

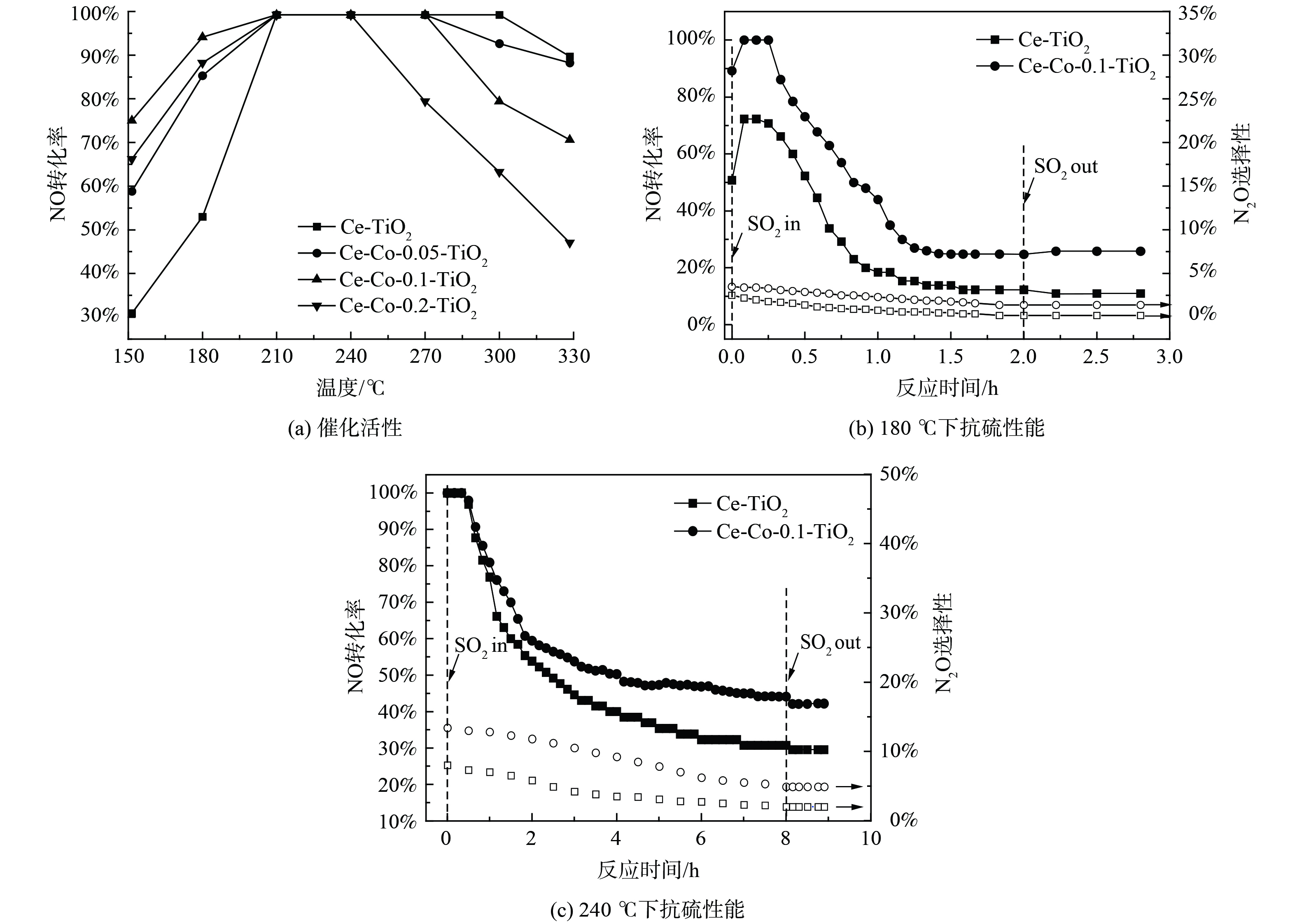
图 1
Ce-(Co)-TiO2催化剂的NO转化率及抗硫性能
Figure 1.
NO conversion and sulfur resistance of Ce-(Co)-TiO2 catalyst
| Citation: |
WANG Jianhui, ZHAO Yueju, SUN Wei, TENG Jilin, WANG Guogang, MIAO Wenhua. Experiment and its device for artificial simulated fog-haze environment[J]. Chinese Journal of Environmental Engineering, 2017, 11(9): 5130-5137. doi: 10.12030/j.cjee.201609073

|
使用NH3的选择性催化还原 (NH3-SCR) 已被广泛用于去除固定污染源烟气中的氮氧化物[1]。基于VO5-WO3/TiO2的商用SCR催化剂最常被用于NH3-SCR系统。然而,由于安装空间有限,高工作温度 (>300 ℃) 限制了这种催化剂在工业窑炉系统中的使用。因此,低温SCR催化剂已被广泛研究。在这类催化剂中,氧化铈具有高储氧能力 (oxygen storage capacity) ,且Ce4+与Ce3+之间能实现可逆转换[2],因此是一种重要的脱硝催化剂活性组分。近年来,已有多种低温NH3-SCR的氧化铈基催化剂被开发,包括CeO2/TiO2基[3]、CeO2/Al2O3基[4-5]、CeO2-MnO2 [6-7]。这些催化剂均可在中低温 (<300 ℃) 下表现出较高的脱硝活性和N2选择性。
烟气中SO2组分的存在会在短时间内对低温脱硝活性造成不可逆转的毒害作用,使其失活。许多研究者对SO2对Ce/TiO2催化剂在中低温下的中毒机理进行了深入研究。ZHANG等[8]发现CeO2的硫酸化减少了Ce-O-Ti活性位数量,进一步导致催化剂在300 ℃下活性降低;XU等[9]使用DRIFT、SO2-TPD等表征技术对Ce/TiO2在300 ℃下的中毒机理进行研究,发现前12 h内活性下降是由于硫酸铵盐的沉积导致,后36 h是由于Ce(SO4)2和Ce2(SO4)3的生成;DONG等[10]在250 ℃下对V/Sb/TiO2和V/Ce/Sb/TiO2催化剂进行了的抗硫中毒研究,发现Ce2(SO4)3的生成减少了表面硫酸铵盐的沉积,故其抗硫性能得以提升;XU等[11]发现在200 ℃下CeO2-WO3/TiO2催化剂的失活是由于硫酸铵盐和硫酸铈的沉积共同导致。因此,硫酸铵盐和硫酸铈的沉积是铈基催化剂在含硫SCR气氛下失活的主要原因。少量研究者对不同温度下的SO2中毒进行了研究,HUANG等[12]研究了在SO2和H2O共同存在的气氛下,不同反应温度下Mn-Fe/MPS催化剂的失活情况,发现在170 ℃时脱硝活性比190 ℃时下降得更快,但详细的机制还未被揭示。MA等[13]研究了Fe-Cu /CNTs-TiO2催化剂在不同温度下的失活情况,发现在150、200和250 ℃下不同的活性演变可归结为(NH4)2SO4的沉积,并降低了NH3的吸附、NO的吸附和氧化能力。 XU等[14]也在300和350 ℃下进行了SO2气氛下的NH3-SCR实验,结果表明较低的反应温度增强了SO2对脱硝活性的负面影响,但是其机理并未被进一步揭示。
为探究Ce-TiO2催化剂在不同温度下的不同中毒机制和反应机理,采用溶胶-凝胶法制备了Ce-TiO2 (Ce/Ti摩尔比为0.25) ,并在SO2存在的气氛下对催化剂进行不同温度 (180 ℃、240 ℃) 下的抗硫中毒测试,结合不同时间下的中毒活性演变、硫组分定量测试及一系列的表征测试,得出催化剂在不同温度下的不同中毒机制以及反应机理,以期为了解SO2中毒效应与反应温度之间的内在联系,以及Ce-TiO2催化剂在SO2气氛下的失活机理及其活性改善提供参考。
采用溶胶凝胶法制备Ce-TiO2及Ce-Co-TiO2催化剂,具体流程如下:取适量硝酸铈、硝酸钴、无水乙醇、硝酸、去离子水倒入烧杯,配置成A液并充分搅拌;取适量钛酸四丁酯、无水乙醇、冰醋酸倒入烧杯,配置成B液并充分搅拌,随后将A液缓缓滴入B液并持续搅拌,滴定完成后继续搅拌3 h;随后室温静置12 h后形成透明凝胶,之后置于80 ℃的烘箱中干燥24 h直至形成固体粉末;最后置于450 ℃的马弗炉煅烧6 h,研磨至40~80目,装样密封保存。
催化剂的活性评价与抗SO2中毒实验都在内径为18 mm的固定床反应管内进行。反应温度为150~330 ℃,称取0.2 g催化剂粉末并置于反应管内床层上方。本实验模拟烟气组成为:1 340 mg∙L−1 NO、760 mg∙L−1 NH3、570 mg∙L−1 SO2 (抗硫测试) 、5% O2、N2作为平衡气、气体总流量为700 mL∙min−1 ,空速为42 000 h−1,各路气体首先经过预混器并进行充分混合,然后进入反应管内进行反应。经过反应后的NOx、N2O通过Testo350烟气分析仪、BedfontG200分析仪进行测试,催化剂的脱硝活性、N2O选择性由公式 (1) 和 (2) 计算。
| η=[NOx]out[NOx]in×100% | (1) |
| S=2[N2O]out[NH3]in−[NH3]out+[NOx]in−[NOx]out×100% | (2) |
式中:η表示脱硝活性,S表示N2O选择性。[NOx]out和[NOx]in分别代表反应器出口、入口测得的NOx质量浓度,[N2O]out代表反应器出口所测得的N2O质量浓度,[NH3]in和[NH3]out分别代表入口、出口的NH3质量浓度。Fresh-Ce-TiO2表示新鲜未中毒的Ce-TiO2催化剂;P-180-Ce-TiO2表示在180 ℃下SO2氛围中进行 NH3-SCR反应2 h后的V/TiO2催化剂;P-240-Ce-TiO2表示在240 ℃下SO2氛围中进行NH3-SCR反应8 h后的Ce-TiO2催化剂。
BET测试使用JW-BK112仪器在-196 ℃的整个相对压力范围内获得N2的吸附-解吸等温线,并通过BET方程和BJH方程从吸附/解吸等温线计算出比面积、孔体积和平均孔径。NH3-TPD测试使用彼奥德化学吸附分析仪:在常温下通入1%NH3/N2吸附1 h,再进行1 h的He吹扫,最后在He流中以10 ℃·min−1的升温速率将温度提高到900 ℃,利用热导检测器监测从催化剂上解吸的NH3。H2-TPR分析仍使用彼奥德化学吸附分析仪进行:通入10%H2/Ar气流并以10 ℃·min−1升温速率升至900 ℃,通过检测器检测H2消耗量。XPS测试使用ESCALAB Mark II光谱仪,用Al KR辐射(1486.6 eV)观察表面成分的含量和化学状态,使用被污染的碳 (BE=284.6 eV) 对结合能进行了校正。XRD测试使用Philips X pert Pro衍射仪,扫描速度为5(°)·min−1;TG-DTG测试使用Netzsch热分析仪STA449C,50 mL·min−1氮气氛围下,升温速率为10 ℃·min−1。ICP测试采用 NexION1000G仪器进行测试:首先称取50 mg中毒后样品置于200 mL离心管中,加入一定量去离子水,经过1 h超声、过滤、定容后进行ICP测试。原位漫反射红外傅里叶变换光谱 (in-situ DRIFTS) 测试是在NicoletNexus 5700 FTIR光谱仪上进行的,该光谱仪配备了1个Harrick IR池和1个MCT检测器。
图1为Ce-TiO2催化剂及不同含量的Co掺杂改性后催化剂的活性和抗硫性。Ce-TiO2催化剂在150 ℃下脱硝活性仅30.8%,随着反应温度升高至210 ℃,脱硝活性上升至100%,进一步升高至330 ℃,脱硝活性反而下降至89.7%。这可能是由于高温下NH3发生了过度氧化,进一步产生NO,降低了脱硝反应活性[15]。当Co掺杂比例为Co/Ti为0.1时,150 ℃下脱硝活性升高至75%,低温活性大大增加。随着温度升高至300 ℃,脱硝活性下降至79.4%。分析在不同反应温度下,SO2对Ce-TiO2和Ce-Co-0.1-TiO2催化剂的活性和N2O选择性影响:在180 ℃下,未通入SO2时,Ce-TiO2催化剂的脱硝效率达到50.7%,N2O选择性为2.5%,通入1 h质量浓度为570 mg∙L−1的SO2后,NO转化率下降至18.4%,并在接下来的1 h内缓慢降低至12.5%,N2O选择性缓慢降低至0.2%,且切断SO2后活性无明显变化。在Co掺杂后,1 h内活性下降至44.1%,并在接下来的1 h内降至24.8%,N2O选择性缓慢降至1.4%。在240 ℃下,SO2通入8 h后,Ce-TiO2催化剂活性缓慢降至30.9%,N2O选择性从8.1%降至2.0%,Co掺杂后催化剂活性下降至44.2%,N2O选择性从13.4%降低至4.9%,SO2切断后脱硝活性无明显变化。以上结果表明,SO2与NO的在催化剂表面的竞争吸附不是活性降低的主要原因,催化剂物化特性的改变导致了催化剂活性的永久丧失。同时,对比不同温度下的活性变化发现,反应温度越低,催化剂活性下降越迅速。这表明SO2对催化剂脱硝活性的中毒作用与反应温度有着密切联系。为更深入地研究反应温度与SO2中毒之间的关系,对不同温度中毒后的催化剂进行表征。
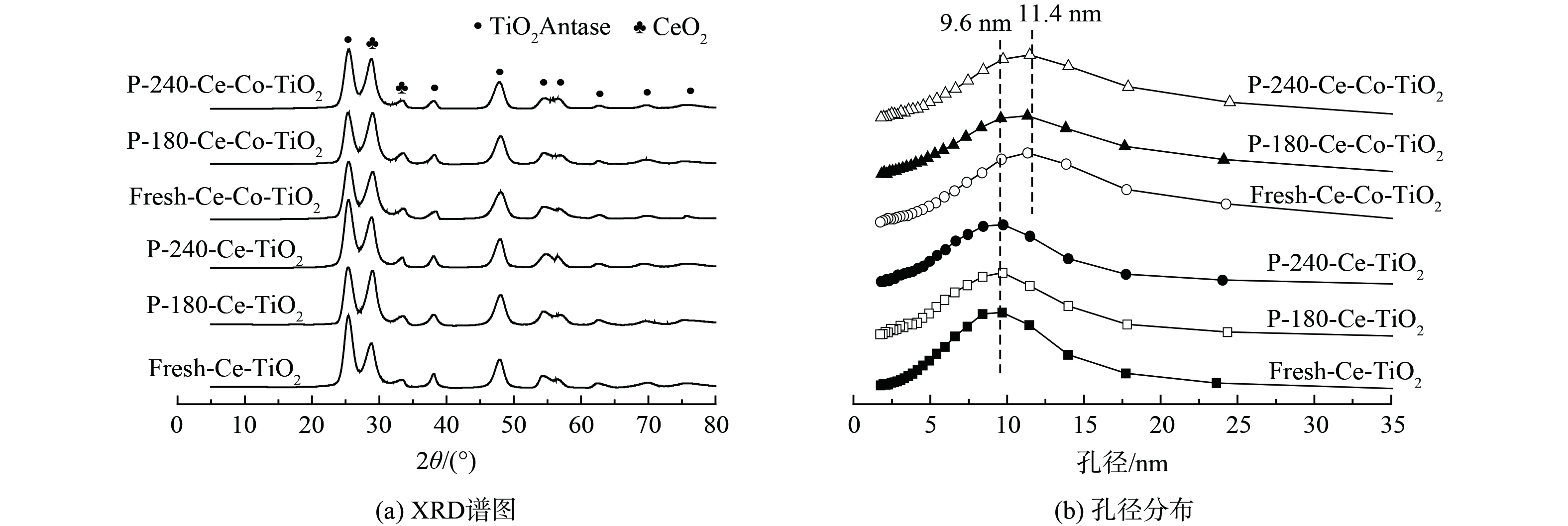
1) XRD和BET分析。XRD和BET结果见图2。新鲜Ce-TiO2催化剂含有锐钛型TiO2、CeO2晶相,经过180 ℃和240 ℃中毒后催化剂的晶相并未发生变化,无新的含硫晶相产生。Co改性后催化剂晶相并未发生变化,Co3O4晶相并未发现。这可能是由于Co3O4较为均匀分布在TiO2载体上,无法被X射线检测到,且中毒后催化剂晶相无明显变化。新鲜Ce-TiO2催化剂的最可几孔径为9.6 nm,中毒后孔径分布无明显变化;Co掺杂后催化剂的最可几孔径为11.4 nm,且中毒后无明显变化。表1为催化剂的结构参数。Co掺杂后催化剂比表面积从120.9 m2·g−1降至98.7 m2·g−1,且中毒后催化剂比表面积下降,孔容也下降。以上结果表明,Ce-TiO2及Co改性后催化剂中毒后部分介孔被堵塞,硫酸铵盐的沉积或硫酸铈的生成均有可能导致这一现象。
| 样品名称 | Sbet/(m2·g−1) | Vt/(cm3·g−1) |
| Fresh Ce-TiO2 | 120.9 | 0.24 |
| P-180-Ce-TiO2 | 109.4 | 0.20 |
| P-240-Ce-TiO2 | 95.1 | 0.18 |
| Fresh Ce-Co-TiO2 | 98.7 | 0.22 |
| P-180-Ce-Co-TiO2 | 91.6 | 0.20 |
| P-240-Ce-Co-TiO2 | 90.1 | 0.20 |
 DownLoad:
CSV
DownLoad:
CSV
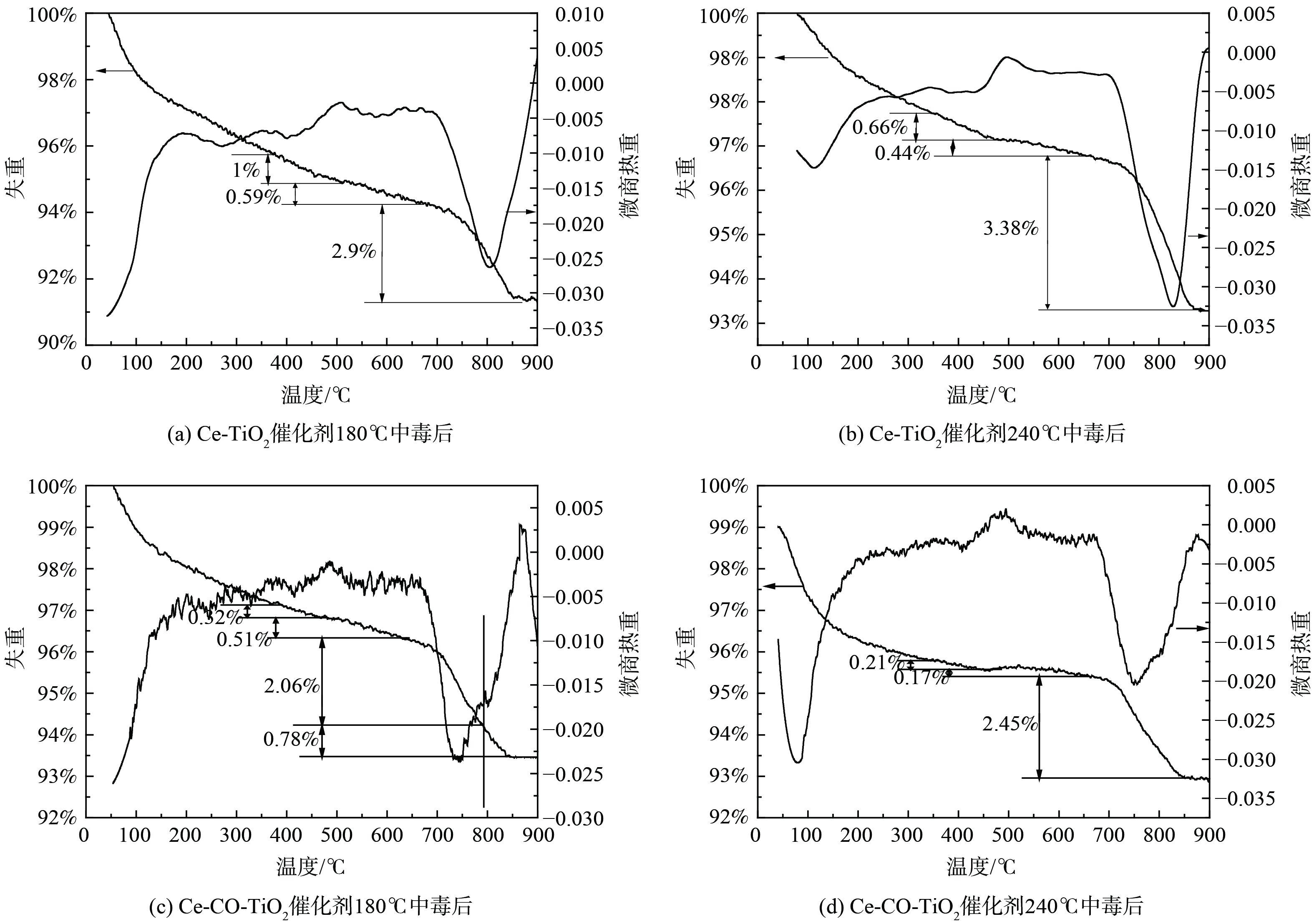
2) TG-DTG分析。为进一步获得不同温度下SO2中毒后催化剂上硫酸铵盐、硫酸铈的定量信息,不同温度中毒后催化剂进行了TG-DTG分析,结果如图3所示。中毒后催化剂出现了3个明显的失重峰,分别始于350 ℃、490 ℃和695 ℃。纯(NH4)2SO4在280 ℃开始分解为NH4HSO4,而纯NH4HSO4在380 ℃开始分解[16]。图3中第1个失重可归结为沉积的(NH4)2SO4的分解,第2个失重峰可以归结为NH4HSO4的分解,第3个失重峰可归结为硫酸铈、硫酸钴的分解。此外,在180 ℃下Ce-TiO2催化剂沉积的硫酸铵盐失重率为1.59%,随着反应温度上升至240 ℃,硫酸铵盐失重率降低至1.1%。该结果表明,铵盐的生成随着反应温度的增加而降低。随着Co的掺杂,催化剂沉积铵盐量在从1.59%降至0.83%。该结果表明,Co的掺杂抑制了硫酸铵盐的沉积。催化剂表面沉积的铵盐在升温过程中能释放SO2,并进一步与金属氧化物反应生成硫酸盐[17]。因此,中毒后TG-DTG的高温失重峰无法为硫酸铈含量提供定量信息。
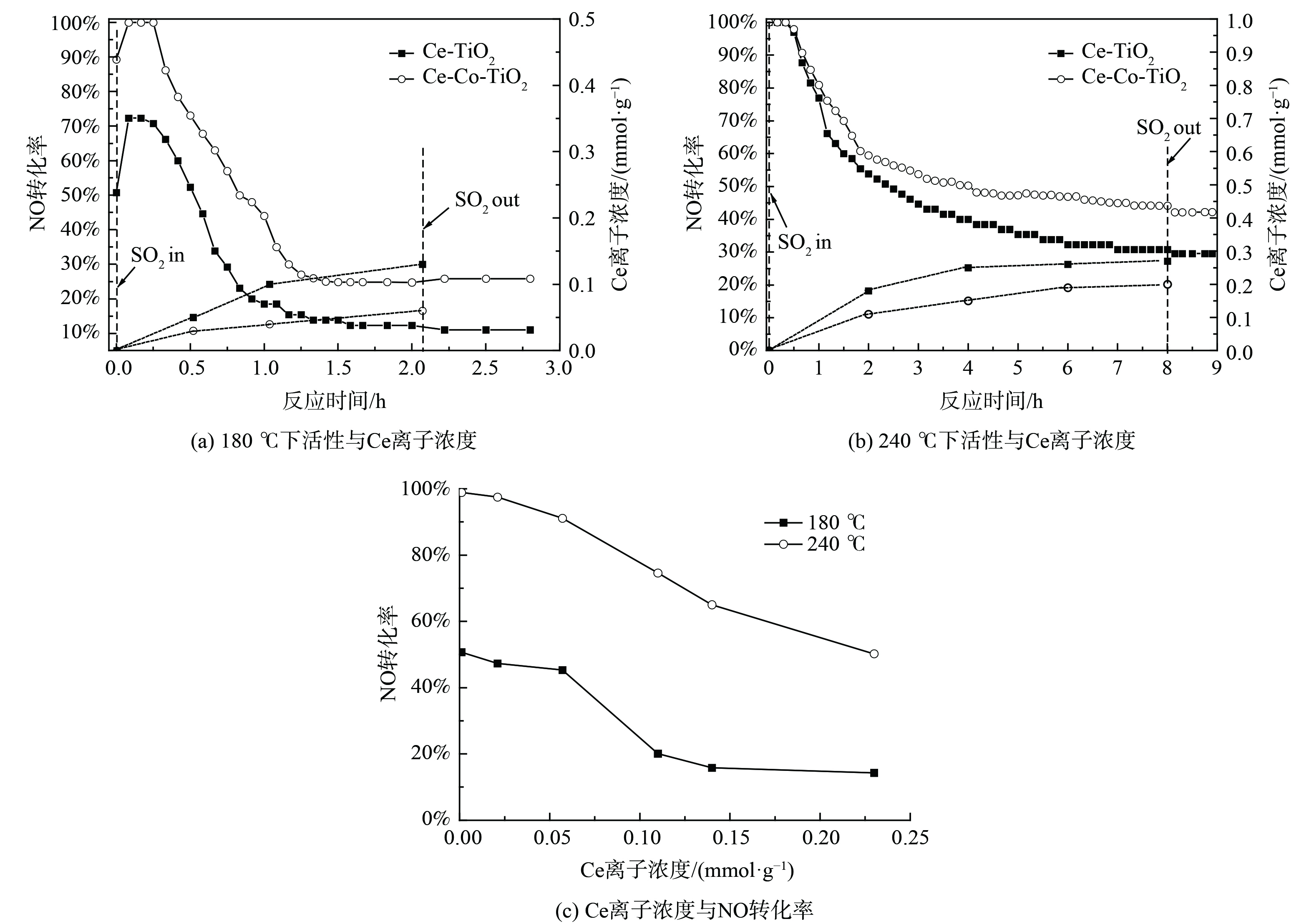
3) ICP分析。为进一步获得Ce-TiO2及Ce-Co-TiO2催化剂在不同温度下SO2中毒后催化剂上硫酸铈的定量信息,对水洗后溶液进行ICP-AES测试,结果如图4所示。在180 ℃下进行抗SO2测试,催化剂表面在最初1 h内迅速生成硫酸铈,ICP测试其Ce离子含量为0.1 mmol·g−1,经过Co掺杂后,1 h中毒后催化剂Ce离子含量下降至0.04 mmol·g−1。在240 ℃下,Ce离子含量明显增加,2 h后中毒后Ce离子含量为0.18 mmol·g−1,经过Co掺杂后,Ce离子含量下降至0.11 mmol·g−1。以上结果表明,随反应温度升高,Ce-TiO2催化剂生成的硫酸铈含量增加,Co掺杂后硫酸铈的生成被抑制。活性演变曲线表明,在180 ℃下催化剂活性迅速下降,在240 ℃下催化剂活性缓慢下降。不同温度下不同的活性演变可能揭示了不同的中毒机理及反应机理。为进一步探究180 ℃和240 ℃下Ce-TiO2催化剂不同的活性演变机理,对Ce-TiO2催化剂进行了不同时间的预硫化处理,并对预硫化后催化剂进行了ICP-AES测试和活性测试。在180 ℃ (图4 (a) 下,当硫酸铈含量达到0.1 mmol·g−1时,NO转化率对硫酸铈的沉积极其敏感,脱硝活性从50.7%迅速降至20.1%。当硫酸铈含量为0.1、0.13 mmol·g−1时,催化剂脱硝活性分别为18.5%、12.3%。因此,在180 ℃下,TG-DTG结果表明催化剂表面生成了大量的硫酸铵盐,但硫酸铵盐对Ce-TiO2催化剂低温脱硝活性的影响较小;随着1 h后硫酸铈的生成量达到0.1 mmol·g−1,低温脱硝活性快速降低;随后硫酸铈含量仍逐渐增加,低温脱硝活性则缓慢下降并最终保持在11%。随着温度升高至240 ℃,脱硝活性下降较为平缓。这可能是由于中温脱硝活性对硫酸铈的生成不是十分敏感,预硫后活性结果表明随硫酸铈含量的增加,催化剂中温活性逐渐降低,故推测在240 ℃下含硫气氛中活性的下降是由于硫酸铈的不断累积造成的。
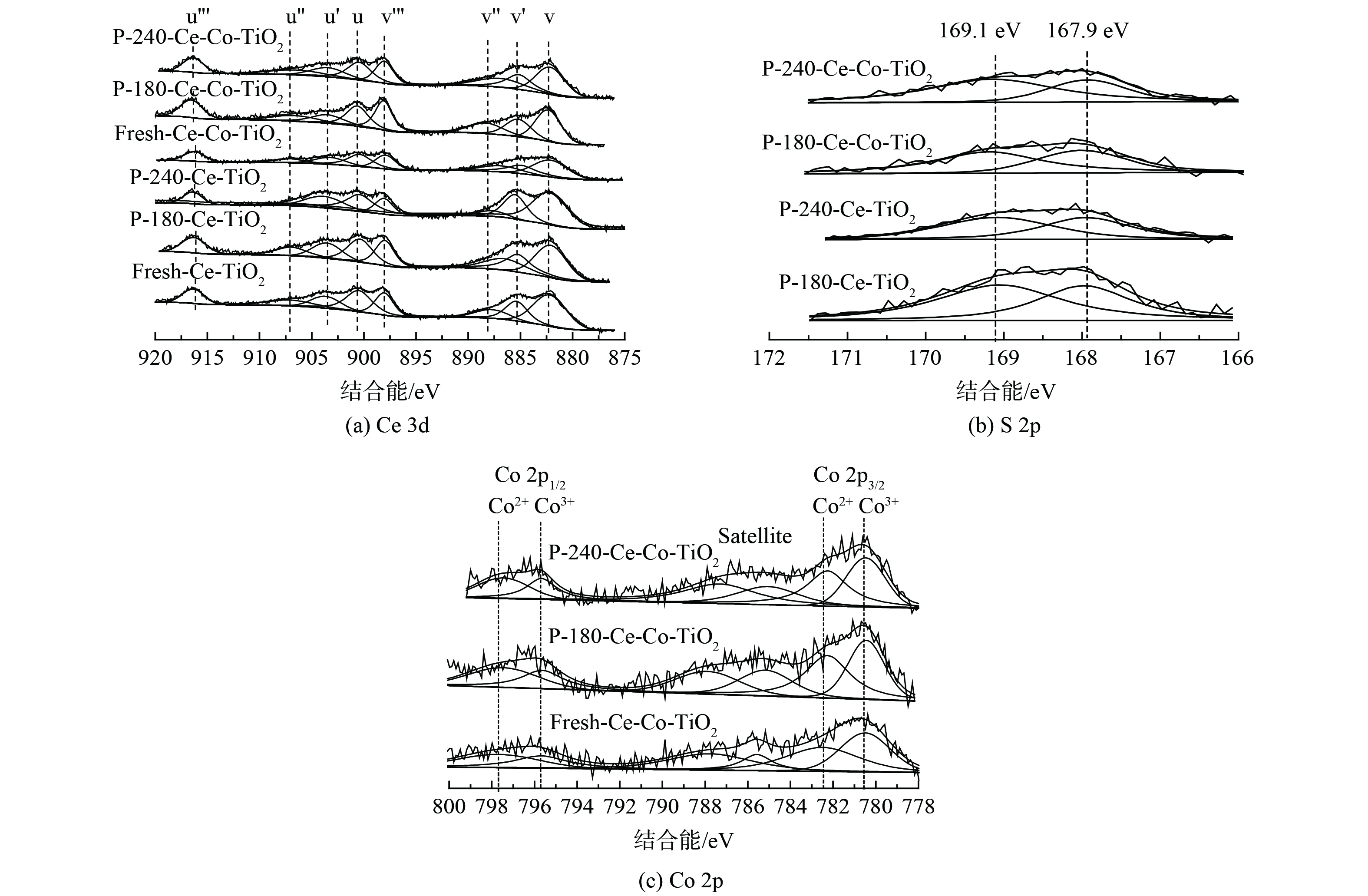
3) XPS分析。XPS图谱可确定催化剂表面元素形态与浓度。为探究不同温度中毒对催化剂元素含量及价态的影响,对中毒前后的Ce/TiO2催化剂进行了XPS测试。图5 (a) 为不同温度中毒前后Ce 3d XPS图谱,Ce 3d XPS图谱可被拟合为8个XPS谱峰,4对Ce 3d5/2/Ce 3d3/2谱峰,从高结合能到低结合能依次标记为u’’’ (916.7 eV) 、u’’ (907.3 eV) 、u’ (903.9 eV) 、u (900.9 eV) 、v’’’ (898.3 eV) 、v’’ (888.6 eV) 、v’ (885.8 eV) 、v (882.2 eV) 。其中,u’和v’峰归属于Ce3+,其余谱峰归属于Ce4+。经过SO2中毒后,v' 和u'所对应的峰相对强度逐渐增加,而v''和 u''所对应的峰相对强度逐渐减少。随着反应温度的升高,这种现象更加明显,该现象表明,随着中毒温度升高,催化剂表面Ce3+含量逐渐升高,这可能是由于表面Ce2(SO4)3的大量生成。图5 (b) 为不同温度中毒后催化剂的S 2p图谱。167.9 eV和169.1 eV的XPS峰分别对应于S 2p3/2和S 2p1/2,这表明S6+出现在中毒后的催化剂表面。进一步对催化剂表面元素组分进行定量分析,结果见表2。随着Co的掺杂,催化剂表面Ce3+含量从25.6%增至29.1%,中毒后表面S含量则分别从3.48%、4.23%降至3.02%、3.48%。该结果表明Co掺杂后催化剂表面的S含量降低,且Ce3+/Ce比例增加量也较小,这说明Co掺杂后表面Ce2(SO4)3的生成量减少。图5 (c) 为中毒前后Co 2p的XPS谱图,根据Co 2p1/2和2p3/2轨道分裂结合能差值15.2 eV及1比2面积比例进行分峰拟合,Co3+/Co比例见表2。新鲜Ce-Co/TiO2催化剂中Co3+/Co比值为48.1%,经过抗硫中毒实验后该比例下降至46.9%和45.8%。这可能是由于部分三价Co被硫酸化生成CoSO4。
| 样品名称 | S元素含量 | 比例 | |
| Ce3+/Ce | Co3+/Co | ||
| Fresh Ce-TiO2 | — | 25.6% | — |
| P-180-Ce-TiO2 | 3.48% | 34.5% | — |
| P-240-Ce-TiO2 | 4.23% | 41.9% | — |
| Fresh Ce-Co-TiO2 | — | 29.1% | 48.1% |
| P-180-Ce-Co-TiO2 | 3.02% | 32.4% | 46.9% |
| P-240-Ce-Co-TiO2 | 3.48% | 34.7% | 45.8% |
DownLoad:
CSV
脱硝活性尤其是低温活性依赖于催化剂的氧化还原性能。H2-TPR测试结果如图6 (a) 所示。Ce-TiO2催化剂只显示一个还原峰,始于270 ℃且还原峰中心在455 ℃,该峰归属于CeO2的还原[18]。在Co掺杂后,2个还原峰开始出现,分别位于356 ℃和537 ℃。该结果表明Co掺杂后促进了催化剂的氧化还原性能,并因此进一步促进了催化剂的低温脱硝活性。NH3的吸附长期以来被认为是脱硝反应的第一步,NH3-TPD测试结果如图6 (b) 所示。Ce-TiO2催化剂具有2个NH3脱附峰,分别位于235 ℃和575 ℃.一般认为200 ℃以下的脱附归属于弱酸位点,300 ℃以上脱附归属于强酸位点[19]。该结果表明Ce-TiO2催化剂表面具有较强的酸性。在Co掺杂后,催化剂的NH3脱附温度和脱附强度均无明显变化,说明酸性未发生明显改变。为进一步探究SO2在Co改性前后的Ce-TiO2催化剂的吸附情况,SO2-TPD测试结果如图6 (c) 所示。Ce-TiO2催化剂具有2个SO2脱附峰,分别位于200 ℃和585 ℃。在Co掺杂后,SO2脱附温度未发生改变,但峰强度明显降低。该结果表明Co掺杂后催化剂对SO2的吸附能力明显降低。图6 (d) 为Co改性前后催化剂的O1s图谱。O1s图谱在529.3 eV、531.1 eV左右两处均具有特征峰,分别归属于晶格氧Oβ、吸附氧Oɑ。值得注意的是,吸附氧Oɑ由于其更高的活动性在氧化还原反应中表现出比Oβ更高的活性,从而促进NO氧化为NO2,有利于反应活性的提高。因此,对Oɑ/(Oɑ+Oβ)比值进行计算,结果表明Co掺杂前Oɑ/(Oɑ+Oβ)为0.31,Co掺杂后该比值增加至0.43。文献[20]表明,吸附氧Oɑ可被归结于氧缺陷位或表面羟基基团,结合上述Co掺杂后Ce3+含量的增加,推断Co的掺杂导致了Ce4+向Ce3+的还原,在这一过程中晶格氧的去除导致了缺陷和氧空位的形成。

为进一步探究低温下硫酸铈的生成对催化剂反应机理的影响,利用原位红外技术 (in situ DRIFT) 探究催化剂反应机理的变化。首先Ce-TiO2和Ce-Co-TiO2催化剂经过180 ℃下的预硫化处理,然后放置于原位红外反应池中,预吸附NH3直至饱和后通入NO+O2,红外图谱如图7 (a) 和 (b) 所示。其中,1 184 cm−1处的振动峰可归结于吸附在L酸位点上的NH3,1 689 cm−1处的振动峰可归结于吸附在B酸位点上的NH3,1 600 cm−1可归结于NH3的过度氧化导致的硝酸盐中间产物的振动峰[21]。图7 (a) 表明,当通入NO+O2 10 min后,在1 305 cm−1处出现了新的吸附峰,这可以归因于吸附后形成的NO3−[22]。随NO+O2通入时间的增加,1 184 cm−1处的振动峰强度未发生变化。该结果表明预硫化后的Ce-TiO2表面吸附的NH3不具反应活性,脱硝反应E-R路径无法进行。图7 (b) 表明,对于预硫后的Ce-Co-TiO2催化剂,随NO+O2通入时间的增加,1 184 cm−1处的振动峰强度明显降低。该结果表明预硫后的Ce-Co-TiO2催化剂表面吸附的NH3具有反应活性,遵循E-R脱硝反应路径。

为进一步探究2种预硫后催化剂的L-H脱硝反应路径,预吸附NO+O2直至饱和随后通入NH3的红外实验被进行。图7 (c) 表明预硫后Ce-TiO2催化剂吸附NO+O2出现了1 238 cm−1、1 566 cm−1、1 600 cm−1的吸附峰,分别归属于单齿硝酸盐 (O-N-O) [23]、双齿硝酸盐 (-O-N-O-) [24]、吸附态NO2。随着NH3的通入时间增加,表面硝酸盐振动峰强度无明显变化,NH3的吸附峰开始出现。该结果表明吸附态的NH3与亚硝酸盐/硝酸盐在预硫后催化剂表面可以共存,预硫后Ce-TiO2催化剂不遵循L-H脱硝反应路径。如图7 (d) 所示,对于预硫后的Ce-Co-TiO2催化剂,1 331 cm−1和1 620 cm−1处的振动峰可分别归属于单齿硝酸盐 (O-N-O) 和NO2,当通入20 min的NH3进行反应,1 331 cm−1和1 620 cm−1处的振动峰完全消失。这表明预硫后Ce-Co-TiO2催化剂表面上吸附态的硝酸盐能与NH3反应生成NH4NO3,并进一步分解生成N2和H2O[25],预硫后Ce-Co-TiO2催化剂能够遵循L-H路径进行脱硝反应。
1) 在180 ℃下,由于低温脱硝活性对催化剂氧化还原性能具有较高的要求,较低含量硫酸铈的生成就会导致低温脱硝活性的急剧下降。随着反应温度升高至240 ℃,催化剂表面硫酸铈生成量明显增加,催化活性缓慢下降,中温脱硝活性对硫酸铈的生成敏感性较低,持续的硫酸铈生成导致了催化活性的持续缓慢下降。
2) Co改性能在一定程度上提升Ce-TiO2催化剂的脱硝活性及中低温抗硫活性。这主要是由于Co掺杂提升了催化剂的氧化还原性能,且抑制了SO2在催化剂表面的吸附,进而维持了催化剂E-R、L-H脱硝反应路径的进行。
| [1] | 宿志一.雾霾天气对输变电设备外绝缘的影响[J].电网技术,2013,37(8):2284-2290 |
| [2] | 赵莹.雾霾天气对复合绝缘子憎水性的影响[J].电瓷避雷器,2015(1):45-48 |
| [3] | 赵洁.雾霾天气对铁塔的腐蚀影响及防护实验研究[D].保定:华北电力大学,2015 |
| [4] | 张丹丹.模拟大气气溶胶对Q345钢腐蚀影响的研究[D].沈阳:沈阳理工大学,2015 |
| [5] | 刘腾宇. 汽油车尾气二次有机气溶胶生成的烟雾箱模拟[D].广州:中国科学院研究生院(广州地球化学研究所),2015 |
| [6] | 马军. 模拟雾霾对输电线外绝缘的影响及监测装置的设计[D]. 武汉:华中科技大学,2012 |
| [7] | 任凯锋, 李建军, 王文丽, 等.光化学烟雾模拟实验系统[J]. 环境科学学报, 2005, 25(11):1431-1435 |
| [8] | 武山, 吕子峰, 郝吉明,等. 大气模拟烟雾箱系统的研究进展[J].环境科学学报,2007,27(4):529-536 |
| [9] | BEHNKE W, HOLLÄNDER W, KOCH W, et al. A smog chamber for studies of the photochemical degradation of chemicals in the presence of aerosols[J]. Atmospheric Environment,1988, 22(6):1113-1120 |
| [10] | ALFARRA M R, PAULSEN D, GYSEL M, et al. A mass spectrometric study of secondary organic aerosols formed from the photooxidation of anthropogenic and biogenic precursors in a reaction chamber[J].Atmospheric Chemistry and Physics, 2006, 6(12):5279-5293 |
| [11] | 刘佳,尚静. 小型室内烟雾箱的设计和搭建[J]. 环境科学与技术,2015,38(8):191-195 |
| [12] | LIU C, CHU B W, MA Q X, et al. Effect of mineral dust on secondary organic aerosol yield and aerosol size inα-pinene/NOx photo-oxidation[J]. Atmospheric Environment, 2013,77:781-789 |
| [13] | 王坤,葛茂发,王炜罡. 大气光化学反应烟雾箱的建立及应用研究[C].第五届全国环境化学大会摘要集.2009:63 |
| [14] | 潘玉.广西南宁市邕检科技有限公司气溶胶发生器:CN203525677U[P].2013-08-02 |
| [15] | 王黎明,刘动,陈枫林,等. 雾霾模拟方法及其装置研究[J].高电压技术,2014,40(11):3297-3304 |
| [16] | 张小曳.中国不同区域大气气溶胶化学成分浓度、组成与来源特征[J].气象学报, 2014,72(6):1108-1117 |
| [17] | 周杨,华北地区气溶胶理化特性、来源解析及实验室模拟[D].济南:山东大学,2012 |
| [18] | 樊文雁,胡波,王跃思,等. 北京雾、霾天细粒子质量浓度垂直梯度变化的观测[J]. 气候与环境研究,2009,14(6):631-638 |
| [19] | 吴兑. 霾与雾的识别和资料分析处理[J]. 环境化学,2008,27(3):327-330 |
| [20] | 常清,杨复沫,李兴华,等. 北京冬季雾霾天气下颗粒物及其化学组分的粒径分布特征研究[J]. 环境科学学报,2015,35(2):363-370 |
| [21] | 贺克斌,杨复沫,段凤魁,等. 大气颗粒物与区域复合污染[M]. 北京:科学出版社,2011:201-374 |
| [22] | 王跃思,王莉莉. 大气霾污染来源、影响与调控[J]. 科学与社会,2014,4(2):9-18 |
| [23] | 赵秀娟,蒲维维,孟伟,等.北京地区秋季雾霾天PM2.5污染与气溶胶光学特征分析[J].环境科学,2013,34(2):416-423 |

Abstract: Effect of fog-haze on the work and human life was received more and more attention, however the research work was limited by the uncertainty of the actual fog-haze weather. To overcome this difficulty, this paper analyzed the haze ingredients in the typical regions, and got the haze formation method by tracing the roots, designed four unit simulation apparatuses of generating haze-fog, such as emissions of burning coal, emissions of burning oil(for example of motor vehicle exhaust), earth and dust, humidity of certain conductivity. A cylindrical chamber made of plexiglass was produced, which is three point two meters high and four meters in diameter. Six monitors were set up at different positions of chamber. Temperature, humidity, particle distribution and concentration were monitored by temperature and humidity sensors, particle spectromter, airborne particle counter and so on. In short, the system could achieve haze environments of different concentrations through adjusting simulation parameters, and could be used to the accelerated aging test.
WANG Jianhui, ZHAO Yueju, SUN Wei, TENG Jilin, WANG Guogang, MIAO Wenhua. Experiment and its device for artificial simulated fog-haze environment[J]. Chinese Journal of Environmental Engineering, 2017, 11(9): 5130-5137. doi: 10.12030/j.cjee.201609073
| 样品名称 | Sbet/(m2·g−1) | Vt/(cm3·g−1) |
| Fresh Ce-TiO2 | 120.9 | 0.24 |
| P-180-Ce-TiO2 | 109.4 | 0.20 |
| P-240-Ce-TiO2 | 95.1 | 0.18 |
| Fresh Ce-Co-TiO2 | 98.7 | 0.22 |
| P-180-Ce-Co-TiO2 | 91.6 | 0.20 |
| P-240-Ce-Co-TiO2 | 90.1 | 0.20 |
DownLoad:
CSV
| 样品名称 | S元素含量 | 比例 | |
| Ce3+/Ce | Co3+/Co | ||
| Fresh Ce-TiO2 | — | 25.6% | — |
| P-180-Ce-TiO2 | 3.48% | 34.5% | — |
| P-240-Ce-TiO2 | 4.23% | 41.9% | — |
| Fresh Ce-Co-TiO2 | — | 29.1% | 48.1% |
| P-180-Ce-Co-TiO2 | 3.02% | 32.4% | 46.9% |
| P-240-Ce-Co-TiO2 | 3.48% | 34.7% | 45.8% |
DownLoad:
CSV
| 样品名称 | Sbet/(m2·g−1) | Vt/(cm3·g−1) |
| Fresh Ce-TiO2 | 120.9 | 0.24 |
| P-180-Ce-TiO2 | 109.4 | 0.20 |
| P-240-Ce-TiO2 | 95.1 | 0.18 |
| Fresh Ce-Co-TiO2 | 98.7 | 0.22 |
| P-180-Ce-Co-TiO2 | 91.6 | 0.20 |
| P-240-Ce-Co-TiO2 | 90.1 | 0.20 |
| 样品名称 | S元素含量 | 比例 | |
| Ce3+/Ce | Co3+/Co | ||
| Fresh Ce-TiO2 | — | 25.6% | — |
| P-180-Ce-TiO2 | 3.48% | 34.5% | — |
| P-240-Ce-TiO2 | 4.23% | 41.9% | — |
| Fresh Ce-Co-TiO2 | — | 29.1% | 48.1% |
| P-180-Ce-Co-TiO2 | 3.02% | 32.4% | 46.9% |
| P-240-Ce-Co-TiO2 | 3.48% | 34.7% | 45.8% |
All rights reserved: Reesearch Center for Eco-Environmental Sciences,Chinese Academy of Sciences Copyright © 1997-2016
Address: Postcode: 100085 Phone: 010-62941074 E-mail: cjee@rcees.ac.cn
Supported by: Beijing Renhe Information Technology Co. Ltd Technical support: info@rhhz.net

