


 下载:
下载:


-
砷来源广泛,包括火山喷发、岩石风化等自然来源以及采矿、冶金等人为来源[1-2]。在全球范围内,土壤中砷的平均质量分数为1.8 mg·kg−1,而我国土壤中砷平均质量分数达到9.2 mg·kg−1,超过世界水平的5倍[3]。我国云南、贵州、四川等西南地区的土壤中砷背景值远超全国土壤背景值[4]。土壤中的砷通过食物链进入人体后,可引发色素沉着、慢性肺病、心血管疾病和神经系统紊乱等健康问题[5]。因此,对砷污染土壤的修复十分迫切。
电动修复是常用的一种砷污染土壤修复方法,其利用电渗析、电迁移等电动效应使砷酸根和亚砷酸根定向迁移,从而降低土壤中砷的总量[6-7]。但常规电动修复技术对砷的修复效果有限,KARACA等[8]对沉积物中的砷进行电动修复时发现,运行18 d后砷几乎没有被去除。电极逼近法为电动修复的一种,其在电动过程中每隔一段时间将电极向某一方向移动一定距离,以此来影响土壤pH、氧化还原电位 (Eh) 等环境因子,而砷的溶解性和迁移性与环境因子密切相关。YAO等[9]发现,相比于固定电极法 (FE-EK) ,阴极逼近法 (AC-EK) 通过提高阴极区域pH可将砷的去除率提高4倍。付博等[10]发现,当pH<4时,随着pH的降低,粗砂和细砂中砷的溶出量不断增加。周一敏等[11]发现,当Eh较低时,五价砷[As(V)]会转化为移动性更高的三价砷[As(III)],另外还能驱动土壤中砷的释放。由此可见,电动逼近技术对提高砷污染土壤修复效果具有很大潜力。
目前,常采用向土壤中加入化学药剂[2,7]、增设渗透反应墙[6,12]等方式提高砷去除率,但基于电极逼近技术对砷污染土壤进行修复的研究尚很缺乏。基于此,本研究采用不同的电极逼近方式对砷污染土壤进行修复。研究不同逼近方式对总砷[As(T)]的分布以及As(III)、As(V)迁移转化的影响,探究捕集室土壤中砷赋存形态的转化,以期为砷污染场地修复提供技术参考和理论依据。
-
供试土壤采自辽宁大连某污染场地,经风干并研磨后过20目标准筛备用。供试土壤基本理化性质为:pH为7.17,Eh为273.5 mV,电导率为2 012.5 μS·cm−1,Al、Fe、Ca质量分数分别为40.13、44.17、102.55 g·kg−1,As(T)、As(III)、As(V)质量分数分别为355.08、120.32、234.76 mg·kg−1。其中,As(T)质量分数超过《土壤环境质量 建设用地土壤污染风险管控标准 (试行) 》 (GB 36600-2018) [13]第一类用地筛选值 (20 mg·kg−1) 的17倍。
-
如图1(a)所示,实验装置主体由土壤室和捕集室组成,捕集室置于实验装置中部,可自由取出,2侧为土壤室。取样点位置如图1(b)所示,从阳极到阴极划分为阳极区 (S1~S3) 、捕集室 (S4) 、阴极区 (S5~S7) 3部分,S1~S7每个区域设置3个取样点,将3个取样点的土壤混合后作为该区域的代表性土壤。
-
实验共设置4个电动处理组,分别为FE-EK、AC-EK、阳极逼近处理组 (AA-EK) 、两极逼近处理组 (AAC-EK) 。其中,FE-EK处理组不移动电极;AC-EK处理组的阴极电极每隔10 d向阳极方向移动4 cm,共移动2次;AA-EK处理组的阳极电极每隔10 d向阴极方向移动4 cm,共移动2次;AAC-EK处理组的阳极电极和阴极电极每隔10 d相向各移动4 cm,共移动2次。各电动处理组土壤室内均填装1 600 g污染土壤,捕集室内填装400 g混有质量分数为20% Fe2O3的污染土壤。另取400 g混有Fe2O3的污染土壤,不通电,作为电动处理组的对照组 (CK) 。
实验以不锈钢电极为电极,电压恒定为24 V,处理时间30 d。实验过程中每隔4~5 d采用重量法补充去离子水,保持土壤含水率为30%。取样间隔为10 d,移动电极后的无电场区域不再继续取样。
-
本研究中总能耗和单位修复能耗的计算方法见式(1)和式(2)[14]。
式中:E为总能耗,kWh;U为实验电压,V;I为电流,A;t为修复时间,h。
式中:E0为单位修复能耗,kWh·mg−1;c0和c30为第0 天和第30天时捕集室中As(T)质量分数,mg·kg−1;m为捕集室中土壤质量,kg。
-
电流使用电流监控装置监测并记录。pH和电导率使用pH计 (PHS-3C型,上海仪电科学仪器股份有限公司) 和电导率仪 (CON700,美国Eutech公司) 测定[6]。Eh参考《土壤 氧化还原电位的测定 电位法》 (HJ 746—2015) [15],使用便携式ORP测定仪 (TR-901型,上海仪电科学仪器股份有限公司) 测定。As(T)质量分数利用HNO3-HF-HClO4对土壤进行分步消解[16],并用电感耦合等离子体质谱仪 (ICAPRQ,美国Thermo Fisher Scientific公司) 测定。As(III)质量分数参考ZHENG等[17]以及张静等[18]的提取方法,并用原子荧光光谱仪 (AFS-9700A,北京海光仪器有限公司) 测定[19]。As(V)质量分数为As(T)与As(III)的差值。砷赋存形态参考XU等[7]的方法依次提取可交换态砷 (Ex-As) 、铝结合态砷 (Al-As) 、铁结合态砷 (Fe-As) 以及钙结合态砷 (Ca-As) ,并用ICP-OES (Avio 220 Max,美国PerkinElmer公司) 测定。残渣态砷 (Res-As) 测定方法同As(T)。
-
如图2所示,各处理组在移动电极前的电流值相似,表明各处理组间的平行性较好。通电后电流在短时间内即达到最大值,约为100 mA;随后电流值迅速下降,至第5 d时仅为9.42~14.04 mA;第5 d补水后电流值又迅速上升。这是因为,电动初始时土壤中含有大量可移动离子;而后电解水产生的H+和OH−被不断中和,孔隙水中的离子强度降低[9];补水后土壤中的可移动离子数量又有所增加 [20-21]。电导率常用来表示土壤孔隙液中溶解离子的数量[22]。各处理组的电导率变化如图3所示,表现为两极高、中间低。这归因于阴离子和阳离子不断迁往两极[6],降低了中间区域可溶性离子数量。各处理组电导率在S1、S4、S5区域存在显著差异 (p<0.05) 。
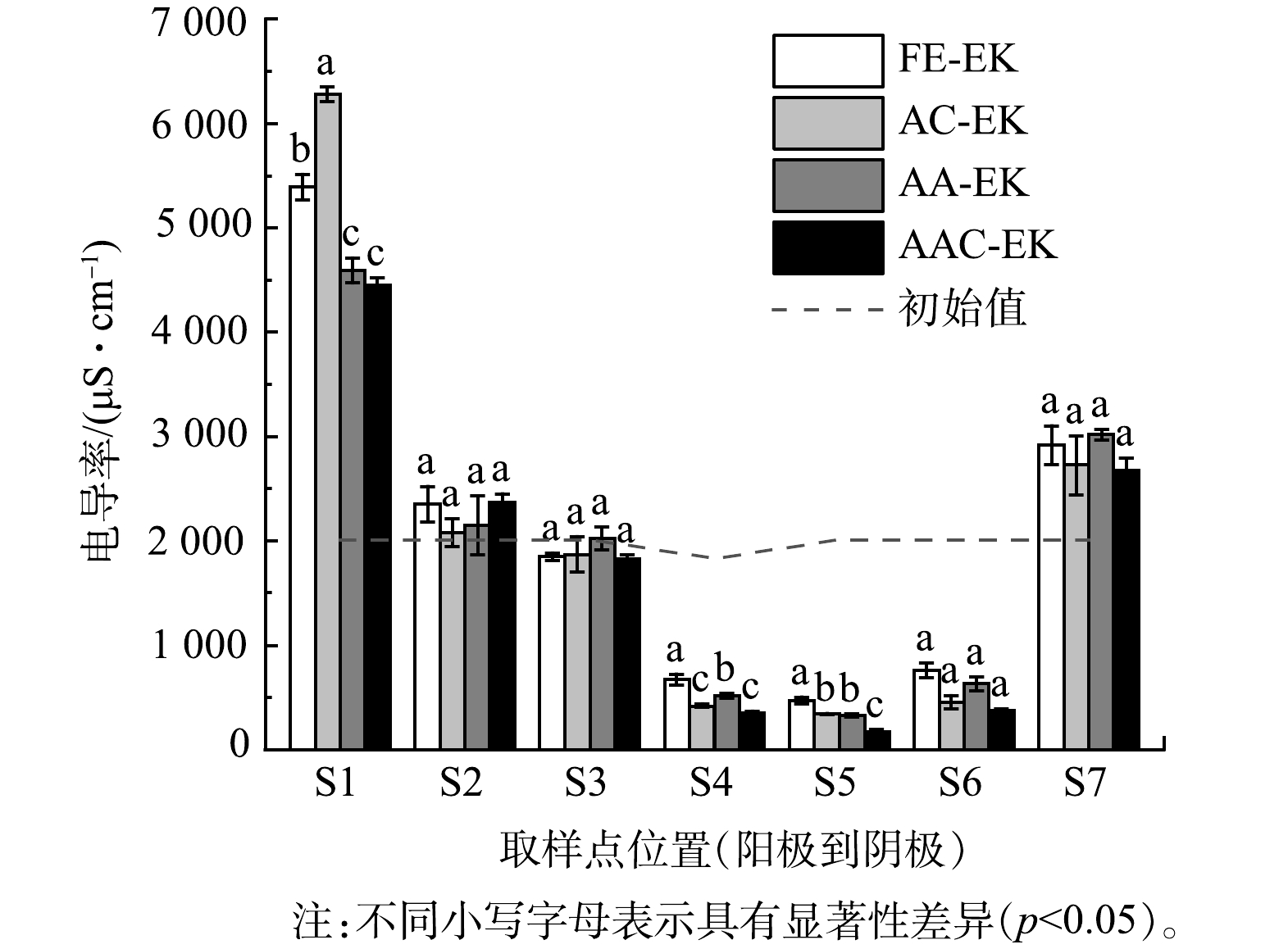
运行10 d后,电极逼近处理组的电流值高于固定电极处理组,以第20 d为例,FE-EK、AC-EK、AA-EK以及AAC-EK的电流值分别为27.36、42.64、50.74、57.68 mA。这主要是因为,随着电极的移动,土壤有效长度缩短,提高了系统电流[9]。因AAC-EK的两极间距最短,所以AAC-EK的电流值又高于AC-EK和AA-EK。AC-EK的电流值低于AA-EK主要是因为AC-EK能提高阴极区pH,容易生成氢氧化物、碳酸盐等不溶性和非导电物质,降低系统电流[21]。
-
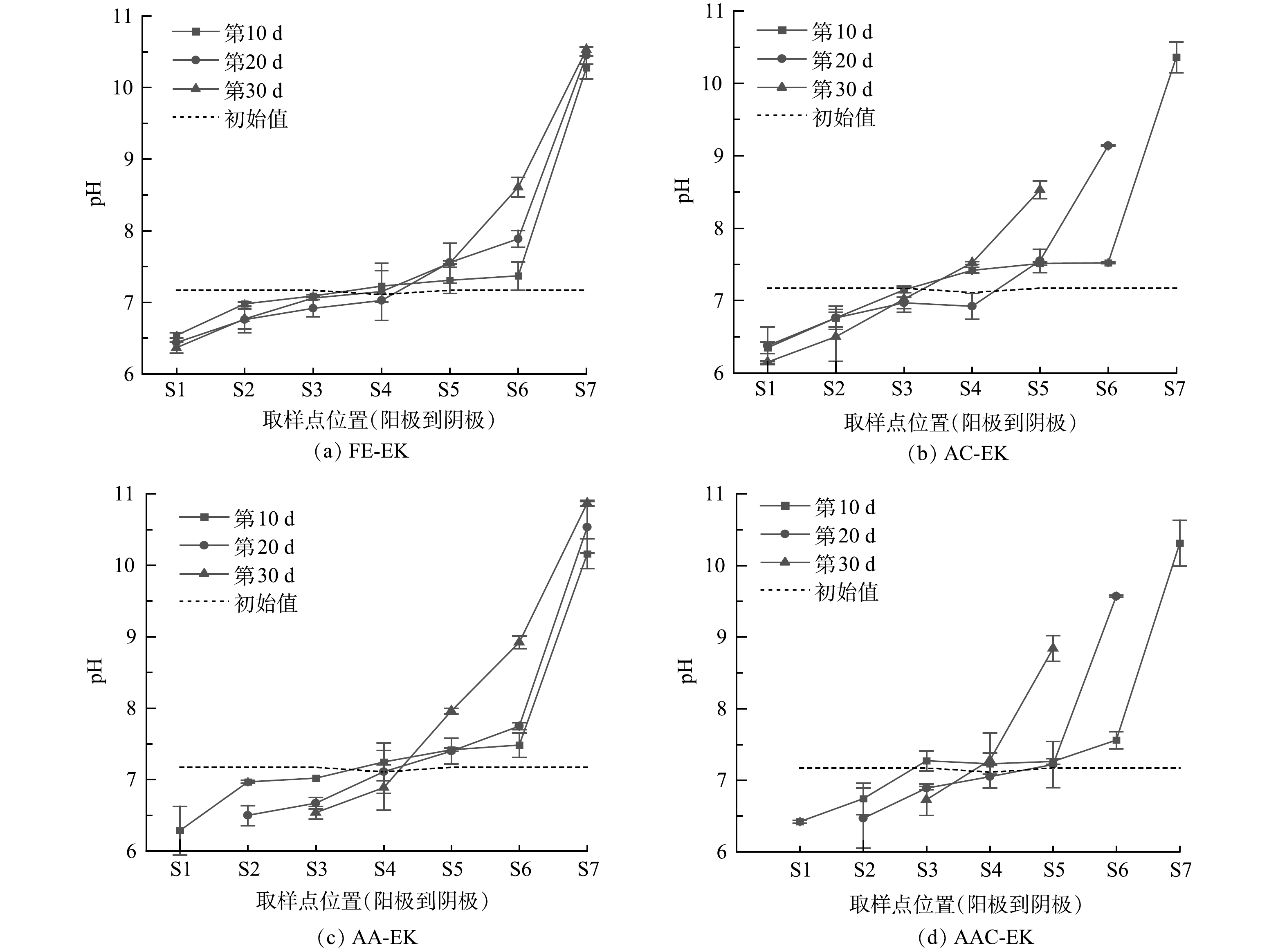
土壤室土壤初始pH为7.17,捕集室土壤初始pH为7.11。如图4所示,土壤pH从阳极至阴极呈逐渐增大趋势,且阴极区变化幅度高于阳极区。这是因为,在外加电场作用下,阳极和阴极因发生水解反应分别生成H+和OH−[9]。AA-EK能够促进阳极区pH降低,例如其S2区域在10~20 d降低0.47,高于FE-EK下降幅度,但AA-EK并未能阻止阴极区的pH升高,这可能是由于土壤的酸缓冲性能较高,向阴极移动的H+在到达阴极区前就被消耗殆尽。反之,AC-EK的阴极电极不断向阳极靠近,使其阴极区pH随时间的推移逐渐升高。由于AAC-EK电流值最高,导致其S2~S6区域的pH变化幅度一般高于AC-EK、AA-EK或FE-EK。
-
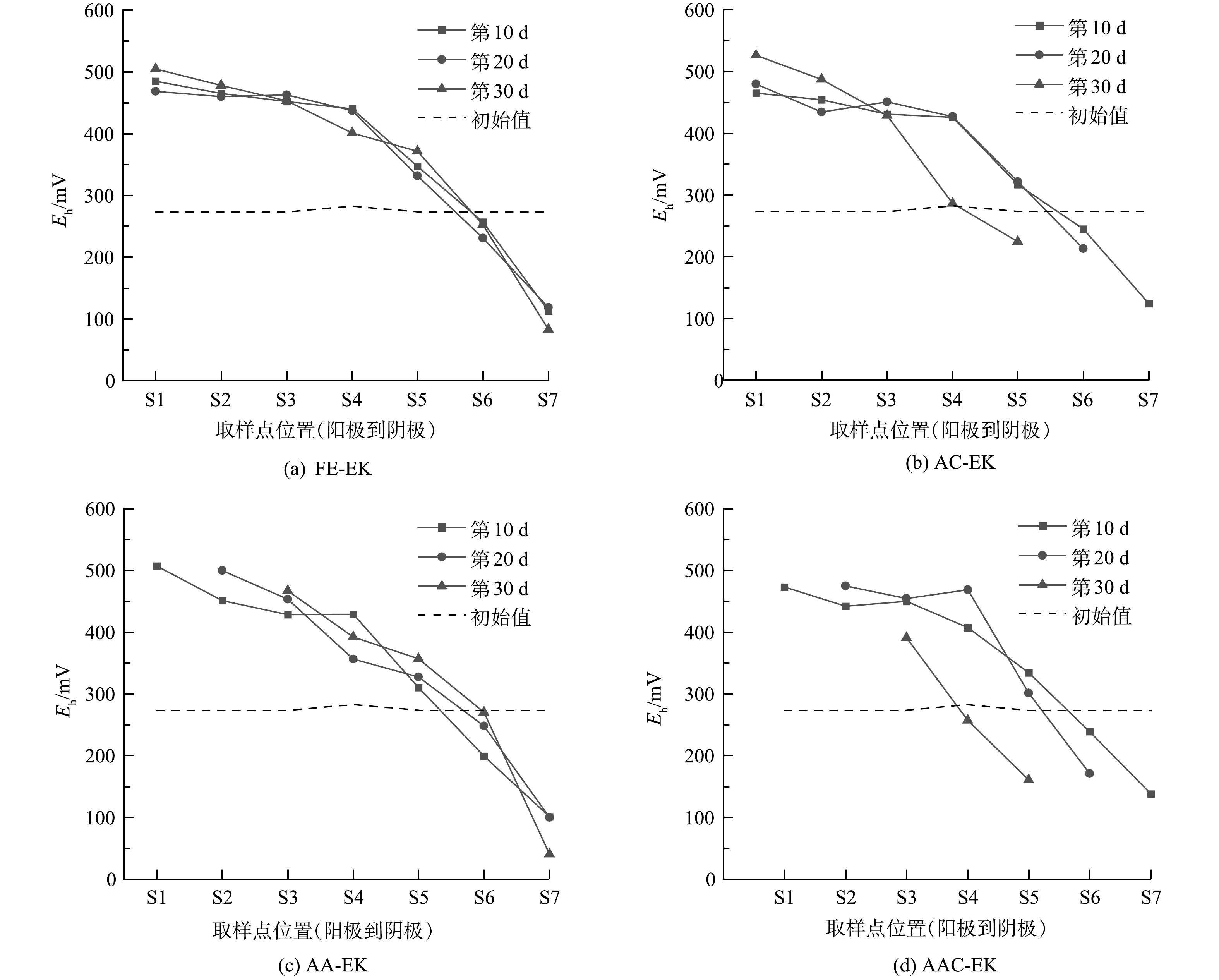
如图5所示,土壤室土壤初始Eh为273.5 mV,捕集室土壤初始Eh为282.5 mV。电动结束后,土壤Eh表现为从阳极到阴极逐渐降低的分布趋势。其中,S1~S5区域的Eh一般高于初始值,S6~S7区域的Eh低于初始值。阳极Eh的升高主要源于水电解反应产生的氧气及活性自由基;而阴极Eh的降低主要源于水解反应产生氢气,使阴极土壤处于还原气氛。与FE-EK相比,阳极电极的移动促进阳极区Eh升高,而阴极电极的移动促进阴极区Eh降低。以AC-EK为例,其第30 d时S5区域的Eh比FE-EK低147 mV,与SHEN等[23]的研究结果一致。
-
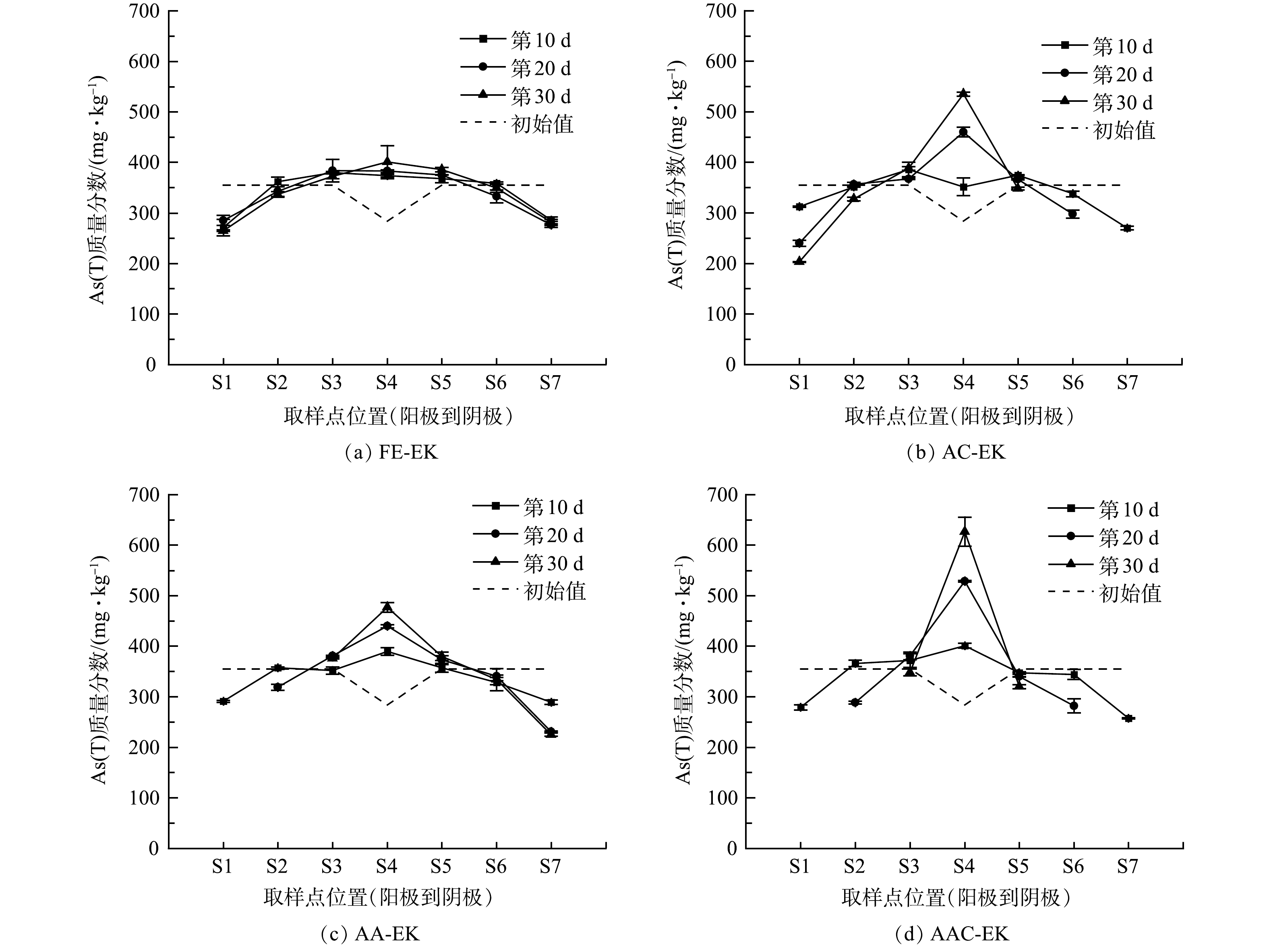
如图6所示,土壤室土壤初始As(T)质量分数为355.08 mg·kg−1,捕集室土壤初始As(T)质量分数为283.97 mg·kg−1。修复过程中,土壤中As(T)从两极区域向中间区域聚集,并最终呈现两极低、中间高的分布趋势。As(T)分布的变化是因为,As(T)在电场作用下同时受到电迁移和电渗析作用,一方面,带负电荷的H2AsO4−、HAsO42−、H2AsO3−等随电迁移迁往阳极;另一方面,溶解于土壤孔隙水中的砷随电渗流迁往阴极[24],导致两极及其附近区域As(T)质量分数降低。由于土壤中对砷吸附能力较强的铝、铁、钙等元素较多,可与砷形成不可移动的沉淀,导致砷移动性显著降低;此外,捕集室中Fe2O3对砷具有很强的吸附能力,迁移至此的砷难以继续向两极迁移,使得捕集室中As(T)质量分数不断升高。运行30 d后,AC-EK、AA-EK以及AAC-EK捕集室中As(T)质量分数与初始值相比显著升高 (p<0.05) ,S1、S7区域As(T)质量分数显著性降低 (p<0.05),以AAC-EK处理组As(T)质量分数显著性降低点位最多 (S1、S2、S5、S6、S7) ,而FE-EK捕集室中As(T)质量分数与初始值相比无显著性差异 (p>0.05) ,仅S1区域As(T)显著性降低 (p<0.05) ,这表明电极逼近对As(T)的迁移具有显著促进作用。
运行30 d后,FE-EK的As(T)整体迁移率最低 (15.38%) ,AAC-EK的As(T)整体迁移率最高 (31.50%) ,AC-EK与AA-EK居于2者之间 (27.25%、21.65%) 。AC-EK之所以能促进砷的迁移主要因为以下几个方面:首先,电极间距的缩短增大了系统电流,加速了砷的迁移;其次,阴极电极的移动增大了阴极区土壤pH,使土壤对带负电荷的砷酸根和亚砷酸根吸附能力减弱[2],且OH−能置换出以含氧阴离子形式存在的砷[25];此外,阴极电极的移动还降低了土壤Eh,使Fe(III)向Fe(II)转化,Fe(OH)3等铁系物因此发生溶解[26],砷因失去吸附相被释放到土壤溶液中,有利于砷的迁移。AA-EK因电极间距的缩短增大了系统电流,同样能促进As(T)的迁移;但因为其阳极区域pH不断降低,增强了土壤对砷的吸附,导致促进效果不明显。虽然AAC-EK阳极区pH也较低,但它的电流值最高,且其阴极区pH最高,Eh最低,有利于砷的解吸,所以AAC-EK对As(T)的迁移效果优于AC-EK和AA-EK。
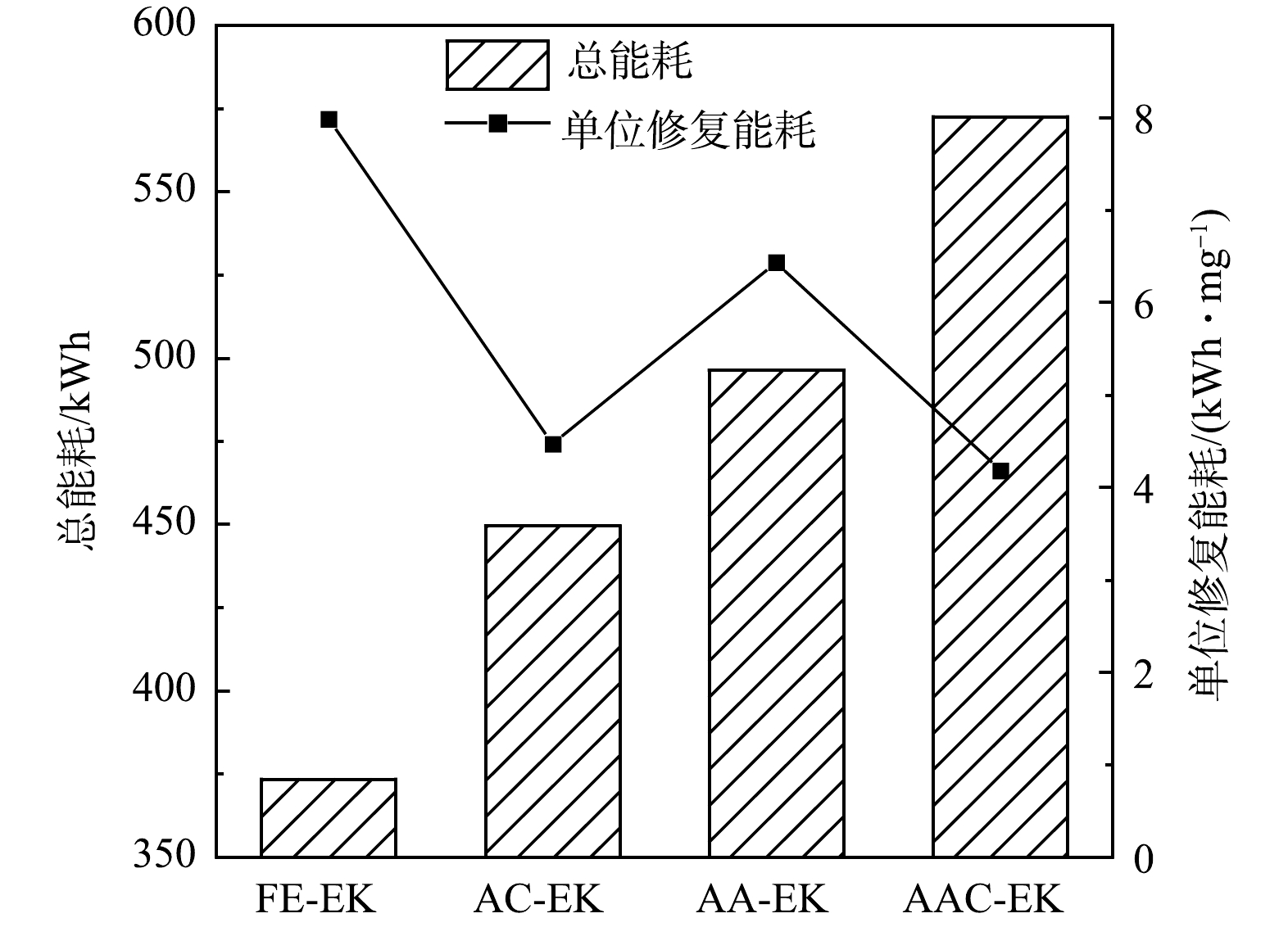
如图7所示,FE-EK、AC-EK、AA-EK、AAC-EK的总电能耗依次为373.46、449.59、496.46、572.64 kWh,单位修复能耗依次为7.98、4.47、6.44、4.18 kWh·mg−1。可见,总电能耗最高的AAC-EK的单位修复能耗最低。这是因为,当电压一定时,单位修复能耗除了与电流强度有关还与污染物迁移量有关,AAC-EK捕集室中的As(T)的增加量为FE-EK的2.93倍。
-
初始土壤中,As(V)为无机砷的主要形式,约为As(III)的1.95倍。电动处理30 d后As(V)的分布如图8(a)所示。As(V)表现为中间高、两极低的分布趋势,FE-EK、AC-EK、AA-EK、AAC-EK捕集室中As(V)质量分数依次升高60.62%、120.61%、93.99%、162.86%。这是因为,阴极带负电荷的As(V)不断移向阳极,在迁移过程中,pH逐渐降低,As(V)迁移能力随之下降;且中间区域的Fe2O3对As(V)有强亲和力,导致As(V)移动至捕集室后难以继续移动,并最终停滞在捕集室;另外,由于电渗析流会带动部分溶解于土壤间隙液中As(V)向阴极迁移,导致阳极区的As(V)也有不同程度的降低。各处理组间As(V)分布差异主要集中在阴极区,AC-EK和AAC-EK能够升高阴极区pH,进而提高砷的移动性,所以这2个处理组阴极区的As(V)质量分数低于AA-EK和FE-EK;又因为AA-EK电流较大,所以其阴极区的As(V)质量分数又低于FE-EK。

As(III)的分布如图8(b)所示。As(III)与As(V)分布趋势一致,为中间高、两极低。这是因为,阳极区土壤pH<9.2,As(III)以不带电的分子形式 (H3AsO3) 存在,主要受电渗析作用迁往阴极[27];在阴极区,越接近阴极土壤pH越高,As(III)又以分子形式向含氧酸根形式转化,带负电荷的亚砷酸根 (H2AsO3−、HAsO32−、AsO33−) 逐渐增多,并随电迁移迁往阳极,导致S6、S7区域的As(III)质量分数低于S5区域。对比来看,各处理组阳极区As(III)质量分数从低到高依次为AAC-EK、AA-EK、AC-EK、FE-EK。处理组间的差异可能与电流强度有关,当电流较大时电渗析作用较强,更多的As(III)受电渗析作用迁移向阴极,所以电流越大阳极区的As(III)残留量越低,同时使得捕集室中As(III)质量分数越高。
由于土壤Eh普遍升高,导致部分As(III)转化为As(V)。运行30 d后,FE-EK、AC-EK、AA-EK、AAC-EK各点位As(III)平均质量分数较初始值分别降低9.78%、7.81%、13.65%、4.09%。与此同时,As(V)质量分数随之升高。有研究指出,As(III)的毒性高于As(V)[3],因此,经电动修复土壤中砷的毒性被降低。比较而言,AA-EK因能提高阳极区Eh,所以对As(III)的削减量最高;AAC-EK虽然也能提高阳极区Eh,但其阴极区Eh明显降低,所以对As(III)的总体削减效果较差。
-
各处理组捕集室中砷的形态分布如图9所示。初始土壤中各形态砷占比从低到高依次为Ex-As (0.84%)、Al-As (5.16%)、Fe-As (9.05%)、Res-As (40.88%)、Ca-As (44.07%)。砷在电场的作用下不断向捕集室中迁移,并在Fe2O3的作用下发生赋存形态的明显转化,表现为Ex-As、Al-As、Ca-As占比下降,Fe-As和Res-As占比上升。对比各处理组砷赋存形态占比可知,FE-EK处理组的Ex-As最终占比最高,AA-EK、AAC-EK处理组的Ex-As最终占比较低,分别为0.44%和0.36%;FE-EK处理组的Res-As最终占比最低,AAC-EK处理组Res-As占比最终最高,达64.98%,为CK的1.42倍。
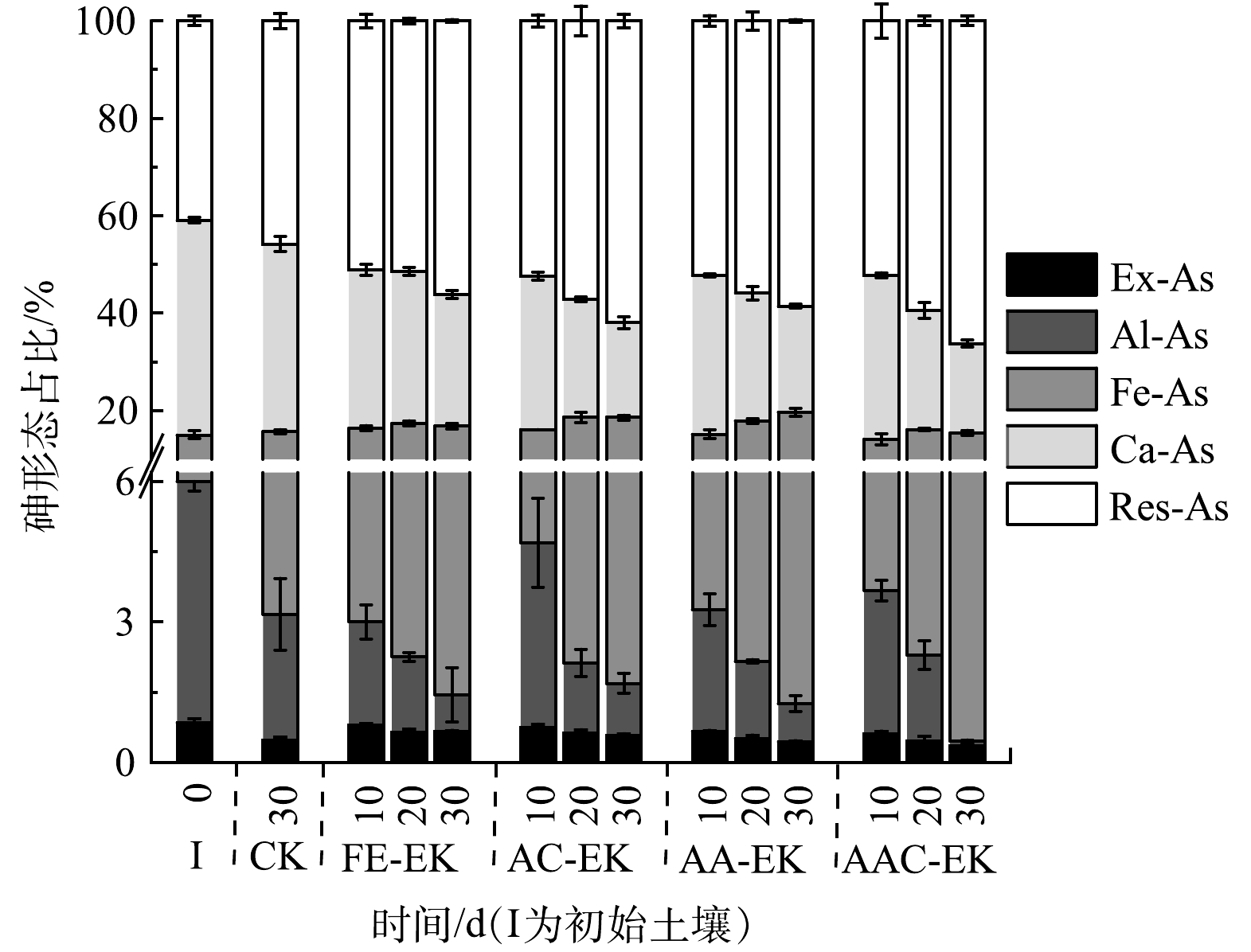
Ex-As占比的降低是因为Fe2O3的加入为砷提供了更多的吸附位点,使Ex-As转化为Fe-As。由于AA-EK、AAC-EK的电流较大,且阳极区pH相对较低,电极腐蚀后产生的Fe2+/Fe3+在随电迁移迁往阴极的过程中因pH逐渐增大而被沉淀于捕集室中,进一步加强了对Ex-As的吸附,导致其Ex-As占比较低。Al-As占比的降低也可能是受Fe2O3的影响。胡丽琼等[28]研究发现,当向砷污染水稻土中加入Fe2O3的量达到0.5 mg·kg−1时,Al-As已降至检测限以下。Res-As占比的升高一方面是由于Al-As、Ca-As向Res-As转化;另一方面,砷被铁吸附后形成Fe-As双核或单基配体化合物,或通过发生化学反应使沉淀于铁氧化物表面的砷酸盐形成砷酸铁沉淀,进而生成Res-As [29-31]。不同赋存形态砷的生物有效性从大到小依次为Ex-As>Ca-As>Al-As>Fe-As>Res-As[29]。可见,经电动修复后,砷的生物有效性大幅度降低。因AAC-EK处理组Ex-As占比最低,Res-As占比最高,所以处理效果最好。
-
1) 相比于固定电极,3种电极逼近方式通过影响环境因子 (pH、Eh) 以及系统电流,对As(T)的迁移具有促进作用,以AAC-EK的As(T)整体迁移率最高 (31.50%) ,且单位修复能耗最低。
2) 砷的价态转化受Eh影响,电动修复后,各处理组As(III)平均质量分数较初始值有所降低,As(V)平均质量分数较初始值有所升高。
3) 电动联合Fe2O3施用可使砷的形态从Ex-As、Al-As、Ca-As向Fe-As、Res-As转化,降低捕集室中的砷的生物有效性,以AAC-EK的稳定化效果最佳。由此可见,AAC-EK在修复砷污染土壤方面具备很大潜力。

图 1
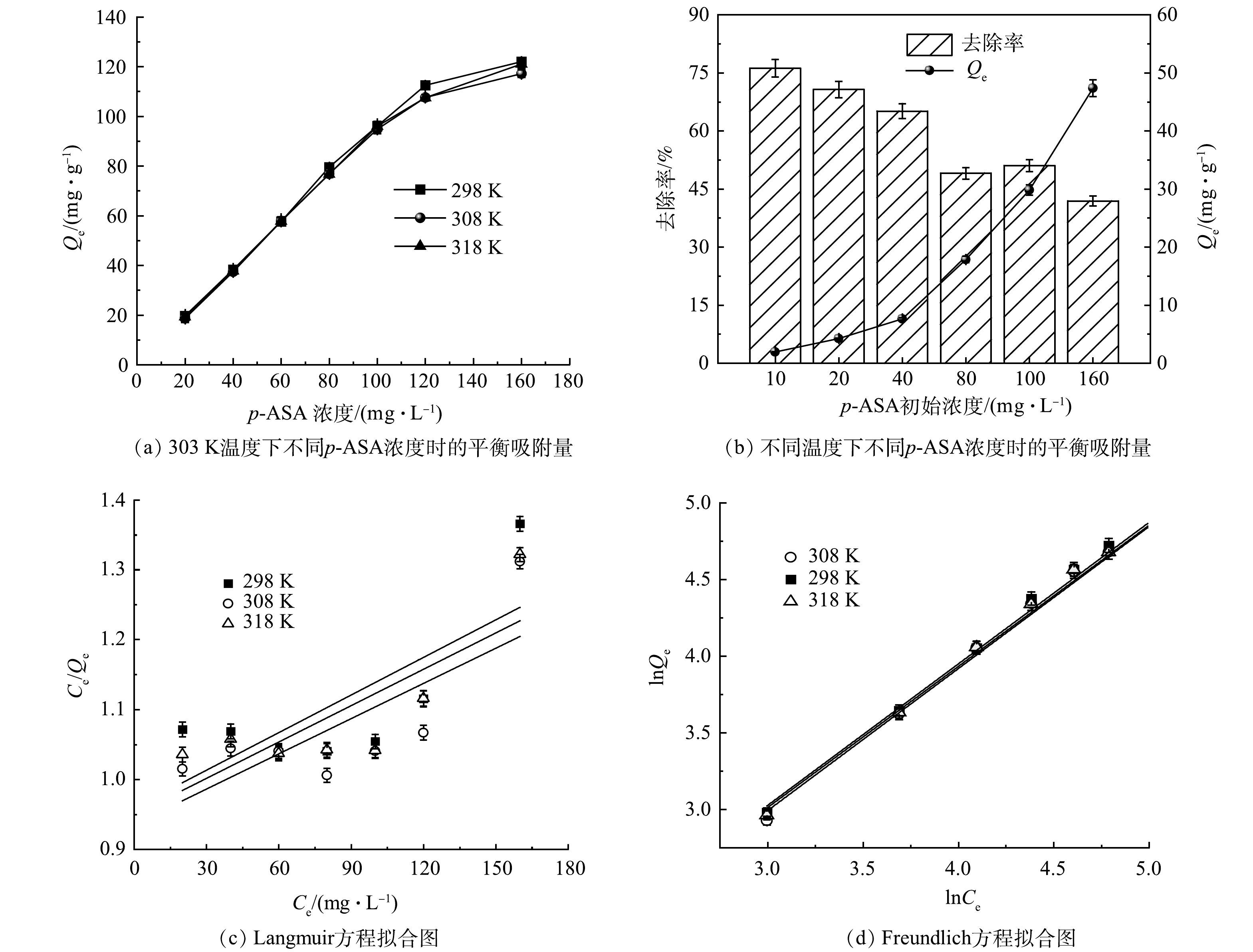
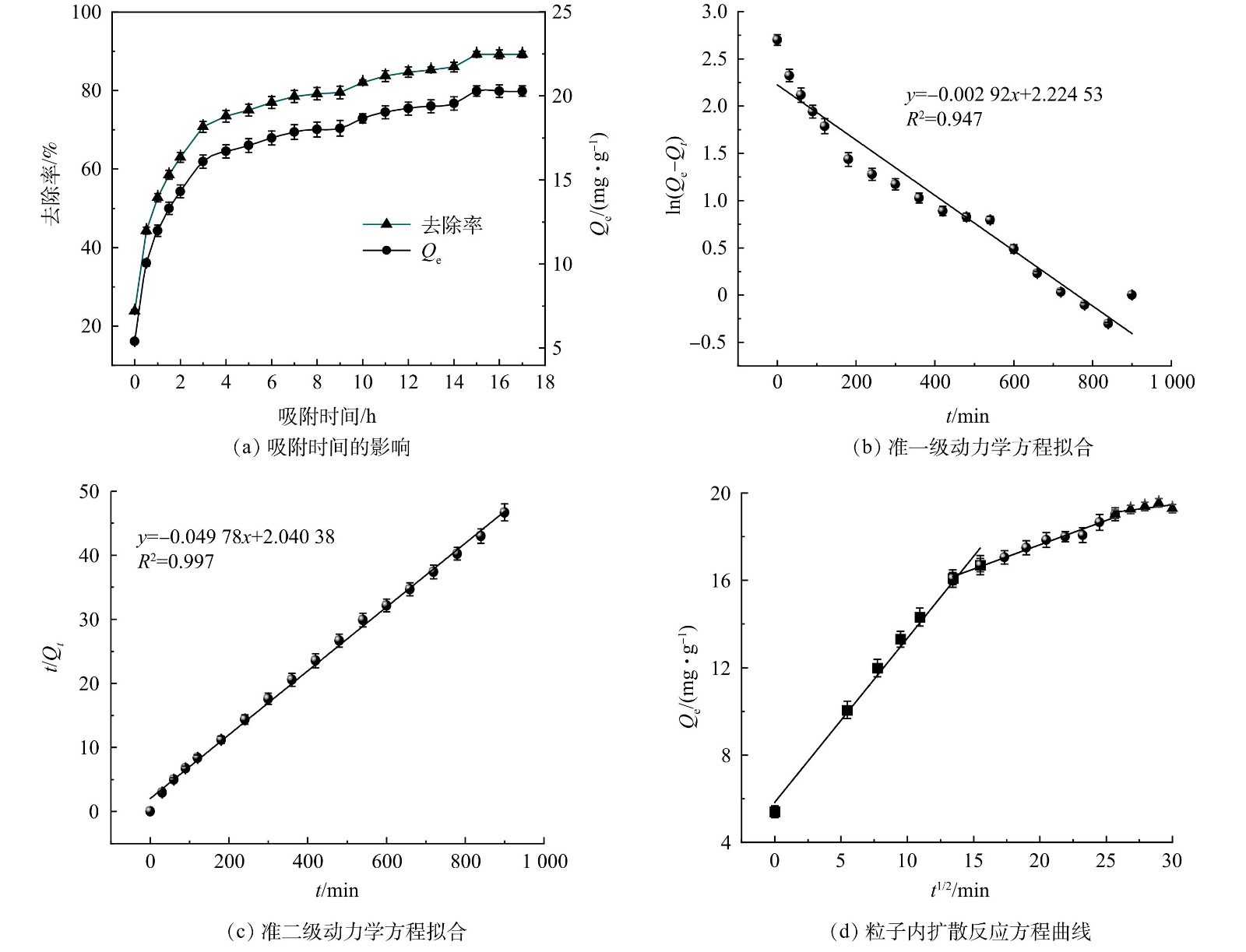
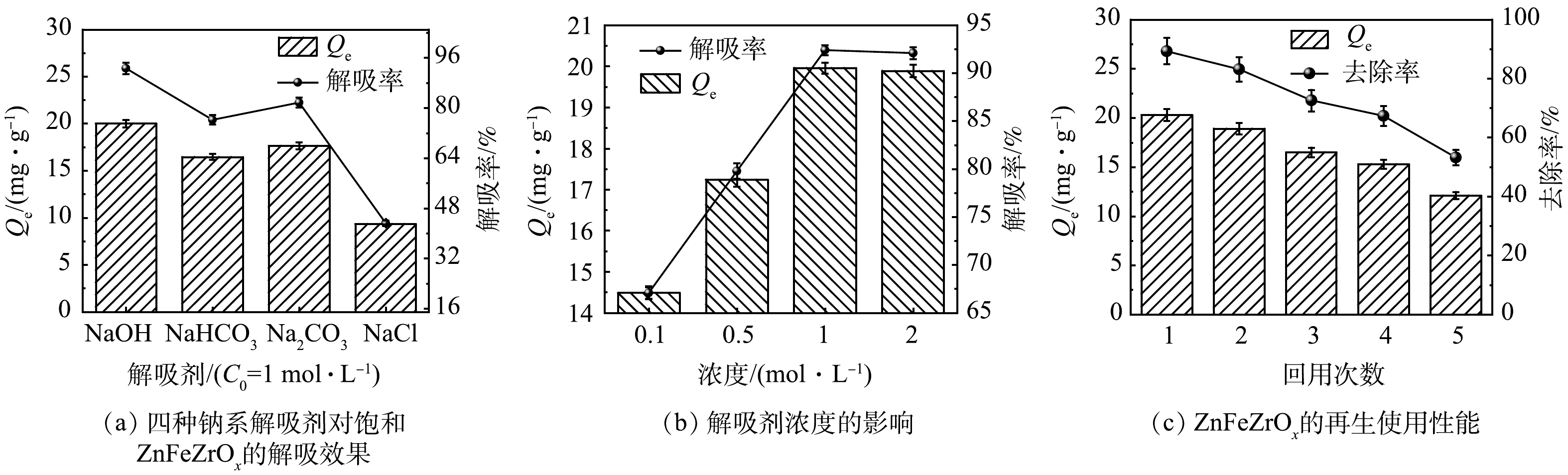
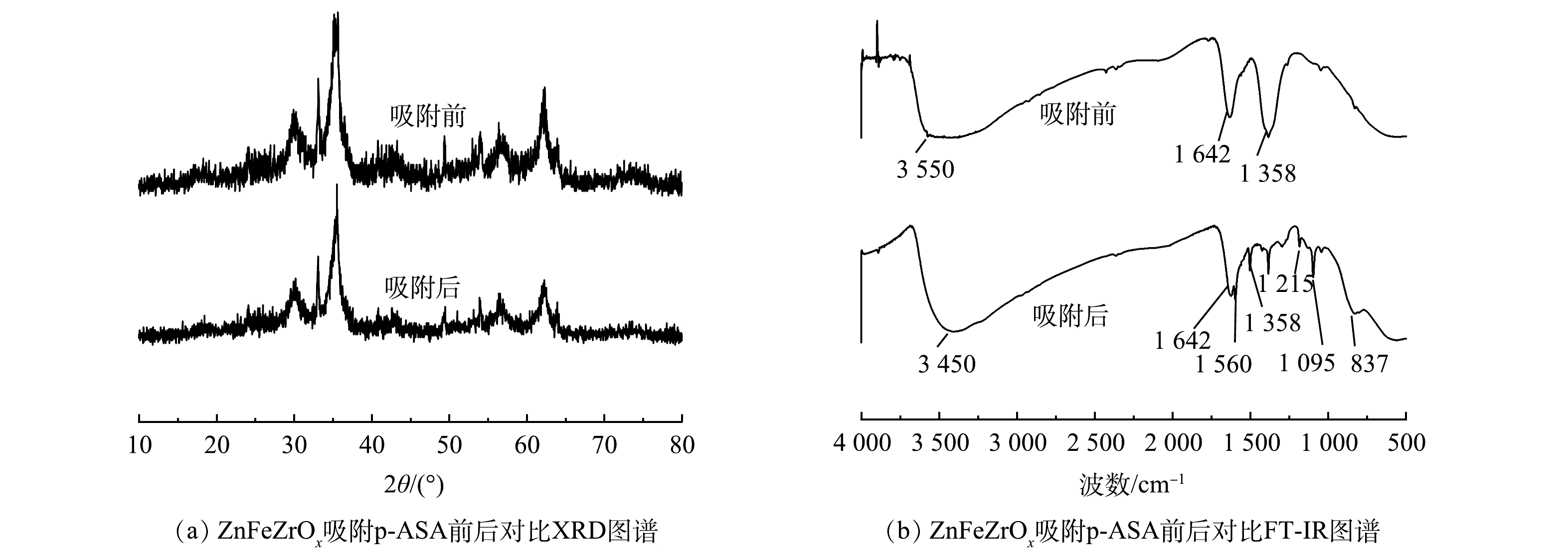
不同因素对ZnFeZrOx吸附p-ASA的影响
Figure 1.
Effect of different factors on adsorption of p-ASA onto ZnFeZrOx
