















-
随着合成化学工业的发展,水体中检出的各类微量有机污染物也日渐增多,包括不同种类的农药、内分泌干扰物、药物和个人护理品等。传统的饮用水和污水处理工艺通常无法有效去除这些微量污染物。基于紫外(UV)的高级氧化技术(UV/H2O2)是去除微量有机污染物的有效方法[1-2],目前在国内饮用水厂和污水处理厂已有一定的应用。然而,常规UV光源(254 nm)下的高级氧化工艺依赖于化学氧化剂的投加来产生HO·等活性物种,这涉及到氧化剂的储存、运输以及残余药剂的去除等问题,增加了操作的复杂性和处理成本。真空紫外线(VUV)技术是近年来的一个研究热点。与常规UV高级氧化工艺相比,VUV/UV高级氧化的优点在于不需要化学氧化剂,水分子在VUV辐照下的光解就可以产生高浓度的HO·,是一种很有前景的绿色高级氧化技术。另一方面,由于VUV能量高,仅5 mm的水层即可使VUV辐射强度衰减约90%,因此产生的HO·也主要集中在灯管附近很薄的水层内。此时,HO·的利用率及相关影响因素决定了VUV/UV高级氧化技术的实际效率。在已有研究中,大量基于VUV/UV工艺的研究均是在序批式反应器内进行的,其中自由基可与目标污染物及背景物质充分接触反应。然而,实际应用中的VUV/UV反应器通常为过流式,这可能导致HO·在整个反应器内分布不均匀,表现出与序批式反应器不同的效果[3]。目前,在过流式条件下水中常见的背景物质对VUV/UV工艺降解微量污染物的影响研究还较少。此外,不同于常规高级氧化工艺,VUV光解水产生HO·的过程受到可吸收185 nm辐射的无机离子的显著影响,但对该蔽光效应的量化分析还比较缺乏。基于此,本研究探究了过流式VUV/UV反应器中氯离子(Cl–)、碳酸氢根离子(HCO3–)、硝酸盐(NO3–)和溶解性有机物(dissolved organic matter,DOM)对典型微量污染物阿特拉津(atrazine)降解的影响,并计算不同水质条件下VUV/UV工艺降解目标污染物所需的单位能耗。本研究可为VUV/UV技术在实际条件下的推广应用提供参考。
-
阿特拉津(ATZ)购于梯希爱(上海)化成工业发展有限公司,纯度大于98%。氯化钠、碳酸氢钠和硝酸钠购于国药集团化学试剂有限公司,均为分析纯。甲酸、乙腈购于西格玛-阿拉丁公司,纯度为色谱纯。用于配制溶解性有机物溶液的腐殖酸(HA)购于北京伊诺凯科技有限公司,纯度大于90%。目标污染物ATZ的初始浓度固定为2.5 μmol·L−1,考察的Cl–和HCO3–浓度设置为1.0、2.5和5.0 mmol·L−1,NO3–浓度设置为10.0和20.0 mg·L−1(以氮计),DOM浓度设置为1.0、5.0和10.0 mg·L−1(以碳计)。反应溶液均用去离子水配制。
-
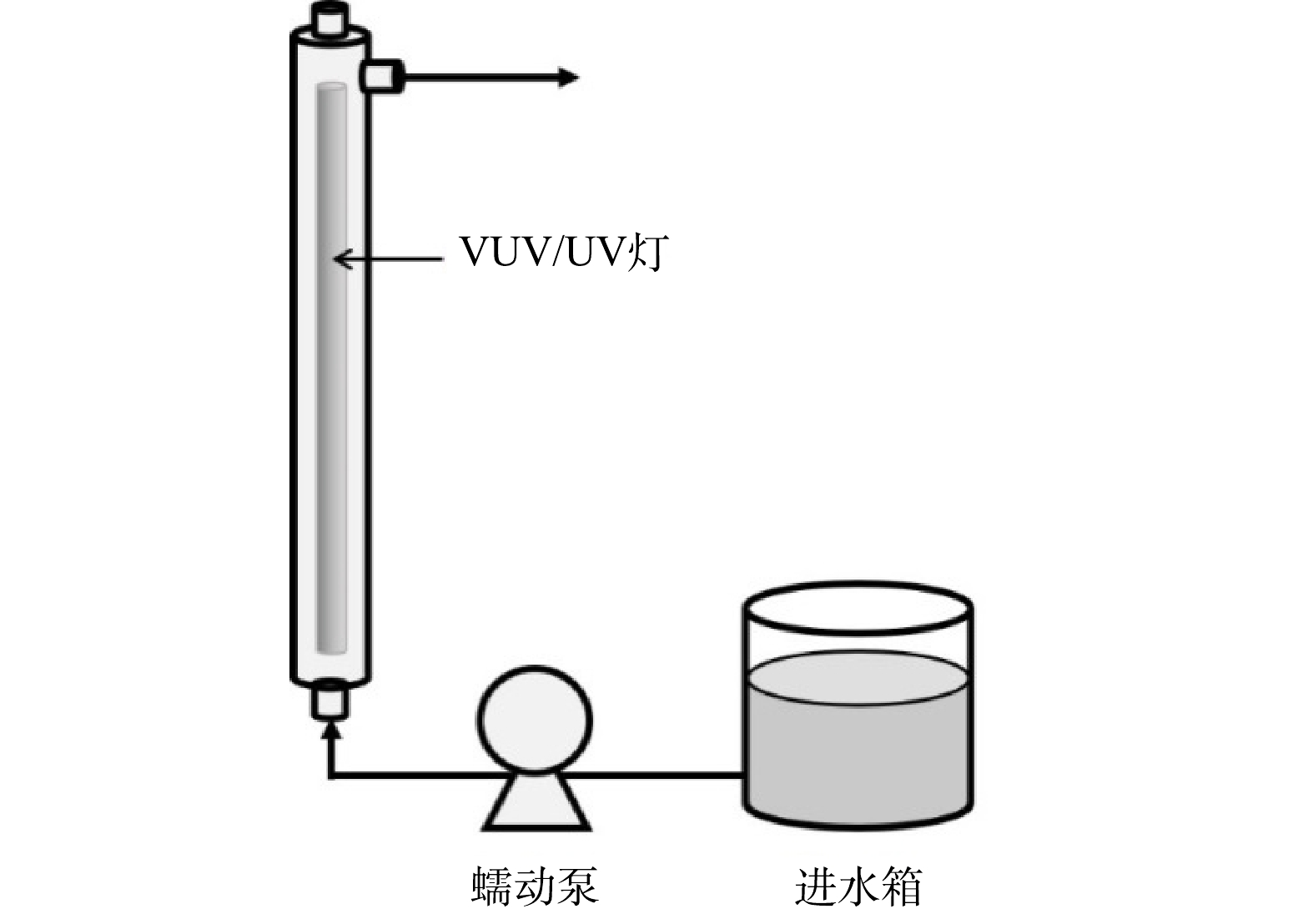
污染物的降解实验在高为450 mm、内径为50 mm、有效体积为750 mL的管式反应器中进行。反应器竖直放置,中心固定一根长为436 mm、外径为20.5 mm、功率为21 W的VUV/UV灯(GCL436T5VH/4,莱劭思,美国)。整个反应装置示意图如图1。
在进行辐照实验前,至少提前10 min开启VUV/UV灯,使其输出达到稳定,并同时在反应器内循环冷却水以防灯管发热影响输出功率。进水箱中的反应溶液通过蠕动泵与反应器下端的进水口相连,进水模式为过流式,即溶液只通过1次反应器后从上端出口流出,在此处取样,进行后续分析。通过控制蠕动泵的转速来改变进入反应器的流量(1.5~9.0 L·min−1),进而改变每次进水在反应器内的水力停留时间(HRT)(5~30 s)以及接受的UV辐射剂量(70.5~423 mJ·cm−2)。每次调整流量后先将反应器内的溶液排空,再进行新流量下的降解实验。在每个进水流量下,将该流量所对应的HRT认定为溶液通过反应器时所接收辐照的时间。实验温度控制在室温(25±2 ℃)。以2 μmol·L−1的ATZ为感光剂在同型号的UV灯(GCL436T5L/4,莱劭思,美国)下直接光解,确定反应器内的平均UV强度为14.1 mW·cm−2。
-
目标污染物ATZ的浓度由装有C18色谱柱(安捷伦,150 mm×2.1 mm,3 μm,美国)的高效液相色谱HPLC(安捷伦,1200系列,美国)来测定,测定条件如下:流动相由乙腈和0.2%甲酸水组成(体积比为75:25),流速为0.8 mL·min−1,柱温为40 ℃,检测波长为234 nm,进样体积为50 μL。NO3–和NO2–的浓度用哈希试剂测定(方法编号分别为10020和10019) 。DOM的浓度用TOC分析仪(岛津,TOC-VCPH,日本)测定。pH由pH计(赛多利斯,PB-10,德国)测定。
-
水和不同溶质对185 nm VUV有不同的吸收能力,根据式(1)可以计算水和溶质对VUV光子的吸收比例。
式中:fi为物质对185 nm VUV光子的吸收比例;ai为物质i在185 nm处的单位吸光度,cm−1;εi为物质在185 nm处的摩尔吸收系数,L·(mol·cm)−1;Ci为物质的浓度,mol·L−1;atotal为水中所有物质在185 nm处的单位吸光度,cm−1。
-
过流式VUV/UV反应器中Cl–浓度对ATZ降解的影响结果如图2所示。由图2可见,水中Cl–的存在可抑制VUV/UV工艺对ATZ的降解,且Cl–浓度越高,对ATZ降解抑制程度越高。当辐照时间为30 s时,在Cl–浓度为1.0、2.5和5.0 mmol·L−1的溶液中,ATZ的去除率由空白组中的57%分别降至42%、39%和33%(图2(a))。对各Cl–浓度下不同辐照时间时ATZ的降解率进行拟一级反应动力学拟合,发现拟合程度较好(R2 > 0.96)。拟一级降解速率常数kapp,VUV/UV从空白组中的0.029 4 s−1分别降低至0.021 5、0.017 7和0.015 0 s−1;而ATZ在UV下发生直接光降解的速率常数kapp,UV为0.012 5 s−1。这表明1.0、2.5和5.0 mmol·L−1 Cl–对VUV贡献的ATZ降解速率常数kapp,VUV(即kapp,VUV/UV–kapp,UV)的抑制程度分别为47%、69%和85%。
Cl–对ATZ降解的抑制可归因于以下2点。一方面,Cl–在185 nm处的摩尔吸收系数较大(ε=3 500 L·(mol·cm−1))[4],根据式(1)计算可得1.0、2.5和5.0 mmol·L−1的Cl–在185 nm处的单位吸光度分别为3.5、8.8和17.5 cm−1,而水的单位吸光度为1.8 cm−1(25 ℃)[5]。这表明,当水溶液中含有1.0、2.5和5.0 mmol·L−1的Cl–时,VUV/UV发出的185 nm处VUV光子分别只有34%、17%和9%被水吸收(图2(b)),VUV光解水产生的HO·浓度相对于空白组中的浓度显著降低,这是导致ATZ降解受到抑制的主要原因。另一方面,Cl–在VUV辐照下会产生Cl·(式(2)),而Cl·又会与Cl–快速反应生成Cl2–·(式(3)),Cl·氧化性强,而Cl2–·氧化性较弱,二者与ATZ的二级反应速率常数分别为6.9×109 L·(mol·s)−1和1.0×107 L (mol·s)−1 [6],Cl·与ATZ的反应活性甚至超过HO·与ATZ的反应活性(后者的二级反应速率常数2.3×109 L (mol·s)−1 [7],表明ATZ可能会因被Cl·氧化而降解。由图2(b)可见,水溶液中Cl–存在时由VUV贡献的ATZ降解速率常数kapp,VUV的下降程度低于VUV光子被Cl–吸收的比例(如5.0 mmol·L−1时的85% vs.空白组的91%),说明Cl–在VUV辐射下产生的活性氯自由基(Cl·、Cl2–·等)确实在ATZ降解过程中发挥了作用。需要注意的是,虽然Cl–可能会与水中的HO·结合生成ClHO–·,但这个反应是可逆的,且逆反应速率高于正反应速率(式(4)),所以二者的结合产物ClHO–·在产生后会立即解离而再次生成HO·,因此Cl–对HO·的影响可以忽略[8-12]。本研究中VUV/UV辐照下Cl–对ATZ降解的抑制程度要略高于YANG等在细管流反应器内得到的结果[13],表明反应器内的流态对降解可能也有一定的影响。总体而言,Cl–对VUV/UV工艺降解水中微量污染物的影响程度主要取决于污染物自身特性。当目标污染物的UV直接光解速率低且与活性氯自由基反应慢时,Cl–对VUV光子的竞争可能导致污染物(如三氯乙基磷酸酯[14])降解速率超过90%以上的降幅。而当目标污染物与活性氯自由基的反应速率快且超过其与HO·的反应速率时,Cl–在VUV辐射下产生的活性氯自由基会促进污染物(如尿素[15])的降解。
-
过流式VUV/UV反应器中HCO3–浓度对ATZ的降解影响结果如图3所示。与上述Cl–存在时的结果类似,当水中HCO3–浓度较高时,ATZ的降解受到了明显的抑制。当HCO3–浓度为1.0、2.5和5.0 mmol·L−1、辐照时间为30 s时,ATZ的去除率分别为49%、35%和29%(图3(a))。对上述各HCO3–浓度下不同辐照时间的降解数据进行拟一级动力学拟合,得到的降解速率常数kapp,VUV/UV分别为0.024 0、0.014 8和0.011 5 s−1 (R2 > 0.98)。
HCO3–对VUV/UV辐照下ATZ降解的影响可从蔽光效应和自由基反应两个方面解释。HCO3–在185 nm处的摩尔吸光系数(ε = 290 L·(mol·cm)−1)[4]远低于Cl–,相应地,1.0、2.5和5.0 mmol·L−1的HCO3–在185 nm处的单位吸光度分别为0.29、0.73和1.45 cm−1,会分别吸收14%、29%和45%的VUV光子(图3(b)),导致水分子在VUV辐射下产生HO·的途径受到抑制。但不同于Cl–存在的情况,高浓度(如5.0 mmol·L−1)HCO3–对VUV光子的竞争吸收程度远小于其对VUV贡献的ATZ降解的抑制程度(分别为45%和100%),表明此时HCO3–对HO·的清除作用不可忽略。事实上,HCO3–对HO·的二级反应速率常数为8.5×106 L·(mol·s)−1,当HCO3–浓度为1.0、2.5和5.0 mmol·L−1时,HCO3–对HO·的消耗速率为8.5×103、2.1×104和4.3×104 s−1,分别为ATZ对HO·消耗速率(5.8×103 s−1)的1.5、3.7和7.4倍。这表明当HCO3–浓度较低(如1.0 mmol·L−1)时,ATZ降解速率的降低可以主要用HCO3–对VUV的蔽光效应来解释;而当HCO3–浓度较高(如5.0 mmol·L−1以上)时,除了蔽光效应外,HCO3–对HO·较强的清除作用也是导致ATZ降解速率显著降低的重要原因。5.0 mmol·L−1的HCO3–可以完全抑制由VUV贡献的ATZ降解,此时ATZ只有在UV下的直接光降解途径。虽然HCO3–在VUV辐射下以及在清除HO·的过程中会产生新自由基CO3–·(式(5)),但CO3–·与ATZ的二级反应速率常数为4.0×106 L·(mol·s)−1 [16],与HO·的相比低了1 000倍以上,因此CO3–·与ATZ的反应在该体系中可以忽略。此外,水体碱度的增加也会带来pH的升高。当HCO3–的浓度分别为1.0、2.5和5.0 mmol·L−1时,溶液的pH分别为7.8、8.2和8.5,而HO·的氧化还原电位在高pH条件下有所下降[17],这也是导致ATZ降解速率降低的原因之一。本研究中HCO3–对VUV/UV工艺降解ATZ的影响程度同样高于YANG等在细管流反应器中的结果[13],但HCO3–对不同种类的微量有机污染物的降解均体现出明显的抑制效果,究其原因在于CO3–·与大部分的目标污染物的反应活性都偏低[18],难以表现出其额外的氧化作用。
-
在过流式VUV/UV反应器中,NO3–浓度对ATZ的降解影响结果如图4所示。NO3–同样可抑制ATZ的降解。当辐照时间为30 s、NO3–为10 mg·L−1和20 mg·L−1时,ATZ的降解率从空白组中的57%分别降至42%和40%(图4(a))。对上述2个浓度(10 mg·L−1和20 mg·L−1)下不同辐照时间的降解率进行拟一级动力学拟合,得到的降解速率常数kapp,VUV/UV从空白组中的0.029 4 s−1分别降低至0.021 1 s−1和0.019 0 s−1 (R2>0.95)。
NO3–在VUV辐照下对ATZ降解的影响归因于一下几点。首先,NO3–在185 nm处的摩尔吸光系数(ε = 480 0 L·(mol·cm)−1)[4]高于Cl–,10 mg·L−1和20 mg·L−1 NO3–对VUV光子的吸收比例分别为66%和79%(图4(b)),高于相应条件下VUV贡献的ATZ降解速率常数kapp,VUV的下降程度(分别为49%和62%),说明ATZ除了被VUV辐照水分子产生的HO·氧化外还存在其他的降解途径。事实上,NO3–在UV辐照下可以产生NO2·和HO·(式(6)),但该反应在254 nm辐照下的量子产率较小,而在185 nm辐照下的量子产率较大,所以VUV辐照下NO3–产生的NO2·和HO·会在一定程度上补充水中氧化性自由基。已有研究[19-20]表明,UV高级氧化过程中NO3–的存在可能会促进水中目标污染物的降解,其原因同样在于NO3–在低波长(< 240 nm)辐照下产生了氧化性自由基。本研究中NO3–对VUV/UV辐照下ATZ降解的抑制程度低于YANG等的研究结果[13],其原因主要在于后者采用的NO3–浓度更高。
另一方面,VUV辐照水分子除了产生HO·外,还会同时产生还原性物种如水合电子eaq–,进而将NO3–还原为NO2–。另外,NO3–的直接VUV光解也会产生NO2–(式(7))。但NO2–在产生的同时又能被HO·快速氧化为NO3–(式(8))。因此在探究NO3–影响ATZ降解的过程中,监测了NO3–和NO2–的浓度变化。由图5可见,反应溶液中NO2–的浓度随辐照时间的延长而线性增加。10.0 mg·L−1 NO3–在VUV/UV辐照30 s后,NO2–浓度从反应起始的0.008 mg·L−1升高至0.040 mg·L−1,净生成量为0.032 mg·L−1;而20.0 mg·L−1的NO3–在相同的辐照条件下,NO2–浓度从反应起始的0.010 mg·L−1升高至0.047 mg·L−1,净生成量为0.037 mg·L−1。而NO3–的浓度基本没有发生变化,10.0 mg·L−1和20.0 mg·L−1 NO3–在被VUV/UV辐照30 s (相应的UV剂量为423 mJ·cm−2)后,两者的浓度均只降低了0.1 mg·L−1。这一结果表明在NO3–浓度为10.0 mg·L−1和20.0 mg·L−1时,NO2–生成速率的限制因素并不是NO3–的浓度,而可能是反应体系中还原性物种和氧化性物种的浓度。溶液中的eaq–可以和溶解氧快速发生反应被清除,HO·则容易将NO2–氧化为NO3–。这两方面原因共同导致在上述反应条件下NO2–的生成量不高。本研究中NO2–的生成量要显著低于HAN等在平行光束仪下反应皿中得到的结果[21],说明实际反应器内溶液的流态可能会加快NO2–向NO3–的转化。亚硝酸盐是一种致癌物,在多国的饮用水标准中都有明确的限值。例如,我国饮用水标准中规定NO2–浓度不得超过1.0 mg·L−1(以N计),而在欧盟的标准中,这一限值更低至0.1 mg·L−1(以N计)。本实验中选用的NO3–浓度为我国饮用水标准中规定的NO3–上限。实验结果表明,将VUV/UV作为饮用水深度处理工艺以去除水中微量有机污染物时,在UV剂量为400 mJ·cm−2左右时,生成的NO2–不会有超标的风险。
-
在过流式VUV/UV反应器中,DOM浓度对ATZ降解的影响结果如图6所示。与空白组相比,DOM浓度为1.0 mg·L−1时,对ATZ的降解几乎没有影响,而当DOM浓度增大至5.0和10.0 mg·L−1时,ATZ的降解明显受到抑制。且DOM浓度越高,ATZ降解被抑制的程度越大。VUV/UV辐照30 s,在1.0、5.0和10.0 mg·L−1 DOM的影响下,ATZ的去除率分别为53%、39%和35%(图6(a))。对上述3个DOM浓度不同HRT时的降解数据进行拟一级动力学拟合,得到的降解速率常数kapp,VUV/UV分别为0.027 6、0.018 7和0.014 8 s−1(R2 > 0.96)。
DOM对VUV/UV工艺降解微量有机污染物的影响也有多条途径的参与。首先是蔽光效应,DOM成分(如腐殖酸、黄腐酸等)中通常含有大量的共轭和芳香结构,在254 nm波长下通常具有较高的吸光度,本研究中测得1.0、5.0和10.0 mg·L−1 DOM溶液对UV(254 nm)的单位吸光度分别为0.022、0.073和0.109 cm−1。溶液吸光度的增大会导致反应器内的平均UV强度降低,因此,引入水介质因子FW(式(9))对具有一定吸光能力的溶液所接收的平均UV强度进行校正(式(10))。在DOM为1.0、5.0和10.0 mg·L−1时,溶液的平均UV强度分别降低至空白组的97%、92%和88%,这会导致ATZ的UV直接光降解速率常数kapp,UV降低。而DOM在185 nm处的单位吸光度要高于其在254 nm处的单位吸光度,有文献报道作为DOM标准物质的SRNOM在185 nm处的摩尔吸光系数为1402 L·(mol·cm)−1(25 ℃)[22],即0.117 L·mg−1·cm−1。参考该数值,本研究中1.0、5.0和10.0 mg·L−1的DOM在185 nm下的单位吸光度分别为0.117、0.585和1.170 cm−1,分别可以竞争吸收6%、25%和39%的VUV光子(图6(b)),因此VUV光解水产生的HO·浓度降低。值得一提的是,部分DOM在UV辐照下可以转变为激发态DOM*,并同时产生单线态氧1O2等活性物种[23-24],可能会促进目标污染物的降解[25-26]。本研究中DOM浓度为1.0 mg·L−1时,ATZ的降解并未受到抑制,可能与DOM在UV辐照下激发产生上述活性氧化物种的促进作用有关。而另一方面,DOM是一种反应容量极大的HO·清除剂,其二级反应速率常数一般在104 L·(mg·s)−1级别[27-28],具体数值与DOM的结构及组成有关。因此DOM的存在会使得溶液中HO·浓度降低,从而导致ATZ被HO·氧化降解的途径受到一定程度的抑制。
式中:A为实际水样在254 nm处的吸光度;Eavg为高透光率溶液条件下反应器内的平均UV强度,mW·cm−2;E'avg为实际水样条件下反应器内的平均UV强度,mW·cm−2。
-
采用目标污染物降解90%所需的电能(EEO,kWh·m−3)对VUV/UV工艺在不同水质条件下的能耗进行评估,其计算公式如式(11)所示,计算结果如表1所示。
式中:P表示紫外灯的输出功率,kW;Q表示进水溶液的体积流速,m3·h−1;kapp表示目标污染物的降解速率常数,s−1;V表示反应器有效辐照体积,L。
在空白组中,单独UV辐照下ATZ降解的速率常数为0.012 5 s−1,计算得到相应的EEO为1.43 kWh·m−3,而在VUV/UV辐照下ATZ降解的速率常数提高至0.029 4 s−1,相应的EEO降低至0.61 kWh·m−3。计算可知UV和VUV对ATZ降解的贡献率分别为43%和57%。由表1可见,VUV/UV辐照下,5.0 mmol·L−1 HCO3–对ATZ降解效率的影响最大,具体而言,5.0 mmol·L−1 HCO3–对HO·较强的清除作用使得VUV对ATZ降解的贡献几乎可以忽略不计,如ATZ在5.0 mmol·L−1 HCO3–的背景下降解的EEO为1.56 kWh·m−3,与ATZ在单独UV辐照下降解的EEO相当。Cl–和DOM也会显著影响VUV/UV工艺降解ATZ的效率,在5.0 mmol·L−1 Cl–和10.0 mg·L−1 DOM的背景下,ATZ降解的EEO分别升高至1.19 kWh·m−3和1.21 kWh·m−3,与ATZ在空白组中降解的EEO相比,增加了95%和98%。我国生活饮用水卫生标准(GB 5749-2022)中规定的氯离子、硝酸盐和总有机碳的质量浓度上限分别为250(即7.0 mmol·L−1)、20和5.0 mg·L−1。上述3个上限条件中,硝酸盐的影响最弱,在20 mg·L−1 NO3–的背景下,ATZ降解的EEO为0.94 kWh·m−3,与空白组中的EEO相比增加了54%。我国北方地区水源水中的无机离子浓度要高于南方地区,因此,VUV/UV工艺在基础水质良好而微量污染物风险较高的南方地区饮用水处理中具有更好的应用前景。
-
1)过流式VUV/UV反应器中ATZ的降解受到水中Cl–、HCO3–、NO3–和DOM的抑制,当这些背景物质分别为5.0 mmol·L−1、5.0 mmol·L−1、20.0 mg·L−1和10.0 mg·L−1时,在辐照时间为30 s的条件下,ATZ的去除率由空白组中的57%分别下降至33%、29%、40%和35%。
2) 3种无机阴离子(Cl–、HCO3–和NO3–)均对185 nm的VUV辐射有一定的吸收能力,因此,会对VUV产生蔽光效应,其中NO3–>Cl–>HCO3–。但Cl–和NO3–在VUV辐照下产生的活性自由基如Cl·和Cl2–·、NO2·和HO·能在一定程度上补充水中氧化性自由基,使得由VUV贡献的ATZ降解速率常数kapp,VUV被抑制的比例低于VUV光子被相应阴离子竞争吸收的比例,且在本研究中NO3–在VUV辐照下产生的NO2–不超过国家生活饮用水标准限值。HCO3–对VUV的蔽光作用较弱,而HCO3–对ATZ降解的抑制作用主要是由于HCO3–对HO·较强的清除作用导致的。低浓度DOM对ATZ的降解几乎没有抑制作用,这与DOM在VUV/UV辐照下可能产生活性物种有关,而高浓度DOM对ATZ的降解有显著抑制主要是由于DOM对HO·的清除作用导致的。
3)水中复杂的背景物质会使VUV/UV工艺降解微量有机污染物时的能耗增加,在5 mmol·L−1 Cl–、5 mmol·L−1 HCO3–、20 mg·L−1 NO3–和10 mg·L−1 DOM的影响下,ATZ降解的EEO从空白组中的0.61 kWh·m−3分别升高至1.19、1.56、0.94和1.21 kWh·m−3。因此VUV/UV工艺更适合用于基础水质良好而微量污染物风险较高的饮用水深度处理中。
背景水质对过流式VUV/UV反应器降解水中阿特拉津的影响
Effect of water matrices on atrazine degradation in a flow-through VUV/UV reactor
-
摘要: 为探究连续流进水模式下水中复杂的背景物质对真空紫外/紫外 (VUV/UV) 高级氧化工艺效率的影响,采用过流式VUV/UV反应器,考察了水中不同浓度的氯离子 (Cl–) 、碱度 (HCO3–) 、硝酸盐 (NO3–) 和溶解性有机物 (DOM) 对微量污染物阿特拉津 (ATZ) 降解的影响。结果表明,上述背景组分对ATZ的VUV/UV降解均表现出一定的抑制作用,辐照时间为30 s时,ATZ去除率从空白组中的57%分别最多下降至33%、29%、40%和35%,且过流式条件下的抑制程度与文献中序批式反应器中的略有不同。污染物去除率下降的原因在于,一方面,三种无机阴离子都对VUV辐射有一定的蔽光效应,NO3–强于Cl–强于HCO3–;另一方面,VUV辐照下Cl–产生的Cl·和Cl2•–、NO3–产生的NO2·和HO·都能补充水中氧化性自由基浓度,使得VUV贡献的ATZ降解速率常数的抑制程度低于VUV光子被阴离子竞争吸收的比例,但HCO3–对HO·较强的清除作用则导致了ATZ的降解速率常数的快速下降。低浓度DOM在VUV/UV辐照下可能产生的活性物种抵消了其对VUV/UV辐射的蔽光效应,但高浓度DOM对HO·的清除作用仍使其对ATZ降解产生了显著的抑制。在所有考察的水质条件下,ATZ降解所需的单位能耗EEO介于0.61~1.56 kWh·m-3。Abstract: In order to explore the impact of complex water matrices on VUV/UV advanced oxidation efficiency under continuous flow mode, the degradation of atrazine (ATZ) in water with additional chloride (Cl–), bicarbonate (HCO3–), nitrate (NO3–) and dissolved organic matter (DOM) at different concentrations were investigated in a flow-through VUV/UV reactor. The results showed that all these water constituents had some inhibition effects on ATZ degradation. The ATZ removal decreased from 57% in deionized water to 33%, 29%, 40% and 35% under the largest investigated matrix concentrations at an irradiation time of 30 s. These inhibition extents were somewhat different from those reported in batch reactors in literatures. The reduction of ATZ removal could be ascribed to following reasons. On the one hand, all the three anions have a certain shielding effect on VUV irradiation, following the order: NO3– > Cl– > HCO3–. On the other hand, reactive species under VUV irradiation, such as Cl· and Cl2•– produced from chloride, NO2· and HO· formed by nitrate, could supplement the concentration of oxidative radicals in water and contribute to ATZ degradation, which resulted in a lower reduction of ATZ degradation contributed by VUV than the proportion of VUV photons absorbed by the anions. In contrast, the strong scavenging of HO· by HCO3– led to a significant reduction of ATZ degradation. The reactive species generated from DOM at low contents could counteract the shielding effect, while DOM at high concentration still had a prominent inhibition on ATZ degradation due to its remarkable scavenging of HO·. The specific energy consumption under the investigated water matrices ranged between 0.61~1.56 kWh·m−3.
-
降雨径流是城市面源污染物空间迁移的主要载体。在降雨过程中,雨水及其形成的地表径流冲刷地面污染物,通过排水沟渠或分流制排水系统直接进入江河湖泊中,对受纳水体的水质保障带来较大压力。为保护水资源、改善水生态、优化水环境、确保水安全,海绵城市建设理念应运而生。为海绵城市主流措施之一的生物滞留系统凭借其应用灵活、径流调控成效突出的优势得到了广泛应用。本课题组前期的研究结果[1]表明,回填填料、种植植物对生物滞留系统的氮磷去除效果具有显著影响,海绵铁的添加有助于提升硝态氮的去除效果。生物滞留系统的填料组成和填料层的深度也会影响污染物的去除效率[2]。同时,植物的栽种可以提升系统对有机物和营养性污染物的去除效率[3]。
为进一步探明填料和植物对生物滞留系统滞净效能的强化情况,国内外学者开展了一系列相关研究。高晓丽等[4]、王书敏等[5]对生物滞留设施填料研究进展和生物滞留介质土理化性质进行了综述分析,介绍了国内外相关研究中填料的组成及配比、去污效果、渗透性能、填料的改良及填料深度,分析了介质土的粒径时空分布特点,可为生物滞留系统填料的筛选提供依据。仇付国等[2, 6-7]尝试用铝污泥和沸石对传统基质填料进行改良以提高系统对氨氮和磷的吸附效果,并在系统底部设置或增加淹没区高度,创造缺氧环境以提高系统对硝态氮的去除效果。为研究季节及植物对生物滞留系统径流污染物去除的影响,HERMAWAN等[8]研究了热带条件下生物滞留系统的季节特性,评估了其性能并筛选出适宜条件生长的植物。余雪花等[3]和陈韬等[9]对生物滞留系统中能有效去除多种污染物的最佳植物进行了研究,比较了雨水中各类污染物在不同种植植物下的去除效果,结果表明,植物对TP的去除影响不显著,对TN去除率良好,对硝态氮去除有一定效果,但并不稳定。
尽管诸多业界学者已在生物滞留系统回填介质填料方面做了一定程度的研究,然而,生物滞留系统滞净效果影响因素众多,研究基础仍然薄弱,在区域环境背景下开展生物滞留系统滞净效能的研究仍需加强。鉴于此,本研究以重庆地区常用的海绵设施种植植物为供试对象,以改良回填介质为装填基质,在模拟径流条件下开展了场次降雨实验,旨在阐明径流污染物在改良填料生物滞留系统中不同季节的去除情况,获得改良填料系统污染物去除的季节分布特征,以期为生物滞留系统优化建设提供参考。
1. 实验设计
1.1 构建生物滞留系统
如图1所示,设计生物滞留是实验装置为长方体柱状,柱子高约1.2 m,底面长宽均为36 cm,前后两面从上到下设置5个出水阀门,左右两面设置4个出水阀门;综合设计生物滞留装置内填料由下往上依次为 21 cm海绵铁、7 cm高度的3~5 mm火山岩、9 cm高度的5~8 mm火山岩、5 cm高度的河砂和50 cm高度的沙土比7∶3的人工配土[1, 5];栽种植物为金叶女贞(Ligustrum × vicaryi Rehder)和麦冬(Ophiopogon japonicus (Linn. f.) Ker-Gawl.)。土壤取自重庆文理学院校内实验外场地旁,自然风干后,打磨成100目左右的细土。
1.2 进出水设计及取样
模拟雨水径流实验工况如表1所示,参考王书敏等[10-11]山城雨水径流相关研究,计算出各污染物进水浓度。实验模拟雨水采用磷酸二氢钾、硫酸铵、硝酸钾等药剂配制。TN进水浓度为4.76~10.04 mg·L−1,硝态氮进水浓度为0.3~2.64 mg·L−1,氨氮进水浓度为1.98~3.96 mg·L−1,TP进水浓度为1.51~2.53 mg·L−1,磷酸盐进水浓度为1.33~2.88 mg·L−1,TOC进水浓度为7.9~21.68 mg·L−1。进水开始后约2 h,打开最下面的阀门,其连接的高度与出水高度(30 cm/40 cm/50 cm)一致的导管开始出水,而后不间断接水,取5~10 L水样后摇匀从中取500 mL左右,现场测pH、氧化还原电位等参数,冷藏备用。
表 1 实验工况Table 1. Test conditions季节 降雨时长/h 前期干旱天数/d 淹没区高度/cm 降雨强度/(mm·h−1) 气温/℃ 进水量/L 出水量/L 春 8 5 40 1.2 23 140 132 8 5 40 1.2 23 140 123 8 5 40 1.6 19.5 187 160.5 8 5 30 1.2 23 140 134.5 夏 8 5 30 1.2 23 140 126.5 8 5 30 1.2 23 140 128 8 5 40 1.2 35 140 131.2 8 5 50 1.2 35 140 139.5 8 5 50 1.6 39 187 160 8 5 50 1.6 39 140 134 8 5 50 0.8 34 94 76 秋 8 5 40 0.8 24 94 85 8 5 30 0.8 25 94 88 8 5 30 1.6 24 187 180.5 8 5 40 0.8 24 94 82 冬 8 5 40 1.2 9 140 138.5 8 5 40 1.6 9 187 175.5 8 5 40 1.6 10 140 134 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.3 分析方法
水样使用0.45 μm的滤膜过滤处理,氨氮和硝态氮分别采用紫外分光光度计(岛津UV-2600, 日本)和全自动间断化学分析仪(CleverChem380, 德国)进行测定;水样中TN用紫外分光光度计检测;TOC测定采用TOC分析仪(Analytikjena Multi N/C 2100 s,德国);总磷用分光光度计测定;磷酸盐则添加抗坏血酸和钼酸铵后,用分光光度计测定。数据处理和绘图采用软件Excel 2016和Origin 2018。
2. 结果与讨论
2.1 季节对污染物浓度去除效果影响
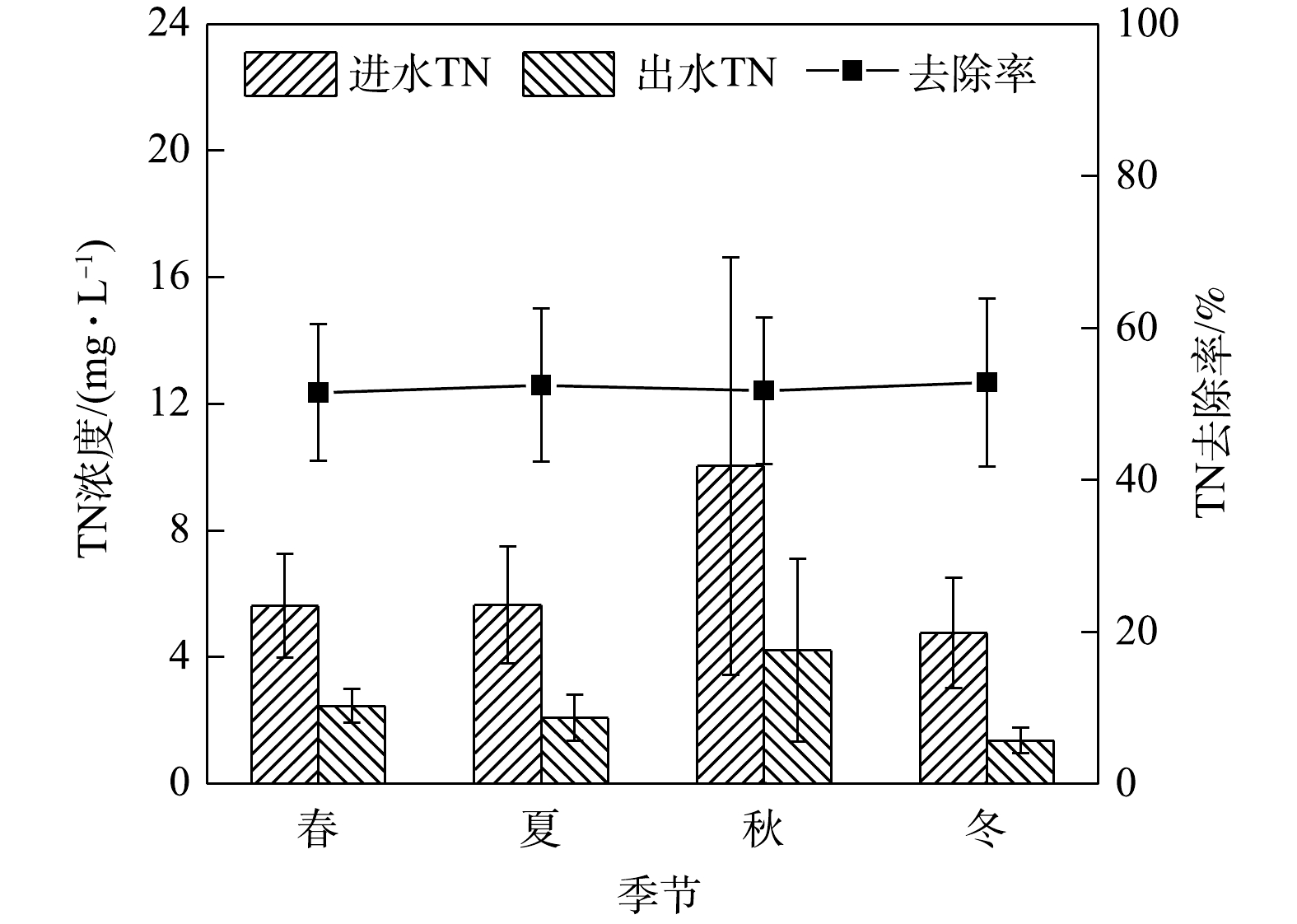
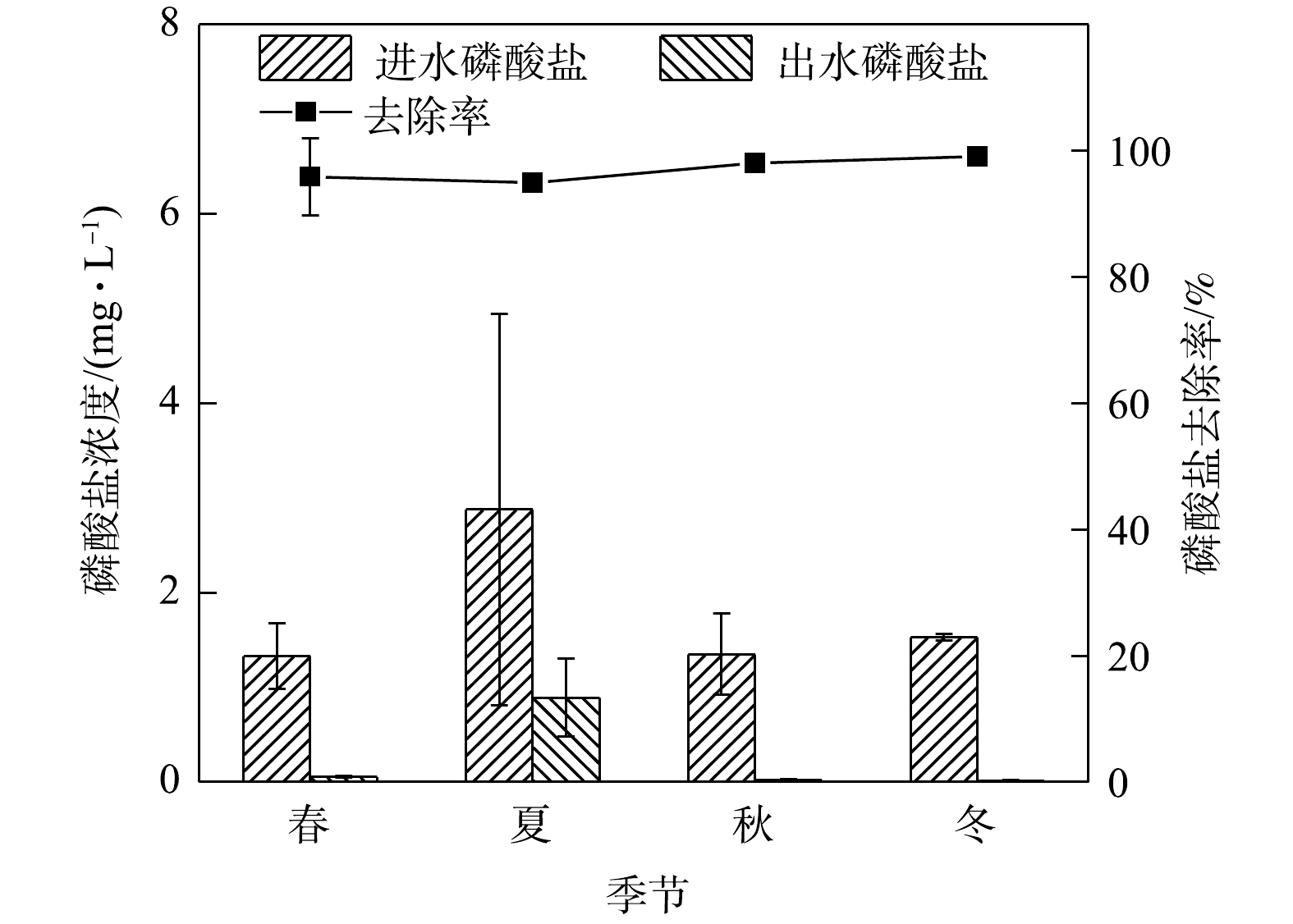
1)季节变化对TN去除效果的影响。由图2可知,在春夏秋冬四季,秋季径流的总氮平均进水浓度最高为10.04 mg·L−1,平均出水浓度为4.21 mg·L−1;冬季平均进水浓度为4.76 mg·L−1,出水浓度为1.37 mg·L−1,夏冬两季出水水质均满足地表水环境质量标准Ⅴ类限值,春秋两季不满足。生物滞留系统对总氮的去除率冬季表现最高,夏季,秋季与春季表现略差,整体在20%以内变化。
TN在冬季去除率较高的原因可能是因为春季开始种植的麦冬生长到冬季,根系较其他季节多。GOH等[12]发现,繁盛的根系在吸收TN、保持介质的导水性以及减少系统径流方面发挥着重要作用。生物滞留系统对氮污染物的去除机理主要有吸附、过滤、硝化反硝化、微生物同化和植物吸收等作用。TN去除率高则说明系统的反硝化能力较强。有研究[1]表明,海绵铁对TN的去除有良好效果。海绵铁促进硝化作用的机理主要是:硝化细菌的细胞膜具有复杂的内褶结构,铁离子通过加大对其的渗透性进而加快硝化的进行;同时由于海绵铁对
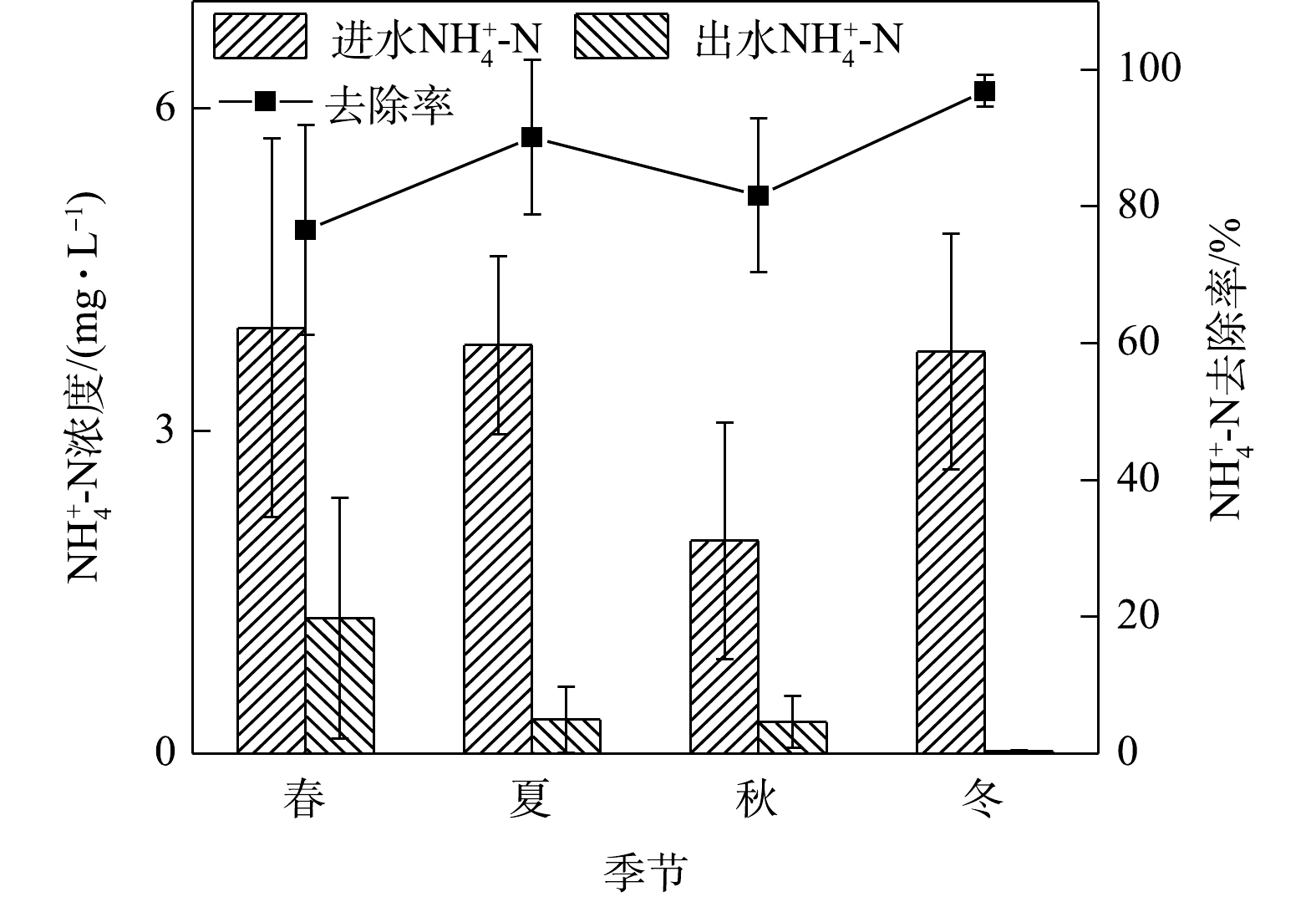
NO−3 NO−3 NH+4 2)季节变化对氨氮去除效果的影响。如图3所示,在春夏秋冬四季,出水氨氮平均浓度在冬季最低,春季最高。春季平均进水浓度为6.08 mg·L−1,出水浓度平均为1.26 mg·L−1,冬季平均进水浓度为3.74 mg·L−1,平均出水浓度为0.02 mg·L−1,春夏秋冬平均出水浓度均满足地表水环境质量标准Ⅳ类限值。在实验系统中,氨氮最高去除率在冬季,为96.88%,最低在春季,为69.34%。
硝化作用是指氨氮在有氧条件下,经亚硝酸细菌和硝酸细菌的作用转化为硝酸的过程。植物通过根系储氧,在根毛周围形成好氧区,离根毛较远的区域则呈现缺氧状态,更远的区域则是完全厌氧,进而形成内部好氧、缺氧、厌氧区的多串联单元,使硝化反硝化作用同时发生[17]。城市径流中的有机氮可经过吸附、沉淀等物理作用被介质截留,在微生物作用下转化为
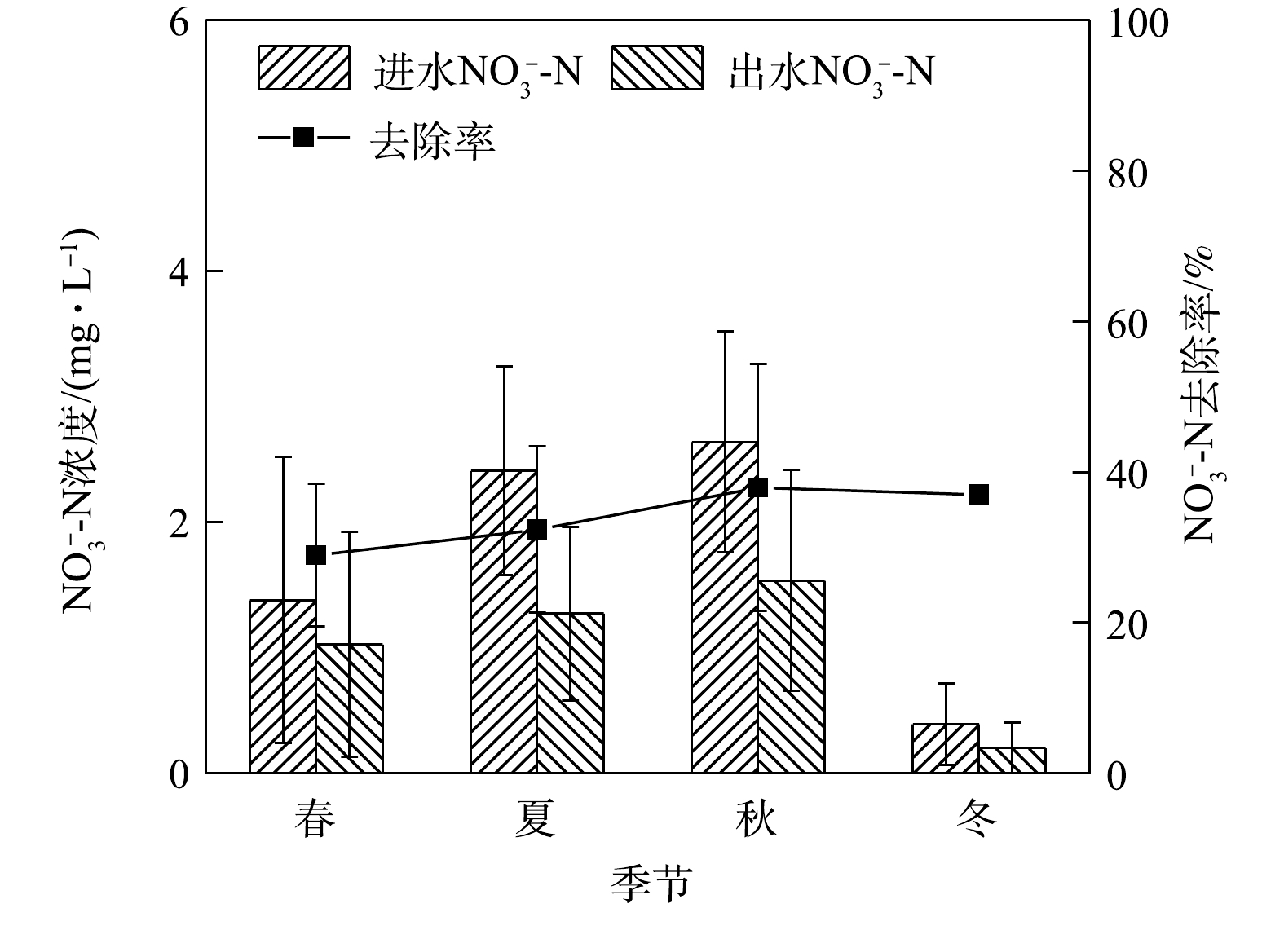
NH+4 NO−3 NH+4 NO−3 NO−3 3)季节变化对硝态氮去除效果的影响。硝态氮的去除是生物滞留系统的一大技术难点。其原因是系统中硝态氮的去除不稳定或者说去除效能过低。如图4所示,硝态氮在秋季平均进水浓度最高为2.64 mg·L−1,平均出水浓度为1.54 mg·L−1,不满足地表水环境质量标准限值。本研究系统对硝态氮的去除率较低,在春季去除率最低为28.98%,夏、秋、冬三季则在5%左右浮动,且相对稳定,全年整体去除效能偏低。
有研究[21-22]表明,硝态氮在春季的去除与植物无关,而可能是由微生物群落驱动的,这是因为土壤微生物量C、N、P在春季和冬季含量较高。在实验系统中,硝态氮在冬季出水浓度最低,这可能是因为植物在种植几个月后达到一定的生物量水平,而根质量增长与硝态氮去除率息息相关[23]。此外,增加饱和带深度可以显著提高
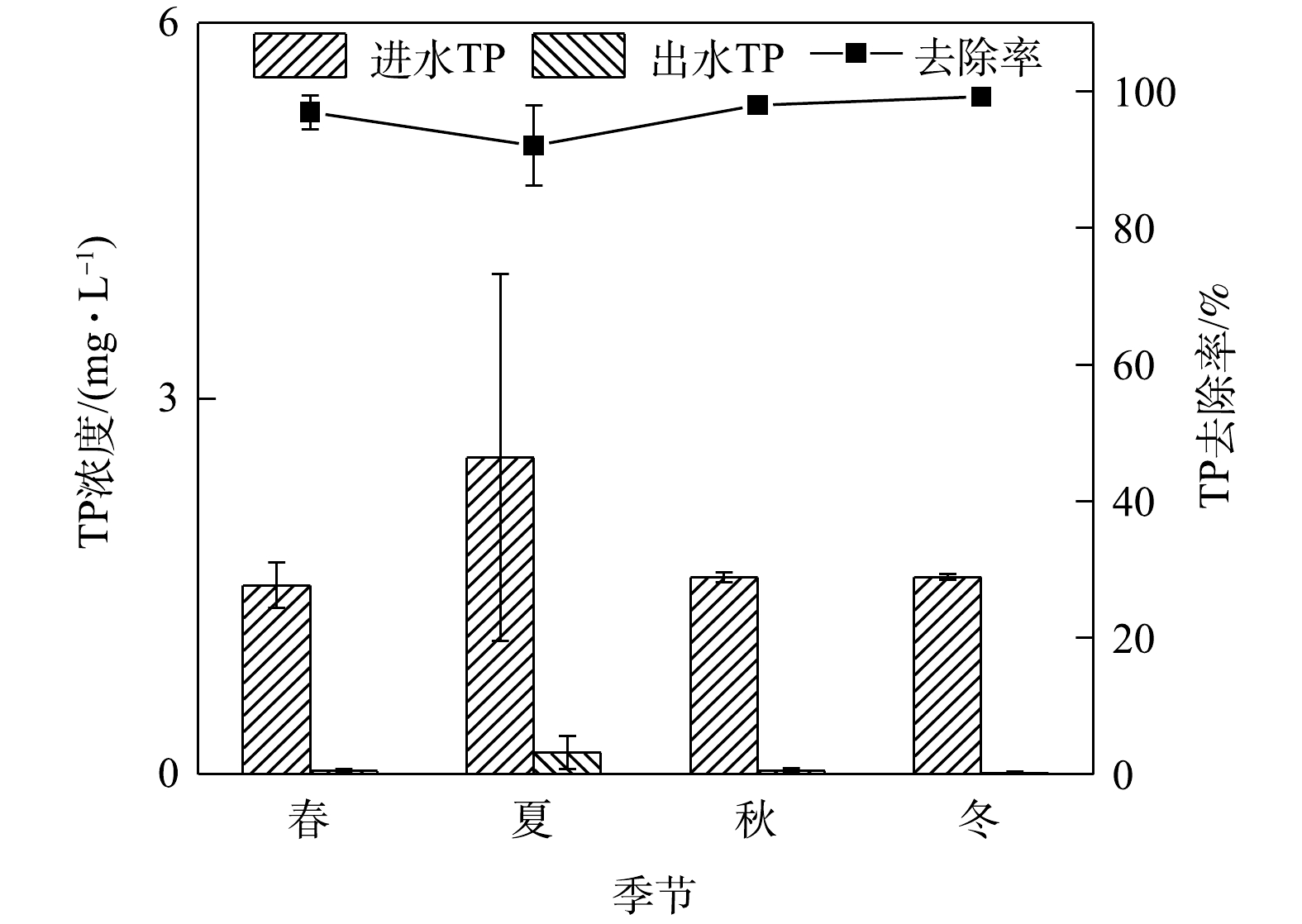
NO−3 NO−3 NH+4 4)季节变化对TP去除效果的影响。如图5所示,在春夏秋冬四季,秋季最低平均进水浓度为1.57 mg.L−1,平均出水浓度为0.01 mg.L−1,春夏秋冬平均出水浓度均满足地表水环境质量标准Ⅲ类限值。实验系统对雨水径流中总磷的去除效率全年处于高水平状态,对TP去除率较为理想,全年最高去除率为99.24%。
结果表明,TP的去除率与季节关系不大,这可能是因为TP的去除主要由填料层介质驱动的。AHMAD等[28]对土壤中磷的组分进行季节性分析发现,各磷组分的定位-季节互作效应不显著,但从夏季到冬季土壤中的TP含量会增加。有研究[3, 8, 23]表明,TP的去除与植物种类无关或者关联较小。TP在不同降雨期迁移到土壤中,然后在土壤中转化为不同的磷形态,植物在根系和微生物的作用下,不断地转化、吸收和利用磷,实现水-土-根系/生物系统中磷的动态平衡[29]。通常生物滞留系统对径流雨水中TP的去除率在80%以上,而改变填料类型、配比及添加改良剂则去除率增至90%以上[8, 29-32]。本研究的实验系统在淹没层设置海绵铁的情况下,还设有2种粒径火山岩,此设计填料层极好地提高了TP的去除率,这与匡颖等[33]、林修咏[1]的研究结果一致,且系统TP的去除率受季节影响较小。
5)季节变化对磷酸盐去除效果的影响。如图6所示,实验系统对磷酸盐的去除率极为理想。在春夏秋冬四季,夏季平均进水浓度最高为2.88 mg.L−1,相应平均出水浓度为0.89 mg.L−1,去除率高达96%。磷酸盐去除率同TP情况相似,全年去除率整体水平较高,各季度去除率变化差别不大。磷酸盐是生物滞留系统去除的主要营养元素之一。城市径流中的磷主要分布在溶解态(一般为有机磷和磷酸盐)和附属于颗粒物的磷之间[34]。溶解态磷中最主要的正磷酸盐通过填料的吸附和植物的吸收而去除[35]。实验系统选用的火山岩填料,其主要成份为硅、铝、锰、铁等几十种矿物质和微量元素,而Al和Fe与
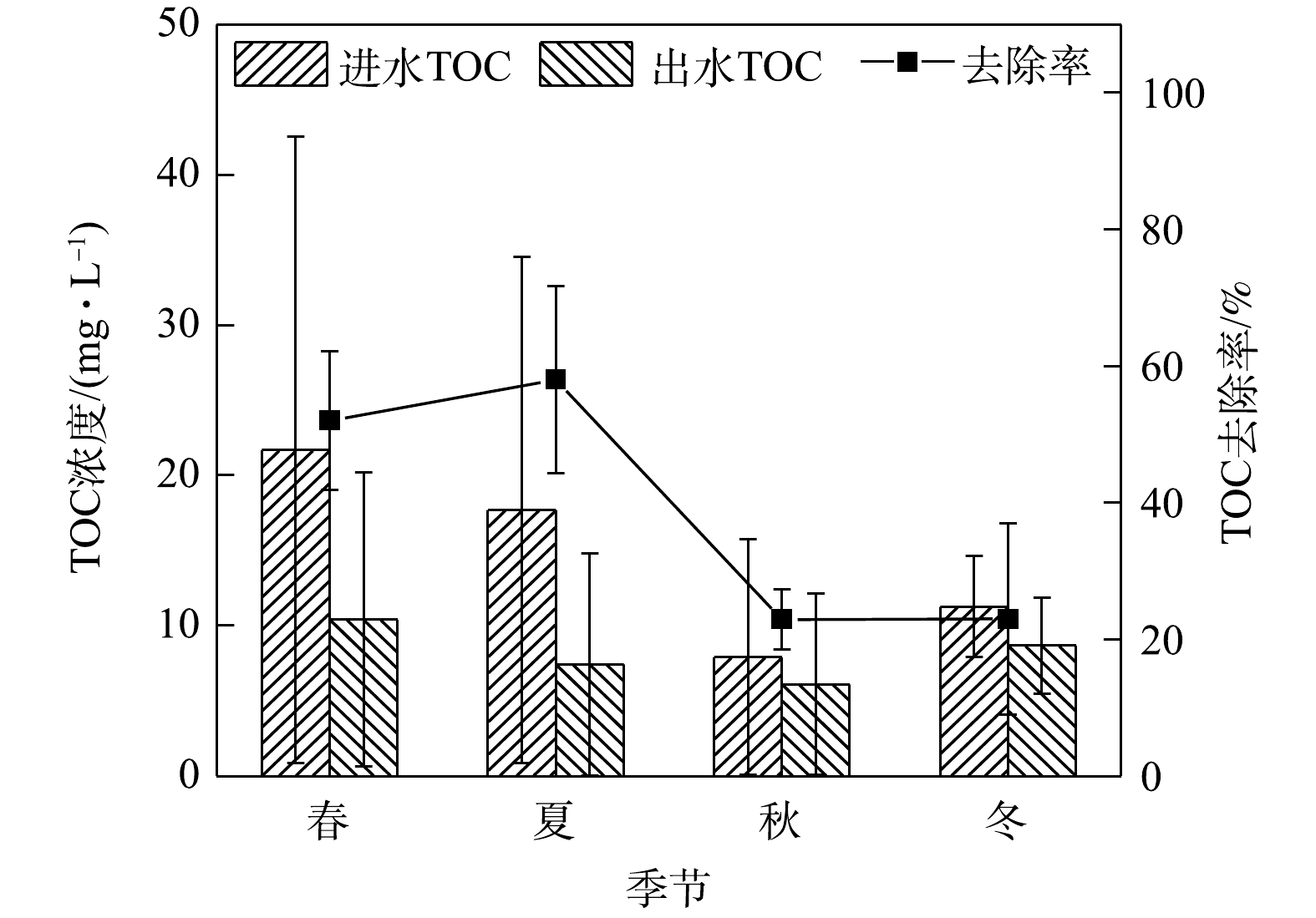
PO3−4 6)季节变化对TOC去除效果的影响。如图7所示,在实验系统中,TOC在春夏两季进水浓度高,春季平均进水浓度最高为21.68 mg·L−1,平均出水浓度为10.41 mg·L−1,相应去除率为52.01%,较秋冬两季高。这可能是因为秋季植物枯萎凋谢增加了土壤中的有机碳[38]。整体来看,系统对TOC的去除率并不理想。据了解,不同种植基质对雨水径流中的TOC浓度具有非常显著的影响[39]。总有机碳(TOC)是以碳的含量表示水中有机物的总量,水的TOC值越高,说明水中有机物含量越高。雨水径流中TOC的来源主要有2个方面:一是自然界本身,如腐烂的动植物;二是人类活动,如工业有机试剂,人畜排泄物等。实验系统中的种植层是沙土比为7∶3的人工配土,土壤则选取当地土壤以便于植物生长以及适应当地气温环境。有研究[40]表明,在土壤中掺入木质有机质后,土壤水分、肥力、有机碳含量均可得到改善进而改善微生物活性和植物生长。TOC去除率越高,说明其中的微生物对有机物的需求越高[1]。可能是系统中微生物活性的缘故,出水TOC浓度在实验系统中四季均不稳定,尤其是在春季达到极端异常值。
2.2 填料对污染物去除的对比分析
生物滞留系统对径流污染物的去除情况除了受季节影响外,填料基质也是其主要影响因素之一。有研究[4, 14, 41-42]表明,填料的类型、填料层的深度、填料组成及配比等均会影响TN的去除效果。本实验系统以火山岩矿物为填料,通过设置内部缺氧段,采用海绵铁强化除磷,可以提高其脱氮除磷能力[33]。就本实验系统的实验结果来说,其TN和TP去除率同文献报道的其他实验结果[1, 26]基本一致,且磷酸盐去除率也很理想。生物滞留系统填料层中采用的火山岩具有天然蜂窝多孔性结构,孔径间隙分布不均、表面粗糙且亲水性强,适宜微生物在其表面繁殖生长形成生物膜,有利于硝化菌的增殖生长,提高介质层的硝化能力,从而使填料层能去除更多的氨氮[18]。因为硝化菌增殖生长,对反硝化菌可能造成了一定影响,故系统硝态氮去除效果不是很理想。有研究[1]表明,海绵铁填料对TOC(约94.53%)的去除率较高。本研究实验系统未能达到理想去除效果,这可能是因为pH过高和水温过低,从而影响了TOC的去除[43]。
对比了本研究与其他改良填料基质下径流污染物的去除情况,结果如表2所示。由表2可知,本研究实验系统相比其他研究,在脱氮除磷方面同其他单一填料较占优势。但同其他改良填料相比,氮污染物及TOC去除效果相对不是很好,磷污染物去除效果与其他改良填料相差无几甚至普遍偏高。
表 2 本实验和其他改性改良(改性)实验径流污染物去除情况统计Table 2. Statistics of runoff pollutant removal in this experiment and other modified experiments采用填料基质 栽种植物 污染物去除率/% 来源 TN NO3-N NH4-N TP 磷酸盐 TOC 建筑废料 麦冬 68.4 — — — 85 — [44] 砂土基质+水厂铝污泥 马莲 44.1 50 96.5 99.6 — — [30] 铝污泥+沸石+淹没区设置 马莲 80 79 93 98 — — [6] 砂土+给水厂污泥 山麦冬 74 78 90 99 — — [45] 红壤+细沙+淹没层(碎砖块) 麦冬草 83.99 72.27 80.45 — — 78.82 [1] 红壤+细沙+淹没层(海绵铁) 87.82 94.38 97 — — 94.54 红壤+细沙+淹没层(火山岩) 84.65 76.55 — — 81.27 中砂+表层土+剩余肥料+废报纸 灌木木槿 80.4 — — 91.8 — — [12] 中砂+表层土+剩余肥料+轮胎屑 50 — — 80 — — 生物炭+C33混凝土砂+粉土和粘土 — — — 50 ~90 — — — [46] 含水层+黄土+碎石 黑眼花 −119.3~85.06 −583.5 ~58.65 40.84 ~94.22 −467.4 ~48.89 — — [36] 种植土+矿渣填料层(2~5 mm)+砾石层 草带 −1.85 — — 53.79 — — [47] 培养基层+粗层+排水层 变叶木 50~70 — — 82~97 — — [26] 砂+粉煤灰 — — — — 84.33 — — [48] C33混凝土+普通生物炭(SWT) — — — — — 49.75 — [31] C33混凝土+铁改良生物炭(GXT) — — — — — 97.86 — [30] 海绵铁+火山岩(3~5 mm)+火山岩(5~8 mm)+河砂+人工配土 金叶女贞+麦冬 60.58 41.20 56.29 96.56 97.02 38.97 本研究 | Show TableDownLoad:
CSV
3. 结论
1)生物滞留系统对总氮和氨氮的去除效果趋势均为夏冬两季高于春秋两季,去除率均在冬季最高,分别达到69.53%和96.88%;而对于硝态氮来说,整体去除率较低,在28.98%~47.18%内波动,且受季节的影响较大。
2)生物滞留系统对总磷和磷酸盐的去除效果受到季节的影响最小,四季去除率波动幅度不大,且长时间稳定保持在90%以上;TOC全年平均去除率为38.96%,夏季去除率最高为58.01%,春夏两季去除率较秋冬两季去除率高,受季节影响较大。
3)本研究的实验系统的脱氮除磷性能较其他单一填料更有优势,但同其他改良填料相比,氮污染物及TOC去除效果并不是很好,磷污染物去除情况与其相差不大且去除率较高。
-

图 2 不同氯离子浓度下过流式VUV/UV反应器中ATZ降解的浓度变化和速率常数以及水吸收VUV比例
Figure 2. Concentration changes and rate constants of ATZ degradation and the ratios of VUV absorbed by water in the flow-through VUV/UV reactor with Cl– of different concentrations

图 3 不同碳酸氢根离子浓度下过流式VUV/UV反应器中ATZ降解的浓度变化和速率常数以及水吸收VUV比例
Figure 3. Concentration changes and rate constants of ATZ degradation and the ratios of VUV absorbed by water in the flow-through VUV/UV reactor with HCO3– of different concentrations

图 4 不同硝酸盐浓度下过流式VUV/UV反应器中ATZ降解的浓度变化和速率常数以及水吸收VUV比例
Figure 4. Concentration changes and rate constants of ATZ degradation and the ratios of VUV absorbed by water in the flow-through VUV/UV reactor with NO3– of different concentrations
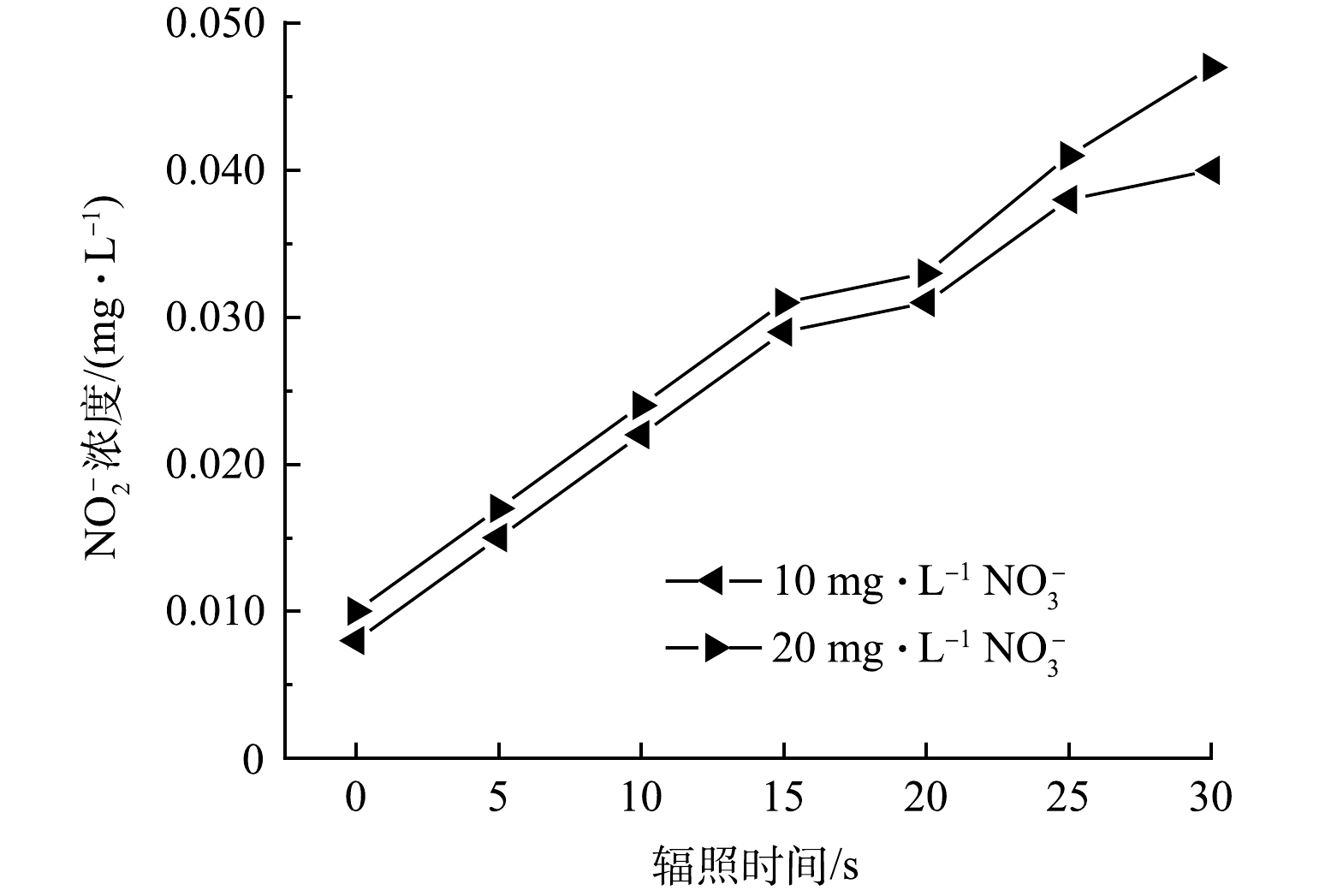
图 5 不同硝酸盐浓度下过流式VUV/UV反应器中亚硝酸盐的浓度变化
Figure 5. Concentration changes of NO2– in the flow-through VUV/UV reactor under NO3– of different concentrations
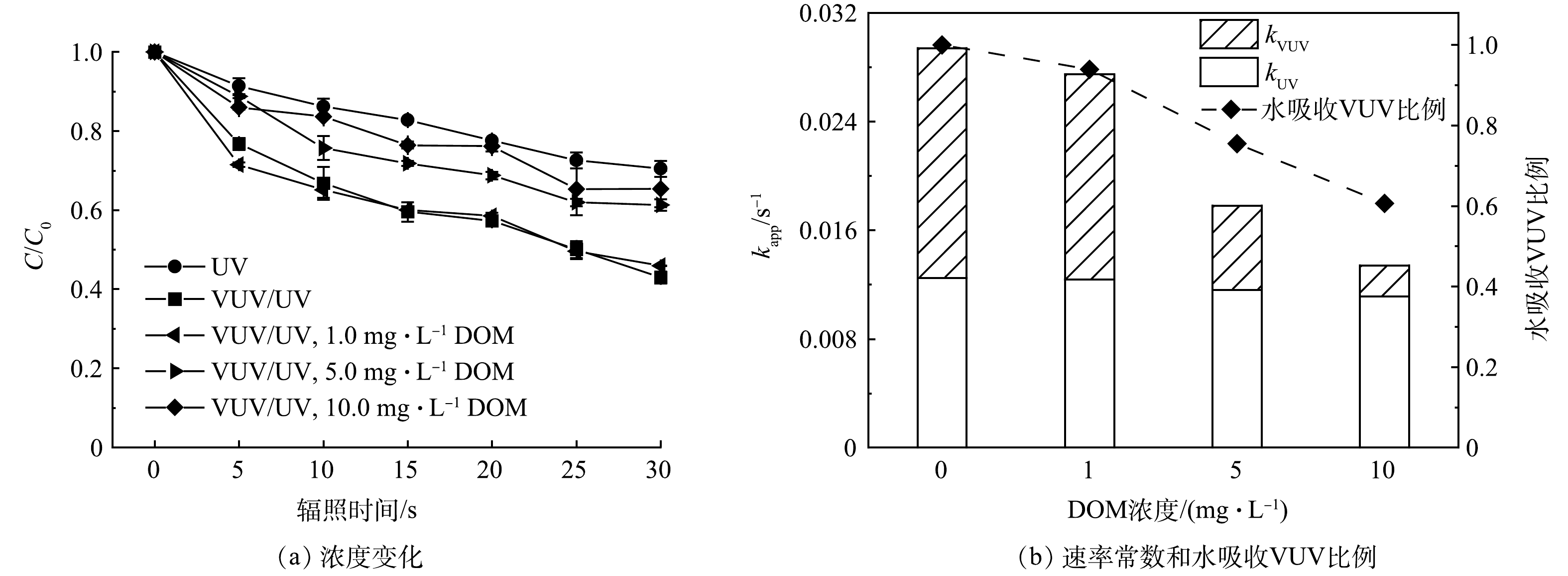
图 6 不同溶解性有机物浓度下过流式VUV/UV反应器中ATZ降解的浓度变化和速率常数以及水吸收VUV比例
Figure 6. Concentration changes and rate constants of ATZ degradation and the ratios of VUV absorbed by water in the flow-through VUV/UV reactor with DOM of different concentrations
表 1 过流式VUV/UV反应器中ATZ在不同水质条件下降解的能耗EEO
Table 1. The energy consumption of ATZ degradation under different matrices in the flow-through VUV/UV reactor
背景物质 浓度 kapp/s-1 EEO/(kWh·m-3) Cl– 1.0 mmol·L-1 0.021 5 0.83 2.5 mmol·L-1 0.017 7 1.01 5.0 mmol·L-1 0.015 0 1.19 HCO3– 1.0 mmol·L-1 >0.024 0 0.75 2.5 mmol·L-1 0.014 8 1.21 5.0 mmol·L-1 0.011 5 1.56 NO3– 10.0 mg·L-1 0.021 1 0.85 20.0 mg·L-1 0.019 0 0.94 DOM 1.0 mg·L-1 0.027 6 0.65 5.0 mg·L-1 0.018 7 0.96 10.0 mg·L-1 0.014 8 1.21
下载: 导出CSV
-
[1] 胡晋博, 李梦凯, 蔡恒文, 等. 不同光源UV/H2O2工艺降解四环素动力学[J]. 环境工程学报, 2021, 15(8): 2618-2626. doi: 10.12030/j.cjee.202012105 [2] 詹露梦, 李文涛, 李梦凯, 等. 过流式UV/H2O2反应器中阿特拉津降解动力学的测定及模拟评估[J]. 环境工程学报, 2021, 15(3): 982-991. doi: 10.12030/j.cjee.202008067 [3] 邵婉婷, 王文龙, 杜烨, 等. 双波长紫外线(VUV/UV)对有机污染物强化去除特性与原理[J]. 环境科学研究, 2021, 34(6): 1397-1406. doi: 10.13198/j.issn.1001-6929.2020.11.28 [4] FURATIAN L, MOHSENI M. Influence of major anions on the 185 nm advanced oxidation process - Sulphate, bicarbonate, and chloride[J]. Chemosphere, 2018, 201: 503-510. doi: 10.1016/j.chemosphere.2018.02.160 [5] WEEKS J L, MEABURN G, GORDON S. Absorption coefficients of liquid water and aqueous solutions in far ultraviolet[J]. Radiation Research, 1963, 19(3): 559-567. doi: 10.2307/3571475 [6] YE B, LIU Z Y, ZHU X Q, et al. Degradation of atrazine (ATZ) by ammonia/chlorine synergistic oxidation process[J]. Chemical Engineering Journal, 2021, 415(14): 128841. [7] DE LAAT J, BERGER P, POINOT T, et al. Modeling the oxidation of atrazine by H2O2/UV. Estimation of kinetic parameters[J]. Ozone-Science & Engineering, 1997, 19(5): 395-408. [8] JAYSON G G, PARSONS B J, SWALLOW A J. Some simple, highly reactive, inorganic chlorine derivatives in aqueous-solution - their formation using pulses of radiation and their role in mechanism of fricke dosimeter[J]. Journal of the Chemical Society-Faraday Transactions, 1973, 1(9): 1597-1607. [9] ANASTASIO C, MATTHEW B M. A chemical probe technique for the determination of reactive halogen species in aqueous solution: Part 2 - chloride solutions and mixed bromide/chloride solutions[J]. Atmospheric Chemistry and Physics, 2006, 6: 2439-2451. doi: 10.5194/acp-6-2439-2006 [10] GUAN Y H, MA J, LI X C, et al. Influence of pH on the formation of sulfate and hydroxyl radicals in the UV/peroxymonosulfate system[J]. Environmental Science & Technology, 2011, 45(21): 9308-9314. [11] LIAO C H, KANG S F, WU F A. Hydroxyl radical scavenging role of chloride and bicarbonate ions in the H2O2/UV process[J]. Chemosphere, 2001, 44(5): 1193-1200. doi: 10.1016/S0045-6535(00)00278-2 [12] GREBEL J E, PIGNATELLO J J, MITCH W A. Effect of halide ions and carbonates on organic contaminant degradation by hydroxyl radical-based advanced oxidation processes in saline waters[J]. Environmental Science & Technology, 2010, 44(17): 6822-6828. [13] YANG L X, LI M K, LI W T, et al. Bench- and pilot-scale studies on the removal of pesticides from water by VUV/UV process[J]. Chemical Engineering Journal, 2018, 342: 155-162. doi: 10.1016/j.cej.2018.02.075 [14] CHEN Y J, YE J S, CHEN Y, et al. Degradation kinetics, mechanism and toxicology of tris(2-chloroethyl) phosphate with 185 nm vacuum ultraviolet[J]. Chemical Engineering Journal, 2019, 356: 98-106. doi: 10.1016/j.cej.2018.09.007 [15] LONG L C, BU Y A, CHEN B Y, et al. Removal of urea from swimming pool water by UV/VUV: The roles of additives, mechanisms, influencing factors, and reaction products[J]. Water Research, 2019, 161: 89-97. doi: 10.1016/j.watres.2019.05.098 [16] CANONICA S, KOHN T, MAC M, et al. Photosensitizer method to determine rate constants for the reaction of carbonate radical with organic compounds[J]. Environmental Science & Technology, 2005, 39(23): 9182-9188. [17] XIAO Y J, ZHANG L F, ZHANG W, et al. Comparative evaluation of iodoacids removal by UV/persulfate and UV/H2O2 processes[J]. Water Research, 2016, 102: 629-639. doi: 10.1016/j.watres.2016.07.004 [18] WOJNAROVITS L, TOTH T, TAKACS E, et al. Rate constants of carbonate radical anion reactions with molecules of environmental interest in aqueous solution: A review[J]. Science of the Total Environment, 2020, 717: 137219. doi: 10.1016/j.scitotenv.2020.137219 [19] SUN P Z, PAVLOSTATHIS S G, HUANG C H. Photodegradation of veterinary ionophore antibiotics under UV and solar irradiation[J]. Environmental Science & Technology, 2014, 48(22): 13188-13196. [20] WANG Y F, RODDICK F A, FAN L H. Direct and indirect photolysis of seven micropollutants in secondary effluent from a wastewater lagoon[J]. Chemosphere, 2017, 185: 297-308. doi: 10.1016/j.chemosphere.2017.06.122 [21] HAN M Q, MOHSENI M. Impact of organic and inorganic carbon on the formation of nitrite during the VUV photolysis of nitrate containing water[J]. Water Research, 2019, 168: 115169. [22] SERRANO M A, MOHSENI M. Temperature dependence of the absorbance of 185 nm photons by water and commonly occurring solutes and its influence on the VUV advanced oxidation process[J]. Environmental Science:Water Research & Technology, 2018, 4(9): 1303-1309. [23] LESTER Y, SHARPLESS C M, MAMANE H, et al. Production of photooxidants by dissolved organic matter during UV water treatment[J]. Environmental Science & Technology, 2013, 47(20): 11726-11733. [24] MAIZEL A C, REMUCAL C K. Molecular composition and photochemical reactivity of size-fractionated dissolved organic matter[J]. Environmental Science & Technology, 2017, 51(4): 2113-2123. [25] DALRYMPLE R M, CARFAGNO A K, SHARPLESS C M. Correlations between dissolved organic matter optical properties and quantum yields of singlet oxygen and hydrogen peroxide[J]. Environmental Science & Technology, 2010, 44(15): 5824-5829. [26] WENK J, von GUNTEN U, CANONICA S. Effect of dissolved organic matter on the transformation of contaminants induced by excited triplet states and the hydroxyl radical[J]. Environmental Science & Technology, 2011, 45(4): 1334-1340. [27] GOLDSTONE J V, PULLIN M J, BERTILSSON S, et al. Reactions of hydroxyl radical with humic substances: bleaching, mineralization, and production of bioavailable carbon substrates[J]. Environmental Science & Technology, 2002, 36: 362-372. [28] WESTERHOFF P, MEZYK S P, COOPER W J, et al. Electron pulse radiolysis determination of hydroxyl radical rate constants with suwannee river fulvic acid and other dissolved organic matter isolates[J]. Environmental Science & Technology, 2007, 41(13): 4640-4646. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3681
- HTML全文浏览数: 3681
- PDF下载数: 44
- 施引文献: 0
