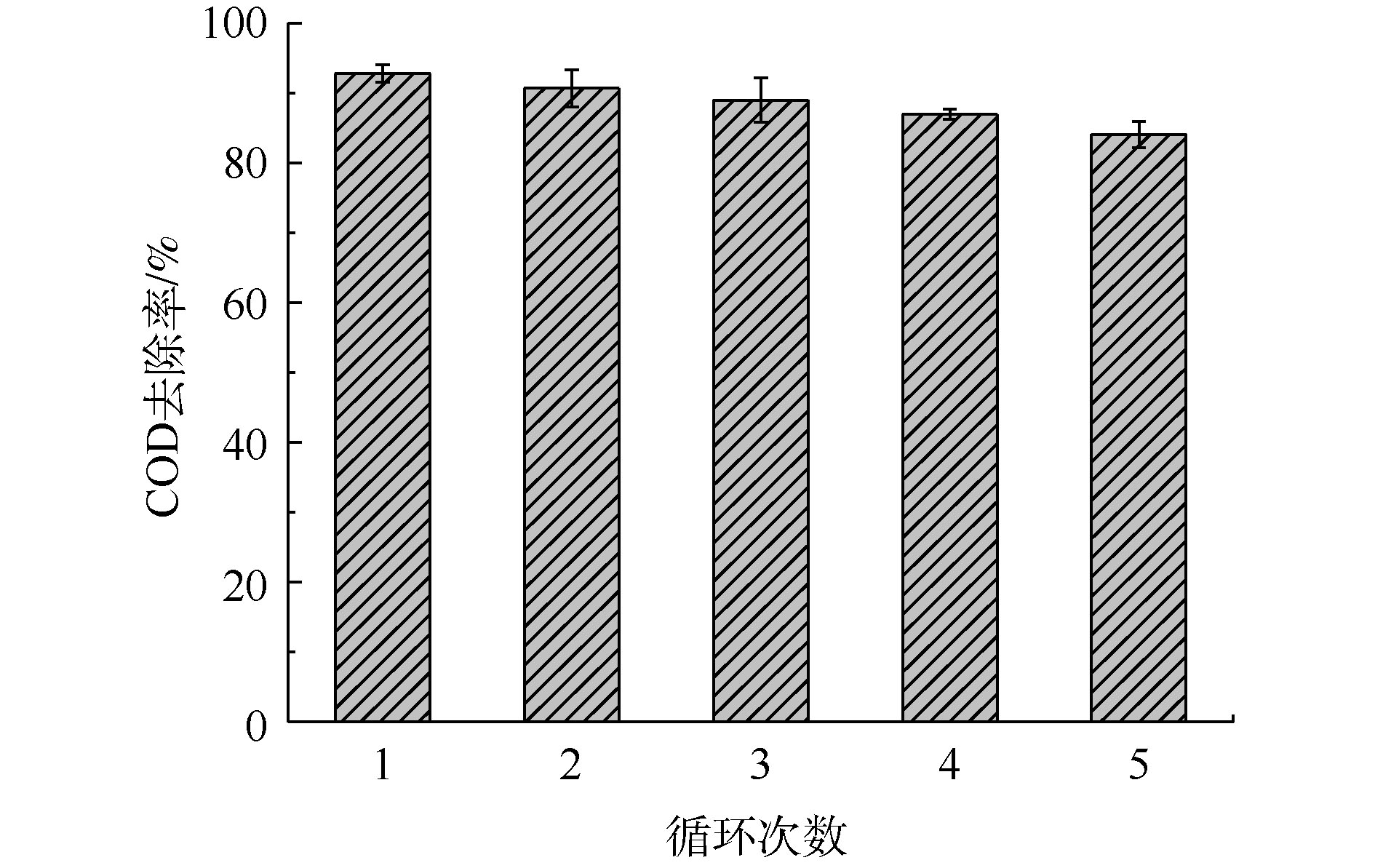
在实际应用中,催化剂的稳定性十分重要。催化剂的多次循环使用可以节约催化剂成本,并且减少固废。本实验对催化剂进行了5次循环,每次使用后对催化剂进行过滤、洗涤、干燥后进行循环使用。如图8所示,催化剂在每一次使用时的催化效率分别是92.78%、90.67%、88.98%、86.94%和84.04%。经过多次循环,催化剂活性下降,但每次循环均没有显著下降,表明该催化剂具有良好的循环性能。
为了探讨催化剂失活原因以及催化剂活化的方式,对使用前后的催化剂进行了表征,研究催化剂使用前后的形貌、组成以及比表面积变化。图9(a)显示的是催化剂使用前的形貌,图9(b)和图9(c)表示催化剂使用1次和3次后的形貌。可以看出,使用后的催化剂较使用前的催化剂不易分散,产生的团聚现象更为严重,从而导致催化剂的性能降低。
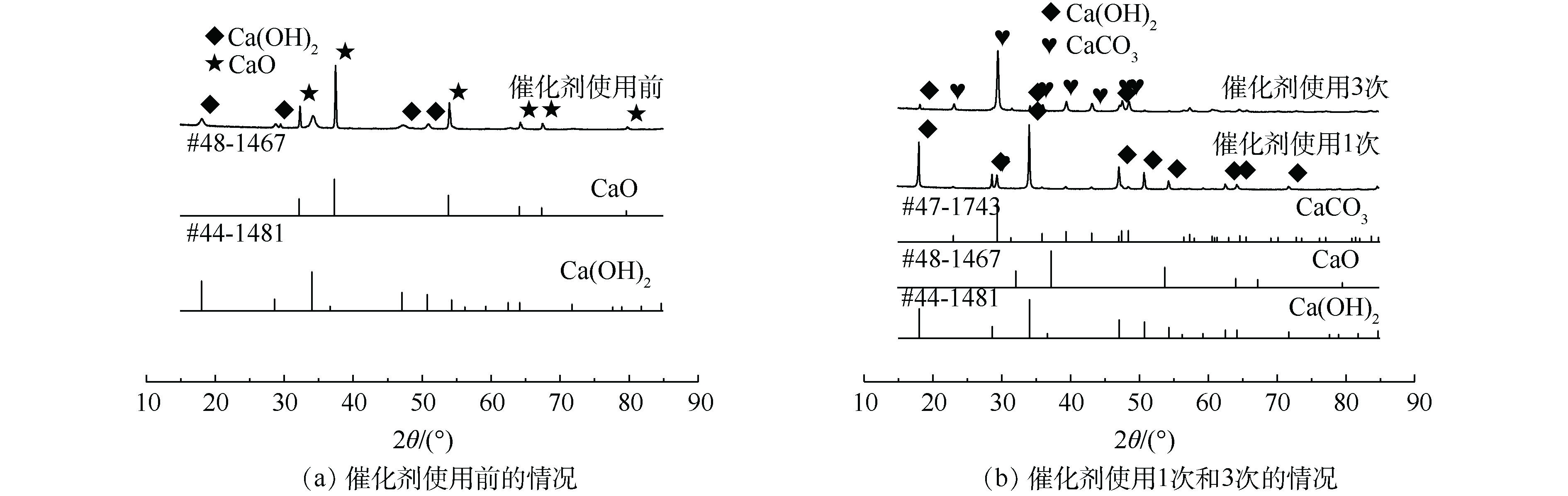
图10显示的是催化剂使用前后的XRD谱图。图10(a)得到的XRD谱图与标准JCPDS对照可知,2θ为32.3°、37.4°、53.9°对应的是CaO的特征吸收峰,2θ为18°、28.7°、34.1°、47.1°和50.9°对应的是Ca(OH)2的特征峰,表明在使用前的催化剂中Ca(OH)2的特征峰也有一定的吸收强度。使用前的催化剂中CaO的特征峰十分清晰且强度较大,表明其主要成分是CaO,但也含有少量的Ca(OH)2,说明催化剂在存放过程中或者样品测试时受到空气中水分的影响。
图10(b)为催化剂使用1次和使用3次后的XRD图。催化剂使用1次后的XRD谱图在2θ为18°、28.7°、34.1°、47.1°和50.9°处存在较强的峰,这些峰与Ca(OH)2的特征峰一致,表明其主要成分是Ca(OH)2。而使用3次的催化剂测出的2θ为23°、29.4°、39.4°、43.2°和47.1°对应的峰是CaCO3的特征峰,说明催化剂的失活过程主要是:经过多次循环后的催化剂与水结合变成了Ca(OH)2,形成的Ca(OH)2与矿化产物CO2结合从而变为CaCO3,最终由于多次反应后CaCO3含量不断增加从而导致催化剂催化效率逐渐降低。
-
有研究[41-42]表明,pH变化对催化效率有很大影响,这可能会影响催化剂的表面性质和活性自由基的产生。废液深度的变化会影响O3分子与废液的接触时间,从而影响废液的降解率。O3在催化臭氧氧化过程起氧化作用。臭氧用量的增加,可以促进活性自由基的产生并且可以增加臭氧与废液的接触面积,从而促进废液中有机物的降解[43]。因此,废液pH、CaO质量(m)、废液深度(h)、降解时间(t)、臭氧用量等工艺条件的优化就显得尤为重要。考虑到CaO会造成固废以及时间成本,因此,综合考虑了CaO质量、降解时间和COD去除率(η)三者的关系,以0.7η+0.1/t+0.2/m为考察指标进行单纯形优化实验。表4显示各因素的初点和步长,即各个因素的初始值以及变化值。表5为根据均匀设计表U6(65)得到的初始实验条件,经过优化之后得到的优化结果如表6所示。可以看出,pH为12.6~13.2,降解时间为150~180 min以及臭氧量为120 ~200 mg·min−1时会取得较好的催化降解率。综合考虑固废以及时间成本,pH为12.97、CaO质量为1.0 g、废液深度为11 cm、降解时间为150 min、臭氧用量为120 mg·min−1时,COD去除率可达到90.045%,并且0.7η+0.1/t+0.2/m综合效率为0.870 3,能够满足在较短时间、较少催化剂用量下取得较高的降解率,可以应用于高浓度难降解有机废水的处理。
-
图11显示了CaO催化臭氧氧化处理工厂的实际PCB清槽剂废液的处理效果,其工艺条件如下:pH为13.0、CaO质量为2.0 g、废液深度为11 cm、降解时间为180 min、臭氧用量为180 mg·min−1。可以看出,处理180 min后,废液COD去除率达到了94.67%,比单独臭氧氧化过程COD去除率高26.92%,CaO作为催化剂加入到臭氧氧化过程大幅度提高了有机物的降解率。上述结果表明CaO催化臭氧氧化处理实际高浓度难降解废液具有可行性,并且对有机物的降解效果显著,具有广阔的应用前景。
-
1)自由基淬灭实验和水杨酸羟基化实验结果表明,CaO催化臭氧氧化体系中存在 · OH,主要是遵循羟基自由基机理。
2)通过GCMS检测,废液降解后检测出了中间体,如羟基丙酮、乙二醇、正丁醇、乙酸等。因此,有机物可能降解途径是:有机物主要是先与 · OH结合,再进一步被氧化形成酮,然后被氧化为乙酸,最后形成CO2和H2O。
3)催化剂稳定性测试表明,CaO具有优良的循环稳定性,经过5次循环后,催化剂的催化效率可以达到84.04%。SEM、XRD测试结果表明,使用后的催化剂团聚现象明显增大,其主要成分由CaO变为了Ca(OH)2和CaCO3,从而导致催化效率降低。
4)单纯形优化实验表明,在优化条件下可以满足在较短时间,使用较少催化剂情况下,催化效率达到90.04%。最后,将CaO催化臭氧氧化技术应用到实际PCB废液中,废液COD去除率可以达到94.67%,表明CaO催化臭氧氧化技术可应用于实际高浓度难降解废液。
催化臭氧氧化降解PCB有机废液及其机理
Performance and mechanism of PCB organic effluents degradation by catalytic ozonation
-
摘要: 为了高效、快速治理高浓度难降解PCB(printed circuit board)有机废液,研究了氧化钙非均相催化臭氧氧化降解PCB废液的催化机理和催化性能。采用叔丁醇淬灭自由基实验和水杨酸羟基化实验探究催化机理;通过GC/MS研究了PCB废液中有机物可能降解途径;通过单纯形优化法对实验因素进行优化,并通过XRD和BET探究催化剂的循环稳定性。结果表明:氧化钙非均相催化臭氧氧化过程遵循羟基自由基机理;在pH为12.97、CaO质量为1.0 g、废液深度为11 cm、降解时间为150 min、臭氧用量为120 mg·min−1时,COD去除率可达到90.045%;氧化钙经过5次循环后,废液的COD去除率没有显著降低,从92.78%降低至84.04%。CaO应用于催化臭氧氧化过程处理高浓度且难降解的PCB废液,能维持良好的催化性能和循环稳定性,具有良好的应用前景。Abstract: In order to high efficiently and rapidly treat refractory organic effluent generated during manufacturing printed circuit boards (PCB) with high concentration, the performance and mechanism of PCB organic effluents degradation by heterogeneously catalytic ozonation with CaO were studied. The catalytic mechanism was investigated by radical quenching test with tert-butanol and hydroxylation test of salicylic acid. The possible degradation pathway of organic matter was studied by GC/MS. The experimental factors were optimized by simplex optimization method, and the cycle stability of the catalyst was explored by XRD and BET. The results showed that the catalytic ozonation process in the presence of calcium oxide followed hydroxyl radicals mechanism. The COD removal efficiency could reach 90.045% at the pH of 12.97, CaO mass of 1.0 g, the solution height of 11 cm, the degradation time of 150 min, and the ozone dosage of 120 mg·min−1. After five cycles, the COD removal ratio of organic matter decreases insignificantly, which changes from 92.78% to 84.04%. CaO can maintain a good catalytic performance and recycle stability during treating refractory organic industrial wastewater with high concentration, which confirms its good prospect in this field.
-
Key words:
- catalytic ozonation /
- calcium oxide /
- PCB effluents /
- hydroxyl radicals
-
生物质在部分或完全缺氧的条件下通过高温热解产生的富碳、具有高度芳香环分子结构和多孔性副产物称之为生物炭[1]。生物炭可以通过微生物作用、氧化还原、表面沉淀、离子交换以及表面络合作用等一系列物理化学和生物作用影响土壤理化特性及重金属的形态、生物毒性、迁移和转化过程,以期实现土壤修复的目标[2-4]。采用生物炭来实施的重金属污染场地修复技术具有经济性、稳定性、环境友好等优势[5],因此,可以将生物炭用作为重要的土壤重金属修复改良剂。
水可提取的有机质(water extracted organic matter,WEOM)作为生物炭中最具活性的组分,对土壤中重金属的环境地球化学行为的影响同样不容忽视。WEOM中含有众多的荧光的有机质,诸如类蛋白物质、类富里酸和类腐殖酸等[6-10],这些荧光物质能够与土壤中的重金属发生络合作用,从而影响土壤重金属的环境化学行为。而荧光光谱方法是研究荧光有机质组分与相对含量的重要手段。荧光光谱结合荧光猝灭滴定已经广泛的用于研究这些水溶性的荧光有机物与重金属的络合作用[11-12],结合荧光猝灭络合模型能够计算出荧光组分与重金属的络合常数,以及参与络合过程中荧光物质的比例,如修正的Stern-Volmer模型[13]、非线性Ryan-Weber模型[14]。一些有机质的荧光信息经常被强的荧光基团掩盖而无法识别,而导数荧光也能够很好地用于识别有机质光谱中被掩盖的荧光组分,从而获得更多的光谱信息[7, 15]。同时,二维相关光谱方法也已经广泛用于研究WEOM与重金属络合的异质性[16-17]。
然而,不同的热解温度影响了生物炭中WEOM的含量、组分及结构特性,这些荧光组分对土壤重金属的环境化学行为起着非常重要的作用,尤其是低温热解时会产生的生物炭具有较高的WEOM含量,这些WEOM中的有机组分能够与重金属发生络合作用[18]。因此,为了进一步研究不同热解温度下生物炭中的WEOM对重金属的影响,本研究针对300℃和600℃热解下生成的生物炭提取的WEOM与重金属Cu(Ⅱ)的络合特性进行研究,采用导数荧光和二维相关光谱分析结合荧光猝灭滴定方法研究高温和低温热解条件下,生物炭WEOM中荧光组分变化及其与Cu(Ⅱ)的络合参数,解释两种温度下生物炭WEOM对Cu(Ⅱ)环境化学行为的影响,为土壤改良与重金属修复提供理论指导。
1. 材料和方法(Materials and method)
1.1 棉杆生物炭制备
将某农场收集的棉杆在105°C的温度下进行烘干后,采用粉碎机进行破碎后过100目筛。过筛后的棉杆置于管式炉中进行热解,将50 g棉秆置于管式炉中,在N2气氛中由室温分别加热至300℃和600℃温度下热解30 min。
1.2 WEOM的提取
将上述两种热解生物炭以水炭比(V/M)为10∶1进行混合,置于水平振荡仪连续振荡24 h后,采用7000 r∙min-1转速进行离心过滤,取上清液过0.45 μm有机滤膜,滤液即为WEOM。以溶解性有机碳(DOC)作为WEOM的浓度,采用总有机碳分析仪(Multi N/C 2100,德国耶拿)测定DOC的浓度。
1.3 荧光猝灭滴定实验
荧光猝灭滴定实验在50 mL离心管中进行。分别在一系列装有10 mL WEOM溶液的离心管中,添加Cu(Ⅱ)离子,使得WEOM溶液中Cu(Ⅱ)离子的浓度为0、20、40、60、80、120、150、200 μmol∙L−1,添加Cu(Ⅱ)离子溶液的体积未超过WEOM体积的2%。荧光猝灭滴定前将WEOM溶液pH值调至7.0±0.2,WEOM的浓度调整为10 mg∙L−1。将上述溶液置于水平振荡仪,在避光的条件下振荡24h以确保络合达到平衡,反应完成后立即进行同步荧光光谱测定(SFS)。
1.4 光谱分析
采用F-7000分子荧光光度计(日本,日立)测定不同Cu(Ⅱ)浓度下WEOM的同步荧光光谱,波长扫描范围为250—600 nm,扫描波长差为30 nm,扫描速度设置为Fast,超纯水作为空白背景被扣除。采用origin软件对同步荧光光谱数据进行二阶求导,获得WEOM同步荧光光谱的二阶导数荧光,用于识别被掩盖的荧光峰。
二维相关光谱分析能够解释因外界环境扰动而引起的光谱特征细微变化,从而提高对WEOM-Cu(Ⅱ)络合过程的解释能力。本研究将不同Cu(Ⅱ)浓度下的WEOM同步荧光光谱用于分析WEOM-Cu(Ⅱ)络合的二维相关光谱特性。采用“2D Shige”软件绘制WEOM-Cu(Ⅱ)络合过程的二维同步和异步相关光谱图。
1.5 络合模型
WEOM-Cu(Ⅱ)的络合参数采用线性修正Stern-Volmer方程进行计算[13],该模型假定Cu(Ⅱ)离子与配位基按照1∶1进行配位作用,方程如下:
F0/(F0−F)=1/(f×K×CM)+1/f (1) 式中,F0和F分别为未添加和添加了金属Cu(Ⅱ)离子时的荧光强度;K为条件稳定常数,通常采用对数条件稳定常数lgK进行分析;CM为WEOM中金属Cu(Ⅱ)离子的浓度;f为参与配位的荧光基团的比例.
2. 结果与讨论(Results and discussion)
2.1 不同温度热解生物炭WEOM的荧光组分变化
将同步荧光光谱分为类蛋白(250—310 nm)、类富里酸(310—380 nm)和类腐殖酸(380—600 nm)的3个区域[19],图1为3个区域积分面积。结果表明,低温热解生物炭中WEOM的荧光物质以类腐殖酸为主,占总荧光物质的51.05%,其次是类富里酸组分,占36.86%,而类蛋白荧光物质最低,仅占12.09%。高温热解生物炭中WEOM荧光物质明显要低于低温热解生物炭,表明在高温条件下大部分的荧光有机质能够被分解。高温热解生物炭WEOM中荧光组分以类富里酸为主,占总荧光物质的42.11%,其次为类腐殖酸组分,占33.93%,类蛋白荧光组分为23.96%。相比于低温热解生物炭,高温热解生物炭中的类蛋白、类富里酸和类腐殖酸分别减少了24.76%、56.62%和74.73%,表明长波方向的类腐殖酸和类富里酸荧光物质更容易被高温热解。两种生物炭中WEOM的含量分别为20172.3 mg·kg−1和5.9 mg·kg−1,高温热解后WEOM的含量极低。因此,低温热解能够增加生物炭中WEOM含量,但相对于吸附重金属,高温热解生物炭具有更高的比表面积和孔隙结构,表现出较好的吸附能力[19]。而低温生物炭WEOM中较高的类富里酸和类腐殖酸有利于改良土壤结构,提高土壤的腐殖化程度。通常用类腐殖酸与类富里酸的比值(HLR/FLR)来表征WEOM的腐殖化程度,结果表明低温热解生物炭WEOM的HLR/FLR比值为1.38,而高温热解生物炭WEOM的HLR/FLR比值仅为0.81,降低了41.74%,可知低温热解生物炭WEOM具有较高的腐殖化程度。
 图 1 不同温度热解生物炭WEOM荧光区域积分Figure 1. Fluorescence regional integration of biochar-derived WEOM at different pyrolysis temperatures
图 1 不同温度热解生物炭WEOM荧光区域积分Figure 1. Fluorescence regional integration of biochar-derived WEOM at different pyrolysis temperatures2.2 不同温度热解生物炭WEOM的导数荧光
图2为两种温度下热解生物炭WEOM同步光谱经过二阶导数后的光谱。图2a显示,低温热解WEOM的同步光谱中4个荧光峰能够被识别,分别在309 nm(类蛋白荧光)、367 nm(类富里酸荧光)、409 nm(类腐殖酸荧光)和500 nm(类腐殖酸荧光)处,其中500 nm处的类腐殖酸荧光峰以肩峰的形式出现。低温热解WEOM二阶导数荧光仅能识别一个掩盖荧光峰(图2c),为270 nm处的类蛋白荧光峰,表明低温热解生物炭具有较少的荧光基团,但其荧光基团的强度较高。总类腐殖酸强度与总类富里酸强度比值(IHLR/FLR)结果表明,低温热解生物炭WEOM的IHLR/FLR值为1.32,而高温热解生物炭WEOM的IHLR/FLR值为0.94,也预示高温热解降低了生物炭WEOM的腐殖化程度。
图3b显示4个明显的荧光峰,分别在303 nm、338 nm、372 nm和500 nm处,其中303 nm处的荧光峰为类蛋白荧光峰,338 nm和372 nm处为类富里酸荧光物质所产生的荧光峰,500 nm处的荧光峰为类腐殖酸荧光物质引起的[20]。类蛋白和类富里酸荧光物质具有较高的荧光强度,而500 nm处的类腐殖酸荧光峰强度较低。图2d显示,二阶导数荧光能够显示8个明显的荧光峰,其中4个荧光峰在同步荧光光谱中未曾发现,分别在272 nm(类蛋白荧光)、290 nm(类蛋白荧光)、 418 nm(类腐殖酸荧光)和460 nm(类腐殖酸荧光),表明二阶导数荧光能够识别处更多的被掩盖的荧光峰[15]。图1也表明,高温热解导致类富里酸和类腐殖酸区域的荧光物质分解,从而产生更多的荧光组分。
2.3 Cu(Ⅱ)与生物炭WEOM络合2D-SFS-COS分析
图3为生物炭WEOM-Cu(Ⅱ)络合的二维同步荧光相关光谱分析,同步光谱图关于主对角线对称,异步光谱图关于主对角线反对称[21]。结果表明,300℃热解生物炭WEOM的二维同步光谱图显示3个自动峰和3个交叉峰(图3a)。3个自动峰峰位分别在309 nm(类蛋白峰),367 nm(类富里酸)和409 nm(类腐殖酸),3个交叉峰位分别在367/309、409/309和409/367处。根据Noda规则[21],高的相关系数表明该位置的荧光峰对外界环境干扰具有较高的敏感性。因此,WEOM中荧光物质对因Cu(Ⅱ)离子的添加而导致环境扰动的敏感程度次序为:367 → 409/367 →367/309 →409 →409/309 →309,表明类富里酸荧光物质对Cu(Ⅱ)离子添加引起的环境变化具有较高的敏感性,其次是类腐殖酸荧光物质。而600℃热解生物炭WEOM二维同步光谱图显示1个明显自动峰和1个交叉峰(图3c),峰位分别在303 nm和327 nm处。根据Noda规则,303 nm处的类蛋白荧光比327 nm处的类富里酸荧光对Cu(Ⅱ)的添加引起的环境变化具有更高的敏感性。
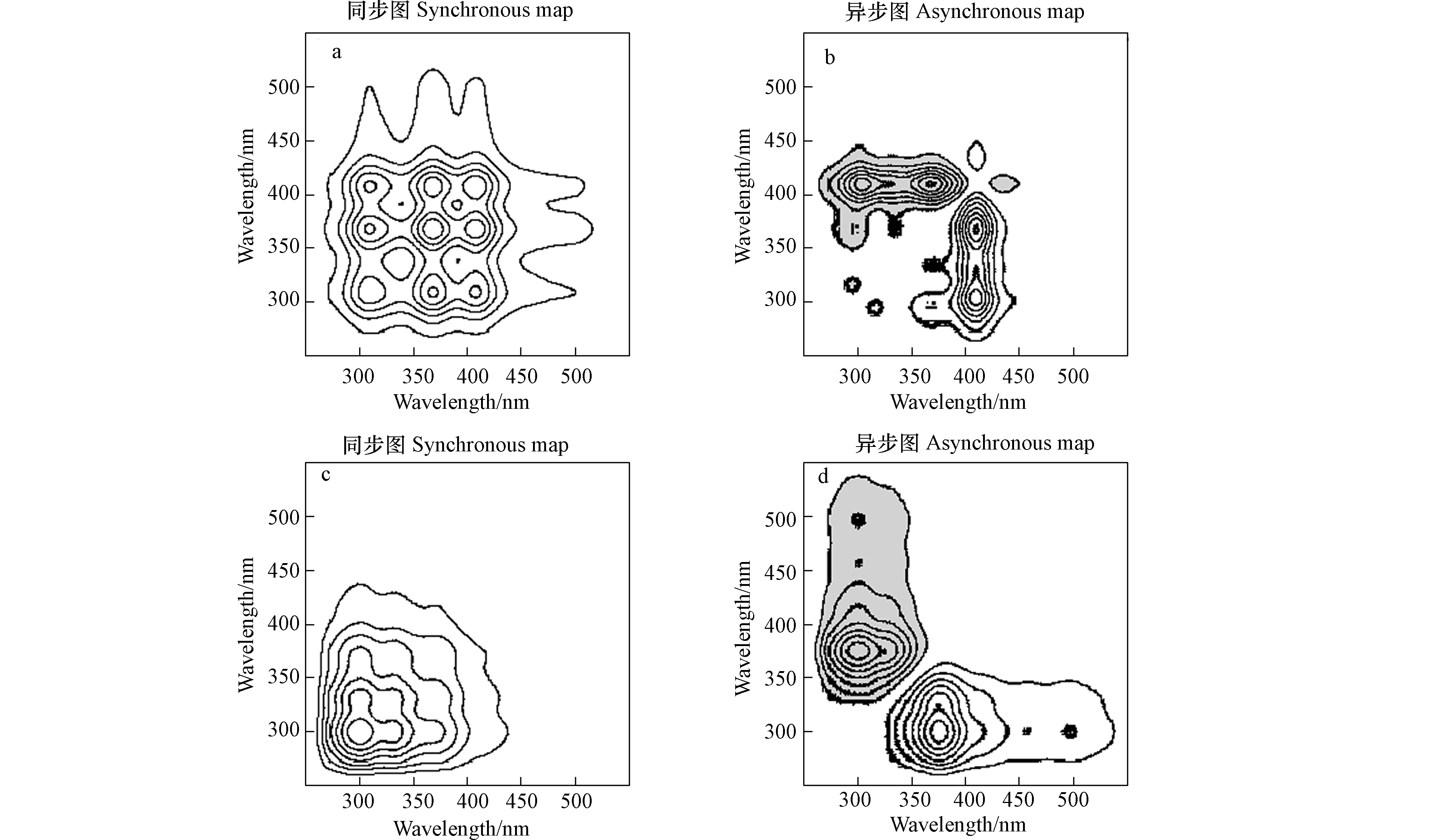 图 3 不同温度热解生物炭WEOM-Cu(Ⅱ)的2D-SFS-COS分析(a, b) 300℃; (c,d) 700℃Figure 3. 2D-SFS-COS analysis of biochar-derived WEOM-Cu(Ⅱ) at different pyrolysis temperatures
图 3 不同温度热解生物炭WEOM-Cu(Ⅱ)的2D-SFS-COS分析(a, b) 300℃; (c,d) 700℃Figure 3. 2D-SFS-COS analysis of biochar-derived WEOM-Cu(Ⅱ) at different pyrolysis temperatures图3b为低温热解生物炭WEOM-Cu(Ⅱ)的异步光谱图,4个正相关峰(白色)和1个负相关峰(灰色)能够被识别,分别在309 nm(类蛋白峰)、367 nm(类富里酸)、409 nm(409/290和409/367,类腐殖酸)和431 nm(类腐殖酸)。根据Noda规则,异步光谱中正的相关峰优先与Cu(Ⅱ)发生络合作用,而负的相关络合作用滞后,且高的相关系数优先发生络合作用。因此,低温热解WEOM中荧光物质与Cu(Ⅱ)络合的次序为:409→309→367→431,表明409 nm处的类腐殖酸荧光物质能够优先与Cu(Ⅱ)络合,其次是类蛋白荧光物质和类富里酸荧光物质。高温热解生物炭WEOM-Cu(Ⅱ)的异步光谱图识别出3个明显的荧光峰(图3d),分别在372 nm、460 nm和500 nm处。372 nm处的类富里酸荧光物质具有极强的相关性,表明类富里酸荧光物质优先与Cu(Ⅱ)离子发生络合作用,其次为500 nm处的类腐殖酸荧光物质。根据Noda规则,高温热解WEOM中荧光物质与Cu(Ⅱ)络合的次序为:372→500→460。
2.4 络合常数
图4为不同温度热解生物炭WEOM中各荧光组分与Cu(Ⅱ)离子络合的荧光猝灭曲线。
 图 4 WEOM组分与Cu(Ⅱ)络合的猝灭曲线Figure 4. Quenching curves of biochar-derived WEOM components with Cu(Ⅱ)
图 4 WEOM组分与Cu(Ⅱ)络合的猝灭曲线Figure 4. Quenching curves of biochar-derived WEOM components with Cu(Ⅱ)由图4可知,类蛋白荧光组分并未呈现明显的猝灭趋势,随着Cu(Ⅱ)离子浓度的增加,组分的荧光强度呈现降低或增加的情况。对于这种现象先前的研究也已经给出相应的解释,如由于Cu(Ⅱ)离子浓度的增加,类蛋白分子结构三维结构变化导致蛋白荧光的量子产量变化[22]。另外,类蛋白荧光可能与无机或其他有机组分相互作用发生猝灭作用,Cu(Ⅱ)离子的加入取代了这些猝灭剂而导致荧光强度的增加[22],进而形成了更稳定的WEOM-Cu(Ⅱ)络合物。类富里酸和类腐殖酸荧光强度随着Cu(Ⅱ)离子浓度的增加呈现出逐级降低的趋势,随着Cu(Ⅱ)离子浓度的继续增加,荧光强度逐级趋于稳定。因此,本研究采用修正的Stern–Volmer方程来计算WEOM中类富里酸和类腐殖酸荧光物质与Cu(Ⅱ)的络合常数和荧光配位比。
由表1可知,类蛋白物质与Cu(Ⅱ)离子络合的Stern–Volmer方程拟合失败。而类富里酸和类腐殖酸组分能够与Cu(Ⅱ)离子的络合具有较好的拟合效果。而327 nm处的类富里酸峰因为荧光猝灭效果不明显,也不能被拟合。表1结果表明,300℃热解生物炭WEOM-Cu(Ⅱ)的lgK值在4.85—5.30之间,类富里酸物质表现出较高的lgK值,其次是500 nm处的类腐殖酸物质。WEOM中荧光物质与Cu(Ⅱ)络合的荧光配位比在26.15%—57.81%之间。尽管367 nm处的类富里酸能够与Cu(Ⅱ)形成较高化学稳定性的络合,但其参与络合的荧光物质比例较低,仅有26.15%。而生成化学稳定性较低的类腐殖酸(409 nm)表现出了较高的荧光配位比(57.81%)。图2a显示,367nm处的类富里酸和409 nm处的类腐殖酸具有较高的荧光强度,表明300℃低温热解棉杆生物炭WEOM与Cu(Ⅱ)络合过程中,367nm处的类富里酸和409 nm处的类腐殖酸扮演更为重要的作用。尽管431nm和500nm处的类腐殖酸也表现出较高的lgK值和荧光配位比,但其荧光物质的含量相对较少。然而,但低温热解生物炭用于土壤修复与改良时,随着这些荧光物质的不断积累,也能够在土壤重金属的迁移和转化过程中起到极为重要的作用。
表 1 不同温度热解WEOM中各组分与Cu(Ⅱ)离子的络合参数Table 1. Binding parameters of biochar-derived WEOM components with Cu(Ⅱ)生物炭Biochar 荧光峰Peak 对数稳定常数lgK 荧光配位比f/% 决定系数R2 300℃ 270c FM FM FM 309abc FM FM FM 367ab 5.30 26.15 0.80* 409abc 4.85 57.81 0.93* 431b 5.08 43.41 0.88* 500ac 5.25 44.97 0.91* 600℃ 272c FM FM FM 290c FM FM FM 303abc FM FM FM 327b FM FM FM 338ac 4.23 38.18 0.98* 372abc 4.82 71.00 0.95* 418c 4.94 74.56 0.94* 460bc 5.02 79.13 0.94* 500abc 5.19 77.76 0.91* a, b, c分别为同步荧光光谱、二维相关光谱和二阶导数光谱识别的荧光峰; * p= 0.01 level (2-tailed). FM, not modeled. | Show Table DownLoad:
CSV
DownLoad:
CSV
600℃热解棉杆生物炭WEOM与Cu(Ⅱ)的lgK值在4.23—5.19之间,随着波长的增加,lgK值呈现逐级增大的趋势。长波处的类腐殖酸物质与Cu(Ⅱ)能够形成较高化学稳定性的络合物。600℃热解棉杆生物炭WEOM中372 nm左右的类富里酸与Cu(Ⅱ)的络合常数明显要低于300℃时367 nm类富里酸与Cu(Ⅱ)的络合常数,而两种温度下类腐殖酸组分与Cu(Ⅱ)的络合常数值变化不大。此外,高温热解WEOM-Cu(Ⅱ)的荧光配位比在38.18%—79.13%之间,明显要高于300℃时的荧光配位比,说明高温热解导致参与Cu(Ⅱ)络合的荧光物质比例增加。然而图1显示,高温热解生物炭WEOM中荧光物质的含量相对较低,同时生物炭WEOM的含量也非常低,因此认为WEOM与金属的络合作用对高温热解生物炭用于土壤重金属修复过程的影响相对较小,而吸附可能成为影响重金属化学行为的主要因素。
3. 结论(Conclusion)
不同温度热解影响生物炭WEOM中荧光物质的组分及含量,生物炭热解实验结果表明,与高温生物炭相比,低温生物炭具有较高的WEOM含量,而高温热解生物炭经热解后的WEOM存在较多荧光组分。导数荧光能够更好的识别处被强的荧光基团掩盖的荧光组分,光谱分析结合荧光猝灭及二维相关分析能够很好的解释生物炭WEOM与重金属的络合特性及络合物的化学稳定性。结果表明,低温热解生物炭WEOM中367 nm处的类富里酸组分与Cu(Ⅱ)形成较高化学稳定性的络合物,而高温热解生物炭WEOM中荧光物质与Cu(Ⅱ)形成络合物的化学稳定性随着波长的增加而增加。300℃热解生物炭WEOM中,与Cu(Ⅱ)形成低化学稳定性的类腐殖酸组分(409 nm)能够优先于其他组分与Cu(Ⅱ)发生络合反应;而600℃热解生物炭WEOM中,372 nm处的类富里酸组分能够优先于其他组分与Cu(Ⅱ)形成有机络合物。高温热解与热处理生物炭增加了WEOM中参与Cu(Ⅱ)配位的荧光物质比例。不同温度热解能够改变生物炭孔隙结构、比表面积及WEOM的含量和荧光组分,这些影响生物炭土壤修复与改良效果,对WEOM与重金属的络合能够为生物炭土壤修复与改良过程中重金属的环境化学行为提供指导依据。
-
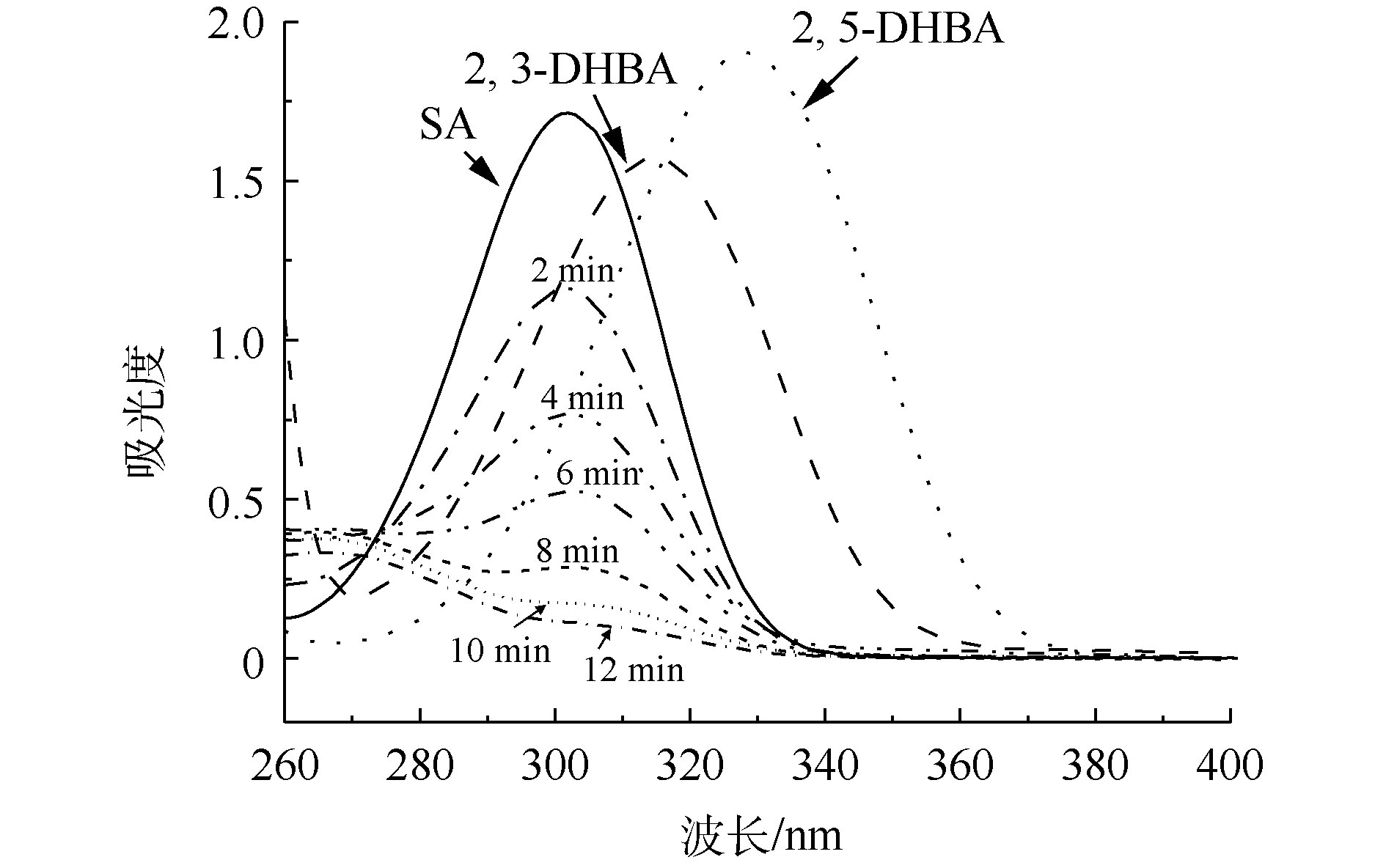
图 2 SA、2,3-DHBA、2,5-DHBA以及催化臭氧氧化处理SA后混合溶液的紫外-可见吸收光谱
Figure 2. Absorption spectra of SA, 2,3-DHBA, 2,5-DHBA and their mixture solution after SA degradation by catalytic ozonation
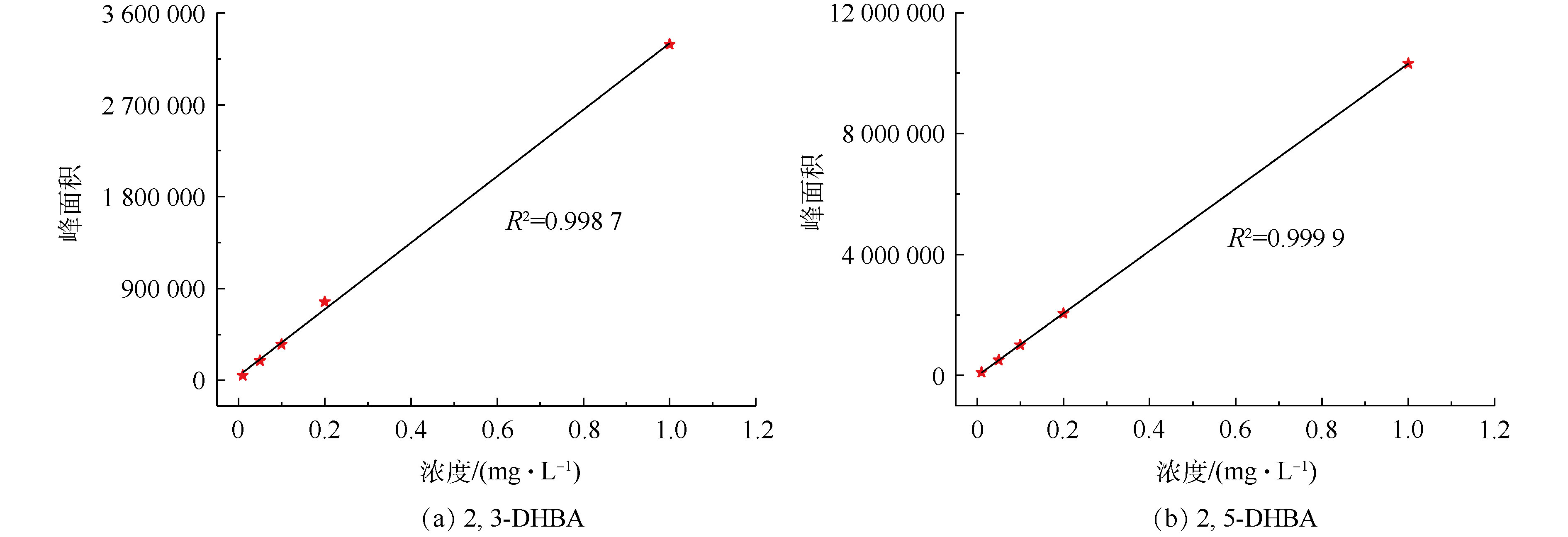
图 3 2,3-DHBA和2,5-DHBA的液相色谱标准曲线
Figure 3. Liquid chromatography standard curves for 2,3-DHBA and 2,5-DHBA
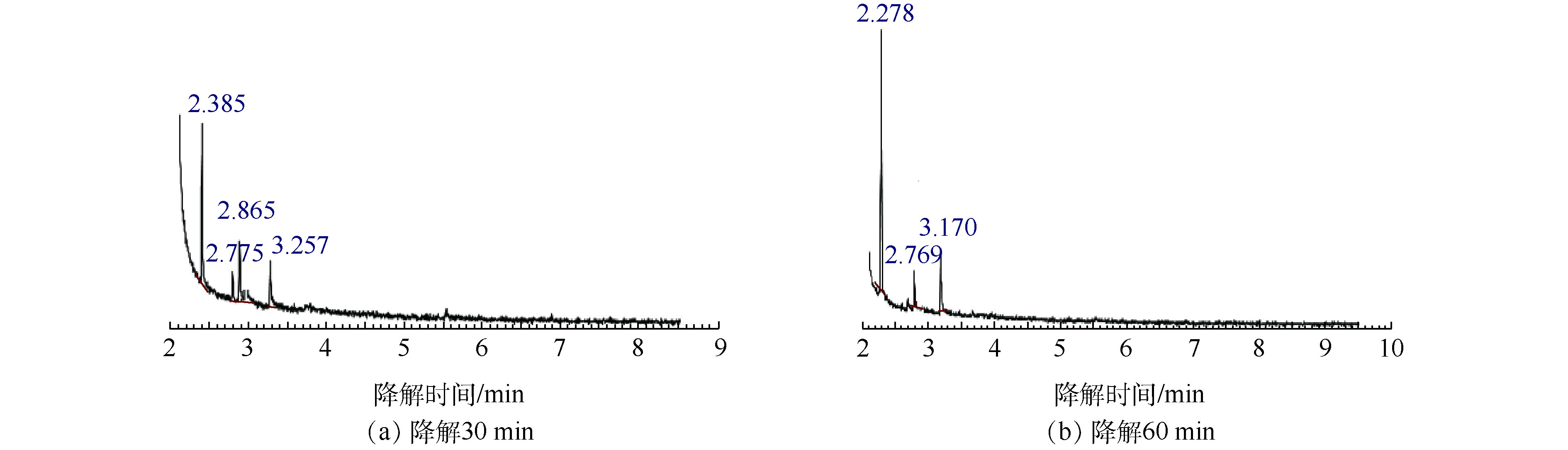
图 4 异丙醇降解30 min和60 min总离子流色谱图
Figure 4. GC chromatograms of isopropanol effluents at 30 min and 60 min degradation
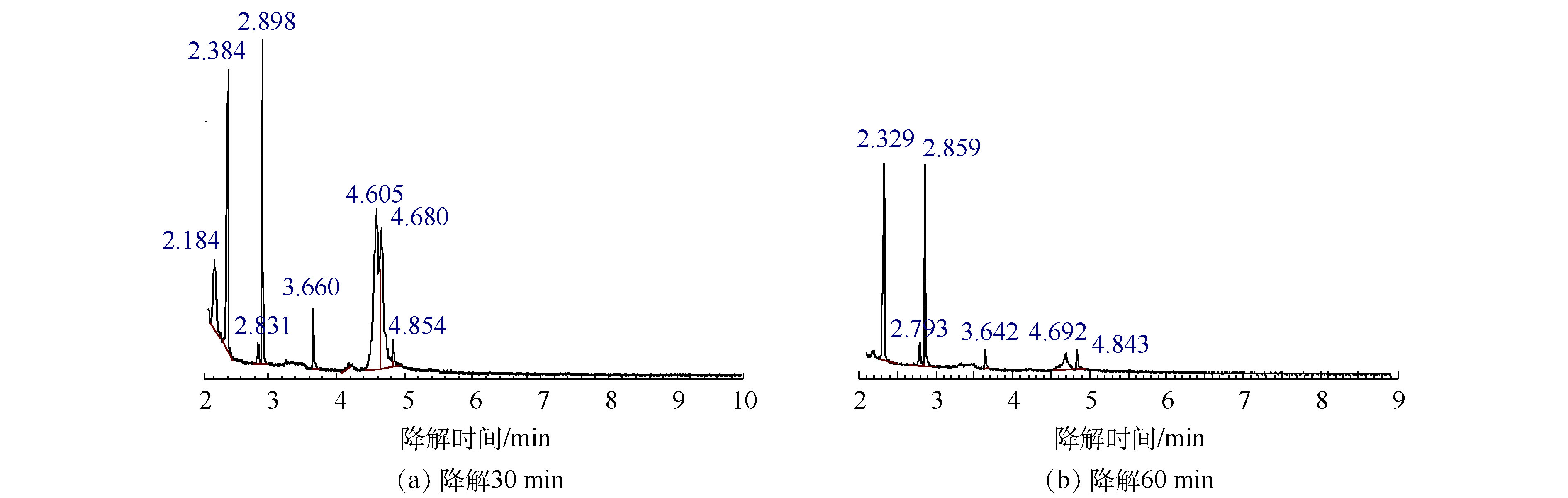
图 6 乙二醇单丁醚降解液总离子流色谱图
Figure 6. GC chromatograms of 2-butoxyethanol effluents at 30 min and 60 min degradation
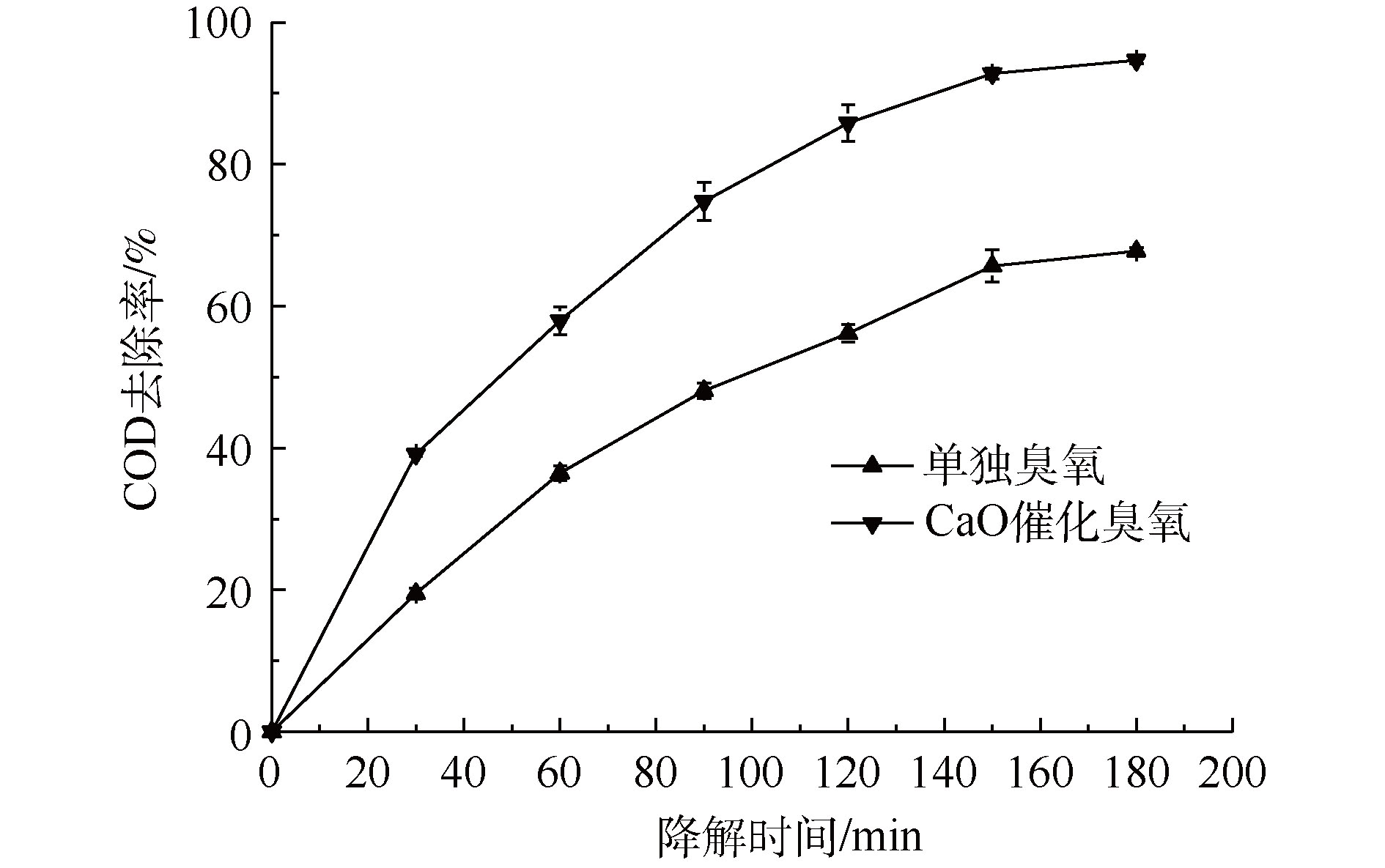
图 11 CaO在实际PCB废液中的催化降解率
Figure 11. Catalytic degradation efficiency in actual PCB effluents by CaO
表 1 2,3-DHBA和2,5-DHBA不同时间的含量
Table 1. Contents of 2,3-DHBA and 2,5-DHBA at different times
降解时间/min 2,3-DHBA浓度/(mg·L−1) 2,5-DHBA浓度/(mg·L−1) 4 0.037 3 0.015 5 8 0.022 1 0.014 4 12 0.020 0 0.013 7
下载: 导出CSV
表 2 异丙醇降解30 min和60 min的降解产物
Table 2. Products of IPA at 30 min and 60 min degradation
异丙醇降解30 min质谱结果 异丙醇降解60 min质谱结果 保留时间/min 分子式 相对分子质量 相对峰面积/% 保留时间/min 分子式 相对分子质量 相对峰面积/% 2.385 C2H4O2 60.05 46.014 2.278 C2H4O2 60.05 68.57 2.775 C2H8O2Si 92.169 9.246 2.769 C2H8O2Si 92.169 2 11.175 2.865 C3H6O2 74.08 22.436 3.170 C3H8O2 76.09 20.255 3.257 C3H8O2 76.09 22.304
下载: 导出CSV
表 3 乙二醇单丁醚降解30 min和60 min的降解产物
Table 3. Products of EB at 30 min and 60 min degradation
乙二醇丁醚降解30 min质谱结果 乙二醇丁醚降解60 min质谱结果 保留时间/min 分子式 相对分子质量 相对峰面积/% 保留时间/min 分子式 相对分子质量 相对峰面积/% 2.184 C4H10O 74.12 7.624 2.329 C2H4O2 60.05 42.980 2.384 C2H4O2 60.05 19.742 2.793 C2H8O2Si 92.169 7.158 2.831 C2H8O2Si 92.169 2 1.453 2.859 (CH2OH)2 62.068 29.959 2.898 (CH2OH)2 62.068 12.649 3.642 C4H8O2 88.105 3.316 3.660 C4H8O2 88.105 1 2.856 4.692 C6H14O2 118.17 12.300 4.605 C6H14O2 118.17 28.126 4.843 C6H14O2 118.17 4.287 4.680 C6H14O2 118.17 25.537 4.854 C4H6O2 86.09 2.013
下载: 导出CSV
表 4 因素的初始值和变化值
Table 4. Initial and change values of factors
初始值与变化值 pH CaO质量/g 废液深度/cm 降解时间/min 臭氧用量/(mg·min−1) 初始值 12.0 0 7 60 80 变化值 0.2 1.0 2 20 20
下载: 导出CSV
表 5 初始实验条件
Table 5. Initial experimental conditions
实验序号 pH CaO质量/g 废液深度/cm 降解时间/min 臭氧用量/(mg·min−1) 1 12.2 2.0 11 140 200 2 12.4 4.0 13 80 180 3 12.6 6.0 9 160 160 4 12.8 1.0 13 100 140 5 13.0 3.0 9 180 120 6 13.2 5.0 11 120 100
下载: 导出CSV
表 6 单纯形优化结果
Table 6. Result of simplex optimization
实验序号 pH CaO质量/g 废液深度/cm 降解时间/min 臭氧用量/(mg·min−1) COD去除率/% 标准差 0.7η+0.1/t+0.2/m 1 12.2 2.0 11 140 200 66.31 0.987 6 0.607 1 2 12.4 4.0 13 80 180 66.97 0.120 2 0.552 1 3 12.6 6.0 9 160 160 83.21 0.720 8 0.653 3 4 12.8 1.0 13 100 140 64.26 2.716 2 0.709 7 5 13.0 3.0 9 180 120 83.4 1.419 3 0.683 8 6 13.2 5.0 11 120 100 62.36 1.007 0 0.526 5 7 12.2 1.4 11 144 200 67.97 1.689 9 0.660 3 8 12.7 1.36 9 180 150 87.24 1.225 3 0.791 1 9 13.2 3.38 9 172 100 91.81 1.565 4 0.736 7 10 12.9 1.0 11 150 120 90.04 0.431 3 0.870 3 11 13.2 2.77 9 175 100 86.8 0.671 7 0.714 3 12 13.2 1.0 11 130 120 75.72 2.517 3 0.776 2 13 13.2 2.804 9 180 100 88.51 2.026 4 0.724 2
下载: 导出CSV
-
[1] GHUGE S P, SAROHA A K. Catalytic ozonation for the treatment of synthetic and industrial effluents: Application of mesoporous materials: A review[J]. Journal of Environmental Management, 2018, 211: 83-102. [2] LOU J C, HUANG Y J, HAN J Y. Treatment of printed circuit board industrial wastewater by ferrite process combined with Fenton method[J]. Journal of Hazardous Materials, 2009, 170(2): 620-626. [3] WANG Y, LI X, ZHEN L, et al. Electro-Fenton treatment of concentrates generated in nanofiltration of biologically pretreated landfill leachate[J]. Journal of Hazardous Materials, 2012, 229-230: 115-121. doi: 10.1016/j.jhazmat.2012.05.108 [4] BIANCO B, MICHELIS I D, VEGLIO F. Fenton treatment of complex industrial wastewater: Optimization of process conditions by surface response method[J]. Journal of Hazardous Materials, 2011, 186(2/3): 1733-1738. [5] MINH D P, GALLEZOT P, AZABOU S, et al. Catalytic wet air oxidation of olive oil mill effluents: Treatment and detoxification of real effluents[J]. Applied Catalysis B: Environmental, 2008, 84(3): 749-757. [6] JING G, LUAN M, CHEN T. Progress of catalytic wet air oxidation technology[J]. Arabian Journal of Chemistry, 2016, 9: 1208-1213. doi: 10.1016/j.arabjc.2012.01.001 [7] MEHRJOUEI M, MULLER S, MOLLER D. A review on photocatalytic ozonation used for the treatment of water and wastewater[J]. Chemical Engineering Journal, 2015, 263(1): 209-219. [8] MAMAGHANI A H, HAGHIGHAT F, LEE C S. Photocatalytic oxidation technology for indoor environment air purification: The state-of-the-art[J]. Applied Catalysis B: Environmental, 2017, 203: 247-269. doi: 10.1016/j.apcatb.2016.10.037 [9] WANG Y, XIE Y, SUN H, et al. Efficient catalytic ozonation over reduced graphene oxide for p-hydroxylbenzoic acid (PHBA) destruction: Active site and mechanism[J]. ACS Applied Materials & Interfaces, 2016, 8(15): 9710-9720. [10] LI S, TANG Y, CHEN W, et al. Heterogeneous catalytic ozonation of clofibric acid using Ce/MCM-48: Preparation, reaction mechanism, comparison with Ce/MCM-41[J]. Journal of Colloid & Interface Science, 2017, 504: 238-246. [11] CHEN C, YOZA B A, CHEN H, et al. Manganese sand ore is an economical and effective catalyst for ozonation of organic contaminants in petrochemical wastewater[J]. Water, Air & Soil Pollution, 2015, 226(6): 182. [12] CHAO S, YOU X, MING H, et al. Mesoporous Ce-Ti-Zr ternary oxide millispheres for efficient catalytic ozonation in bubble column[J]. Chemical Engineering Journal, 2018, 338: 261-270. doi: 10.1016/j.cej.2018.01.046 [13] EINAGA H, FUTAMURA S. Oxidation behavior of cyclohexane on alumina-supported manganese oxides with ozone[J]. Applied Catalysis B: Environmental, 2005, 60(1): 49-55. [14] CAO H, XING L, WU G, et al. Promoting effect of nitration modification on activated carbon in the catalytic ozonation of oxalic acid[J]. Applied Catalysis B: Environmental, 2014, 146(1): 169-176. [15] UMAR M, RODDICK F, FAN L, et al. Application of ozone for the removal of bisphenol A from water and wastewater: A review[J]. Chemosphere, 2013, 90(8): 2197-2207. doi: 10.1016/j.chemosphere.2012.09.090 [16] KATSOYIANNIS I A, CANONICA S, GUNTEN U V. Efficiency and energy requirements for the transformation of organic micropollutants by ozone, O/HO and UV/HO[J]. Water Research, 2011, 45(13): 3811-3822. doi: 10.1016/j.watres.2011.04.038 [17] ZHAO L, MA J, SUN Z, et al. Mechanism of heterogeneous catalytic ozonation of nitrobenzene in aqueous solution with modified ceramic honeycomb[J]. Applied Catalysis B: Environmental, 2009, 89(3): 326-334. [18] IKHLAQ A, BROWN D R, KASPRZYK-HORDERN B. Catalytic ozonation for the removal of organic contaminants in water on alumina[J]. Applied Catalysis B: Environmental, 2014, 154-155(14): 110-122. [19] NAWROCKI J. Catalytic ozonation in water: Controversies and questions[J]. Applied Catalysis B: Environmental, 2013, 142-143(5): 465-471. [20] NAWAZ F, XIE Y, CAO H, et al. Catalytic ozonation of 4-nitrophenol over an mesoporous α-MnO2 with resistance to leaching[J]. Catalysis Today, 2015, 258: 595-601. doi: 10.1016/j.cattod.2015.03.044 [21] NAWAZ F, CAO H, XIE Y, et al. Selection of active phase of MnO2 for catalytic ozonation of 4-nitrophenol[J]. Chemosphere, 2016, 168: 1457-1466. [22] HAO Z, MA W, HAN H, et al. Catalytic ozonation of quinoline using Nano-MgO: Efficacy, pathways, mechanisms and its application to real biologically pretreated coal gasification wastewater[J]. Chemical Engineering Journal, 2017, 327: 91-99. doi: 10.1016/j.cej.2017.06.025 [23] MOUSSAVI G, MAHMOUDI M. Degradation and biodegradability improvement of the reactive red 198 azo dye using catalytic ozonation with MgO nanocrystals[J]. Chemical Engineering Journal, 2009, 152(1): 1-7. doi: 10.1016/j.cej.2009.03.014 [24] MASHAYEKH-SALEHI A, MOUSSAVI G, YAGHMAEIAN K. Preparation, characterization and catalytic activity of a novel mesoporous nanocrystalline MgO nanoparticle for ozonation of acetaminophen as an emerging water contaminant[J]. Chemical Engineering Journal, 2017, 310: 157-169. doi: 10.1016/j.cej.2016.10.096 [25] YUAN X, YAN X, XU H, et al. Enhanced ozonation degradation of atrazine in the presence of nano-ZnO: Performance, kinetics and effects[J]. Journal of Environmental Sciences, 2017, 61: 3-13. doi: 10.1016/j.jes.2017.04.037 [26] BASHIRI H, RAFIEE M. Kinetic monte carlo simulation of 2, 4, 6-thrichloro phenol ozonation in the presence of ZnO nanocatalyst[J]. Journal of Saudi Chemical Society, 2016, 20(4): 474-479. doi: 10.1016/j.jscs.2014.11.001 [27] YANG Y, CAO H, PENG P, et al. Degradation and transformation of atrazine under catalyzed ozonation process with TiO2 as catalyst[J]. Journal of Hazardous Materials, 2014, 279: 444-451. doi: 10.1016/j.jhazmat.2014.07.035 [28] GUPTA V K, FAKHRI A, AGARWAL S, et al. Preparation and characterization of TiO2 nanofibers by hydrothermal method for removal of benzodiazepines (Diazepam) from liquids as catalytic ozonation and adsorption processes[J]. Journal of Molecular Liquids, 2017, 249: 1033-1038. [29] VITTENET J, ABOUSSAOUD W, MENDRET J, et al. Catalytic ozonation with γ-Al2O3 to enhance the degradation of refractory organics in water[J]. Applied Catalysis A: General, 2015, 504: 519-532. doi: 10.1016/j.apcata.2014.10.037 [30] IKHLAO A, BROWN D R, KASPRZYK-HORDERN B. Mechanisms of catalytic ozonation on alumina and zeolites in water: Formation of hydroxyl radicals[J]. Applied Catalysis B: Environmental, 2012, 123-124: 94-106. doi: 10.1016/j.apcatb.2012.04.015 [31] DAI Q, WANG J, JIE Y, et al. Catalytic ozonation for the degradation of acetylsalicylic acid in aqueous solution by magnetic CeO2 nanometer catalyst particles[J]. Applied Catalysis B: Environmental, 2014, 144(2): 686-693. [32] ROSAL R, GONZALO M S, RODRIGUEZ A, et al. Catalytic ozonation of atrazine and linuron on MnOx/Al2O3 and MnOx/SBA-15 in a fixed bed reactor[J]. Chemical Engineering Journal, 2010, 165(3): 806-812. doi: 10.1016/j.cej.2010.10.020 [33] GOMES J F, BEDNARCZYK K, GMUREK M, et al. Noble metal-TiO2 supported catalysts for the catalytic ozonation of parabens mixtures[J]. Process Safety and Environmental Protection, 2017, 111: 148-159. doi: 10.1016/j.psep.2017.07.001 [34] LIU Z Q, TU J, WANG Q, et al. Catalytic ozonation of diethyl phthalate in aqueous solution using graphite supported zinc oxide[J]. Separation & Purification Technology, 2018, 200: 51-58. [35] SUI M, XING S, SHENG L, et al. Heterogeneous catalytic ozonation of ciprofloxacin in water with carbon nanotube supported manganese oxides as catalyst[J]. Journal of Hazardous Materials, 2012, 227-228: 227-236. doi: 10.1016/j.jhazmat.2012.05.039 [36] LIAO M, CHEN J, LI L, et al. Effective degradation of nitrotoluenes in wastewater by heterogeneous catalytic ozonation in the presence of calcium oxide[J]. AIP Conference Proceedings, 2017, 1890(1): 020008. [37] BROSEUS R, VINCET S, ABOULFADL K, et al. Ozone oxidation of pharmaceuticals, endocrine disruptors and pesticides during drinking water treatment[J]. Water Research, 2009, 43(18): 4707-4717. doi: 10.1016/j.watres.2009.07.031 [38] HUANG X, LI X, PAN B, et al. Self-enhanced ozonation of benzoic acid at acidic pHs[J]. Water Research, 2015, 73: 9-16. doi: 10.1016/j.watres.2015.01.010 [39] ZHANG S, QUAN X, ZHENG J F, et al. Probing the interphase " HO zone” originated by carbon nanotube during catalytic ozonation[J]. Water Research, 2017, 122: 86-95. doi: 10.1016/j.watres.2017.05.063 [40] WANG Q, DING F, ZHU N, et al. Determination of hydroxyl radical by capillary zone electrophoresis with amperometric detection[J]. Journal of Chromatography A, 2003, 1016(1): 123-128. doi: 10.1016/S0021-9673(03)01294-9 [41] WANG J, BAI Z. Fe-based catalysts for heterogeneous catalytic ozonation of emerging contaminants in water and wastewater[J]. Chemical Engineering Journal, 2016, 312: 79-98. [42] BAI Z, QI Y, WANG J. Catalytic ozonation of sulfamethazine using Ce0.1Fe0.9OOH as catalyst: Mineralization and catalytic mechanisms[J]. Chemical Engineering Journal, 2016, 300: 169-176. doi: 10.1016/j.cej.2016.04.129 [43] HUANG Y, CUI C, ZHANG D, et al. Heterogeneous catalytic ozonation of dibutyl phthalate in aqueous solution in the presence of iron-loaded activated carbon[J]. Chemosphere, 2015, 119: 295-301. doi: 10.1016/j.chemosphere.2014.06.060 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4988
- HTML全文浏览数: 4988
- PDF下载数: 85
- 施引文献: 0
