










































-
丁基黄药(C4H9OCS2Me,Me为Na+或K+)是一种捕收能力较强的有机浮选药剂,被广泛应用于各种硫化矿及硫铁矿的混合浮选中。浮选废水中残留的丁基黄药有一定的生物毒性和稳定性[1],并因其自身的还原性极易导致水体COD值超标,若不进行有效处理,将对受纳水体及其周边的生态环境造成严重影响。因此,寻找可有效同步去除丁基黄药及COD的处理技术十分必要。
目前,用于处理丁基黄药废水的工艺方法中,相较于物化法(化学沉淀法、吸附法[2]等)易产生二次污染、生物法[3]反应周期长的缺点,高级氧化法(臭氧[4]、Fenton氧化法[5]等)具有处理效率高、无二次污染等明显优势[6]。臭氧作为高效氧化剂已常见于各类有机废水的降解处理过程中[7],但相关研究结果指出单一臭氧处理是无法进一步将氧化过程中产生的中间产物彻底氧化和矿化,进而导致COD去除率不高[8-9]。与此同时,活性炭 (AC) 工艺因操作简便、成本低及无二次污染等优点在选矿废水处理中得到广泛应用[10-11],但活性炭也存在长时间使用处理效果易下降、吸附饱和后难以分离回收等缺陷[12]。而O3/AC耦合工艺可实现AC吸附和O3氧化在系统中同时进行,在反应过程中有机污染物首先通过吸附作用富集至AC表面,而后通过O3氧化作用得到降解,在提升O3处理的COD去除能力的同时,使得AC吸附能力得到再生以延长其使用周期[13]。因此,O3/AC耦合工艺逐渐受到有机废水处理领域的广泛关注[14],但基于O3/AC耦合工艺处理含丁基黄药的选矿废水尚鲜见报道,且以往同类研究主要关注于对丁基黄药的去除而未进一步研究对COD的同步去除且其降解路径也尚不明确。因此,本文以含丁基黄药废水为研究对象,通过探讨单独O3氧化、AC吸附和O3/AC耦合工艺对丁基黄药及其COD的同步去除效果,并结合反应动力学、傅里叶红外 (FT-IR) 光谱、紫外-可见(UV-vis)光谱及GC-MS鉴定等多角度分析,揭示其表观反应过程及可能的降解路径,以期为今后类似废水的处理提供新的参考。
-
丁基黄药,分析纯,武汉拉那白医药化工;椰壳活性炭,粒径≥ 60目,河南中聚;硫酸、氢氧化钠,分析纯,汕头西陇科学;二氯甲烷,分析纯,天津恒兴试剂;无水硫酸钠,分析纯,上海阿拉丁试剂;聚醚砜(PES)滤头,成都摩尔科学仪器。FG-L型臭氧发生器,广州飞歌环保科技。
-
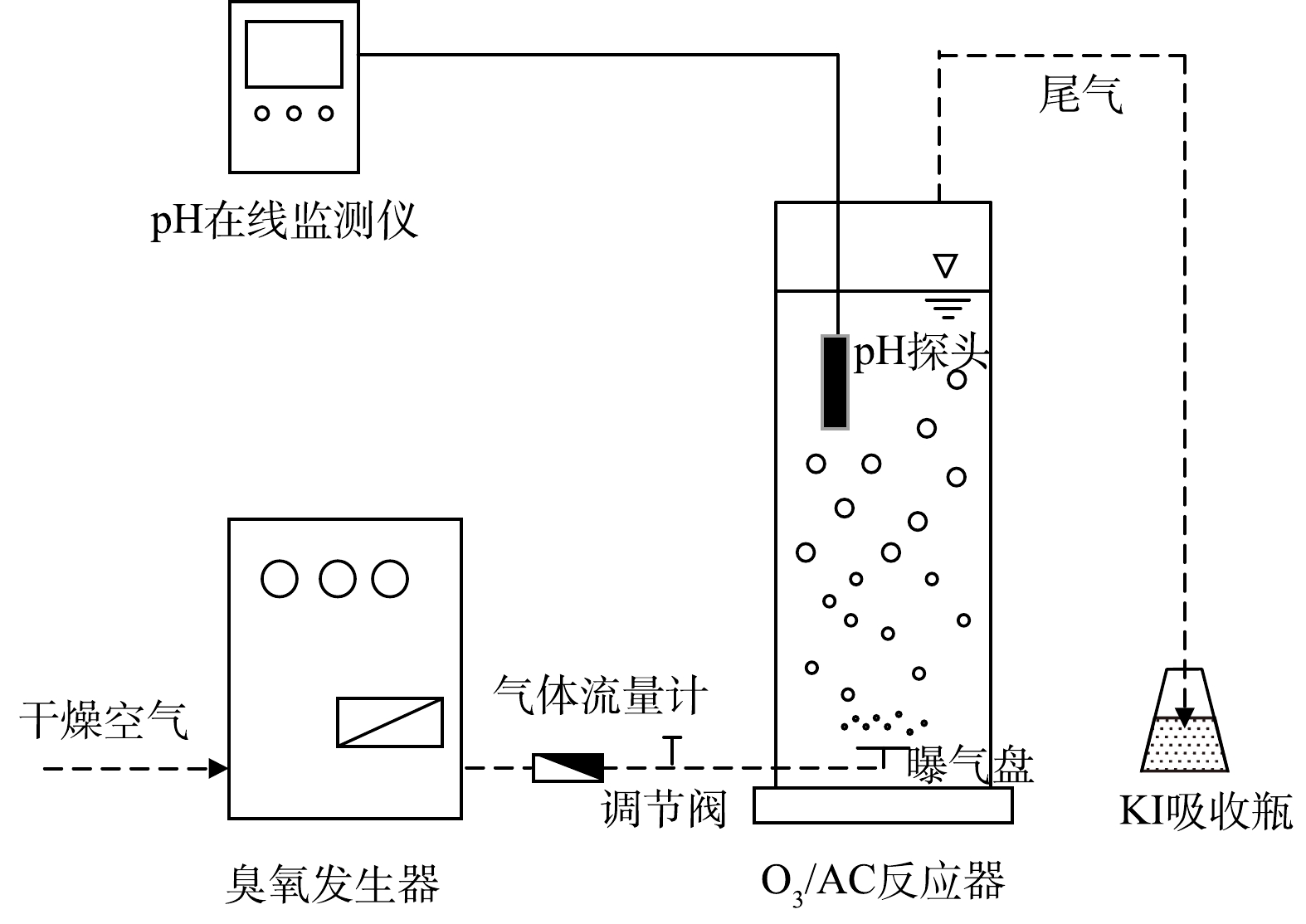
实验装置如图1所示,反应器为有机玻璃材质,有效容积2.2 L(内径85 mm,高度400 mm)。实验时在该反应器中加入1.5 L 400 mg·L−1(基于以往相关研究[15]及前期预实验)的人工配制丁基黄药废水,采用1%H2SO4或5%NaOH溶液调节pH。具体实验过程:通过单独投加定量的臭氧(O3)、活性炭(AC)进行实验以确定基础条件后,同步投加定量的臭氧-活性炭(O3/AC)进行协同降解实验,在控制气体流量为1 L·min−1的条件下,反应80 min,间隔10 min取上清液8 mL过0.45 μm PES滤头(分离待测液中少量的AC以减小其对实验结果的影响)后测定丁基黄药和COD的同步去除率。多余臭氧通过后端KI溶液吸收以减少对周边环境的影响。
-
丁基黄药采用紫外分光光度法测定(SP-1920型紫外可见分光光度计,上海光谱仪器),测定波长为301 nm;COD采用快速消解紫外分光光度法测定(5B-6C(V8)型多参数水质测定仪,北京连华科技);pH采用电极法测定(pH在线监测仪,杭州米科);FT-IR光谱采用傅里叶变换红外光谱仪(FTIR-7600,Lambda Scientific)进行测定,测试方法:将1 mg干燥后的AC样品与200 mg KBr混合,用压片机压成薄片维持1 min后测试,扫描范围为4 000~400 cm−1 [13];反应中间产物采用GC-MS(7890B-5977A,美国安捷伦)测定,测试条件:HP-5MS弹性石英毛细管柱,柱始温40 ℃,以4 ℃·min−1升至100 ℃,再以10 ℃·min−1升至250 ℃;以氦气为载气,流速为0.75 mL·min−1;MS为电子电离源(EI),离子化能量70 eV,源温度200 ℃,接口温度280 ℃;质量扫描范围为全扫描;样品测定前进行预处理,用100 mL二氯甲烷分3次萃取50 mL水样,萃取液经装有无水硫酸钠(105 ℃干燥2 h)的层析柱进行除水处理后待测[16-17]。
-
1) 单独O3氧化实验。在丁基黄药初始质量浓度为400 mg·L−1(对应COD为(634.4±33.8) mg·L−1),溶液初始pH为7.5(未调节,自然状态),O3投加量分别为0.9、1.2、1.5和1.8 g·h−1的条件下,考察单独O3处理时其投加量对丁基黄药及COD的去除效果,结果如图2示。
由图2(a)可知,反应前期(20 min前),O3投加量与丁基黄药的去除率呈正相关;反应后期(60 min后),两者的相关性不明显,均能达到降解平衡状态(去除率达98.4%~99.5%)。说明O3对丁基黄药有明显的去除效果。但达到平衡所用时间随着O3投加量增加(0.9~1.8 g·h−1)而降低(60~30 min),而平衡所需时间长短直接影响反应设施容量大小及产臭氧的能耗,因此,需从能源利用效率角度筛选合理O3投加量。同时结合COD去除率可见(图2(b)),COD的去除趋势与图2(a)中丁基黄药相似,随着O3投加量增加而增加。具体表现在,当O3投加量不高于1.5 g·h−1时,对COD去除率随着O3投加量的增加呈明显上升趋势(由0.9 g·h−1时的36.3%最大去除率提升至1.5 g·h−1时的42.9%);当O3投加量高于1.5 g·h−1,随着O3投加量增加,其对应COD不在明显提升,当O3投加量为1.8 g·h−1,COD去除率为43.6%,较1.5 g·h−1时(42.9%)仅提升0.7%。由此可见,当达到降解平衡状态时,高浓度O3更有利于COD去除,但当浓度高于一定值之后,并不利于进一步提升对COD的去除,究其原因可能是过高浓度O3易发生淬灭反应进而消耗部分氧化剂(O3、各类自由基),导致其氧化效率无法持续提升[18]。据此可知,从上述能效、丁基黄药及COD去除等多方面分析可知,以1.5 g·h−1 O3投加量开展后续试验是相对高效且经济的。另一方面,基于1.5 g·h−1 O3投加量在反应60 min达到丁基黄药去除平衡时的最大去除率98.0%,而COD去除率仅为42.9%,说明丁基黄药易被O3氧化降解,而其生成的有机中间产物却无法被进一步降解[19]。因此,有必要探索将O3氧化同AC吸附相耦合以实现对丁基黄药及COD的高效同步地去除。
2) 单独AC吸附实验。在丁基黄药初始质量浓度为400 mg·L−1,溶液初始pH为7.5,曝气条件与O3处理相同时,控制AC投加量分别为1.00、1.25、1.50和1.75 g·L−1,考察单独AC处理时其投加量对丁基黄药及相应COD同步去除效果,结果如图3所示。
由图3可知,丁基黄药和COD的去除率均随着AC投加量的增加而逐渐升高,且表现出明显的正相关关系及时间效应,但二者直至反应80 min也未达到吸附平衡,其原因可能为单位时间内AC的吸附效率受制于其表面活性位点的数量[20]。当AC投加量由1.00 g·L−1增至1.50 g·L−1时,反应80 min,丁基黄药的去除率由56.1%显著提升至73.7%,对应COD的去除率也由53.0%显著提升至73.5%,而继续增加AC投加量至1.75 g·L−1时,丁基黄药和对应COD的去除效果分别为79.0%和78.4%,较1.50 g·L−1条件下均仅有小幅提升。其原因同2.5中AC处理过程中紫外-可见光谱(分析所述:该体系中COD的去除是基于AC对丁基黄药的吸附作用,这也表明AC可有效协同去除丁基黄药及COD,但要获得较理想的去除效果需基于AC投加量相对较大的基础之上。因此,在保证丁基黄药和COD高效同步去除效果及AC投加量经济合理的前提下,需进一步探索将AC吸附同O3氧化相耦合以缩短反应达到平衡所需时间,从而提升整个反应体系的处理效率。
-
1) AC投加量的影响。基于上述实验结果,将AC吸附与O3氧化二者联用对丁基黄药进行处理,在丁基黄药初始质量浓度为400 mg·L−1,溶液初始pH为7.5,O3投加量为1.5 g·h−1时,控制AC投加量分别为1.00、1.25、1.50和1.75 g·L−1,考察AC投加量对于O3/AC耦合工艺对丁基黄药及COD同步去除效果的影响,结果如图4所示。
由图4(a)可知,单独O3处理丁基黄药显示了出较理想的降解效果(平衡时最大去除率达98.0%),而通过同步投加AC则可进一步提升对丁基黄药的处理效率。在整个反应过程中,尤其是在反应初期(前10 min),单独O3处理仅达46.7%,随着AC的增加,丁基黄药去除也显著提升,当AC投加量达到1.75 g·L−1时,去除率增至78.0%,提升约31.3%,可见该阶段主要是O3/AC耦合作用的结果;在反应后期,O3/AC耦合作用对丁基黄药的去除,整体趋势和最终去除效率与图2(a)反应出的单独O3作用相似,但达到平衡的时间得到明显缩短(较单独O3作用的40 min,缩短至20~30 min)且最大去除效率也得到一定提升(较单独O3作用的98.0%,提升至99.9%),而这充分说明是O3/AC耦合作用所致。结合图4(b),对COD的去除,O3/AC耦合作用试验组的去除效果均明显高于单独O3处理组,且表现出随着AC投加量的增加而提升(较单独O3作用的42.9%,分别提升约24.9%、27.6%、34.3%和36.3%)。究其原因,可能是在AC表面活性位点进行的吸附和催化等共同作用促进了臭氧的氧化[21-22],从而实现O3/AC耦合工艺对丁基黄药及COD的高效同步去除。相较于“O3/H2O2”工艺处理同类废水仅能达到60.25%的COD去除率[15],这将更大限度提升COD的去除效果。
2) O3投加量的影响。在丁基黄药初始质量浓度为400 mg·L−1,溶液初始pH为7.5,AC投加量为1.50 g·L−1时,考察了O3投加量(0.9、1.2、1.5和1.8 g·h−1)对O3/AC耦合工艺同步去除丁基黄药和COD效果的影响,结果如图5示。
由图5(a)可知,O3/AC耦合作用对丁基黄药的去除效果显著高于单独AC处理,其最终的去除率处于99.9%,较单独AC处理的73.7%高出26.2%,且在反应的前20 min内,丁基黄药去除率表现出与O3投加量呈明显正相关,随着反应的持续进行,所有O3组中的丁基黄药均被快速、高效地去除;在反应时间方面,O3/AC耦合体系达到最大去除效果的时间为30~40 min,较单独AC处理所需的80 min明显缩短,极大地提升了整个体系的反应效率。由图5(b)可见,O3/AC耦合作用较单独AC处理,其最大的去除率由73.5%提升至74.7%~78.7%。可见其在一定程度上可继续提升对COD的去除效果,且达到较佳去除效果的时间由原来的80 min缩减至60 min,去除率提升约25%。而分析其原因可能是AC表面活性位点吸附富集的丁基黄药被其固-液相、液相中的氧化剂(O3、·OH等)氧化分解从而提升了AC、O3的吸附、氧化效率[23-24],并体现出对丁基黄药及COD的同步高效去除。
3) 初始pH的影响。由于初始pH是影响O3氧化与AC吸附效果的重要因素[25]。因此,在丁基黄药初始质量浓度为400 mg·L−1,控制初始pH分别为5.0、7.5、10和12.5,反应60 min,分析对比初始pH对O3(1.5 g·h−1)、AC(1.50 g·L−1)及O3/AC(1.5 g·h−1/1.50 g·L−1)3种处理工艺同步去除效果的影响,结果如图6所示。
由图6可知,当反应体系初始pH由5升至7.5时,O3处理丁基黄药和COD的去除率逐渐升高,其主要归因于酸性条件下O3处理是以直接氧化为主,当体系中OH−升高至一定浓度时则以氧化能力更强的羟基自由基(·OH)氧化为主[26];但当初始pH继续升高至12.5时,二者的去除率均逐渐下降,造成该现象的主要原因可能是pH过高时生成各类自由基也会过多,其相互之间易发生淬灭反应而导致氧化效率降低[15];对于AC处理而言,丁基黄药及其对应COD的去除率与溶液初始pH呈正相关关系,其原因可能主要在于AC表面的碱性官能团对阴离子的吸附能力随pH的升高而增强[27];当采用O3/AC耦合工艺进行处理时,体系初始pH的变化对于丁基黄药与COD的去除率无明显影响,其主要是由于在酸性、中性条件下O3/AC耦合去除丁基黄药及COD是以O3氧化作用为主导,而碱性条件下则以AC吸附作用为主导,这进一步说明在酸性至较强碱性条件下(5~12.5)O3/AC耦合工艺对于同步去除丁基黄药及COD均具有较强的处理性能。
-
为进一步探讨表观反应过程以对比O3、AC及O3/AC耦合工艺的处理性能,在丁基黄药初始质量浓度为400 mg·L−1,溶液初始pH为7.5,反应趋于平衡(50 min)的条件下,对O3(1.5 g·h−1)、AC(1.50 g·L−1)及O3/AC(1.5 g·h−1/1.50 g·L−1)3种工艺进行反应动力学分析。同时结合相关研究可知,当污染物初始浓度大于臭氧初始浓度时可近似将反应过程中污染物浓度视为常数,可将二级反应转化为拟一级反应[28],其反应式由式(1)表示。因此,为便于比较,AC、O3/AC工艺亦采用拟一级反应进行拟合,具体拟合图和参数表,如图7和表1所示。
式中:C0表示丁基黄药或COD的初始浓度,mg·L−1,Ct表示t时刻丁基黄药或COD的浓度,mg·L−1,k表示表观反应速率常数,min−1。
由图7及表1可知,三种工艺处理丁基黄药及COD的线性拟合相关系数R2、调整后R2均大于0.8,说明其反应过程较符合拟一级动力学模型。对丁基黄药去除,O3、AC及O3/AC耦合工艺的反应速率常数分别为0.078、0.020及0.165 min−1,表现为
k(O3/AC) >kO3 >kAC;对COD去除,O3、AC及O3/AC的反应速率常数分别为0.011、0.020及0.026 min−1,表现为k(O3/AC) >kAC>kO3 ;整体而言,O3/AC耦合工艺去除丁基黄药及COD的表观反应速率常数均大于两种单独处理工艺,说明O3/AC耦合工艺可更直接高效地同步去除丁基黄药及COD。 -
为了探讨反应前后AC表面化学特性变化,对初始AC、单独处理丁基黄药80 min后的AC以及O3/AC耦合处理丁基黄药80 min后的AC进行FT-IR光谱分析,结果如图8所示。由图8可见,初始AC在3 400、1 500~1 800、1 250 cm−1处存在特征吸收峰,3 400 cm−1处的吸收峰归属于羧基中的O—H键伸缩振动,而1 500~1 800 cm−1和1 250 cm−1处的吸收峰则分别是由C=O键和C—O键伸缩振动所产生[29]。AC单独处理丁基黄药80 min后O—H键吸收峰强度有一定程度的减弱,而O3/AC耦合处理丁基黄药80 min后O—H键、C=O键、C—O键的吸收峰强度均显著增强。其原因可能是O3和其在OH−和AC的作用下快速分解形成的各类氧化剂(·OH等),对AC进行氧化使得其表面酸性官能团增多[13]。
-
1)紫外-可见(UV-vis)光谱分析。为进一步探究3种不同处理工艺对丁基黄药去除的降解路径,针对2.3中相同反应条件下的3种处理方法,分别提取前50 min的反应水样,稀释20倍后进行UV-vis光谱扫描,结果如图9示。由图9(a)可看出,丁基黄药有2个吸收峰,分别在227 nm和301 nm(特征峰)处,与相关研究结果相似[30]。随着O3处理时间的延长,特征峰强度明显减弱直至消失,且均在345 nm处出现新的吸收峰,说明在O3的作用下丁基黄药可能被快速降解为其他中间产物,这也与图2中丁基黄药及COD的降解趋势相吻合。由图9(b)可见,在AC的作用下,其特征峰亦随着反应时间的延长快速减弱但并未消失,且未出现移动,说明AC对丁基黄药及COD的去除为单纯吸附作用。由图9(c)可知,在O3/AC的耦合作用下,其特征峰减弱的速度明显快于单独O3和AC处理,且反应10 min时于345 nm处出现明显吸收峰而后随着时间延长快速消失,结合COD的去除趋势(图4(b))表明丁基黄药在处理过程中可能先生成了过黄药(C4H9OCS2O−)[1, 31-32],而后转化为其他有机中间产物。因此,需采用其它方法对其进一步鉴定分析。
2)中间有机产物的GC-MS分析。采用GC-MS对O3/AC耦合工艺处理丁基黄药所产生的有机中间产物进行定性分析,总离子色谱图共分离出5个峰,结果表明,除溶剂峰外其他3个峰未核对出可能的物质,其中1个峰可能为丁酸(C3H7COOH),该峰的质谱扫描分析结果如图10所示。
3)降解路径推测。在以往研究者提出的臭氧氧化黄药的反应路径[33]和吸附-臭氧氧化法处理硝基酚的反应模型[34-35]基础上,结合上述结果(单独O3处理、O3/AC耦合工艺对丁基黄药和COD去除率分别为98.0%和42.9%、99.9%、77.2%)和紫外-可见光谱及GC-MS分析结果,推测O3/AC耦合工艺同步去除丁基黄药及COD的潜在路径可能为:一部分丁基黄药(C4H9OCS2−)可通过臭氧氧化作用被直接矿化为CO2、H2O等无机物(R1);另一部分则被氧化降解为过黄药、丁酸(C4H9OCS2O−、C3H7COOH)等对COD值产生贡献的有机中间产物(R2),而后少部分继续被矿化降解(R3);与此同时,C4H9OCS2− 和生成的C4H9OCS2O−、C3H7COOH等中间产物也会通过吸附作用被吸附至活性炭表面(R4、R5),而后通过吸附-氧化作用被进一步降解去除(R6、R7)。综上所述,丁基黄药及其在反应过程中生成的中间产物在臭氧氧化、活性炭吸附以及吸附-臭氧氧化多种作用途径下被降解,可能的降解路径如图11所示。
-
1) 臭氧(O3)单独作用可有效去除丁基黄药但COD去除率不佳,活性炭(AC)单独作用对丁基黄药及其COD同步去除效果好但整体去除率偏低,两种单独处理工艺均不能同时实现对二者的高效同步去除。而将二者耦合形成O3/AC耦合工艺,在丁基黄药初始质量浓度400 mg·L−1,初始pH为7.5,O3、AC投加量分别为1.5 g·h−1、1.50 g·L−1,反应60 min时,O3/AC耦合工艺处理丁基黄药和COD的去除率便可达99.9%、77.2%,其COD去除率较单独O3处理提升了34.3%,丁基黄药和COD去除率较单独AC处理提升31.8%、8.1%。与此同时,其达到最大去除效果所用的反应时间也由单独AC处理所需的80 min缩减至30~40 min,在保证去除率的同时极大地提升了反应效率,展现了O3/AC耦合工艺高效同步去除丁基黄药及COD的性能。
2) O3、AC及O3/AC耦合工艺处理丁基黄药的反应过程满足拟一级反应动力模型,丁基黄药及COD去除的线性拟合相关系数R2及调整后R2均大于0.8,且丁基黄药的反应速率常数表现为
k(O3/AC) >kO3 >kAC,COD的反应速率常数表现为k(O3/AC) >kAC>kO3 。综合而言,O3/AC耦合工艺去除丁基黄药及COD的反应速率常数均优于两种单独处理法。3) AC表面酸性官能团在O3的作用下有所增加。AC对丁基黄药的去除为单纯吸附作用;而其在O3、O3/AC的作用下可能生成了过黄药等其他对COD值产生贡献的中间有机产物,GC-MS鉴定该中间有机产物为丁酸等物质。
4) O3/AC耦合工艺去除丁基黄药及COD的潜在路径可能为:丁基黄药和生成的有机中间产物在臭氧氧化、活性炭吸附以及吸附-臭氧氧化多种作用途径下被反应降解,最终实现O3/AC耦合工艺对丁基黄药及COD的高效同步去除。
基于O3/AC耦合工艺对丁基黄药与COD的同步去除性能及降解机理
Performance and mechanism analysis of simultaneous removal of butyl xanthate and COD based on O3/AC combined process
-
摘要: 针对臭氧氧化技术处理含丁基黄药废水COD去除率低等问题,提出了O3/AC耦合工艺。在探讨单独、耦合工艺的丁基黄药去除率和COD去除率、反应影响因素和降解路径的基础上,进一步发掘该耦合工艺对丁基黄药及COD的同步去除性能。结果表明:臭氧(O3)及活性炭(AC)的单独作用均对丁基黄药或COD有一定的去除效果,通过O3/AC的耦合作用则可使丁基黄药的去除率达99.9%,亦使COD的去除率由O3单独作用下的42.9%显著提升至77.2%,且实现最高去除率的时间由AC单独处理所需的80 min缩减至30~40 min;拟一级反应动力学分析结果表明,丁基黄药的反应速率常数表现为
k(O3/AC) >kO3 >kAC,COD的反应速率常数表现为k(O3/AC) >kAC>kO3 ,O3/AC耦合工艺去除丁基黄药及COD的反应速率常数均大于两种单独处理工艺;紫外-可见(UV-vis)光谱分析表明丁基黄药在O3、O3/AC作用下生成中间有机产物,结合GC-MS对产物的鉴定结果表明其中间有机产物可能为丁酸等物质,并以此对O3/AC耦合工艺同步去除丁基黄药及COD的降解路径进行了初步推测。Abstract: Aiming at low COD removal rate from butyl xanthate-containing wastewater by ozone oxidation process, a combined O3/AC process was proposed for its treatment. On the basis of exploring the removal rates of butyl xanthate and COD, reaction influencing factors and degradation pathway of the single or coupling process, the simultaneous removal performance of the system on butyl xanthate and COD was further explored. The results showed that: ozone (O3) and activated carbon (AC) alone had certain removal effects for butyl xanthate or COD, and the coupling O3/AC process could remove 99.9% of butyl xanthate, and lead to the increase of COD removal rate from 42.9% under O3 alone to 77.2%, and the time to achieve the maximum removal effect decreased from 80 min under AC alone to 30~40 min. The pseudo-first order kinetic analysis showed that the reaction rate constants of butyl xanthate werek(O3/AC) >kO3 >kAC, and the reaction rate constants of COD werek(O3/AC) >kAC>kO3 , and the reaction rate constants of the O3/AC coupling process for the removal of butyl xanthate and COD were greater than those of the two processes alone. UV-visible spectroscopy analysis showed that the butyl xanthate produced intermediate organic products under the action of O3 and O3/AC, and the GC-MS analysis showed that the intermediate organic products might be butyric acid and other substances, which was used to make a preliminary speculation on the degradation pathway of simultaneous removal of butyl xanthate and COD by O3/AC coupling process.-
Key words:
- O3/AC process /
- butyl xanthate /
- COD /
- simultaneous removal /
- degradation pathway
-
四溴双酚A(TBBPA)是一种溴代阻燃剂,由于其具有高阻燃性和低廉价格等特点,广泛应用于电子电器产品、家具、油漆、纺织品等日用品中. TBBPA在生产和使用过程中可通过多种途径进入环境. 研究表明,TBBPA具有持久性有机污染物的特征,具有生物累积性、高度亲脂性和难分解性,具有内分泌干扰效应、细胞和神经毒性、免疫毒性等,会对生态系统和人类健康构成严重的威胁[1-4]. TBBPA微溶于水且容易在土壤中聚集,土壤是TBBPA最重要的归宿之一. 近年来,如何有效降解和去除土壤中TBBPA的研究备受关注 .
目前,土壤有机污染物修复技术包括物理修复[5]、生物修复[6-8]和化学修复[9-10],其中化学修复因其低投资、高效率、实施周期短的优点应用广泛. 过硫酸盐(PS)高级氧化技术是最常用的一种化学氧化修复技术,该技术的关键是通过热[11]、紫外光[12]和通过添加过渡金属[13]等方式活化PS. 热活化PS体系中虽然操作简单,但能耗较高,不适用大规模的处理. 在过渡金属活化PS中,亚铁离子(Fe2+)和铁离子(Fe3+)由于含量丰富、环境友好、价格低廉而被广泛用于PS活化,然而Fe2+活化PS技术仍然有一些缺陷,过量的Fe2+会抑制PS的活化,Fe3+还原为Fe2+的速率较慢,难以控制氧化体系中的量[14].
为了提高PS活化性能,本文以典型溴代阻燃剂TBBPA为目标污染物,考虑将Fe2+活化与热活化结合,采用Fe2+耦合热活化PS处理土壤中TBBPA,考察温度、Fe2+初始浓度、PS浓度、初始pH、无机阴离子和其他金属离子对Fe2+/热/PS体系降解TBBPA的影响,以寻求土壤体系中TBBPA降解的最佳反应条件,为土壤中持久性有机污染物的修复提供依据.
1. 材料与方法(Materials and methods)
1.1 实验试剂
TBBPA购于上海麦克林生化科技有限公司,甲醇购于美国迈瑞达科技公司,过硫酸钠、五水合硫酸铜、七水合硫酸锌和六水合硫酸镍购于天津津科精细化工研究所,氯化钠、碳酸氢钠、氢氧化钠和七水合硫酸亚铁购于国药集团化学试剂有限公司,浓硫酸购于北京化工厂. 以上药品除甲醇为色谱纯外,其余药品均为分析纯. 实验溶液配制用水为去离子水,高效液相仪器用水为哇哈哈纯净水.
1.2 实验仪器
LC-20AT 高效液相色谱仪(日本Shimadzu公司);Optima-XPN PN-100 高速离心机(BECKMANCOULTER公司);S400-B多参数pH测试仪(梅特勒-托利多仪器有限公司);FD-A10N-50冷冻干燥机(北京博医康实验仪器有限公司);HH-1-5L恒温水浴锅(江苏科析仪器有限公司);旋转蒸发仪(上海申生科技有限公司);SHA-2恒温振荡器(常州菲普实验仪器厂).
1.3 土壤的采集
供试土壤采自安徽省合肥市瑶海区某农田(117°21'53.424"E, 31°51'23.886"N),采集深度0—20 cm. 去除碎石、树枝杂质,经室内自然干燥,研磨后过60目筛网,储存于棕色玻璃瓶中密封保存. 经检验,土壤中无TBBPA检出. 该土壤基本理化性质见表1.
表 1 土壤基本理化性质Table 1. Basic physicochemical properties of soilpH 孔隙比 Porosity ratio 饱和度/% Saturation 颗粒成分组成/% Particle composition <0.005 mm 0.005—0.075 mm 0.075—0.25 mm 0.25—0.5 mm 7.10 1.50 95.1 13.3 17.7 47.7 21.3 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.4 污染土制备
TBBPA配制:精确称取0.250 g TBBPA粉末溶解于500 mL甲醇中,TBBPA浓度为500 mg·L−1.
TBBPA污染土:称取500 g未污染的土壤于2 L烧杯中,加入200 mL甲醇,玻璃棒搅拌均匀后,取30 mL TBBPA(500 mg·L−1)缓缓倒入烧杯中,边搅拌使之充分混匀,接着加甲醇直至液体浸没土壤表面,用玻璃棒搅拌20—30 min,使TBBPA在土壤中均匀混合,置于通风橱中自然风干、老化2周后备用(测定TBBPA约20 mg·kg−1).
1.5 实验方法
实验选用过硫酸钠(PS)氧化剂,过渡金属离子选用Fe2+,无机阴离子选用Cl−、
HCO−3 表 2 单因素实验参数变量Table 2. One-way experimental parameter variables单因素名称 Single factor name 参数变量 Parameter variable 温度/℃ 25 35 45 55 65 PS浓度/(mmol·L−1) 15 25 50 70 100 Fe2+浓度/(mmol·L−1) 15 25 50 70 100 初始pH 3.0 5.0 7.0 9.0 — PS与无机阴离子浓度摩尔比 1:0.1 1:1 1:2 — — Fe2+与其他金属离子浓度摩尔比 1:0.1 1:1 1:2 — — | Show TableDownLoad:
CSV
称取2.000 g TBBPA污染土壤于30 mL棕色玻璃瓶中,加入2 mL去离子水和2 mL 25 mmol·L−1的硫酸亚铁溶液,再加入2 mL 50 mmol·L−1的PS储备溶液,使用1 mol·L−1稀硫酸和氢氧化钠溶液调节初始pH为3.0,随即用橡胶塞密封,置于温度55 ℃的恒温振荡器中(转速200 r·min−1)12 h. 预定时间间隔(0.05、0.25、0.5、1、2、4、6、8、12 h)取出放入冰水中(5 min)并加入0.2 mL甲醇,随后将样品放入-20 ℃的冰箱中冷冻4 h后放入冷冻干燥机中冷冻干燥48 h,经预处理,测定土壤中TBBPA浓度.
1.6 土壤样品的预处理
将10 mL甲醇溶剂加入待测棕色玻璃瓶中,置于25 ℃恒温振荡器中振荡8 h后,在剩余土壤中加入10 mL甲醇超声辅助萃取,萃取1 h,转移至离心管分离土壤与上清液. 随后,将离心管中的上清液经滤纸自然过滤转移至茄形烧瓶中,利用旋转蒸发仪将烧瓶中的萃取液旋转蒸发至1 mL,再用甲醇溶液重复多次提取至5 mL容量瓶,定容到刻度线,摇晃均匀,将容量瓶中的液体通过孔隙为0.22 μm的有机滤膜过滤,收集1.5 mL滤液于棕色进样瓶瓶中,用HPLC进行定量分析.
1.7 分析方法
(1) TBBPA的测定
本实验采用高效液相色谱仪(HPLC)测定土壤中的TBBPA. HPLC分析条件为:Zorbax Eclipse XDB-C18色谱柱(5 μm,250 mm×4.6 mm),流动相为甲醇:超纯水=85:15,检测波长为230 nm,流速为1.0 mL·min−1,进样体积为10 μL,柱温35 ℃. 对标样进行定性分析,TBBPA在该分析条件下的保留时间为(5.5±0.2) min[10].
(2) 数据分析方法
降解率W计算公式如下所示(公式1),采用准一级反应动力学和二级反应动力学模型(公式2、3)对TBBPA降解进行分析拟合.
W=(1−CtC0)×100% (1) lnCtC0=−kt (2) 1Ct−1C0=kt (3) 式中,t为反应时间(h),C0与Ct分别表示0 h和t h时TBBPA的浓度(mg·kg−1),k为拟合得到的反应动力学常数(h−1).
2. 结果与讨论(Results and discussion)
2.1 Fe2+耦合热活化PS降解TBBPA
比较了加热、热/Fe2+、热/PS、Fe2+/PS和Fe2+/热/PS体系对土壤中TBBPA降解效果的影响(图1),TBBPA在加热、热/Fe2+、热/PS和Fe2+/PS体系中12 h内的降解率分别为25.0%、32.0%、47.3%和89.9%. 由图可以看出,升高温度对土壤中TBBPA有一定的影响;在热/Fe2+的体系下,TBBPA降解速率小;热/PS和Fe2+/PS体系下TBBPA降解速率有进一步提升,原因主要是热和Fe2+都能活化PS产生较多的
SO·−4  图 1 TBBPA在不同体系中的降解效果Figure 1. The degradation efficiency of TBBPA in different reaction systems A control experiment was temperature=25 ℃, without Fe2+ and PS对照组条件CK为温度=25 ℃,未添加Fe2+和未添加PS;[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, pH=3.0, T=55 ℃, t=12 h
图 1 TBBPA在不同体系中的降解效果Figure 1. The degradation efficiency of TBBPA in different reaction systems A control experiment was temperature=25 ℃, without Fe2+ and PS对照组条件CK为温度=25 ℃,未添加Fe2+和未添加PS;[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, pH=3.0, T=55 ℃, t=12 h2.2 TBBPA在Fe2+/热/PS体系中降解的影响因素
(1) 温度
不同温度(25 ℃、35 ℃、45 ℃、55 ℃、65 ℃)对TBBPA在Fe2+/热/PS体系中降解的影响(如图2). 结果表明,TBBPA的降解效率随温度的升高而显著增加,在12 h内25 ℃、35 ℃、45 ℃时TBBPA的降解率分别为89.91%、91.35%、96.10%,55 ℃和65 ℃时降解率则在8 h已经达到100%. 这是由于在Fe2+活化过硫酸盐产生
SO·−4 SO·−4 SO·−4 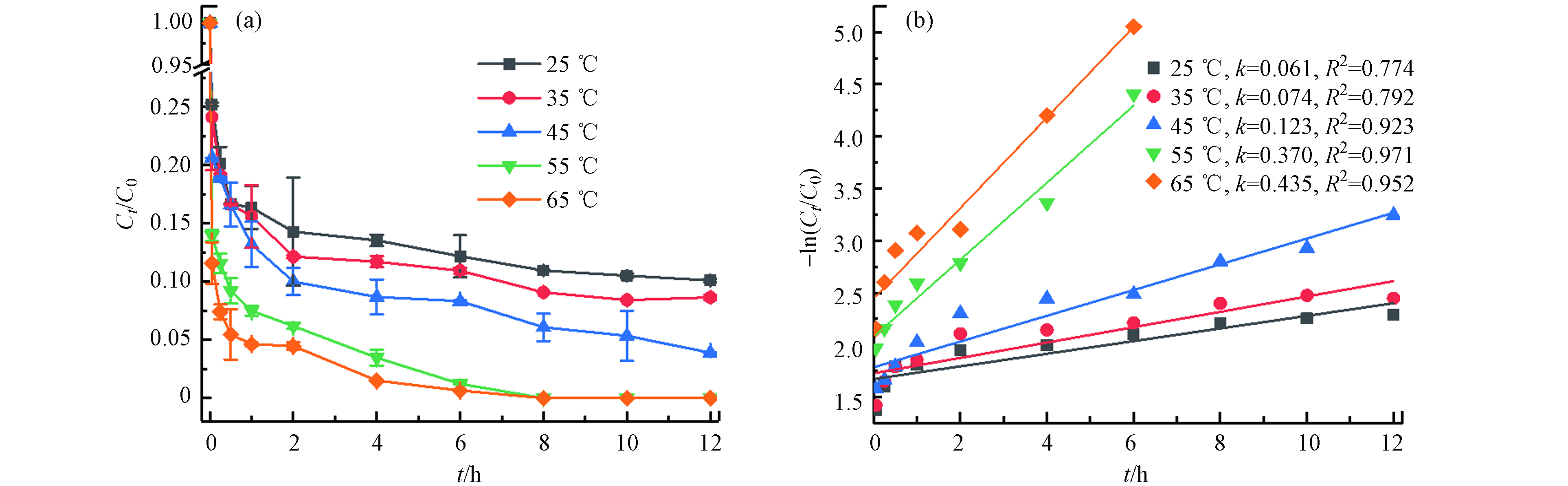 图 2 温度对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, pH=3.0, T=25-55 ℃, t=12 hFigure 2. Effects of temperature on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPA
图 2 温度对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, pH=3.0, T=25-55 ℃, t=12 hFigure 2. Effects of temperature on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPAFe2++S2O2−8→Fe3++SO2−4+SO•−4 (4) S2O2−8+heat→2SO•−4 (5) (2) Fe2+初始浓度
在温度55 ℃、PS浓度为50 mmol·L−1、pH=3的条件下,考察不同初始Fe2+浓度(0、15、25、50、 70、100 mmol·L−1)对土壤中TBBPA降解的影响. 如图3所示,未投加Fe2+时,此时反应仅是热活化反应降解,12 h内TBBPA降解率为47.3%,当Fe2+与PS摩尔比从0.3:1增加到0.5:1,TBBPA降解率从89.6%增加到100%;而继续增大Fe2+与PS摩尔比至2:1,TBBPA的降解效率开始下降,反应结束时仅有31.1%. 由图3(b)可以看出,TBBPA的降解较好拟合二级反应动力学,降解速率常数先增大,然后逐渐减小. 可见,添加Fe2+能够有效活化PS降解土壤中的TBBPA,在Fe2+浓度一定范围内,适当提高Fe2+浓度,能加速TBBPA的降解,在Fe2+与PS摩尔比为0.5:1,降解率最大,随着Fe2+浓度增加,过量的Fe2+也能分解
SO·−4 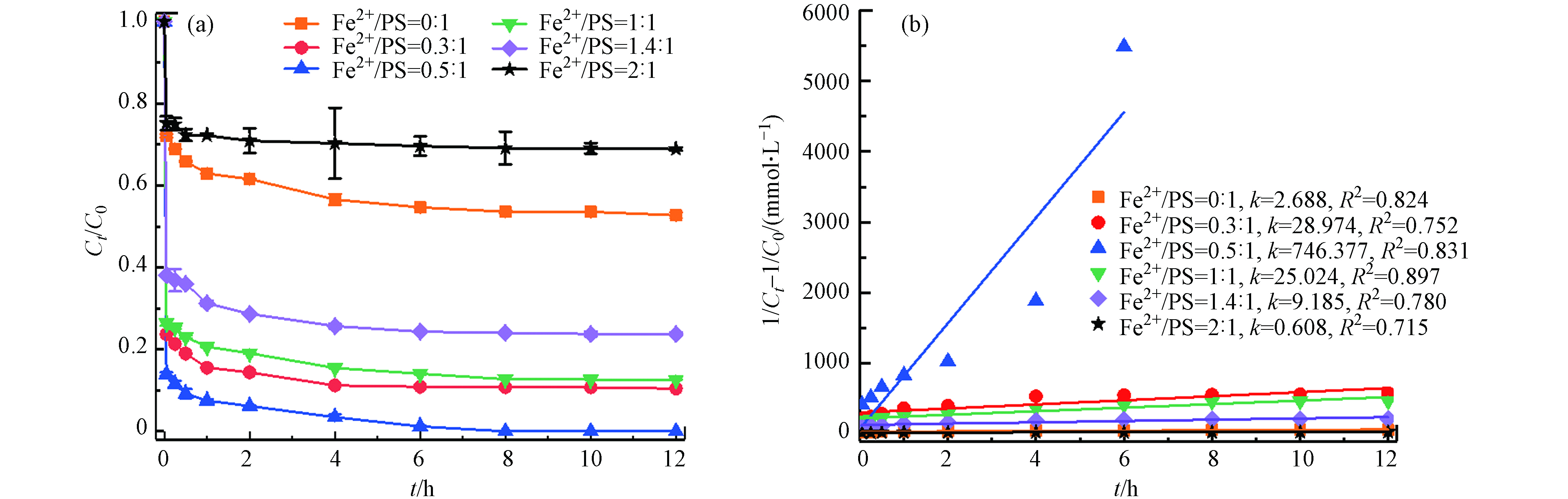 图 3 Fe2+初始浓度对TBBPA降解的影响(a)及二级动力学模型拟合曲线(b)Figure 3. Effects of initial Fe2+ concentration on degradation efficiency (a) and second order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=0—100 mmol·L−1, [PS]0=50 mmol·L−1, pH=3.0, T=55 ℃, t=12 h
图 3 Fe2+初始浓度对TBBPA降解的影响(a)及二级动力学模型拟合曲线(b)Figure 3. Effects of initial Fe2+ concentration on degradation efficiency (a) and second order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=0—100 mmol·L−1, [PS]0=50 mmol·L−1, pH=3.0, T=55 ℃, t=12 hFe2++SO•−4→Fe3++SO2−4 (6) (3) PS浓度
在温度55 ℃、Fe2+浓度为25 mmol·L−1、pH=3的条件下,考察不同PS浓度(15、25、50、70、100 mmol·L−1)对土壤中TBBPA降解的影响. 如图4所示,随着PS浓度的增加,12 h内TBBPA的去除效率从50.1%提高到100%,降解速率常数也从0.014 h−1增至1.135 h−1;TBBPA完全去除所需的时间随PS浓度的增大而缩短,PS浓度50 mmol·L−1、70 mmol·L−1和100 mmol·L−1降解率达到100%的时间分别为8 h、6 h和4 h. 由此可见,增加PS浓度对TBBPA的降解有促进作用. 有研究表明,过量的PS会抑制污染物的降解[18],因为是过量
S2O2−8 SO·−4 SO·−4  图 4 PS浓度对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)Figure 4. Effects of PS concentration on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=15—100 mmol·L−1, pH=3.0, T=55 ℃, t=12 h
图 4 PS浓度对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)Figure 4. Effects of PS concentration on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=15—100 mmol·L−1, pH=3.0, T=55 ℃, t=12 hSO•−4+S2O2−8→•S2O2−8+SO2−4 (7) SO•−4+SO•−4→S2O2−8 (8) (4) 初始pH值
在温度55 ℃,Fe2+与PS最佳摩尔比0.5:1条件下,进一步研究了不同初始pH值(3.0、5.0、7.0和9.0)对Fe2+耦合热活化PS体系降解TBBPA的影响. 如图5所示,当初始pH分别为3.0、5.0、7.0和9.0时,反应12 h后,TBBP降解率分别为100%、99.62%、99.68%和99.66%,随着pH从3增加到9,反应速率常数从0.370 h−1减小到0.316 h−1,酸性条件下的降解率略高于中性和碱性条件,初始pH为5.0、7.0和9.0时降解效率之间相差不大. 出现这种趋势的原因是,在酸性条件下存在一定的酸催化效应(公式9、10)[19];硫酸亚铁溶液和过硫酸盐溶液自身呈酸性;两者在反应开始前3 min时较为迅速,PS很容易氧化Fe2+成为Fe3+,Fe3+水解也会使pH值降低. 总体来说,Fe2+耦合热活化PS体系具有良好的pH适应性,pH=3.0时最为合适.
 图 5 初始pH对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)Figure 5. Effects of initial pH on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, pH=3.0—5.0, T=55 ℃, t=12 h
图 5 初始pH对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)Figure 5. Effects of initial pH on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, pH=3.0—5.0, T=55 ℃, t=12 hS2O2−8+H+→HS2O−8 (9) HS2O−8→SO2−4+SO•−4+H+ (10) 2.3 无机阴离子在Fe2+/热/PS体系中的影响
在场地土壤和地下水的复杂系统中,可能存在无机阴离子对过硫酸盐降解污染物的过程产生一定制约[20]. 因此我们选取氯化钠和碳酸氢钠分别作为Cl−和
HCO−3 HCO−3 (1) Cl−
在最佳反应条件下(温度55 ℃、Fe2+浓度25 mmol·L−1、PS浓度50 mmol·L−1、pH=3),不同浓度的Cl−(0、5、50、100 mmol·L−1)对TBBPA降解的影响(如图6). 反应至4 h时,TBBPA的降解率分别为96.5%、96.2%、98.4%和99.4%;反应至12 h时,降解率分别为100%、99.7%、100%和100%,随着Cl−浓度从0 mmol·L−1增加至100 mmol·L−1,TBBPA完全去除所需时间缩短了2 h. 由图6(b)所示,反应符合准一级动力学模型,其对应的反应动力学常数分别为0.369 h−1、0.367 h−1、0.491 h−1和0.707 h−1. 当Cl−浓度较低时,Cl−与
SO·−4  图 6 Cl-对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)Figure 6. Effects of Cl- on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, [Cl-]0=0—100 mmol·L−1, pH=3.0, T=55 ℃, t=12 h
图 6 Cl-对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)Figure 6. Effects of Cl- on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, [Cl-]0=0—100 mmol·L−1, pH=3.0, T=55 ℃, t=12 hSO•−4+Cl−→Cl•+SO2−4 (11) (2)
HCO−3 在最佳反应条件下(温度55 ℃、Fe2+浓度25 mmol·L−1、PS浓度50 mmol·L−1、pH=3),不同浓度的
HCO−3 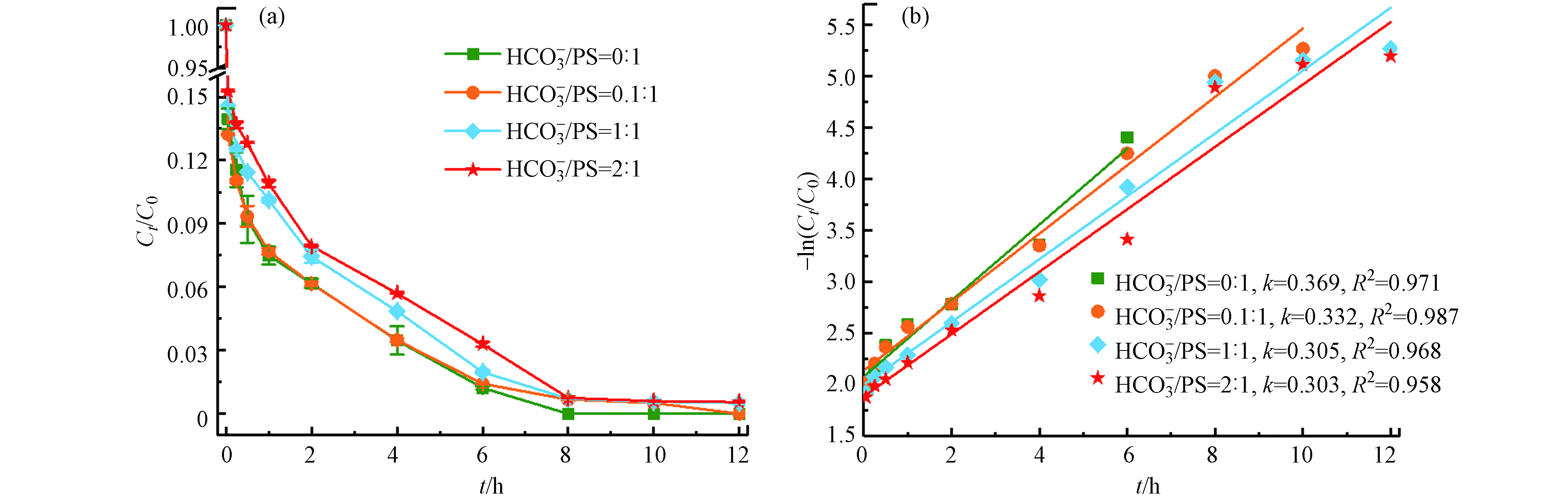 图 7 HCO3-对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)Figure 7. Effects of HCO3- on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, [HCO3-]0=0—100 mmol·L−1, pH=3.0, T=55 ℃, t=12 h
图 7 HCO3-对TBBPA降解的影响(a)及准一级动力学模型拟合曲线(b)Figure 7. Effects of HCO3- on degradation efficiency (a) and pseudo-first-order kinetic plots (b) of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, [HCO3-]0=0—100 mmol·L−1, pH=3.0, T=55 ℃, t=12 h随着
HCO−3 HCO−3 HCO−3 CO2−3 SO⋅−4 HCO⋅3 CO⋅−3 HCO−3 HCO−3→CO2−3+H+ (12) SO⋅−4+HCO−3→SO2−4+HCO⋅3 (13) SO⋅−4+CO2−3→SO2−4+CO⋅−3 (14) 2.4 金属离子Cu2+、Zn2+、Ni2+对Fe2+/热/PS体系催化活性的影响
土壤中存在着大量的金属矿物,多种金属的存在也是影响应用的重要因素[23-24]. 本文选择土壤中的几种过渡金属(Cu2+、Zn2+、Ni2+)考察其对Fe2+/热/PS体系降解土壤中TBBPA的影响. 在体系最佳反应条件下(温度55 ℃、Fe2+浓度25 mmol·L−1、PS浓度50 mmol·L−1、pH=3),分别加入M2+/Fe2+摩尔比为0:1、0.1:1、1:1、2:1(M=Cu、Zn、Ni)的过渡金属离子,反应12 h后计算TBBPA降解效率. 结果如图8所示,Cu2+对Fe2+/热/PS体系表现出较好的促进作用,且随着Cu2+浓度的增大,TBBPA完全去除所需的时间从8 h(Cu2+/Fe2+摩尔比0:1)缩短到1 h(Cu2+/Fe2+摩尔比2:1);当增加Ni2+与Fe2+的摩尔比时,Ni2+显现出的促进作用由弱变强;而逐渐增大Zn2+与Fe2+的摩尔比时,对反应速率的影响出现由抑制转变成促进的双重作用. 这可能与M2+/M3+的氧化还原电位有关,△Ep(Cu2+/Cu3+)<△Ep(Ni2+/Ni3+)<△Ep(Zn2+/Zn3+),电位越低意味着催化剂越容易得到电子,有利于PS的活化[25];同时由于金属离子的氧化性Cu2+>Ni2+>Fe2+>Zn2+,Cu2+和Ni2+则更容易促使PS产生更多
SO⋅−4 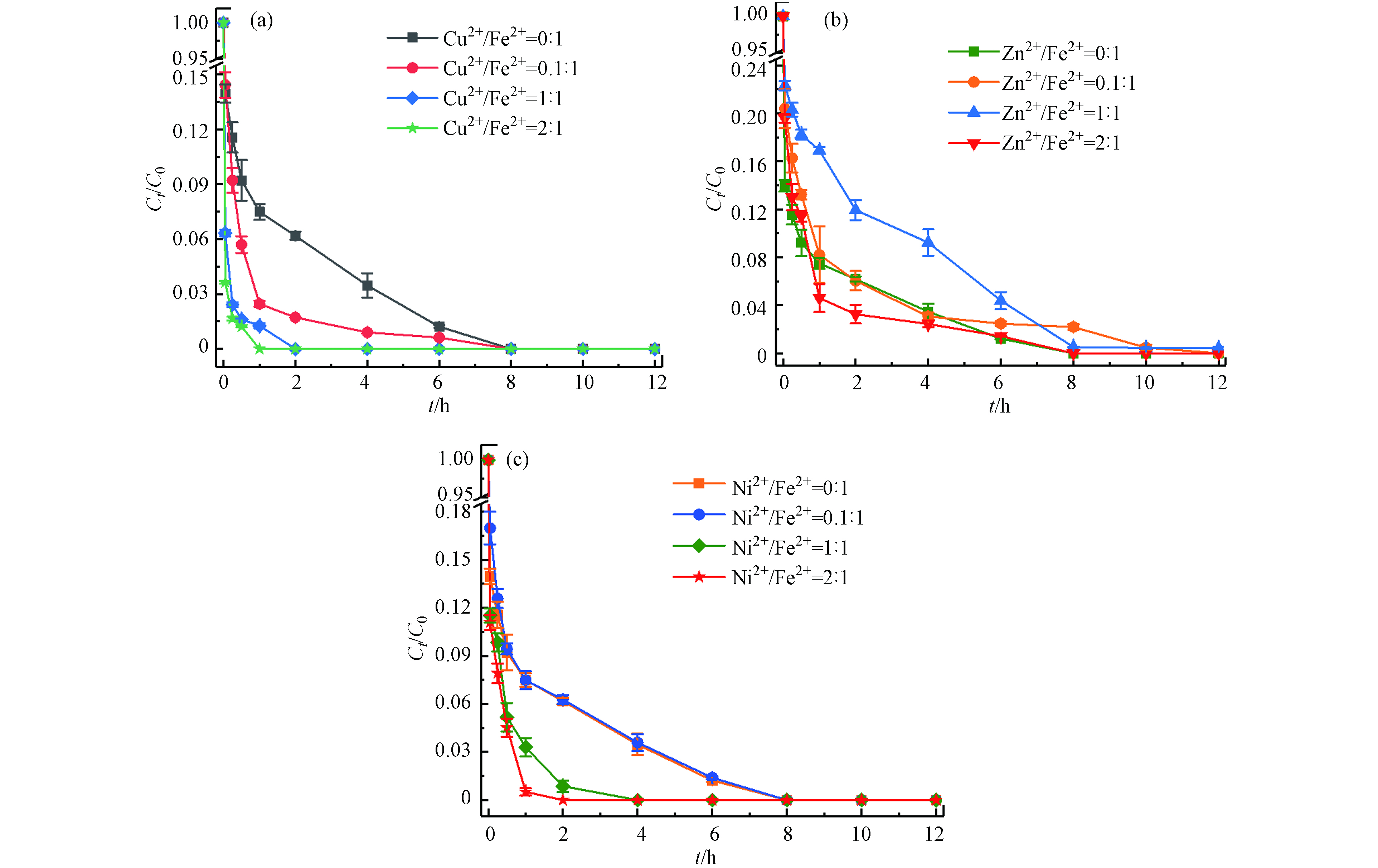 图 8 Cu2+(a)、Zn2+(b)和Ni2+(c)分别对TBBPA降解的影响Figure 8. Effects of Cu2+(a)、Zn2+(b) and Ni2+ (c)on degradation efficiency of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, [Cu2+]0=0-50 mmol·L−1, [Zn2+]0=0-50 mmol·L−1, [Ni2+]0=0—50 mmol·L−1, pH=3.0, T=55 ℃, t=12 h
图 8 Cu2+(a)、Zn2+(b)和Ni2+(c)分别对TBBPA降解的影响Figure 8. Effects of Cu2+(a)、Zn2+(b) and Ni2+ (c)on degradation efficiency of TBBPA[TBBPA]0=20 mg·kg−1, [Fe2+]0=25 mmol·L−1, [PS]0=50 mmol·L−1, [Cu2+]0=0-50 mmol·L−1, [Zn2+]0=0-50 mmol·L−1, [Ni2+]0=0—50 mmol·L−1, pH=3.0, T=55 ℃, t=12 h3. 结论(Conclusion)
(1)Fe2+耦合热活化过硫酸盐的方法能高效降解土壤中TBBPA. Fe2+耦合热活化过硫酸盐降解土壤中TBBPA相比于单一热活化和Fe2+活化方式,降解率分别提升52.7%和10.1%.
(2)对Fe2+耦合热活化PS降解TBBPA的影响因素研究表明,TBBPA的降解率随着温度的增加而升高;增大亚铁离子浓度,TBBPA的降解速率呈现出先增加后降低的趋势,Fe2+与PS的最佳摩尔比为0.5:1;增大PS浓度能够显著提高TBBPA的降解率,缩短TBBPA去除时间;Fe2+耦合热活化PS体系具有较宽的pH应用范围且在酸性条件下更有利于降解. 最优条件:温度55 ℃、Fe2+浓度25 mmol·L−1、PS浓度50 mmol·L−1、pH=3.
(3)较高浓度的Cl-对Fe2+/热/PS体系中TBBPA的降解起促进作用,而HCO3-在研究浓度范围内均呈现抑制作用;Cu2+和Ni2+促进Fe2+/热/PS体系对TBBPA的降解,而随着Zn2+浓度的增加,TBBPA的降解速率呈现出先减小后增大的变化.
-
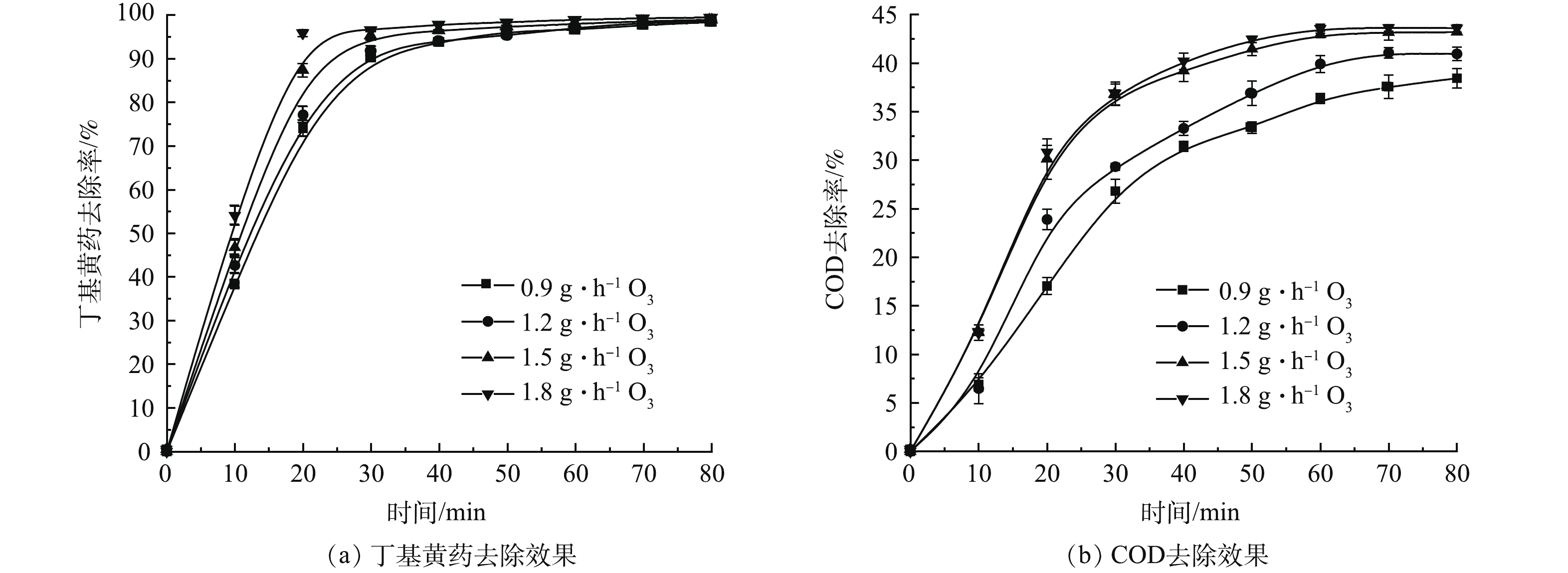
图 2 单独O3处理对丁基黄药及COD的去除效果
Figure 2. Effect of O3 treatment alone on butyl xanthate and COD removal
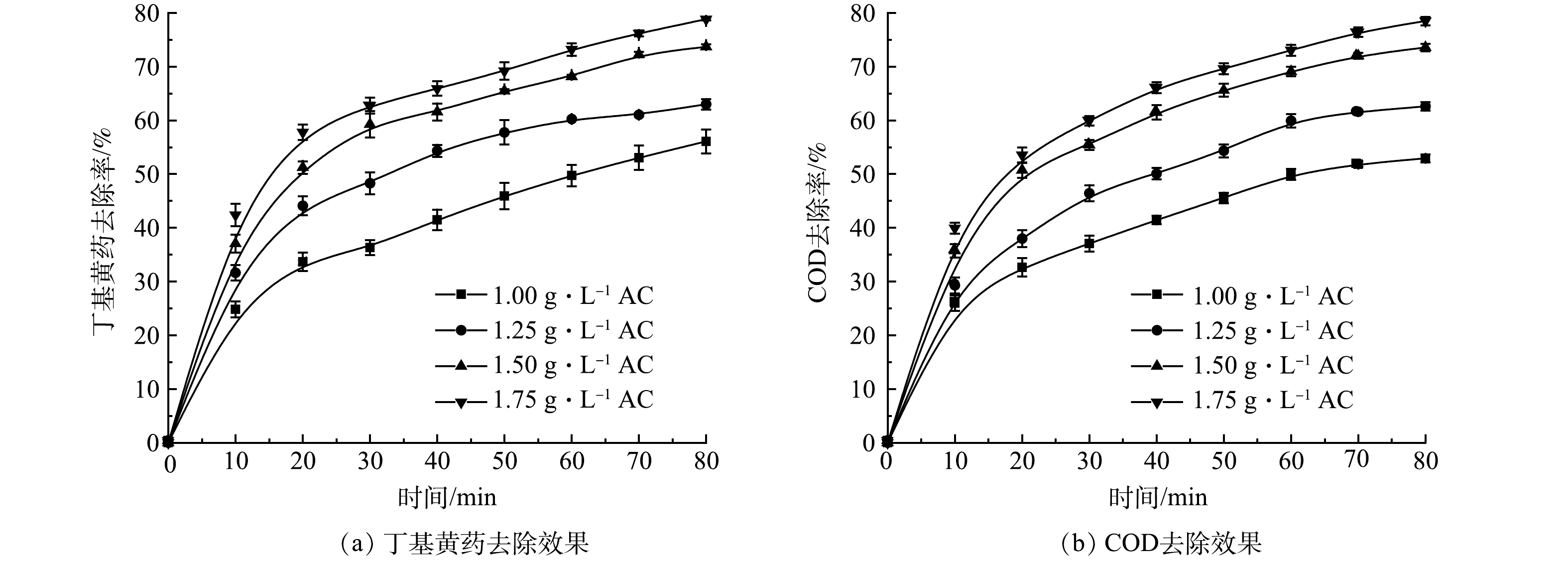
图 3 单独AC处理对丁基黄药及COD的去除效果
Figure 3. Effect of AC treatment alone on butyl xanthate and corresponding COD removal
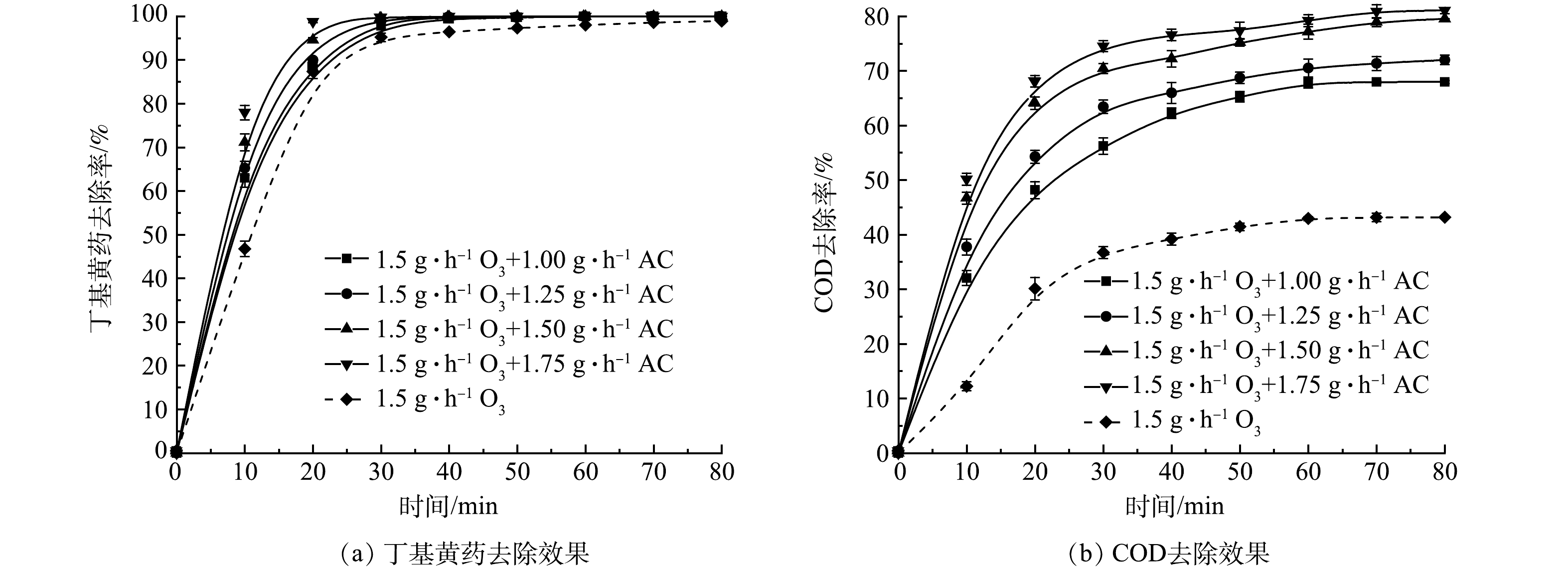
图 4 AC投加量对O3/AC耦合工艺同步去除效果的影响
Figure 4. Effect of AC dosage on the co-removal effect by the O3/AC coupling process
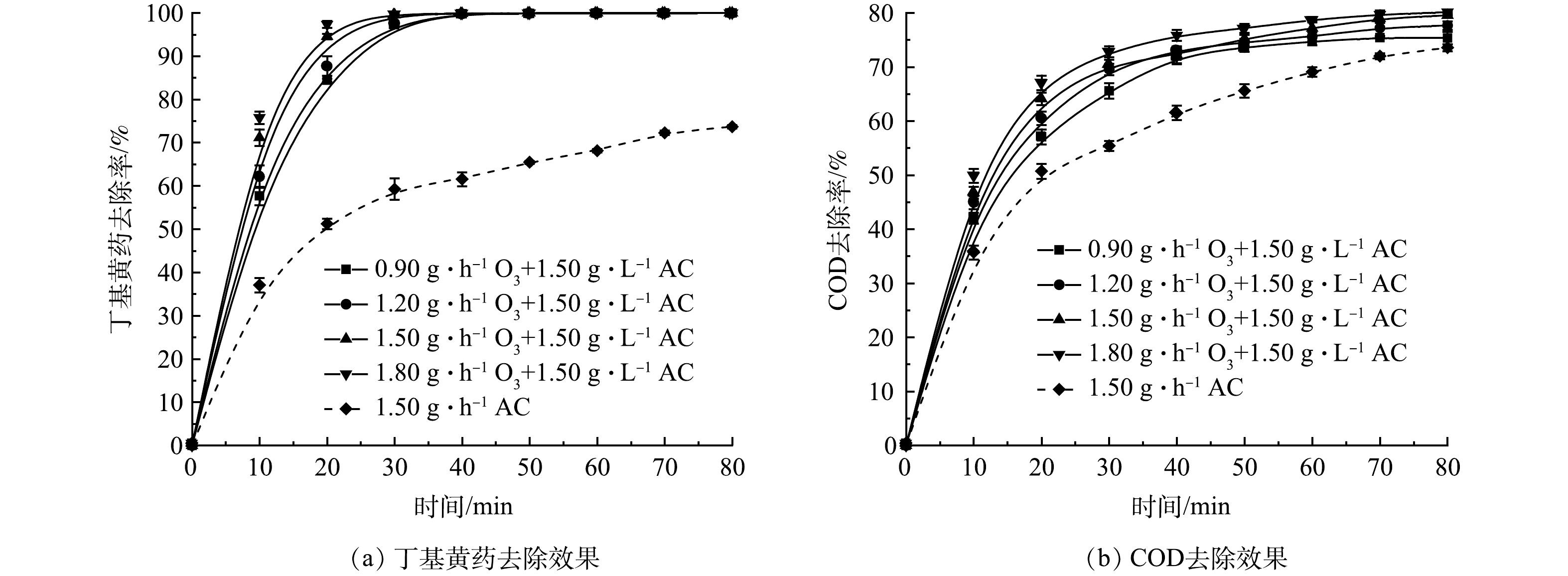
图 5 O3投加量对O3/AC耦合工艺同步去除效果的影响
Figure 5. Effect of O3 dosage on the co-removal effect by the O3/AC coupling process
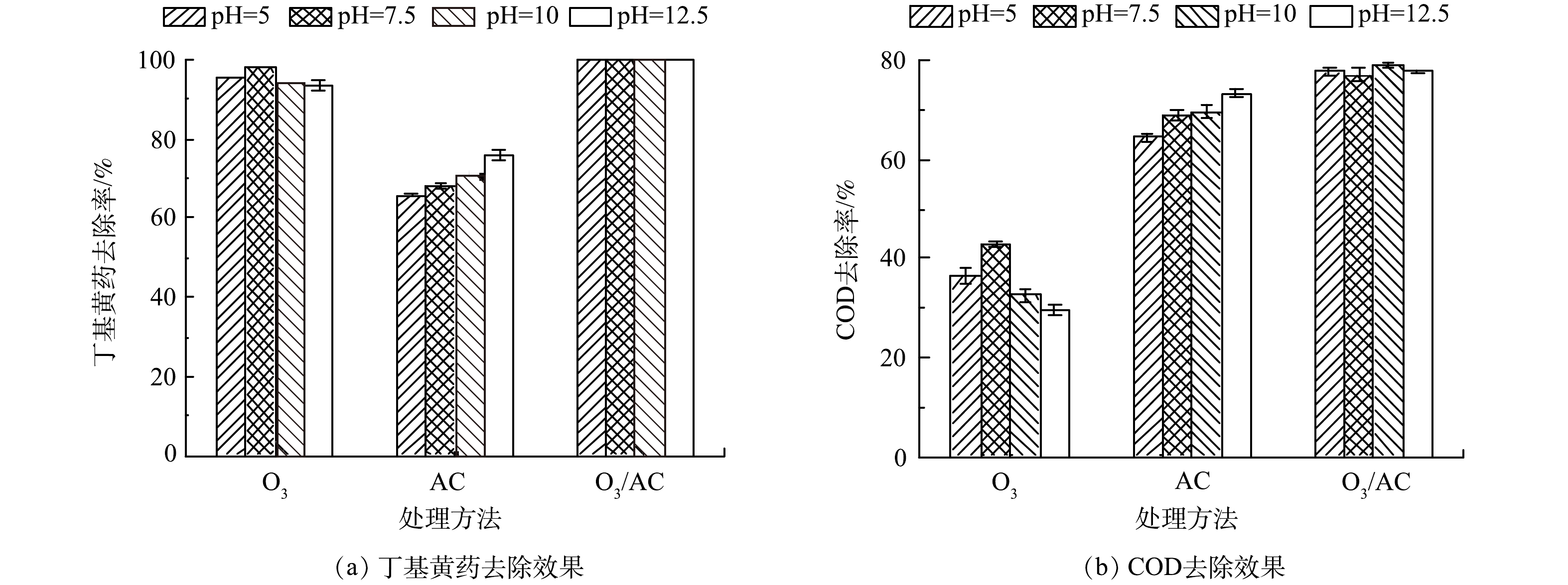
图 6 初始pH对O3、AC及O3/AC工艺同步去除效果的影响
Figure 6. Effect of initial pH on the co-removal effect by O3, AC and O3/AC process
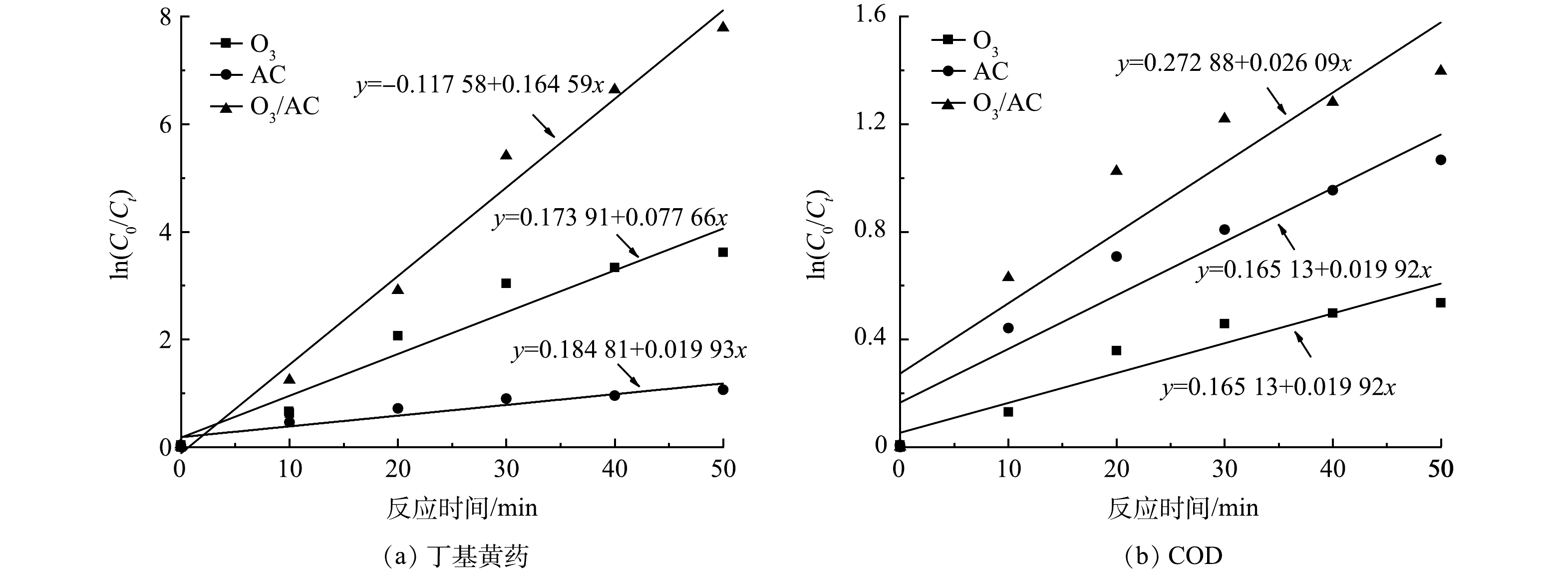
图 7 O3、AC及O3/AC处理丁基黄药与COD的拟一级反应动力学拟合图
Figure 7. Fitting curve of quasi-first-order reaction kinetics of O3, AC and O3/AC treating butyl xanthate and COD
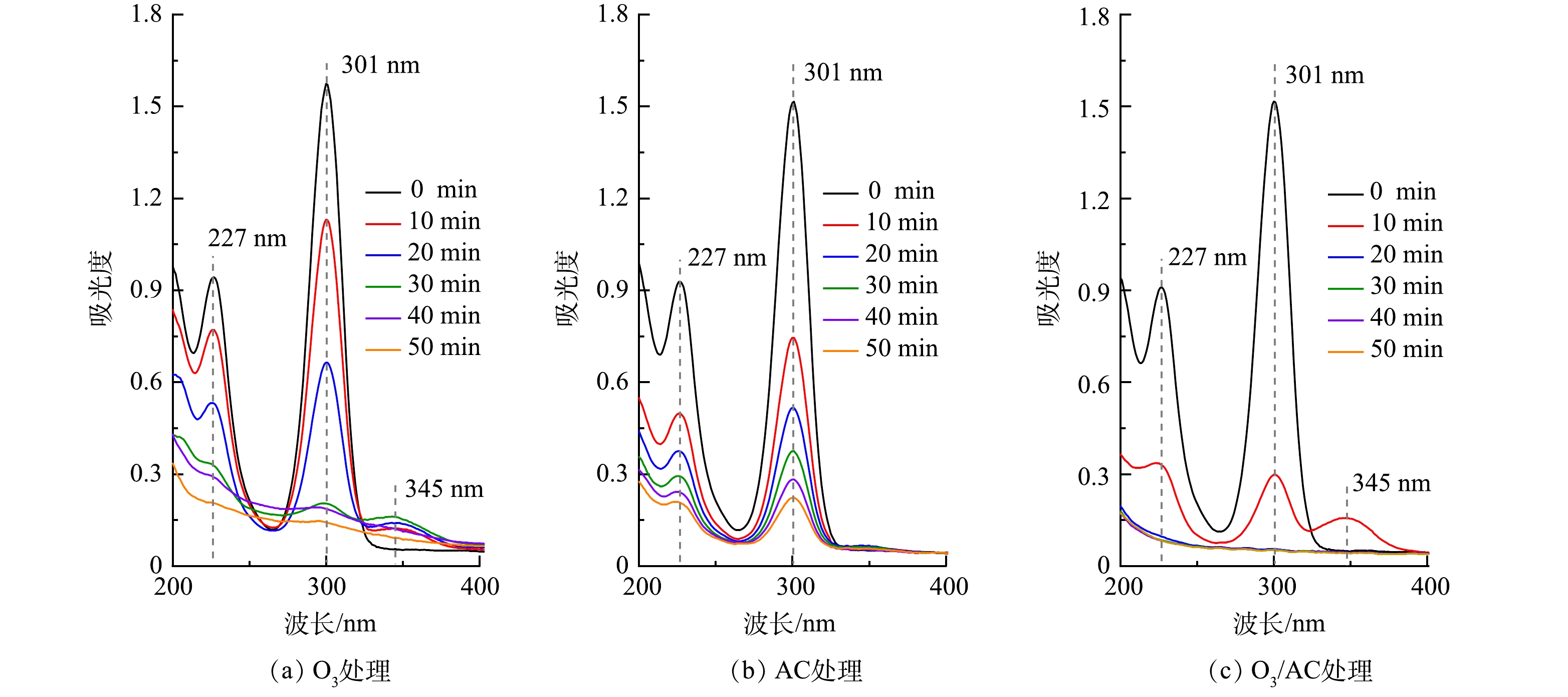
图 9 不同方法处理丁基黄药过程中紫外-可见光谱
Figure 9. UV-Vis spectra during butyl xanthate removal by different methods
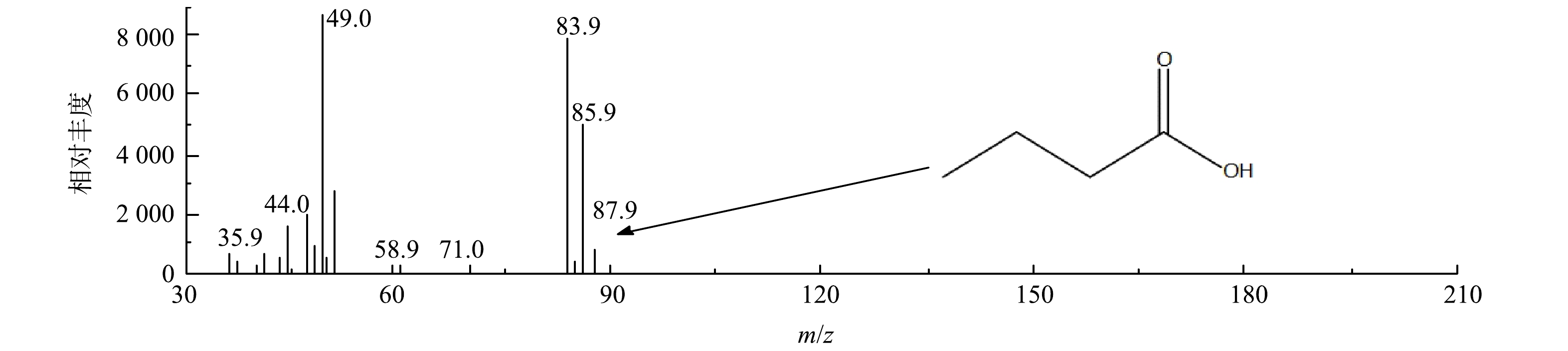
图 10 O3/AC耦合工艺处理丁基黄药反应中间产物质谱扫描分析图
Figure 10. Scanning analysis of mass spectrum of intermediate production of butyl xanthate reaction treated by O3/AC coupling process
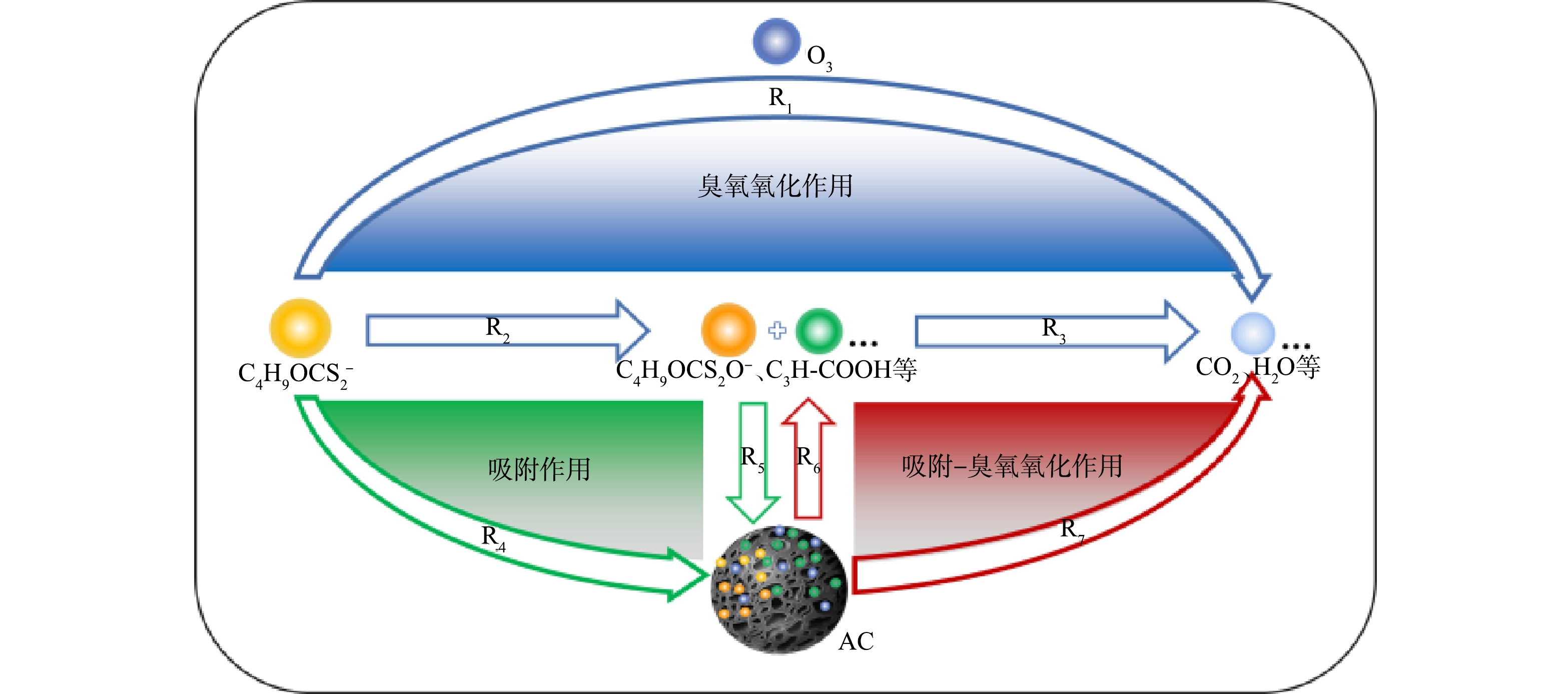
图 11 O3/AC耦合工艺处理丁基黄药降解路径推测图
Figure 11. Estimation of degradation pathway of butyl xanthate treated by O3/AC coupling process
表 1 反应动力学参数表
Table 1. Reaction kinetics parameter table
处理方法 k/min−1 R2 调整后R2 O3 丁基黄药 0.078 0.935 0.919 COD 0.011 0.910 0.887 AC 丁基黄药 0.020 0.889 0.862 COD 0.020 0.914 0.893 O3/AC 丁基黄药 0.165 0.987 0.983 COD 0.026 0.858 0.823
下载: 导出CSV
-
[1] 陈运双, 马瑞雪, 蒋潇宇, 等. TiO2/蒙脱土复合材料光催化降解丁基黄药性能研究[J]. 金属矿山, 2022(5): 212-220. [2] AMROLLAHI A, MASSINAEI M, MOGHADDAM A Z. Removal of the residual xanthate from flotation plant tailings using bentonite modified by magnetic nano-particles[J]. Minerals Engineering, 2019, 134: 142-155. doi: 10.1016/j.mineng.2019.01.031 [3] HAI L, KANGJIA Q, YINGBO D, et al. A newly-constructed bifunctional bacterial consortium for removing butyl xanthate and cadmium simultaneously from mineral processing wastewater: Experimental evaluation, degradation and biomineralization[J]. Journal of Environmental Management, 2022, 316: 115304. doi: 10.1016/j.jenvman.2022.115304 [4] FU P, FENG J, YANG H, et al. Degradation of sodium n-butyl xanthate by vacuum UV-ozone (VUV/O3) in comparison with ozone and VUV photolysis[J]. Process Safety and Environmental Protection, 2016, 102: 64-70. doi: 10.1016/j.psep.2016.02.010 [5] GARCÍA-LEIVA B, TEIXEIRA L A C, TOREM M L. Degradation of xanthate in waters by hydrogen peroxide, fenton and simulated solar photo-fenton processes[J]. Journal of Materials Research and Technology, 2019, 8(6): 5698-5706. doi: 10.1016/j.jmrt.2019.09.037 [6] 梁锐, 李明阳, 高翔鹏, 等. 选矿废水中残留黄药光催化处理及降解效率改进方式研究进展[J]. 过程工程学报, 2022, 22(1): 1-13. [7] JUDITH G, MICHAEL S, PATRICK B. Influence of chemical structure of organic micropollutants on the degradability with ozonation[J]. Water Research, 2022, 222: 118866. doi: 10.1016/j.watres.2022.118866 [8] 马宏涛, 孙水裕, 许明鑫. 臭氧联合混凝沉淀法去除浮选废水中有机磷[J]. 环境工程学报, 2017, 11(1): 285-290. doi: 10.12030/j.cjee.201508150 [9] 张萌, 柳建设. 臭氧降解选矿药剂丁基黄药的实验研究[J]. 环境工程学报, 2011, 5(12): 2712-2716. [10] 付凯, 刘志红, 耿超, 等. 活性炭吸附磷矿浮选废水中十二胺的研究[J]. 水处理技术, 2019, 45(3): 93-96. doi: 10.16796/j.cnki.1000-3770.2019.03.019 [11] 程伟, 张覃, 马文强. 活性炭对浮选废水中黄药的吸附特性研究[J]. 矿物学报, 2010, 30(2): 262-267. doi: 10.16461/j.cnki.1000-4734.2010.02.017 [12] 刘楚玉, 黄自力, 袁晨光, 等. 磁性活性炭的制备及其对选矿废水中丁基黄药的去除研究[J]. 矿冶工程, 2022, 42(3): 70-75. doi: 10.3969/j.issn.0253-6099.2022.03.016 [13] 刘冰, 郑煜铭, 陈燕敏, 等. 臭氧-活性炭处理高浓度制药废水作用机制研究[J]. 环境科学与技术, 2021, 44(2): 122-130. doi: 10.19672/j.cnki.1003-6504.2021.02.016 [14] 杜明辉, 毕莹莹, 董莉, 等. 活性炭粒径对O3-AC处理有机废水的影响机制[J]. 环境工程, 2022, 40(4): 22-28. [15] 彭然, 张汉泉, 张亚平, 等. O3/H2O2去除选矿废水中丁基黄药的研究[J]. 金属矿山, 2011(11): 155-158. [16] 张萌, 田世烜, 张诚, 等. O3/H2O2法去除浮选药剂丁基黄药[J]. 环境工程学报, 2012, 6(3): 729-733. [17] 陈港权. 丁基二硫代碳酸钾臭氧氧化过程中C与S迁移特征的研究[D]. 广州: 广东工业大学, 2015. [18] 邓磊, 蒋姝, 黄文章, 等. 臭氧—Fenton联合氧化处理钻井液废水研究[J]. 工业水处理, 2018, 38(2): 44-47. doi: 10.11894/1005-829x.2018.38(2).044 [19] YAN P, CHEN G, YE M, et al. Oxidation of potassium n -butyl xanthate with ozone: Products and pathways[J]. Journal of Cleaner Production, 2016, 139: 287-294. doi: 10.1016/j.jclepro.2016.08.027 [20] ALEJANDRO A E, IRENE S G, JOSÉ V G P, et al. Efficacy of atrazine pesticide reduction in aqueous solution using activated carbon, ozone and a combination of both[J]. Science of the Total Environment, 2021, 764: 144301. doi: 10.1016/j.scitotenv.2020.144301 [21] 操家顺, 赵俊宇, 李超. 碳基材料在臭氧催化氧化法中的研究应用[J]. 应用化工, 2019, 48(12): 2951-2956. doi: 10.16581/j.cnki.issn1671-3206.20190919.029 [22] 王雪清, 赵越, 蒋广安, 等. 改性活性炭复合催化剂催化臭氧氧化处理石化污水的研究[J]. 现代化工, 2020, 40(2): 172-176. doi: 10.16606/j.cnki.issn0253-4320.2020.02.036 [23] 朱秋实, 陈进富, 姜海洋, 等. 臭氧催化氧化机理及其技术研究进展[J]. 化工进展, 2014, 33(4): 1010-1014. [24] 李根, 朱雷, 陈天翼, 等. 粒径与前驱体对活性炭催化臭氧氧化的影响[J]. 环境科学学报, 2018, 38(4): 1494-1500. doi: 10.13671/j.hjkxxb.2017.0397 [25] 张静, 杜亚威, 茹星瑶, 等. pH对微气泡臭氧氧化处理染料废水影响[J]. 环境工程学报, 2016, 10(2): 742-748. doi: 10.12030/j.cjee.20160236 [26] MALIK S N, GHOSH P C, VAIDYA A N, et al. Hybrid ozonation process for industrial wastewater treatment: Principles and applications: A review[J]. Journal of Water Process Engineering, 2020, 35: 101193. doi: 10.1016/j.jwpe.2020.101193 [27] 曹龙. 臭氧-活性炭-超滤工艺去除水源水邻苯二甲酸酯的效能研究[D]. 广州: 广州大学, 2019. [28] 张萌, 柳建设. 臭氧氧化浮选药剂丁基黄原酸钾反应动力学研究[J]. 环境科学学报, 2011, 31(3): 511-517. doi: 10.13671/j.hjkxxb.2011.03.009 [29] 孙秋红. 吸附—臭氧氧化处理对硝基苯酚废水[D]. 大连: 大连理工大学, 2011. [30] ARACELI E M, ALEJANDRO U, SIMÓN B. Chemical stability of xanthates, dithiophosphinates and hydroxamic acids in aqueous solutions and their environmental implications[J]. Ecotoxicology and Environmental Safety, 2021, 207: 111509. doi: 10.1016/j.ecoenv.2020.111509 [31] CHEN X, HU Y, PENG H, et al. Degradation of ethyl xanthate in flotation residues by hydrogen peroxide[J]. Journal of Central South University, 2015, 22(2): 495-501. doi: 10.1007/s11771-015-2548-0 [32] 杨艳娟. 机械活化改性膨润土基铁铋氧体高级氧化技术催化处理黄药废水[D]. 南宁: 广西大学, 2021. [33] LIU R, SUN W, OUYANG K, et al. Decomposition of sodium butyl xanthate (SBX) in aqueous solution by means of OCF: Ozonator combined with flotator[J]. Minerals Engineering, 2015, 70: 222-227. doi: 10.1016/j.mineng.2014.09.020 [34] 吉励. 吸附—催化臭氧氧化协同降解液相有机污染物的研究[D]. 杭州: 浙江大学, 2009. [35] GU L, ZHANG X, LEI L. Degradation of aqueous p-nitrophenol by ozonation integrated with activated carbon[J]. Industrial & Engineering Chemistry Research, 2008, 47(18): 6809-6815. 期刊类型引用(3)
1. 廖用开,钟雅琪,沈悦,林姗娜,曹鸿健,蔡超. 核壳结构Fe~0@Fe_3O_4复合材料活化过硫酸盐降解土壤中菲. 环境科学学报. 2025(02): 239-249 .  百度学术
百度学术
2. 林雨,张瑾,常通达,时佳蕊,王蔚,蔡纯. 可见光协同氧掺杂石墨相氮化碳活化亚硫酸盐降解水中双酚A. 安全与环境工程. 2024(03): 254-264 . 百度学术
3. 韩莹莹,方乐,孙宇晴,刘新会. 污泥共热解制备污泥基生物炭及其在土壤回用中的改良效益. 环境化学. 2024(05): 1670-1683 . 本站查看
其他类型引用(6)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 2688
- HTML全文浏览数: 2688
- PDF下载数: 79
- 施引文献: 9








