





























-
芬顿技术(Fenton)是一种高级氧化技术,产生的羟基自由基(·OH)氧化能力极强(E=2.8 V),能同时降解有机和无机污染物,在环境领域具有良好的应用前景[1-3]. 但由于使用的试剂过氧化氢(H2O2)在水体环境中不能稳定存在,易分解成H2O和O2,利用率较低[4-5]. 为克服传统Fenton技术的不足,学者们提出利用过氧化钙(CaO2)代替H2O2参与反应的类Fenton技术. CaO2可以在环境水体中缓慢释放H2O2,具有试剂有效期长、H2O2利用率高、pH适用范围广等传统Fenton技术没有的优点,目前已被学者和工程人员广泛研究和应用(如反应式1—4所示)[6]. 然而,现有研究大多是针对Fe2+和CaO2的反应体系,关于Fe3+和CaO2反应的研究相对较少[7-8]. 而且CaO2缓释H2O2的过程中会产生Ca(OH)2,提高溶液的pH,使铁离子沉淀析出影响催化效果,因此铁离子利用率较低. 如何促进铁离子溶解,强化Fe3+/Fe2+循环,对实现铁离子高效利用、增强体系降解性能具有重要的实际意义. 一些学者发现,Fe3+与乙二胺四乙酸盐(EDTA)、三聚磷酸盐(STPP)、草酸盐(OA)等配体络合可以抑制铁沉淀的生成[9-11]. 此外,引入配体也可以有效降低Fe3+/Fe2+的氧化还原电位,增强电子传递速率,促进Fe2+催化CaO2产生更多的·OH,提高反应效果[12].
EDTA是一种十分常用的代表性螯合剂,能和碱金属、稀土元素和过渡金属等形成稳定的水溶性络合物,其与Fe3+形成络合物后可以抑制铁离子沉淀,提高铁离子的利用率. 与其他配体相比,EDTA具有更强的螯合能力,其与Fe2+和Fe3+络合的稳定常数可以达到14.27和24[13]. 本文选用EDTA作为CaO2类Fenton体系的配体,以苯酚为目标污染物,研究了不同因素(EDTA浓度、Fe3+浓度、CaO2投加量、初始pH值)对Fe3+/EDTA/CaO2体系降解苯酚的影响及作用机理,揭示了EDTA增强铁离子溶解及循环、CaO2加快反应体系启动的重要作用,并确定了体系中活性自由基的产生及作用机制,为CaO2类Fenton体系在水环境修复中的应用提供一定的理论依据.
-
六水合三氯化铁、乙二胺四乙酸二钠、过氧化氢、钼酸铵、二水合磷酸二氢钠、十二水合磷酸氢二钠、邻菲啰啉、乙酸、乙酸铵(分析纯,天津光复精细化工研究所);过氧化钙、抗坏血酸(分析纯,国药集团化学试剂有限公司);苯酚(分析纯,北京化工).
-
分析天平(PL203,梅特勒-托利多仪器公司);pH计(PHB-4,上海雷磁);磁力搅拌器(HJ-6A,金坛市医疗仪器厂);紫外-可见分光光度仪(EVOLUTION 201,赛默飞世尔科技公司);高效液相色谱仪(Agilent1100,美国Agilent);气相色谱-质谱联用仪(Agilent 6890/5973,美国Agilent).
-
实验在250 mL血清瓶中进行,向200 mL 50 mmol·L−1的磷酸缓冲溶液(pH=6.5)中加入一定量的苯酚,使溶液中的苯酚浓度为50 mg·L−1. 投加不同体积100 mmol·L−1 Fe3+-EDTA(物质的量比为1:0.5)混合溶液(反应溶液的体积变化可以忽略不计),混合均匀后,投加一定量的CaO2,实验计时开始. 反应过程中,在既定时间取出0.5 mL样品,然后迅速加入0.5 mL无水乙醇终止反应,用0.22 µm滤头过滤到安捷伦样品瓶中. 如无特殊说明,体系中Fe3+浓度为1 mmol·L−1,EDTA浓度为0.5 mmol·L−1,CaO2浓度为0.75 g·L−1,反应过程中体系的pH保持在6.5. 每组设3个平行样取平均值,实验数据的标准误差不超过5%.
-
运用高效液相色谱法测定苯酚的含量[14]. 色谱柱型号为SB-C18(ϕ4.6 mm×250 mm,5 µm),采用等度洗脱,流动相体积比为乙腈:水=40:60. 进样量为10 µL,流速为1 mL·min−1,柱温为30 ℃,VWD波长为270 nm,出峰时间为3.1 min左右.
-
采用气相色谱-质谱联用法(GC-MS)以及液相色谱-质谱联用法(LC-MS) 测定中间产物,样品前处理过程如下[15]:在不同的反应时间(0、10、30、60、120 min)收集100 mL样品,加入乙酸乙酯进行萃取,将上层清液取出加入适量无水硫酸钠干燥脱水,将脱水后的溶液迅速倒入65 ℃的旋转蒸发仪中,蒸发浓缩后用0.22 µm滤膜过滤得到GC-MS所需的样品. 测试条件如下: 以不分流模式注入1 μL样品,色谱柱温度以20 ℃·min−1的速度从40 ℃升到200 ℃保持4 min,以10 ℃·min−1的速度从200 ℃升到250 ℃保持5 min. 在EI模式下获得质谱,电子能量为70 eV; 将待测样品以 20 μL 的进样量注入LC-MS系统,洗脱液为乙腈,流速为0.2 mL·min−1,采用负离子扫描模式在m/z范围30—150获得质谱.
-
选用钼酸铵法测定溶液中的H2O2浓度[16]. 取出适量样品后用0.22 µm滤头过滤,将1 mL过滤后的样品和1 mL 2.4 mmol·L−1的钼酸铵溶液加入到10 mL离心管,用去离子水稀释至10 mL. 显色15 min后用紫外-可见分光光度计在450 nm处检测样品的吸光度. 根据标准曲线可以得到H2O2的浓度.
-
Fe2+浓度采用1,10-菲咯啉显色法测定[17]. Fe2+在pH=3—9时与1,10-菲咯啉反应生成稳定的橙红色络合物,橙红色络合物的吸光度与浓度的关系符合朗伯-比尔定律,可通过测定橙红色络合物在510 nm处的吸光度换算出溶液中Fe2+的浓度. 加入抗坏血酸将Fe3+还原为Fe2+,由此可得到总铁离子的浓度.
-
·OH探针实验[18]:测定反应过程中·OH的累积浓度. 苯甲酸与·OH反应生成邻-羟基苯甲酸、间-羟基苯甲酸、对-羟基苯甲酸的3种异构体,且它们的产量比为邻-羟基苯甲酸∶间-羟基苯甲酸∶对-羟基苯甲酸=1.7∶2.3∶1.2,利用高效液相色谱仪检测对-羟基苯甲酸的产量,进而可知体系生成·OH的产量. 掩蔽实验:分别利用氯仿(CHCl3,157 mmol·L−1)、叔丁醇(TBA,63 mmol·L−1)分别作·O2−、·OH的掩蔽剂. 后续通过高效液相色谱仪检测反应过程中苯酚浓度的变化情况.
-
为了探究体系中各组分对苯酚降解的作用,考察了不同组分体系中苯酚的降解效果,结果如图1(a)所示. 在单独CaO2、EDTA/CaO2和Fe3+/CaO2的条件下,苯酚浓度在120 min内均没有明显变化,说明单独CaO2的氧化能力不足以氧化苯酚,添加EDTA不能直接对CaO2起到作用,而缺少EDTA的Fe3+/CaO2也因为铁离子易沉淀、电子传递速率慢的原因,而无法有效降解苯酚;在Fe3+/EDTA/CaO2中,苯酚的降解分为初始缓慢降解阶段和随后的快速降解阶段,120 min时苯酚的降解率在95%以上. 结合不同组分条件下H2O2浓度的变化(图1(b))可知,在单独CaO2、EDTA/CaO2和Fe3+/CaO2的条件下H2O2浓度在前20 min内迅速升高,之后几乎没有变化,而在Fe3+/EDTA/CaO2中前20 min内H2O2浓度的上升速度明显减缓,这说明有一部分H2O2是被缓慢消耗的,20 min后H2O2消耗速率加快,苯酚降解速率也随之加快,直到120 min时苯酚完全降解,说明苯酚降解与H2O2有关,H2O2消耗速率越快苯酚降解效果越好. 综合以上结果可以分析出:只有在Fe3+、EDTA、CaO2 等3种组分均在时,CaO2水解释放的H2O2才能与Fe3+快速地发生类Fenton反应氧化降解污染物.
-
图2(a)显示了EDTA浓度对Fe3+/CaO2降解苯酚的影响:没有投加EDTA时,Fe3+/CaO2几乎不降解苯酚,而分别投加了0.125、0.25、0.5、1、2 mmol·L−1的EDTA后,120 min时苯酚去除率分别提高至55.7%、72.2%、100%、100%和87.2%. 由此可见,投加EDTA可以有效地促进Fe3+/CaO2氧化降解苯酚,当EDTA浓度小于0.5 mmol·L−1时EDTA浓度越高苯酚的降解效果越好,但随着EDTA浓度从0.5 mmol·L−1增加至2 mmol·L−1,苯酚的降解速率并没有进一步加快甚至反而受到抑制,这可能是因为过量的EDTA会与苯酚竞争·OH,导致苯酚的降解效果变差,因此在本研究中选取0.5 mmol·L−1为EDTA的最佳浓度. 对体系中EDTA浓度为0.125、0.25、0.5 mmol·L−1时的苯酚降解曲线进行动力学拟合(图2(b)),拟合结果表明,3种EDTA浓度条件下的苯酚降解曲线均符合拟一级动力学,且随着EDTA浓度增大,拟合曲线斜率(kobs)也显著增大,说明体系内EDTA浓度是反应的限制性因素.
图3显示了Fe3+/CaO2和Fe3+/EDTA/CaO2不同组分条件下,降解苯酚的过程中溶解态Fe2+和总溶解态铁离子(TFe)的浓度变化. 在Fe3+/CaO2中,10 min内TFe浓度迅速降低至0.34 mmol·L−1,30 min 时TFe仅剩0.02 mmol·L−1,几乎检测不到溶解态Fe2+,这是由于初始条件处于中性环境,且CaO2水解会产生大量Ca(OH)2,游离态铁离子会迅速转化为沉淀,因此该组分条件下不易发生类Fenton反应,无法有效降解苯酚.
而在Fe3+/EDTA/CaO2中,整个反应过程中Fe3+浓度始终维持在0.85 mmol·L−1以上,同时也能测到少量的溶解态Fe2+,这是因为EDTA与Fe3+形成稳定的络合物会抑制Fe3+沉淀,促进类Fenton反应顺利进行.
因此,EDTA的加入可以明显增加铁离子在中性pH环境中的溶解度,促使铁离子高效催化CaO2降解污染物.
-
为了进一步探究CaO2促进苯酚降解的原因,对比了Fe3+/EDTA/H2O2和Fe3+/EDTA/CaO2两种体系对苯酚的降解效果,结果如图4(a)所示. 在Fe3+/EDTA/H2O2体系中,前30 min苯酚几乎没有降解,30 min后苯酚开始被缓慢降解,120 min时苯酚降解率达到48%;在Fe3+/EDTA/CaO2体系中,120 min时苯酚的降解率在95%以上,与Fe3+/EDTA/H2O2体系相比,Fe3+/EDTA/CaO2体系在反应前期能更快地启动反应,随后对体系中苯酚的降解速率也明显更高. 结合不同组分体系中H2O2的浓度变化(图4(b))可以发现,Fe3+/EDTA/CaO2体系在前20 min缓慢消耗H2O2,20 min后H2O2消耗速率加快,苯酚降解速率也随之加快,而在Fe3+/EDTA/H2O2体系中前30 min几乎没有消耗H2O2,30 min后H2O2才被缓慢消耗. 对比两个体系在反应120 min时的H2O2消耗量以及苯酚降解率可以发现,Fe3+/EDTA/CaO2体系的消耗量更多,苯酚降解率更高. 在Fe3+/EDTA/CaO2体系中用H2O2酶掩蔽H2O2后还可以测到少量的Fe2+产生(图5),而掩蔽了Fe3+/EDTA/H2O2体系中的H2O2后却未检测到Fe2+产生,说明在Fe3+/EDTA/CaO2体系中更易产生Fe2+,这可能是因为Fe3+和EDTA形成的络合物Fe3+-EDTA易于吸附到CaO2的固体表面,EDTA起到桥接Fe3+和CaO2的作用,促进电子从CaO2转移到Fe3+,将Fe3+还原为Fe2+. 因此,引入EDTA的Fe3+/EDTA/CaO2体系可以加快Fe3+/Fe2+循环,促进类Fenton反应的进行和苯酚的降解. Pan等[19]在Fe3+-EDTA促进CaO2类Fenton反应的研究中也发现,Fe3+-EDTA倾向于吸附在CaO2的固体表面,形成[Fe3+-EDTA···CaO2]−的过渡态,电子从CaO2转移到Fe3+-EDTA,促进Fe3+-EDTA还原,与本文结论一致.
-
为确定反应过程中活性自由基的种类,在0 min和30 min时利用ERP光谱和DMPO自旋捕获加合物进行分析,结果如图6所示. 0 min时未出现特征峰,而在30 min时检测到·OH 的1:2:2:1四重态的特征峰(图6(a)),表明体系中有·OH产生;30 min时也观察到1:1:1:1的四重态特征峰(图6(b)),证明体系中存在
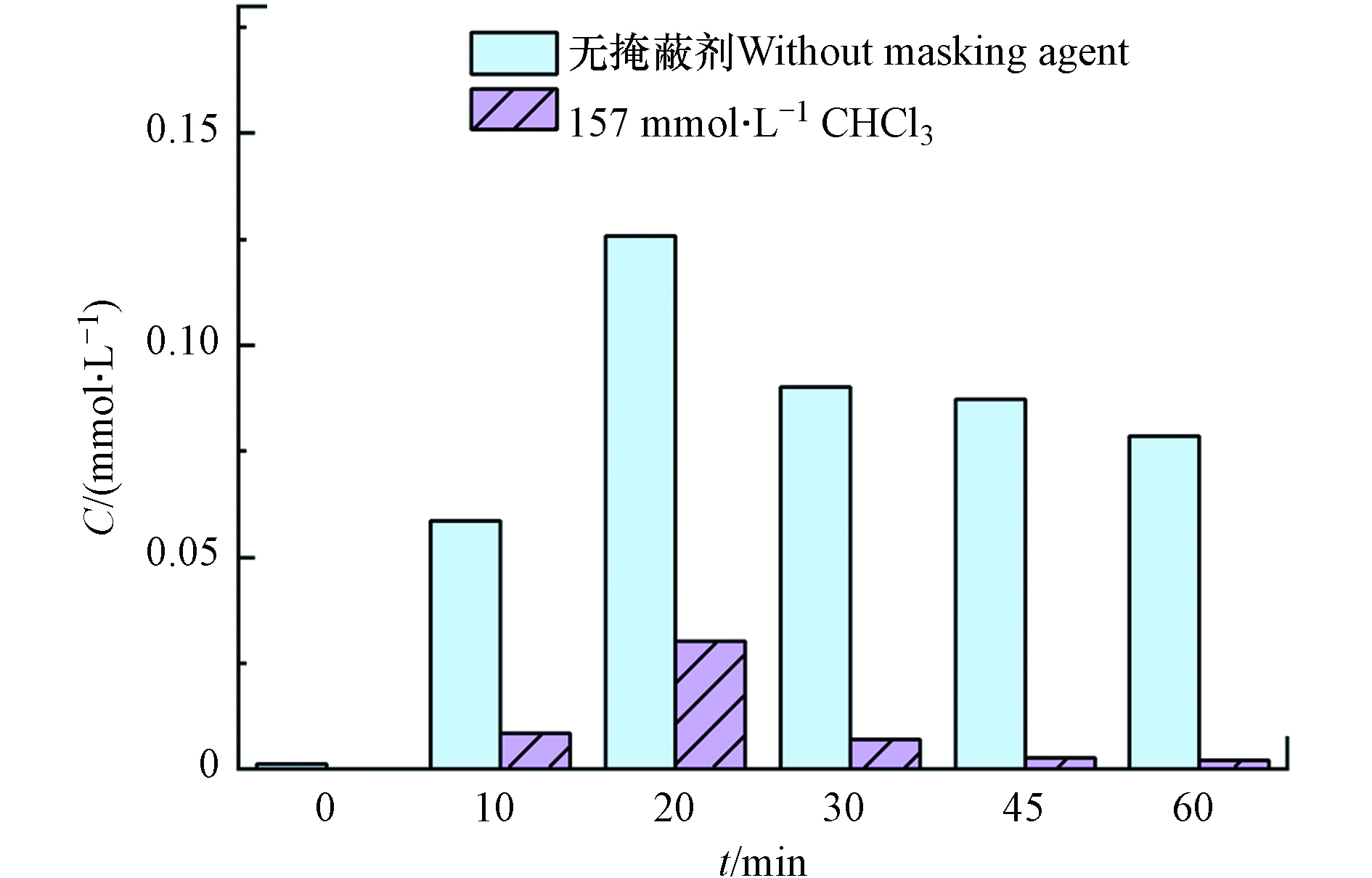
⋅O−2 . 分别利用叔丁醇(TBA)和氯仿(CHCl3)掩蔽·OH和⋅O−2 后,苯酚的降解效果如图7(a)所示,掩蔽·OH后体系几乎没有降解效果;掩蔽⋅O−2 后,反应过程中苯酚的降解速率比无掩蔽体系的降解速率慢,但120 min时苯酚降解率也能达到82.2%. 由此可知,·OH对体系中苯酚的降解起主要作用,⋅O−2 在降解过程中起到一定的促进作用. 反应过程中·OH的累积浓度如图7(b)所示,·OH的产生速率与苯酚的降解速率变化一致,证明是由·OH直接氧化降解苯酚.图8为无掩蔽体系和掩蔽
⋅O−2 体系中Fe2+浓度的变化,掩蔽了⋅O−2 后产生的Fe2+浓度低于无掩蔽体系中的浓度,说明⋅O−2 可以将体系中Fe3+还原为Fe2+. 本实验在Pan等[19]的研究结果基础上又进一步发现在体系中⋅O−2 的作用: Fe3+/EDTA/CaO2体系中除CaO2之外,⋅O−2 也起到了还原Fe3+的作用,二者共同加速Fe3+向Fe2+的转化,促进苯酚降解. -
固定EDTA∶Fe3+物质的量比为0.5∶1,不同Fe3+浓度条件下苯酚的降解效果如图9(a)所示. 随着Fe3+浓度由0.25 mmol·L−1提高到2 mmol·L−1,在120 min的反应时间内苯酚的降解率从20.1%上升至100%. 可见Fe3+浓度对苯酚降解效果至关重要.
-
图9(b)为CaO2投加量对苯酚降解的影响. 由图9可知,随着CaO2投加量不断增加,苯酚的降解率也随之增加,当CaO2投加量增加至0.75 g·L−1时,反应120 min后苯酚的降解率增加至100%. 这主要是由于提高CaO2投加量会释放更多的H2O2,但CaO2投加量进一步増加至1.5 g·L−1时,苯酚的降解会受到抑制,反应120 min时降解率从100%下降至43.4%. 这是因为过量的CaO2与水反应后生成大量的Ca(OH)2,体系中的pH值会逐渐上升超过11,超过磷酸盐的缓冲能力,铁离子沉淀,类Fenton反应受到抑制,影响苯酚的降解效果.
-
图9(c)所示为不同初始pH条件下Fe3+/EDTA/CaO2体系对苯酚的降解效果,结果表明,体系在初始pH=3—7内均有很好的降解效果,在pH=8时苯酚降解效果较差,这可能是因为CaO2在碱性环境中易分解成O2而不是H2O2,且·OH在碱性环境中的活性较低,因此体系在酸性和中性环境下的降解效果更好.
-
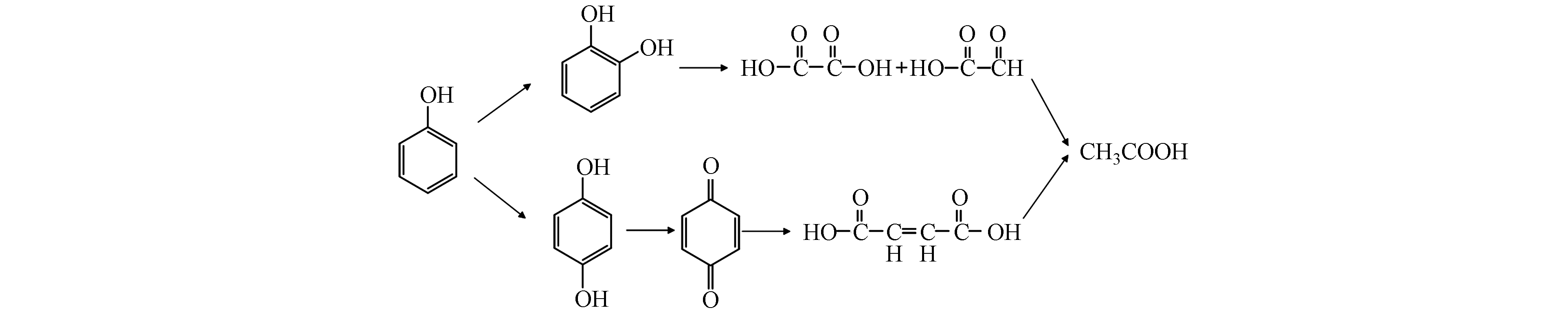
通过GC-MS对Fe3+/EDTA/CaO2体系降解苯酚的氧化产物进行测定,结果如表1所示,测得的苯酚降解中间产物主要包括对苯二酚、邻苯二酚、对苯醌、乙醛酸. 通过LC-MS对氧化产物进行进一步测定,检测到草酸(乙二酸)、马来酸(顺丁烯二酸)、乙酸,通过以上产物推测体系可能发生的降解途径如图10所示,苯酚被氧化成邻苯二酚、对苯二酚和对苯醌,中间产物进一步开环形成小分子酸.
-
(1)Fe3+/EDTA/CaO2体系可以高效降解苯酚,EDTA的加入可以明显增加铁离子在中性环境中的溶解度,而且EDTA可以桥接CaO2和Fe3+,加快二者之间的电子转移速率,促进Fe3+/Fe2+循环,改善苯酚的降解效果. 与Fe3+/EDTA/CaO2体系相比,Fe3+/EDTA/CaO2体系能更快地启动反应,提高反应速率.
(2)Fe3+/EDTA/CaO2体系产生活性自由基降解苯酚,起主要作用的是·OH,
⋅O−2 起到还原Fe3+的作用,加快体系中苯酚的降解速率.(3)Fe3+/EDTA/CaO2体系中EDTA: Fe3+的最佳比例为0.5:1;Fe3+浓度越高,苯酚降解速率越快;随着CaO2投加量的增大,苯酚的降解效果越好,但过量反而会抑制苯酚降解.
Fe3+-EDTA催化CaO2类Fenton体系降解苯酚的机制及效果
Mechanism and effect of phenol degradation by Fe3+-EDTA catalyzing CaO2 Fenton-like system
-
摘要: 基于CaO2的类Fenton体系具有缓释H2O2、高效降解污染物、有效作用周期长等优点,在环境修复领域得到广泛应用. 但目前研究主要针对Fe2+/CaO2体系,关于Fe3+/CaO2的研究较少,而如何提高Fe3+活化CaO2的能力,是铁离子高效循环利用、污染物持续降解的关键. 本研究有针对性地建立了Fe3+/EDTA/CaO2体系,对其降解能力、作用机制、关键因素进行了深入分析,结果表明,Fe3+/EDTA/CaO2体系在中性条件下对苯酚的降解率在95%以上,EDTA的加入可以明显增加铁离子在中性环境中的溶解度,而且EDTA可以桥接CaO2和Fe3+,加快二者之间的电子转移速率,促进Fe3+/Fe2+循环,改善苯酚的降解效果;活性自由基测定的实验表明,降解苯酚起主要作用的活性自由基是羟基自由基(·OH),
⋅O−2 起到还原Fe3+的作用,促进Fe3+/Fe2+循环. 本研究对CaO2类Fenton体系的应用具有一定的理论意义.-
关键词:
- 过氧化钙(CaO2) /
- 三价铁(Fe3+) /
- 乙二胺四乙酸二钠(EDTA) /
- 类芬顿 /
- 苯酚
Abstract: CaO2-based Fenton-like systems are widely applied in environmental remediation with the advantages of slow release of H2O2, high efficiency for pollutant degradation, and long term of effectiveness. However, the current studies focus on Fe2+/CaO2 system, and limited studies were reported regarding to Fe3+/CaO2 system. Whereas the enhancement of Fe3+ on the activation of CaO2 is critical in Fe cyclic utilization and continuous degradation of pollutants. This study established Fe3+/EDTA/CaO2 system, and evaluated the degradation ability, interaction mechanisms, and key factors. The results indicate the degradation efficiency of phenol was greater than 95% by Fe3+/EDTA/CaO2 system under neutral pH conditions. The addition of EDTA enhanced the solubility of Fe3+ under neutral pH conditions; EDTA also bridged CaO2 and Fe3+ and enhanced the circulation of Fe2+/Fe3+ to improve the degradation of phenol. The analysis of the active free radicals indicate hydroxyl radical (·OH) played a key role in phenol degradation, and⋅O−2 enhanced Fe2+/Fe3+ circulation through the reduction of Fe3+. The study provides theoretical foundation in the application of Fenton-like systems.-
Key words:
- calcium peroxide /
- ferric ion /
- ethylenediaminetetraacetic acid disodium salt /
- fenton-like /
- phenol.
-
2,4-二氯苯氧乙酸(2,4-D)是一种含有苯氧乙酸基团的选择性除草剂,由于其具有廉价易得、高选择性等优点广泛用于去除农业和林业中的阔叶杂草[1]. 但是由于其具有急性神经毒性和慢性毒性,对皮肤和眼睛有刺激作用;慢性毒性表现为对血液、肝、肾的毒性及抑制某些酶的活力,抑制某些蛋白质的合成[2]. 并且其在环境中难以降解、具有生物累积性,会对人体和自然环境造成潜在危害.
因此2,4-D及其衍生物被世界卫生组织(WHO)定义为中度毒物,并且规定饮用水当中的2,4-D浓度不得超过100 mg·L−1 [3],因此探究高效安全的技术去除环境中残留的2,4-D是十分必要的.
高级氧化技术是一种使用强氧化物质与水中溶解性污染物反应的方法,目前高级氧化技术主要包括芬顿法和类芬顿法、臭氧类高级氧化法、光催化氧化技术、电化学氧化技术和基于硫酸根自由基(
SO⋅−4 由于传统的芬顿技术存在Fe2+利用率低、反应pH要求严格、易产生二次污染等缺点,基于硫酸根自由基的高级氧化技术能够解决传统芬顿技术的诸多问题[5]. 氧化剂过一硫酸盐(PMS)具有不对称结构,更容易被活化产生更多的活性物质,同时其标准氧化电位E0=2.5—3.1V,高于常用氧化剂的氧化性,在反应中pH适用范围更广并且硫酸根自由基比羟基自由基稳定时间更长[6],因此基于硫酸根自由基的高级氧化技术成为近年来处理有机污染物的研究热点.
本文采用一步水热法合成了NPF-FeCo2O4催化剂,该催化剂能够高效活化PMS产生硫酸根自由基降解2,4-D. 此外,分别采用了TEM、XPS等对材料进行表征分析,同时研究了不同pH、不同阴离子等条件下对2,4-D的去除效率,最后通过淬灭试验和EPR测试对反应机理进行探究.
1. 材料与方法(Materials and methods)
1.1 试验材料
2,4-二氯苯氧乙酸(2,4-D)、2,4-二氯苯酚(2,4-DCP)、活性红、磺胺甲恶唑(SMX)、过硫酸氢钾(PMS)、六水合硝酸钴(Co(NO3)2·6H2O)、九水合硝酸铁(Fe(NO3)3·9H2O)、柠檬酸钾、尿素、聚丙烯酰胺(PAM)、1-丁基-3-甲基咪唑磷酸二丁酯盐、1-丁基-3-甲基咪唑六氟磷酸盐、L-组氨酸、对苯醌、无水乙醇、甲醇、叔丁醇,试验用水为去离子水.
1.2 催化剂的制备
NPF三掺杂的FeCo2O4由一步水热法合成[7]. 首先,将0.45 g 1-丁基-3-甲基咪唑六氟磷酸盐溶解在含有0.6549 g CO(NO3)2·6H2O,0.4539 g Fe(NO3)3·9H2O,2.064 g柠檬酸钾,0.6 g尿素,0.525 g PAM的60 mL去离子水中,充分搅拌后将得到的混合物转移至100 mL高压釜,在200 ℃下保持24 h,冷却至室温后,用蒸馏水和乙醇洗涤产物,最终在真空条件下80 ℃干燥12 h,得到N、P、F三掺杂的催化剂NPF-FeCo2O4,简称NPF-FCO,用同样的方法制作了不加离子液体和加1-丁基-3-甲基咪唑磷酸二丁酯盐作为杂原子掺杂剂的催化剂FeCo2O4和NP-FeCo2O4,分别简称FCO和NP-FCO.
1.3 催化剂表征
通过透射电子显微镜(TEM,JEM-2100,日本JEOL公司)对 NPF-FeCo2O4催化剂进行形貌表征,通过X射线光电子能谱仪(美国ThermoFischer,ESCALAB Xi+)对催化剂的元素组成以及价态进行分析,通过X射线衍射仪(日本理学rigaku Ultima IV)对催化剂反应前后晶型变化进行测试,采用傅立叶红外光谱(赛默飞IN10)确定样品中的官能团,通过电子自旋共振波谱仪(Bruker EMXPLUS)测定体系中产生的自由基.
1.4 试验及分析方法
在室温条件下,取100 mL的浓度为0.1 mmol·L−1的2,4-D溶液于150 mL的烧杯中,加入一定量的催化剂,磁力搅拌30 min,加入一定量的PMS启动反应. 在一定时间间隔取样,取样时通过0.45 μm滤膜过滤,并提前加入100 μL甲醇淬灭样品中剩余的自由基.
采用配有285 nm紫外检测器的高效液相色谱仪(Thermofisher,Ultimate 3000)测定2,4-D的浓度,流动相种类及比例为水(含有0.5%的乙酸):甲醇 = 40:60,检测波长为 285 nm,流速为 1 mL·min−1.
2. 结果与讨论(Results and discussion)
2.1 催化剂的表征
2.1.1 TEM分析
图1(a)为不掺杂离子液体的FeCo2O4 TEM图像,可以看出原始的FeCo2O4 催化剂呈现出均匀的球形形态. 图1(b)−(c)表示随着离子液体的掺杂,催化剂的平均尺寸逐渐减小,并且NP-FeCo2O4和NPF-FeCo2O4催化剂开始形成表面粗糙的不规则球形. 表明在水热过程中离子液体的掺杂影响了FeCo2O4的形态和尺寸. 图1(d)为循环5次使用后的NPF-FeCo2O4催化剂,可以看出经过5次循环后催化剂仍然呈现较规则的圆球状.
 图 1 不同催化剂的TEM图Figure 1. TEM images of different catalysts(a)FeCo2O4,(b)NP-FeCo2O4,(c)NPF-FeCo2O4,(d)5次循环后的NPF-FeCo2O4(a) FeCo2O4, (b) NP-FeCo2O4, (c) NPF-FeCo2O4, (d) NPF-FeCo2O4 after five cycles
图 1 不同催化剂的TEM图Figure 1. TEM images of different catalysts(a)FeCo2O4,(b)NP-FeCo2O4,(c)NPF-FeCo2O4,(d)5次循环后的NPF-FeCo2O4(a) FeCo2O4, (b) NP-FeCo2O4, (c) NPF-FeCo2O4, (d) NPF-FeCo2O4 after five cycles2.1.2 傅里叶红外光谱分析
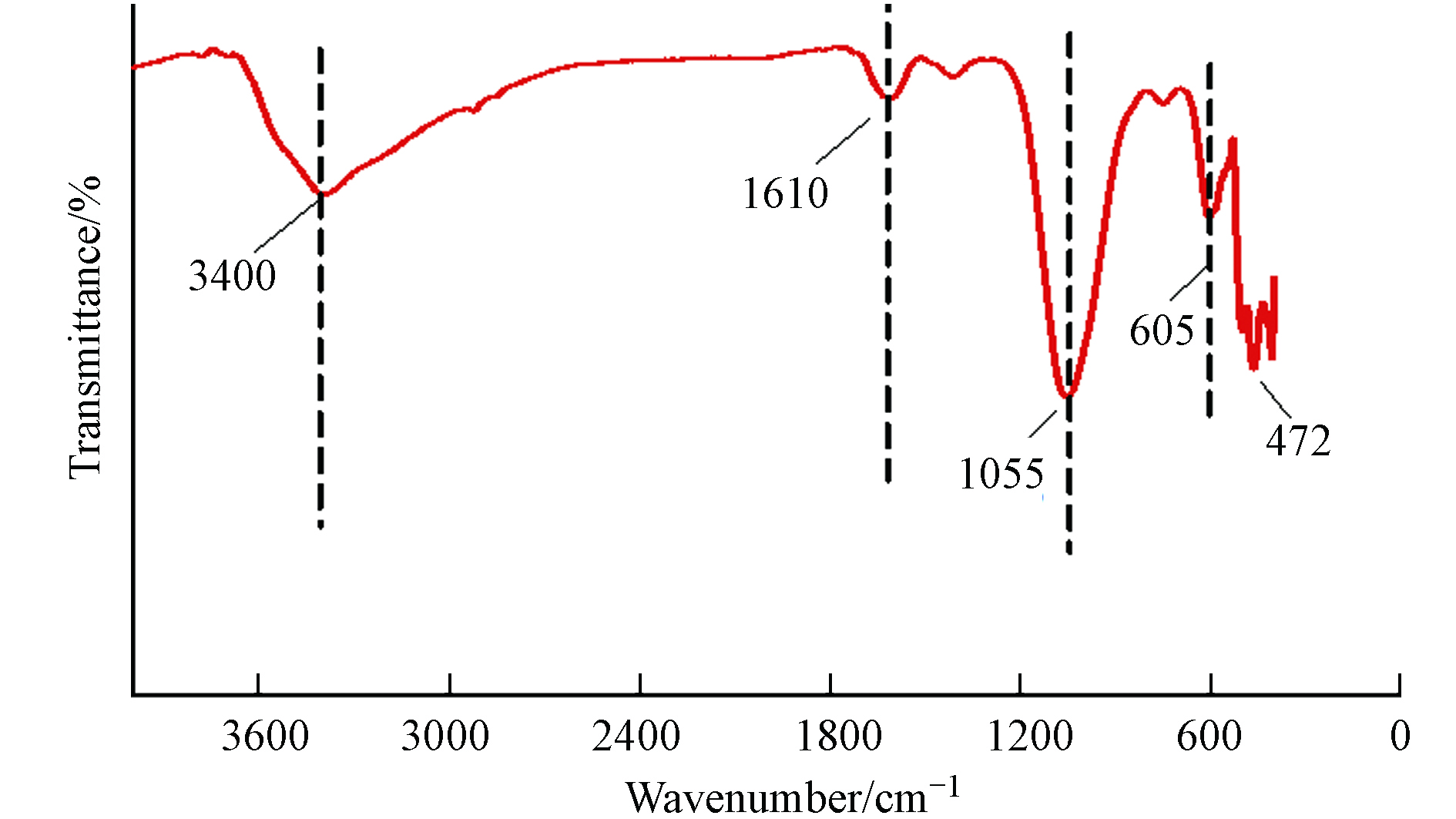
为了确定金属和氧之间的键合状态以及样品中的官能团,对NPF-FeCo2O4催化剂进行了傅里叶红外光谱表征. 如图2所示,傅里叶红外光谱证明材料中存在尖晶石结构. 红外光谱在400—700 cm−1区域的两个吸收带对应于尖晶石FeCo2O4结构. 500 cm−1附近的吸收峰对应尖晶石结构四面体体系中金属-氧键的伸缩振动,即Fe—O. 600—700 cm−1间的吸收峰对应于八面体系统中金属键的拉伸[8],1610 cm−1和3400 cm−1处的拉伸和弯曲模式是羟基(OH)引起的[9].
2.1.3 XPS分析
如图3所示,从拟合的XPS光谱图可以看出,合成的催化剂含有Co、Fe、O、C、N、P和F元素. 位于781.3 eV和783.5 eV的峰属于Co 2p3/2,797.4 eV处的峰属于Co 2p1/2,同时也产生了787.7 eV和803.0 eV的卫星峰. 结合能为781.3 eV和783.5 eV的峰说明Co2+氧化物物种的存在,而797.4 eV的峰属于Co3+氧化物种[10]. 715.7 eV和724.2 eV的峰分别属于Fe3+的2p3/2和2p1/2,表明存在Fe3+阳离子,712.1 eV的峰属于Fe2+的2p3/2[11].
 图 3 NPF-FeCo2O4的XPS谱图Figure 3. XPS spectra of NPF-FeCo2O4(a)Co 2p谱图,(b)Fe 2p谱图,(c)O1s谱图,(d)N 1s谱图,(e)P 2p谱图,(f)总谱图(a) Co 2p, (b) Fe 2p, (c) O1s, (d) N 1s, (e) P 2p, (f) Survey
图 3 NPF-FeCo2O4的XPS谱图Figure 3. XPS spectra of NPF-FeCo2O4(a)Co 2p谱图,(b)Fe 2p谱图,(c)O1s谱图,(d)N 1s谱图,(e)P 2p谱图,(f)总谱图(a) Co 2p, (b) Fe 2p, (c) O1s, (d) N 1s, (e) P 2p, (f) Survey由NPF-FeCo2O4的O1s谱图可以看出有3个峰,530.7 eV的峰值属于晶格氧(O1),结合能为 532.3 eV的峰属于表面羟基(O2),531.3 eV处的峰对应于氧空位(O3)[7]. 可以看出,相较于FeCo2O4和NP-FeCo2O4,NPF-FeCo2O4催化剂的氧空位含量最高,而表面羟基含量最低. 相较于NP-FeCo2O4,NPF-FeCo2O4在P 2p谱图中出现了在133.1 eV处的P—O3(氧空位缺陷)新峰[12],表明N/P/F的共掺杂能够通过表面羟基的损失将氧空位结合到NPF-FeCo2O4中. 同时,在N 1s谱图中,相较于NP-FeCo2O4催化剂,NPF-FeCo2O4出现了峰值为398.4 eV的吡啶N,Co—N的含量高于NP-FeCo2O4,在Fe的2p谱图中,Fe3+/Fe2+的相对面积比随着掺杂元素的增加在逐渐增加,表明Co—N和Fe—N物种的形成能够促进Co/Fe向N原子的电荷转移[11].
因此,由XPS图可以得出,加入含有N、P、F元素的离子液体,能够增加Fe3+/Fe2+氧化还原对、催化剂的氧空位和Co—N、Fe—N、P—O、吡啶N等表面活性位点,他们之间的相互作用有利于提高催化剂的催化性能[7].
2.2 改性FeCo2O4催化PMS降解2,4-D的效率
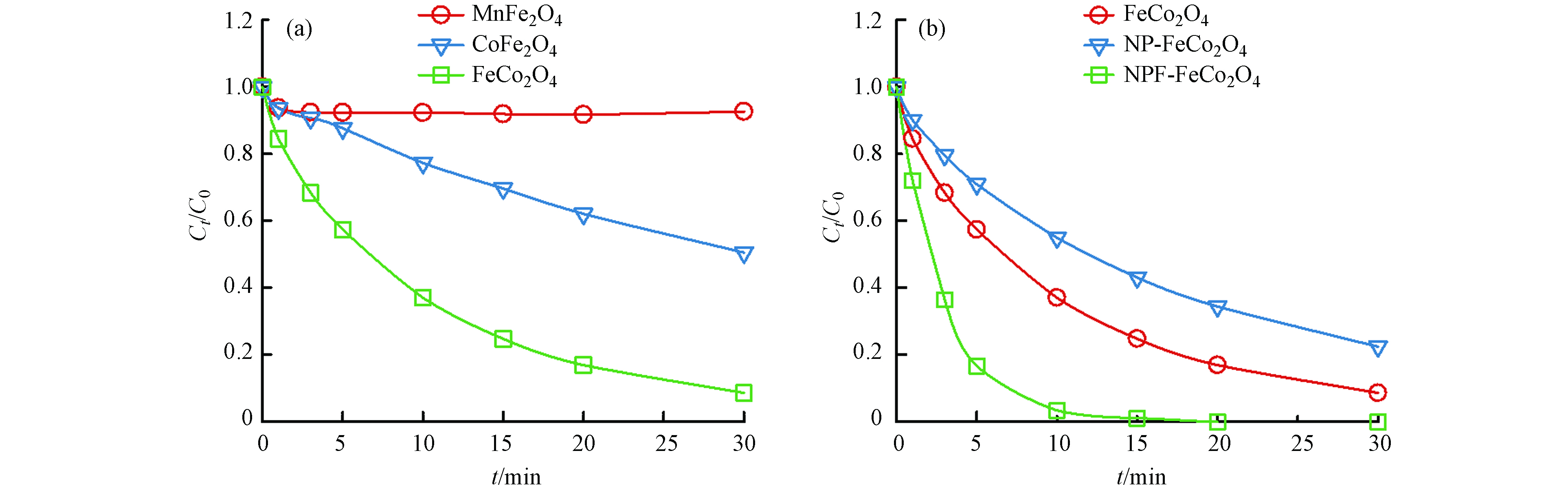
分别对Mn、Fe、Co的3种过渡金属采用同样的方法合成了不同的催化剂,通过催化PMS降解2,4-D的效率来判断催化活性.
如图4所示,在相同条件下,FeCo2O4作为催化剂活化PMS降解2,4-D的效果最好,30 min内去除率能达到90%,但是不能完全降解2,4-D. 因此选择FeCo2O4进行后续的改性研究,如图4(b)中,改性后的NPF-FeCo2O4催化剂在20 min内活化PMS对2,4-D的去除率可达到100%,表明离子液体的加入能够提高FeCo2O4催化性能.
如图5(a)所示,研究了不同体系下2,4-D的降解效率. 在2,4-D浓度为0.1 mmol·L−1,催化剂的投加量为0.1 g·L−1,PMS浓度为1 mmol·L−1的反应条件下,单独加入PMS时,2,4-D几乎不降解. 表明PMS的自分解性差. 同时,在没有PMS的情况下,NPF-FeCo2O4对2,4-D的吸附作用也可以忽略不计. 相比之下,当反应体系达到吸附饱和后加入PMS,20 min内2,4-D的降解率可达到100%. 而由图5(b)NPF-FeCo2O4/PMS体系中2,4-D的浓度和PMS浓度变化对比可知,在该体系中,PMS浓度的变化与2,4-D的降解趋势高度吻合,结果表明NPF-FeCo2O4可有效活化PMS产生活性氧物种促进2,4-D的降解.
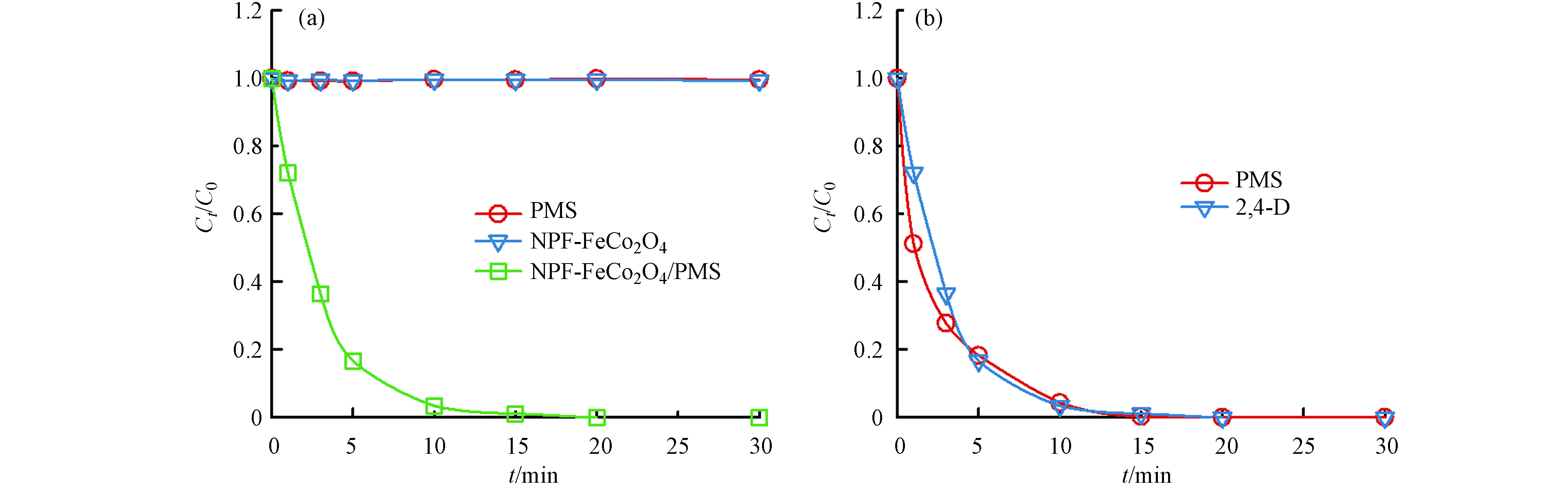 图 5 NPF-FeCo2O4/PMS体系的探究Figure 5. Study on NPF-FeCo2O4/PMS system(a)不同体系下2,4-D的降解效率,(b)2,4-D的降解趋势和PMS变化趋势比较(a) degradation efficiency of 2,4-D under different systems, (b) comparison of the degradation trend of 2,4-D and the change trend of PMS
图 5 NPF-FeCo2O4/PMS体系的探究Figure 5. Study on NPF-FeCo2O4/PMS system(a)不同体系下2,4-D的降解效率,(b)2,4-D的降解趋势和PMS变化趋势比较(a) degradation efficiency of 2,4-D under different systems, (b) comparison of the degradation trend of 2,4-D and the change trend of PMS2.3 反应pH的影响
在2,4-D浓度为0.1 mmol·L−1,催化剂的投加量为0.1 g·L−1,PMS浓度为1 mmol·L−1的反应条件下,考察初始pH对2,4-D降解效率的影响,由于常用的缓冲盐如
PO3−4 HCO−3 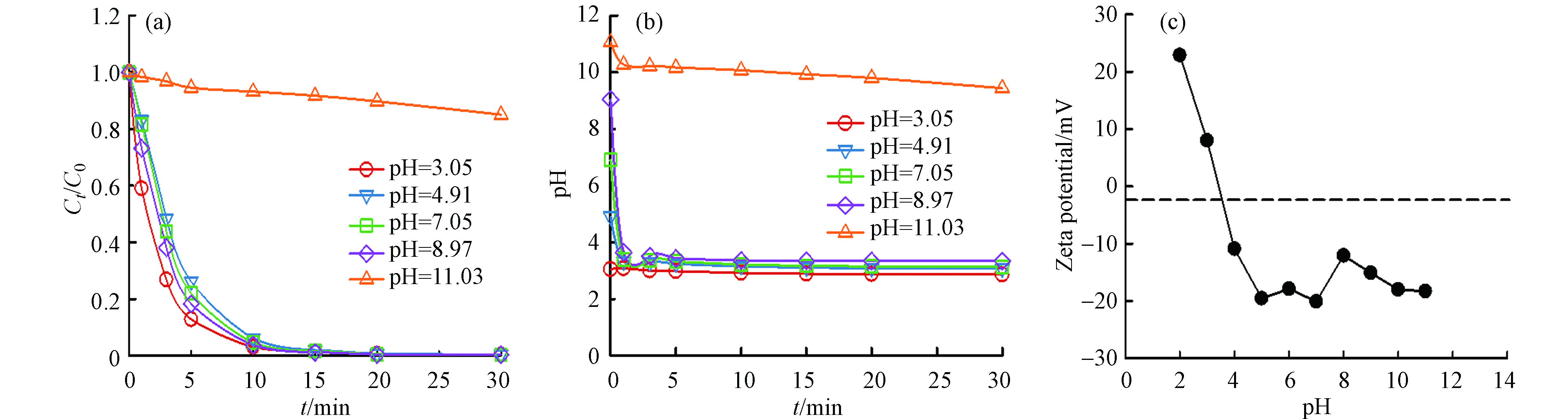 图 6 pH对NPF-FeCo2O4/PMS体系的影响Figure 6. Effect of pH on NPF-FeCo2O4/PMS system(a)不同初始pH条件下2,4-D降解效率,(b)反应过程中体系的pH变化,(c)NPF-FeCo2O4的pHPZC(a) the Effect of pH on the NPF-FeCo2O4/PMS system, (b) the change of pH in system during the reaction, (c) the pHPZC of NPF-FeCo2O4
图 6 pH对NPF-FeCo2O4/PMS体系的影响Figure 6. Effect of pH on NPF-FeCo2O4/PMS system(a)不同初始pH条件下2,4-D降解效率,(b)反应过程中体系的pH变化,(c)NPF-FeCo2O4的pHPZC(a) the Effect of pH on the NPF-FeCo2O4/PMS system, (b) the change of pH in system during the reaction, (c) the pHPZC of NPF-FeCo2O4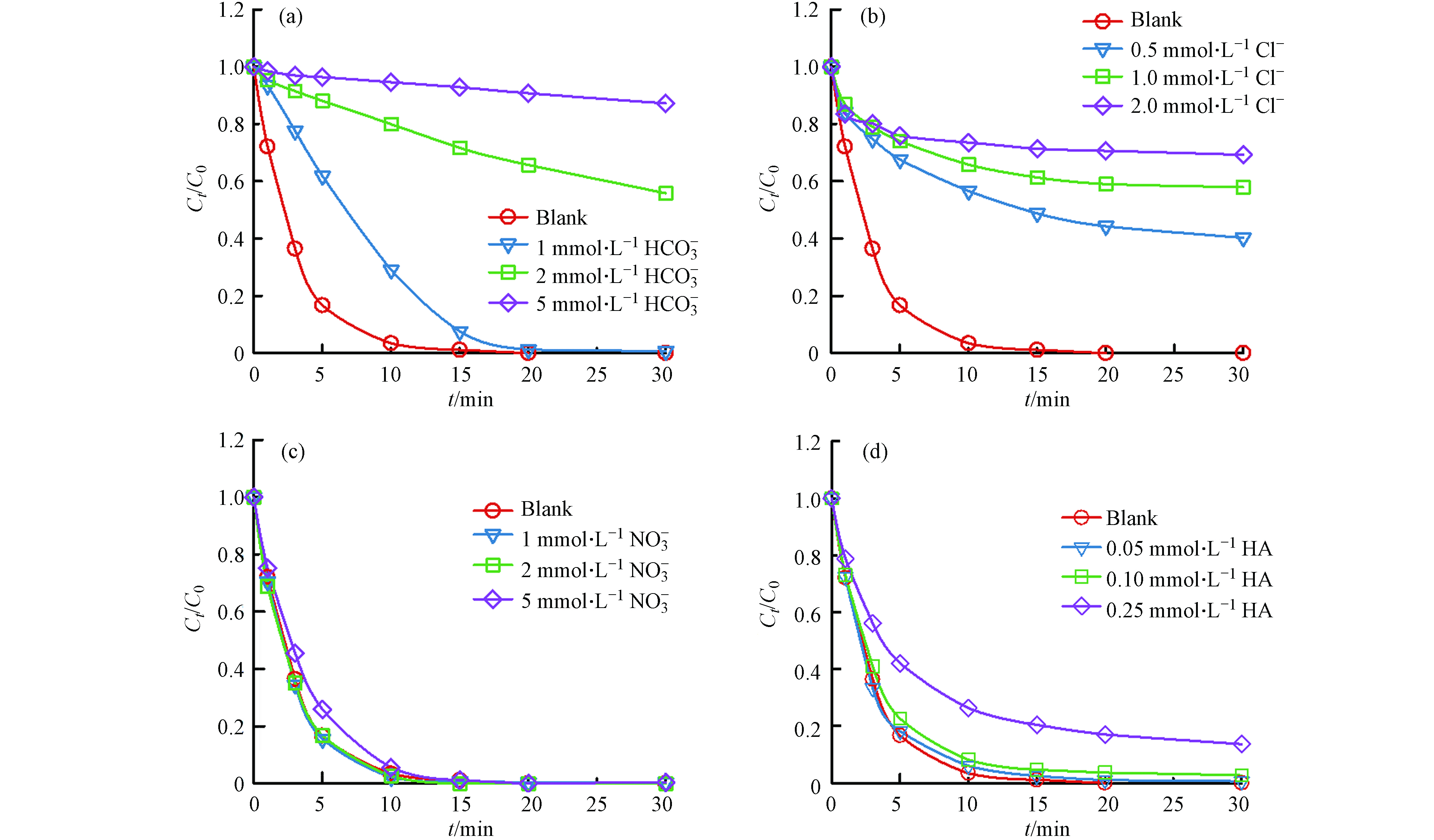 图 7 不同阴离子和HA条件下2,4-D降解效率Figure 7. Degradation efficiency of 2,4-D under different anions and HA(a)
图 7 不同阴离子和HA条件下2,4-D降解效率Figure 7. Degradation efficiency of 2,4-D under different anions and HA(a)HCO−3 NO−3 2.4 水中共存物质的影响
在实际水体中,无机阴离子能够通过改变pH、捕获自由基、抑制PMS分解等途径来影响污染物的降解[16]. 因此选取了水体中常见的几种无机阴离子(Cl−、
NO−3 HCO−3 HCO−3 SO⋅−4 SO⋅−4 CO⋅−3 NO−3 NO−3 NO−3 ⋅OH+Cl−→ClOH− (1) ClOH−+H+→Cl⋅+H2O (2) SO⋅−4+Cl−→Cl⋅+SO2−4 (3) 水中的天然有机物 (NOM) 是一种普遍存在的含有羧基和酚羟基的物质. NOM 可以通过淬灭溶液中的自由基来抑制底物降解,并阻断催化剂表面的活性位点[19]. 如图8(d)所示,5 mg腐殖酸(HA)的加入使2,4-D的去除率下降到87%,这是因为HA作为一种有机物,与2,4-D存在着竞争∙OH和
SO⋅−4 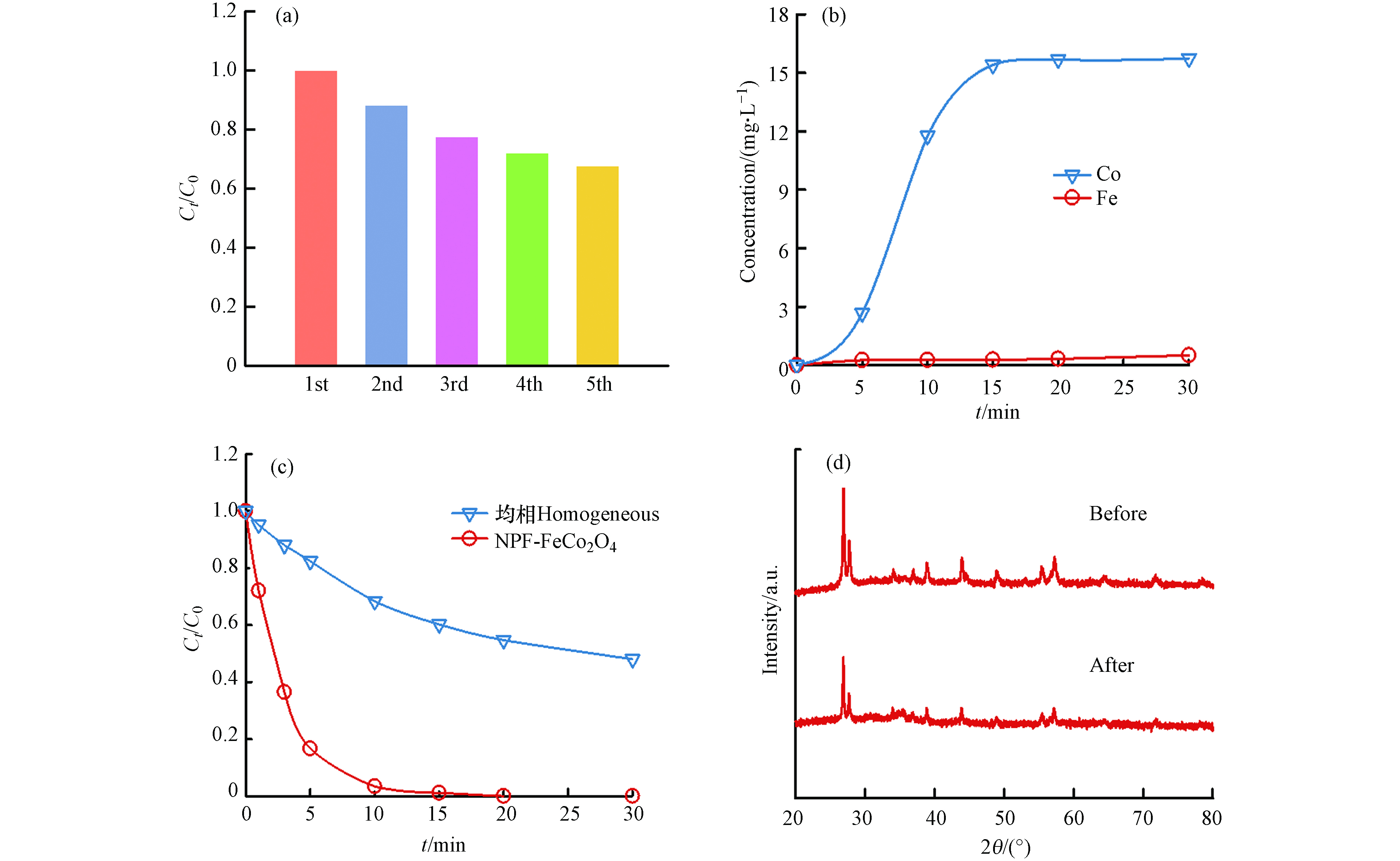 图 8 催化剂的稳定性实验Figure 8. Stability experiment of catalyst(a)NPF-FeCo2O4催化剂的循环试验,(b)体系中金属离子的析出量,(c)均相与非均相体系降解2,4-D,(d)反应前后催化剂的XRD图(a) the cycle test of NPF-FeCo2O4 catalyst, (b) the dissolved amount of metal ions in the system, (c) degradation of 2,4-D in homogeneous and heterogeneous systems, (d) XRD diagram of the catalyst before and after the reaction.
图 8 催化剂的稳定性实验Figure 8. Stability experiment of catalyst(a)NPF-FeCo2O4催化剂的循环试验,(b)体系中金属离子的析出量,(c)均相与非均相体系降解2,4-D,(d)反应前后催化剂的XRD图(a) the cycle test of NPF-FeCo2O4 catalyst, (b) the dissolved amount of metal ions in the system, (c) degradation of 2,4-D in homogeneous and heterogeneous systems, (d) XRD diagram of the catalyst before and after the reaction.⋅OH+HCO−3→CO⋅−3+H2O (4) SO⋅−4+HCO−3→SO2−4+HCO⋅3 (5) HCO⋅3→H++CO⋅−3 (6) 2.5 催化剂的稳定性
为了验证NPF-FeCo2O4催化剂的可重复使用性,进行了5个循环的实验. 如图8(a)所示,在循环反应中催化剂活性逐渐降低,在第5次反应中2,4-D的去除率为68%. 采用原子发射光谱测定了反应30 min溶液中的金属离子析出量,如图8(b)所示,在反应30 min后的溶液里检测到Co离子析出量为15 mg·L−1,Fe离子析出量为0.5 mg·L−1. 进一步测量浸出的金属离子对反应体系中降解2,4-D的贡献,如图8(c)所示,在NPF-FeCo2O4/PMS体系中反应15 min后2,4-D降解率即高于99%,而溶液中Co离子的贡献仅为40%,表明溶液中2,4-D的去除主要是催化剂NPF-FeCo2O4的作用. 对比了反应5次前后NPF-FeCo2O4催化剂的XRD图,如图8(d)所示,催化剂反应前后的特征峰强度有轻微的减小,催化剂的形态结构有所消耗,表明反应过程中Co离子的浸出和催化剂结构的消耗损失是催化剂失活的原因.
为了探究NPF-FeCo2O4/PMS体系对不同污染物的去除效率,选择了2,4-二氯苯氧乙酸(2,4-D),磺胺甲恶唑(SMX)、2,4-二氯苯酚(2,4-DCP)和活性红为代表的不同官能团污染物. 如图9所示,在反应30 min后,对4种污染物的去除率均能达到100%. 表明 NPF-FeCo2O4/PMS体系对不同种类污染物降解的广泛适用性.
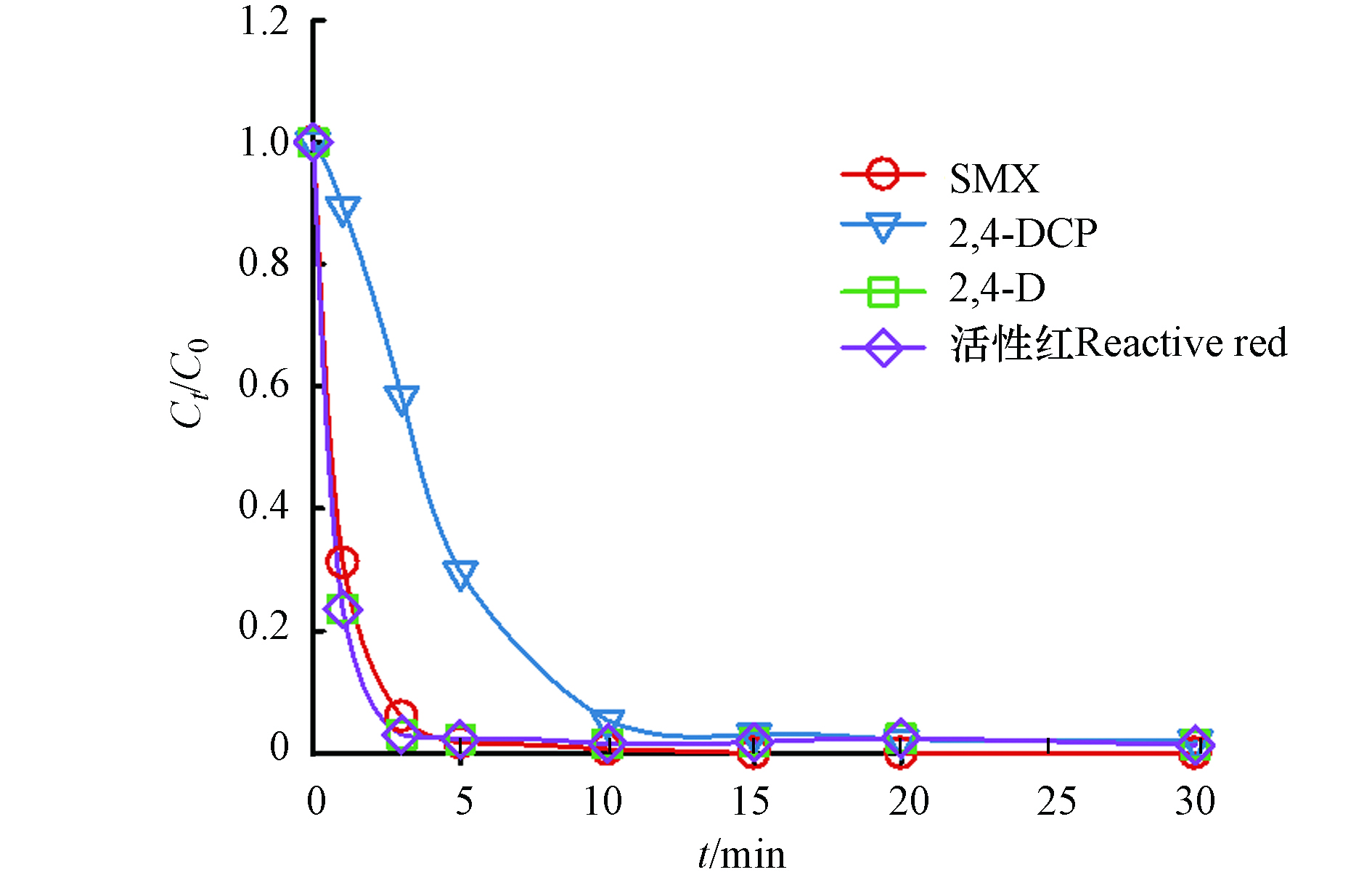 图 9 催化剂对不同污染物的去除效率Figure 9. The removal efficiency of catalysts for different pollutants
图 9 催化剂对不同污染物的去除效率Figure 9. The removal efficiency of catalysts for different pollutants2.6 催化机理探究
应用EPR技术原位捕获反应体系中产生的活性氧物种,使用DMPO作为∙OH、
SO⋅−4 O⋅−2 SO⋅−4 SO⋅−4 SO⋅−4 O⋅−2  图 10 (a)不同体系中
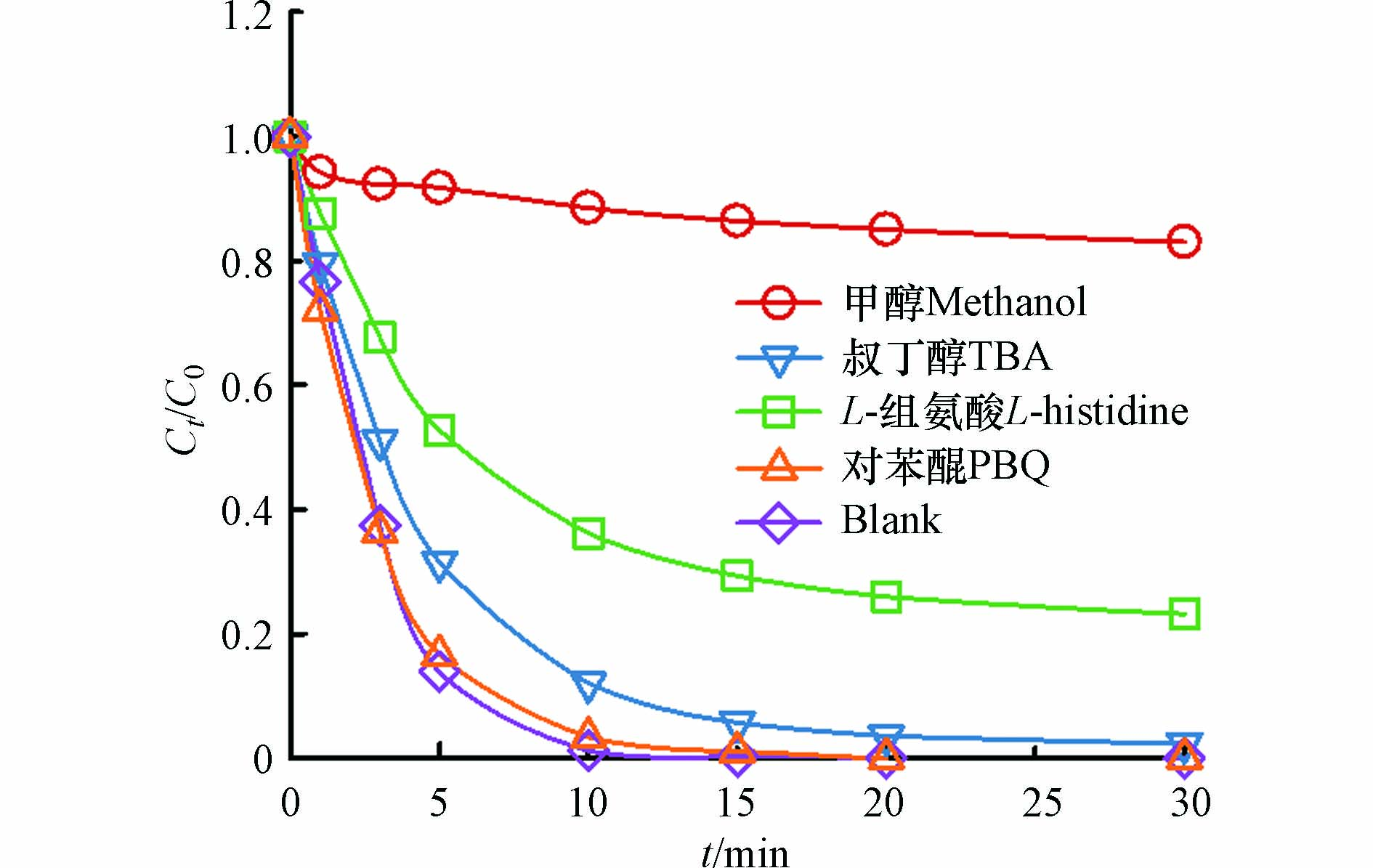
图 10 (a)不同体系中SO⋅−4 Figure 10. Study on the mechanism of catalysis: (a) identification ofSO⋅−4 为了进一步确定NPF-FeCo2O4/PMS体系中主要的活性氧物种,进行了自由基淬灭实验. 如图10所示,分别选择了甲醇、叔丁醇、L-组氨酸、对苯醌作为自由基淬灭剂. 其中,甲醇能够同时淬灭反应体系中的∙OH和SO4∙-,叔丁醇则用于淬灭体系中的∙OH,对苯醌和L-组氨酸能够有效的淬灭体系中的
O⋅−2 3. 结论(Conclusion)
(1)通过简便的一步水热法利用离子液体合成了N、P、F 3种元素掺杂的NPF-FeCo2O4催化剂,该催化剂能够高效活化PMS去除水中的2,4-D. 在催化剂投加量为0.1 g·L−1,2,4-D浓度为0.1 mmol·L−1,PMS投加量为0.3 g·L−1时,30 min内体系能去除100%的2,4-D.
(2)相较于未掺杂离子液体的FeCo2O4和只掺杂了N、P两种元素的NP/ FeCo2O4催化剂,掺杂了N、P、F的3种元素使得催化剂能够有更多氧空位、Co—N、Fe—N、P—O和吡啶N等表面活性位点,从而提高了催化性能.
(3)NPF-FeCo2O4/PMS体系可在pH值为3—9的范围内对2,4-D有高效的去除效果,同时该体系对不同官能团的污染物都能达到一定的去除率,水中低浓度的阴离子Cl-、HCO3-和腐殖酸对反应有轻微的抑制,通过EPR测试和淬灭试验共同结果可以得知NPF-FeCo2O4/PMS体系中SO4∙-是降解2,4-D的主要活性氧物种.
(4)本文为双金属催化剂在活化PMS降解水中有机污染物的应用提供了参考,但是在实际处理中还应考虑到Co离子的浸出问题. 今后的研究重点,需提高催化剂中金属Co的利用率如制备成单原子催化剂等,可显著减少催化剂中金属Co的含量,同时保持较高的催化效率. 另外,还可将Co基催化剂与碳材料结合,利用碳基材料稳定金属纳米颗粒,减少金属离子浸出对环境造成的潜在危害.
-
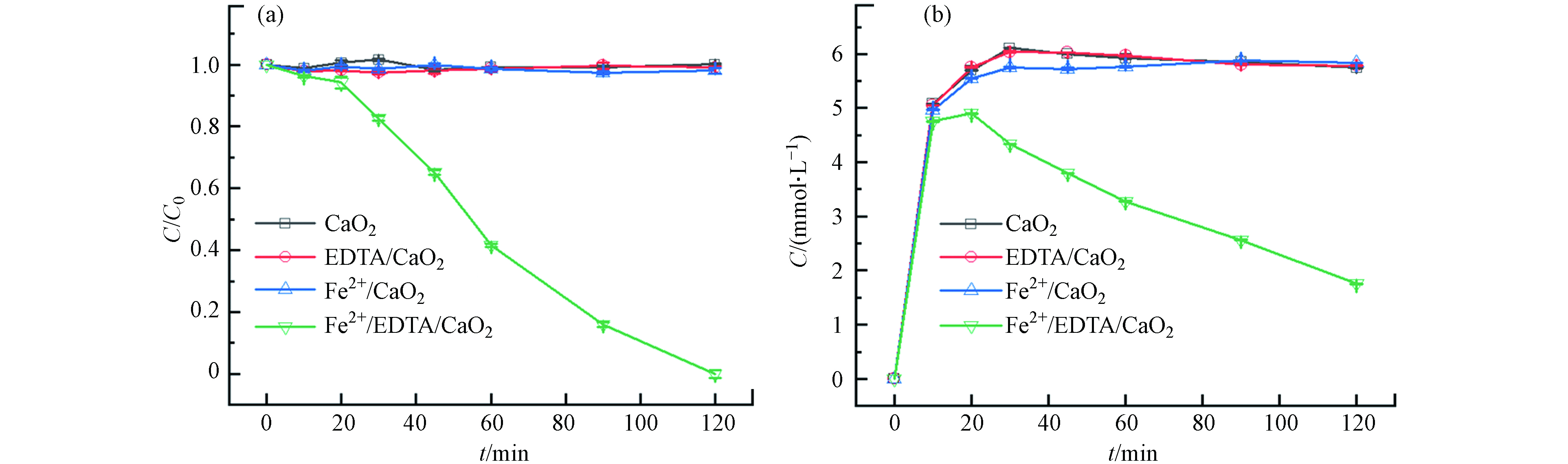
图 1 不同组分体系中苯酚的降解效果(a)及H2O2的浓度变化(b)
Figure 1. The degradation of phenol(a)and the change of H2O2 concentration(b)in different component systems
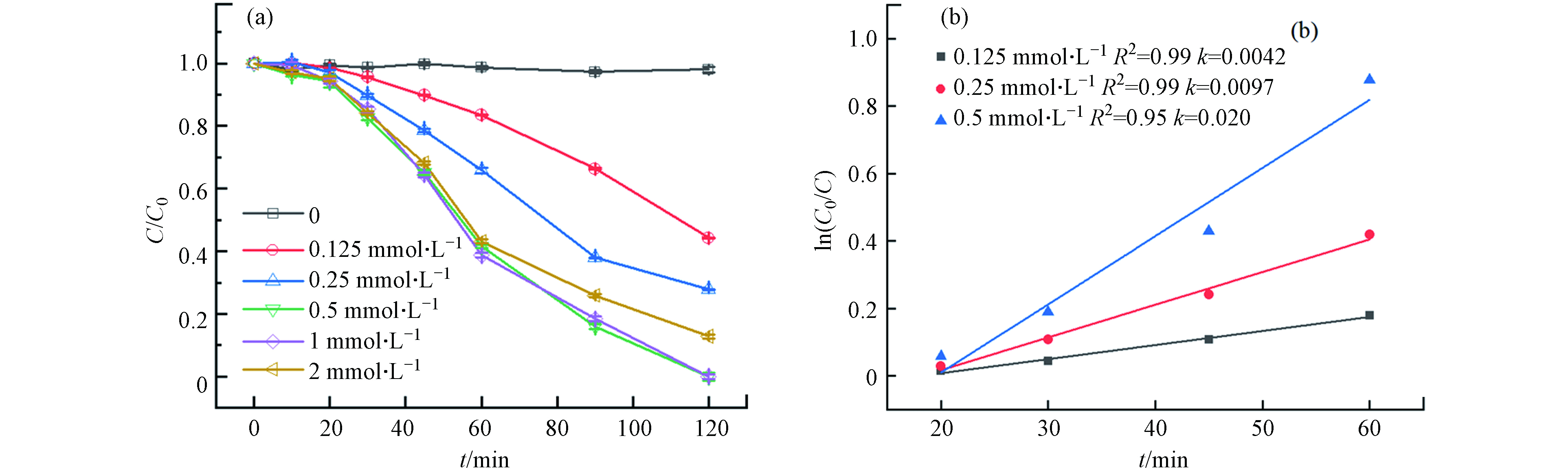
图 2 EDTA浓度对Fe3+/EDTA/CaO2体系降解苯酚的影响(a)及动力学拟合(b)
Figure 2. Effect of EDTA concentration on the degradation of phenol(a)by Fe3+/EDTA/CaO2 system and kinetic fitting(b)
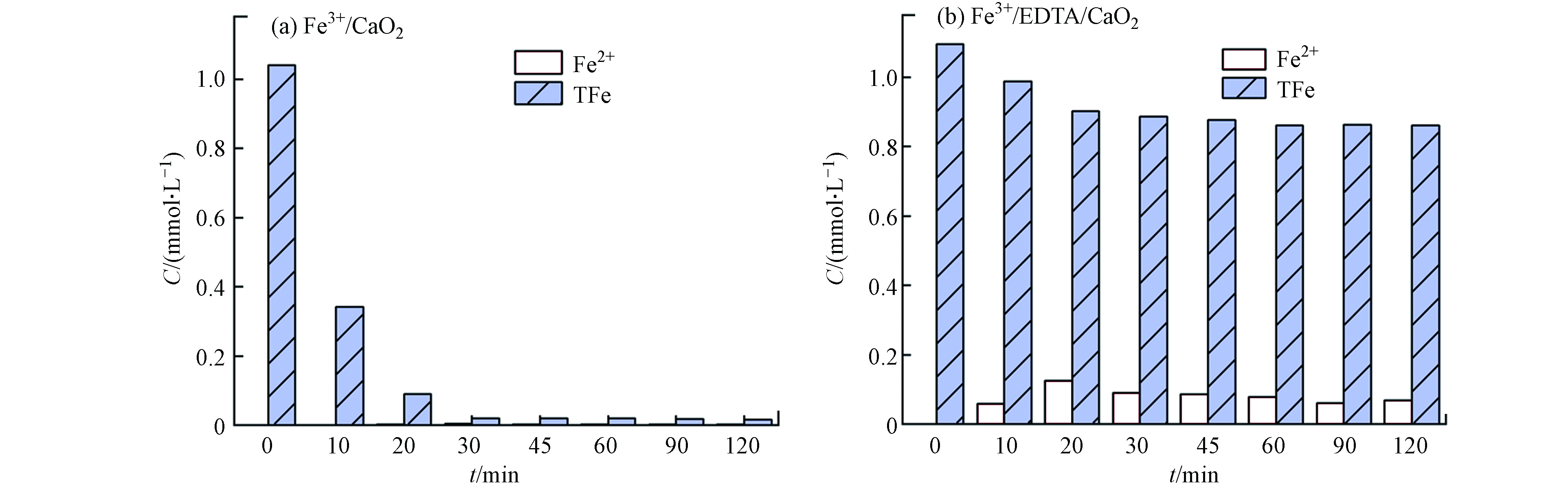
图 3 Fe3+/CaO2和Fe3+/EDTA/CaO2体系中游离态铁离子的浓度变化
Figure 3. Changes in the concentration of free state iron ions in the Fe3+/CaO2 and Fe3+/EDTA/CaO2 systems
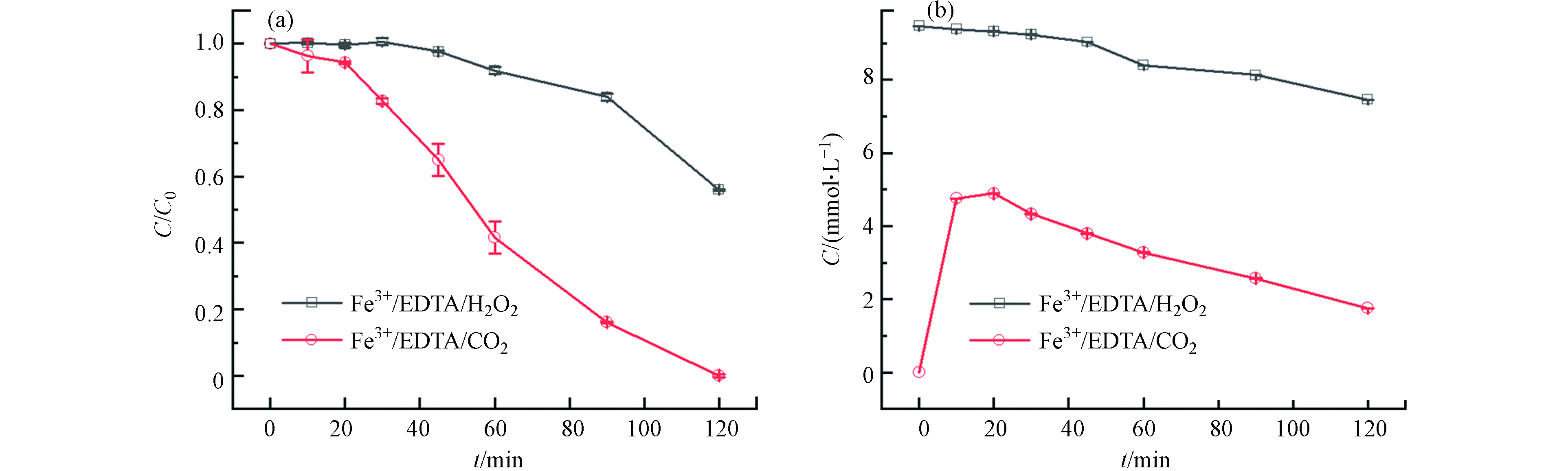
图 4 不同组分体系中的苯酚降解效果(a)及H2O2浓度变化(b)
Figure 4. The degradation of phenol(a)in different component systems(b)
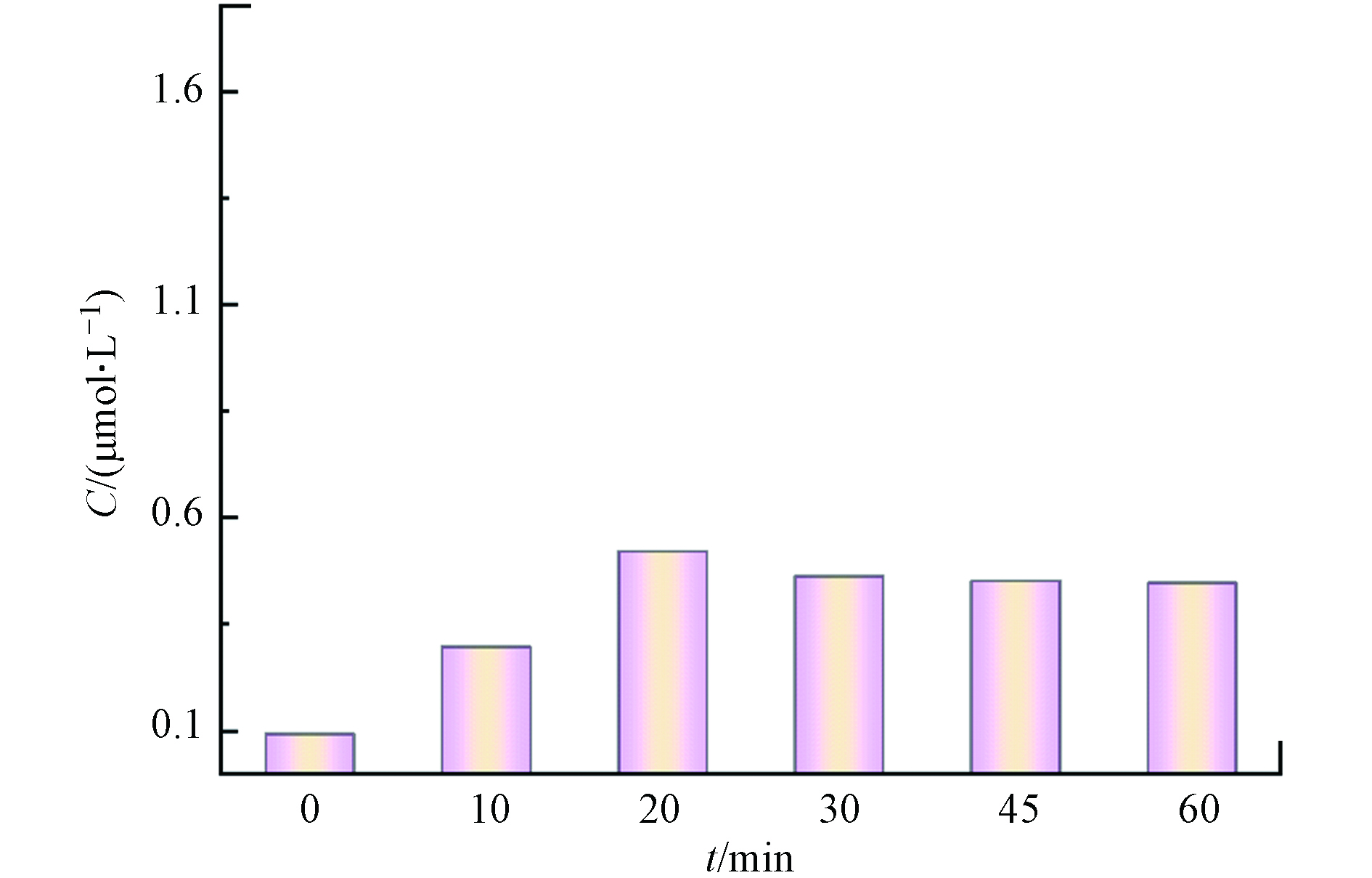
图 5 掩蔽H2O2后Fe3+/EDTA/CaO2体系中Fe2+的浓度变化
Figure 5. The change of Fe2+ concentration after clearing H2O2 in the Fe3+/EDTA/CaO2 system
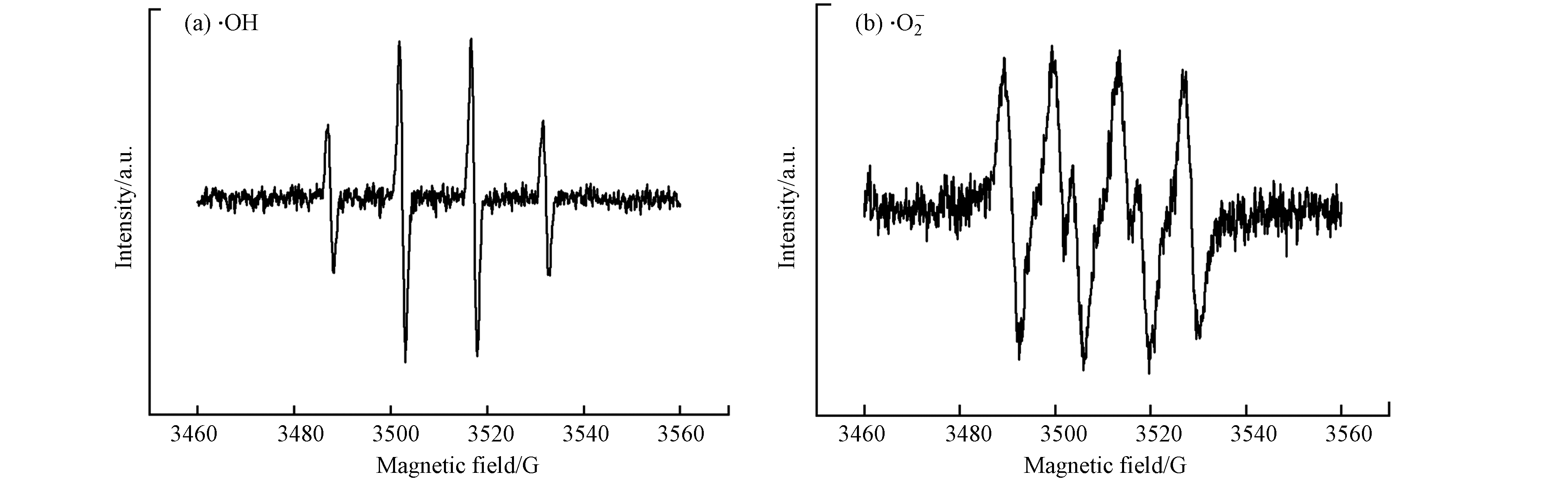
图 6 30 min时体系中活性自由基的EPR图
Figure 6. EPR of reactive free radicals in the system at 30 min
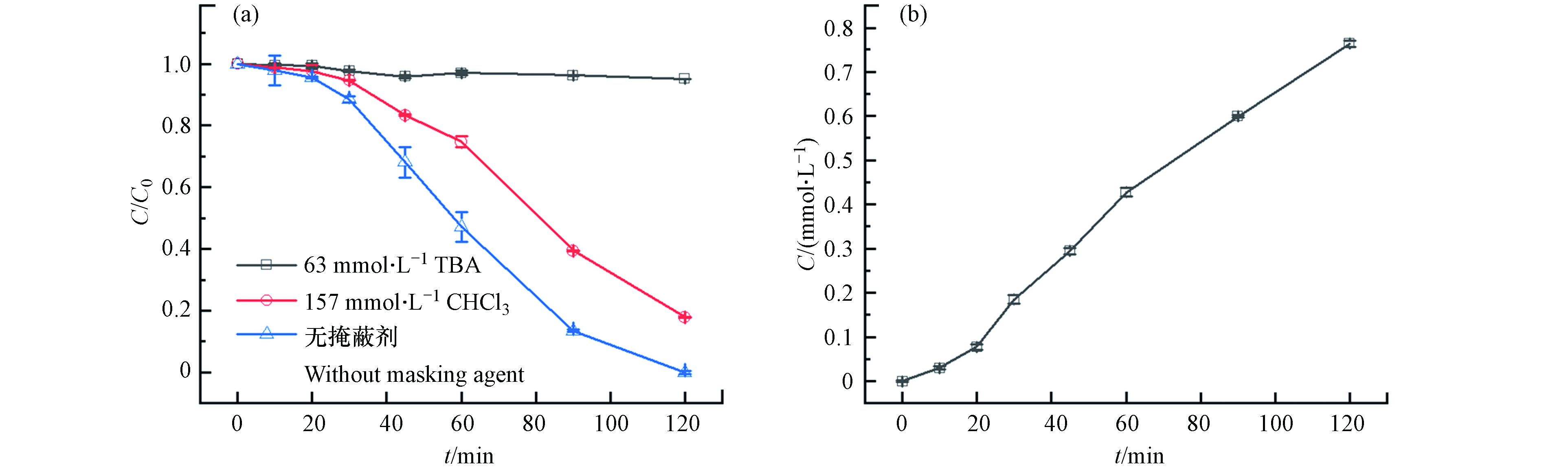
图 7 活性自由基的掩蔽(a)与探针实验(b)
Figure 7. Clearing (a) and probe experiments (b) of reactive free radicals
表 1 GC-MS测得苯酚降解的中间产物
Table 1. Intermediate products of phenol degradation measured by GC-MS
仪器出峰时间/min Peak time 化合物 Compound 分子结构 Molecular Structure 取样时间/min Sampling time 0 10 30 60 120 6.73 苯酚 
√ √ √ √ 12.5 对苯二酚 √ √ √ √ 12.1 邻苯二酚 √ √ √ √ 6.7 对苯醌 √ √ √ 3.5 乙醛酸 √ √  下载: 导出CSV
下载: 导出CSV
-
[1] FENTON H J H. Oxidation of tartaric acid in presence of iron [J]. Journal of the Chemical Society, Transactions, 1894, 65: 899-910. doi: 10.1039/CT8946500899 [2] NEYENS E, BAEYENS J. A review of classic Fenton's peroxidation as an advanced oxidation technique [J]. Journal of Hazardous Materials, 2003, 98(1/2/3): 33-50. [3] ALEKSIĆ M, KUŠIĆ H, KOPRIVANAC N, et al. Heterogeneous Fenton type processes for the degradation of organic dye pollutant in water—The application of zeolite assisted AOPs [J]. Desalination, 2010, 257(1/2/3): 22-29. [4] SAFARZADEH-AMIRI A, BOLTON J R, CATER S R. The use of iron in advanced oxidation processes [J]. Journal of Advanced Oxidation Technologies, 1996, 1(1): 18-26. [5] KOCHANY J, LIPCZYNSKA-KOCHANY E. Application of the EPR spin-trapping technique for the investigation of the reactions of carbonate, bicarbonate, and phosphate anions with hydroxyl radicals generated by the photolysis of H2O2 [J]. Chemosphere, 1992, 25(12): 1769-1782. doi: 10.1016/0045-6535(92)90018-M [6] 方兴斌. CaO2催化氧化水中双酚A及化学缓释供氧的研究[D]. 上海: 华东理工大学, 2017. FANG X B. Bisphenol A degradation using Fe3+-activated CaO2 and O2 supply through CaO2 slow relasing in microbial systems[D]. Shanghai: East China University of Science and Technology, 2017(in Chinese).
[7] 章琴琴, 丁世敏, 封享华, 等. Fenton法降解邻苯二甲酸二乙酯的动力学特征及其影响因素研究 [J]. 环境化学, 2020, 39(11): 3009-3016. doi: 10.7524/j.issn.0254-6108.2019082201 ZHANG Q Q, DING S M, FENG X H, et al. Study on the degradation kinetic characteristics and influencing factors of diethyl phthalate by Fenton treatment [J]. Environmental Chemistry, 2020, 39(11): 3009-3016(in Chinese). doi: 10.7524/j.issn.0254-6108.2019082201
[8] 钱婧, 李威, 张银龙, 等. Fenton法/草酸钠-Fenton法降解典型抗癌药5-氟尿嘧啶 [J]. 环境化学, 2014, 33(7): 1229-1234. doi: 10.7524/j.issn.0254-6108.2014.07.016 QIAN J, LI W, ZHANG Y L, et al. Degradation of anticancer drug 5-fluorouracil by Fenton and oxalic-Fenton process [J]. Environmental Chemistry, 2014, 33(7): 1229-1234(in Chinese). doi: 10.7524/j.issn.0254-6108.2014.07.016
[9] LI T Y, ZHANG C W, ZHANG J Y, et al. Remediation of 2, 4-dichlorophenol-contaminated groundwater using nano-sized CaO2 in a two-dimensional scale tank [J]. Frontiers of Environmental Science & Engineering, 2021, 15(5): 87. [10] ZHANG X, GU X G, LU S G, et al. Degradation of trichloroethylene in aqueous solution by calcium peroxide activated with ferrous ion [J]. Journal of Hazardous Materials, 2015, 284: 253-260. doi: 10.1016/j.jhazmat.2014.11.030 [11] WANG H F, ZHAO Y S, SU Y, et al. Fenton-like degradation of 2, 4-dichlorophenol using calcium peroxide particles: Performance and mechanisms [J]. RSC Advances, 2017, 7(8): 4563-4571. doi: 10.1039/C6RA26754H [12] LI Y M, WANG J, ZHANG A, et al. Enhancing the quantity and quality of short-chain fatty acids production from waste activated sludge using CaO2 as an additive [J]. Water Research, 2015, 83: 84-93. doi: 10.1016/j.watres.2015.06.021 [13] ENGELMANN M D, BOBIER R T, HIATT T, et al. Variability of the Fenton reaction characteristics of the EDTA, DTPA, and citrate complexes of iron [J]. Biometals, 2003, 16(4): 519-527. doi: 10.1023/A:1023480617038 [14] 陈东洋, 冯家力, 张昊, 等. 固相萃取/高效液相色谱法测定饮用水中苯并(a)芘及双酚A [J]. 分析测试学报, 2015, 34(7): 848-851. doi: 10.3969/j.issn.1004-4957.2015.07.017 CHEN D Y, FENG J L, ZHANG H, et al. Determination of benzo(a) pyrene and bisphenol A in drinking water by solid phase extraction/high performance liquid chromatography [J]. Journal of Instrumental Analysis, 2015, 34(7): 848-851(in Chinese). doi: 10.3969/j.issn.1004-4957.2015.07.017
[15] 张成武. 基于Fe(Ⅱ)-STPP配合物活化分子氧的高级氧化体系降解对硝基酚的效果及机理[D]. 长春: 吉林大学, 2019. ZHANG C W. Reserch on the effect and mechanism of P-nitrophenol degradation by advanced oxidation technology based on activation of molecular oxygen by Fe(Ⅱ)-STPP complex[D]. Changchun: Jilin University, 2019(in Chinese).
[16] LING Y H, LONG M C, HU P D, et al. Magnetically separable core-shell structural γ-Fe2O3@Cu/Al-MCM-41 nanocomposite and its performance in heterogeneous Fenton catalysis [J]. Journal of Hazardous Materials, 2014, 264: 195-202. doi: 10.1016/j.jhazmat.2013.11.008 [17] BIAGLOW J E, KACHUR A V. The generation of hydroxyl radicals in the reaction of molecular oxygen with polyphosphate complexes of ferrous ion [J]. Radiation Research, 1997, 148(2): 181-187. doi: 10.2307/3579576 [18] JOO S H, FEITZ A J, SEDLAK D L, et al. Quantification of the oxidizing capacity of nanoparticulate zero-valent iron [J]. Environmental Science & Technology, 2005, 39(5): 1263-1268. [19] PAN Y, SU H R, ZHU Y T, et al. CaO2 based Fenton-like reaction at neutral pH: Accelerated reduction of ferric species and production of superoxide radicals [J]. Water Research, 2018, 145: 731-740. doi: 10.1016/j.watres.2018.09.020 期刊类型引用(1)
1. 彭建彪,常宇,吴佳琪,郭欣婷,司宁,马霖轩,刘海津,曹治国,高士祥. Co-Cu双金属氧化物活化过一硫酸盐去除水中磺胺甲噁唑. 环境化学. 2024(02): 642-649 .  本站查看
本站查看
其他类型引用(1)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 3885
- HTML全文浏览数: 3885
- PDF下载数: 88
- 施引文献: 2










