


-
多环芳烃是由2个或2个以上的苯环排列而成的多环化合物,不含杂质原子或者取代基[1]。这类化合物不易被生物降解,具有致癌、致畸和致突变的特性,对生物体有着很强的毒性[2]。随着社会工业的不断发展,许多工业废水中都含有多环芳烃[3]。这些废水排放后,对河流、湖泊和海洋造成污染,进而污染整个生态环境[4]。菲是一种能对人体健康造成严重危害的多环芳烃化合物,已被许多国家和地区列为20多种优先预防和消除的多环芳烃污染物[5]。目前去除多环芳烃的技术有吸附、光催化降解、氧化技术和生物修复等[6-9],其中吸附法由于选择性强、绿色、操作简单等特点而备受青睐,而其他的方法存在去除成本高、去除率低、二次污染严重等缺点。因此,开发易于分离、回收、快速和高效的吸附剂具有重要意义。
DNA由碱基、脱氧核糖和磷酸组成,并通过碱基对互补的氢键连接形成双螺旋结构[10]。DNA双链的特殊结构使其能够与平面小分子物质结合,1961年,LERMAN发现吖啶通过疏水作用、范德华力等非共价作用嵌入到DNA双链的碱基对中[11]。相关研究表明,一些平面小分子如黄曲霉毒素类[12-13]和多环芳烃类[14-16]以嵌入的方式进入DNA双链的碱基对中。吕嘉楠等[17]发现菲借助范德华力和氢键嵌入到DNA双链的碱基对中,在体外以嵌入的方式与DNA相互作用形成DNA-菲复合物。但DNA是水溶性分子,菲与其结合后不易将复合物从水中分离。而磁性纳米材料表面可修饰,并且具有磁响应性和生物安全性,常被用于DNA的分离和纯化[18-19]。因此,基于DNA与菲之间存在的嵌入作用以及磁性纳米颗粒与DNA的结合作用,将DNA磁性纳米颗粒作为一种新的吸附剂去除污水中的菲。
本研究考察了时间、菲的初始质量浓度和温度等因素对DNA结合菲的影响,以及对DNA磁性纳米颗粒去除菲的影响。优化了吸附参数,并且通过吸附动力学、等温线和热力学模型进一步分析DNA磁性纳米颗粒对菲的吸附机理。
-
菲(99%)标准品(坛墨质检科技股份有限公司);鲱鱼精子DNA(北京索莱宝生物科技有限公司);磁性纳米颗粒(MSi100-DNA-0507,英芮诚生化科技有限公司);本实验中使用的其他化学品均为分析纯级。使用三级蒸馏水制备溶液。
-
荧光分光光度计(Cary Eclipse,美国安捷伦科技有限公司);温控振荡器(IS-RDV3,美国晶体技术有限公司);紫外分光光度计(Cary-60,美国安捷伦科技有限公司);荧光显微镜(DM4B,德国徕卡公司);涡旋混合器(V2,中国沙海一恒有限公司)。
-
取20 μL 50 mg·mL−1磁性纳米颗粒悬浮液于EP管中,利用磁性分离架取出清液保留沉淀,加入500 μL 0.10 mg·mL−1 DNA溶液,与磁性纳米颗粒混合均匀。将其放入40 ℃,转速为300 r∙min−1的恒温摇床中混合240 min,之后利用磁性分离架将沉淀与溶液分离,得到DNA磁性纳米颗粒。DNA溶液的浓度根据式(1)计算。
式中:A为DNA在260 nm处的吸光度;ε为常数,6 600 L∙(mol∙cm)−1;b为比色皿的厚度,cm;c为DNA溶液的摩尔浓度,mol∙L−1。
根据初始DNA的浓度减去结合后清液中DNA的浓度,得到负载DNA的容量。计算得到DNA负载到磁性纳米颗粒的容量为3.45 μg·mg−1。
-
1)菲与DNA结合的实验。用无水乙醇制备菲母液(100 mg·L−1),用母液和Tris-HCl缓冲溶液(pH=7.4)制备不同质量浓度的菲溶液,用Tris-HCl缓冲溶液维持体系的pH。将菲溶液(3 mL)与DNA溶液(1 mL)置于EP管中,放入300 r∙min−1的温控振荡器中振荡90 min,加入磁性纳米颗粒,使其结合溶液中的DNA-菲复合物,用磁性分离架将负载复合物的磁性纳米颗粒与溶液分离。将溶液转移至比色皿中,用荧光分光光度计测定溶液中菲的质量浓度。实验重复3次,以确保实验数据的准确性。
2)DNA磁性纳米颗粒对菲的吸附实验。取一定量的吸附剂放入EP管中,加入菲溶液(1 mL),移至300 r∙min−1的温控振荡器中振荡90 min。之后用磁性分离架将DNA磁性纳米颗粒与溶液分离,将溶液转移至比色皿中,用荧光分光光度计测定溶液中菲的质量浓度。实验重复3次。菲的去除率和吸附容量分别使用式(2)和式(3)计算。
式中:R为去除率,%;C0和Ci分别为菲的初始质量浓度和吸附后的质量浓度,μg·L−1;V0和Vi分别为吸附前和吸附后溶液的体积,L;m为吸附剂的质量,g;q为吸附剂的吸附容量,mg·g−1。
-
1)吸附动力学模型。用准一级动力学(式(4))、准二级动力学(式(5))和粒子内扩散模型(式(6))描述DNA磁性纳米颗粒对菲的吸附过程[20]。
式中:qe为平衡吸附量,mg·g−1;qt为t时刻的吸附量,mg·g−1;k1为准一级吸附速率常数,min−1;k2为准二级吸附速率常数,g·(mg·min)−1。
式中:ki为粒子内扩散速率常数,mg·(g·min0.5)−1;C为与边界厚度有关的常数。
2)吸附等温线模型。使用Langmuir(式(7))、Freundlich(式(8))和Dubinin-Radushkevich(式(9))模型进行了吸附等温线研究[21]。
式中:qe为平衡吸附量,mg·g−1;qm为最大吸附量,mg·g−1;Ce为吸附平衡时的质量浓度,mg·L−1;KL为Langmuir吸附平衡常数,L·mg−1;KF为Freundlich吸附平衡常数;n为无量纲因子;Kad为Dubinin-Radushkevich平衡常数,mol2·kJ−2;qs为理论最大吸附量,mg·g−1;ε为Polanyi电势,kJ·mol−1,通过式(10)计算。
式中:T为吸附绝对温度,K;R为理想气体常数,8.314 J·(mol·K)−1;根据Dubinin-Radushkevich等温线方程,可以得到平均吸附能E (kJ·mol−1),通过式(11)计算。
3)吸附热力学模型。为了有效的评价吸附剂的吸附性能,根据热力学参数:吉布斯自由能(ΔG)、焓变(ΔH)和熵变(ΔS)提供的信息,了解吸附机理。吸附热力学模型如式(12)~式(15)所示[22]。
式中:ΔG为吉布斯自由能,kJ·mol−1;ΔH为焓变,kJ·mol−1;ΔS为熵变,J·(mol·K)−1。
-
1)时间对结合率的影响。如图1(a)所示,随着时间的增加,DNA与菲的结合率不断升高,在菲初始质量浓度为150、200、250 μg∙L−1的体系中,50 min时结合率分别为95.47%、93.46%、91.14%,之后趋于平衡。这是因为在反应前50 min溶液中DNA的质量浓度较高,之后大部分的DNA与菲结合,其结合菲的能力有所降低,结合率变化不明显,结合达到平衡。因此,50 min为DNA结合菲的最佳时间。
2)温度对结合率的影响。由图1(b)可知,温度为15 ℃时,菲的结合率最低,在35 ℃时增至最大,之后略微下降。这是因为温度较低时,体系中的分子活性被抑制,DNA与菲的结合效率下降。随着温度的不断升高,体系中分子活性得到提升,DNA与菲更容易结合[23]。体系温度高于35 ℃后,可能使DNA与菲之间的结合能力减弱,结合率下降。因此,DNA结合菲的最佳温度为35 ℃。
3)菲初始质量浓度对结合率的影响。如图1(c)所示,菲的初始质量浓度由100 μg∙L−1增加至350 μg∙L−1,结合率由96.41%降至79.92%。菲的初始质量浓度较低时,体系中DNA的结合能力较强,结合率较高。菲的初始质量浓度增加,DNA的结合能力不足,当体系中菲与大部分DNA结合后,剩余的菲与DNA的结合变得困难,结合率降低。
4)DNA质量浓度对结合率的影响。随着体系中DNA质量浓度的增加,结合率不断升高。如图1(d)所示,结合率从0.5 mg∙mL−1时的89.81%增长到了2.5 mg∙mL−1时的97.17%。DNA质量浓度较低时,体系中DNA的结合能力较低,溶液中未结合的菲较多,结合率低。DNA质量浓度增加,结合能力增强,结合菲的容量随之增加。
-
包括菲在内的大多数多环芳烃均具有荧光效应,当在荧光显微镜下被紫外线照射时,能观察到荧光斑点。用荧光显微镜观察DNA磁性纳米颗粒吸附菲前和吸附后的荧光变化情况,结果如图2所示。在吸附菲后(图2(b))的DNA磁性纳米颗粒上可以观察到明显的荧光斑点,说明菲已经吸附到DNA磁性纳米颗粒上。表明DNA磁性纳米颗粒能够有效吸附菲。
-
1)时间对吸附效果的影响。在35 ℃,pH为7.4的条件下吸附90 min后,实验结果如图3(a)所示。前10 min DNA磁性纳米颗粒对菲的吸附速度较快,随后去除率不断升高,50 min后吸附趋于平衡。吸附初始阶段,体系中有较多的嵌入位点。随着时间的增加,大部分嵌入位点被菲占据,体系中的位点不断减少,吸附速率变缓,吸附逐渐达到平衡[24]。因此,最佳的吸附时间为50 min。
2)温度对吸附效果的影响。在温度从15 ℃升高至35 ℃的过程中,菲的去除率不断增加,35 ℃后去除率降低,如图3(b)所示。温度升高,体系中的分子活性增加,吸附剂吸附菲的能力增强,去除效果较好[23]。温度高于35 ℃时,溶液中分子的运动速率加快且混溶性变高,可能引起菲的解吸,导致去除率下降[25]。因此,35 ℃为吸附的最佳温度。
3)菲初始质量浓度对吸附效果的影响。当菲的质量浓度由100 μg·L−1增加到350 μg·L−1,去除率由96.47%降至61.79%,如图3(c)所示。菲的质量浓度较低时,吸附体系中有充足的嵌入位点,能够有效的吸附菲。菲的质量浓度增加后,体系中的嵌入位点不足,当大部分位点被占据后,菲与剩余位点结合的概率降低,导致去除效果下降[26]。
4) pH对吸附效果的影响。当溶液pH为中性时,吸附剂对菲的去除效果最佳,偏酸或偏碱时,去除率降低,如图3(d)所示。溶液中pH的变化对DNA的磷酸骨架结构有一定的影响,进而影响对菲的去除效果[27]。当pH较低时,溶液中的氢离子含量增加,可能会导致吸附剂发生质子化的现象,导致去除效果下降[28]。pH较高时,溶液中较多的氢氧根离子可能会削弱吸附剂与吸附质之间的静电吸引力[29]。无论是在酸性或者碱性条件下,DNA磁性纳米颗粒对菲的去除效果均会降低。因此,吸附的最佳pH为6.9~7.4。
5)环境共存物质对吸附效果的影响。环境共存物质的存在会影响DNA磁性纳米颗粒对菲的吸附效果。吸附体系中环境共存物质的质量浓度见表1。一些常见的离子,如Ca2+、Mg2+和Cl−等对去除效果没有影响。但在吸附体系中加入十六烷基三甲基溴化铵(CTAB)和十二烷基硫酸钠(SDS)这类表面活性剂后,去除率大幅降低。可能是表面活性剂在溶液中产生胶束,菲在其胶束内核中聚集,从而影响DNA磁性纳米颗粒对菲的吸附效果[30]。并且表面活性剂对DNA的结构具有一定的破坏作用,其能改变溶液的界面张力,使DNA由拉伸状态被压缩成小球状态,这些变化使菲与DNA之间的结合变得困难[31]。在吸附体系中加入葡萄糖、尿素和氨基酸这类物质,则对去除效果没有影响。
6)与已报道吸附剂的吸附性能比较。已报道的其他吸附剂的吸附性能如表2所示。有机膨润土、石墨烯-生物炭复合材料等吸附剂对菲均有较好的去除效果,DNA磁性纳米颗粒对菲的去除率为96.47%,与氧化钙-活性炭纳米复合材料的去除率接近,但高于石墨烯-生物炭复合材料、TiO2-石墨烯复合材料等其他吸附剂。DNA磁性纳米颗粒达到吸附平衡的时间也远小于聚环糊精、石墨烯-生物炭复合材料等吸附剂。因此,DNA磁性纳米颗粒作为一种去除菲的吸附剂,具有潜在的应用价值。
-
1)吸附动力学。绘制了准一级动力学和准二级动力学模型曲线,如图4(a)所示。表3为动力学模型的相关参数。可知,准二级动力学(R2≥0.996 5)的R2优于准一级(R2≤0.952 8),并且由准二级动力学得到的平衡吸附量与实验实际测得的十分接近。因此,DNA磁性纳米颗粒吸附菲的过程遵循准二级动力学模型。粒子内扩散模型拟合如图4(b)所示,参数见表4。吸附剂吸附不同质量浓度菲的qt和t0.5的关系均为线性,且均没有通过原点,说明粒子内扩散不是唯一的限速步骤,还涉及到孔扩散和膜扩散过程[36]。
2)吸附等温线。等温线拟合如图4(c)所示,参数见表5。可知,Langmuir模型的(R2≥0.997 4)相关系数高于Freundlich模型(R2≤0.815 6),并且Langmuir模型在15、25、35 ℃得到的理论吸附量与实际测得的较为吻合。因此,DNA磁性纳米颗粒对菲的吸附符合Langmuir模型,表明吸附剂具有均匀的表面,吸附发生在单分子层[24]。Dubinin-Radushkevich模型拟合如图4(d)所示,参数见表6。平均吸附能E的大小反映了吸附的机制,如物理吸附(E<8 kJ·mol−1)、离子交换(8<E<16 kJ·mol−1)和化学吸附(E>16 kJ·mol−1)。实验得到的E≤3.44 kJ·mol−1,表明吸附剂吸附菲的过程存在物理吸附[21]。
3)吸附热力学。图5(a)为DNA磁性纳米颗粒吸附菲的lnKd与1/T的线性图。在35 ℃时,根据线性图5(a)的斜率和截距计算得到吸附剂吸附不同质量浓度菲的焓变(ΔH)和熵变(ΔS),结果如表7所示。可见,吸附过程中的ΔG为负值,说明吸附为自发进行,吸附实验具有可行性。随着菲的初始质量浓度不断增加,ΔG不断减小,表明体系中菲的质量浓度越大越不利于吸附的进行。吸附剂与吸附质之间主要存在物理和化学2种吸附方式,当ΔH在5~40 kJ·mol−1时主要是物理吸附,在40~125 kJ·mol−1时为化学吸附[37]。ΔH为正值表明吸附过程是吸热的,ΔH≤33.96 kJ·mol−1,小于40 kJ·mol−1,表明吸附过程涉及物理吸附。并且ΔS>0,表明固体吸附界面的随机性增加,菲被DNA磁性纳米颗粒吸附后,吸附体系更加混乱[38]。
-
碱性缓冲溶液能够破环DNA和磁性纳米颗粒之间的离子平衡,将DNA与磁性纳米颗粒分离,使磁性纳米颗粒得到重复利用。实验使用磷酸盐缓冲溶液(pH=12)洗涤DNA磁性纳米颗粒,用磁性分离架去除洗涤后的清液,重复洗涤5~6次。将洗涤后的磁性纳米颗粒重新与DNA结合,进行吸附实验。如图5(b)所示,重复进行6次后,制备的DNA磁性纳米颗粒对菲的去除率在90%以上,表明磁性纳米颗粒具有较好的重复利用性。
-
本研究基于DNA与菲之间存在的嵌入作用,证明了DNA结合菲的能力较强,随后将DNA与磁性纳米颗粒结合,制备了DNA磁性纳米颗粒,得出如下结论。
1)在35 ℃、pH=7.4、结合时间为50 min、DNA的质量浓度为0.1 mg∙mL−1、菲的初始质量浓度分别为150、200、250 μg∙L−1时,DNA与菲的结合率分别为95.47%、93.46%、91.14%,结合量分别为4.296、5.067、6.836 mg∙g−1。
2)其他条件相同时,在吸附剂用量为1 mg、菲的初始质量浓度分别为100、150、200、250 μg∙L−1时,DNA磁性纳米颗粒对菲的去除率分别为96.47%、95.61%、93.46%、88.03%,吸附量分别为96、143、187、220 μg∙g−1。水中的一些天然离子和其他物质对吸附剂去除菲的效果影响较小,但表面活性剂对去除效果影响较大。
3) DNA磁性纳米颗粒对菲的吸附符合准二级动力学模型和Langmuir模型,说明吸附剂表面均匀,吸附发生在单分子层。粒子内扩散模型表明吸附不仅仅受限于粒子内扩散。热力学参数表明吸附过程是自发和吸热的。
4)磁性纳米颗粒具较好的重复利用性,经过6次的吸附解吸后,再次制备的DNA磁性纳米颗粒对菲的去除率仍能达到90%以上。
DNA磁性纳米颗粒对水中菲的吸附性能
Adsorption performance of DNA magnetic nanoparticles to phenanthrene in water
-
摘要: 多环芳烃(polycyclic aromatic hydrocarbons, PAHs)作为一种致癌污染物,在水体中分布广泛且容易在生物体内富集,对人体健康有着严重的威胁。本研究将DNA与磁性纳米颗粒结合作为吸附剂,利用DNA与多环芳烃化合物的嵌入结合原理去除水体中的菲,分别考察了时间、菲的初始质量浓度、温度等因素对DNA结合菲的影响,以及对DNA磁性纳米颗粒去除菲的影响。结果表明:在35 ℃、pH=7.4、时间为50 min的条件下,在DNA的质量浓度为0.1 mg∙mL−1、菲的初始质量浓度分别为150、200、250 μg∙L−1时,结合率分别为95.47%、93.46%、91.14%。在相同条件下,DNA磁性纳米颗粒用量为1 mg、菲的初始质量浓度分别为100、150、200、250 μg∙L−1时,去除率分别为96.47%、95.61%、93.46%、88.03%,吸附量分别为96、143、187、220 μg∙g−1。DNA磁性纳米颗粒对菲的吸附过程符合准二级动力学模型和Langmuir模型,热力学参数表明吸附过程是自发和吸热的。DNA磁性纳米颗粒作为吸附剂可用于去除污水中的菲,以上研究结果可为基于PAHs-DNA嵌入结合作用处理污水中的菲提供参考。Abstract: Polycyclic aromatic hydrocarbons (polycyclic aromatic hydrocarbons, PAHs) are carcinogenic pollutants that are widely distributed in water bodies and easily enriched in living organisms, which are detrimental to human health. In the present study, DNA combined with magnetic nanoparticles was used as an adsorbent to remove phenanthrene from wastewater on the basis the principle of embedded binding of DNA with polycyclic aromatic compounds. The effects of time, initial mass concentration of phenanthrene, and temperature on the DNA-bound phenanthrene and phenanthrene removal by DNA magnetic nanoparticles were investigated. The results demonstrated that under the conditions of 35 ℃, pH=7.4, and 50 min, 0.1 mg∙mL−1 of DNA could lead to the binding rates of 95.47%, 93.46%, and 91.14% for phenanthrene with the initial mass concentrations of 150, 200, and 250 μg∙L−1, respectively. Under the same conditions, 1 mg of DNA magnetic nanoparticles could result in the removal rates of 96.47%, 95.61%, 93.46%, 88.03% and the adsorption amounts of 96, 143, 187, 220 μg∙g−1 for phenanthrene with the initial mass concentrations of 100, 150, 200, and 250 μg∙L−1, respectively. The adsorption of phenanthrene by DNA magnetic nanoparticles accorded with the pseudo-second-order kinetic model and the Langmuir model. The thermodynamic parameters indicated that the adsorption process was a spontaneous and endothermic one. DNA magnetic nanoparticles as an adsorbent can be used to remove phenanthrene from wastewater. This study can provide a reference for the treatment of phenanthrene in wastewater based on PAHs-DNA embedded binding interaction.
-
Key words:
- PAHs /
- phenanthrene /
- DNA magnetic nanoparticles /
- adsorption performance
-
抗生素应用于多个领域,主要涉及医药和畜牧饲料行业。由于抗生素的滥用,导致环境中抗生素污染问题普遍存在[1-3],目前,在水环境[4-6]、土壤[7-9]、水产动物[10]和植物[11]中均检测到了多种抗生素。青霉素G(PCN)是由青霉菌产生的一种β-内酰胺类水溶性抗生素[12],其可阻止肽聚糖的产生从而破坏细菌细胞壁的合成[13],是最具抗菌活性的抗生素,现已被广泛用于治疗人类和动物的疾病中[14]。PCN具有难以降解且含有生物毒性的特性,传统水处理方法难以完全对其产生作用,如果直接将其排放到水环境中,将会对生态环境以及人类构成较大威胁[15-16],因此,探索去除水环境中PCN的新方法十分必要。
O3氧化是一种清洁的水处理技术,且具有无二次污染和经济可行等特点[17],可作为强氧化剂,对污水中的难降解有机物进行降解[18]。有学者用O3氧化降解垃圾渗滤液[19]、有机氯农药[20]和布洛芬[21]等难降解有机物,结果表明,降解效果均十分明显。有研究[22-24]表明,将H2O2与O3联合时,H2O2会促进HO·的产生,从而使O3的利用率以及降解效果均可得到显著提升。陈炜鸣等[23]在采用O3降解垃圾渗滤液浓缩液的过程中,发现添加0.13 mol·L−1 H2O2能显著提升有机物的去除效果,且O3利用率提升了22.29%,同时废水可生化性得到了明显改善,BOD5/COD值由0.01提高到0.43。LI等[25]采用O3预处理氢化可的松制药废水,在H2O2/O3的摩尔比为0.3的条件下,反应15 min后,COD去除率可达67%,COD去除率相对于单一O3氧化体系提升了23%,证明添加适量H2O2可显著提高降解效果。虽然众多研究已经证明了O3和O3/H2O2法对难降解有机物的降解效果显著,但目前许多研究倾向于对工艺条件的优化,而对降解过程中的中间产物分析和降解规律的研究却相对较少。
基于此,本研究以难降解有机物PCN为目标,对其在O3/H2O2体系中的降解规律及其相关的机理进行研究,对降解过程中的中间产物及可能的降解路径进行探讨,并根据实验数据对降解动力学过程进行分析,为该法处理水中PCN的工程应用提供参考。
1. 实验材料与方法
1.1 实验试剂
实验试剂包括PCN(1 650 U·mg−1,阿拉丁)、H2O2(分析纯)、淀粉(分析纯)、甲酸(色谱级)、乙腈(色谱级)、NaOH(分析纯)、Na2S2O3(分析纯)、KI(分析纯)。
1.2 实验设备与仪器
自制反应器、微波快速消解COD测定仪(GZ-WXJ-Ⅲ)、pH计(pHS-3C)、液相色谱仪(Agilent-1200,美国Agilent公司)、液质联用色谱仪(WATERS TQD,美国waters公司)、精密分析天平(FA1004)、傅里叶红外光谱仪器(Nicolet Nexus 410,美国Nicolet公司)、真空冷冻干燥机(LFD-56D10S)等。
自制有机玻璃材质反应器高度为200 mm,内径为90 mm,O3由臭氧发生器(JZ110B-SJG)供给,采用微孔石英砂芯底层曝气,通过转子流量计控制流量,同时O3产量使用碘量法进行测量。利用2个串联的吸收瓶组成尾气收集装置,对溢出尾气进行收集,定时在反应器中部取样。
1.3 实验方法
将PCN溶液加入反应器中均匀混合,在通入O3前,加入适量H2O2并控制反应温度和pH,待臭氧发生器稳定工作后,调节气体流量为1.2 L·min−1(臭氧产量为492 mg·h−1),反应开始后按时取样,然后用Na2S2O3终止反应。样品经0.22 μm滤膜过滤后,测定其COD和ρ(PCN)。每次均设计重复实验,每个样品都进行平行测定,然后取其均值。
1.4 分析方法
使用HPLC对PCN的降解产物进行检测。具体实验条件为:Hypersil BDS C18色谱柱;流动相为超纯水(含0.1%甲酸)∶乙腈=50∶50(体积比);流速为1.0 mL·min−1;柱温为30 ℃;进样量为20 μL[26]。质谱检测采用电喷雾电离源,在负离子模式下进行检测,扫描的质荷比m/z为100~700。O3气相浓度采用碘量法(CJ/T 3028.2-1994)测定,COD采用重铬酸钾快速消解法进行测定。
2. 结果与讨论
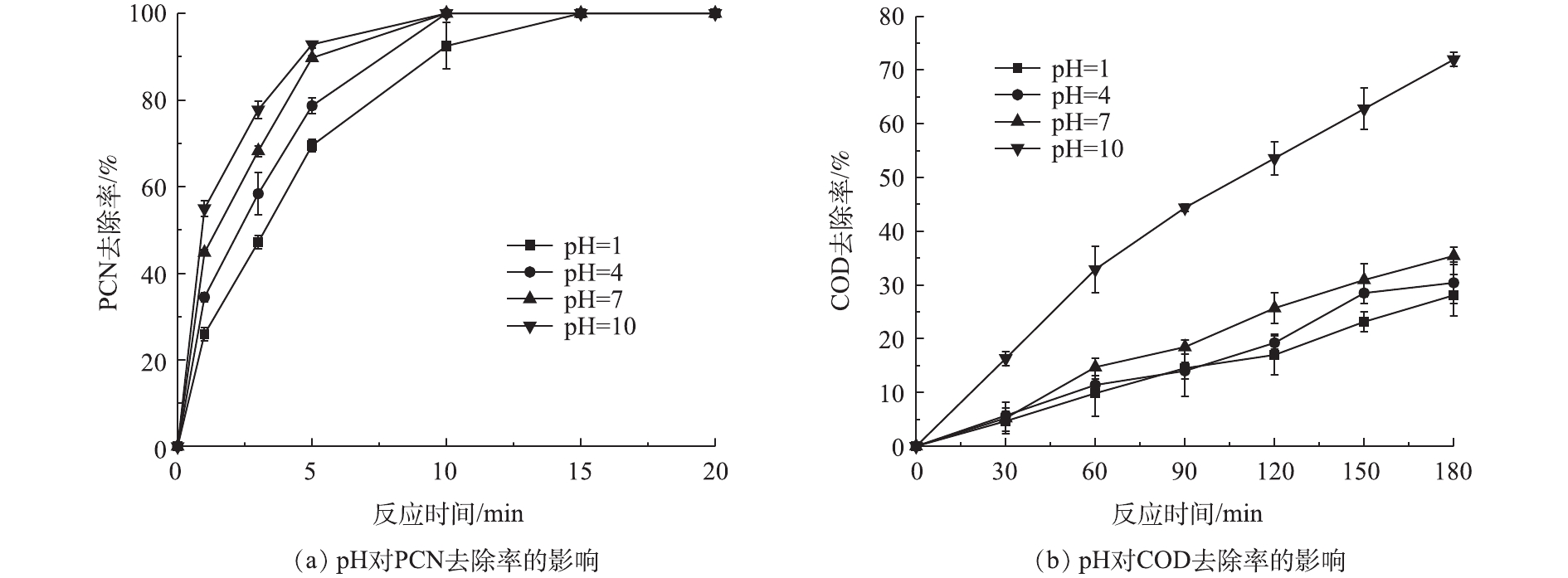
2.1 pH对PCN和COD去除率的影响
在温度为20 ℃、ρ(PCN)为25 mg·L−1、O3气体流量为1.2 L·min−1、H2O2投加量为7.84 mmol·L−1的反应条件下,考察pH对PCN和COD去除效果的影响,结果如图1所示。由图1可知,在不同pH下,COD和PCN的去除效果差异明显,在酸性和中性条件下,COD去除效果相对较差,PCN去除速率缓慢,当pH升高时,反应去除速率也相应加快;在碱性反应环境下,去除效果显著提升,在反应5 min后,PCN去除率为92.5%,在反应3 h后,COD去除率为71.9%。这是因为pH会影响O3/H2O2体系中HO·的产生效率,在酸性条件下,主要是O3分子的氧化,而在碱性情况下,溶液中OH-会促进HO·的生成,此时主要以HO·氧化为主,反应速率得到了提升,具体反应如式(1)~式(3)所示。
O3+OH−→HO−2+O2 (1) H2O2↔HO−2+H+ (2) O3+HO−2→HO⋅+O−2+O2 (3) 此外,在碱性环境中,H2O2更容易离解生成
HO−2 ,而HO−2 又是HO·的诱发剂,所以可促进HO·的生成,进而加快氧化速率[23]。2.2 O3投加量对PCN和COD去除率的影响
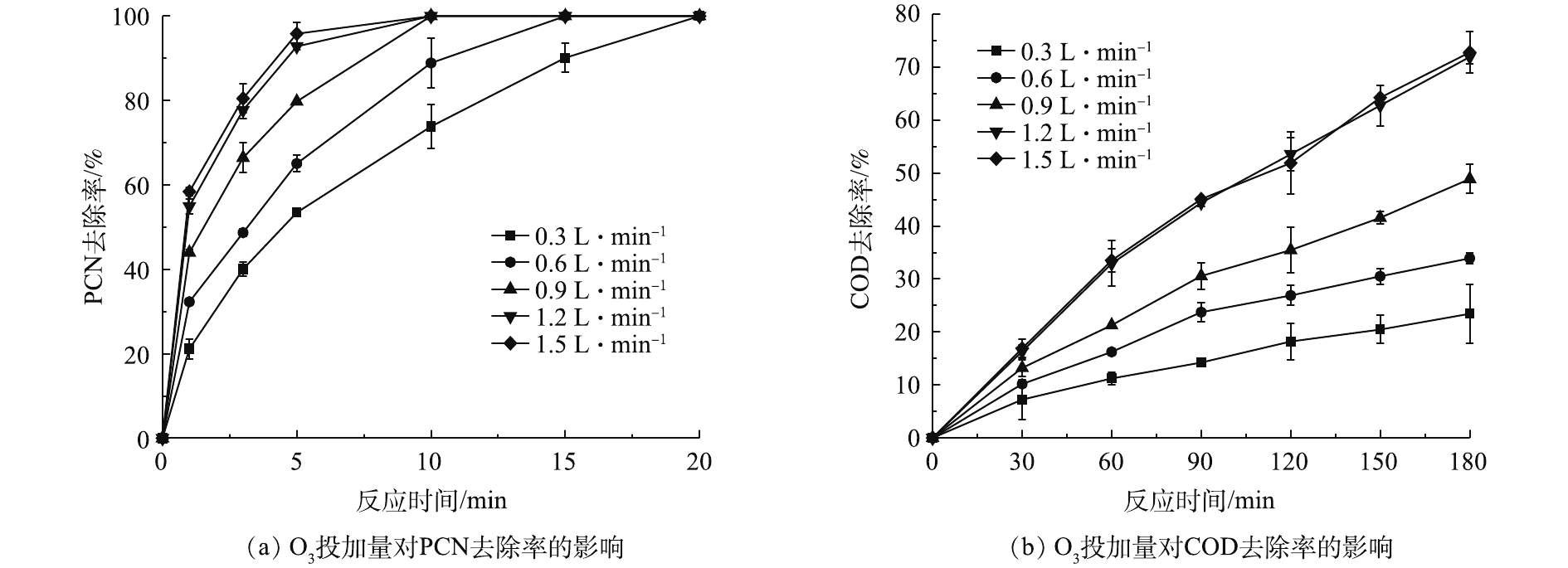
在温度为20 ℃、ρ(PCN)为25 mg·L−1、pH=10,H2O2投加量为7.84 mmol·L−1的反应条件下,考察O3投加量对PCN和COD去除效果的影响,结果如图2示。由图2可知,O3投加量对去除PCN和COD的影响较大,当流量由0.3 L·min−1(O3产量为123 mg·h−1)升至1.5L·min−1(O3产量为615 mg·h−1)时,随着O3投加量的不断增加,PCN和COD的去除率也不断提升,当O3流量为1.5 L·min−1时,PCN和COD去除效果达到最佳。在反应5 min后,PCN去除率为95.83%,在反应3 h后,COD去除率为72.8%。由图2还可看出,在1.2 L·min−1(O3产量为492 mg·h−1)和1.5 L·min−1反应条件下,PCN和COD的去除效果无明显差异,PCN和COD的去除率增幅明显降低,原因可能是,当水中O3溶解度达到最大时,O3的利用率将会降低,未参加反应的O3分子将会直接从液相转移至气相中,故导致无法继续提高降解效能。所以本实验最佳O3流量设定为1.2 L·min−1,以避免造成O3的浪费。
2.3 H2O2投加量对PCN和COD去除率的影响
在温度为20 ℃、ρ(PCN)为25 mg·L−1、pH=10、O3气体流量为1.2 L·min−1的条件下,考察H2O2投加量对PCN和COD去除效果的影响,结果如图3所示。由图3可知,在O3/H2O2体系氧化PCN的过程中,PCN能在较短时间内快速被氧化成中间产物,而中间产物的氧化速率则较为缓慢,但H2O2的促进效果明显。当H2O2的投加量由0升至7.84 mmol·L−1时,PCN和COD的去除率也相应随之升高。在反应5 min后,PCN去除率为100%,增幅为37.4%;在反应3 h后,COD去除率为71.9%,增幅为26.3%。相比于单独的O3体系,添加双氧水能显著提升COD和PCN的去除率,这是由于O3和H2O2之间存在协同机制,适量双氧水可促进氧化过程中HO·的生成,从而提升反应效果[3]。具体反应如式(4)所示。
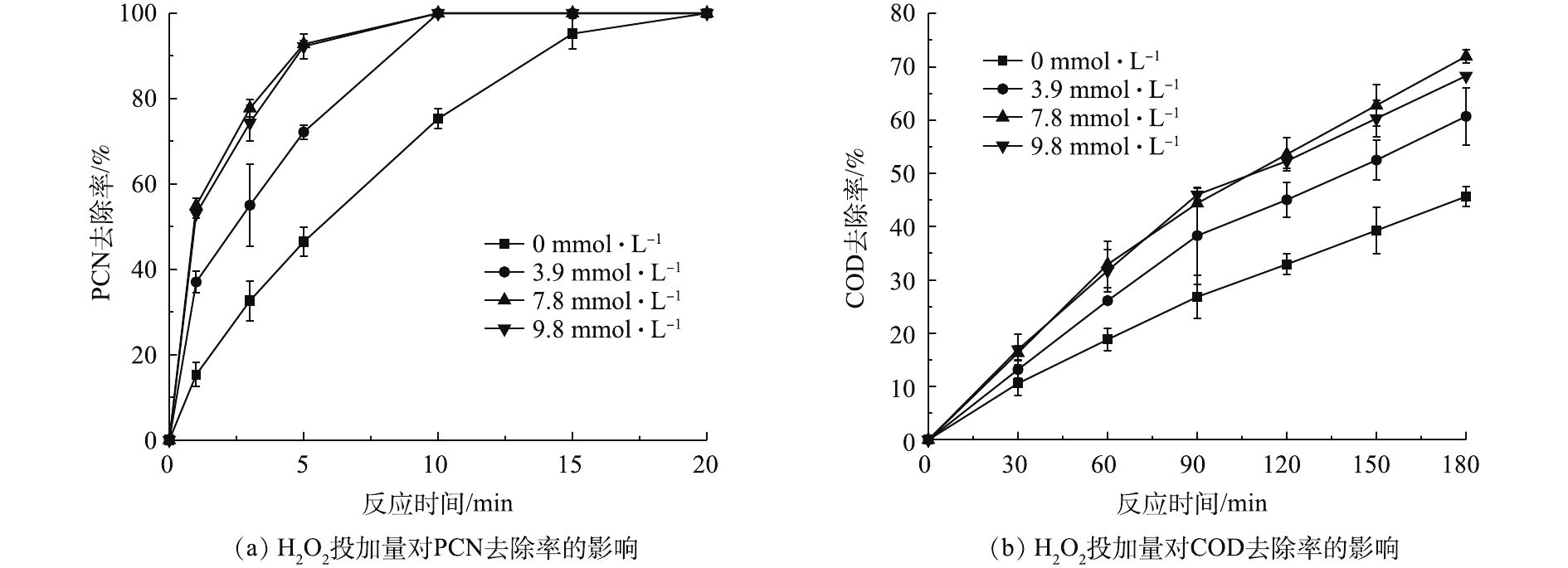 图 3 H2O2投加量对PCN、COD去除率的影响Figure 3. Effect of H2O2 dosage on the removal rates of PCN and COD
图 3 H2O2投加量对PCN、COD去除率的影响Figure 3. Effect of H2O2 dosage on the removal rates of PCN and CODO3+2H2O2→2HO⋅+2O2 (4) 由图3可看出,当H2O2投加量大于7.84 mmol·L−1时,COD去除率略微下降。这可能是由于反应体系中多余的H2O2成为了HO·的捕获剂,从而降低了HO·氧化有机物的效率[22-23]。具体反应机理如式(5)所示。
H2O2+2HO⋅→2H2O+O2 (5) 2.4 温度对PCN去除率的影响
在ρ(PCN)为25 mg·L−1、pH为10、H2O2投加量为7.84 mmol·L−1、O3气体流量为1.2 L·min−1的条件下,考察温度对PCN去除效果的影响,结果如图4所示。由图4可知:在10~30 ℃时,随着温度的上升,PCN去除速率也逐渐加快,去除速率由8.11 mg·(L·min)−1增至17.34 mg·(L·min)−1;但当温度为40 ℃时,PCN去除速率明显降低。原因可能是:当反应温度升高时,加快了分子之间的运动,加速了HO·的生成和O3在水中的扩散速率,从而提升了PCN去除速率。但当温度继续升高时,H2O2的自分解效果加剧,且O3在溶液中的溶解度也有所降低,导致去除速率明显减缓。
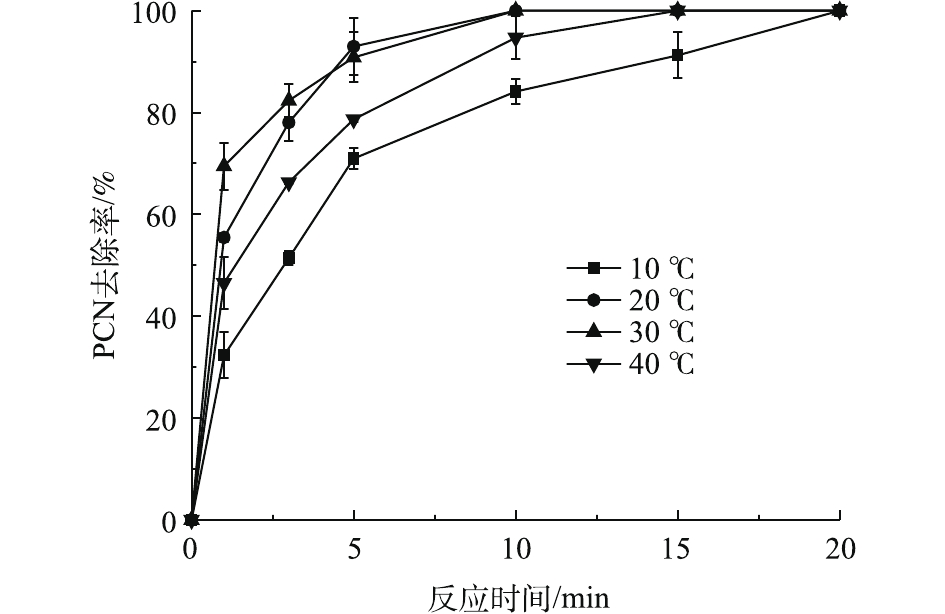 图 4 不同温度对PCN去除率的影响Figure 4. Effect of different temperature on the removal rate of PCN
图 4 不同温度对PCN去除率的影响Figure 4. Effect of different temperature on the removal rate of PCN2.5 反应体系中pH的变化
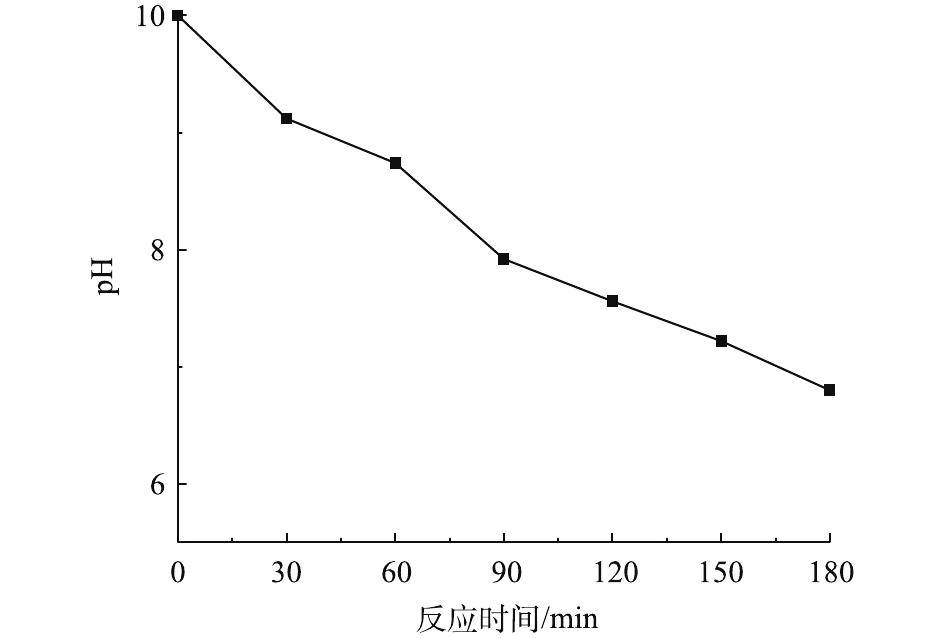
在温度为20 ℃、ρ(PCN)为25 mg·L−1、pH为10,O3气体流量为1.2 L·min−1、H2O2投加量为7.84 mmol·L−1的条件下,考察O3/H2O2反应体系中pH的变化趋势,结果如图5所示。由图5可知,在O3氧化PCN的过程中,反应体系pH随反应时间的延长呈下降趋势,在反应3 h后,pH由10下降至6.8,最终反应溶液呈弱酸性,这可能是由于在降解过程中产生了酸性中间产物,从而导致pH的下降。这与红外光谱和LC-MS的分析结论相一致。体系pH的降低不利于反应的进行,这可能也是反应过程中反应速率均呈先快后慢的变化趋势的原因。
2.6 表观动力学方程的建立
通过大量的实验得出最优pH和温度,研究O3、H2O2和PCN初始浓度对氧化过程中PCN浓度衰减的影响,结果如表1所示,降解动力学方程见式(6)。
表 1 不同反应物的初始浓度对反应速率的影响Table 1. Effect of initial concentration of different reactants on reaction rate序号 反应物初始浓度/(mg·L−1) T/K 初始速率/(mg·(L·min)−1) 拟合方程 PCN O3 H2O2 1 25 8.2 266.4 303.15 13.87 y=0.358 9x−2.901 9R2=0.996 1 2 50 8.2 266.4 303.15 17.76 3 75 8.2 266.4 303.15 20.22 4 100 8.2 266.4 303.15 23.29 5 25 2.05 266.4 303.15 5.31 y=0.697 9x−1.756 4R2=0.997 6 6 25 4.1 266.4 303.15 8.25 7 25 6.15 266.4 303.15 11.4 8 25 8.2 266.4 303.15 13.87 9 25 8.2 66.6 303.15 8.84 y=0.323 3x−3.701 1R2=0.999 8 10 25 8.2 133.2 303.15 11.12 11 25 8.2 199.8 303.15 12.6 12 25 8.2 266.4 303.15 13.87 13 25 8.2 266.4 283.15 8.11 — 14 25 8.2 266.4 293.15 13.87 15 25 8.2 266.4 303.15 17.34 | Show Table DownLoad:
CSV
DownLoad:
CSV
−dCPCN/dt=k0exp(−Ea/RT)⋅CαPCN⋅CβO3⋅CγH2O2 (6) 式中:α、β、γ分别为PCN、O3、H2O2的反应级数;CPCN、CO3、CH2O2分别为PCN、O3、H2O2的初始浓度,mol·L−1;Ea为反应活化能,kJ·mol−1;k0为指前因子,mol·(L·s)−1;R为气体常数,取值8.314 J·(mol·K)−1;T为反应温度,℃。
根据表1的数据并结合表观动力学计算原理,可计算出PCN、O3和H2O2反应物的反应级数,其数值分别为α=0.367、β=0.697 3、γ=0.323 3。
由于总反应速率常数k=k0exp(-Ea/RT),两侧一起取对数可得式(7)。据T和k相应值可得图6。计算得到Ea=27.59 kJ·mol−1,k0=0.052 mol·(L·s)−1,因此,得出总动力学方程,见式(8)。
lnk=−(Ea/R)(1/T)+lnk0 (7) −dCPCN/dt=0.052exp(−27594.9/RT)⋅C0.367PCN⋅C0.6973O3⋅C0.3233H2O2 (8) 本动力学模型是依据反应物初始浓度对降解速率的影响而建立的,对于整个降解过程而言,模型可能会高估反应速率。由式(8)可知,O3的反应级数为0.697 9,高于PCN (0.358 9)和H2O2 (0.335 4)的反应级数,说明降解过程中O3初始浓度对反应速率的影响最大。原因可能是,在O3氧化降解PCN的过程中,存在O3分子直接氧化和HO·氧化2种氧化方式,反应过程中只要有O3就能氧化有机物,而H2O2与O3反应只能加快HO·的生成。此外,反应活化能 (Ea=27.59 kJ·mol−1)较低,说明该反应容易发生。
2.7 PCN降解前后红外光谱分析
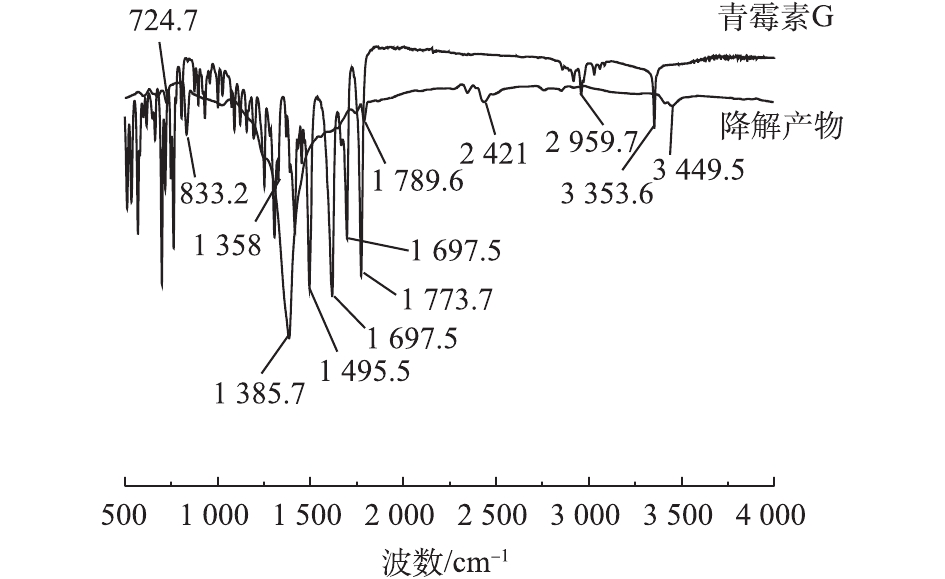
将PCN溶液及其氧化降解的最终产物进行冷冻干燥后进行红外光谱检测,结果如图7所示。在PCN红外光谱图中,1 773.7 cm−1为—COOH中的C=O的伸缩振动峰,3 353.6 cm−1为—COOH中的O—H的伸缩振动峰;1 495.5、1 617.9和2 959.7 cm−1为苯环结构对应的吸收峰,650~1 000 cm−1为苯环上的C—H取代伸缩振动峰;而1 697.5 cm−1为酰胺结构的C=O的伸缩振动;1 307 cm−1处为—(CH3)2的吸收峰。
由图7可知,PCN在氧化前后的谱图有着明显差异,在1 450~1 620 cm−1和3 000 cm−1处苯环骨架吸收峰消失,这说明氧化破坏了PCN的苯环结构。在2 421 cm−1处出现了新的吸收峰,这说明在氧化过程中可能有含叁键或者累积双键的物质产生。在1 697.5 cm−1处的酰胺结构吸收峰消失不见,说明氧化反应破坏了PCN的抑菌结构β-内酰胺环,从而使PCN的抑菌性减弱[27-28]。在1 385.7 cm−1处的峰强度有明显增大,这说明原—(CH3)2结构仍存在,吸收峰在3 449.5 cm−1处出现,有可能是伯胺官能团的不对称伸缩振动与—COOH上O—H的伸缩振动,说明最终产物中可能含有胺类化合物。在1 789.5 cm−1和833.2 cm−1处分别出现羧酸的C=O的伸缩振动和O—H的弯曲振动,这表明最终产物中可能含有酸类化合物,这是导致反应中pH下降的原因。
2.8 PCN降解产物分析
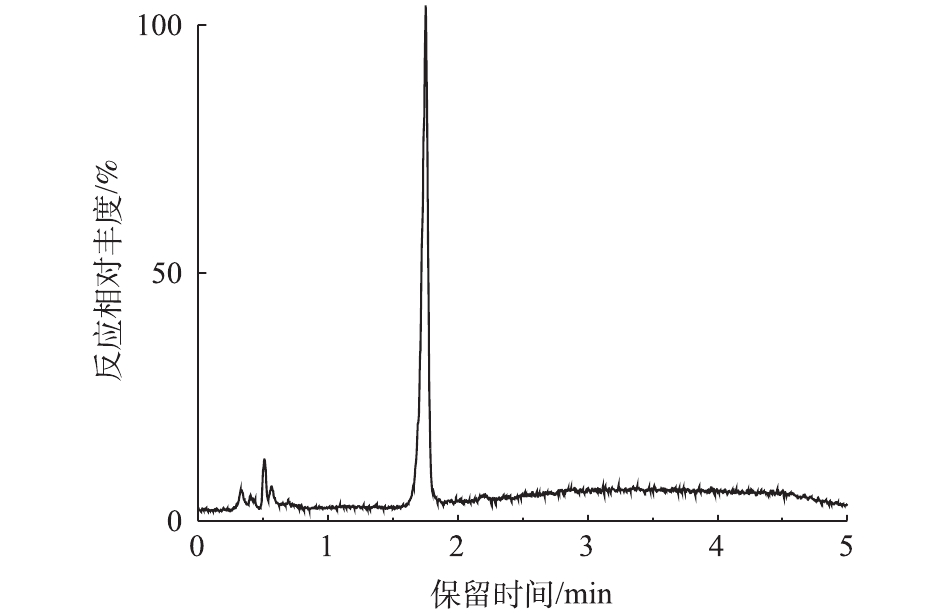
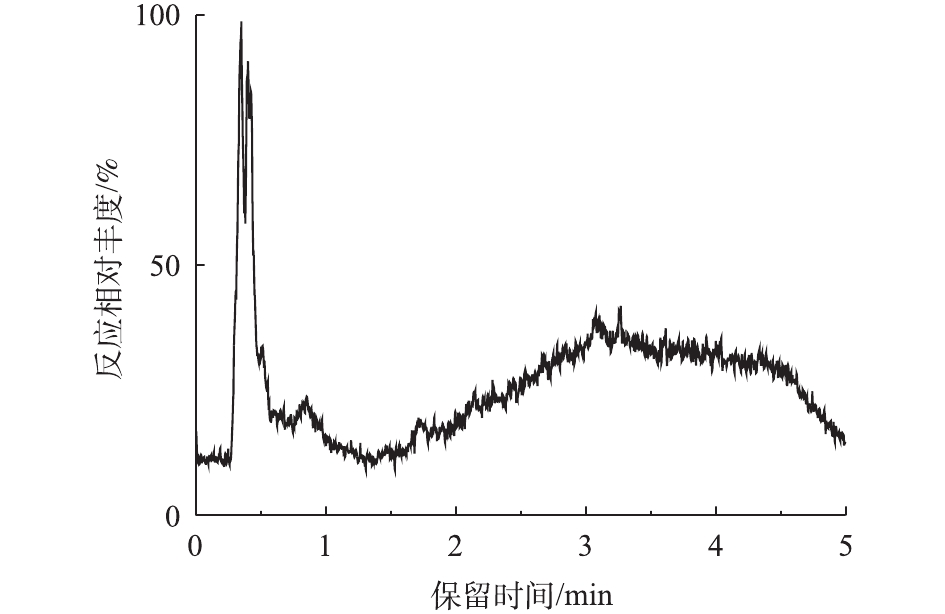
对PCN的降解产物进行LC-MS检测,PCN及其降解产物的总离子流图如图8和图9所示。结果表明,PCN及其降解产物得到了较好的分离,降解后没有检测到PCN的出峰,说明PCN已被完全降解,离子流图显示了PCN降解产物的变化;综合FT-IR的表征结果,对降解产物进行了质谱分析,推测出PCN降解产物可能的分子结构(表2)。
表 2 PCN及其降解产物的质谱数据Table 2. Mass spectrometry data of PCN and its degradation products物质 分子式 保留时间/min 离子质荷比 青霉噻唑酸 C16H21N2O5S 0.841 352 青霉素钠 C16H18N2O4S 1.753 334 去羧青霉素噻唑酸 C15H20N2O3S 0.505 308 6-氨基青霉噻唑酸 C8H14N2O4S 0.407 234 青霉胺 C5H11NO2S 0.488 149 化合物1 C10H11NO3 0.515 193 化合物2 C8H16N2O6S 0.488 267 化合物3 C7H15NO5S 0.339 225 | Show TableDownLoad:
CSV
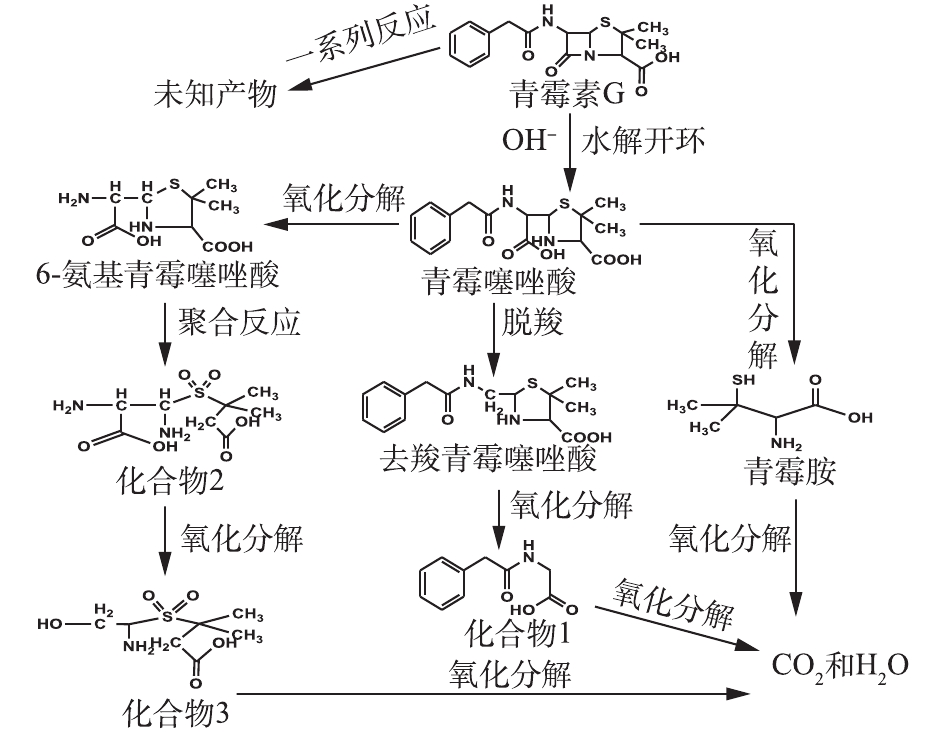
在O3降解PCN的过程中,可能有HO·氧化以及水解等非常复杂的反应存在。在碱性条件下,PCN的β-内酰胺环容易水解打开生成青霉噻唑酸;经脱酸反应后,可能生成去羧青霉噻唑酸;同时在HO·的强氧化能力下,青霉噻唑酸可能进一步被氧化降解成6-氨基青霉噻唑酸、青霉胺和其他未知产物;中间产物也可能最终矿化成为CO2和H2O。根据中间产物分析,推测PCN可能的降解路径如图10所示。
根据LC-MS对产物的分析结果,并结合红外光谱表征结果可知,PCN降解前后的官能团结构发生了较大的变化,氧化使PCN的β-酰胺环被破坏,这也解释了PCN及其降解产物的抑菌性消失或者减弱的原因。
3. 结论
1) O3和H2O2有显著的协同作用,能明显加快反应速率,显著提升COD和PCN的去除率。在初始ρ(PCN):25 mg·L−1,pH=10、O3投加量为1.48 g·L−1、H2O2投加量为7.84 mmol·L−1、温度为20 ℃的条件下,反应10 min后,PCN被完全去除,反应3 h后,COD去除率为71.9%。这说明O3/H2O2体系能有效氧化降解PCN和降解过程中产生的中间产物。
2)通过数据的拟合,得到了O3/H2O2降解PCN的反应动力学方程,O3的反应级数为0.697 3,高于PCN(0.367)和H2O2(0.323 3)的反应级数,说明在降解过程中,O3初始浓度对反应速率的影响最大;此反应的活化能(Ea=27.59 kJ·mol−1)较低,说明此反应容易发生。
3)根据LC-MS和红外光谱检测结果得出,PCN分子结构在降解前后发生了明显变化,PCN的抑菌结构β-内酰胺环被破坏。此外,降解产物中含有酸性物质,这会导致反应体系pH下降,从而不利于O3反应的进行。
-
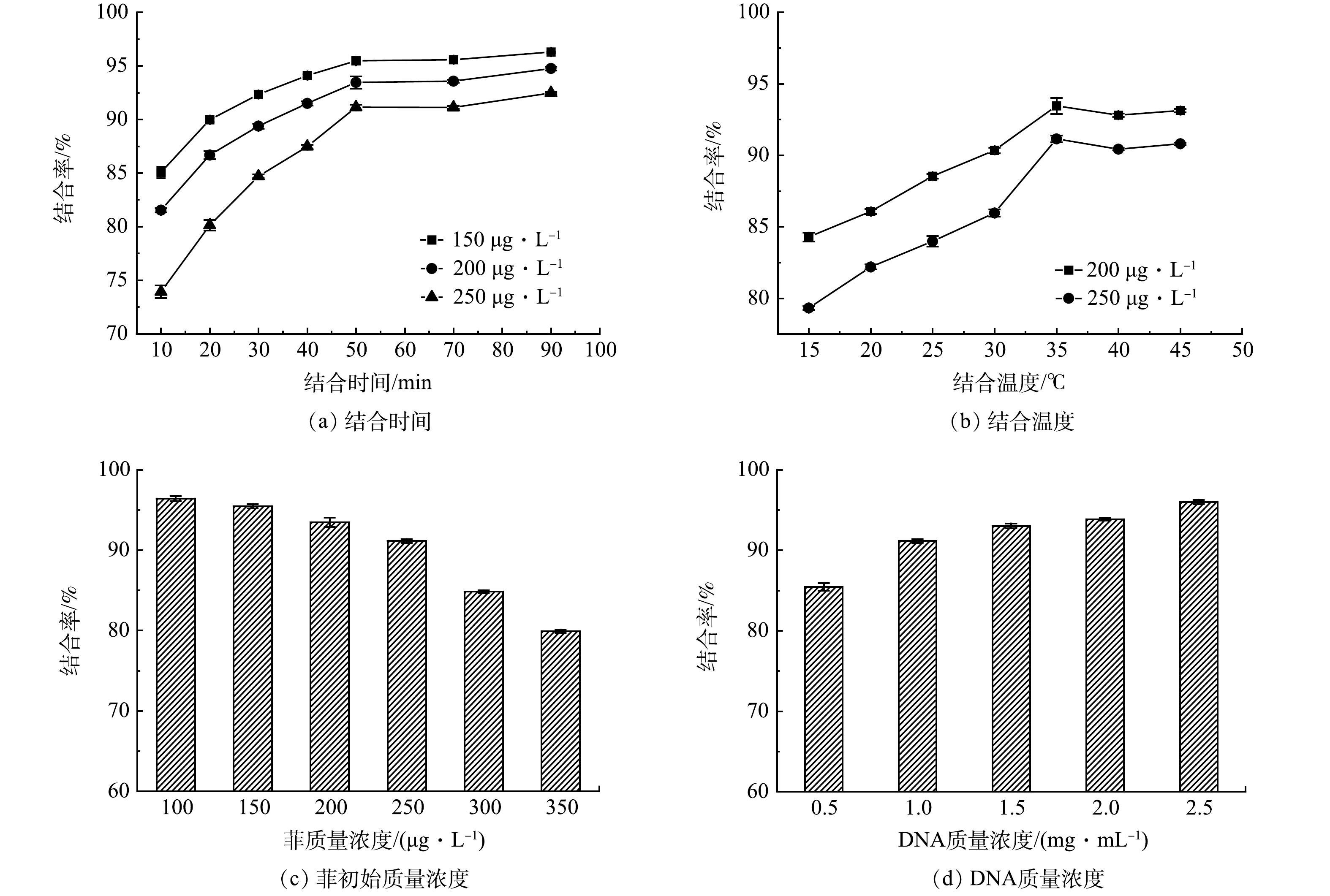
图 1 不同影响因素对DNA结合菲的影响
Figure 1. Effects of different factors on DNA-binding phenanthrene

图 2 DNA磁性纳米颗粒吸附菲前和吸附后的荧光图
Figure 2. Fluorescence images of DNA magnetic nanoparticles before and after adsorption of phenanthrene
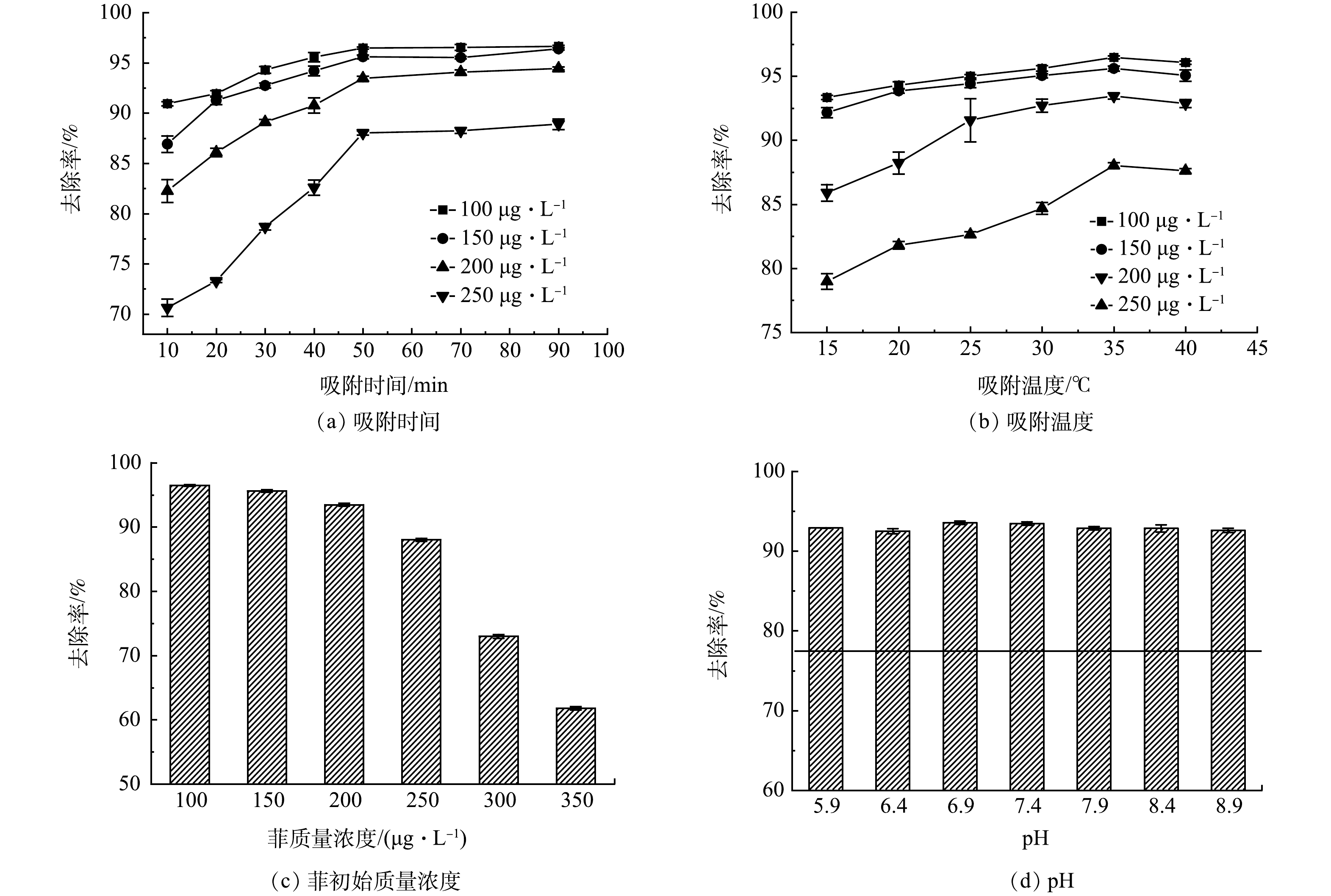
图 3 不同影响因素对DNA磁性纳米颗粒去除菲的影响
Figure 3. Effects of different factors on phenanthrene removal by DNA magnetic nanoparticles
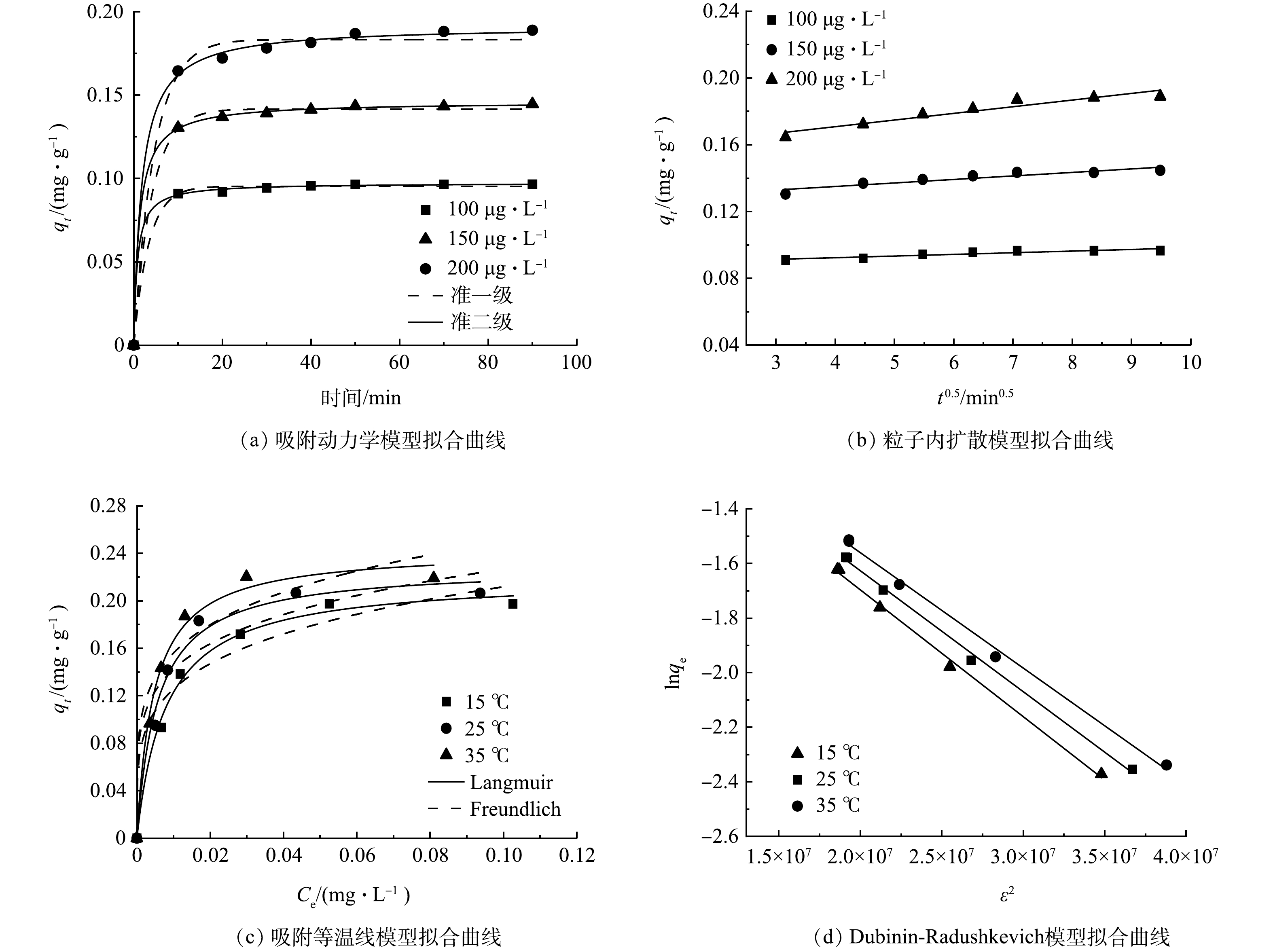
图 4 吸附动力学和吸附等温线模型拟合曲线
Figure 4. Fitting curves of the adsorption kinetics and isotherm model
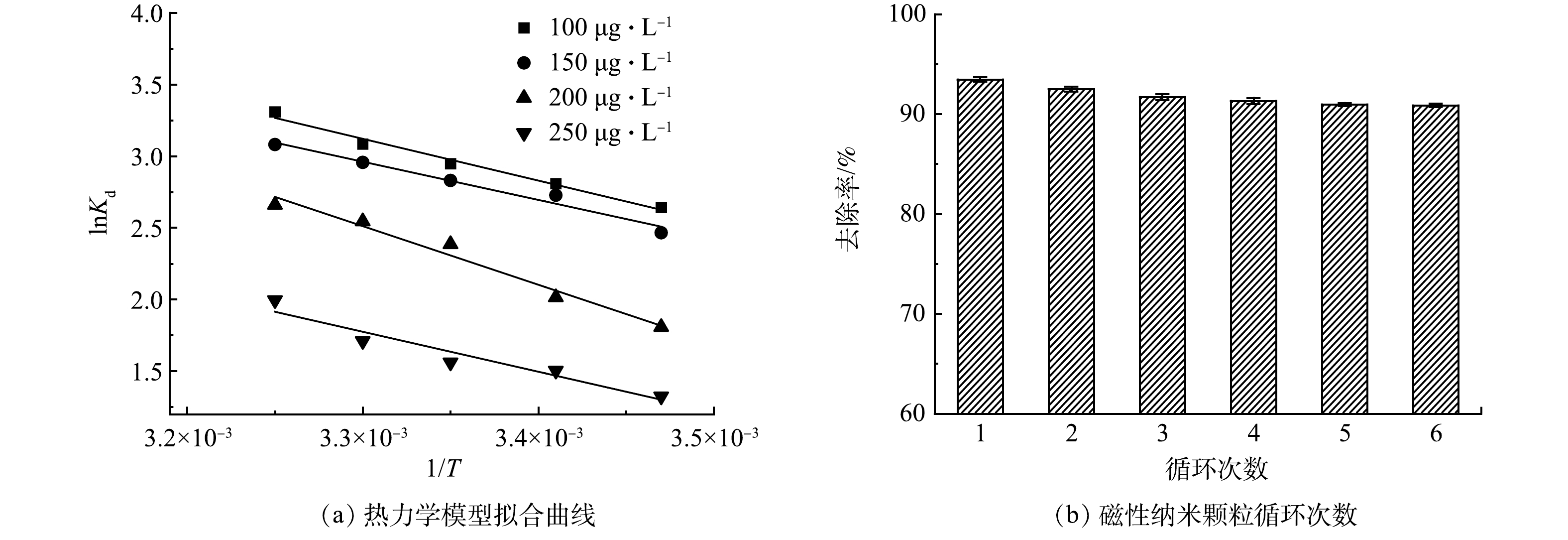
图 5 吸附热力学模型和磁性纳米颗粒循环次数
Figure 5. Thermodynamic modeling of phenanthrene adsorption and recycling number of magnetic nanoparticle
表 1 环境共存物质的质量浓度
Table 1. Mass concentration of environmentally coexisting substances
共存物质 质量浓度/(mg·mL−1) 去除率/% 无 ― 93.46 NaCl 1.17 93.03 CaCl2 2.22 92.84 MgSO4 2.34 93.22 KCl 1.49 93.50 葡萄糖 1.64 92.63 尿素 1.53 93.29 SDS 1.45 81.68 CTAB 1.64 62.85 L-天冬氨酸 0.67 93.89 L-亮氨酸 0.67 92.73 甘氨酸 0.67 93.29
下载: 导出CSV
表 2 现有已报道的吸附剂对菲的吸附性能比较
Table 2. Adsorption capacities of phenanthrene on previously reported adsorbents
下载: 导出CSV
表 3 吸附动力学模型参数
Table 3. Adsorption kinetic model parameters
菲质量浓度/(μg·L−1) q实 /(mg·g−1) 准一级动力学 准二级动力学 K1/min−1 qe /(mg·g−1) R2 K2/(g·(mg·min)−1) qe /(mg·g−1) R2 100 0.098 0.076 0.017 0.914 0 9.759 0.098 0.999 9 150 0.146 0.043 0.019 0.869 0 5.187 0.146 0.999 9 200 0.194 0.061 0.056 0.952 8 2.228 0.194 0.999 6
下载: 导出CSV
表 4 粒子内扩散模型参数
Table 4. Intraparticle diffusion model parameters
菲质量浓度/(μg·L−1) ki/(mg·(g·min0.5)−1) C R2 100 0.001 0.088 0.819 6 150 0.002 0.127 0.836 4 200 0.004 0.155 0.898 3
下载: 导出CSV
表 5 吸附等温线模型参数
Table 5. Adsorption isotherm model parameters
温度/ ℃ q实 /(mg·g−1) Langmuir模型 Freundlich模型 KL/(L·mg−1) qm/(mg·g−1) R2 n KF R2 15 0.197 149.6 0.213 0.997 4 3.77 0.41 0.815 6 25 0.207 227.5 0.218 0.997 4 4.08 0.42 0.717 9 35 0.219 282.3 0.231 0.997 6 3.94 0.48 0.746 4
下载: 导出CSV
表 6 Dubinin-Radushkevich模型参数
Table 6. Dubinin-Radushkevich model parameters
温度/ ℃ Kad/(mol2·kJ−2) qs/(mg·g−1) R2 E/(kJ·mol−1) 15 4.65ⅹ10−8 0.464 0.995 9 3.28 25 4.43ⅹ10−8 0.477 0.996 3 3.26 35 4.23ⅹ10−8 0.489 0.994 8 3.44
下载: 导出CSV
表 7 在35 ℃下的吸附热力学参数
Table 7. Thermodynamic parameters at 35 °C
菲质量浓度/(μg·L−1) ΔG/(kJ·mol−1) ΔH/(kJ·mol−1) ΔS/(J·(mol·K)−1) 100 −8.48 24.22 105.87 150 −7.89 22.16 97.74 200 −6.82 33.96 132.95 250 −5.11 23.18 91.25
下载: 导出CSV
-
[1] REIZER E, VISKOLCZ B, FISER B. Formation and growth mechanisms of polycyclic aromatic hydrocarbons: A mini-review[J]. Chemosphere, 2022, 291: 132793. doi: 10.1016/j.chemosphere.2021.132793 [2] RAVANBAKHSH M, YOUSEFI H, LAK E, et al. Effect of polycyclic aromatic hydrocarbons (PAHs) on respiratory diseases and the risk factors related to cancer[J]. Polycyclic Aromatic Compounds, 2023, 43(9): 8371-8387. doi: 10.1080/10406638.2022.2149569 [3] SHI W J, GONG H, ZHOU W Q, et al. Distribution and ecological risk of polycyclic aromatic hydrocarbons in wastewater treatment plant sludge and sewer sediment from cities in middle and lower Yangtze River[J]. Science of the Total Environment, 2023, 881: 163212. doi: 10.1016/j.scitotenv.2023.163212 [4] MONTUORI P, DE R E, DI D F, et al. Estimation of polycyclic aromatic hydrocarbons pollution in Mediterranean Sea from Volturno River, Southern Italy: Distribution, risk assessment and loads[J]. International Journal of Environmental Research and Public Health, 2021, 18(4): 1383. doi: 10.3390/ijerph18041383 [5] CAI Y, WANG Z C, CUI L J, et al. Distribution, source diagnostics, and factors influencing polycyclic aromatic hydrocarbons in the Yellow River Delta wetland[J]. Regional Studies in Marine Science, 2023, 67: 103181. doi: 10.1016/j.rsma.2023.103181 [6] PATHAK S, SAKHIYA A K, ANAND A, et al. A state-of-the-art review of various adsorption media employed for the removal of toxic polycyclic aromatic hydrocarbons (PAHs): An approach towards a cleaner environment[J]. Journal of Water Process Engineering, 2022, 47: 102647. [7] 李庆华, 张丽, 杨懿, 等. g-C3N4/TiO2复合薄膜光催化降解石油采出水中多环芳烃[J]. 环境工程学报, 2023, 17(6): 1788-1798. [8] RAYAROTH M P, MARCHEL M, BOCZKAJ G. Advanced oxidation processes for the removal of mono and polycyclic aromatic hydrocarbons: A review[J]. Science of the Total Environment, 2023, 857: 159043. doi: 10.1016/j.scitotenv.2022.159043 [9] THACHARODI A, HASSAN S, SINGH T, et al. Bioremediation of polycyclic aromatic hydrocarbons: An updated microbiological review[J]. Chemosphere, 2023, 328: 138498. doi: 10.1016/j.chemosphere.2023.138498 [10] Watson J D, Crick F H C. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid[J]. Nature, 1953, 171: 737-738. doi: 10.1038/171737a0 [11] LERMAN L S. Structural considerations in the interaction of DNA and acridines[J]. Journal of Molecular Biology, 1961, 3(1): 18-30. doi: 10.1016/S0022-2836(61)80004-1 [12] HUANG G X, MA J, LI J S, et al. Study on the interaction between aflatoxin M1 and DNA and its application in the removal of aflatoxin M1[J]. Journal of Molecular Liquids, 2022, 355: 118938. doi: 10.1016/j.molliq.2022.118938 [13] LI J S, WANG J T, FAN J F, et al. Binding characteristics of aflatoxin B1 with free DNA in vitro[J]. Spectrochimica Acta - Part A:Molecular and Biomolecular Spectroscopy, 2020, 230: 118054. doi: 10.1016/j.saa.2020.118054 [14] MA J, HUANG G X, MO C X, et al. Insights into the intercalative binding of benzo [b] fluoranthene with herring sperm DNA in vitro and its application[J]. Journal of Molecular Liquids, 2023, 378: 121628. doi: 10.1016/j.molliq.2023.121628 [15] XIONG Y N, LI J S, HUANG G X, et al. Interacting mechanism of benzo(a)pyrene with free DNA in vitro[J]. International Journal of Biological Macromolecules, 2021, 167: 854-861. doi: 10.1016/j.ijbiomac.2020.11.042 [16] HUANG G X, MA J, LI J S, et al. Removal of 1, 2-benzanthracene via the intercalation of 1, 2-benzanthracene with DNA and magnetic bead-based separation[J]. Nucleosides Nucleotides & Nucleic Acids, 2021, 40(2): 137-156. [17] 吕嘉楠, 李军生, 黄国霞, 等. 光谱法对菲与游离DNA的结合模式研究[J]. 大众科技, 2021, 23(7): 21-24. [18] FAN Q Q, GUAN Y P, ZHANG Z, et al. A new method of synthesis well-dispersion and dense Fe3O4@SiO2 magnetic nanoparticles for DNA extraction[J]. Chemical Physics Letters, 2019, 715: 7-13. doi: 10.1016/j.cplett.2018.11.001 [19] FORT A, GUIRY M D, SULPICE R. Magnetic beads, a particularly effective novel method for extraction of NGS-ready DNA from macroalgae[J]. Algal Research, 2018, 32: 308-313. doi: 10.1016/j.algal.2018.04.015 [20] VAREDA J P. On validity, physical meaning, mechanism insights and regression of adsorption kinetic models[J]. Journal of Molecular Liquids, 2023, 376: 121416. doi: 10.1016/j.molliq.2023.121416 [21] YEO J Y J, KHAERUDINI D S, SOETAREDJO F E, et al. Experimental and modelling study of adsorption isotherms of amoxicillin, ampicillin and doripenem on bentonite-chitosan composite[J]. South African Journal of Chemical Engineering, 2023, 43: 38-45. doi: 10.1016/j.sajce.2022.09.013 [22] KONG L J, ZHANG M Y. Adsorption of methylene blue on chestnut shell-based activated carbon: Calculation of thermodynamic parameters for solid–liquid interface adsorption[J]. Catalysts, 2022, 12(8): 813. doi: 10.3390/catal12080813 [23] 刘立华, 刘爽, 沈玉龙, 等. 改性柚子皮对镉离子的吸附及动力学研究[J]. 南开大学学报(自然科学版), 2020, 53(2): 38-44. [24] NAHAR A, AKBOR M A, PINKY N S, et al. Waste newspaper driven activated carbon to remove polycyclic aromatic hydrocarbon from wastewater[J]. Heliyon, 2023, 9(7): e17793. doi: 10.1016/j.heliyon.2023.e17793 [25] YUE R Y, AN C J, YE Z B, et al. A pH-responsive phosphoprotein washing fluid for the removal of phenanthrene from contaminated peat moss in the cold region[J]. Chemosphere, 2023, 313: 137389. doi: 10.1016/j.chemosphere.2022.137389 [26] ARAVIND K J, KRITHIGA T, VIJAI A K, et al. Kinetics and regression analysis of phenanthrene adsorption on the nanocomposite of CaO and activated carbon: Characterization, regeneration, and mechanistic approach[J]. Journal of Molecular Liquids, 2021, 334: 116080. doi: 10.1016/j.molliq.2021.116080 [27] GE Z Q, SUN T T, XING J F, et al. Efficient removal of ethidium bromide from aqueous solution by using DNA-loaded Fe3O4 nanoparticles[J]. Environmental Science and Pollution Research, 2019, 26(3): 2387-2396. doi: 10.1007/s11356-018-3747-7 [28] KONG X C, ZHANG J H, JI Q Y, et al. Insights into adsorption mechanisms of nitro polycyclic aromatic hydrocarbons on common microplastic particles: Experimental studies and modeling[J]. Chemosphere, 2023, 320: 138050. doi: 10.1016/j.chemosphere.2023.138050 [29] LI S S, LUO J Q, FAN J X, et al. Aflatoxin B1 removal by multifunctional membrane based on polydopamine intermediate layer[J]. Separation and Purification Technology, 2018, 199: 311-319. doi: 10.1016/j.seppur.2018.02.008 [30] MASRAT R, MAJID K. Solubilization of pyrene by mixed polymer-cationic/nonionic surfactant systems: Effect of polymer concentration[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2022, 653: 129974. doi: 10.1016/j.colsurfa.2022.129974 [31] XU L, FENG L, HAO J C, et al. Compaction and decompaction of DNA dominated by the competition between counterions and DNA associating with cationic aggregates[J]. Colloids Surfaces B:Biointerfaces, 2015, 134: 105-112. doi: 10.1016/j.colsurfb.2015.06.038 [32] ZHOU W J, WANG X H, CHEN C P, et al. Removal of polycyclic aromatic hydrocarbons from surfactant solutions by selective sorption with organo-bentonite[J]. Chemical Engineering Journal, 2013, 233: 251-257. doi: 10.1016/j.cej.2013.08.040 [33] TOPUZ F, UYAR T. Poly-cyclodextrin cryogels with aligned porous structure for removal of polycyclic aromatic hydrocarbons (PAHs) from water[J]. Journal of Hazardous Materials, 2017, 335: 108-116. doi: 10.1016/j.jhazmat.2017.04.022 [34] TANG J C, LV H H, GONG Y Y, et al. Preparation and characterization of a novel graphene/biochar composite for aqueous phenanthrene and mercury removal[J]. Bioresource Technology, 2015, 196: 355-363. doi: 10.1016/j.biortech.2015.07.047 [35] BAI H Z, ZHOU J, ZHANG H J, et al. Enhanced adsorbability and photocatalytic activity of TiO2-graphene composite for polycyclic aromatic hydrocarbons removal in aqueous phase[J]. Colloids and Surfaces B:Biointerfaces, 2017, 150: 68-77. doi: 10.1016/j.colsurfb.2016.11.017 [36] DAI W J, WU P, LIU D, et al. Adsorption of polycyclic aromatic hydrocarbons from aqueous solution by organic montmorillonite sodium alginate nanocomposites[J]. Chemosphere, 2020, 251: 126074. doi: 10.1016/j.chemosphere.2020.126074 [37] TAN K L, HAMEED B H. Insight into the adsorption kinetics models for the removal of contaminants from aqueous solutions[J]. Journal of the Taiwan Institute of Chemical Engineers, 2017, 74: 25-48. doi: 10.1016/j.jtice.2017.01.024 [38] SUN Z W, HUANG D, DUAN X H, et al. Functionalized nanoflower-like hydroxyl magnesium silicate for effective adsorption of aflatoxin B1[J]. Journal of Hazardous Materials, 2020, 387: 121792. doi: 10.1016/j.jhazmat.2019.121792 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1205
- HTML全文浏览数: 1205
- PDF下载数: 82
- 施引文献: 0
