

-
锑污染是湖南省资江流域典型水体污染问题。锑具有破坏DNA并改变细胞内mRNA水平、影响体内酶和器官运转的危害,对人体毒性较大[1]。我国长期以来的锑矿采选冶炼带来了较严重的水环境污染[2]。严重影响了当地生态环境以及周边地区居民的身体健康[3]。因而去除水体中过量的锑十分必要。目前已有的除锑方法主要有:吸附法[4-7]、混凝沉淀法[8-9]、电化学法[10-11]、膜过滤法[12-13]、离子交换法等[14-15]。
水体净化中最常用的吸附材料是碳材料。NAVARRO等[16]在使用活性炭吸附铜离子电解液中的锑时发现活性炭在水中含有带正电的表面基团,推测活性炭吸附金属离子的机理为静电作用。SALAM等[17]在研究合成多壁碳管吸附锑的干扰因素和机理时,认为Sb(Ⅲ)在碳管上的吸附机制为液膜扩散和进一步的颗粒内扩散。虽然碳材料对Sb有一定的吸附效果,但对于Sb质量浓度较低的水体处理效果一般,难以达到水质标准要求,而金属基材料对Sb的吸附效果更好。QI等[18]合成了Ce-Fe3O4复合材料并对Sb进行的吸附实验取得了较好的效果。SHAN等[19]使用赤铁矿颗粒对Sb进行处理,吸附容量达到了36.7 mg·g−1。除铁基材料外还有其他金属材料应用于锑的吸附,如活性氧化铝对锑也有较好的吸附去除效果[20],锰氧化物[21],镍铁氧化物复合材料[22]、钴氧化物对水中锑吸附效果都较好[23],可达饮用水标准。但这些金属基材料吸附除锑都存在一些缺点,如部分金属材料具有磁性易团聚不易分散[24],有些金属具有毒性对水体安全有影响。而将金属材料与活性炭结合制成复合材料用于吸附除锑是不错的选择。LUO等[25]使用活性炭负载氧化锆去除水中锑,在pH=7时复合材料对Sb(Ⅲ)和Sb(Ⅴ)的最大吸附容量为70.83 mg·g−1和57.17 mg·g−1。YU等[26]使用FeCl3改性颗粒活性炭,在pH=7.0、温度25 ℃的条件下对Sb的去除效果是普通活性炭的3.5倍。但上述研究尚未对复合材料的除锑机理进行深入的探讨,且氧化锆不经济、在饮用水中还存在安全隐患。
基于此,本研究采用共沉淀法将将水处理中常用且经济、安全的铁氧化物负载于颗粒活性炭上制成复合吸附材料用于去除水中锑,并对复合材料的理化特性进行了分析表征,深入探究了其对锑的吸附机理,以期为锑微污染水体的饮用水除锑提供参考。
-
锑元素标准溶液(500 mg·L−1 Sb(Ⅴ))由国家钢铁材料测试中心提供。硼氢化钾(优级纯)由科密欧公司生产。其他试剂如活性炭、焦锑酸钾(本实验Sb均由此药剂配制成Sb(Ⅴ))、氢氧化钠、氢氧化钾、盐酸、各类铁盐等均由国药集团提供。活性炭为椰壳炭,过30~50目筛,用去离子水清洗数次,洗去表面的灰分和杂质,烘干至恒重。
-
采用液相沉积法制备复合材料。称取一定量的二价与三价铁盐溶于100 mL的溶剂中,溶剂由水与乙醇组成,使用磁力搅拌至铁盐溶解后加入5 g活性炭(提前用硝酸处理并洗净烘干)并滴入NaOH溶液调整pH,持续搅拌一段时间后放入烘箱静置一段时间使材料陈化,待反应完全后取出材料用纯水冲洗掉多余物质,后置于烘箱内干燥即得铁氧化物/活性炭复合吸附材料(Fe2O3@AC)。
-
采用AFS-8220型原子荧光光度计测量锑的质量浓度。用扫描电子显微镜SEM(MIRA LMS)观察相关材料表面的形貌特征。运用傅里叶红外光谱仪(赛默飞 Niolet iN10)和X射线衍射仪(Ultima IV)表征相关材料所含的官能团与化学键等微观性质。
-
配置一定质量浓度的锑原水后,取100 mL锑原水和一定质量的吸附剂装入锥形瓶,置于水浴振荡器中,调节速率120 r·min−1、温度25 ℃,必要时使用盐酸和NaOH调节pH,反应一段时间后用注射器抽取上清液并用孔径为22 μm的滤膜过滤,使用原子荧光光度计测量水中锑的剩余的质量浓度。
-
本研究分别探讨了在Fe2O3@AC材料制备过程中所用溶剂组成比例、二价铁与三价铁比例及总铁浓度、制备液pH等对所制得的复合材料吸附除锑效果的影响,实验中吸附剂投加量为0.1 g·L−1,原水锑质量浓度为38 μg·L−1,原水体积为100 mL,pH=7.5,吸附时间240 min。
考虑到乙醇作为有机溶剂时具有改变材料界面结构,控制材料一定程度的膨胀或收缩作用[27]。本实验以纯水和乙醇按照不同比例配置制备溶剂,以探讨制备过程中不同溶剂组成比例对Fe2O3@AC吸附除锑效果的影响。所用纯水:乙醇体积比分别为1:4、2:3、3:2、4:1、5:0,将制备的吸附材料进行吸附锑的实验,结果如图1(a)。纯水:乙醇体积比为4∶1时Fe2O3@AC除锑效果最好。这是因为乙醇作为溶剂对材料的结构与铁氧化合物的晶型具有修饰作用,可在铁氧化物晶核生成后的颗粒生长过程中产生螯合作用进而抑制其生长的过大,形成粒径分布较为均匀的铁氧化物颗粒,拥有更好的吸附结构[28]。乙醇比例进一步升高后吸附效果则会降低,推测是因为水含量减少使铁盐的溶解性受影响,在调节pH时容易迅速形成沉淀堵塞活性炭的孔隙,反而不利于其吸附性能。
由于材料制备的共沉淀反应过程中Fe2+与Fe3+的摩尔比对于颗粒的组成、形貌、磁性均有影响[29]。当Fe2+:Fe3+摩尔比分别为0.3、0.5、1、2、3时,Fe2O3@AC除锑效果如图1(b)所示。含有不同 Fe2:Fe3+摩尔比的复合材料除锑效果有所不同,Fe2+:Fe3+摩尔比为1:1时吸附材料除锑效果最好。有研究表明,当Fe2+:Fe3+比值较小时,共沉淀法生成的铁氧化物颗粒主要由γ—FeO(OH)组成,当Fe2+:Fe3+比值超过0.7时,产物的尺寸较为均匀,成分以Fe3O4为主,且会因O2的存在或pH的变化而被氧化为Fe2O3。本研究表明在Fe2+:Fe3+摩尔比为1:1时形成的铁氧化物种类和晶型具有最佳除锑效果。
材料制备过程中铁盐的投加量将影响铁氧化物颗粒的生成与负载数量,进而影响对锑的吸附位点数量和吸附效果。在铁盐浓度分别为0.099、0.198、0.297、0.396、0.495、0.594、0.693 mol·L−1时,Fe2O3@AC吸附锑的结果如图1(c)所示。随着铁盐浓度的升高,锑去除率逐渐增加,铁盐浓度为0.594 mol·L−1时,去除率达到最高,约为95%,后续继续增加铁盐浓度反而会使锑的去除率略微下降。这可能是投加量过多导致生成的颗粒过多堵塞了活性炭的孔隙。
活性炭表面的官能团种类和带电特性受环境pH的影响较大,铁氧化物的最终形态成分也与pH有关,并且负载在活性炭上的颗粒的平均粒径与分布范围也与pH有关[30]。为此,本研究考察了pH分别为1.67、1.88、4.16、5.98、12.85时Fe2O3@AC对锑的吸附性能。如图1(d)所示,酸性条件下制备出来的复合材料的除锑效果优于碱性条件,推测可能是酸性条件下活性炭表面的官能团以羧基和酚羟基为主,这些官能团有助于锑的吸附,且低pH下铁氧化物的形态也优于高pH。当pH=1.88时除锑效果最佳。
综上所述,本研究制备Fe2O3@AC复合吸附材料的最佳制备条件为:溶剂采用纯水:乙醇体积比4:1,制备用铁盐采用Fe2+:Fe3+摩尔比1:1,总铁浓度为0.594 mol·L−1,制备液pH=1.88。该条件下制备得到的Fe2O3@AC呈黑褐色,粒径为0.35~0.6 mm,总比表面积为883 m2·g−1,总孔容为0.548 cm3·g−1,孔结构以毛细孔为主。本研究所得Fe2O3@AC对Sb(Ⅴ)的最大吸附容量为1.79 mmol·g−1。有研究表明,其他材料对Sb(Ⅴ)吸附容量为:赤铁矿[19]为0.3 mmol·g−1,FeOOH[6]为0.83 mmol·g−1,MnO2[6]为0.81 mmol·g−1,Co3O4@rGO [23]为0.47 mmol·g−1,Ce-Fe3O4[18]为1.03 mmol·g−1,NiFe2O4[22]为0.76 mmol·g−1,AC为0.19 mmol·g−1。通过对比可见, Fe2O3@AC的最大Sb(Ⅴ)吸附容量显著优于其他单一金属氧化物和活性炭。
进一步使用正交实验对材料制备条件进行验证。如表1所示,考虑4个因素:水:乙醇、Fe2+:Fe3+、铁离子总浓度、pH,各因素选取三水平。4个影响因素的最佳指标值分别为k3、k2、k3、k2,对应的最佳制备条件分别为:水:乙醇=4:1、Fe2+:Fe3+=1:1、总铁浓度为0.6 mol·L−1、pH为1.9。这与上述单因子实验结果基本一致。极差分析结果表明,4因素中铁离子总浓度影响最大,其次为Fe2+:Fe3+和水:乙醇,pH的影响最小。
-
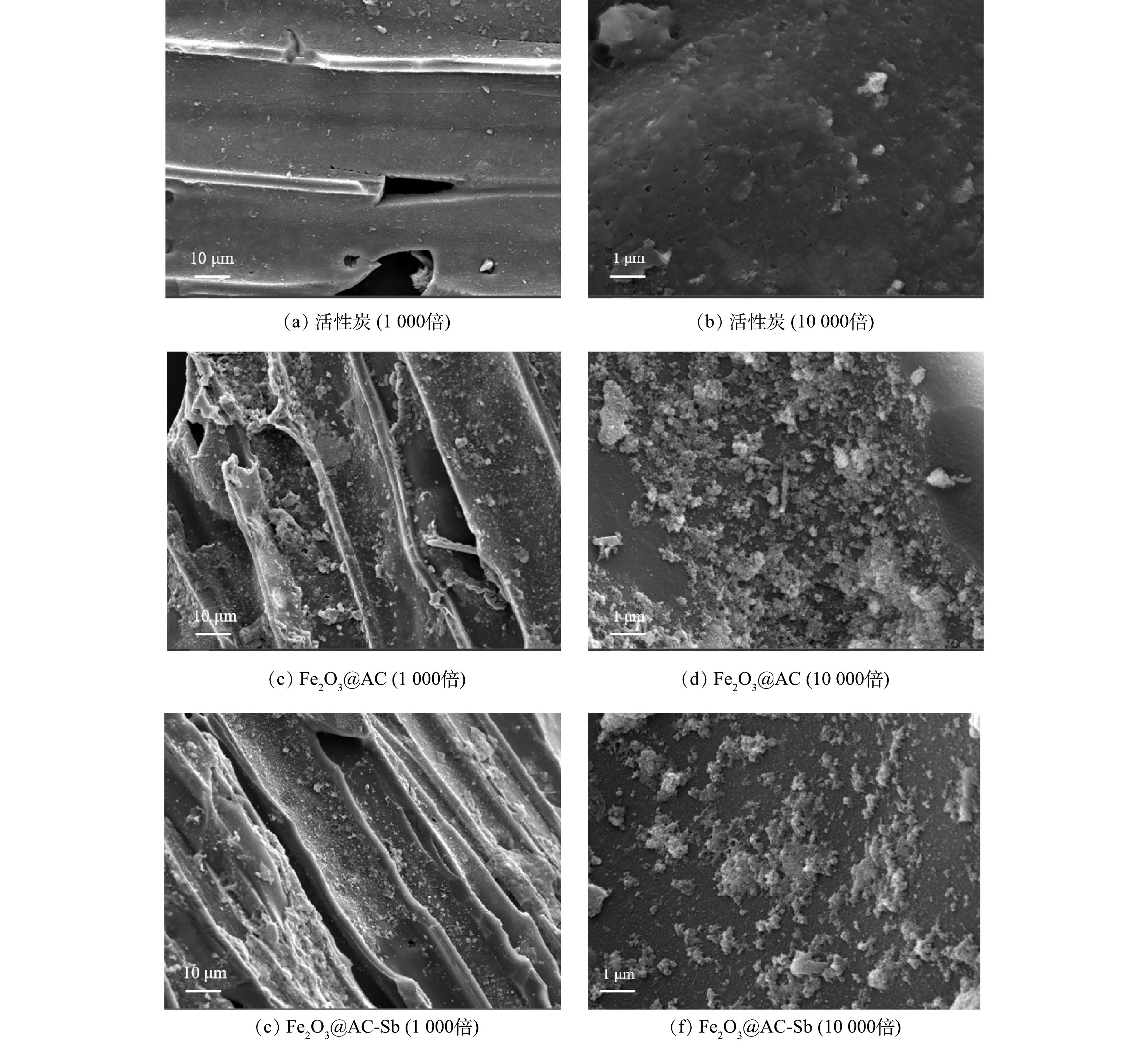
1) SEM表征分析。活性炭与Fe2O3@AC表面微观形态的SEM表征结果见图2。普通活性炭表面除少许灰分杂质颗粒外无其他附着物。而经过负载改性后的Fe2O3@AC表面明显可见大量的铁氧化合物颗粒,总体看呈现不均匀的形态,且大多数形状为多面体或准球形,铁氧化合物颗粒的尺寸分布较宽。这与后续XRD的表征结果一致。此外,铁氧化合物对活性炭表面的覆盖率较高,且无明显团聚堵塞孔隙的现象,可以印证本研究在前述制备过程中对浓度、前体盐比例、溶剂成分等变量的控制较为成功,生成的颗粒物形状和分散附着均较为良好。吸附反应前后Fe2O3@AC表面的物质结构没有明显变化,含量略微减少,推测可能是表面铁氧化物颗粒在反应过程中有一部分被消耗所致。
2) FTIR表征分析。铁氧化物粉末、普通活性炭、吸附锑前后铁氧化物/活性炭材料的红外光谱见图3,特征峰对比见表2。由图3可见,在铁氧化物粉末的红外光谱图中存在412 cm−1和682 cm−1处的吸收峰(—Fe—OH键),836 cm−1处的吸收峰(—Fe—O—Fe键),之后位于1 383、3 333、3 373 cm−1处的峰值则是铁氧化物粉末在制备过程中乙醇的加入而加强的—OH和—OOH的弯曲和拉伸震动。普通活性炭粉末的特征峰位于1 586 cm−1和3 438 cm−1,代表活性炭表面的—OH与—COOH [31]。而Fe2O3@AC复合材料代表—Fe—OH键的特征峰位于411 cm−1和679 cm−1处,—Fe—O—Fe键的特征峰位于879 cm−1处,特征峰略微有偏移。而位于1 257~1 586 cm−1的4个小峰和3 417 cm−1处的峰值则对应于—OH和—COOH,其来源于活性炭自带的表面羟基和羧基以及铁氧化物负载改性时所带的—OH,因此,峰值相较于单独的铁氧化物粉末和活性炭分布范围更宽。说明铁氧化物已成功负载于活性炭上,并对其表面的物化性质有一定的影响[32]。
3) XRD与XPS分析。采用X射线衍射图谱(XRD)对吸附材料的微观结构进行表征,结果如表3和图4(a)。Fe2O3@AC的衍射峰(存在于26.66°、35.06°、39.12°、46.22°、56.06°、61.2°处)与铁氧化物粉末的衍射峰(存在于27.5°、35.38°、39°、47.93°、56.46°、61.64°处)很相似,上述衍射峰分别为(220)、(311)、(400)、(422)、(511)和(440)反射,对应于磁铁矿的立方尖晶石结构。说明铁氧化物颗粒负载前后其本身的晶体结构以及纯度并没有被改变,且已成功负载于活性炭之上。此外,XRD光谱峰的宽度与颗粒的大小成反比,即光谱中较宽的峰意味着颗粒尺寸较小[32]。由图4(a)可以明显看出,Fe2O3@AC的铁氧化物特征峰比铁氧化物粉末的铁氧化物特征峰更宽。这说明铁氧化物经过负载后具有了比分散状态时更小的颗粒尺寸,能更好的分布于活性炭上,不易堵塞孔隙进而可发挥更好的吸附效果。Fe2O3@AC吸附前后的XRD图谱未发生变化,表明吸附与铁氧化物颗粒表面的锑并不改变铁氧化合物颗粒的晶型[33]。Fe2O3@AC吸附Sb(Ⅴ)后的XPS窄扫描如图4(b)所示。可见,检测到529 eV处的峰表明Fe2O3@AC表面存在锑元素,并且Sb(Ⅴ)已被Fe2O3@AC吸附。
-
将上述2.1节最佳条件下制备的Fe2O3@AC用于水源水中微量锑的去除研究,以探讨吸附除锑的最佳条件与效果,并与活性炭等其他材料的吸附除锑效果进行了比较。
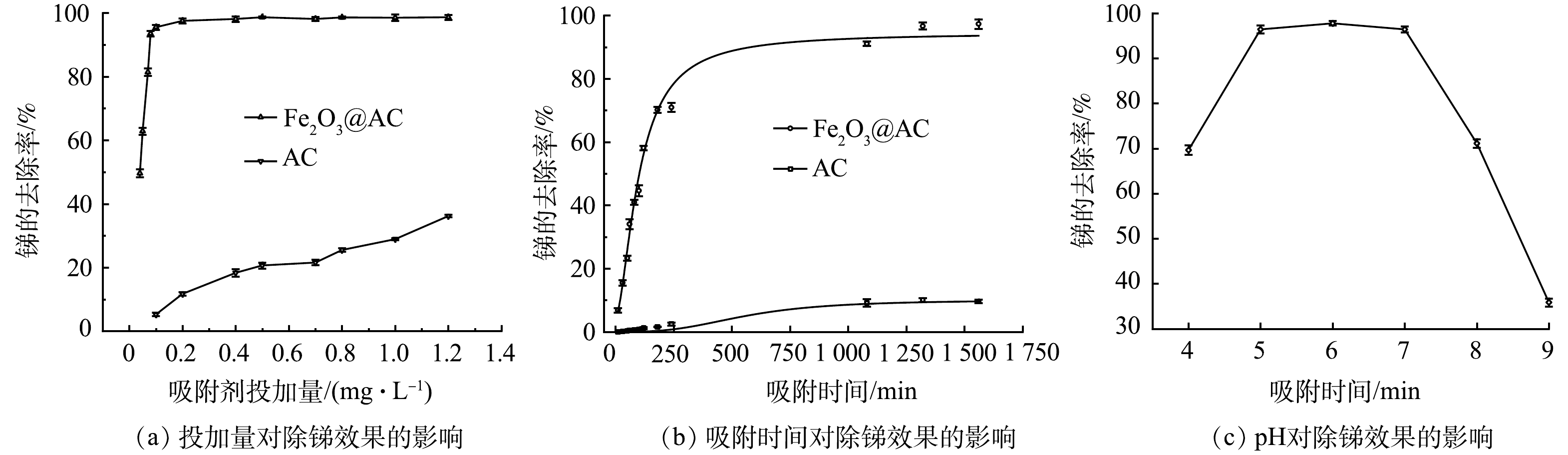
1)吸附材料投加量对除锑效果的影响。本实验比较在不同吸附剂投加量下,Fe2O3@AC和普通活性炭吸附除锑的效果。原水锑质量质量浓度为38 μg·L−1,吸附时间为720 min,实验结果如图5(a)所示。图中锑的去除率随着2种吸附剂投量的增加而逐渐升高。Fe2O3@AC投加量为0.08 g·L−1时,锑去除率即高达93.4%,剩余锑质量浓度仅为2.51 μg·L−1,达到了国家饮用水标准要求,继续增加投加量,锑去除率还会继续缓慢增加。但普通活性炭投量为0.1 g·L−1时去除率极低(仅为5%),增加投加量到1.2 g·L−1时,锑去除率也仅为36.6%,剩余锑质量浓度仍高达24.09 g·L−1。可见Fe2O3@AC对锑的去除效果远高于普通活性炭。
2)吸附时间对除锑效果的影响。以含有38 μg·L−1锑的水样为原水,在吸附剂投加量为0.08 g·L−1时进行吸附实验,探讨吸附时间对除锑效果的影响,以评估吸附过程速率。如图5(b)所示,在Fe2O3@AC和普通活性炭上锑的去除率均随吸附时间的延长而升高,并在前200 min内吸附量增加较快,后期吸附量增加缓慢,并在1 000 min后基本达到吸附平衡。Fe2O3@AC的吸附速率显著优于普通活性炭。吸附前200 min,Fe2O3@AC的锑吸附去除率达70.1%,而普通活性炭仅为1.6%。达到吸附平衡时,Fe2O3@AC吸附后溶液中剩余锑质量浓度为1.06 μg·L−1(去除率达97%),达到了生活饮用水卫生标准的要求,而普通活性炭吸附后的剩余锑质量浓度高达29.7 μg·L−1。这进一步说明Fe2O3@AC对锑具有较好的吸附性能。这主要是因为活性炭经过酸处理和铁氧化物改性后,具有更多丰富的表面官能团(—OH、—COOH,—Fe—OH,—Fe—O—Fe),因而具有较强的金属螯合能力[34]。
3)溶液pH对除锑效果的影响。在原水中锑质量浓度为38 μg·L−1,Fe2O3@AC投加量为0.08 g·L−1条件下,考察了不同pH条件下的除锑效果。如图5(c)所示,当pH在5~7时除锑效果最好。这是因为不同pH下锑离子在溶液中的存在形态和吸附剂的表面电荷有所不同。有研究[35]表明,Fe2O3@AC的pHzpc为6.5,故而在pH大于7时吸附剂表面带电荷,且在pH大于7时Sb(Ⅴ)以H2SbO4−和Sb(OH)6−形态存在,所以此时材料表面与锑离子存在静电排斥不利于吸附反应。而当环境pH降低至5~7时,Fe2O3@AC表面的负电荷数减少甚至带正电荷,而Sb(Ⅴ)此时依然以H2SbO4−和Sb(OH)6−形态存在(Sb(Ⅴ)在pH>2.7时粒子都带负电荷)[35],因此,材料表面与锑粒子在pH处于5~7时电荷的互斥作用减弱甚至存在电荷的吸引,所以此时吸附效果较好。当pH进一步降低至5以下时吸附效果却有所降低。这可能是因为,pH的下降可影响Fe2O3@AC表面铁氧化物的稳定性能,使其部分被酸溶解,进而影响对Sb(Ⅴ)的吸附。
-
在原水中锑质量浓度为33 μg·L−1,Fe2O3@AC投加量为0.08 g·L−1的条件下进行吸附动力学实验,采用准一级动力学[36]、准二级动力学[37]、Bangham动力学[38]和颗粒内扩散模型[39]对其进行拟合,结果见图6和表4。由图6(a)~(b)和表4可知,准一级与准二级动力学模型对Fe2O3@AC吸附锑的反应过程均有较好的拟合效果,但准二级动力学拟合结果优于准一级动力学。以上结果表明锑在Fe2O3@AC上的附着很大程度上是以化学吸附为主[40],其中涉及到吸附剂与吸附质之间的电子共用或电子转移。
对于多孔粉末状吸附剂而言,吸附类型主要有液膜扩散、颗粒内扩散、质量扩散。由图6(c)~(d)和表4可知,颗粒内扩散对于Fe2O3@AC吸附锑的过程拟合效果很差。这说明整个过程主要吸附类型不是颗粒内扩散为主导的,可能是多种模式的叠加状态。根据以上结果推测:100 min之前斜率大,吸附速率快,此时以液膜扩散为主,锑迅速随着Fe2O3@AC表面的液膜分布在各个吸附面上;在100~200 min,吸附速率放缓,此时以颗粒内扩散为主,锑在Fe2O3@AC表面和内部的孔隙中扩散附着。在200 min后则是以质量扩散为主,此时水中锑质量浓度大大降低,吸附速率也随之放缓,最终逐渐达到平衡。
-
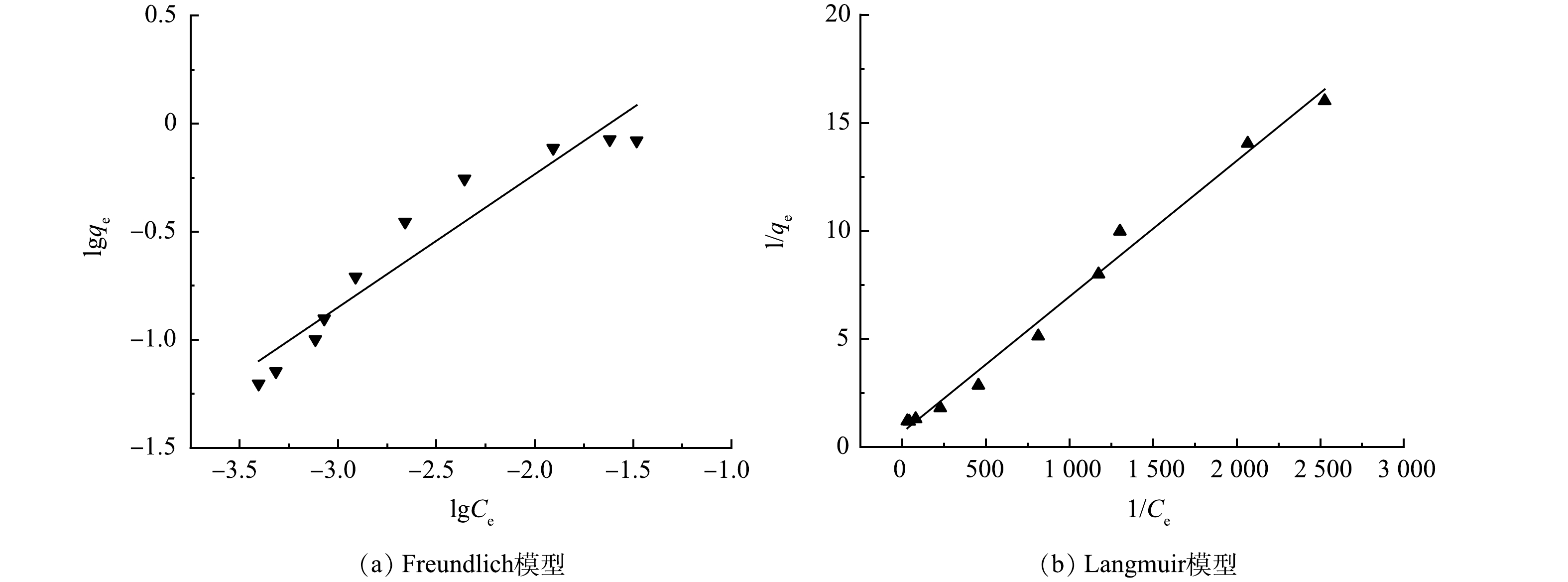
在吸附剂投加量为0.5 g·L−1,锑质量浓度为30~500 μg·L−1条件下进行等温吸附实验,并采用Langmuir和Freundlich 2种模型进行拟合,其结果如图7和表5所示。可看出Langmuir模型的拟合效果更好,拟合系数R2>0.99,表明Fe2O3@AC吸附锑为单层吸附[41]。
-
通过吸附等温线的拟合表明Fe2O3@AC吸附锑是单层吸附;通过吸附动力学的模拟表明锑在Fe2O3@AC上的附着很大程度上是以化学吸附为主;通过对比图3中Fe2O3@AC吸附锑前后的红外图谱可见, 411 cm−1和679 cm−1处代表—Fe—OH键的特征峰、879 cm−1处代表—Fe—O—Fe键的特征峰经吸附反应后分别拉伸至420、695、891 cm−1,说明吸附反应中—Fe—OH键和—Fe—O—Fe键均有参与反应;而代表羟基(—OH)和羧基(—COOH)的吸收峰1 257、1 319和3 417 cm−1在吸附后移至1 269、1 315和3 382 cm−1,表明羟基(—OH)和羧基(—COOH)对于锑的吸附去除也有着重要作用,即参与吸附锑反应的基团有—OH,—COOH,—Fe—OH,—Fe—O—Fe,进一步说明吸附反应中活性炭及铁氧化物表面官能团与锑之间发生了化学吸附反应。由此推断Fe2O3@AC吸附锑为单层吸附+化学吸附。
共沉淀的定义为可溶形态的杂质被固相物质封闭或吸附后共同沉淀。本研究中Fe2O3@AC吸附锑的过程为共沉淀,即溶解态锑与固相铁氧化物形成络合物。Sb(Ⅴ)在水中存在形态通常为H3SbO4和Sb(OH)6−:—Fe(OH)可与H3SbO4反应生成Fe3(SbO4)2(式(1))[42];—Fe—O—Fe可与Sb(OH)6−反应生成FeOSb(OH)5−(式(2))[43]。
-
1) Fe2O3@AC的最佳制备条件为:溶剂采用纯水:乙醇体积比4:1,铁盐采用Fe2+:Fe3+摩尔比1:1,总铁质量浓度为0.594 mol·L−1,制备液pH为1.88,在此条件下获得的铁氧化物/活性炭除锑效果最佳。
2) Fe2O3@AC除锑最佳条件为:原水锑质量浓度38 μg·L−1,吸附剂投加量为0.08 g·L−1,pH为5~7时,铁氧化物/活性炭吸附除锑效率可达97%,处理出水锑含量低于5 μg·L−1,满足国家饮用水水质标准的要求。且Fe2O3@AC性能优于其他活性炭、金属基吸附剂。
3)铁氧化物以颗粒形式成功负载于活性炭上且晶体结构完好,且吸附前后晶体结构不变。参与吸附除锑反应的基团包括—OH、—COOH、—Fe—OH、—Fe—O—Fe。
4) Fe2O3@AC对锑的吸附符合Langmuir模型,吸附动力学符合准二级动力学,表明其对锑的吸附以单层化学吸附为主,锑在吸附剂表面的扩散方式是多种方式的叠加,吸附前期以液膜扩散为主,中期以颗粒内扩散为主,后期以质量扩散为主,吸附剂中的基团与锑之间存在螯合反应,且最终通过共沉淀去除锑。
铁氧化物/活性炭对饮用水中Sb(V)的吸附性能及机理
Adsorption performance and mechanism of iron oxide/activated carbon on Sb(V) in drinking water
-
摘要: 采用液相沉积法制备了铁氧化物/活性炭复合材料(Fe2O3@AC),通过单因素实验和正交实验优化了材料的制备条件,使用SEM、FTIR、XRD、XPS等分析方法对材料的形貌和性质进行了表征分析,通过吸附实验探究了Fe2O3@AC吸附除锑的效果及影响因素,并进一步对吸附除锑的机理进行了深入探讨。结果表明:最佳制备条件为纯水:乙醇:=4:1,Fe2+:Fe3+=1:1,总铁浓度为0.594 mol·L−1,制备液pH=1.88。Fe2O3@AC吸附除锑的能力较其他金属基材料和活性炭有明显提高,锑原水质量浓度为38 μg·L−1,Fe2O3@AC投加量为0.08 g·L−1,吸附平衡后水中锑的去除率达97%,剩余锑质量浓度为1.06 μg·L−1,满足国家饮用水卫生标准要求。微观表征显示铁氧化物颗粒成功负载于活性炭上,且铁氧化物晶体的结构完好。吸附反应符合准二级动力学和Langmuir等温模型,吸附反应以单层化学吸附为主,吸附类型为液膜扩散,颗粒内扩散,质量扩散的叠加形式。吸附方式为共沉淀,及溶解态锑与固相铁氧化物形成络合物,参与反应的官能团为—OH、—COOH、—Fe—OH、—Fe—O—Fe。Abstract: The iron oxide/activated carbon composites(Fe2O3@AC) were prepared by liquid-phase deposition, and their preparation conditions were optimized by the single factor and orthogonal experiments. SEM, FTIR, XRD, XPS were used to characterize the morphology and properties. The adsorption experiments were conducted to investigate antimony removal effect by Fe2O3@AC adsorption and the corresponding influencing factors, and antimony removal mechanism was discussed in depth. The results showed that the optimal preparation conditions were following: pure water:ethanol of 4:1, Fe2+:Fe3+ of 1:1, total iron concentration of 0.594 mol·L−1 and the preparation solution pH of 1.88. Compared with other metal-based materials and activated carbon, The ability of antimony removal by Fe2O3@AC adsorption increased significantly. When the concentration of antimony in the raw water was 38 μg·L−1 and Fe2O3@AC dosage was 0.08 g·L−1, the removal rate of antimony could reach 97% after adsorption equilibrium, and the remaining antimony concentration was 1.06 μg·L−1, which met the requirements of National Drinking Water Sanitation Standards. Microscopic characterization showed that the iron oxide particles were successfully loaded on the activated carbon and the iron oxide crystals were structurally intact. The adsorption reaction conformed to the quasi-secondary kinetic and Langmuir isotherm models, and the adsorption reaction was dominated by monolayer chemisorption. The adsorption types were superposition of liquid film diffusion, intraparticle diffusion and mass diffusion forms. The adsorption was co-precipitated and complexes were formed between dissolved antimony and solid phase iron oxides, the functional groups involved in the reaction were —OH,—COOH,—Fe—OH,—Fe—O—Fe.
-
Key words:
- iron oxides /
- activated carbon /
- adsorption /
- antimony removal
-
制定水质标准的科学依据是水质基准[1],但目前的研究中,很少有针对我国水域的水质基准研究报道[2],我国现行水质标准的确定主要是依据国外的水质基准数值[3-4],但不同的生物区系与水质状况各不相同,这会导致水质基准产生明显差异[5-6]. 因此,基于我国水环境管理的迫切需求,应依据我国国情开展水质基准研究[7].
荧蒽(fluoranthene,FLU),是多环芳烃中4环芳烃的代表性化合物[8],也是水体中多环芳烃污染物的一个主要成分[9]. 然而,目前我国尚缺乏有关本土水生生物荧蒽的生态毒性数据,因此通过实验开展荧蒽的水生生物基准阈值研究工作,获得有关数据是十分必要的,同时对荧蒽在我国的水生生物基准的建立以及水环境相关工作等具有一定的现实意义与科学意义. 本研究进行了急性与慢性毒理学试验,参考美国的水质基准“指南”要求(“三门八科”最低毒性数据),以及对我国淡水生物区系特征和影响因素等具体分析,最终选取了符合物种筛选规定的9种具有典型代表性的中国本土水生生物进行试验. 根据实验结果得出荧蒽本土水生生物基准阈值,同时,本文分析比较了本土物种与美国物种毒性敏感度是否一致,其中美国物种毒性敏感度基于美国水生生物毒性数据,分析结果可表明在进行我国本土水生生物基准研究时,是否可以直接采用非本土水生生物毒性数据.
1. 材料与方法(Materials and methods)
1.1 实验材料
一般来说,复杂水生态系统的功能、基本结构特征可通过鱼类、底栖动物、浮游生物和植物所表征[10-11]. 在推导我国水质基准时,试验生物的确定可依据美国水质基准“指南”中的规定,即“三门八科”最低毒性数据要求,同时参考《中国脊椎动物大全》[12]和《中国生物多样性国情研究报告》[13]等文献资料,试验生物必须包括鲤科鱼类;另外鉴于我国鲤科种类丰富、数量庞大[14],因此本文选用两种鲤科鱼. 如上所述,物种选择应具体分析中国淡水生物群的特点,同时根据源自欧盟和美国水质基准的物种选择原则,本次荧蒽基准研究初步选择了9个本地水生生物物种作为受试物种,它们分别是锦鲤、麦穗鱼、泥鳅、泽蛙蝌蚪,大型溞、青虾、摇蚊幼虫,霍甫水丝蚓,水螅属. 其中锦鲤和麦穗鱼为脊索动物门鲤科,其余依次为鳅科、蛙科、节肢动物门溞科、虾科、摇蚊科、环节动物门颤蚓科和腔肠动物门水螅虫科,共“四门八科”. 另外对照毒性数据筛选原则[15],本研究在搜集荧蒽的水生植物毒性数据数据的过程中,发现所得结果很少能够满足上述筛选原则.
在“朝来春及大森林水产市场”购入试验所需的9种本土水生动物(既包括淡水,也包括海水),正式试验前均在实验室驯养至少7 d;在中国环境科学研究院环境基准与风险评估国家重点实验室可获得试验所需的大型溞(Daphnia magna),其龄期在24 h之内[15]. 水生生物培养条件为:pH 8.0±0.2,DO (8.3±0.3) mg·L−1,温度(22±2)℃,试验类型采用换水式,每24 h进行一次换水,急性试验不喂食,慢性试验按照0.1%生物重量一天喂食2次;溞类光照周期为12 h:12 h.
在筛选受试物种时,不同种类的受试生物对其龄期有着不同的要求,其中对于水溞或其他水溞类动物要求为24 h内,蚊类,要求为第二代、三代幼虫,对于鱼类或其他物种,要求为至少先于性腺发育前60 d的幼龄阶段生物[15].
在“美国Sigma Aldrich化学品公司”购入实验所用荧蒽,其化学式为C16H10,纯度≥98%(HPLC)为色谱纯;其他试剂均为分析纯. 为了确保浓度配置的准确性,在第一次实验之前,通过中国计量学院校准了用于制备蒽溶液的移液枪,对每个浓度组进行了气相色谱测试. 为了避免试验过程中的任何干扰,实验者严格遵循操作规范,在气相色谱分析之前,所有未使用的玻璃器皿都用高价的酸溶液进行冲洗. 在3组空白实验中加入内标荧蒽-d10,96 h后测定其回收率,结果显示,荧蒽-d10的回收率为74.3%—89.6%. US EPA[15]公布的回收率为70%—130%,试验结果满足其要求,证明了本实验结果的可靠性和准确性.
1.2 实验方法
根据美国材料与试验协会[16]标准方法[17-18]进行急性实验,设空白对照组、助溶剂(二甲基亚砜, DMSO)对照组、浓度组,每组3个重复. 急性实验的时间设置为:溞类48 h,其余水生生物96 h. 具体实验信息如表1所示.
表 1 FLU 9种本土水生生物急性毒性实验信息Table 1. Acute toxicity tests information of FLU to nine resident aquatic organisms分类Classification 生物科Families 物种Species 生长阶段Growth phase 体重/gWeight 体长/cmLength 实验时间/hTime 浓度设置/ (mg·L−1)Concentration 脊索动物门 鲤科 锦鲤(Rhodens sinensis) 幼龄期(<30 d) 0.25 ± 0.05 3.0 ± 0.5 96 0.00, 2.40, 3.10, 4.00, 5.20, 6.80, 8.80, 11.40 麦穗(Pseudorasbora parva) 幼龄期(<30 d) 0.25 ± 0.02 2.5 ± 0.2 96 0.00, 2.40, 3.10, 4.00, 5.20, 6.80, 8.80, 11.40 鳅科 泥鳅(Misgurnus anguillicaudatus) 幼龄期(<30 d) 0.70 ± 0.05 6.0 ± 0.5 96 0.00, 0.96, 1.16, 1.39, 1.67, 2.00, 2.40, 2.88 蛙科 泽蛙蝌蚪(Rana limnocharis) 幼龄期(<10 d) 0.20 ± 0.02 1.6 ± 0.2 96 0.00, 3.50, 4.60, 5.50, 7.80, 10.10, 13.20, 17.10 节肢动物门 溞科 大型溞(Daphnia magna) <24 h — — 48 0.00, 2.70, 3.60, 4.70, 6.20, 8.00, 10.40, 13.50 虾科 青虾(Macrobrachium nipponense) 幼龄期(<10 d) 0.25 ± 0.05 3.0 ± 0.2 96 0.00, 2.40, 3.10, 4.00, 5.20, 6.80, 8.80, 11.40 摇蚊科 摇蚊幼虫(Chironomus plumosus) 第3代(幼虫) 0.03 ± 0.01 1.0 ± 0.2 96 0.00, 2.70, 3.60, 4.70, 6.20, 8.00, 10.40, 13.50 环节动物门 颤蚓科 霍甫水丝蚓(Limnodrilus hoffmeisteri) 幼龄期(<10 d) 0.05 ± 0.01 1.5 ± 0.2 96 0.00, 3.40, 4.40, 5.70, 7.40, 9.60, 12.50, 16.20 腔肠动物门 水螅虫科 水螅属(Hydra sp.) 幼龄期(<10 d) 0.03 ± 0.01 1.0 ± 0.2 96 0.00, 1.39, 1.67, 2.00, 2.40, 2.88, 3.74, 4.87 | Show Table DownLoad:
CSV
DownLoad:
CSV
在急性毒性实验结束后,参考荧蒽对麦穗、泥鳅和大型溞的急性实验结果开展慢性实验,设空白对照组、助溶剂(二甲基亚砜, DMSO)对照组、浓度组,每组3个重复. 慢性实验的组次中,其最高浓度不得高于急性毒性LC50值. 受试液和喂食的频率为1 d,采用静态更新试液法. 慢性毒理学实验时间设置为:溞类不得少于21 d,麦穗、泥鳅不得少于8 d. 具体实验浓度设置如下:麦穗为0.00、0.42、0.55、0.72、0.94、1.22、1.59 mg·L−1 FLU;泥鳅为0.00、0.35、0.46、0.60、0.78、1.01、1.32 mg·L−1 FLU;大型溞为0.00、0.42、0.55、0.72、0.94、1.22、1.59 mg·L−1 FLU. 试验中需要记录的内容如下:大型溞的产卵数量、时间(第一窝及总数)、麦穗和泥鳅的生长指标(体重、体长等),以上数据每天记录一次.
1.3 数据搜集与分析
数据选自ELSEVIER数据库(http://www.sciencedirect.com)、US EPA ECOTOX 毒性数据库(http://cfpub.epa.gov/ecotox/)、和CNKI数据库(http://www.cnki.net)等. 在进行筛选数据时应满足以下要求:①数据无法使用的情况有:使用受权限、信息保密、测试信息不完全、由于其他因素而无法传播、试验缺少对照组、试验生物受污染、试验设计不科学等;②可以使用具有不稳定性质(易挥发、易水解、易降解)的物质进行试验,但一般仅可采用流水式试验的结果;③若慢性NOEC和LOEC值均出现在测试终点中,NOEC值的选取具有优先权[19]. 另外,对于水生植物,前人研究发现FLU对淡水藻[20](Scenedesmus subspicatus)的96-h EC50为7.447 mg·L−1,本文选用此数据.
另外,实验数据的正态分布检验方法有如下两种:K-S检验、T检验. 本文选用前者检验表2中数据是否符合正态分布,检验结果表明美国水生生物毒性数据对FLU毒性敏感的总体分布情况是大致吻合的,符合正态分布(P =0.08>0.05).
表 2 FLU美国水生生物急性毒性数据Table 2. Acute toxicity data of FLU to American aquatic organisms排序 Rank 物种 Species (LC50/EC50)/(mg·L−1) t/h 1 美洲海螯虾 0.6 96 2 浅绛单孔蚓 0.7 96 3 俄勒冈虾 0.8 96 4 蓝鳃太阳鱼 20.9 96 5 斑点叉尾鮰 36.0 96 6 端足虫 44.0 96 7 非洲爪蟾 52.0 96 8 隐居蜾嬴蜚 54.0 96 9 沙蟹 74.0 96 10 虹鳟 91.0 96 11 呆头鲦鱼 118.3 96 12 宽纹北箭蜓 139.9 96 13 伸展摇蚊 250.0 96 | Show TableDownLoad:
CSV
EC/LCx可通过概率单位直线回归法和95%置信区间来计算,按照测试标准所规定的浓度规定,若实测浓度与名义浓度的差值在20%以内,即可采用名义浓度,x值分别取50(急性毒性实验)和10(慢性毒性实验),通过在试验开始的前后,对溶液中的FLU浓度进行检测,对比检测结果后计算实测和名义浓度的比率,结果为90.03%—99.15%,满足要求[21],因此本文在计算EC/LCx时,浓度采用名义浓度.
2. 结果与讨论(Results and discussion)
2.1 急性毒性实验
表3显示了9种本土水生生物的急性毒性试验结果,其中空白对照组和助溶剂对照组在试验期间中没有出现死亡现象. 窗体顶端结果表明泥鳅对FLU暴露最为敏感,其96-h LC50为1.887 mg·L−1, 其次是水螅属、青虾、麦穗鱼、大型溞、锦鲤、霍甫水丝蚓和摇蚊幼虫,对FLU最不敏感的是泽蛙蝌蚪,其96-h LC50为8.695 mg·L−1.
表 3 FLU本土水生生物急性试验结果Table 3. Results of acute toxicity tests of FLU to resident aquatic organisms物种 Species 暴露时间/h Exposure time 公式 Formula R2 P LC50/(mg·L−1) 锦鲤 96 y = 3.4458x + 2.2589 0.9268 <0.01 6.251 (5.748—6.443) 泥鳅 96 y = 1.4976x + 4.5875 0.9436 <0.01 1.887 (1.594—2.258) 大型溞 48 y = 3.8675x –2.0147 0.9850 <0.01 5.487 (5.012—5.897) 青虾 96 y = 2.5612x + 3.7744 0.9089 <0.01 3.011 (2.412—3.647) 霍甫水丝蚓 96 y = 1.5825x + 3.7347 0.9065 <0.01 6.313 (5.688—6.578) 麦穗 96 y = 1.6443x + 3.8262 0.854 <0.01 5.177 (4.231—5.593) 摇蚊幼虫 96 y = 2.3053x + 2.9664 0.9243 <0.01 7.628 (7.390—8.159) 水螅属 96 y = 1.3879x + 1.9976 0.9017 <0.01 2.032 (1.785—3.074) 泽蛙蝌蚪 96 y = 1.9267x + 3.1911 0.9044 <0.01 8.695(8.393—90.145) | Show TableDownLoad:
CSV
2.2 慢性毒性数据
在慢性毒性实验中各试验浓度组均未出现死亡现象. 对于慢性毒性数据(表4),Barata等[22]研究表明大型溞的存活率21-d NOEC为0.937 mg·L−1,这一结果与本试验中大型溞总繁殖数量的21-d EC10为0.881 mg·L−1十分接近. 根据表4,对于大型溞,对FLU暴露的产卵总数量试验终点相比于其存活率,表现出更为敏感的趋势. 在慢性毒性试验中,3种水生生物中对FLU暴露的敏感性最低的为大型溞,最高的为泥鳅. 结果表明泥鳅在急性与慢性实验中,对FLU暴露都表现出最高的敏感性.
表 4 FLU本土水生生物慢性试验结果Table 4. Results of chronic toxicity tests of FLU to resident aquatic organisms.物种 Species 暴露时间/d Exposure time 终点 Endpoint 公式 Formula R2 P EC10/( mg·L−1) 锦鲤 28 生长(t/d) y = 4.5521x +2.8004 0.9930 <0.01 0.798 泥鳅 28 生长(t/d) y = 5.3131x –1.989 0.9743 <0.01 0.269 大型溞 28 生长(t/d) y = 4.7745x+ 1.667 0.8971 <0.05 1.187 第一窝时间 (t/d) y = 2.8499x+ 1.525 0.9572 <0.01 1.243 第一窝数量 (n) y = 3.8261x+ 0.657 0.9315 <0.01 0.987 总数量 (n) y= 3.7966x+ 1.0801 0.9203 <0.01 0.881 总窝数 (n) y= 3.7450x+ 1.4623 0.9364 <0.01 1.471 | Show TableDownLoad:
CSV
2.3 基于本土与美国生物毒性数据拟合SSD曲线比较
Davies等提出“灵活使用毒性数据”的设想[23],即在某一地区进行生态风险评估时,可使用其他地区的生物毒性数据. 但由于不同地区的水中溶解氧浓度、水温差别较大,且不同种类生物对于相同有害物质的敏感程度也各不相同[24],因此这一设想在提出后受到多方面质疑. 本研究将本地(表2)和美国(表3)的生物毒性数据进行SSD曲线拟合,并进行比较讨论,探讨 "灵活使用毒性数据 "这一概念的可行性. 受限于相对缺少慢性数据,本研究仅采用急性毒性数据. 本研究以本土、美国、本土+美国的FLU毒性数据为基础,共得出3条SSD拟合曲线(图1). 在对FLU暴露的敏感性方面,图中本土数据(黑)、总体数据(蓝)与美国数据(红)从左到右存在显著的位移分布,意味着美国生物比本土生物表现出更加敏感的趋势. 经过计算,总体数据、本土数据及非本土数据三条曲线的HC5值分别是1.527、1.869、2.142 mg·L−1.
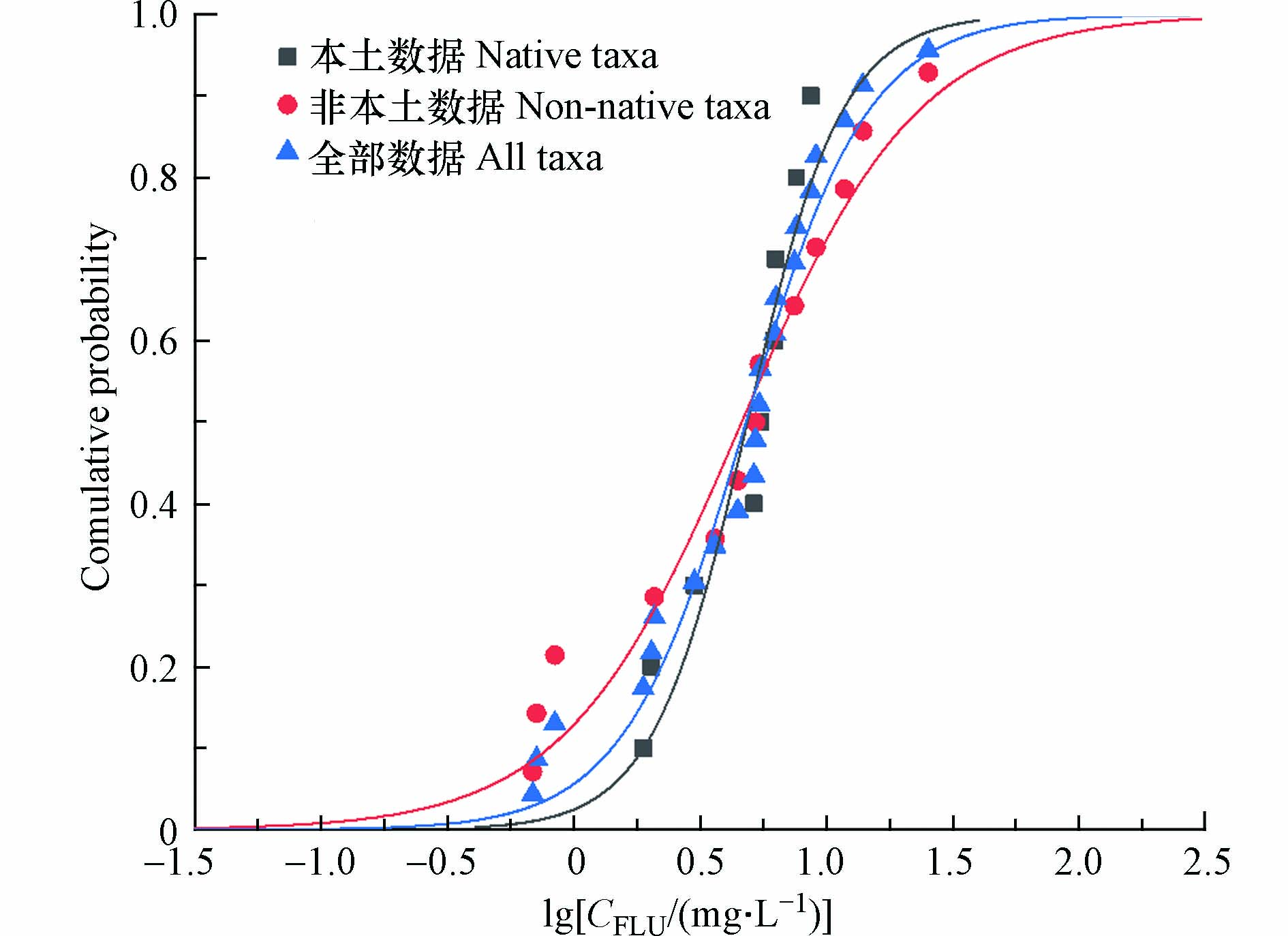 图 1 FLU本土、美国及全部生物毒性数据SSD曲线拟合比较Figure 1. Species sensitivity distribution of native, American and all species toxicity data for FLU
图 1 FLU本土、美国及全部生物毒性数据SSD曲线拟合比较Figure 1. Species sensitivity distribution of native, American and all species toxicity data for FLUTwo-sample Kolmogorov-Smirnov(K-S test)检验常用于数学统计中,可用于对两样本总体分布差异性的检验[24]. 利用此方法,研究美国毒性数据和本土毒性数据是否存在较为明显的不同具有十分重要的学术和科学意义:若二者差异明显,则在本土进行相关的基准阈值研究时,应选取本土水生生物;若二者较为一致,那么证明美国水生生物毒性数据可用于本土基准阈值的推导过程,从而降低受试生物的消耗以及节省资源的投入. K-S结果如下:(ks = 1.342, n1 = 9, n2 = 13, P = 0.01). P= 0.0114<0.05,按a=0.05水准,结果表明两组数据在FLU毒性敏感的总体分布上并为呈现较高的一致性,两组数据之间存在的差异十分显著(图1). 另外,由于浮游类不被包含在非本土生物毒性数据中,因此在本土生物数据中浮游类大型溞的毒性数据暂时去除之后,对数据再次进行K-S检验,两者之间仍然存在较为显著的差异(ks= 0.830, n1 = 8, n2= 13, P = 0.0183). 因此我们可以初步判断本土对FLU的敏感性与非本土生物有着较大差别,需要进一步开展针对本土化的相关研究工作(图1).
2.4 FLU水生生物水质基准阈值推导
以实测数据为基础,在计算FLU水生生物基准阈值时,本文采用了USEPA“指南”推荐的SSR方法[15],9种本土物种的SMAV值和GMAV值及其排序如表5所示,CMC(FLU的急性基准阈值)=FAV/评价因子,其中FAV取1.141 mg·L−1,评价因子通常取值为2,最后计算出我国FLU的急性基准阈值结果为0.570 mg·L-1[25-26]. 鉴于我国FLU慢性毒性数据较少,FCV值可通过“指南”所推荐的方法计算,即FCV=FAV/FACR,FACR的值是3种水生生物SACR的几何平均值(如表所示,3种水生生物的SAVR值分别为:大型溞6.23、麦穗6.49、泥鳅7.01),最终计算得出FACR值为6.57,FCV值为0.174 mg·L−1,已知淡水藻的FLU植物毒性值为7.447 mg·L−1,远远大于计算所得的FCV值,因此对植物的保护作用得到验证[27]. CCC的确定按“指南”所指出的方法,即由FPV、FCV和FRV三者中最小的值所确定[28]. 相比于动物,植物的敏感性要低得多,且我国缺少相应的BCF和MPC值,因而CCC可直接通过FCV值计算,在很多情况下FRV则不被考虑,最后计算出我国FLU的慢性基准阈值CCC为0.174 mg·L−1.
表 5 FLU水生生物种平均值及急慢性比Table 5. Ranked GMAVs with SACRs.排序 Rank 物种 Species SMAVs/(mg·L−1) GMAVs/(mg·L−1) SACRs 来源 Source 1 泥鳅 1.887 1.887 7.01 本研究 2 水螅属 2.032 2.032 本研究 3 青虾 3.011 3.011 本研究 4 麦穗 5.177 5.177 6.49 本研究 5 大型溞 5.487 5.487 6.23 本研究 6 锦鲤 6.251 6.251 本研究 7 霍甫水丝蚓 6.313 6.313 本研究 8 摇蚊幼虫 7.628 7.628 本研究 9 泽蛙蝌蚪 8.695 8.695 本研究 植物 淡水藻 7.447 7.447 [20] | Show TableDownLoad:
CSV
另外,本文也对比了FLU对美国水生生物的毒性. 例如,本研究中本土代表性鱼类敏感性相对较高,泥鳅、麦穗鱼和锦鲤的96-h LC50分别为1.887、5.177、6.251 mg·L−1 FLU,而作为非本土鱼类的蓝鳃太阳鱼[29]、斑点叉尾鮰[30]、虹鳟[31]和呆头鲦鱼[32]的96-h LC50分别为20.90、36.00、91.00、118.3 mg·L−1 FLU,数值不在一个数量级,本土鱼类更加敏感;已经报道的非本土两栖类动物非洲爪蟾[33]的96-h LC50为52.00 mg·L−1FLU,远远高于本土两栖类泽蛙蝌蚪的实测值(8.695 mg·L−1 FLU);本土底栖动物节肢动物门的摇蚊幼虫96-h LC50为7.628 mg·L−1 FLU,而已报道的非本土昆虫类生物是端足虫[30]和伸展摇蚊[34],96-h LC50分别为44.00 mg·L−1和250.00 mg·L−1 FLU,差异较为明显,本土节肢动物门相对敏感. 综上所述,可以确定本土水生生物中鱼类、两栖类对FLU和底栖节肢动物门比非本土同类生物更加敏感.
本土底栖动物中环节动物门受试生物为霍甫水丝蚓,96-h LC50为6.313 mg·L−1 FLU,非本土浅绛单孔蚓[30]96-h LC50为0.700 mg·L−1 FLU,此外还有底栖甲壳类生物差异较大,本土青虾96-h LC50为3.011 mg·L−1FLU,而非本土底栖甲壳类动物美洲海螯虾[35]和俄勒冈虾[35]96-h LC50相对较低,为0.600 mg·L−1及0.800 mg·L−1 FLU. 因此可以确定非本土水生生物中底栖环节动物及甲壳类动物对FLU比本土生物更加敏感.
另外,借助荷兰RIVM公布的ETX2.0软件及逻辑斯蒂SSD曲线求出HC5值,分别是1.657 mg·L−1和1.869 mg·L−1. 因此当评价因子取值为2时,CMC在这两种SSD曲线下,计算的结果分别为0.829 mg·L−1和0.935 mg·L−1,上文采用US EPA“指南”中推荐的SSR法计算的CMC值为0.570 mg·L−1,由此可见,计算结果都同在一个数量级.
3. 结论(Conclusion)
(1) 基于US EPA“指南”推荐的方法,荧蒽本土水生生物急性基准阈值(CMC)为0.570 mg·L−1,慢性基准阈值(CCC)为0.174 mg·L−1;另外,通过荷兰RIVM公布的ETX2.0软件及欧盟推荐的SSD方法所得的基准阈值与本文所得结果(SSR方法得出)在数量级上一致,本文结果得到验证.
(2) 在敏感性方面,通过比较分析本土与美国物种的一致性较低,说明在进行我国荧蒽水生生物基准阈值的推导时,可利用美国水生生物毒性数据的概率很小.
致谢:感谢中国环境科学研究院刘征涛研究员在文章修改中给予的帮助.
-
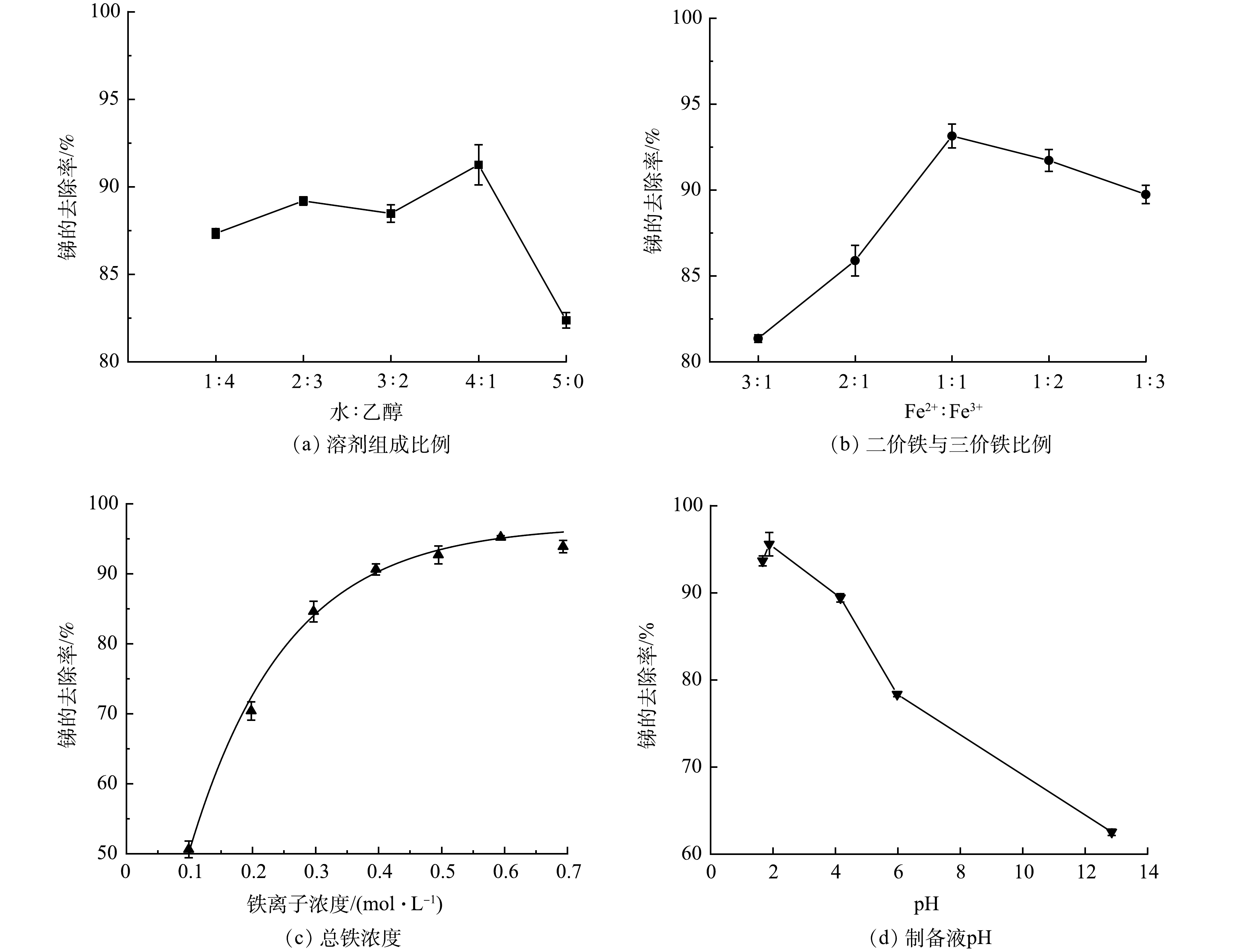
图 1 不同制备条件下的Fe2O3@AC对锑的去除率
Figure 1. Antimony removal rate by Fe2O3@AC under different preparation conditions
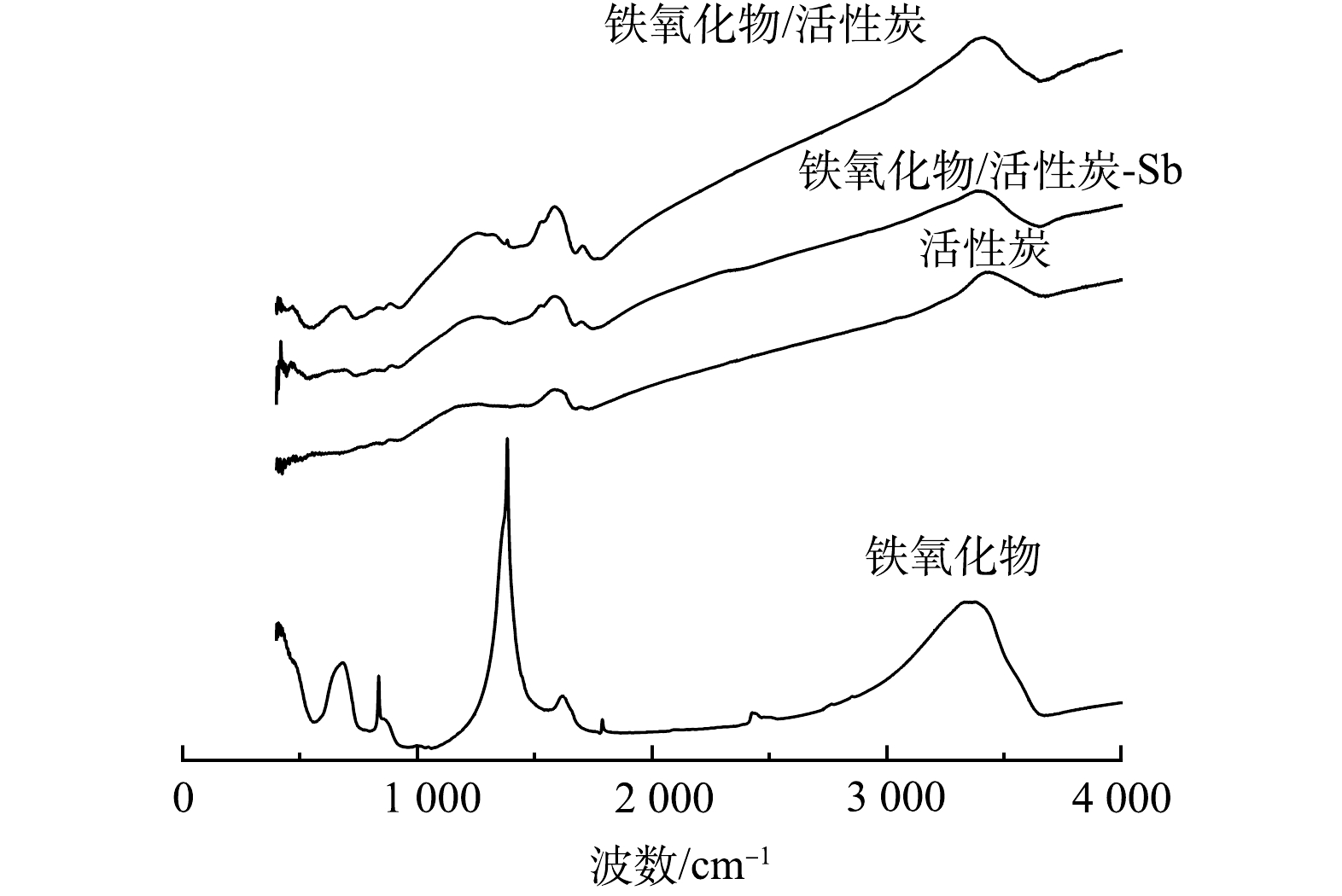
图 3 铁氧化物、活性炭、Fe2O3@AC吸附锑前后的FTIR图
Figure 3. FTIR spectra of iron oxide, activated carbon, Fe2O3@AC and Fe2O3@AC after antimony adsorption
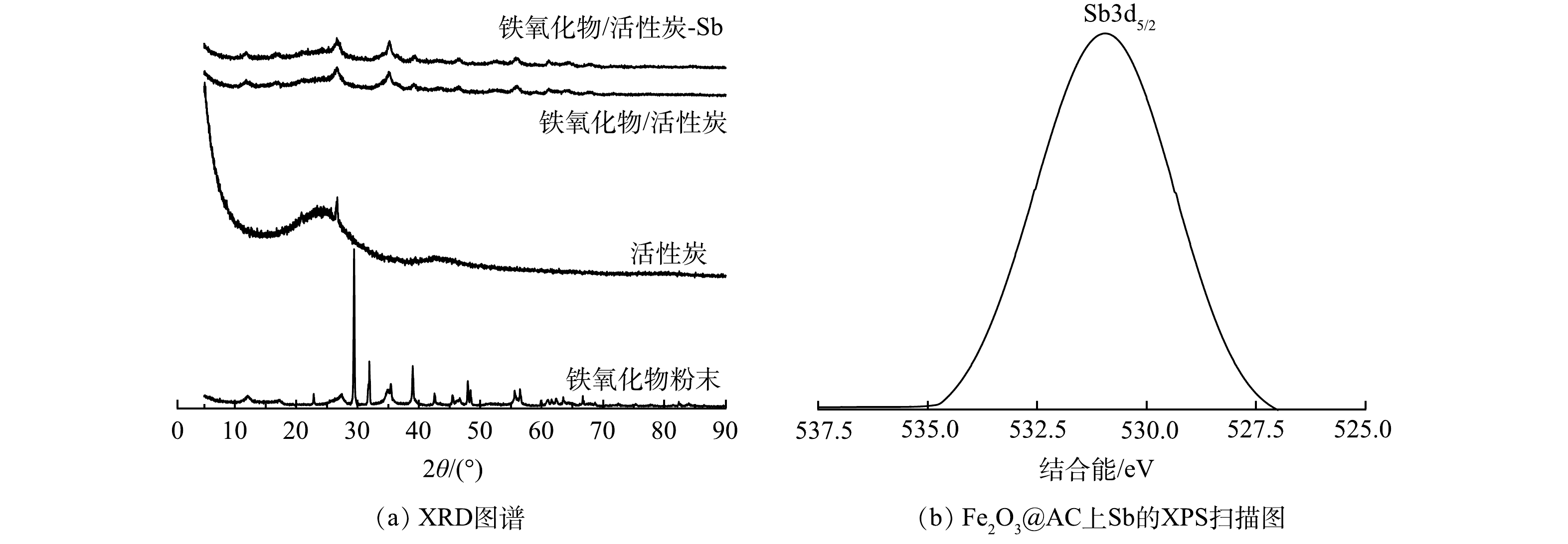
图 4 Fe2O3@AC、活性炭、铁氧化物的XRD、XPS图
Figure 4. XRD patterns and XPS spectra of Fe2O3@AC, activated carbon, and iron oxide

图 6 Fe2O3@AC对锑的吸附动力学拟合曲线
Figure 6. Fitting curve of adsorption kinetics of antimony by Fe2O3@AC
表 1 Fe2O3@AC优化制备条件的正交表
Table 1. Orthogonal table of optimized Fe2O3@AC preparation conditions
编号 水:乙醇 Fe2+:Fe3+ 铁离子浓度/(mol·L−1) pH 锑去除率/% 1 2:3(1) 2:1(1) 0.2(1) 1.7(1) 62 2 2:3(1) 1:1(2) 0.6(3) 1.9(2) 91 3 2:3(1) 1:2(3) 0.4(2) 4.1(3) 73 4 3:2(2) 2:1(1) 0.6(3) 4.1(3) 79 5 3:2(2) 1:1(2) 0.4(2) 1.7(1) 82 6 3:2(2) 1:2(3) 0.2(1) 1.9(2) 77 7 4:1(3) 2:1(1) 0.4(2) 1.9(2) 85 8 4:1(3) 1:1(2) 0.2(1) 4.1(3) 83 9 4:1(3) 1:2(3) 0.6(3) 1.7(1) 88 k1 75.3 75.3 74 77.3 k2 79.3 85.3 80 84.3 k3 85.3 79.3 86 78.3 极差 10 10 12 7 注:表中编号(1)、(2)、(3)为正交实验中各因素选取的水平数。
下载: 导出CSV
表 2 各材料的FTIR特征峰对比
Table 2. Comparison of FTIR characteristic peaks of each material
材料 —OH/cm−1 —OOH/—COOH/cm−1 —Fe—OH/cm−1 —Fe—O—Fe/cm−1 铁氧化物粉末 1 383 3 333, 3 373 412,682 836 活性炭 1 586 3 438 —— —— Fe2O3@AC 1 257, 1 319, 1 383, 1 586 3 417 411, 679 879 Fe2O3@AC-Sb 1 269, 1 315, 1 383, 1 587 3 382 420, 695 891
下载: 导出CSV
表 3 XRD特征峰对比
Table 3. Comparison of XRD characteristic peaks
材料 磁铁矿/(°) 铁氧化物/活性炭 26.66、35.06、39.12、46.22、56.06、61.2 铁氧化物 27.5、35.38、39、47.93、56.46、61.64
下载: 导出CSV
表 4 吸附动力学模型参数拟合结果
Table 4. Results of parameter fitting of adsorption kinetic model
吸附剂 吸附量 准一级动力学 准二级动力学 q/(μg·g−1) qe/(μg·g−1) K1 R2 qe/(μg·g−1) K2 R2 Fe2O3@AC 401.775 296.19 0.003 06 0.953 1 434.783 1.73×10−5 0.997 4
下载: 导出CSV
表 5 吸附等温线模型参数拟合结果
Table 5. Results of parameter fitting of adsorption isotherm model
吸附剂 Langmuir等温线 Freundlich等温线 b qmax/(μg·g−1) R2 Kf n R2 Fe2O3@AC 107.9 1.48 0.99 9.95 1.62 0.92
下载: 导出CSV
-
[1] SHANAWANY S E, FODA N, HASHAD D I, et al. The potential DNA toxic changes among workers exposed to antimony trioxide[J]. Environmental Science and Pollution Research, 2017, 24(13): 12455-12461. doi: 10.1007/s11356-017-8805-z [2] 尤翔宇, 谭爱华, 苏艳蓉, 等. 锑冶炼行业污染防治现状及对策[J]. 湖南有色金属, 2015, 31(6): 5. doi: 10.3969/j.issn.1003-5540.2015.06.019 [3] WU F, FU Z, LIU B, et al. Health risk associated with dietary co-exposure to high levels of antimony and arsenic in the world's largest antimony mine area[J]. Science of the Total Environment, 2011, 409(18): 3344-3351. doi: 10.1016/j.scitotenv.2011.05.033 [4] XI J, HE M, WANG K, et al. Adsorption of antimony(III) on goethite in the presence of competitive anions[J]. Journal of Geochemical Exploration, 2013, 132: 201-208. doi: 10.1016/j.gexplo.2013.07.004 [5] WU Z, HE M, GUO X, et al. Removal of antimony (III) and antimony (V) from drinking water by ferric chloride coagulation: Competing ion effect and the mechanism analysis[J]. Separation and Purification Technology, 2010, 76(2): 184-190. doi: 10.1016/j.seppur.2010.10.006 [6] XU W, WANG H, LIU R, et al. The mechanism of antimony (III) removal and its reactions on the surfaces of Fe–Mn binary oxide[J]. Journal of Colloid and Interface Science, 2011, 363(1): 320-326. doi: 10.1016/j.jcis.2011.07.026 [7] LEUZ A K, MOENCH H, JOHNSON C A. Sorption of Sb(Ⅲ) and Sb(Ⅴ) to Goethite: Influence on Sb(Ⅲ) Oxidation and Mobilization[J]. Environmental Science & Technology, 2006, 40(23): 7277-7282. [8] GUO X, WU Z, HE M. Removal of antimony(V) and antimony(III) from drinking water by coagulation-flocculation-sedimentation (CFS)[J]. Water Research, 2009, 43(17): 4327-4335. doi: 10.1016/j.watres.2009.06.033 [9] KANG M, KAMEI T, MAGARA Y. Comparing polyaluminum chloride and ferric chloride for antimony removal[J]. Water Research, 2003, 37(17): 4171-4179. doi: 10.1016/S0043-1354(03)00351-8 [10] SONG P, YANG Z, ZENG G, et al. Optimization, kinetics, isotherms, and thermodynamics studies of antimony removal in electrocoagulation process[J]. Water, Air, & Soil Pollution, 2015, 226(11): 1-12. [11] ZHU J, WU F, PAN X, et al. Removal of antimony from antimony mine flotation wastewater by electrocoagulation with aluminum electrodes[J]. Journal of Environmental Sciences, 2011, 23(7): 1066-1071. doi: 10.1016/S1001-0742(10)60550-5 [12] DU X, QU F, LIANG H, et al. Removal of antimony (III) from polluted surface water using a hybrid coagulation–flocculation–ultrafiltration (CF–UF) process[J]. Chemical Engineering Journal, 2014, 254: 293-301. doi: 10.1016/j.cej.2014.05.126 [13] SAITO T, KAWAKITA H, UEZU K, et al. Introduction process of n-methylglucamino groups for binding antimony (III) to a polymer brush[J]. Ars Separatoria Acta, 2006, 4: 8-17. [14] GUIN R, DAS S K, SAHA S K. The anion exchange behavior of Te and Sb[J]. 1998, 230(1/2): 269-272. [15] RIVEROS P A. The removal of antimony from copper electrolytes using amino-phosphonic resins: Improving the elution of pentavalent antimony[J]. Hydrometallurgy, 2010, 105(1/2): 110-114. [16] NAVARRO P, ALGUACIL F J. Adsorption of antimony and arsenic from a copper electrorefining solution onto activated carbon[J]. Hydrometallurgy, 2002, 66(1): 101-105. [17] SALAM M A, MOHAMED R M. Removal of antimony (III) by multi-walled carbon nanotubes from model solution and environmental samples[J]. Chemical Engineering Research and Design, 2013, 91(7): 1352-1360. doi: 10.1016/j.cherd.2013.02.007 [18] QI Z, JOSHI T P, LIU R, et al. Synthesis of Ce (III)-doped Fe3O4 magnetic particles for efficient removal of antimony from aqueous solution[J]. Journal of Hazardous Materials, 2017, 329: 193-204. doi: 10.1016/j.jhazmat.2017.01.007 [19] SHAN C, MA Z, TONG M. Efficient removal of trace antimony (III) through adsorption by hematite modified magnetic nanoparticles[J]. Journal of Hazardous Materials, 2014, 268: 229-236. doi: 10.1016/j.jhazmat.2014.01.020 [20] XU Y H, OHKI A, MAEDE S. Adsorption and removal of antimony from aqueous solution by an activated alumina: 1. Adsorption capacity of adsorbent and effect of process variables[J]. Toxicological & Environmental Chemistry, 2001, 80(3-4): 133-144. [21] WANG X, HE M, LIN C, et al. Antimony (III) oxidation and antimony (V) adsorption reactions on synthetic manganite[J]. Geochemistry, 2012, 72: 41-47. doi: 10.1016/j.chemer.2012.02.002 [22] KARAKA Z K, BONCUKCUOLU R, KARAKA B H. Antimony removal from aqueous solutions using magnetic nickel ferrite (NiFe2O4) nanoparticles[J]. Separation Science and Technology, 2018, 54(7): 1-18. [23] HJA B, LEI T, PCA B, et al. Efficient antimony removal by self-assembled core-shell nanocomposite of Co3O4@rGO and the analysis of its adsorption mechanism[J]. Environmental Research, 2020: 187. [24] HEY T, WAN J, TOKUNAGA T. Kinetic stability of hematite nanoparticles: The effect of particle sizes[J]. Journal of Nanoparticle Research, 2008, 10(2): 321-332. doi: 10.1007/s11051-007-9255-1 [25] LUO J, LUO X, CRITTENDEN J, et al. Removal of antimonite (Sb (III)) and antimonate (Sb (V)) from aqueous solution using carbon nanofibers that are decorated with zirconium oxide (ZrO2)[J]. Environmental Science & Technology, 2015, 49(18): 11115-11124. [26] YU T, WANG X, LI C. Removal of antimony by FeCl3-modified granular-activated carbon in aqueous solution[J]. Journal of Environmental Engineering, 2014, 140(9): A4014001. doi: 10.1061/(ASCE)EE.1943-7870.0000736 [27] GERGOVA K, ESER S. Effects of activation method on the pore structure of activated carbons from apricot stones[J]. Carbon, 1996, 34(7): 879-888. doi: 10.1016/0008-6223(96)00028-0 [28] WU W, WU Z, YU T, et al. Recent progress on magnetic iron oxide nanoparticles: synthesis, surface functional strategies and biomedical applications[J]. Science and Technology of Advanced Materials, 2015, 16(2): 023501. doi: 10.1088/1468-6996/16/2/023501 [29] JOLIVET J P, HENRY M. De la solution à l'oxyde-Condensation des cations en solution aqueuse. Chimie de surface des oxyde[M]. EDP Sciences, 2012. [30] LAURENT S, FORGE D, PORT M, et al. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications[J]. Chemical Reviews, 2008, 108(6): 2064-2110. doi: 10.1021/cr068445e [31] TORRES-GOMEZ N, NAVA O, ARGUETA-FIGUEROA L, et al. Shape tuning of magnetite nanoparticles obtained by hydrothermal synthesis: Effect of temperature[J]. Journal of Nanomaterials, 2019: 2019. [32] YADAV N, SINGH A, KAUSHILK M. Hydrothermal synthesis and characterization of magnetic Fe3O4 and APTS coated Fe3O4 nanoparticles: Physicochemical investigations of interaction with DNA[J]. Journal of Materials Science:Materials in Medicine, 2020, 31(8): 1-11. [33] 许光眉, 施周, 邓军. 石英砂负载氧化铁吸附除锑, 磷的 XRD, FTIR 以及 XPS 研究[J]. 环境科学学报, 2007, 27(3): 402-407. doi: 10.3321/j.issn:0253-2468.2007.03.008 [34] FAN H, MA X, ZHOU S, et al. Highly efficient removal of heavy metal ions by carboxymethyl cellulose-immobilized Fe3O4 nanoparticles prepared via high-gravity technology[J]. Carbohydrate Polymers, 2019, 213: 39-49. doi: 10.1016/j.carbpol.2019.02.067 [35] KANG M, KAWASAKI M, TAMADA S, et al. Effect of pH on the removal of arsenic and antimony using reverse osmosis membranes[J]. Desalination, 2000, 131(1/2/3): 293-298. [36] MATHIALAGAN T, VIRARAGHAVAN T. Adsorption of cadmium from aqueous solutions by perlite[J]. Journal of Hazardous Materials, 2002, 94(3): 291-303. doi: 10.1016/S0304-3894(02)00084-5 [37] YUAN P, FAN M, YANG D, et al. Montmorillonite-supported magnetite nanoparticles for the removal of hexavalent chromium [Cr (VI)] from aqueous solutions[J]. Journal of Hazardous Materials, 2009, 166(2/3): 821-829. [38] 陈云, 王营茹, 孙家寿, 等. 改性累托石吸附处理亚甲基蓝机理研究[J]. 武汉工程大学学报, 2011, 33(9): 68-71. doi: 10.3969/j.issn.1674-2869.2011.09.017 [39] MALL I D, SRIVASTAVA V C, AGARWAL N K, et al. Removal of congo red from aqueous solution by bagasse fly ash and activated carbon: kinetic study and equilibrium isotherm analyses[J]. Chemosphere, 2005, 61(4): 492-501. doi: 10.1016/j.chemosphere.2005.03.065 [40] MALL I D, SRIVASTAVA V C, AGARWAL N K. Removal of Orange-G and Methyl Violet dyes by adsorption onto bagasse fly ash: Kinetic study and equilibrium isotherm analyses[J]. Dyes and Pigments, 2006, 69(3): 210-223. doi: 10.1016/j.dyepig.2005.03.013 [41] YILMAZ M, BAYRAMOGLU G, ARICA M Y. Separation and purification of lysozyme by Reactive Green 19 immobilised membrane affinity chromatography[J]. Food Chemistry, 2005, 89(1): 11-18. doi: 10.1016/j.foodchem.2004.01.072 [42] LONG X, WANG X, GUO X, et al. A review of removal technology for antimony in aqueous solution[J]. Journal of Environmental Sciences, 2020, 90: 189-204. doi: 10.1016/j.jes.2019.12.008 [43] UNGUREANU G, SANTOS S, BOAVENTURA R, et al. Arsenic and antimony in water and wastewater: Overview of removal techniques with special reference to latest advances in adsorption[J]. Journal of Environmental Management, 2015, 151: 326-342. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5343
- HTML全文浏览数: 5343
- PDF下载数: 204
- 施引文献: 0
