


 下载:
下载:


-
汞 (Hg) 是环境中生物毒性很强的金属污染物,具有持久性、易迁移性和高度的生物富集性等特点[1]。汞可通过地球化学循环和食物链富集,给人类和生态环境造成极大危害[2]。目前,燃煤电厂排放的汞是最大的人为汞排放源。作为以煤为主的能源消费大国,我国汞污染较为严重[3-5],每年煤炭燃烧向大气中排放810 t汞,占各种人为源汞排放总量的35%[6]。2013年,《关于汞的水俣公约》规定了汞的长期减排控制措施。我国现行的《火电厂大气污染物排放标准》也对汞的排放控制提出了明确限值[7],即自2015年起全面执行火电厂汞排放质量浓度不超过30 μg·m−3的规定。煤炭燃烧产生的汞有单质汞 (Hg0) 、氧化汞 (Hg2+) 和颗粒汞 (Hgp) 3种形态。Hg2+易溶于水,故可通过湿法脱硫设备去除[8];HgP易吸附在尘粒、飞灰颗粒表面,可通过除尘装置捕获去除。然而,Hg0因具有较高的挥发性 (2.46×10−1 Pa,25 ℃) 和较低的水溶性 (6×10−5 g·L−1,25 ℃) ,极易在大气中通过长距离运输而造成全球性汞污染,为最难控制的汞形态[9]。因此,有效控制Hg0是实现汞污染减排的关键。
催化脱除Hg0是一种行之有效的方法。传统选择性催化还原 (selective catalytic reduction,SCR) 催化剂 (V2O5-MoO3/TiO2和V2O5-WO3/TiO2) 在催化还原NOx的同时,可将Hg0氧化为Hg2+,并进一步利用后续脱硫装置进行协同脱除,从而提高设备经济性,因此被认为是应用前景良好的控制技术[10-11]。然而,SCR脱硝系统通常布置在高温、高尘、高酸性气体环境中,会降低催化剂的使用寿命[12]。由于其较高的工作温度,因而适用于非电行业的中低温催化氧化技术受到关注。目前,中低温钒钛SCR催化剂催化氧化Hg0,易受烟气组分 (如O2、NO、NH3、HCl、SO2、H2O) 和温度影响[13-16]。烟气中O2和NO可提供活性氧物种,从而促进Hg0的氧化。NH3会与Hg0竞争吸附催化剂活性位点,进而抑制Hg0氧化[17-20]。然而,烟气中SO2对Hg0氧化的影响机理还存在较大争议。有研究表明,SO2对Hg0的氧化表现为促进作用[21-22]。一方面,在O2存在的情况下,低浓度SO2会氧化生成SO3,与Hg0反应生成HgSO4;另一方面,SO2吸附在催化剂表面生成硫酸盐,可为Hg0氧化提供活性中心。然而,在某些情况下,SO2会与催化剂表面晶格氧反应生成硫酸盐和亚硫酸盐,使得催化剂表面活性氧位点减少,从而抑制Hg0氧化[23-26]。除此之外,多烟气组分共存时,Hg0的脱除机理尚不明确。
现有研究中,Cu作为活性组分,具有良好的氧化还原性[27]和抗硫性[28],添加到催化剂表面可极大地提升Hg0氧化效率。本研究以低温V2O5-MO3/TiO2脱硝催化剂为基础配方、Cu2O为改性组分,采用浸渍法制备Cu2O-V2O5-MoO3/TiO2催化剂,通过固定床反应器考察O2、NO、NH3、HCl、SO2、H2O等烟气组分对Hg0氧化性能的影响;并在此基础上,进一步探讨多烟气组分共存条件下Hg0的脱除机理,以期为SCR脱硝催化剂协同汞氧化提供参考。
-
所用催化剂由质量分数为3%的V2O5、质量分数为6% MoO3、Cu2O (质量分数为0~10%) 和TiO2组成。催化剂的制备步骤:称取定量偏钒酸铵、草酸、磷酸铵、钼酸铵、氧化亚铜和钛白粉溶于50 mL去离子水中,恒温水浴搅拌2 h,所得浆液置于105 ℃烘箱中3 h烘干水分。随后,将样品放入马弗炉中,在250 ℃空气气氛下焙烧1 h,之后在490 ℃下焙烧3 h,得到Cu2O负载量分别为0、1%、2%、6%、10%的Cu2O-V2O5-MoO3/TiO2催化剂,将其分别标记为0CuVMT、1CuVMT、2CuVMT、6CuVMT、10CuVMT。所有样品过60~80目 (0.180~0.250 mm) 筛备用。
-
使用美国Micromeritics公司生产的ASAP2020低氮吸附仪测定催化剂比表面积、孔容和孔径。其中,比表面积通过Brunauer-Emmett-Teller (BET)方法计算获得,孔容和孔径采用Barret-Joyner-Halenda (BJH)方法计算获得。
使用美国赛默飞世尔生产的Thermo Scientific K-Alpha型X射线光电子能谱仪进行X射线光电子能谱(XPS)分析,射线光源采用单色化AlKa源 (Mono AlKa,能量为1486.6eV) 。
H2程序升温还原(H2-TPR)实验在AutoChem II 2920型化学吸附仪(Micrometritics Co.)上进行。实验步骤如下:取50 mg样品(40~60目),在纯氧气氛下400 ℃预处理30 min;降至室温后用He吹扫15 min;然后通入体积分数为10% H2/Ar混合气,待仪器基线平稳后,以10 ℃·min−1速率升温至600 ℃,采用TCD检测耗氢量。
-
催化剂性能评价装置主要由模拟烟气、固定床反应装置、汞检测系统和尾气处理4部分组成 (见图1) 。其中,模拟烟气包括Hg0、O2、NO、NH3、HCl、SO2及平衡气Ar,烟气总流量为1 L·min−1。气体流量采用质量流量计精确控制。汞蒸气由置于恒温水浴锅中的汞渗透管产生,经载气Ar带出。固定床反应器使用内径4 mm石英管,中层添加石英砂作为支撑,采用管式电炉控制催化剂层反应温度。进出口Hg0浓度使用测汞仪 (RA-915+、LUMEX、美国) 进行监测,进出口NO和N2O体积分数由质谱仪 (DECRA、Hiden Analytical Ltd.,英国) 进行监测,SO2体积分数采用Testo 350烟气分析仪(Testo Co., Germany)检测。实验尾气经净化装置处理后排空。为避免汞蒸气沉积于管壁及水蒸气冷凝,实验管路均使用聚四氟乙烯管连接,并用伴热带加热至120 ℃。
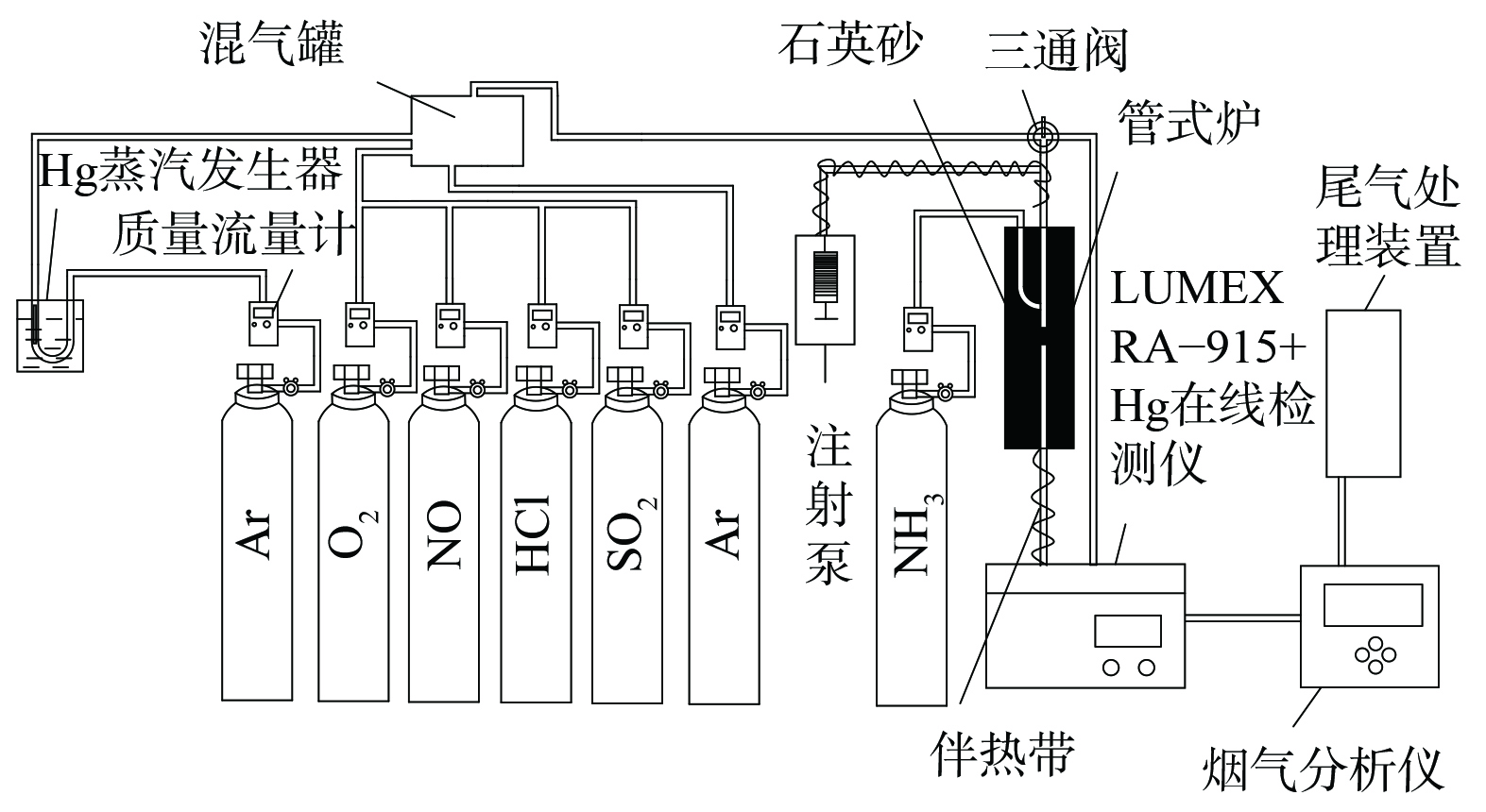
活性评价实验过程:称取50 mg催化剂置于石英管内,用石英棉固定两端;使用Ar吹扫管路,待基线稳定后,将模拟烟气切换至旁路,检测反应器进口Hg0的初始浓度;15 min后将模拟烟气切换至反应器,检测出口Hg0浓度。Hg0氧化率(EHg)和NO转化率(ENO)计算方法见式 (1)~(2) 。
式中:[Hg0]in和[Hg0]out分别为固定床反应器进口和出口Hg质量浓度,μg·m−3;[NO]in和[NO]out分别表示固定床反应器进口和出口NO的体积分数,%。
-
不同Cu2O负载量催化剂的NO转化率和Hg0氧化率见图2。由于实际工业烟气中Hg0质量浓度较低,SCR净化装置空速为3 000~8 000 h−1。为缩短Hg0和NO在模拟烟气中达到反应平衡的时间,本研究将脱硝实验的空速设置为30 000 h−1,汞氧化实验的空速设置为1 600 000 h−1。图2表明,在200 ℃时,不同Cu2O负载量的CuVMT催化剂的NO转化率表现为:0CuVMT(94.1%)>1CuVMT(91.2%)>2CuVMT(90.9%)>6CuVMT(88.5%)>10CuVMT(76.7%),而Hg0氧化率表现为:0CuVMT(64.1%)<1CuVMT(96.1%)<2CuVMT(99.9%)~10CuVMT(99.9%)。与0CuVMT和1CuVMT催化剂相比,2CuVMT催化剂的NO转化率分别降低了3.4%和0.32%,而N2O生成量没有明显提升,与此同时,Hg0氧化率分别提升了35.8%和3.8%。当Cu2O负载量超过2%之后,NO转化率继续降低,N2选择性也出现不同程度下降,而Hg0氧化率保持不变。因此,综合催化剂的NO转化率、Hg0氧化率、N2选择性及制备成本等几方面因素,最终选定2CuVMT催化剂作为脱硝协同汞氧化的最优配方。

-
工业烟气组分复杂,与Hg0相比,各烟气组分在气体浓度及分子偶极等方面占据优势,会优先与催化材料活性位点发生键合,进而严重影响Hg0的氧化。因此,有必要深入研究不同烟气组分对2CuVMT催化剂脱除Hg0性能的影响,进而阐明复杂组分下Hg0的脱除机理,所有反应时长均为10 h。
-
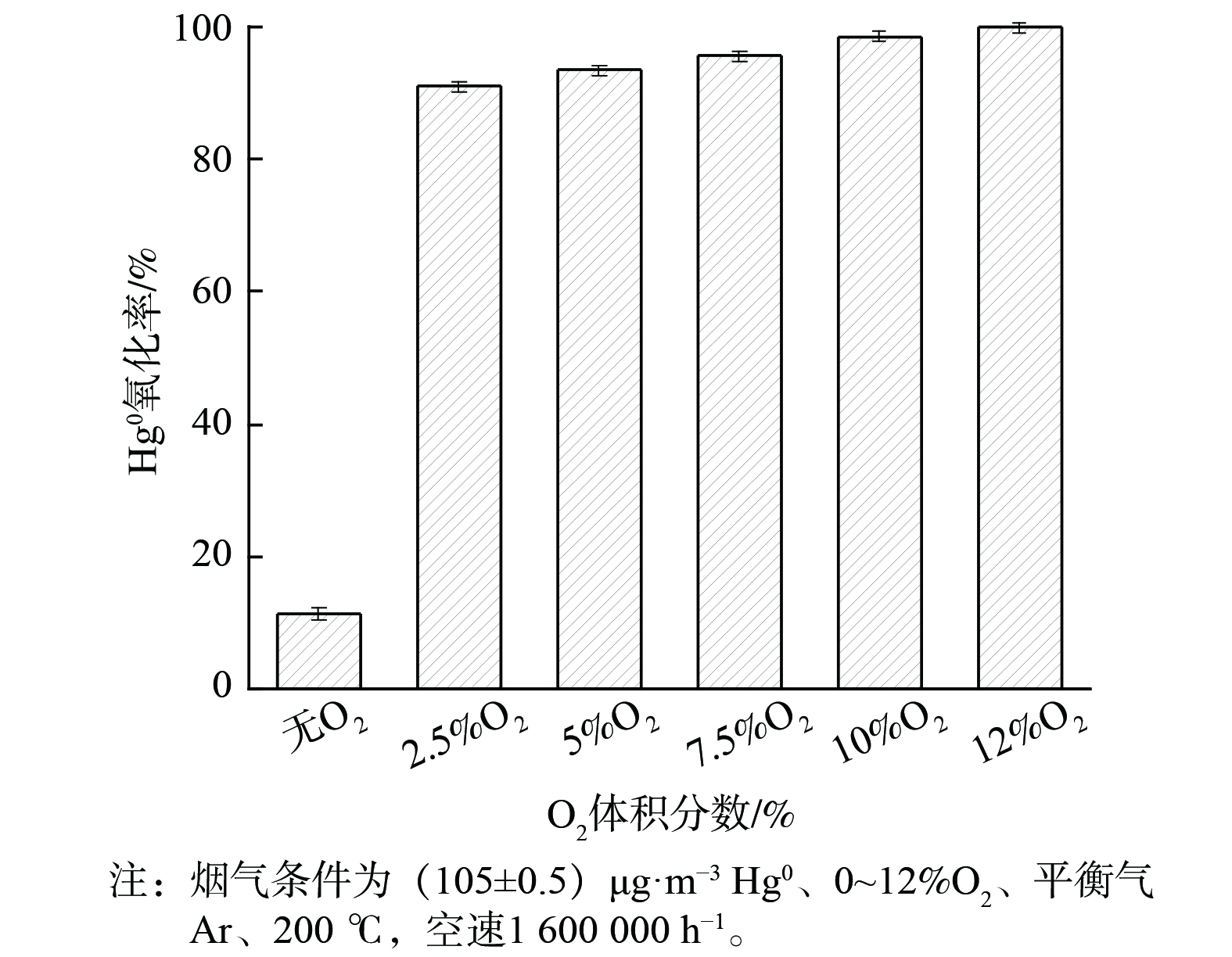
在200 ℃下,O2体积分数对2CuVMT催化剂Hg0氧化率的影响见图3。在无氧条件下,2CuVMT催化剂的Hg0氧化率仅为11.3%。此时,Hg0处于氩气惰性气氛下,物理吸附态的Hg0会与催化剂表面晶格氧结合[14,29],生成HgO。但由于晶格氧含量有限,Hg0氧化性能不高。当向反应体系通入2.5%的O2后,催化剂对Hg0的氧化性能显著提升,达到91.5%。这是由于O2可再生催化剂表面吸附氧和晶格氧,生成新的含氧活性位点,从而促进Hg0的氧化[30]。随着O2体积分数继续增至12%,Hg0氧化率也逐渐增至99.9%。
-
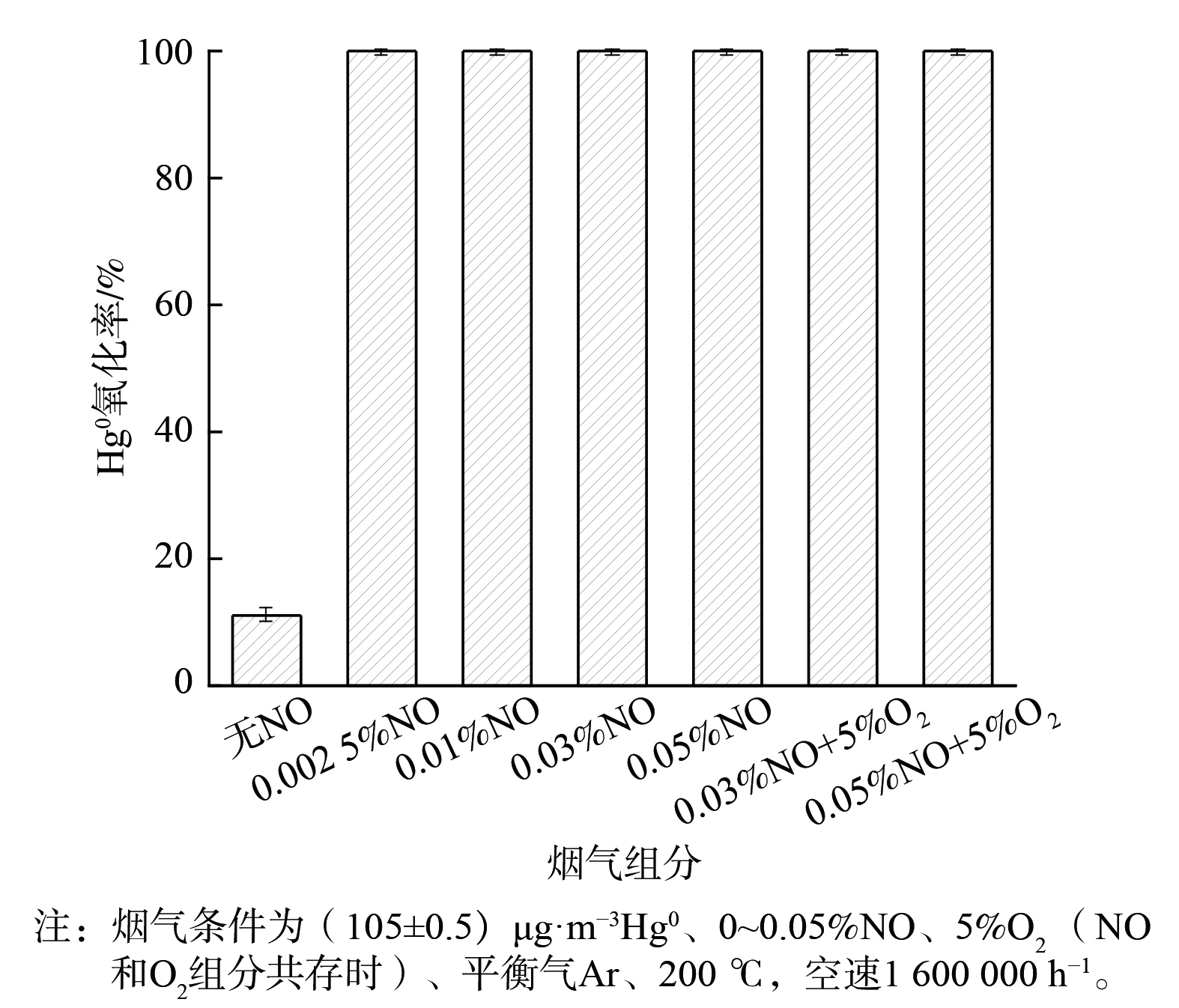
NO体积分数对2CuVMT催化剂Hg0氧化率的影响见图4。当0.002 5% NO通入反应体系后,Hg0氧化率立即升至99.9%;当NO体积分数增至0.05%时,Hg0氧化率依然稳定在99.9%。这可能是由于吸附态NO与催化剂表面活性氧反应生成具有氧化性的NO+、NO2一、NO2和NO3一等活性中间体,可将Hg0氧化为Hg(NO3)2[14,31],进而促进了Hg0氧化。当反应体系中继续添加5% O2后,O2可补充NO消耗掉的催化剂表面晶格氧[32],故Hg0的氧化率依然保持在99.9%。
-
NH3体积分数对2CuVMT催化剂Hg0氧化率的影响见图5。当烟气中加入0.005% NH3后,Hg0氧化率基本降至0。继续增加NH3体积分数至0.05%时,Hg0氧化率依然为0。这表明NH3对Hg0的氧化有强烈抑制作用。NH3会与Hg0发生强烈的竞争吸附[33],消耗催化剂表面晶格氧,从而抑制Hg0在催化剂表面发生氧化反应。反应过程如式 (3) 所示。

为探究SCR气氛条件对Hg0氧化率的影响,在Ar+NH3的气氛下,考察向烟气中添加体积分数为5%的O2,或添加混合气体 (含体积分数为5%的O2+体积分数为0.05%的NO) 对催化剂Hg0氧化率的影响。当体积分数为5%的O2加入烟气中时,2CuVMT催化剂Hg0氧化率显著提升。这说明O2补充了NH3消耗的催化剂晶格氧,并部分抵消了NH3对Hg0氧化的负面影响[34]。
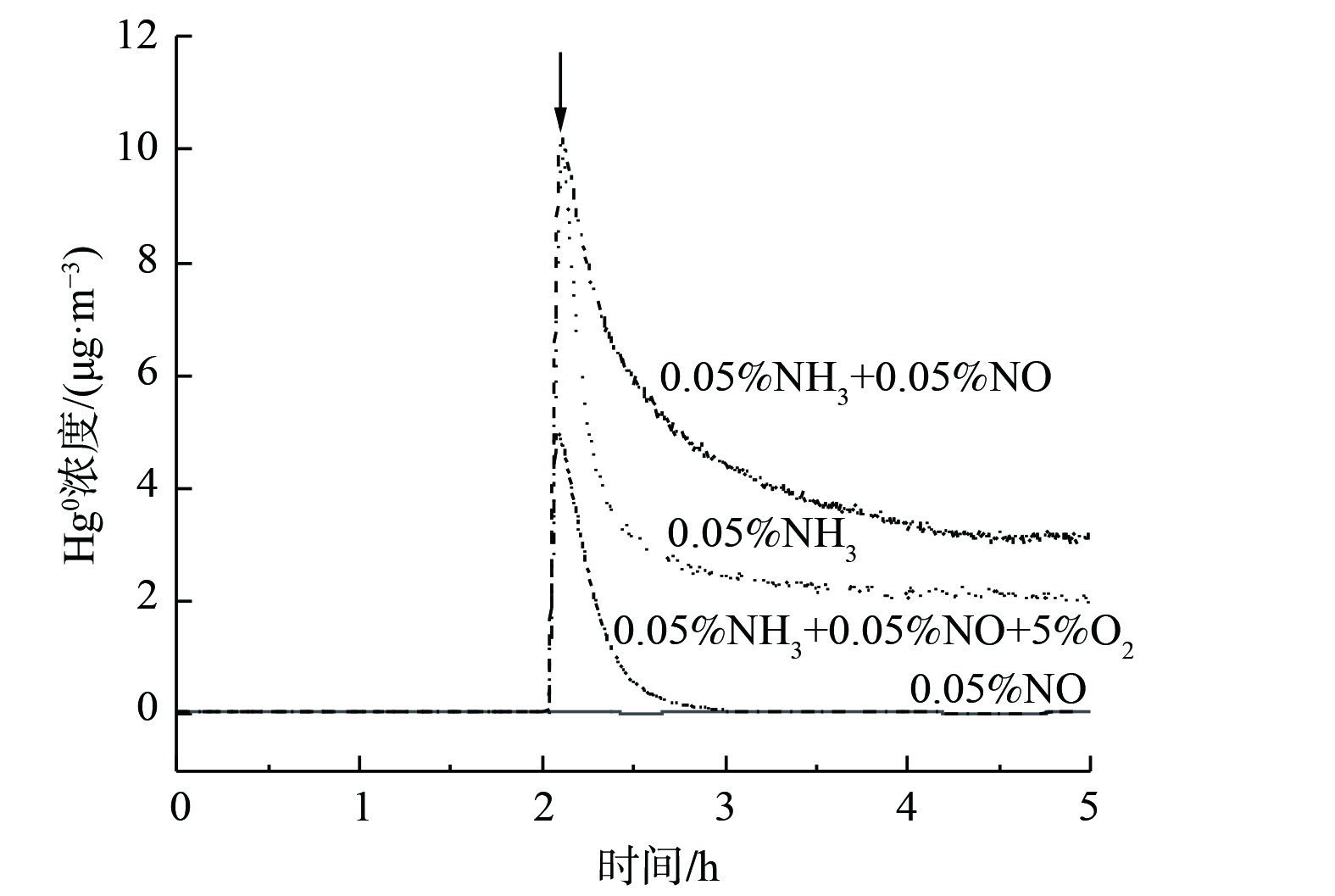
当NO、NH3和O2中的2种或3种组分以不同体积分数加入烟气时,各种添加方式下Hg0的氧化率表现为: (0.05% NO+5% O2) > (0.05% NH3+5% O2) > (0.05% NO+0.05% NH3+5% O2) > (0.05% NO+0.05% NH3) 。当烟气中通入混合气体 (0.05% NH3+0.05% NO) 时,Hg0氧化率为0。这表明此时NH3的抑制作用占主导地位,继续向烟气中加入5% O2后,Hg0氧化率升至57.3%。对比 (NH3+O2) 和 (NO+NH3+O2) 2种混合气体,反应体系中通入NO后,Hg0氧化率反而下降。这表明在此气氛下,发生了Hg2+被还原为Hg0的副反应。为探究副反应的发生机制,在200 ℃下通入混合气体 (10% O2+105 μg·m−3 Hg0+Ar) 3 h,使得催化剂表面沉积HgOx。之后用Ar吹扫,当Hg0质量浓度降为0后 (图6中箭头处) ,分别在烟气中通入0.05% NO、0.05% NH3、 (0.05% NH3+0.05% NO) 和 (0.05% NH3+0.05% NO+5% O2) 。这表明NH3和 (NO+NH3) 对HgOx存在还原作用,并且NO+NH3的还原性更强,而加入O2可在一定程度上抑制HgOx还原反应。因此,烟气中通入混合气体 (0.05% NH3+0.05% NO+5% O2) 后,仅有少量HgOx被还原。反应过程如式 (4)~(5) 所示。其中,x为1或1/2。
-
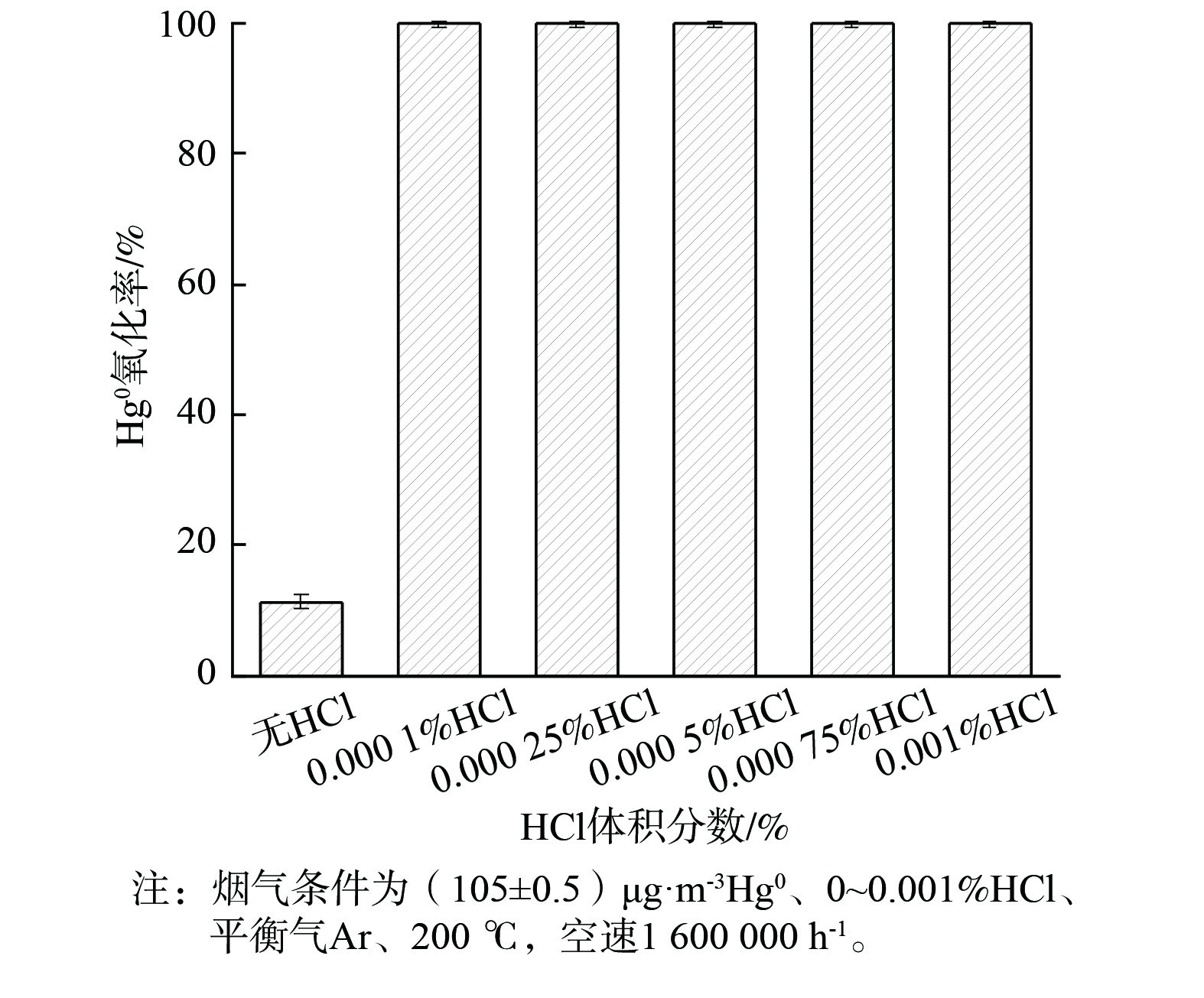
HCl是影响Hg0氧化反应的重要因素[35]。HCl的体积分数对2CuVMT催化剂Hg0氧化率的影响见图7。当体积分数为0.000 1%的HCl添加到烟气中时,Hg0氧化率即可达到99.9%。随着HCl体积分数逐渐增至0.001%时,Hg0氧化率稳定在99.9%。这表明HCl对Hg0氧化有明显促进作用。这可能是由于HCl在催化剂表面形成化学吸附态的活性Cl物种,可直接将Hg0氧化生成HgCl2[36]。
-
SO2和H2O对2CuVMT催化剂Hg0氧化率的影响见图8。当烟气中添加0.001% SO2时,Hg0氧化率可达到37.5%。随着SO2体积分数增至0.01%时,Hg0氧化率提升至52.1%。这表明SO2对Hg0氧化有一定促进作用。这可能是由于SO2和Hg0在催化剂表面发生竞争吸附的同时,在晶格氧的作用下,SO2和O* (晶格氧) 反应生成SO3和硫酸根,进而与吸附态Hg0反应生成HgSO3或HgSO4[23-24],从而有利于Hg0的去除。当烟气中同时存在SO2和O2时,Hg0氧化率明显下降,此时,SO2和O2反应生成SO3,加快了与催化剂活性组分生成CuSO4,使得催化剂硫酸化,最终导致催化剂活性位点数量减少、Hg0氧化率下降[27-28]。在实际SCR工况下,体系还会含有一定量水分。因此,考察在烟气中 (SCR条件:Ar+5% O2+0.05% NO+0.05% NH3+0.001%HCl) 加入H2O和SO2后对2CuVMT催化剂Hg0氧化率的影响。当烟气中加入体积分数为5%的H2O后,Hg0的氧化效率由99.9%降为97.3%。这是由于H2O和Hg0会竞争吸附催化剂表面活性位点,导致氧化率下降。继续向烟气中添加0.05% SO2后,催化剂Hg0的氧化效率降至95.1%。

-
反应温度是影响催化剂活性的重要因素。不同温度 (150~350 ℃) 对2CuVMT催化剂Hg0氧化率的影响见图9。随着反应温度的升高,2CuVMT催化剂Hg0氧化率呈现先平稳后降低的趋势。在150~250 ℃烟温范围内,Hg0氧化率保持在99.9%。然而,当温度升至300 ℃时,Hg0氧化率开始下降;在350 ℃时,Hg0氧化率进一步降至64.1%。参考文献[37]的研究结果,低温条件有利于Hg0吸附在催化剂表面参与氧化反应,而高温 (≥300 ℃) 则不利于Hg0在催化剂表面吸附,会使得参与氧化反应的Hg0质量浓度降低,从而导致Hg0的氧化被抑制。

-
1) BET分析。0CuVMT和2CuVMT催化剂的比表面积、孔容和介孔平均孔径见表1。在负载Cu2O后,催化剂的比表面积和孔容均呈下降趋势。其中,比表面积由0CuVMT的68.4 m2·g−1降至2CuVMT的57.3 m2·g−1,孔容由0.36 cm3·g−1 (0CuVMT) 略微降至0.33 cm3·g−1 (2CuVMT)。这表明添加Cu2O会堵塞催化剂部分孔道,导致催化剂比表面积和孔容降低。由于Cu2O堵塞了催化剂微孔,催化剂介孔平均孔径由0CuVMT催化剂的19 nm增至2CuVMT催化剂的21.3 nm。XU等[38]的研究结果表明,CuO/TiO2催化剂的比表面积、孔容、孔径与催化剂活性没有明显相关关系。
2) XPS表征。为确定催化剂表面O、Cu和V元素的化学价态,对CuVMT催化剂进行了XPS光谱分析,测定结果见图10。不同催化剂反应前后样品的O 1s XPS光谱 (图10 (a) ) 显示出2种特征峰。其中,结合能为530~530.3 eV的特征峰为晶格氧 (Oβ) [39],而在532~532.3 eV的特征峰归属于化学吸附氧 (Oα) [40]。当负载2%Cu2O后,催化剂表面的化学吸附氧 (Oα) 占比 (Oα/ (Oα+Oβ) ) 由31.9%逐渐升至36.5%。这表明化学吸附氧对于Hg0的氧化具有更高的活性[41]。
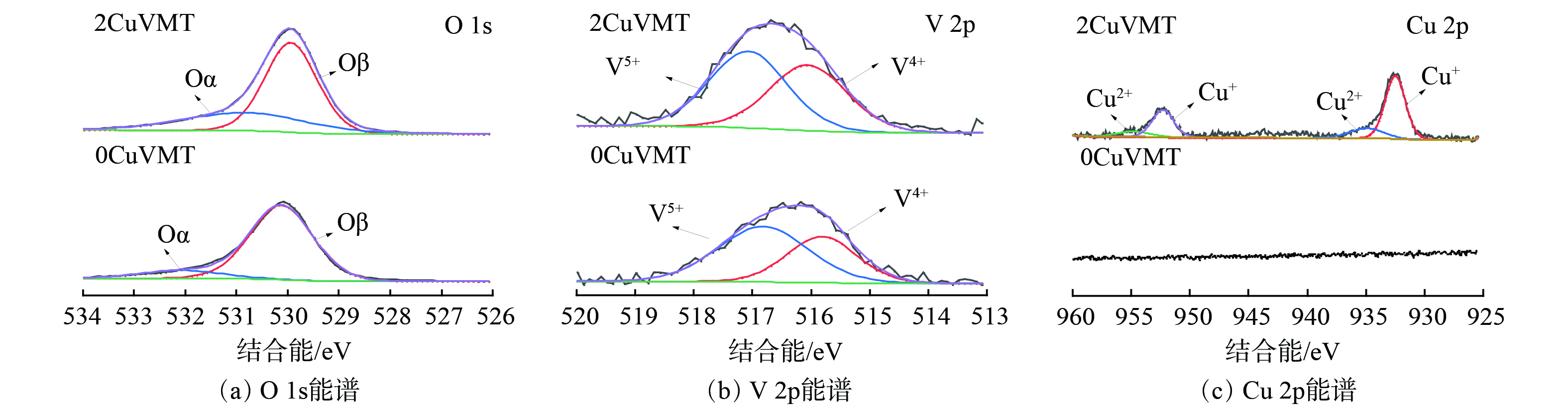
CuVMT催化剂样品的V 2p XPS光谱 (图10 (b) ) 可分为516.4 eV和517.3 eV两个峰,分别对应V4+和V5+的特征峰[42-43]。随着Cu负载量的增加,催化剂中V5+/V4+的比例由155% (0CuVMT) 降至117% (2CuVMT) 。这说明Cu2O改性提高了催化剂表面V4+的含量。这可能是由于Cu2O将部分V5+还原为了V4+。
CuVMT催化剂反应前后样品的Cu2p XPS光谱 (图10 (c) ) 表明,催化剂表面Cu的主要价态为Cu+和Cu2+。其中,Cu+为Cu元素主要的存在形态,其对应的结合能为932.5eV和952.2eV,而结合能在935.7eV和954.6eV的特征峰归属于Cu2+[44]。结合V 2p XPS结果,Cu2O改性使得催化剂表面V4+增加。这说明在制备过程中催化剂表面的Cu+和V5+确实存在相互作用,发生反应V4++Cu2+↔V5++Cu+,使得催化剂表面产生不饱和化学键和氧空位[41],可有效增加催化剂表面化学吸附氧含量,与O 1s XPS结果相一致。
3) H2-TPR表征。为阐明Cu改性对催化剂氧化还原性能的影响,对CuVMT催化剂进行了H2-TPR表征分析,测定结果见图11。0CuVMT催化剂在400~600 ℃出现了V5+和V4+的还原峰[45-46]。添加体积分数为2%的Cu2O改性后,催化剂原先V物种的还原峰大幅向低温方向偏移,且在154~263 ℃和300~400 ℃出现新的还原峰。其中,154~263 ℃的峰归属于Cu2+和Cu+的还原[47-48],300~400 ℃的峰为Cu-O-V物种的还原[47]。这表明添加Cu物种可增强催化剂表面Cu与V物种的交互作用,这与XPS结果相一致。总体来看,Cu改性后催化剂的还原峰整体向低温区迁移,H2的消耗量由0.036 mmol·g−1 (0CuVMT) 增至0.054 mmol·g−1 (2CuVMT) ,使得催化剂的氧化还原性能提高。

-
基于上述研究结果,本研究中Hg0的脱除过程主要分为吸附和催化氧化2个阶段。在吸附阶段,气态Hg0物理吸附在催化剂表面,反应过程如式 (6) 所示。Ads表示物质的吸附态。
Hg0(ads)与催化剂活性组分 (CuOx、V2O5) 中的晶格氧发生反应,生成HgO(ads)。与此同时,催化剂表面形成可由氧气补充的氧空位,反应过程如式 (7)~(8) 所示(M表示Cu或V)。
当反应体系中通入NO时,NO可与催化剂晶格氧反应生成NO2等具有氧化性的活性中间产物,从而促进Hg0的氧化。反应过程如式 (9)~(12) 所示。
当HCl通入反应体系时,HCl与催化剂活性组分反应生成活性Cl物种,进而与催化剂表面吸附态的Hg0(ads)反应,生成HgCl2。反应过程如式 (13)~(17) 所示。
当反应体系中存在SO2时,SO2可与晶格氧反应生成SO3,进而在催化剂表面与Hg0 (ads)发生催化氧化反应,生成HgSO4。反应过程如式 (18)~(20) 所示。
-
1) 通过对传统V2O5-MoO3/TiO2催化剂掺杂Cu2O改性,提高了催化剂在低温条件下Hg0的氧化率,Cu2O负载量为2%时,催化剂具有较好的脱硝协同氧化Hg0性能。
2) 不同烟气组分对Hg0氧化率的影响分析发现,O2、NO、HCl、SO2对Hg0的氧化具有促进作用,而NH3会消耗催化剂表面晶格氧,从而抑制Hg0的氧化。在多组分烟气条件下,Hg0氧化率表现为ENO+O2>ENH3+O2>ENO+NH3+O2>ENO+NH3。在NH3和 (NO+NH3) 两种气氛下,会将HgOx还原为Hg0,使得Hg0氧化率降低。
3) 随着温度由150 ℃升至250 ℃,Hg0氧化率一直维持在99.9%,进一步升温至350 ℃,Hg0氧化率则大幅下降。
4) 结合BET、XPS和H2-TPR分析,经过Cu2O改性后部分催化剂表面微孔被堵塞,催化剂表面存在Cu和V的相互作用,使得催化剂表面产生不饱和化学键和氧空位,有效提升催化剂表面化学吸附氧含量和低温氧化还原性能,促进Hg0氧化。此时催化剂表面的氧化反应遵循Mars-Maessen机理,即Hg0优先吸附在催化剂表面与晶格氧发生反应。
图 1
催化剂活性评价装置
Figure 1.
Catalyst activity evaluation device
