

-
全氟和多氟烷基物质(Per- and polyfluoroalkyl substances,PFASs)是一组人工合成的有机化合物,具有高稳定性、表面活性、疏水疏油性、长距离迁移性和难降解性等独特的理化性质,被广泛应用于电子、食品、化工、家具、农药和医疗等众多领域[1]. 在生产和使用过程中,PFASs可通过多种途径进入到环境中. 研究表明,PFASs在全球环境中无处不在,在大气[2]、水体[3]、沉积物[3]及生物体内[4]均有检出. PFASs中的C—F键具有很强的极性,是自然界中键能最大的共价键之一(键能约460 kJ·mol−1 )[5],其优良的物理化学性质使其在环境介质中难以被物理、化学及生物作用降解[6]. 近年来对环境中PFASs污染及其潜在生态风险研究成为学术界和环境管理领域重点关注的问题. 2009年5月,联合国环境规划署正式将PFOS及其盐类等9种物质列为新型持久性有机污染物并纳入斯德哥尔摩公约[7]. 2014年,我国环境保护部印发了关于“全氟辛基磺酸及其盐类”等10种持久性有机污染物禁止生产、流通、使用和进出口公告.
目前有关PFASs的研究多集中于湖泊、河流和海洋,PFASs在这些水体中广泛存在. C—F链末端的亲水基团使PFOS和PFOA具有良好的水溶性(溶解度570 mg·L−1和3400 mg·L−1)[5],而海洋沉积物易与水体发生相互作用,并能为有机污染物提供疏水介质,因此海洋沉积物被认为是PFASs重要的“汇”之一. 北部湾位于中国南部,东起广东雷州半岛和海南岛,西与越南相邻,北至广西壮族自治区沿岸. 深度范围为10 m至60 m,平均深度38 m,海域总面积约为12.8万平方公里. 北部湾开发是国家一带一路重要战略,目前,以石化、浆纸、能源、粮油加工、冶金等产业主,一批相关产业正在加速进入北部湾地区,临海大工业产生的产业聚集效应正在逐步形成,对北部湾生态环境造成了越来越大的压力.
本研究针对北部湾海域共70个调查站点,采集海洋沉积物,测定其中11种PFASs(PFHxA、PFHpA、PFOA、PFNA、PFDA、PFUnDA、PFDoDA、PFTrDA、PFTeDA、PFHxS、PFOS)的含量,通过分析11种PFASs单体浓度水平和分布特征进行初步的生态风险评估,以期了解该地区全氟化合物的污染状况,为实施海洋环境监管与污染防控措施提供支撑.
-
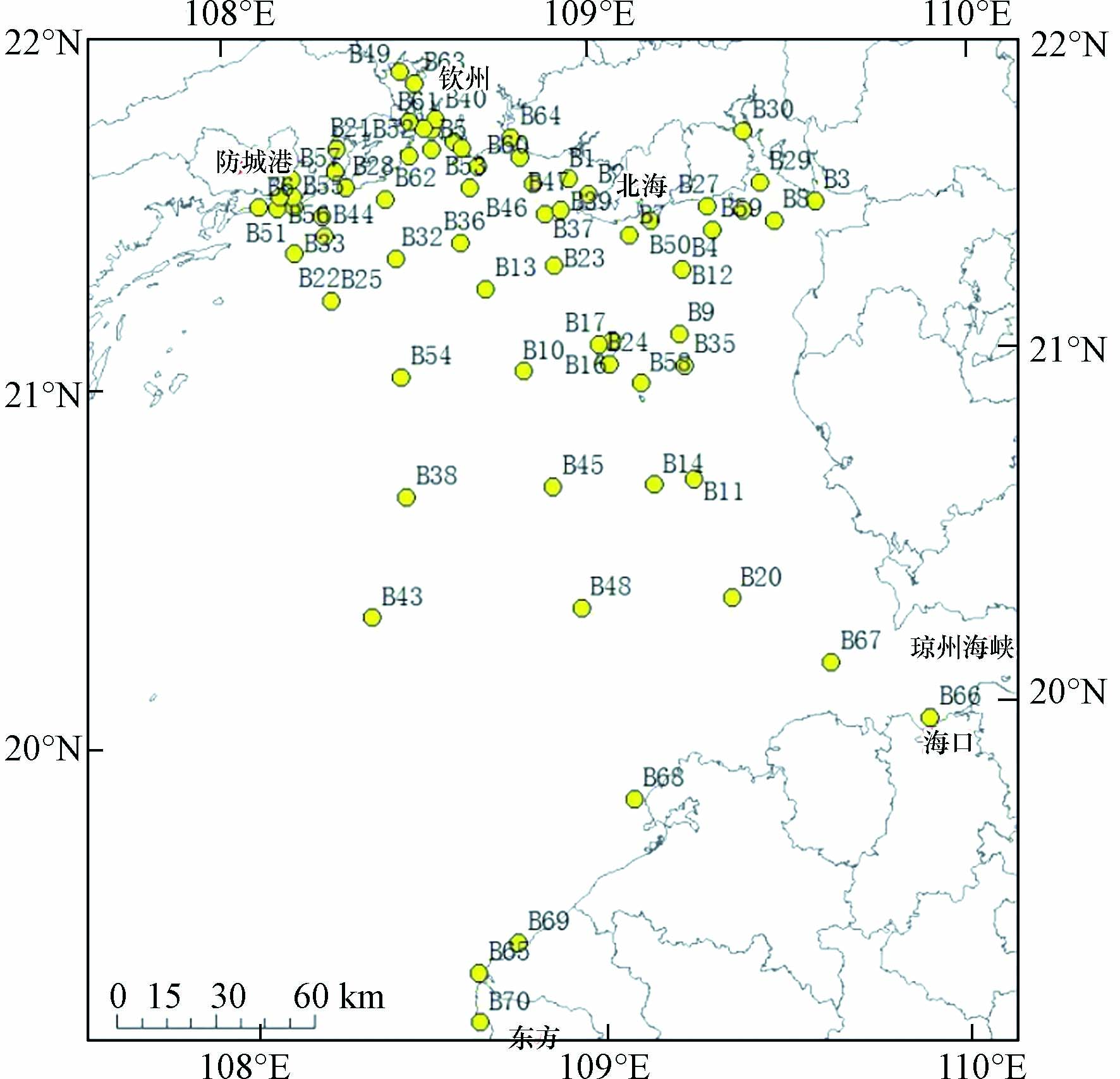
海洋沉积物主要依托当地环境监测部门协助采样获得. 共采集2020年8月的70个表层沉积物样品,采样点位分布见图1. 采用抓斗式采泥器采集沉积物,取上层0—20 cm未受搅动的表层沉积物作为待测样品,放入250 mL棕色试剂瓶中. 沉积物样品的贮存、运输按照《海洋监测规范》(GB 17378-2007)等标准要求执行.
-
沉积物中PFASs的含量分析按照实验室自建方法《海洋沉积物 全氟化合物的测定 液相色谱-串联质谱法》进行检测[8]. 目标化合物包括2种全氟磺酸类物质(PFSAs)及9种全氟羧酸类物质(PFCAs). 11种PFASs单体的类别、名称、CAS号及分子量见表1.
标准样品:全氟己酸(PFHxA)、全氟庚酸(PFHpA)、全氟辛酸(PFOA)、全氟壬酸(PFNA)、全氟癸酸(PFDA)、全氟十一烷酸(PFUnDA)、全氟十二烷酸(PFDoDA)、全氟十三烷酸(PFTrDA)、全氟十四烷酸(PFTeDA)、全氟己烷磺酸(PFHxS)、全氟辛烷磺酸(PFOS)及同位素标记化合物13C4-全氟辛酸(MPFOA)、13C4-全氟辛烷磺酸(MPFOS)和13C2-全氟正癸酸(MPFDA)均加购于加拿大Wellington公司,纯度≥98%. 有机试剂甲醇(色谱纯、Honeywell)、乙腈(色谱纯、Honeywell);无机试剂:冰醋酸(优级纯),氨水(优级纯,25% ),醋酸铵(色谱纯)购于广州化学试剂厂. Oasis WAX固相萃取小柱(Agela,6 mL/500 mg),购于美国.
-
高效液相色谱-三重四极杆串联质谱仪(Agilent 1260/ABSIEX 4000Q): 液相系统,Agilent 1260 Infinity 高效液相色谱(美国);质谱系统,AB SCIEX 4000 Qtrap(美国);ETEL4 MG-2200氮吹仪(日本);24位固相萃取装置(美国、visiprep-24TMPL);超声清洗机SCQ-1000C(中国上海)等.
-
将沉积物样品冷冻干燥,研磨并过0.300 mm孔径筛,取1.00 g于聚丙烯(PP)离心管中,加入内标(MPFOA、MPFOS、MPFDA各2.0 ng),加入5.0 mL甲醇溶液,涡旋1 min,静置30 min后,在30 ℃水浴下进行超声提取,再用5.0 mL甲醇萃取2次,每次萃取后,在4000 r·min−1转速下离心10 min,取上清液,将上清液合并后,旋蒸至 1 mL左右,溶入50.0 mL超纯水中,以每秒1—2 滴过WAX固相萃取柱萃取,WAX小柱预先用4.0 mL含0.5%氨的甲醇溶液、4.0 mL甲醇和4.0 mL超纯水活化,上样完成后的小柱抽真空干燥,干燥后依次用4.0 mL 0.5%氨的甲醇溶液、4.0 mL甲醇洗脱,洗脱液收集于10 mL试管中,氮吹至1 mL,用0.22 μm滤膜过滤后转移至1.5 mL进样瓶中,待上机检测.
所有样品均由高效液相色谱-串联质谱仪进行测定;用5.0 mmol·L−1乙酸铵水溶液作为流动相A,用乙腈为流动相B,流速为0.5 mL·min−1,进样体积为 10μL,柱温为40 ℃.
-
实验过程和分析过程中严格控制和避免使用可能会影响实验结果的材料和试剂. 所有实验装置和容器在使用前均用甲醇进行处理. 为保证数据的可靠性,分析过程中设置运输空白、实验室空白、样品平行和样品加标等操作. 每20个样品做1个实验空白,测定结果中目标物浓度均低于方法检出限. 每20个样品设置1个平行样,平行双样的相对偏差均≤30%. 每20个样品设置了1个实际样品基体加标,基体加标样品回收率范围控制在50%—150%. PFASs单体的检出限LOD、定量下限LOQ及回收率见表2. LOD、LOQ的计算方法为:对标准曲线最低点重复测定7次,计算其标准偏差,将其3.143倍(修约为1位有效数字)作为仪器检出限. 取仪器检出限的5倍作为测试样品添加浓度,分别添加至7个石英砂样品中,按照实验步骤进行样品的前处理,上机测定. 计算7个石英砂样品测定浓度的标准偏差,并取其3.143倍作为方法检出限,再取方法检出限的4倍作为定量下限.
-
本研究基于风险熵(risk quotient, RQ)对PFOA、PFHxA及PFOS3种检出率及浓度较高的典型PFASs进行初步风险评估. RQ为本研究检出环境样品浓度(measured environment concentration, MEC)与预测的无效应浓度(predicted-no-effect concentration, PNEC)的比值. 风险评估分为四个等级:“较小风险”(RQ<0.01);“低风险”(0.01<RQ<0.1);“中等风险”(0.1<RQ<1);“高风险”(RQ>1). 计算公式如下:
由于PFASs在海洋沉积物中关于PNEC值的研究较少,且PNEC值在不同基质中差异很大,因此运用平衡分配法计算沉积物中PNEC值,参考欧盟化学物质风险评价技术指导文件(TGD)[9]中标准环境特征参数获得沉积物中PFOA、PFHxA及PFOS的PNEC值,分别为8.6、1、0.49 ng·g−1.
-
采集的70个沉积物样品∑PFASs浓度范围为ND.—1.50 ng·g−1,平均浓度为0.18 ng·g−1,PFASs检出率为100%. 在11种目标PFASs单体中,除PFHxS未检出外,其余单体均有检出(见表3). 6种单体的检出率(detection frequency, DF)低于10%,PFOA和PFHxA的DF高于50%. PFOA为最主要的单体,DF远远高于其他单体,为85.7%,平均浓度为0.19 ng·g−1,与其他海域沉积物中PFOA的浓度相比处于中等偏低水平(见表4). 其次是PFHxA和PFOS,PFHxA的DF较高,呈现PFHxA(DF为55.7%)>PFOS(DF为24.3%),但PFOS检出样品平均浓度较高,呈现PFOS(平均浓度为0.29 ng·g−1)>PFHxA(平均浓度为0.16 ng·g−1). 总体而言,北部湾的PFASs浓度水平与之前全球研究相比处于相对较低水平(表4),这可能与北部湾地区经济发展水平较低相关,先前的研究表明PFASs的污染与经济发展和行业类型呈正相关[10].
由于近几年短链PFHxA鲜有研究,本研究对比了长链PFOA及PFOS在世界范围内的浓度水平及变化情况. 与世界其他地区相比,北部湾沉积物中PFOA含量(ND.—0.70 ng·g−1,平均值0.19 ng·g−1)低于中国渤海PFOA浓度(0.06—2.70 ng·g−1,平均值0.54 ng·g−1),但和中国东海PFOA浓度相当(ND.—0.87 ng·g−1,平均值0.18 ng·g−1)相当,与2019年北部湾PFOA浓度(0.04—0.25 ng·g−1,平均值0.11 ng·g−1)相比,浓度略有升高. 在北部湾沉积物中PFOS浓度(ND.—1.50 ng·g−1,平均值0.29 ng·g−1)明显高于中国东海PFOS浓度(ND.—0.89 ng·g−1,平均值0.10 ng·g−1),与2019年北部湾PFOS浓度(0.04—0.20 ng·g−1,平均值0.10 ng·g−1)相比,有明显升高趋势. 总体而言,北部湾海域沉积物中PFOA与PFOS浓度与世界其他地区相比处于相对较低水平,但随着经济快速发展,近年来有明显升高趋势.
调查分析的11种PFASs单体依碳链长度分为短链(C<7)、长链(C≥7)2类. 结果表明,沉积物中长链PFASs的浓度水平高于短链PFASs,事实上,先前的研究也检测出类似的结论[1,11-12],与短链PFASs相比,长链PFASs更容易附着在沉积物上[13]. 长链中PFOA占主导地位,在长链检出样品中DF为96.8%;短链中PFHxA占主导地位,在短链检出样品中DF为100%.
-
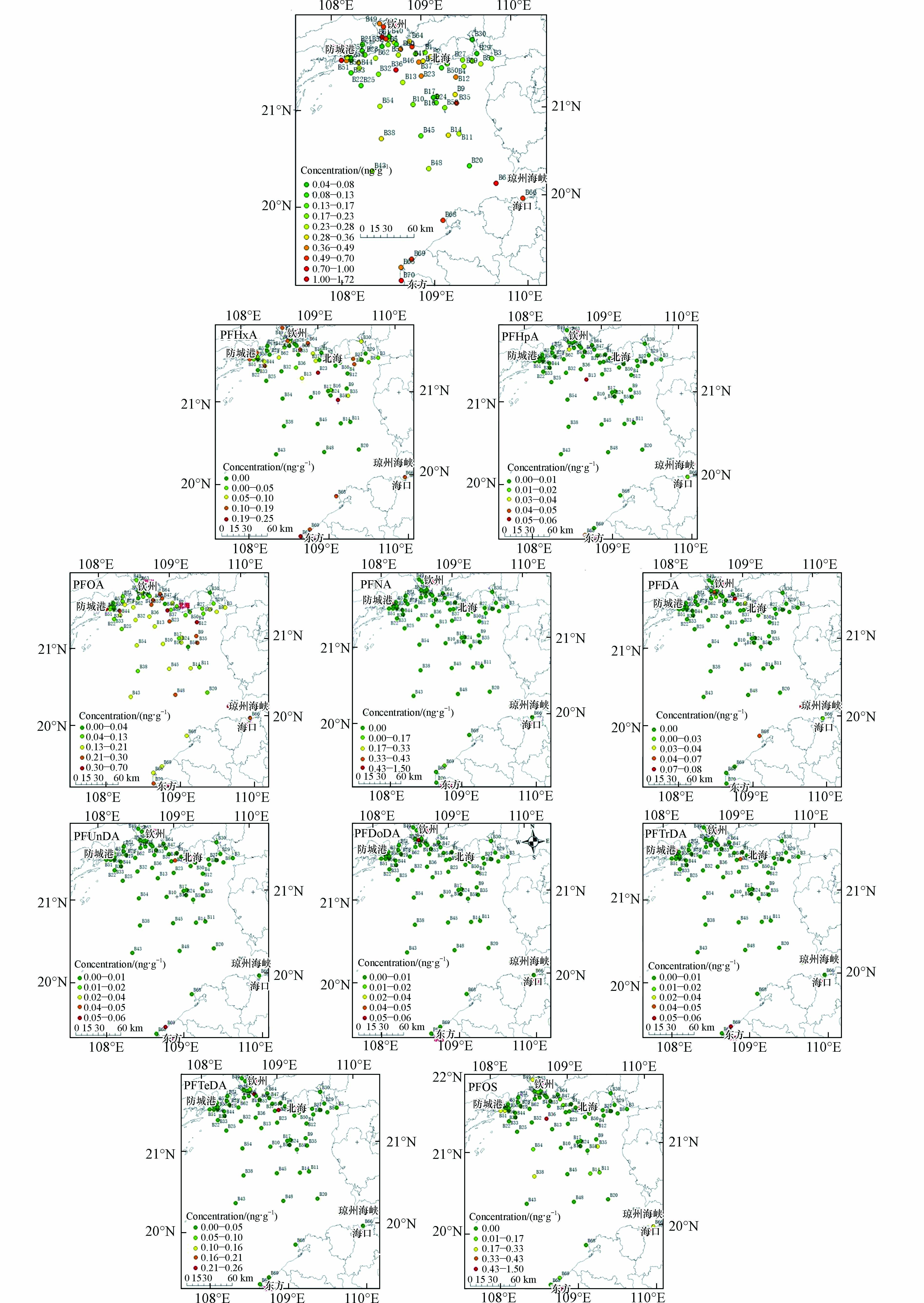
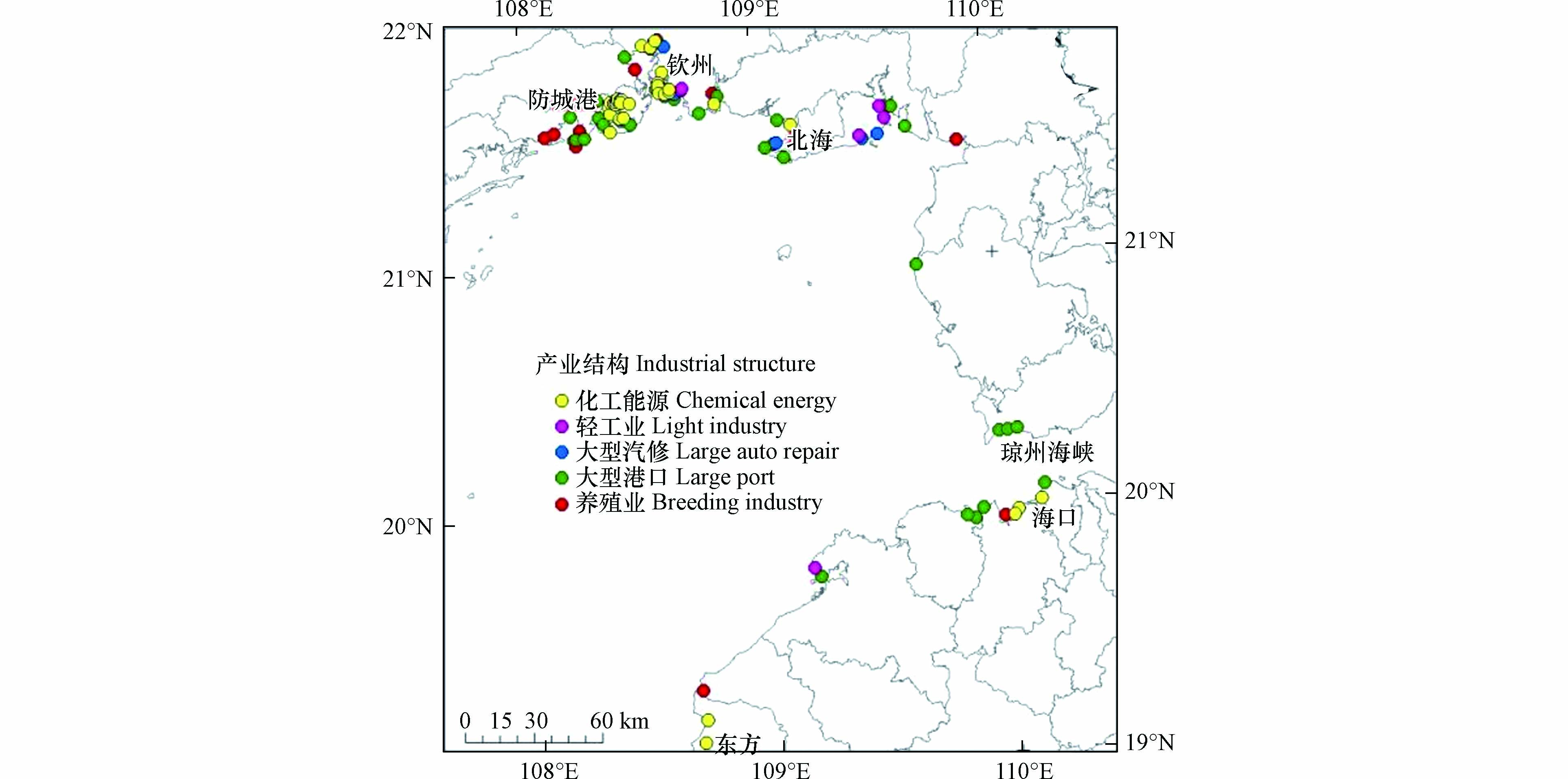
北部湾海域沉积物中∑PFASs浓度及单体浓度都呈现出显著的空间差异性,总体来看呈现靠近城市高于远离城市,轮船运输沿线高于非运输线路(见图2). 其中近钦州的B49、B63、B61及B52点位∑PFASs污染程度较高(均值为0.62 ng·g−1),说明此研究区域内存在点源污染排放,如钦州工厂林立(见图3),受纳工业废水和生活污水,大幅度提高了此研究区域的污染水平,随江流汇入北部湾之后,浓度逐渐降低. 海南近岸海域的B66、B67、B68、B69、B65及B70点位,尤其是靠近琼州海峡的B67点位浓度较高,可能与船舶运输污染及工业废水和生活污水相关. 另一浓度较高的区域为B6、B51及B56位于中越交界近岸海域的点位,由于此研究区域中国境内暂无大型工业企业,可能与越南城市污染相关. 浓度峰值位于北部湾中部的B36点位,平均浓度达到1.72 ng·g−1,经复核样品采集及处理分析过程,均未发现异常,排除实验因素导致的异常高值,经研究,北部湾是我国大西南重要的海上通道,由防城港、钦州港和北海港3座港口组成的北部湾港,经由琼州海峡运往东南亚及南亚各国,这可能与航运线上的船舶污染有关,先前的研究报道了PFASs可以从船舶上的油漆和润滑脂向周围水域扩散[23]. 同时,北部湾季风洋流经琼州海峡由东向西进入湾内形成涡旋,此高值点位于涡旋内部,造成污染物汇聚沉积,可能也是造成此高值点的原因.
PFASs各单体浓度空间分布见图2. 检出率最高的单体PFOA在整个北部湾海域分布广泛,PFOA主要来源于纺织品阻燃、橡胶品乳化、食品包装过程和纸类表面处理,以及灭火剂使用等[24],除与越南交界海域的几处点位浓度较高以外,浓度较高区域主要集中在钦州入海河口的B41、B53、B47及B60点位,河流沿岸多家大型汽修、港船厂及能源公司(见图3),可能与工厂生产排放有关;北海市近岸海域PFOA多处浓度较高,浓度较高的B37定位及B12点位均位于北海市重要港口附近,B8点位附近有多家大型造纸厂(见图3),来自防城港市、钦州市及北海市河流汇聚入海,呈现北部湾中部海域(B35、B9、B23、B32、B36、B13等多点位)PFOA含量中等偏高趋势,需要特别关注;海南省近岸海域位于琼州海峡的B67点位浓度最高,浓度达到0.42 ng·g−1,位于海口市近岸海域的B66点位及位于东方市近岸海域的B70点位浓度也相对较高,这可能与点位近岸多家能源及石化公司有关.
检出率第二高的PFHxA分布也较为广泛,但与PFOA不同的是,PFHxA主要分布在防城港市(B51、B55、B34、B6点位)、钦州市(B63、B61、B64、B60点位)、北海市(B2、B27、B59点位)及海南省近岸海域(B65、B66、B67、B68、B69点位),这可能是因为PFHxA属于短链PFASs,在环境中的积累能力低于长链的原因. PFHxA主要源于纺织、造纸和皮革等行业[25],经研究,PFHxA与PFOA来源行业相似,且近岸分布区域类似,因此推测北部湾沿岸城市正在不同程度上使用短链PFASs取代长链PFASs的生产和使用.
另一需要关注的PFASs单体为PFOS,检出率低于PFOA及PFHxA,但检出浓度较高,主要分布在防城港市、钦州市及海口市近岸海域,PFOS的存在可归因于金属电镀行业的工业排放[26]. 浓度峰值出现在北部湾海域中部的B36点位,浓度达到1.50 ng·g−1,经研究,该区域位于季风洋流涡旋内部及航运必经线路,可能是造成此异常高值点的原因.
-
相关性分析在PFASs的来源解析中应用较广,已证明各种PFASs之间的相关性分析可用于推断PFASs的潜在来源,呈显著正相关且相关系数较大的污染物可能来自相似的源或运输路线[27]. 在本研究中表层沉积物的组分特征表明,PFOA是最主要的全氟化合物单体,具有高浓度及检出率,其次是PFHxA及PFOS. PFASs单体之间的Spearman相关性分析结果见图4,本研究沉积物中PFOA与其余大部分PFASs单体都有显著相关性,各单体相关性呈现PFNA和PFTrDA(R2=0.83,P≤0.01)>PFDA和PFUnDA(R2=0.4,P≤0.01)>PFDA和PFDoDA(R2=0.37,P≤0.01)=PFTeDA和PFTrDA(R2=0.37,P≤0.01)=PFOA和PFHxA(R2=0.37,P≤0.01)>PFOA和PFUnDA(R2=0.35,P≤0.01)>PFDA和PFHxS(R2=0.23,P≤0.01)=PFHxA和PFDA(R2=0.23,P≤0.01)>PFOA和PFDA(R2=0.2,0.01<P<0.05)=PFOA和PFOS(R2=0.2,0.01<P<0.05). 可见,PFHxA与PFOA、PFDA呈显著正相关,PFOA与PFHxA、PFUnDA呈显著正相关,与PFDA、PFOS相关性也相对较高,PFNA与PFTrDA相关性最高(R2=0.83,P≤0.01). 因此可以推断,这些全氟化合物单体可能共享相似的来源或运输路线.
PFASs中有不同来源的典型标志物,PFOA主要来源于纺织品阻燃、橡胶品乳化、食品包装过程和纸类表面处理,以及灭火剂使用等[24];PFNA和PFDA主要源于全氟羧酸生产过程的排放[24];PFHxS主要源于大气PFASs的降解[28];PFHxA主要源于纺织、造纸和皮革等行业[25];PFUnDA主要源于材料中间体的制备[29];PFOS的存在可归因于金属电镀行业的工业排放[26]. 本研究中,PFOA、PFHxA及PFOS为最主要的PFASs污染物(见表3). 经研究,北部湾周边主要能源产业结构见图3,污水经污水处理厂实行管道深海排放,排污区离岸3—6 km. 根据相关性分析结果,结合区域工业特点,因此可以推断,本研究区域内PFASs污染可能来源于纺织品业、造纸业、皮革加工行业及金属电镀行业等.
-
采用熵值法对最主要的污染单体PFOA、PFHxA及PFOS污染程度进行评价(见图5). 86%的采样点PFOA的RQ值高于0.01但小于0.1(低风险),没有高于0.1的点位. 23%的采样点PFHxA的RQ值高于0.01但小于0.1(低风险),31%的采样点PFHxA的RQ值高于0.1但小于1(中风险),主要分布于防城港市、钦州市、北海市及海南省近岸海域. 23%的采样点PFOS的RQ值高于0.1但小于1(中风险),主要分布于钦州市、北海市及海南省近岸海域,有一个采样点位(B36)的RQ值高于1(高风险),需要重点关注. 总体而言,除B36点位外,其他采样点的PFOA、PFHxA及PFOS的RQ值均低于1,表明目前的PFOA、PFHxA及PFOS的浓度水平可能对北部湾水生和低栖生物没有重大风险. 但这3种单体尤其是PFHxA及PFOS以较高风险熵值被广泛检测到,仍需持续关注这些传统PFASs的环境风险.
-
(1)北部湾海域沉积物样品中,∑PFASs浓度范围为ND.—1.50 ng·g−1,平均浓度为0.18 ng·g−1. 单体除PFHxS未检出外,其余单体均被检出,PFOA、PFHxA及PFOS为最主要的单体.
(2)北部湾海域沉积物中∑PFASs浓度及单体浓度都呈现出显著的空间差异性,总体来看呈现靠近城市高于远离城市,轮船运输沿线高于非运输线路. 检出率最高的单体PFOA在整个北部湾海域分布广泛,PFHxA与PFOA不同,主要分布在防城港市、钦州市、北海市及海南省近岸海域,因PFHxA与PFOA有相似的来源,且近岸分布区域类似,因此推测北部湾沿岸城市正在不同程度上使用短链PFASs取代长链PFASs的生产和使用. PFOS检出浓度较高,主要分布在防城港市、钦州市及海口市近岸海域.
(3)相关性分析得出,本研究沉积物中PFOA与其余大部分PFASs单体都有显著相关性,PFHxA与PFOA、PFDA呈显著正相关,PFOA与PFHxA、PFUnDA呈显著正相关,与PFDA、PFOS相关性也相对较高,PFNA与PFTrDA相关性最高(R2=0.83,P≤0.01). 因此可以推断,这些全氟化合物单体可能共享相似的来源或运输路线. 根据3种主要单体的主要来源推断本研究区域内PFASs污染可能来源于纺织品业、造纸业及皮革加工行业.
(4)采用熵值法对PFOA、PFHxA及PFOS的污染程度进行评价,在全部采样点中,PFOA和PFHxA的RQ值均低于1,表明目前的PFOA和PFHxA的浓度水平对南海水生和底栖生物处于中低风险. PFOS有一个点位高于1,处于高风险,需要特别关注. 但这3种PFASs单体在我国北部湾海域,尤其是PFOS及PFHxA以较高风险熵值被广泛检测到,仍需持续关注这些传统PFASs的环境风险.
全氟和多氟烷基物质在北部湾海域表层沉积物中的污染特征及风险评估
Pollution characteristics and risk assessment of perfluoroalkyl substances in surface sediments of the Beibu Gulf
-
摘要: 自21世纪以来,全氟和多氟烷基物质(per- and polyfluoroalkyl substances,PFASs)的环境问题一直受到科学界和公众的广泛关注. PFASs具有难降解、生物富集和长距离迁移等特点,已在大气、土壤和水体等环境介质及生物体中广泛检出. 本研究以北部湾海域70个表层沉积物样品为对象,对其中11种典型PFASs(PFHxA、PFHpA、PFOA、PFNA、PFDA、PFUnDA、PFDoDA、PFTrDA、PFTeDA、PFHxS、PFOS)进行了系统研究. 通过高效液相色谱-三重四极杆串联质谱法对该海域表层沉积物中PFASs污染水平进行分析,利用相关性分析对该海域表层沉积物中PFASs来源进行解析,并运用环境风险熵值法对该海域表层沉积物中PFASs污染进行了风险评估. 结果表明,北部湾海域70个点位中,除全氟己烷磺酸(PFHxS)未被检出外,其余10种PFASs均被检出,全氟己酸(perfluorohextanoic acid,PFHxA)、全氟辛酸(perfluorooctanoic acid,PFOA)及全氟辛烷磺酸(perfluorooctance sulfonic acid,PFOS)是浓度及检出率占绝对优势的单体;北部湾不同城市近岸海域沉积物中PFASs浓度具有明显差异. 相关性分析结果显示沉积物中的PFASs可能主要来自纺织业、造纸业、皮革加工行业及金属电镀行业等. 风险评估结果显示沉积物中PFHxA及PFOA浓度水平对北部湾水生和底栖生物处于中低风险,PFOS有一个点位处于高风险,需要持续关注.Abstract: The environmental problems of per- and polyfluoroalkyl substances (PFASs) have attracted extensive attention since the 21st century. PFASs have the characteristics of refractory, bioaccumulation and long-distance migration, and they have been widely detected in organisms and environmental media such as atmosphere, soil and water. In this study, 70 surface sediment samples were taken from the Beibu Gluf, and the pollution characteristics and potential ecological risks of 11 typical PFASs(PFHxA、PFHpA、PFOA、PFNA、PFDA、PFUnDA、PFDoDA、PFTrDA、PFTeDA、PFHxS、PFOS) were systematically studied. The pollution level of PFASs in the surface sediments of the Beibu Gulf was analyzed by high performance liquid chromatography-triple quadrupole tandem mass spectrometry, and the potential sources of PFASs of the Beibu Gulf were analyzed by correlation analysis. At the same time, the risk level of PFASs pollution in the surface sediments was evaluated by risk quotient method.The results showed that among 70 surface sediment samples, all 10 PFASs were detected except PFHxS. Perfluorohextanoic acid (PFHxA), perfluorooctanoic acid (PFOA) and perfluorooctance sulfonic acid (PFOS) had absolute advantages in concentration and detection rate. The concentration of PFASs in sediments from different city sea areas were significantly different. The results of correlation analysis showed that PFASs in sediments mainly came from textile industry, paper industry, leather processing industry, metal electroplating industry and so on. The results of risk assessment showed that the concentration levels of PFHxA and PFOA were at a lower risk to aquatic and benthic organisms in the Beibu Gulf, and the concentration of PFOS of one point was at a high risk, which need to be continuous attention.
-
全氟和多氟烷基物质(Per- and polyfluoroalkyl substances,PFASs)是一组人工合成的有机化合物,具有高稳定性、表面活性、疏水疏油性、长距离迁移性和难降解性等独特的理化性质,被广泛应用于电子、食品、化工、家具、农药和医疗等众多领域[1]. 在生产和使用过程中,PFASs可通过多种途径进入到环境中. 研究表明,PFASs在全球环境中无处不在,在大气[2]、水体[3]、沉积物[3]及生物体内[4]均有检出. PFASs中的C—F键具有很强的极性,是自然界中键能最大的共价键之一(键能约460 kJ·mol−1 )[5],其优良的物理化学性质使其在环境介质中难以被物理、化学及生物作用降解[6]. 近年来对环境中PFASs污染及其潜在生态风险研究成为学术界和环境管理领域重点关注的问题. 2009年5月,联合国环境规划署正式将PFOS及其盐类等9种物质列为新型持久性有机污染物并纳入斯德哥尔摩公约[7]. 2014年,我国环境保护部印发了关于“全氟辛基磺酸及其盐类”等10种持久性有机污染物禁止生产、流通、使用和进出口公告.
目前有关PFASs的研究多集中于湖泊、河流和海洋,PFASs在这些水体中广泛存在. C—F链末端的亲水基团使PFOS和PFOA具有良好的水溶性(溶解度570 mg·L−1和3400 mg·L−1)[5],而海洋沉积物易与水体发生相互作用,并能为有机污染物提供疏水介质,因此海洋沉积物被认为是PFASs重要的“汇”之一. 北部湾位于中国南部,东起广东雷州半岛和海南岛,西与越南相邻,北至广西壮族自治区沿岸. 深度范围为10 m至60 m,平均深度38 m,海域总面积约为12.8万平方公里. 北部湾开发是国家一带一路重要战略,目前,以石化、浆纸、能源、粮油加工、冶金等产业主,一批相关产业正在加速进入北部湾地区,临海大工业产生的产业聚集效应正在逐步形成,对北部湾生态环境造成了越来越大的压力.
本研究针对北部湾海域共70个调查站点,采集海洋沉积物,测定其中11种PFASs(PFHxA、PFHpA、PFOA、PFNA、PFDA、PFUnDA、PFDoDA、PFTrDA、PFTeDA、PFHxS、PFOS)的含量,通过分析11种PFASs单体浓度水平和分布特征进行初步的生态风险评估,以期了解该地区全氟化合物的污染状况,为实施海洋环境监管与污染防控措施提供支撑.
1. 材料与方法(Materials and methods)
1.1 样品采集
海洋沉积物主要依托当地环境监测部门协助采样获得. 共采集2020年8月的70个表层沉积物样品,采样点位分布见图1. 采用抓斗式采泥器采集沉积物,取上层0—20 cm未受搅动的表层沉积物作为待测样品,放入250 mL棕色试剂瓶中. 沉积物样品的贮存、运输按照《海洋监测规范》(GB 17378-2007)等标准要求执行.
1.2 试剂与材料
1.2.1 标准品及试剂
沉积物中PFASs的含量分析按照实验室自建方法《海洋沉积物 全氟化合物的测定 液相色谱-串联质谱法》进行检测[8]. 目标化合物包括2种全氟磺酸类物质(PFSAs)及9种全氟羧酸类物质(PFCAs). 11种PFASs单体的类别、名称、CAS号及分子量见表1.
表 1 本研究中PFASs的类别、名称、CAS号及分子量Table 1. Category, name, CAS number and molecular weight of PFASs in this study类别Category 中文名称Chinese name 英文名称English name 缩写Abbreviation CAS 分子量/(g·mol−1)Molecular weight 内标物Internal standard 全氟烷基羧酸类 全氟己酸 Perfluorohextanoic acid PFHxA 307-24-4 314.0 MPFOA 全氟庚酸 Perfluoroheptanoic acid PFHpA 375-85-9 364.1 MPFOA 全氟辛酸 Perfluorooctanoic acid PFOA 335-67-1 414.0 MPFOA 全氟壬酸 Perfluorononanoic acid PFNA 375-95-1 464.0 MPFOA 全氟癸酸 Perfluorodecanoic acid PFDA 335-76-2 514.0 MPFDA 全氟十一烷酸 Perfluoroundecanoic acid PFUnDA 2058-94-8 564.1 MPFDA 全氟十二烷酸 Perfluorododecanoic acid PFDoDA 307-55-1 614.0 MPFDA 全氟十三烷酸 Perfluorotridecanoic acid PFTrDA 72629-94-8 664.1 MPFDA 全氟十四烷酸 Perfluorotetradecanoic acid PFTeDA 376-06-7 714.0 MPFDA 全氟烷基磺酸类 全氟己烷磺酸 Perfluorohexane sulfonic acid PFHxS 355-46-4 399.9 MPFOS 全氟辛烷磺酸 Perfluorooctance sulfonic acid PFOS 1763-23-1 499.9 MPFOS | Show Table DownLoad:
CSV
DownLoad:
CSV
标准样品:全氟己酸(PFHxA)、全氟庚酸(PFHpA)、全氟辛酸(PFOA)、全氟壬酸(PFNA)、全氟癸酸(PFDA)、全氟十一烷酸(PFUnDA)、全氟十二烷酸(PFDoDA)、全氟十三烷酸(PFTrDA)、全氟十四烷酸(PFTeDA)、全氟己烷磺酸(PFHxS)、全氟辛烷磺酸(PFOS)及同位素标记化合物13C4-全氟辛酸(MPFOA)、13C4-全氟辛烷磺酸(MPFOS)和13C2-全氟正癸酸(MPFDA)均加购于加拿大Wellington公司,纯度≥98%. 有机试剂甲醇(色谱纯、Honeywell)、乙腈(色谱纯、Honeywell);无机试剂:冰醋酸(优级纯),氨水(优级纯,25% ),醋酸铵(色谱纯)购于广州化学试剂厂. Oasis WAX固相萃取小柱(Agela,6 mL/500 mg),购于美国.
1.2.2 仪器
高效液相色谱-三重四极杆串联质谱仪(Agilent 1260/ABSIEX 4000Q): 液相系统,Agilent 1260 Infinity 高效液相色谱(美国);质谱系统,AB SCIEX 4000 Qtrap(美国);ETEL4 MG-2200氮吹仪(日本);24位固相萃取装置(美国、visiprep-24TMPL);超声清洗机SCQ-1000C(中国上海)等.
1.3 样品前处理
将沉积物样品冷冻干燥,研磨并过0.300 mm孔径筛,取1.00 g于聚丙烯(PP)离心管中,加入内标(MPFOA、MPFOS、MPFDA各2.0 ng),加入5.0 mL甲醇溶液,涡旋1 min,静置30 min后,在30 ℃水浴下进行超声提取,再用5.0 mL甲醇萃取2次,每次萃取后,在4000 r·min−1转速下离心10 min,取上清液,将上清液合并后,旋蒸至 1 mL左右,溶入50.0 mL超纯水中,以每秒1—2 滴过WAX固相萃取柱萃取,WAX小柱预先用4.0 mL含0.5%氨的甲醇溶液、4.0 mL甲醇和4.0 mL超纯水活化,上样完成后的小柱抽真空干燥,干燥后依次用4.0 mL 0.5%氨的甲醇溶液、4.0 mL甲醇洗脱,洗脱液收集于10 mL试管中,氮吹至1 mL,用0.22 μm滤膜过滤后转移至1.5 mL进样瓶中,待上机检测.
所有样品均由高效液相色谱-串联质谱仪进行测定;用5.0 mmol·L−1乙酸铵水溶液作为流动相A,用乙腈为流动相B,流速为0.5 mL·min−1,进样体积为 10μL,柱温为40 ℃.
1.4 质量保证与控制
实验过程和分析过程中严格控制和避免使用可能会影响实验结果的材料和试剂. 所有实验装置和容器在使用前均用甲醇进行处理. 为保证数据的可靠性,分析过程中设置运输空白、实验室空白、样品平行和样品加标等操作. 每20个样品做1个实验空白,测定结果中目标物浓度均低于方法检出限. 每20个样品设置1个平行样,平行双样的相对偏差均≤30%. 每20个样品设置了1个实际样品基体加标,基体加标样品回收率范围控制在50%—150%. PFASs单体的检出限LOD、定量下限LOQ及回收率见表2. LOD、LOQ的计算方法为:对标准曲线最低点重复测定7次,计算其标准偏差,将其3.143倍(修约为1位有效数字)作为仪器检出限. 取仪器检出限的5倍作为测试样品添加浓度,分别添加至7个石英砂样品中,按照实验步骤进行样品的前处理,上机测定. 计算7个石英砂样品测定浓度的标准偏差,并取其3.143倍作为方法检出限,再取方法检出限的4倍作为定量下限.
表 2 海洋沉积物全氟化合物方法检出限Table 2. Detection limit of PFASs in marine sediment化合物Compound 添加量/ngAddition amount 检测平均值/(ng·g−1)AVE 标准偏差/(ng·g−1)SD 方法检出限/(ng·g−1)LOD 定量下限/(ng·g−1)LOQ 回收率/%Rate of recovery PFHxA 0.05 0.051 0.001 0.004 0.017 85.3 PFHpA 0.05 0.044 0.002 0.006 0.022 88.9 PFOA 0.05 0.042 0.002 0.006 0.025 98.7 PFNA 0.05 0.051 0.004 0.012 0.046 105.0 PFDA 0.05 0.051 0.007 0.022 0.086 106.3 PFUnDA 0.05 0.056 0.005 0.016 0.062 116.8 PFDoDA 0.05 0.055 0.010 0.032 0.127 119.3 PFTrDA 0.05 0.077 0.003 0.011 0.042 117.3 PFTeDA 0.05 0.074 0.005 0.015 0.061 97.2 PFHxS 0.05 0.042 0.005 0.014 0.057 118.2 PFOS 0.05 0.052 0.015 0.048 0.191 117.3 | Show TableDownLoad:
CSV
1.5 风险评价方法
本研究基于风险熵(risk quotient, RQ)对PFOA、PFHxA及PFOS3种检出率及浓度较高的典型PFASs进行初步风险评估. RQ为本研究检出环境样品浓度(measured environment concentration, MEC)与预测的无效应浓度(predicted-no-effect concentration, PNEC)的比值. 风险评估分为四个等级:“较小风险”(RQ<0.01);“低风险”(0.01<RQ<0.1);“中等风险”(0.1<RQ<1);“高风险”(RQ>1). 计算公式如下:
stringUtils.convertMath(!{formula.content}) 由于PFASs在海洋沉积物中关于PNEC值的研究较少,且PNEC值在不同基质中差异很大,因此运用平衡分配法计算沉积物中PNEC值,参考欧盟化学物质风险评价技术指导文件(TGD)[9]中标准环境特征参数获得沉积物中PFOA、PFHxA及PFOS的PNEC值,分别为8.6、1、0.49 ng·g−1.
2. 结果与讨论(Results and discussion)
2.1 北部湾海域沉积物中PFASs浓度水平及组成特征
采集的70个沉积物样品∑PFASs浓度范围为ND.—1.50 ng·g−1,平均浓度为0.18 ng·g−1,PFASs检出率为100%. 在11种目标PFASs单体中,除PFHxS未检出外,其余单体均有检出(见表3). 6种单体的检出率(detection frequency, DF)低于10%,PFOA和PFHxA的DF高于50%. PFOA为最主要的单体,DF远远高于其他单体,为85.7%,平均浓度为0.19 ng·g−1,与其他海域沉积物中PFOA的浓度相比处于中等偏低水平(见表4). 其次是PFHxA和PFOS,PFHxA的DF较高,呈现PFHxA(DF为55.7%)>PFOS(DF为24.3%),但PFOS检出样品平均浓度较高,呈现PFOS(平均浓度为0.29 ng·g−1)>PFHxA(平均浓度为0.16 ng·g−1). 总体而言,北部湾的PFASs浓度水平与之前全球研究相比处于相对较低水平(表4),这可能与北部湾地区经济发展水平较低相关,先前的研究表明PFASs的污染与经济发展和行业类型呈正相关[10].
表 3 北部湾海域沉积物中PFASs的浓度(ng·g−1)和检出率(%)Table 3. Concentration (ng·g−1) and detection rate (%) of PFASs in coastal sediments of the Beibu Gulf化合物Compounds 均值Average 最小值Minimum 中位数Median 最大值Maximum 检出样品个数Number of samples detected 检出率/%Detection rate 短链Short-chain PFASs PFHxA 0.16 ND. 0.12 0.84 39 55.7 PFHpA 0.05 ND. 0.04 0.12 6 8.6 长链Long-chain PFASs PFOA 0.19 ND. 0.17 0.70 60 85.7 PFNA ND. ND. ND. 0.04 1 1.4 PFDA 0.05 ND. 0.06 0.08 7 10.0 PFUnDA 0.14 ND. 0.15 0.23 4 5.7 PFDoDA ND. ND. ND. 0.06 1 1.4 PFTrDA 0.05 ND. 0.05 0.06 2 2.9 PFTeDA 0.20 ND. 0.25 0.26 3 4.3 PFHxS ND. ND. ND. ND. 0 0 PFOS 0.29 ND. 0.20 1.50 17 24.3 总计Total PFASs Short-chain PFCs 0.14 ND. 0.11 0.84 39 55.7 Long-chain PFCs 0.19 ND. 0.17 1.50 62 88.6 ∑PFCs 0.18 ND. 0.14 1.50 70 100 ND.,未检出. ND.,not detected. | Show TableDownLoad:
CSV
由于近几年短链PFHxA鲜有研究,本研究对比了长链PFOA及PFOS在世界范围内的浓度水平及变化情况. 与世界其他地区相比,北部湾沉积物中PFOA含量(ND.—0.70 ng·g−1,平均值0.19 ng·g−1)低于中国渤海PFOA浓度(0.06—2.70 ng·g−1,平均值0.54 ng·g−1),但和中国东海PFOA浓度相当(ND.—0.87 ng·g−1,平均值0.18 ng·g−1)相当,与2019年北部湾PFOA浓度(0.04—0.25 ng·g−1,平均值0.11 ng·g−1)相比,浓度略有升高. 在北部湾沉积物中PFOS浓度(ND.—1.50 ng·g−1,平均值0.29 ng·g−1)明显高于中国东海PFOS浓度(ND.—0.89 ng·g−1,平均值0.10 ng·g−1),与2019年北部湾PFOS浓度(0.04—0.20 ng·g−1,平均值0.10 ng·g−1)相比,有明显升高趋势. 总体而言,北部湾海域沉积物中PFOA与PFOS浓度与世界其他地区相比处于相对较低水平,但随着经济快速发展,近年来有明显升高趋势.
调查分析的11种PFASs单体依碳链长度分为短链(C<7)、长链(C≥7)2类. 结果表明,沉积物中长链PFASs的浓度水平高于短链PFASs,事实上,先前的研究也检测出类似的结论[1,11-12],与短链PFASs相比,长链PFASs更容易附着在沉积物上[13]. 长链中PFOA占主导地位,在长链检出样品中DF为96.8%;短链中PFHxA占主导地位,在短链检出样品中DF为100%.
表 4 不同地区海洋沉积物中PFOA及PFOS的浓度Table 4. Concentration of PFOA and PFOS in coastal sediments from different sea area基质Matrix 地点Location PFOA/(ng·g−1) PFOS/(ng·g−1) 采样年份Sampling year 参考文献Reference Range Mean Range Mean 沉积物Sediment Overseas France ND. — ND.—22 1.7 2012 [14] China South Sea ND.—0.017 — ND.-0.026 — 2017—2018 [15] Coastal areas of Bohai 0.436—18.9 2.69 0.68—5.5 1.37 2017 [16] East Chine Sea ND.—0.87 0.18 ND.—0.89 0.10 2012 [17] Chinese Bohai Sea 0.20—1.00 0.46 ND.—0.20 — 2020 [18] Chinese Yellow Sea 0.091—1.826 — 0.007—0.227 — 2018 [19] Charieston, SC 0.02—2.52 0.42 0.09—7.37 1.52 2011 [20] German Baltic Sea 0.067—0.39 0.13 ND.—0.38 0.17 2017 [21] Sea of Beibu Gulf 0.04—0.25 0.11 0.04—0.20 0.10 2019 [22] Sea of Beibu Gulf ND.—0.70 0.19 ND.—1.50 0.29 2020 本研究 ND.,未检出. ND.,not detected. | Show TableDownLoad:
CSV
2.2 PFASs在北部湾海域沉积物中的空间分布
北部湾海域沉积物中∑PFASs浓度及单体浓度都呈现出显著的空间差异性,总体来看呈现靠近城市高于远离城市,轮船运输沿线高于非运输线路(见图2). 其中近钦州的B49、B63、B61及B52点位∑PFASs污染程度较高(均值为0.62 ng·g−1),说明此研究区域内存在点源污染排放,如钦州工厂林立(见图3),受纳工业废水和生活污水,大幅度提高了此研究区域的污染水平,随江流汇入北部湾之后,浓度逐渐降低. 海南近岸海域的B66、B67、B68、B69、B65及B70点位,尤其是靠近琼州海峡的B67点位浓度较高,可能与船舶运输污染及工业废水和生活污水相关. 另一浓度较高的区域为B6、B51及B56位于中越交界近岸海域的点位,由于此研究区域中国境内暂无大型工业企业,可能与越南城市污染相关. 浓度峰值位于北部湾中部的B36点位,平均浓度达到1.72 ng·g−1,经复核样品采集及处理分析过程,均未发现异常,排除实验因素导致的异常高值,经研究,北部湾是我国大西南重要的海上通道,由防城港、钦州港和北海港3座港口组成的北部湾港,经由琼州海峡运往东南亚及南亚各国,这可能与航运线上的船舶污染有关,先前的研究报道了PFASs可以从船舶上的油漆和润滑脂向周围水域扩散[23]. 同时,北部湾季风洋流经琼州海峡由东向西进入湾内形成涡旋,此高值点位于涡旋内部,造成污染物汇聚沉积,可能也是造成此高值点的原因.
PFASs各单体浓度空间分布见图2. 检出率最高的单体PFOA在整个北部湾海域分布广泛,PFOA主要来源于纺织品阻燃、橡胶品乳化、食品包装过程和纸类表面处理,以及灭火剂使用等[24],除与越南交界海域的几处点位浓度较高以外,浓度较高区域主要集中在钦州入海河口的B41、B53、B47及B60点位,河流沿岸多家大型汽修、港船厂及能源公司(见图3),可能与工厂生产排放有关;北海市近岸海域PFOA多处浓度较高,浓度较高的B37定位及B12点位均位于北海市重要港口附近,B8点位附近有多家大型造纸厂(见图3),来自防城港市、钦州市及北海市河流汇聚入海,呈现北部湾中部海域(B35、B9、B23、B32、B36、B13等多点位)PFOA含量中等偏高趋势,需要特别关注;海南省近岸海域位于琼州海峡的B67点位浓度最高,浓度达到0.42 ng·g−1,位于海口市近岸海域的B66点位及位于东方市近岸海域的B70点位浓度也相对较高,这可能与点位近岸多家能源及石化公司有关.
检出率第二高的PFHxA分布也较为广泛,但与PFOA不同的是,PFHxA主要分布在防城港市(B51、B55、B34、B6点位)、钦州市(B63、B61、B64、B60点位)、北海市(B2、B27、B59点位)及海南省近岸海域(B65、B66、B67、B68、B69点位),这可能是因为PFHxA属于短链PFASs,在环境中的积累能力低于长链的原因. PFHxA主要源于纺织、造纸和皮革等行业[25],经研究,PFHxA与PFOA来源行业相似,且近岸分布区域类似,因此推测北部湾沿岸城市正在不同程度上使用短链PFASs取代长链PFASs的生产和使用.
另一需要关注的PFASs单体为PFOS,检出率低于PFOA及PFHxA,但检出浓度较高,主要分布在防城港市、钦州市及海口市近岸海域,PFOS的存在可归因于金属电镀行业的工业排放[26]. 浓度峰值出现在北部湾海域中部的B36点位,浓度达到1.50 ng·g−1,经研究,该区域位于季风洋流涡旋内部及航运必经线路,可能是造成此异常高值点的原因.
2.3 北部湾海域沉积物中PFASs的来源解析
相关性分析在PFASs的来源解析中应用较广,已证明各种PFASs之间的相关性分析可用于推断PFASs的潜在来源,呈显著正相关且相关系数较大的污染物可能来自相似的源或运输路线[27]. 在本研究中表层沉积物的组分特征表明,PFOA是最主要的全氟化合物单体,具有高浓度及检出率,其次是PFHxA及PFOS. PFASs单体之间的Spearman相关性分析结果见图4,本研究沉积物中PFOA与其余大部分PFASs单体都有显著相关性,各单体相关性呈现PFNA和PFTrDA(R2=0.83,P≤0.01)>PFDA和PFUnDA(R2=0.4,P≤0.01)>PFDA和PFDoDA(R2=0.37,P≤0.01)=PFTeDA和PFTrDA(R2=0.37,P≤0.01)=PFOA和PFHxA(R2=0.37,P≤0.01)>PFOA和PFUnDA(R2=0.35,P≤0.01)>PFDA和PFHxS(R2=0.23,P≤0.01)=PFHxA和PFDA(R2=0.23,P≤0.01)>PFOA和PFDA(R2=0.2,0.01<P<0.05)=PFOA和PFOS(R2=0.2,0.01<P<0.05). 可见,PFHxA与PFOA、PFDA呈显著正相关,PFOA与PFHxA、PFUnDA呈显著正相关,与PFDA、PFOS相关性也相对较高,PFNA与PFTrDA相关性最高(R2=0.83,P≤0.01). 因此可以推断,这些全氟化合物单体可能共享相似的来源或运输路线.
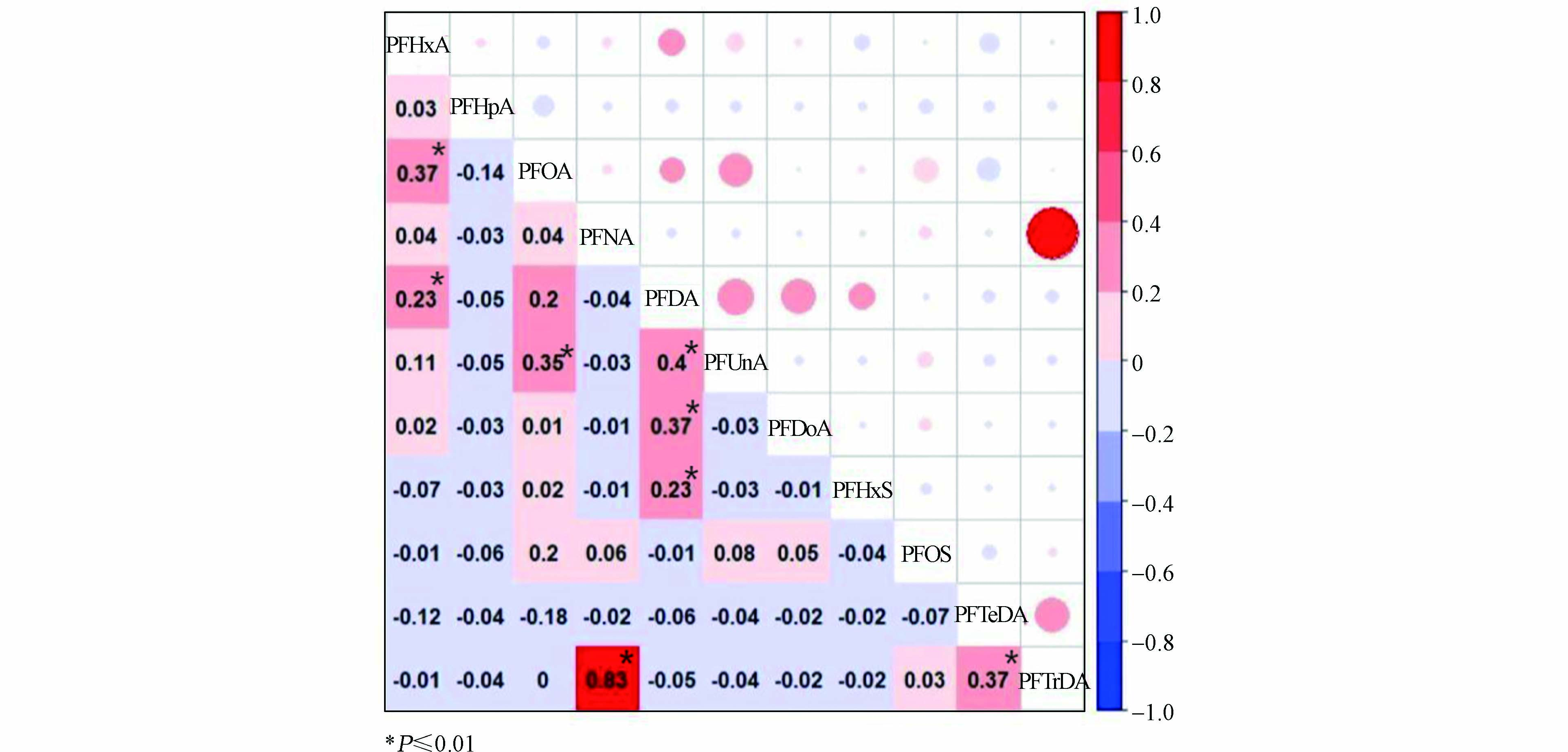 图 4 各PFASs单体之间相关性分析Figure 4. Correlation analysis between PFASs monomers*表述在 0.01 级别,相关性显著
图 4 各PFASs单体之间相关性分析Figure 4. Correlation analysis between PFASs monomers*表述在 0.01 级别,相关性显著PFASs中有不同来源的典型标志物,PFOA主要来源于纺织品阻燃、橡胶品乳化、食品包装过程和纸类表面处理,以及灭火剂使用等[24];PFNA和PFDA主要源于全氟羧酸生产过程的排放[24];PFHxS主要源于大气PFASs的降解[28];PFHxA主要源于纺织、造纸和皮革等行业[25];PFUnDA主要源于材料中间体的制备[29];PFOS的存在可归因于金属电镀行业的工业排放[26]. 本研究中,PFOA、PFHxA及PFOS为最主要的PFASs污染物(见表3). 经研究,北部湾周边主要能源产业结构见图3,污水经污水处理厂实行管道深海排放,排污区离岸3—6 km. 根据相关性分析结果,结合区域工业特点,因此可以推断,本研究区域内PFASs污染可能来源于纺织品业、造纸业、皮革加工行业及金属电镀行业等.
2.4 北部湾海域沉积物中PFASs生态风险评价
采用熵值法对最主要的污染单体PFOA、PFHxA及PFOS污染程度进行评价(见图5). 86%的采样点PFOA的RQ值高于0.01但小于0.1(低风险),没有高于0.1的点位. 23%的采样点PFHxA的RQ值高于0.01但小于0.1(低风险),31%的采样点PFHxA的RQ值高于0.1但小于1(中风险),主要分布于防城港市、钦州市、北海市及海南省近岸海域. 23%的采样点PFOS的RQ值高于0.1但小于1(中风险),主要分布于钦州市、北海市及海南省近岸海域,有一个采样点位(B36)的RQ值高于1(高风险),需要重点关注. 总体而言,除B36点位外,其他采样点的PFOA、PFHxA及PFOS的RQ值均低于1,表明目前的PFOA、PFHxA及PFOS的浓度水平可能对北部湾水生和低栖生物没有重大风险. 但这3种单体尤其是PFHxA及PFOS以较高风险熵值被广泛检测到,仍需持续关注这些传统PFASs的环境风险.
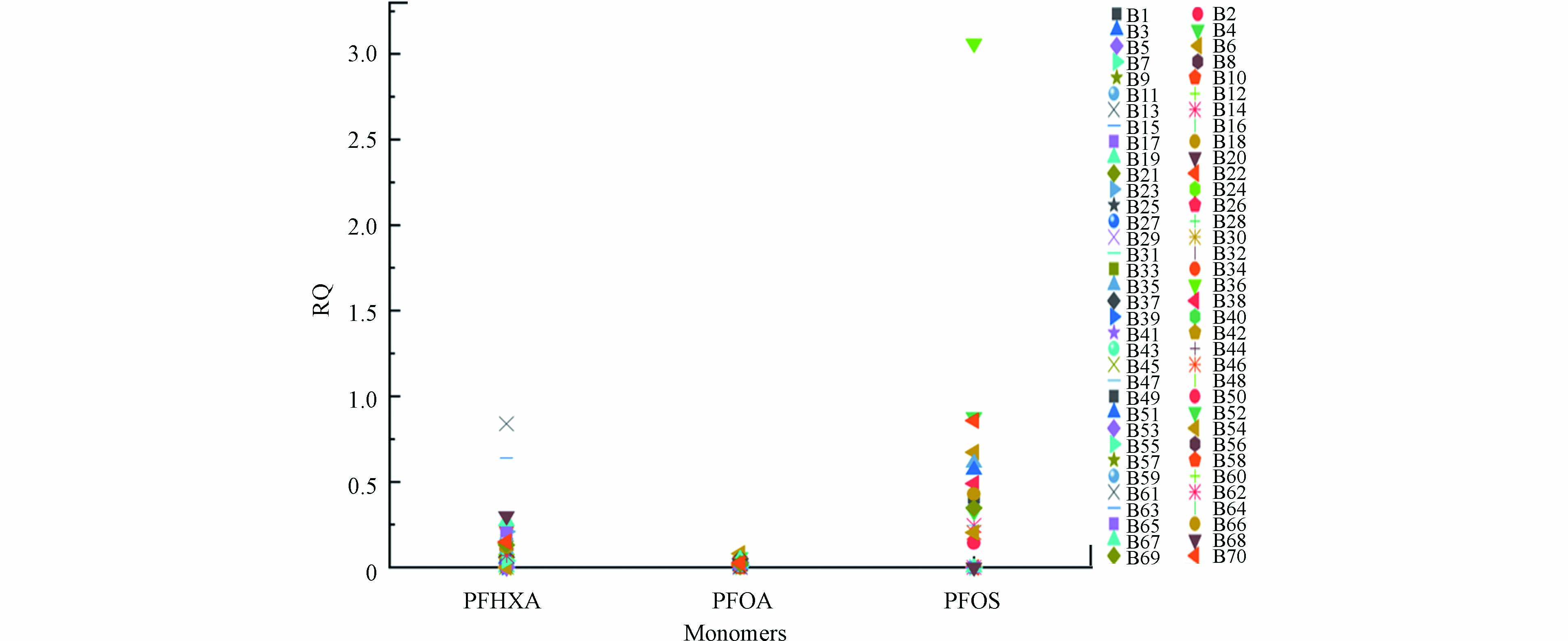 图 5 北部湾海域沉积物中PFOA、PFHxA及PFOS的风险熵(RQ)Figure 5. Risk Entropy (RQ) of PFOA 、PFHxA and PFOS in coastal sediments of the Beibu Gulf
图 5 北部湾海域沉积物中PFOA、PFHxA及PFOS的风险熵(RQ)Figure 5. Risk Entropy (RQ) of PFOA 、PFHxA and PFOS in coastal sediments of the Beibu Gulf3. 结论 (Conclusion)
(1)北部湾海域沉积物样品中,∑PFASs浓度范围为ND.—1.50 ng·g−1,平均浓度为0.18 ng·g−1. 单体除PFHxS未检出外,其余单体均被检出,PFOA、PFHxA及PFOS为最主要的单体.
(2)北部湾海域沉积物中∑PFASs浓度及单体浓度都呈现出显著的空间差异性,总体来看呈现靠近城市高于远离城市,轮船运输沿线高于非运输线路. 检出率最高的单体PFOA在整个北部湾海域分布广泛,PFHxA与PFOA不同,主要分布在防城港市、钦州市、北海市及海南省近岸海域,因PFHxA与PFOA有相似的来源,且近岸分布区域类似,因此推测北部湾沿岸城市正在不同程度上使用短链PFASs取代长链PFASs的生产和使用. PFOS检出浓度较高,主要分布在防城港市、钦州市及海口市近岸海域.
(3)相关性分析得出,本研究沉积物中PFOA与其余大部分PFASs单体都有显著相关性,PFHxA与PFOA、PFDA呈显著正相关,PFOA与PFHxA、PFUnDA呈显著正相关,与PFDA、PFOS相关性也相对较高,PFNA与PFTrDA相关性最高(R2=0.83,P≤0.01). 因此可以推断,这些全氟化合物单体可能共享相似的来源或运输路线. 根据3种主要单体的主要来源推断本研究区域内PFASs污染可能来源于纺织品业、造纸业及皮革加工行业.
(4)采用熵值法对PFOA、PFHxA及PFOS的污染程度进行评价,在全部采样点中,PFOA和PFHxA的RQ值均低于1,表明目前的PFOA和PFHxA的浓度水平对南海水生和底栖生物处于中低风险. PFOS有一个点位高于1,处于高风险,需要特别关注. 但这3种PFASs单体在我国北部湾海域,尤其是PFOS及PFHxA以较高风险熵值被广泛检测到,仍需持续关注这些传统PFASs的环境风险.
-
图 5 北部湾海域沉积物中PFOA、PFHxA及PFOS的风险熵(RQ)
Figure 5. Risk Entropy (RQ) of PFOA 、PFHxA and PFOS in coastal sediments of the Beibu Gulf
表 1 本研究中PFASs的类别、名称、CAS号及分子量
Table 1. Category, name, CAS number and molecular weight of PFASs in this study
类别Category 中文名称Chinese name 英文名称English name 缩写Abbreviation CAS 分子量/(g·mol−1)Molecular weight 内标物Internal standard 全氟烷基羧酸类 全氟己酸 Perfluorohextanoic acid PFHxA 307-24-4 314.0 MPFOA 全氟庚酸 Perfluoroheptanoic acid PFHpA 375-85-9 364.1 MPFOA 全氟辛酸 Perfluorooctanoic acid PFOA 335-67-1 414.0 MPFOA 全氟壬酸 Perfluorononanoic acid PFNA 375-95-1 464.0 MPFOA 全氟癸酸 Perfluorodecanoic acid PFDA 335-76-2 514.0 MPFDA 全氟十一烷酸 Perfluoroundecanoic acid PFUnDA 2058-94-8 564.1 MPFDA 全氟十二烷酸 Perfluorododecanoic acid PFDoDA 307-55-1 614.0 MPFDA 全氟十三烷酸 Perfluorotridecanoic acid PFTrDA 72629-94-8 664.1 MPFDA 全氟十四烷酸 Perfluorotetradecanoic acid PFTeDA 376-06-7 714.0 MPFDA 全氟烷基磺酸类 全氟己烷磺酸 Perfluorohexane sulfonic acid PFHxS 355-46-4 399.9 MPFOS 全氟辛烷磺酸 Perfluorooctance sulfonic acid PFOS 1763-23-1 499.9 MPFOS
下载: 导出CSV
表 2 海洋沉积物全氟化合物方法检出限
Table 2. Detection limit of PFASs in marine sediment
化合物Compound 添加量/ngAddition amount 检测平均值/(ng·g−1)AVE 标准偏差/(ng·g−1)SD 方法检出限/(ng·g−1)LOD 定量下限/(ng·g−1)LOQ 回收率/%Rate of recovery PFHxA 0.05 0.051 0.001 0.004 0.017 85.3 PFHpA 0.05 0.044 0.002 0.006 0.022 88.9 PFOA 0.05 0.042 0.002 0.006 0.025 98.7 PFNA 0.05 0.051 0.004 0.012 0.046 105.0 PFDA 0.05 0.051 0.007 0.022 0.086 106.3 PFUnDA 0.05 0.056 0.005 0.016 0.062 116.8 PFDoDA 0.05 0.055 0.010 0.032 0.127 119.3 PFTrDA 0.05 0.077 0.003 0.011 0.042 117.3 PFTeDA 0.05 0.074 0.005 0.015 0.061 97.2 PFHxS 0.05 0.042 0.005 0.014 0.057 118.2 PFOS 0.05 0.052 0.015 0.048 0.191 117.3
下载: 导出CSV
表 3 北部湾海域沉积物中PFASs的浓度(ng·g−1)和检出率(%)
Table 3. Concentration (ng·g−1) and detection rate (%) of PFASs in coastal sediments of the Beibu Gulf
化合物Compounds 均值Average 最小值Minimum 中位数Median 最大值Maximum 检出样品个数Number of samples detected 检出率/%Detection rate 短链Short-chain PFASs PFHxA 0.16 ND. 0.12 0.84 39 55.7 PFHpA 0.05 ND. 0.04 0.12 6 8.6 长链Long-chain PFASs PFOA 0.19 ND. 0.17 0.70 60 85.7 PFNA ND. ND. ND. 0.04 1 1.4 PFDA 0.05 ND. 0.06 0.08 7 10.0 PFUnDA 0.14 ND. 0.15 0.23 4 5.7 PFDoDA ND. ND. ND. 0.06 1 1.4 PFTrDA 0.05 ND. 0.05 0.06 2 2.9 PFTeDA 0.20 ND. 0.25 0.26 3 4.3 PFHxS ND. ND. ND. ND. 0 0 PFOS 0.29 ND. 0.20 1.50 17 24.3 总计Total PFASs Short-chain PFCs 0.14 ND. 0.11 0.84 39 55.7 Long-chain PFCs 0.19 ND. 0.17 1.50 62 88.6 ∑PFCs 0.18 ND. 0.14 1.50 70 100 ND.,未检出. ND.,not detected.
下载: 导出CSV
表 4 不同地区海洋沉积物中PFOA及PFOS的浓度
Table 4. Concentration of PFOA and PFOS in coastal sediments from different sea area
基质Matrix 地点Location PFOA/(ng·g−1) PFOS/(ng·g−1) 采样年份Sampling year 参考文献Reference Range Mean Range Mean 沉积物Sediment Overseas France ND. — ND.—22 1.7 2012 [14] China South Sea ND.—0.017 — ND.-0.026 — 2017—2018 [15] Coastal areas of Bohai 0.436—18.9 2.69 0.68—5.5 1.37 2017 [16] East Chine Sea ND.—0.87 0.18 ND.—0.89 0.10 2012 [17] Chinese Bohai Sea 0.20—1.00 0.46 ND.—0.20 — 2020 [18] Chinese Yellow Sea 0.091—1.826 — 0.007—0.227 — 2018 [19] Charieston, SC 0.02—2.52 0.42 0.09—7.37 1.52 2011 [20] German Baltic Sea 0.067—0.39 0.13 ND.—0.38 0.17 2017 [21] Sea of Beibu Gulf 0.04—0.25 0.11 0.04—0.20 0.10 2019 [22] Sea of Beibu Gulf ND.—0.70 0.19 ND.—1.50 0.29 2020 本研究 ND.,未检出. ND.,not detected.
下载: 导出CSV
-
[1] BUCK R C, FRANKLIN J, BERGER U, et al. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins [J]. Integrated Environmental Assessment and Management, 2011, 7(4): 513-541. doi: 10.1002/ieam.258 [2] HAN D M, MA Y G, HUANG C, et al. Occurrence and source apportionment of perfluoroalkyl acids (PFAAs) in the atmosphere in China [J]. Atmospheric Chemistry and Physics, 2019, 19(22): 14107-14117. doi: 10.5194/acp-19-14107-2019 [3] 崔文杰, 彭吉星, 谭志军, 等. 全氟烷基物质在胶州湾海水、沉积物及生物中污染特征 [J]. 环境科学, 2019, 40(9): 3990-3999. doi: 10.13227/j.hjkx.201901104 CUI W J, PENG J X, TAN Z J, et al. Pollution characteristics of perfluorinated alkyl substances (PFASs) in seawater, sediments, and biological samples from Jiaozhou Bay, China [J]. Environmental Science, 2019, 40(9): 3990-3999(in Chinese). doi: 10.13227/j.hjkx.201901104
[4] JOUANNEAU W, BÅRDSEN B J, HERZKE D, et al. Spatiotemporal analysis of perfluoroalkyl substances in white-tailed eagle (Haliaeetus albicilla) nestlings from northern Norway-a ten-year study [J]. Environmental Science & Technology, 2020, 54(8): 5011-5020. [5] LAU C, ANITOLE K, HODES C, et al. Perfluoroalkyl acids: A review of monitoring and toxicological findings [J]. Toxicological Sciences, 2007, 99(2): 366-394. doi: 10.1093/toxsci/kfm128 [6] TANIYASU S, KANNAN K, SO M K, et al. Analysis of fluorotelomer alcohols, fluorotelomer acids, and short- and long-chain perfluorinated acids in water and biota [J]. Journal of Chromatography A, 2005, 1093(1/2): 89-97. [7] WANG Z Y, BOUCHER J M, SCHERINGER M, et al. Toward a comprehensive global emission inventory of C4-C10 perfluoroalkanesulfonic acids (PFSAs) and related precursors: Focus on the life cycle of C8-based products and ongoing industrial transition [J]. Environmental Science & Technology, 2017, 51(8): 4482-4493. [8] 刘庆, 贺德春, 许振成, 等. 液相色谱-串联质谱法测定水体、沉积物及土壤中全氟化合物 [J]. 中国环境监测, 2014, 30(4): 134-139. doi: 10.3969/j.issn.1002-6002.2014.04.023 LIU Q, HE D C, XU Z C, et al. Analysis of polyfluoroalkyl chemicals in water, sediments and soil by liquid chromatography-tandem mass spectrometry [J]. Environmental Monitoring in China, 2014, 30(4): 134-139(in Chinese). doi: 10.3969/j.issn.1002-6002.2014.04.023
[9] LEEUWEN K V. Technical guidance document on risk assessment in support of commission directive 93/67/EEC on risk assessment for new notified substances and commission regulation (EC) No1488/94 on risk assessment for existing substances Part II[S]. European Chemicals Bureau (ECB), 2003(4): 204-218. [10] KWOK K Y, WANG X H, YA M L, et al. Occurrence and distribution of conventional and new classes of per- and polyfluoroalkyl substances (PFASs) in the South China Sea [J]. Journal of Hazardous Materials, 2015, 285: 389-397. doi: 10.1016/j.jhazmat.2014.10.065 [11] BRENDEL S, FETTER É, STAUDE C, et al. Short-chain perfluoroalkyl acids: Environmental concerns and a regulatory strategy under REACH [J]. Environmental Sciences Europe, 2018, 30(1): 9. doi: 10.1186/s12302-018-0134-4 [12] URTIAGA A, SORIANO A, CARRILLO-ABAD J. BDD anodic treatment of 6: 2 fluorotelomer sulfonate (6: 2 FTSA). Evaluation of operating variables and by-product formation [J]. Chemosphere, 2018, 201: 571-577. doi: 10.1016/j.chemosphere.2018.03.027 [13] LEE Y M, LEE J Y, KIM M K, et al. Concentration and distribution of per- and polyfluoroalkyl substances (PFAS) in the Asan Lake area of South Korea [J]. Journal of Hazardous Materials, 2020, 381: 120909. doi: 10.1016/j.jhazmat.2019.120909 [14] MUNOZ G, LABADIE P, BOTTA F, et al. Occurrence survey and spatial distribution of perfluoroalkyl and polyfluoroalkyl surfactants in groundwater, surface water, and sediments from tropical environments [J]. Science of the Total Environment, 2017, 607/608: 243-252. doi: 10.1016/j.scitotenv.2017.06.146 [15] WANG Q, TSUI M M P, RUAN Y F, et al. Occurrence and distribution of per- and polyfluoroalkyl substances (PFASs) in the seawater and sediment of the South China sea coastal region [J]. Chemosphere, 2019, 231: 468-477. doi: 10.1016/j.chemosphere.2019.05.162 [16] LIU Y Q, ZHANG Y, LI J F, et al. Distribution, partitioning behavior and positive matrix factorization-based source analysis of legacy and emerging polyfluorinated alkyl substances in the dissolved phase, surface sediment and suspended particulate matter around coastal areas of Bohai Bay, China [J]. Environmental Pollution, 2019, 246: 34-44. doi: 10.1016/j.envpol.2018.11.113 [17] GAO Y, FU J J, ZENG L X, et al. Occurrence and fate of perfluoroalkyl substances in marine sediments from the Chinese Bohai Sea, Yellow Sea, and East China Sea [J]. Environmental Pollution, 2014, 194: 60-68. doi: 10.1016/j.envpol.2014.07.018 [18] ZHAO Z, CHENG X H, HUA X, et al. Emerging and legacy per- and polyfluoroalkyl substances in water, sediment, and air of the Bohai Sea and its surrounding rivers [J]. Environmental Pollution, 2020, 263: 114391. doi: 10.1016/j.envpol.2020.114391 [19] FENG X M, YE M Q, LI Y, et al. Potential sources and sediment-pore water partitioning behaviors of emerging per/polyfluoroalkyl substances in the South Yellow Sea [J]. Journal of Hazardous Materials, 2020, 389(10): 122-124. [20] WHITE N D, BALTHIS L, KANNAN K, et al. Elevated levels of perfluoroalkyl substances in estuarine sediments of Charleston, SC [J]. Science of the Total Environment, 2015, 521/522: 79-89. doi: 10.1016/j.scitotenv.2015.03.078 [21] JOERSS H, APEL C, EBINGHAUS R. Emerging per- and polyfluoroalkyl substances (PFASs) in surface water and sediment of the North and Baltic Seas [J]. The Science of the Total Environment, 2019, 686: 360-369. doi: 10.1016/j.scitotenv.2019.05.363 [22] 肖少可. 传统和替代全氟化合物在北部湾的空间分布、生物富集及营养级迁移[D]. 南宁: 广西大学, 2021. XIAO S K. Spatial distribution, bioaccumulation and trophic transfer of legacy and alternative per-and polyfluoroalkyl substances in the beibu gulf[D]. Nanning: Guangxi University, 2021(in Chinese).
[23] HABIBULLAH-AL-MAMUN M, AHMED M K, RAKNUZZAMAN M, et al. Occurrence and distribution of perfluoroalkyl acids (PFAAs) in surface water and sediment of a tropical coastal area (Bay of Bengal coast, Bangladesh) [J]. Science of the Total Environment, 2016, 571: 1089-1104. doi: 10.1016/j.scitotenv.2016.07.104 [24] AHRENS L, BARBER J L, XIE Z Y, et al. Longitudinal and latitudinal distribution of perfluoroalkyl compounds in the surface water of the Atlantic Ocean [J]. Environmental Science & Technology, 2009, 43(9): 3122-3127. [25] LI J, del VENTO S, SCHUSTER J, et al. Perfluorinated compounds in the Asian atmosphere [J]. Environmental Science & Technology, 2011, 45(17): 7241-7248. [26] XIAO F, HALBACH T R, SIMCIK M F, et al. Input characterization of perfluoroalkyl substances in wastewater treatment plants: Source discrimination by exploratory data analysis [J]. Water Research, 2012, 46(9): 3101-3109. doi: 10.1016/j.watres.2012.03.027 [27] XIE S W, WANG T Y, LIU S J, et al. Industrial source identification and emission estimation of perfluorooctane sulfonate in China [J]. Environment International, 2013, 52: 1-8. doi: 10.1016/j.envint.2012.11.004 [28] 张鸿, 赵亮, 何龙, 等. 不同功能区表层土中全氟化合物污染指纹及其来源解析 [J]. 环境科学, 2014, 35(7): 2698-2704. doi: 10.13227/j.hjkx.2014.07.037 ZHANG H, ZHAO L, HE L, et al. Pollution fingerprints and sources of perfluorinated compounds in surface soil of different functional areas [J]. Environmental Science, 2014, 35(7): 2698-2704(in Chinese). doi: 10.13227/j.hjkx.2014.07.037
[29] 何宗健, 甘甜, 彭希珑, 等. 环鄱阳湖城市污水处理厂污泥中全氟化合物的污染特征 [J]. 南昌大学学报(工科版), 2020, 42(2): 103-108. doi: 10.13764/j.cnki.ncdg.2020.02.001 HE Z J, GAN T, PENG X L, et al. Pollution characteristics of perfluorinated compounds in sludge from urban wastewater treatment plants around Poyang Lake [J]. Journal of Nanchang University (Engineering & Technology), 2020, 42(2): 103-108(in Chinese). doi: 10.13764/j.cnki.ncdg.2020.02.001
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 4672
- HTML全文浏览数: 4672
- PDF下载数: 156
- 施引文献: 0
