










-
汞 (Hg) 是环境中生物毒性很强的金属污染物,具有持久性、易迁移性和高度的生物富集性等特点[1]。汞可通过地球化学循环和食物链富集,给人类和生态环境造成极大危害[2]。目前,燃煤电厂排放的汞是最大的人为汞排放源。作为以煤为主的能源消费大国,我国汞污染较为严重[3-5],每年煤炭燃烧向大气中排放810 t汞,占各种人为源汞排放总量的35%[6]。2013年,《关于汞的水俣公约》规定了汞的长期减排控制措施。我国现行的《火电厂大气污染物排放标准》也对汞的排放控制提出了明确限值[7],即自2015年起全面执行火电厂汞排放质量浓度不超过30 μg·m−3的规定。煤炭燃烧产生的汞有单质汞 (Hg0) 、氧化汞 (Hg2+) 和颗粒汞 (Hgp) 3种形态。Hg2+易溶于水,故可通过湿法脱硫设备去除[8];HgP易吸附在尘粒、飞灰颗粒表面,可通过除尘装置捕获去除。然而,Hg0因具有较高的挥发性 (2.46×10−1 Pa,25 ℃) 和较低的水溶性 (6×10−5 g·L−1,25 ℃) ,极易在大气中通过长距离运输而造成全球性汞污染,为最难控制的汞形态[9]。因此,有效控制Hg0是实现汞污染减排的关键。
催化脱除Hg0是一种行之有效的方法。传统选择性催化还原 (selective catalytic reduction,SCR) 催化剂 (V2O5-MoO3/TiO2和V2O5-WO3/TiO2) 在催化还原NOx的同时,可将Hg0氧化为Hg2+,并进一步利用后续脱硫装置进行协同脱除,从而提高设备经济性,因此被认为是应用前景良好的控制技术[10-11]。然而,SCR脱硝系统通常布置在高温、高尘、高酸性气体环境中,会降低催化剂的使用寿命[12]。由于其较高的工作温度,因而适用于非电行业的中低温催化氧化技术受到关注。目前,中低温钒钛SCR催化剂催化氧化Hg0,易受烟气组分 (如O2、NO、NH3、HCl、SO2、H2O) 和温度影响[13-16]。烟气中O2和NO可提供活性氧物种,从而促进Hg0的氧化。NH3会与Hg0竞争吸附催化剂活性位点,进而抑制Hg0氧化[17-20]。然而,烟气中SO2对Hg0氧化的影响机理还存在较大争议。有研究表明,SO2对Hg0的氧化表现为促进作用[21-22]。一方面,在O2存在的情况下,低浓度SO2会氧化生成SO3,与Hg0反应生成HgSO4;另一方面,SO2吸附在催化剂表面生成硫酸盐,可为Hg0氧化提供活性中心。然而,在某些情况下,SO2会与催化剂表面晶格氧反应生成硫酸盐和亚硫酸盐,使得催化剂表面活性氧位点减少,从而抑制Hg0氧化[23-26]。除此之外,多烟气组分共存时,Hg0的脱除机理尚不明确。
现有研究中,Cu作为活性组分,具有良好的氧化还原性[27]和抗硫性[28],添加到催化剂表面可极大地提升Hg0氧化效率。本研究以低温V2O5-MO3/TiO2脱硝催化剂为基础配方、Cu2O为改性组分,采用浸渍法制备Cu2O-V2O5-MoO3/TiO2催化剂,通过固定床反应器考察O2、NO、NH3、HCl、SO2、H2O等烟气组分对Hg0氧化性能的影响;并在此基础上,进一步探讨多烟气组分共存条件下Hg0的脱除机理,以期为SCR脱硝催化剂协同汞氧化提供参考。
-
所用催化剂由质量分数为3%的V2O5、质量分数为6% MoO3、Cu2O (质量分数为0~10%) 和TiO2组成。催化剂的制备步骤:称取定量偏钒酸铵、草酸、磷酸铵、钼酸铵、氧化亚铜和钛白粉溶于50 mL去离子水中,恒温水浴搅拌2 h,所得浆液置于105 ℃烘箱中3 h烘干水分。随后,将样品放入马弗炉中,在250 ℃空气气氛下焙烧1 h,之后在490 ℃下焙烧3 h,得到Cu2O负载量分别为0、1%、2%、6%、10%的Cu2O-V2O5-MoO3/TiO2催化剂,将其分别标记为0CuVMT、1CuVMT、2CuVMT、6CuVMT、10CuVMT。所有样品过60~80目 (0.180~0.250 mm) 筛备用。
-
使用美国Micromeritics公司生产的ASAP2020低氮吸附仪测定催化剂比表面积、孔容和孔径。其中,比表面积通过Brunauer-Emmett-Teller (BET)方法计算获得,孔容和孔径采用Barret-Joyner-Halenda (BJH)方法计算获得。
使用美国赛默飞世尔生产的Thermo Scientific K-Alpha型X射线光电子能谱仪进行X射线光电子能谱(XPS)分析,射线光源采用单色化AlKa源 (Mono AlKa,能量为1486.6eV) 。
H2程序升温还原(H2-TPR)实验在AutoChem II 2920型化学吸附仪(Micrometritics Co.)上进行。实验步骤如下:取50 mg样品(40~60目),在纯氧气氛下400 ℃预处理30 min;降至室温后用He吹扫15 min;然后通入体积分数为10% H2/Ar混合气,待仪器基线平稳后,以10 ℃·min−1速率升温至600 ℃,采用TCD检测耗氢量。
-
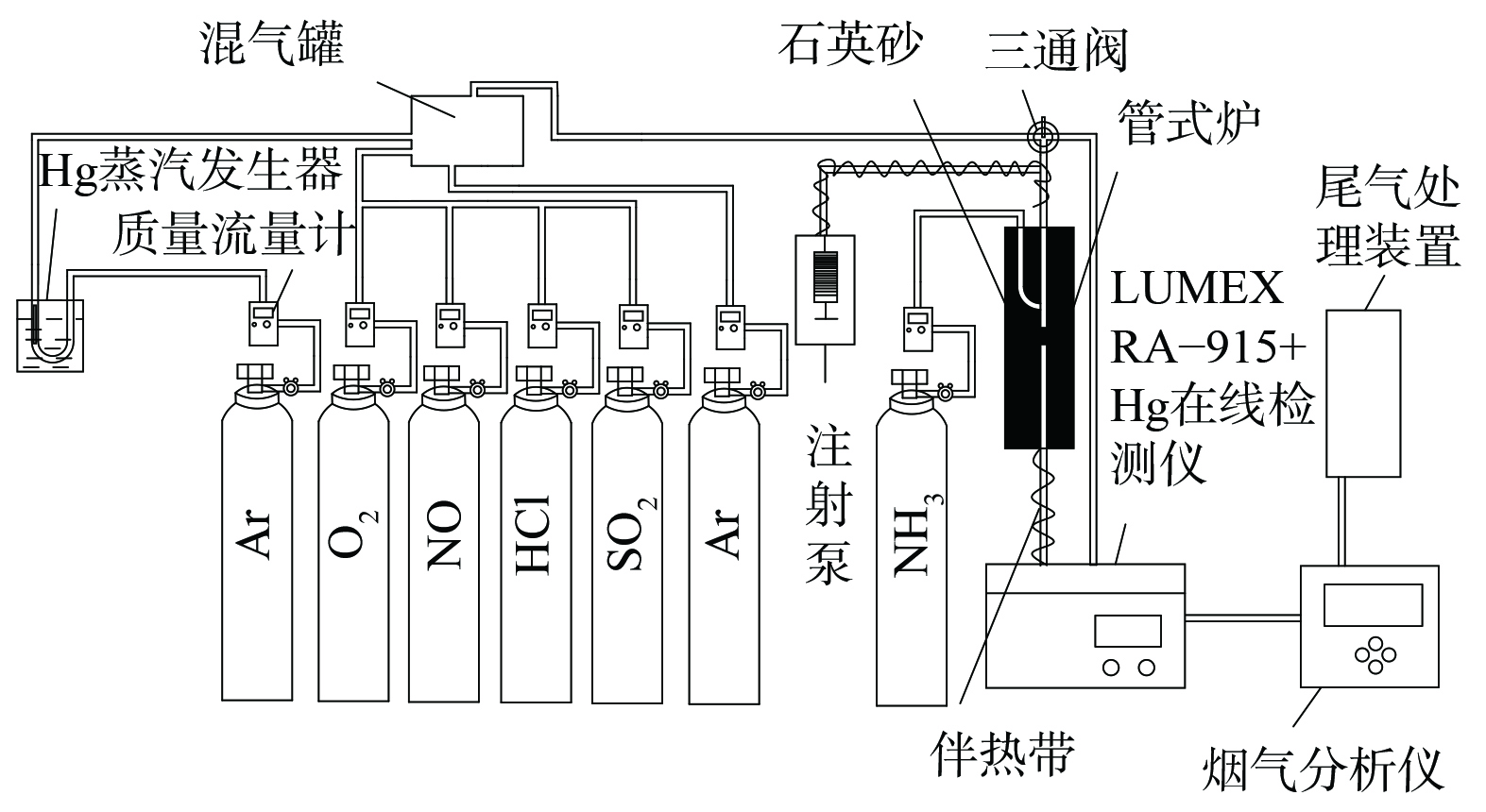
催化剂性能评价装置主要由模拟烟气、固定床反应装置、汞检测系统和尾气处理4部分组成 (见图1) 。其中,模拟烟气包括Hg0、O2、NO、NH3、HCl、SO2及平衡气Ar,烟气总流量为1 L·min−1。气体流量采用质量流量计精确控制。汞蒸气由置于恒温水浴锅中的汞渗透管产生,经载气Ar带出。固定床反应器使用内径4 mm石英管,中层添加石英砂作为支撑,采用管式电炉控制催化剂层反应温度。进出口Hg0浓度使用测汞仪 (RA-915+、LUMEX、美国) 进行监测,进出口NO和N2O体积分数由质谱仪 (DECRA、Hiden Analytical Ltd.,英国) 进行监测,SO2体积分数采用Testo 350烟气分析仪(Testo Co., Germany)检测。实验尾气经净化装置处理后排空。为避免汞蒸气沉积于管壁及水蒸气冷凝,实验管路均使用聚四氟乙烯管连接,并用伴热带加热至120 ℃。
活性评价实验过程:称取50 mg催化剂置于石英管内,用石英棉固定两端;使用Ar吹扫管路,待基线稳定后,将模拟烟气切换至旁路,检测反应器进口Hg0的初始浓度;15 min后将模拟烟气切换至反应器,检测出口Hg0浓度。Hg0氧化率(EHg)和NO转化率(ENO)计算方法见式 (1)~(2) 。
式中:[Hg0]in和[Hg0]out分别为固定床反应器进口和出口Hg质量浓度,μg·m−3;[NO]in和[NO]out分别表示固定床反应器进口和出口NO的体积分数,%。
-
不同Cu2O负载量催化剂的NO转化率和Hg0氧化率见图2。由于实际工业烟气中Hg0质量浓度较低,SCR净化装置空速为3 000~8 000 h−1。为缩短Hg0和NO在模拟烟气中达到反应平衡的时间,本研究将脱硝实验的空速设置为30 000 h−1,汞氧化实验的空速设置为1 600 000 h−1。图2表明,在200 ℃时,不同Cu2O负载量的CuVMT催化剂的NO转化率表现为:0CuVMT(94.1%)>1CuVMT(91.2%)>2CuVMT(90.9%)>6CuVMT(88.5%)>10CuVMT(76.7%),而Hg0氧化率表现为:0CuVMT(64.1%)<1CuVMT(96.1%)<2CuVMT(99.9%)~10CuVMT(99.9%)。与0CuVMT和1CuVMT催化剂相比,2CuVMT催化剂的NO转化率分别降低了3.4%和0.32%,而N2O生成量没有明显提升,与此同时,Hg0氧化率分别提升了35.8%和3.8%。当Cu2O负载量超过2%之后,NO转化率继续降低,N2选择性也出现不同程度下降,而Hg0氧化率保持不变。因此,综合催化剂的NO转化率、Hg0氧化率、N2选择性及制备成本等几方面因素,最终选定2CuVMT催化剂作为脱硝协同汞氧化的最优配方。
-
工业烟气组分复杂,与Hg0相比,各烟气组分在气体浓度及分子偶极等方面占据优势,会优先与催化材料活性位点发生键合,进而严重影响Hg0的氧化。因此,有必要深入研究不同烟气组分对2CuVMT催化剂脱除Hg0性能的影响,进而阐明复杂组分下Hg0的脱除机理,所有反应时长均为10 h。
-
在200 ℃下,O2体积分数对2CuVMT催化剂Hg0氧化率的影响见图3。在无氧条件下,2CuVMT催化剂的Hg0氧化率仅为11.3%。此时,Hg0处于氩气惰性气氛下,物理吸附态的Hg0会与催化剂表面晶格氧结合[14,29],生成HgO。但由于晶格氧含量有限,Hg0氧化性能不高。当向反应体系通入2.5%的O2后,催化剂对Hg0的氧化性能显著提升,达到91.5%。这是由于O2可再生催化剂表面吸附氧和晶格氧,生成新的含氧活性位点,从而促进Hg0的氧化[30]。随着O2体积分数继续增至12%,Hg0氧化率也逐渐增至99.9%。
-
NO体积分数对2CuVMT催化剂Hg0氧化率的影响见图4。当0.002 5% NO通入反应体系后,Hg0氧化率立即升至99.9%;当NO体积分数增至0.05%时,Hg0氧化率依然稳定在99.9%。这可能是由于吸附态NO与催化剂表面活性氧反应生成具有氧化性的NO+、NO2一、NO2和NO3一等活性中间体,可将Hg0氧化为Hg(NO3)2[14,31],进而促进了Hg0氧化。当反应体系中继续添加5% O2后,O2可补充NO消耗掉的催化剂表面晶格氧[32],故Hg0的氧化率依然保持在99.9%。
-
NH3体积分数对2CuVMT催化剂Hg0氧化率的影响见图5。当烟气中加入0.005% NH3后,Hg0氧化率基本降至0。继续增加NH3体积分数至0.05%时,Hg0氧化率依然为0。这表明NH3对Hg0的氧化有强烈抑制作用。NH3会与Hg0发生强烈的竞争吸附[33],消耗催化剂表面晶格氧,从而抑制Hg0在催化剂表面发生氧化反应。反应过程如式 (3) 所示。
为探究SCR气氛条件对Hg0氧化率的影响,在Ar+NH3的气氛下,考察向烟气中添加体积分数为5%的O2,或添加混合气体 (含体积分数为5%的O2+体积分数为0.05%的NO) 对催化剂Hg0氧化率的影响。当体积分数为5%的O2加入烟气中时,2CuVMT催化剂Hg0氧化率显著提升。这说明O2补充了NH3消耗的催化剂晶格氧,并部分抵消了NH3对Hg0氧化的负面影响[34]。
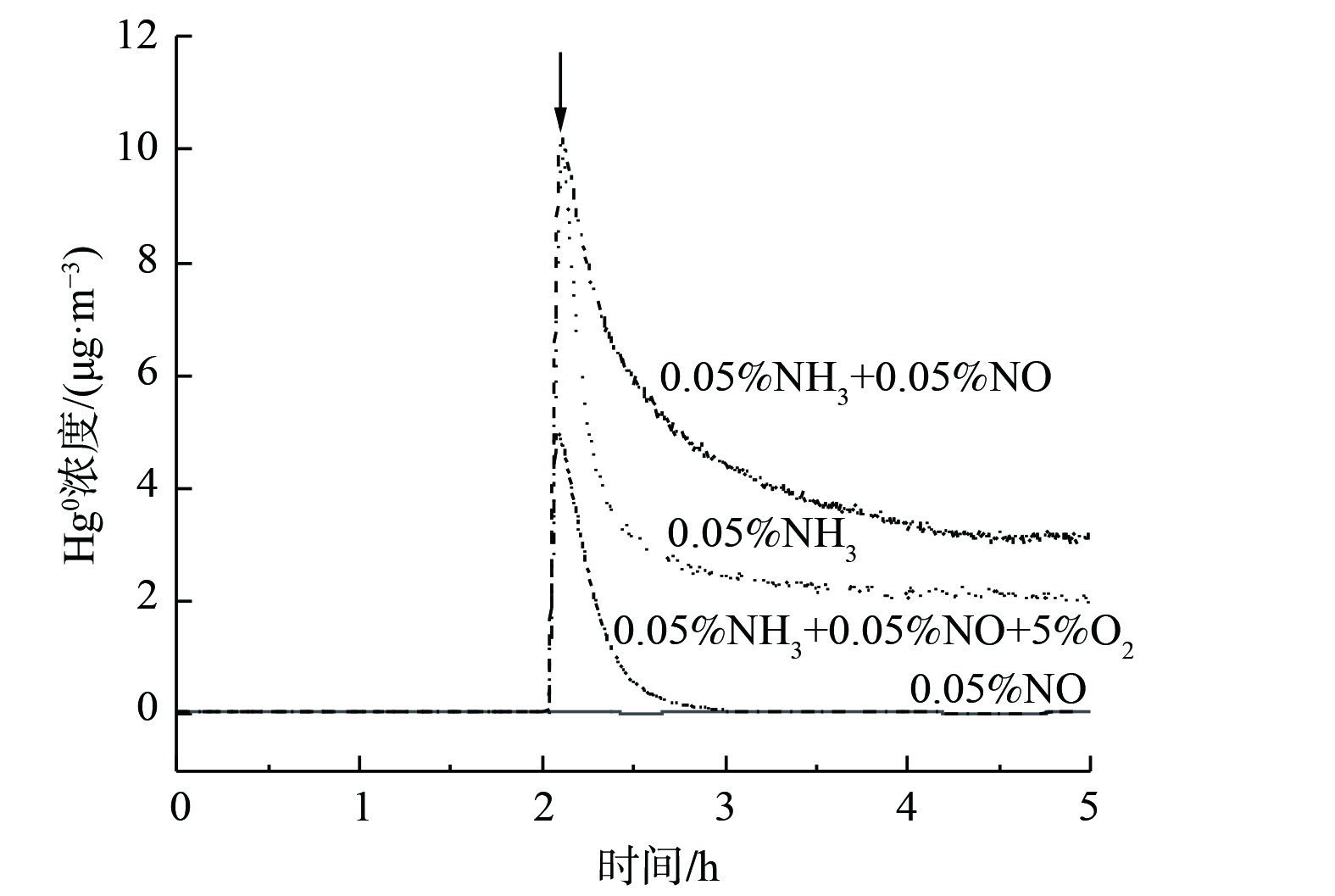
当NO、NH3和O2中的2种或3种组分以不同体积分数加入烟气时,各种添加方式下Hg0的氧化率表现为: (0.05% NO+5% O2) > (0.05% NH3+5% O2) > (0.05% NO+0.05% NH3+5% O2) > (0.05% NO+0.05% NH3) 。当烟气中通入混合气体 (0.05% NH3+0.05% NO) 时,Hg0氧化率为0。这表明此时NH3的抑制作用占主导地位,继续向烟气中加入5% O2后,Hg0氧化率升至57.3%。对比 (NH3+O2) 和 (NO+NH3+O2) 2种混合气体,反应体系中通入NO后,Hg0氧化率反而下降。这表明在此气氛下,发生了Hg2+被还原为Hg0的副反应。为探究副反应的发生机制,在200 ℃下通入混合气体 (10% O2+105 μg·m−3 Hg0+Ar) 3 h,使得催化剂表面沉积HgOx。之后用Ar吹扫,当Hg0质量浓度降为0后 (图6中箭头处) ,分别在烟气中通入0.05% NO、0.05% NH3、 (0.05% NH3+0.05% NO) 和 (0.05% NH3+0.05% NO+5% O2) 。这表明NH3和 (NO+NH3) 对HgOx存在还原作用,并且NO+NH3的还原性更强,而加入O2可在一定程度上抑制HgOx还原反应。因此,烟气中通入混合气体 (0.05% NH3+0.05% NO+5% O2) 后,仅有少量HgOx被还原。反应过程如式 (4)~(5) 所示。其中,x为1或1/2。
-
HCl是影响Hg0氧化反应的重要因素[35]。HCl的体积分数对2CuVMT催化剂Hg0氧化率的影响见图7。当体积分数为0.000 1%的HCl添加到烟气中时,Hg0氧化率即可达到99.9%。随着HCl体积分数逐渐增至0.001%时,Hg0氧化率稳定在99.9%。这表明HCl对Hg0氧化有明显促进作用。这可能是由于HCl在催化剂表面形成化学吸附态的活性Cl物种,可直接将Hg0氧化生成HgCl2[36]。
-
SO2和H2O对2CuVMT催化剂Hg0氧化率的影响见图8。当烟气中添加0.001% SO2时,Hg0氧化率可达到37.5%。随着SO2体积分数增至0.01%时,Hg0氧化率提升至52.1%。这表明SO2对Hg0氧化有一定促进作用。这可能是由于SO2和Hg0在催化剂表面发生竞争吸附的同时,在晶格氧的作用下,SO2和O* (晶格氧) 反应生成SO3和硫酸根,进而与吸附态Hg0反应生成HgSO3或HgSO4[23-24],从而有利于Hg0的去除。当烟气中同时存在SO2和O2时,Hg0氧化率明显下降,此时,SO2和O2反应生成SO3,加快了与催化剂活性组分生成CuSO4,使得催化剂硫酸化,最终导致催化剂活性位点数量减少、Hg0氧化率下降[27-28]。在实际SCR工况下,体系还会含有一定量水分。因此,考察在烟气中 (SCR条件:Ar+5% O2+0.05% NO+0.05% NH3+0.001%HCl) 加入H2O和SO2后对2CuVMT催化剂Hg0氧化率的影响。当烟气中加入体积分数为5%的H2O后,Hg0的氧化效率由99.9%降为97.3%。这是由于H2O和Hg0会竞争吸附催化剂表面活性位点,导致氧化率下降。继续向烟气中添加0.05% SO2后,催化剂Hg0的氧化效率降至95.1%。
-
反应温度是影响催化剂活性的重要因素。不同温度 (150~350 ℃) 对2CuVMT催化剂Hg0氧化率的影响见图9。随着反应温度的升高,2CuVMT催化剂Hg0氧化率呈现先平稳后降低的趋势。在150~250 ℃烟温范围内,Hg0氧化率保持在99.9%。然而,当温度升至300 ℃时,Hg0氧化率开始下降;在350 ℃时,Hg0氧化率进一步降至64.1%。参考文献[37]的研究结果,低温条件有利于Hg0吸附在催化剂表面参与氧化反应,而高温 (≥300 ℃) 则不利于Hg0在催化剂表面吸附,会使得参与氧化反应的Hg0质量浓度降低,从而导致Hg0的氧化被抑制。
-
1) BET分析。0CuVMT和2CuVMT催化剂的比表面积、孔容和介孔平均孔径见表1。在负载Cu2O后,催化剂的比表面积和孔容均呈下降趋势。其中,比表面积由0CuVMT的68.4 m2·g−1降至2CuVMT的57.3 m2·g−1,孔容由0.36 cm3·g−1 (0CuVMT) 略微降至0.33 cm3·g−1 (2CuVMT)。这表明添加Cu2O会堵塞催化剂部分孔道,导致催化剂比表面积和孔容降低。由于Cu2O堵塞了催化剂微孔,催化剂介孔平均孔径由0CuVMT催化剂的19 nm增至2CuVMT催化剂的21.3 nm。XU等[38]的研究结果表明,CuO/TiO2催化剂的比表面积、孔容、孔径与催化剂活性没有明显相关关系。
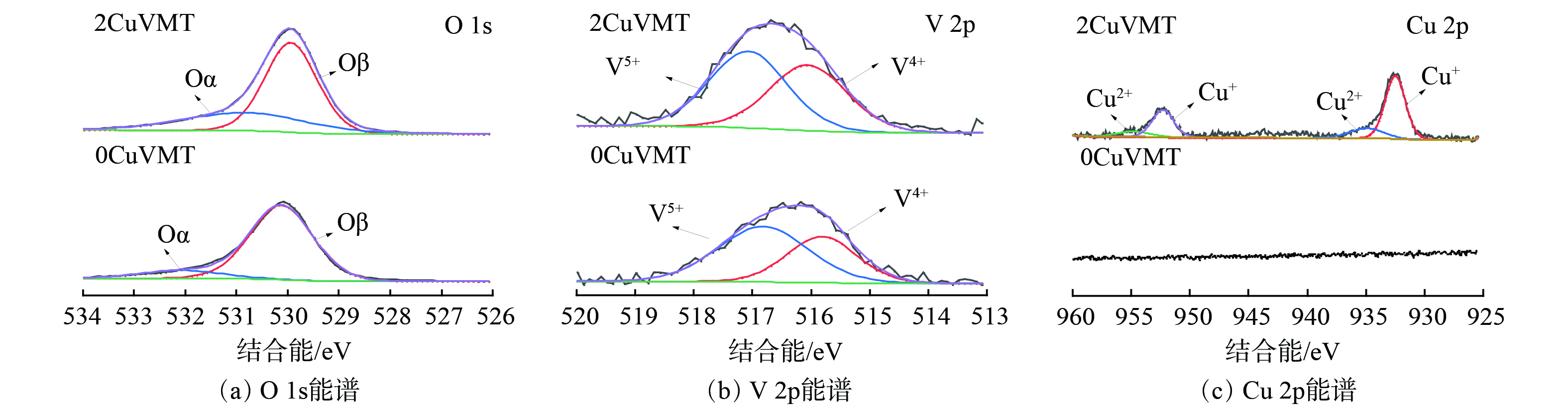
2) XPS表征。为确定催化剂表面O、Cu和V元素的化学价态,对CuVMT催化剂进行了XPS光谱分析,测定结果见图10。不同催化剂反应前后样品的O 1s XPS光谱 (图10 (a) ) 显示出2种特征峰。其中,结合能为530~530.3 eV的特征峰为晶格氧 (Oβ) [39],而在532~532.3 eV的特征峰归属于化学吸附氧 (Oα) [40]。当负载2%Cu2O后,催化剂表面的化学吸附氧 (Oα) 占比 (Oα/ (Oα+Oβ) ) 由31.9%逐渐升至36.5%。这表明化学吸附氧对于Hg0的氧化具有更高的活性[41]。
CuVMT催化剂样品的V 2p XPS光谱 (图10 (b) ) 可分为516.4 eV和517.3 eV两个峰,分别对应V4+和V5+的特征峰[42-43]。随着Cu负载量的增加,催化剂中V5+/V4+的比例由155% (0CuVMT) 降至117% (2CuVMT) 。这说明Cu2O改性提高了催化剂表面V4+的含量。这可能是由于Cu2O将部分V5+还原为了V4+。
CuVMT催化剂反应前后样品的Cu2p XPS光谱 (图10 (c) ) 表明,催化剂表面Cu的主要价态为Cu+和Cu2+。其中,Cu+为Cu元素主要的存在形态,其对应的结合能为932.5eV和952.2eV,而结合能在935.7eV和954.6eV的特征峰归属于Cu2+[44]。结合V 2p XPS结果,Cu2O改性使得催化剂表面V4+增加。这说明在制备过程中催化剂表面的Cu+和V5+确实存在相互作用,发生反应V4++Cu2+↔V5++Cu+,使得催化剂表面产生不饱和化学键和氧空位[41],可有效增加催化剂表面化学吸附氧含量,与O 1s XPS结果相一致。
3) H2-TPR表征。为阐明Cu改性对催化剂氧化还原性能的影响,对CuVMT催化剂进行了H2-TPR表征分析,测定结果见图11。0CuVMT催化剂在400~600 ℃出现了V5+和V4+的还原峰[45-46]。添加体积分数为2%的Cu2O改性后,催化剂原先V物种的还原峰大幅向低温方向偏移,且在154~263 ℃和300~400 ℃出现新的还原峰。其中,154~263 ℃的峰归属于Cu2+和Cu+的还原[47-48],300~400 ℃的峰为Cu-O-V物种的还原[47]。这表明添加Cu物种可增强催化剂表面Cu与V物种的交互作用,这与XPS结果相一致。总体来看,Cu改性后催化剂的还原峰整体向低温区迁移,H2的消耗量由0.036 mmol·g−1 (0CuVMT) 增至0.054 mmol·g−1 (2CuVMT) ,使得催化剂的氧化还原性能提高。
-
基于上述研究结果,本研究中Hg0的脱除过程主要分为吸附和催化氧化2个阶段。在吸附阶段,气态Hg0物理吸附在催化剂表面,反应过程如式 (6) 所示。Ads表示物质的吸附态。
Hg0(ads)与催化剂活性组分 (CuOx、V2O5) 中的晶格氧发生反应,生成HgO(ads)。与此同时,催化剂表面形成可由氧气补充的氧空位,反应过程如式 (7)~(8) 所示(M表示Cu或V)。
当反应体系中通入NO时,NO可与催化剂晶格氧反应生成NO2等具有氧化性的活性中间产物,从而促进Hg0的氧化。反应过程如式 (9)~(12) 所示。
当HCl通入反应体系时,HCl与催化剂活性组分反应生成活性Cl物种,进而与催化剂表面吸附态的Hg0(ads)反应,生成HgCl2。反应过程如式 (13)~(17) 所示。
当反应体系中存在SO2时,SO2可与晶格氧反应生成SO3,进而在催化剂表面与Hg0 (ads)发生催化氧化反应,生成HgSO4。反应过程如式 (18)~(20) 所示。
-
1) 通过对传统V2O5-MoO3/TiO2催化剂掺杂Cu2O改性,提高了催化剂在低温条件下Hg0的氧化率,Cu2O负载量为2%时,催化剂具有较好的脱硝协同氧化Hg0性能。
2) 不同烟气组分对Hg0氧化率的影响分析发现,O2、NO、HCl、SO2对Hg0的氧化具有促进作用,而NH3会消耗催化剂表面晶格氧,从而抑制Hg0的氧化。在多组分烟气条件下,Hg0氧化率表现为ENO+O2>ENH3+O2>ENO+NH3+O2>ENO+NH3。在NH3和 (NO+NH3) 两种气氛下,会将HgOx还原为Hg0,使得Hg0氧化率降低。
3) 随着温度由150 ℃升至250 ℃,Hg0氧化率一直维持在99.9%,进一步升温至350 ℃,Hg0氧化率则大幅下降。
4) 结合BET、XPS和H2-TPR分析,经过Cu2O改性后部分催化剂表面微孔被堵塞,催化剂表面存在Cu和V的相互作用,使得催化剂表面产生不饱和化学键和氧空位,有效提升催化剂表面化学吸附氧含量和低温氧化还原性能,促进Hg0氧化。此时催化剂表面的氧化反应遵循Mars-Maessen机理,即Hg0优先吸附在催化剂表面与晶格氧发生反应。
不同烟气组分对Cu2O改性V2O5-MoO3/TiO2脱硝催化剂汞氧化性能的影响
Effect of different flue gas components on mercury oxidation performance of Cu2O modified V2O5-MoO3/TiO2 De-NOx catalyst
-
摘要: 为提高传统选择性催化还原 (Selective Catalytic Reduction,SCR) 催化剂的低温汞氧化效率,采用Cu2O对钒钛催化剂进行改性,通过浸渍法制备了系列Cu2O-V2O5-MoO3/TiO2催化剂,利用固定床反应器研究催化剂在不同烟气组分条件下对单质汞的氧化特性。结果表明,在200 ℃时,2%Cu2O-V2O5-MoO3/TiO2催化剂的Hg0氧化率稳定在99.9%,NO转化率保持在90.9%,具有较好的脱硝协同汞氧化性能。单独的烟气组分如O2、NO、HCl、SO2均有利于Hg0的氧化,而NH3和NO+NH3会抑制Hg0氧化为Hg2+。随着反应温度升高,Hg0氧化率呈现先平稳后降低的趋势,在350 ℃时,Hg0氧化率仅为64.1%。比表面积测试法 (BET) ,X射线光电子能谱技术 (XPS) 和H2程序升温还原 (H2-TPR) 分析表明,Cu2O改性后的V2O5-MoO3/TiO2催化剂,表面Cu和V存在相互作用,使催化剂表面产生不饱和化学键和氧空位,有利于化学吸附氧的增加,从而促进Hg0的氧化。本研究可为提升SCR脱硝催化剂对汞的协同氧化性能提供参考。
-
关键词:
- Cu2O-V2O5-MoO3/TiO2催化剂 /
- 低温选择性催化还原(SCR) /
- Hg0氧化 /
- 烟气组分
Abstract: To improve the mercury oxidation efficiency over conventional traditional Selective Catalytic Reduction (SCR)catalysts at low temperatures, the vanadium-titanium catalysts were modified by Cu2O. A series of Cu2O-V2O5-MoO3/TiO2 catalysts were prepared by the impregnation method. The effects of different flue gas components on the oxidation of mercury over the catalysts were investigated by using a fixed-bed reactor. The results suggested that the Hg0 oxidation efficiency was stabilized at 99.9% and the NO conversion efficiency was maintained at 90.9% over 2%Cu2O-V2O5-MoO3/TiO2 catalyst at 200 ℃, which showed a good performance of synergistic De-NOx and mercury oxidation. The individual flue gas components, such as O2, NO, HCl, and SO2 were conductive to the oxidation of Hg0, while NH3 and the coexistence of NO and NH3 inhibited the oxidation of Hg0 to Hg2+. With the increase of reaction temperature, the Hg0 oxidation efficiency presented a trend of stability and then decrease.The oxidation rate was only 64.1% when the reaction temperature reached 350 ℃. Specific surface area testing (Brunauer-Emmett-Teller, BET), X-ray photoelectron spectroscopy (XPS) and H2 temperature programmed reduction (H2-TPR) analysis demonstrated that when loaded with Cu2O, the interaction between Cu and V existed over the Cu2O-V2O5-MoO3/TiO2 catalyst surface produced unsaturated chemical bonds and oxygen vacancies on the surface of the catalyst,which was conductive to the increase of chemisorbed oxygen, thus promoting the oxidation of Hg0. This study can provide a reference for improving the co-oxidation performance of SCR denitrification catalyst for mercury. -
硝酸盐(NO3−)是一种在水体中广泛存在的无机污染物。近几十年来,由于人类的工业和农业活动,水体中的NO3−浓度不断升高,已逐渐成为世界各国密切关注的问题[1-2]。我国30个省71条主要河流样品的硝酸盐数据表明,这些样品中有约7.83%的硝酸盐含量超过45 mg·L−1[1]。一般而言,污水厂二沉池出水中的硝酸盐浓度在10 mg·L−1,约占我国污水厂一级A标准中规定出水总氮浓度要求(15 mg·L−1)的67%左右,而一些水环境敏感地区(如滇池、太湖等)的污水厂出水总氮浓度的内控指标低至5 mg·L−1(以氮计),硝态氮去除对污水厂总氮控制造成极大压力。污水厂出水中硝酸盐氮浓度过高导致的总氮超标是城镇污水处理厂中最常见的问题之一[3]。目前,污水中的NO3−主要通过生物反硝化去除,需要外加碳源、延长反硝化停留时间或增大回流比强化脱氮,能耗和药耗高。化学还原法具有还原效率高、操作简单、成本低等优点,如果能利用化学还原方法强化NO3−脱除,将有助于降低厌氧生物脱氮负荷,进一步提高污水中的总氮去除效率。
近年来,基于二氧化碳阴离子自由基 (CO2·−)的高级还原技术逐步引起人们关注。CO2·−分子结构中存在一个未成对的电子,具有很强的还原特性,还原电位E0(CO2/CO2·−)为−1.9 V[4]。CO2·−作为电子供体,通过电子传递、亲核攻击等途径还原降解污染物[5-6]。研究表明,CO2·−能有效去除水中卤代有机物、全氟化合物、溴酸盐和六价铬等污染物[7-10]。近期研究发现,基于CO2·−的高级还原技术能将NO3−定向还原为气态氮,并且水中共存的溶解性有机物对CO2·−驱动的NO3−还原去除影响很小,仅在有机物浓度30 mg·L−1时的NO3−还原去除略有下降[11-12]。但是,目前基于污水体系的CO2·−的高级还原研究较少,其实际应用仍存在诸多挑战[11]。
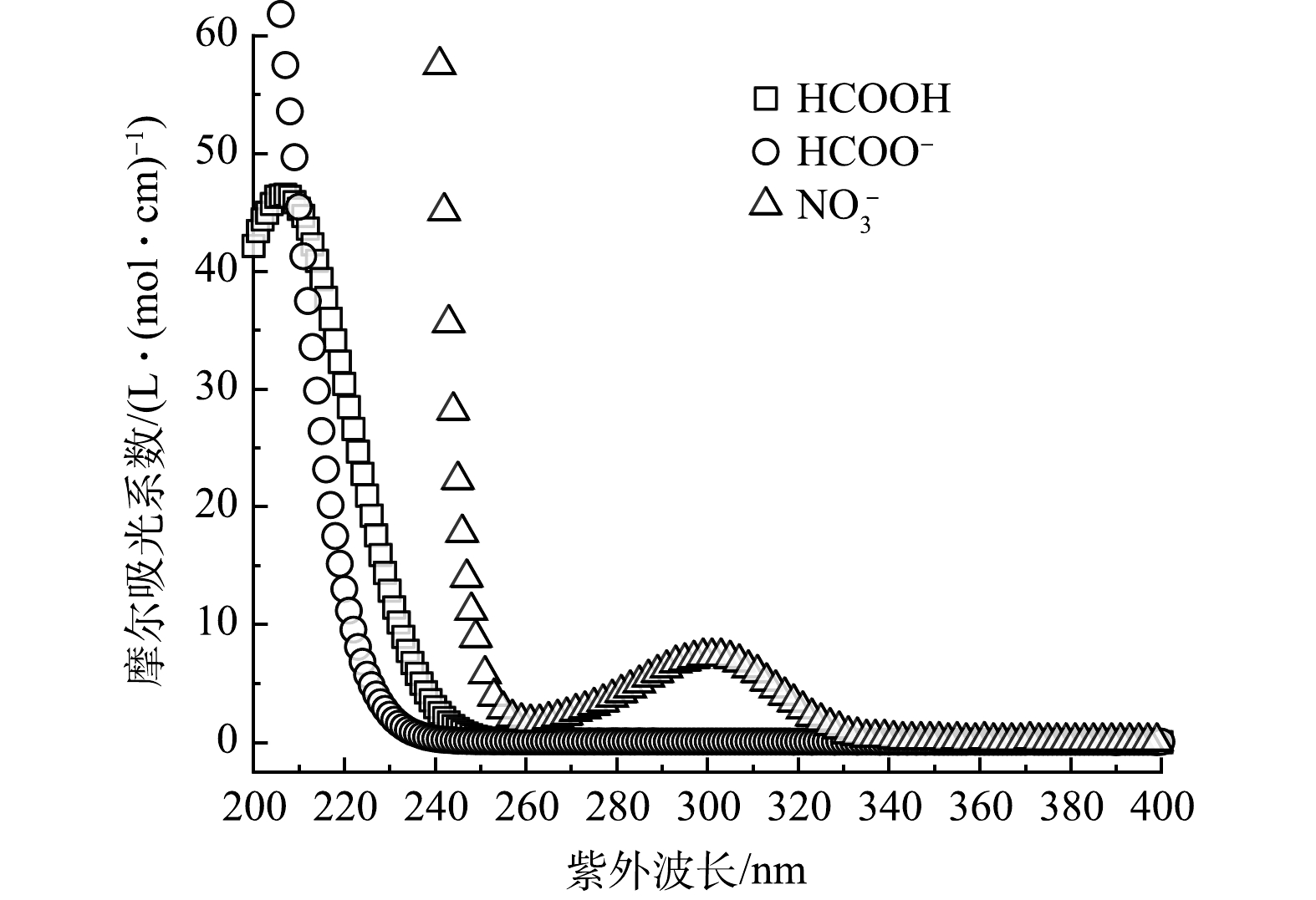
紫外消毒已在污水处理中广泛应用,截至2016年我国已建成使用的5 000多座城镇污水厂中超过50%采用了中压紫外消毒。硝酸盐在波长250~400 nm具有吸光性,特别是在302 nm处具有特征吸收峰,摩尔吸光系数为7.2 L·(mol·cm)−1。同时,甲酸盐(HCOO−)可作为一种新型绿色碳源被投入到二级生物反应池中促进生物脱氮过程[13-14],未被生物利用的甲酸盐则进入紫外消毒阶段。污水中NO3−在紫外辐照下光解产生含氮活性物种(RNS)(式(1))和羟基自由基(HO·)(式(2))[15],HO·与甲酸盐或者甲酸反应生成CO2·−(式(3)~(4))。CO2·−的还原性可以促进进一步硝酸盐还原(式(5))。
NO−3hv→NO⋅2+O⋅− (1) O⋅−+H2O↔HO⋅+HO−pKa=11.9 (2) HO⋅+HCOO−→H2O+CO⋅−2k″HO⋅−HCOO−=3.2×109L⋅(mol⋅s)−1 (3) HO⋅+HCOOH→H++H2O+CO⋅−2k″HO⋅−HCOOH=1.3×108L⋅(mol⋅s)−1 (4) CO⋅−2+NO−3→products (5) 基于此,本研究将围绕污水处理的紫外消毒环节构建中压紫外/甲酸盐反应体系,解析此体系中的NO3−还原效果和关键影响因素;围绕硝酸盐光解以及CO2·−与硝酸盐的反应活性揭示硝酸盐降解原理,并进一步探索硝酸盐强化脱除方法。研究结果将深化CO2·−高级还原体系的基础数据和反应机制,为污水的深度脱氮提供新思路。
1. 材料及方法
1.1 实验药剂
硝酸钠、甲酸钠、甲酸均为分析纯,采购于上海安谱璀世有限公司;过氧化氢为纯度30%,购于广州牌药剂公司。95%~98%浓硫酸(H2SO4)为分析纯,购于福晨(天津)化学试剂有限公司;氢氧化钠(NaOH)为分析纯,购于国药集团化学试剂有限公司。所有试剂在使用前均未经过纯化或其他处理步骤,实验用水均来自Merck-Millipore系统产生的超纯水。
1.2 实验装置及实验过程
硝酸盐的光还原降解实验在平行光紫外辐照装置中进行,由中压紫外光源(500 W,PLS-LAM500,Beijing PerfectLight)、反应石英皿和磁力搅拌装置组成。中压汞灯在225~425 nm的发射光谱,主波长在320 nm和360 nm,与污水处理厂的中压汞灯消毒的特征波长一致,所有实验未采用滤光手段控制光源辐照的波长。由于中压汞灯的特征波长的复杂性,本研究以254 nm处的紫外光强作为指示光强,其值为0.5 mW·cm−2。在研究紫外光强影响时,通过改变中压紫外灯与水样的间距,将光强划分为低、中、高3个水平,其在254 nm处的光强值分别为0.04、0.5和1.3 mW·cm−2。
实验水样中初始NO3−浓度为0.1 mmol·L−1,甲酸或者甲酸盐为0~10 mmol·L−1。此实验中初始硝酸盐浓度低于实际污水中的硝酸盐存在水平,其原因是为了与实验中较低光强的紫外光源相匹配。除研究pH影响的实验外,水样初始pH为7.0±0.2,不另加pH缓冲溶液。在研究pH影响时,水样中加入10 mmol·L−1磷酸盐缓冲溶液,采用硫酸或氢氧化钠调节初始pH。石英皿置于紫外装置下,以加入甲酸/甲酸盐(0~10 mmol·L−1)为反应起始点,反应过程中采用磁力搅拌器搅拌,确保溶液和试剂混合均匀。在一定时间间隔下取样测定硝酸盐及产物浓度。
1.3 CO2·−与硝酸盐的二级反应速率常数测定
采用激光闪光光解技术测定CO2·−与硝酸盐的二级反应速率常数。激光闪光光解系统(LKS80,Applied Photophysics Ltd., United Kingdom)的激发波长为266 nm,激光束截面为0.5 cm2,脉冲激光能量为(25±5) mJ,脉冲持续时间为4~6 ns;使用150 W氙灯作为检测光源。实验水样中含H2O2(50 mmol·L−1),HCOO−(500 mmol·L−1),竞争参考物甲基紫精(MV2+)浓度为100 μmol·L−1,向溶液中投加不同浓度的硝酸盐(0~100 mmol·L−1)。激光实验前,水样由氩气吹脱30 min (溶解氧<0.2 mg·L−1)并调节pH为7.0。测定MV2+反应产物MV·+在600 nm特征吸收峰信号。
1.4 分析与检测方法
硝酸盐、亚硝酸盐、甲酸盐浓度利用离子色谱仪(ThermoFisher, ICS-600)进行测定。离子色谱仪的主要组成部分为:RFC-30淋洗液自动发生器,ChromeLeon色谱工作站,Dionex IonPacTM AG-19保护柱(4 mm×50 mm),Dionex IonPacTM AS-19分析柱(4 mm×250 mm)。KOH淋洗液由RFC-30淋洗液自动发生器在线自动产生,流速为1.0 mL·min−1。样品检测前需经过0.22 μm滤膜过滤。采用纳氏试剂分光光度法测定氨氮的浓度。pH采用pH计(S400,Mettler Toledo)测定。反应溶液所受的中压汞灯辐照光强通过UV254光强测定仪UVC-254A(Lutron electronic enterprise Co; LTD)初步量化,以确定辐照光强的相对大小。
2. 结果与讨论
2.1 MPUV/HCOO−体系对硝酸盐的降解效能
1)甲酸盐浓度对硝酸盐光还原降解的影响。图1所示为不同HCOO−投加量(0~10 mmol·L−1)条件下硝酸盐的光还原降解速率。当体系中不存在甲酸盐时,紫外辐照120分钟后硝酸盐去除率仅为13.7%,NO3−拟一级降解速率kobs为1.0×10−3 min−1。HCOO−投加量为0.1、0.5、1、5和10 mmol·L−1时,反应120min的硝酸盐去除率分别为20.6%、39.4%、44.1%、45.9%和43.9%。由此可见,甲酸盐在0.1~1 mmol·L−1条件下,硝酸盐降解速率kobs由2.41×10−3 min−1逐步增大到5.98×10−3 min−1。随着甲酸浓度继续增大(1~10 mmol·L−1),NO3−的一级降解速率趋于稳定。可见,硝酸盐紫外光解产生的HO·可以促进甲酸盐转化为CO2·−,进而强化硝酸盐的还原降解(式(1)~式(4))。然而,受到硝酸盐光解的HO·生成量限制,高浓度的甲酸盐(大于1 mmol·L−1)不能形成更多的CO2·−,使得硝酸盐不能进一步降解。因此,硝酸盐和甲酸盐的摩尔浓度比在1:5~1:10的促进效果较好。
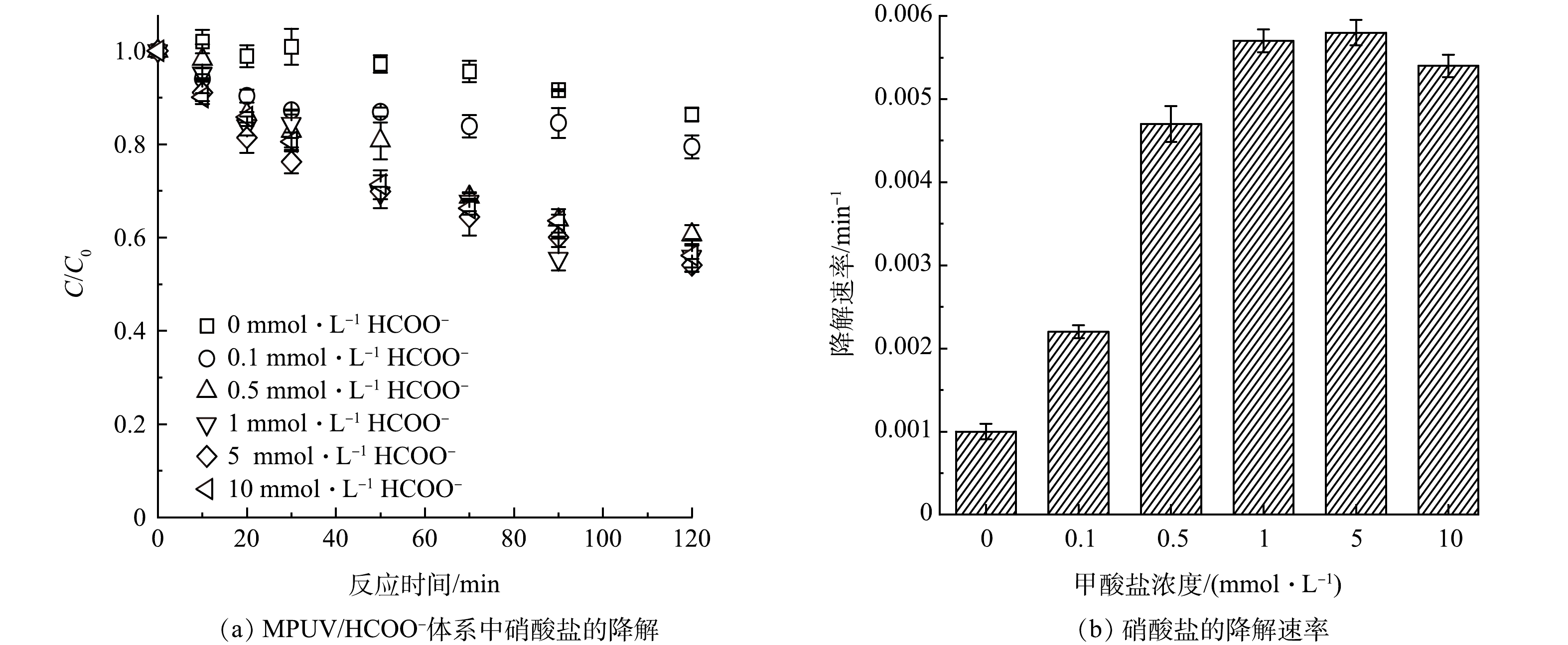 图 1 甲酸盐浓度对MPUV/HCOO−体系降解硝酸盐的影响Figure 1. Effects of formate concentrations on nitrate degradation in MPUV/HCOO− system
图 1 甲酸盐浓度对MPUV/HCOO−体系降解硝酸盐的影响Figure 1. Effects of formate concentrations on nitrate degradation in MPUV/HCOO− system2)紫外光强对硝酸盐光还原降解的影响。图2为紫外光强对MPUV/HCOO−体系降解硝酸盐的影响结果。将甲酸盐投加浓度设为2 mmol·L−1,以防止高光强导致的甲酸盐快速消耗而影响实验结果。通过调节紫外灯与水样间距选定3种光强,其在254 nm处指示的低、中、高光强分别为0.04、0.5和1.3 mW·cm−2。单独紫外光解(无甲酸盐)时,硝酸盐降解速率随着紫外光强增大而缓慢增大,在低、中、高光强条件下,kobs分别为1.8×10−4、1.0×10−3 和1.2×10−3 min−1,表明紫外光强增大能促进硝酸盐光解。当2 mmol·L−1甲酸盐存在时,MPUV/HCOO−体系中硝酸盐降解速率随紫外光强的增加而显著增加,kobs分别为9.9×10−4、3.6×10−3和6.9×10−3 min−1。MPUV/HCOO−体系对紫外强化的依赖性更高,说明增加紫外光强不仅促进了硝酸盐分解,也增大了硝酸盐光解介导的CO2·−生成,从2个途径加速了硝酸盐的光还原降解。因此,紫外光强是重要的硝酸盐光还原影响因素。
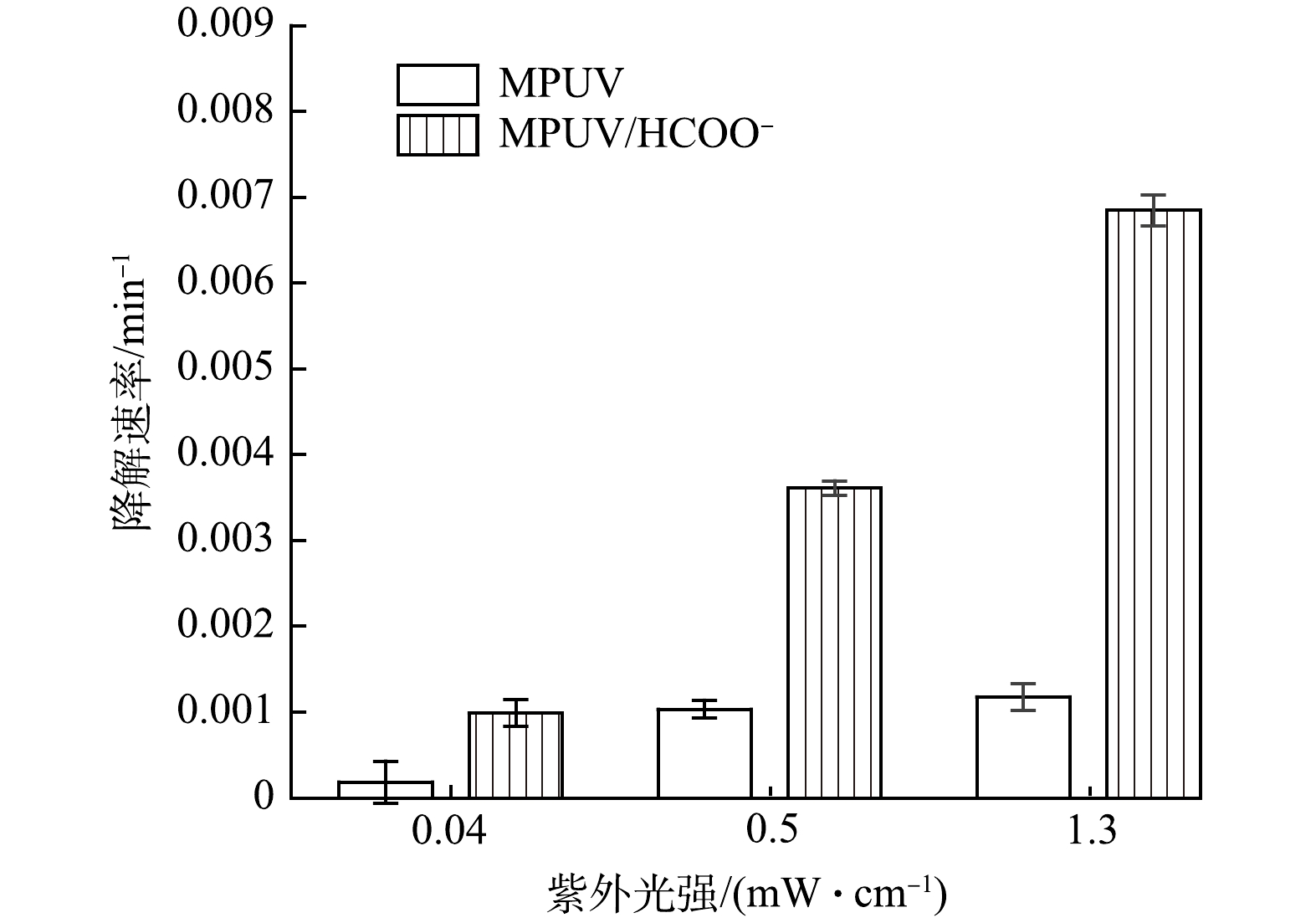 图 2 紫外光强对MPUV/HCOO−体系降解硝酸盐的影响Figure 2. Effects of MPUV intensities on nitrate degradation in MPUV/HCOO− system
图 2 紫外光强对MPUV/HCOO−体系降解硝酸盐的影响Figure 2. Effects of MPUV intensities on nitrate degradation in MPUV/HCOO− system3)pH对硝酸盐光还原降解的影响。图3反映了pH对MPUV/HCOO−体系降解硝酸盐的影响。甲酸盐投加量设为0.5 mmol·L−1,以规避硝酸盐去除能力上限造成的干扰。溶液pH的增大可促进硝酸盐光降解。在pH为2.5、5.0和8.0条件下,MPUV/HCOO−体系中硝酸盐降解的kobs分别1.5×10−3、2.6×10−3和3.6×10−3 min−1。可见,碱性条件更利于MPUV/HCOO−体系的硝酸盐降解。溶液的pH对硝酸盐和甲酸盐的存在形态以及还原性自由基与硝酸盐的反应速率均存在影响。对于甲酸的存在形态,HCOOH/HCOO−的pKa为3.8,pH=2.5时的kobs最低,说明HCOO−形式更助于硝酸盐的光还原降解。对于硝态氮,硝酸盐光敏化生成一种吸光度极高的中间体过氧亚硝酸盐(ONOO−),ONOOH/ONOO−的pKa为6.6,pH=8.0时的kobs高于pH=5.0,意味着以ONOO−形式存在更助于硝酸盐的光解,并促进自由基还原降解途径。以上多重因素叠加下,MPUV/HCOO−体系以去质子化形式存在的CO2·−、HCOO−和ONOO−相比于其质子化形式的HCO2·、HCOOH和HOONO,更有助于硝酸盐的降解,碱性条件下MPUV/HCOO−体系降解硝酸盐效果更佳。
 图 3 pH条件对MPUV/HCOO−体系降解硝酸盐的影响Figure 3. Effects of pH conditions on nitrate degradation in MPUV/HCOO− system
图 3 pH条件对MPUV/HCOO−体系降解硝酸盐的影响Figure 3. Effects of pH conditions on nitrate degradation in MPUV/HCOO− system2.2 MPUV/HCOO−体系对硝酸盐的降解机制
1)硝酸盐的紫外光解。图4为硝酸盐、甲酸和甲酸盐的紫外吸收光谱。可见硝酸盐在300 nm处存在特征吸收峰,而甲酸和甲酸盐不能直接光解。辐照波长(λ)>280 nm条件下,硝酸盐光解主要产生自由基(·NO2和HO·)(式(1)和式(2))。在波长<280 nm的区间内,NO3−异构化形成过氧亚硝酸盐(ONOO−)。并且,·NO2和HO·也可以重组形成过氧亚硝酸(HOONO)通过去质子化生成ONOO− [16]。过氧亚硝酸(盐)被认为是硝酸盐光还原降解过程中的重要活性中间体[17],ONOO−在300 nm处存在强吸收峰,摩尔吸光系数约为1 600 L·(mol·cm)−1。但ONOO−的反应活性极高,在pH<6.6时迅速异构化为硝酸盐(ONOOH→NO3−+H+, k=0.9 s−1),而在pH>6.6时分解为一氧化氮和超氧阴离子(ONOO−→·NO+O2·−, k=0.023 s−1)[18],导致ONOO−仅为一种瞬态反应活性中间体,可能参与了硝酸盐降解,但反应过程难以捕捉。
2)CO2·−与硝酸盐的二级反应动力学常数。基于激光闪光光解技术,建立了以MV2+为竞争参考物的CO2·−与硝酸盐二级反应速率常数(
k″CO⋅−2−NO−3 k″CO⋅−2−NO−3 k″CO⋅−2−NO−3 CO⋅−2+MV2+→CO2+MV⋅+kS1=6.4×109L⋅(mol⋅s)−1 (6) CO⋅−2+NO−3→products (7) CO⋅−2+H2O2→CO2+HO⋅+OH−kS2=7.0×105L⋅(mol⋅s)−1 (8) Y=AA0=k″CO⋅−2−NO−3[NO−3]+kS1[MV2+]+kS2⋅[H2O2]kS1[MV2+]+kS2[H2O2]=k″CO⋅−2−NO−3[NO−3]kS1[MV2+]+kS2[H2O2]+1=k″CO⋅−2−NO−3⋅X+1 (9) 式中
:A0 kS1 kS2 图5(a)反映了在不同NO3−浓度(0~100 mmol·L−1)下,0~2 500 ns内竞争物与CO2·−反应生成的中间体自由基MV·+在600 nm的吸光信号值。可见,随着硝酸盐浓度的逐渐升高,MV·+的信号逐渐减弱,证明了一部分CO2·−被硝酸盐反应消耗,使MV·+生成速率降低。图5(b)为基于式(9)的线性拟合方程,通过拟合曲线得到斜率值
k''CO·−2−NO−3  图 5 MV2+竞争动力学法测定CO2·−与NO3−的二级反应速率常数Figure 5. Determination of second-order reaction rate constant of CO2·− with NO3− using MV2+ competition kinetics method
图 5 MV2+竞争动力学法测定CO2·−与NO3−的二级反应速率常数Figure 5. Determination of second-order reaction rate constant of CO2·− with NO3− using MV2+ competition kinetics methodCO⋅−2+⋅NO2→NO−2+CO2k″CO⋅−2−⋅NO2=6.0×109L⋅(mol⋅s)−1 (10) CO⋅−2+⋅NO→NOCO−2k″CO⋅−2−⋅NO=2.9×109L⋅(mol⋅s)−1 (11) CO⋅−2+NH2OH−+H2O→NH3+2OH−+CO2k″CO⋅−2−NH2OH−=1.0×109L⋅(mol⋅s)−1 (12) 3)还原产物及反应机理分析。进一步分析了MPUV/HCOO−体系降解硝酸盐的主要产物。由图6可见,在初始HCOO−浓度0.5 mmol·L−1时,在pH为2.5、5.0、8.0的条件下,硝酸盐减少量与亚硝酸盐增加量的比值接近1:1,并且未检测到氨氮的生成,说明MPUV/HCOO−体系主要将硝酸盐转化为亚硝酸盐。由二级反应速率常数可知,如果体系中CO2·−充足,可以实现亚硝酸盐的进一步还原。图7为MPUV/HCOO−体系促进硝酸盐的降解过程:步骤1,紫外光解硝酸盐生成含氮活性物种和羟基自由基;步骤2,羟基自由基与甲酸盐反应生成CO2·−;步骤3,CO2·−促进了活性氮中间体还原(主要过程)且其可与硝酸盐直接发生还原反应(次要过程)。因此,硝酸盐的紫外光解及CO2·−主导的高级还原过程共同促进了硝酸盐还原降解。
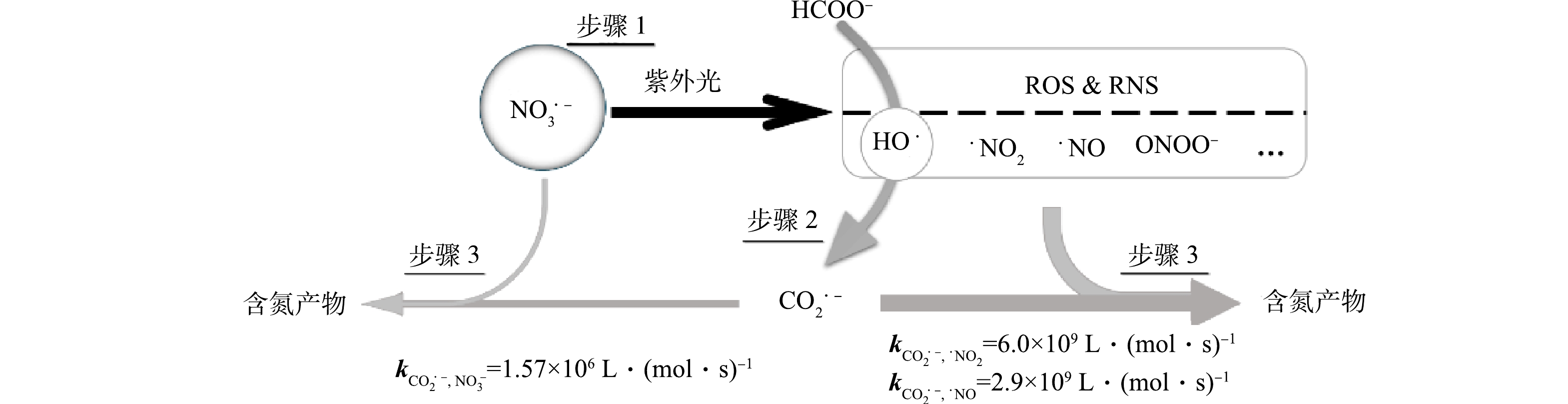 图 7 MPUV/HCOO−体系降解硝酸盐的反应机制Figure 7. Mechanisms of nitrate degradation in the MPUV/HCOO− system
图 7 MPUV/HCOO−体系降解硝酸盐的反应机制Figure 7. Mechanisms of nitrate degradation in the MPUV/HCOO− system2.3 H2O2对MPUV/HCOO−体系降解硝酸盐的强化作用
由于硝酸盐光解的HO·生成量较低,限制了CO2·−的转化生成和硝酸盐的CO2·−还原路径。因此,向MPUV/HCOO−体系中投加过氧化氢(H2O2)提升HO·生成量,从而进一步强化生成CO2·−以促进硝酸盐的脱除。如图8(a)所示,在HCOO−浓度为2 mmol·L−1、光照反应120 min后,H2O2投量为0、0.1、0.2、0.5、0.8和1 mmol·L−1时的硝酸盐去除率分别为32.5%、42.8%、47.7%、42.8%、33.2%和15.8%,在H2O2投量为0.2 mmol·L−1时硝酸盐去除率最高,而H2O2投量增大到1 mmol·L−1反而抑制了硝酸盐降解。相比不加H2O2的MPUV/HCOO−体系降解硝酸盐(k=3.6×10−3 min−1),投加0.2 mmol·L−1的H2O2使硝酸盐降解速率(k=6.2×10−3 min−1)提高了1.7倍。在H2O2投加量为0.2 mmol·L−1时,通过紫外光解H2O2促进HO·产生,进而提升了体系中CO2·−浓度水平并强化了硝酸盐还原。由图8(b)可见,甲酸盐消耗量随着H2O2投量的增大而增大,证明了在中压汞灯/甲酸盐体系中补充H2O2会促进甲酸盐向CO2·−的转化。并且,当体系中存在剩余甲酸盐时可以通过适量投加H2O2淬灭,如1 mmol·L−1 H2O2反应120 min后的甲酸盐基本全部去除。图8(c)显示了H2O2投加量为0.2 mmol·L−1时反应120 min后的硝酸盐向亚硝酸盐的转化率为63.3%且无氨氮被检出,说明36.7%的硝态氮转化为气态氮(如氧化亚氮、氮气等)排出,与CHEN等[21]的研究结果一致。而紫外光解过量的H2O2 (1.0 mmol·L−1)形成了以HO·主导的氧化体系,导致硝酸盐光解产生的活性氮中间体被重新氧化为硝态氮,使硝酸盐浓度在90~120 min反而增加(图8(a))。在本研究紫外条件下,甲酸盐和H2O2的摩尔比为10:1时,硝酸盐还原降解效果最佳。
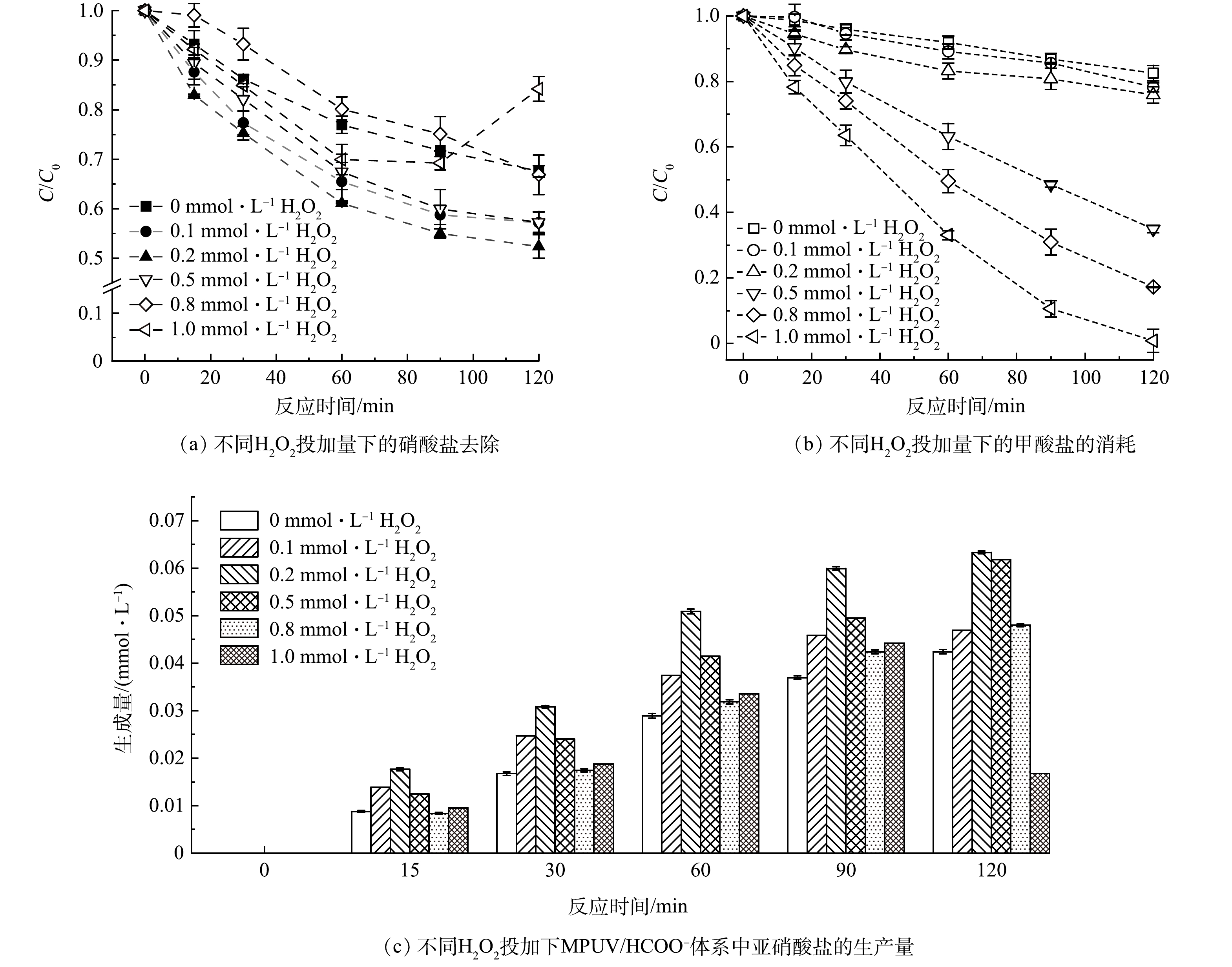 图 8 外加H2O2对MPUV/HCOO−体系降解硝酸盐的影响Figure 8. Effects of H2O2 addition on nitrate degradation in MPUV/HCOO− system
图 8 外加H2O2对MPUV/HCOO−体系降解硝酸盐的影响Figure 8. Effects of H2O2 addition on nitrate degradation in MPUV/HCOO− system3. 结论
1)相较于单独中压紫外(MPUV)光解,MPUV/HCOO−体系促进了硝酸盐的还原降解,CO2·−对促进硝酸盐降解具有重要的贡献。
2)在MPUV/HCOO−体系中,甲酸盐与硝酸盐摩尔浓度比在5:1~10:1条件下的硝酸盐去除效果最好。增大紫外光强和弱碱性(pH=8.0)环境能可有效促进硝酸盐的还原降解。
3)利用激光闪光光解技术量化了CO2·−与NO3−和NO2−反应的二级反应速率常数,分别为1.57×106 L·(mol·s)−1和9.12×107 L·(mol·s)−1。虽然CO2·−与硝酸盐反应活性较低,但可以促进硝酸盐光解产生的活性氮中间体的还原,中压紫外光解和CO2·−的介导还原共同促进了硝酸盐还原效能。
4)在MPUV/HCOO−体系中投加H2O2能进一步促进硝酸盐降解,投加0.2 mmol·L−1的H2O2使硝酸盐降解速率提高了1.7倍,并将36.7%的硝态氮转化为气态氮排出。
-

图 2 Cu2O负载量对CuVMT催化剂脱硝协同汞氧化性能的影响
Figure 2. Effect of Cu2O loading on denitridication and mercury oxidation performance over CuVMT catalysts
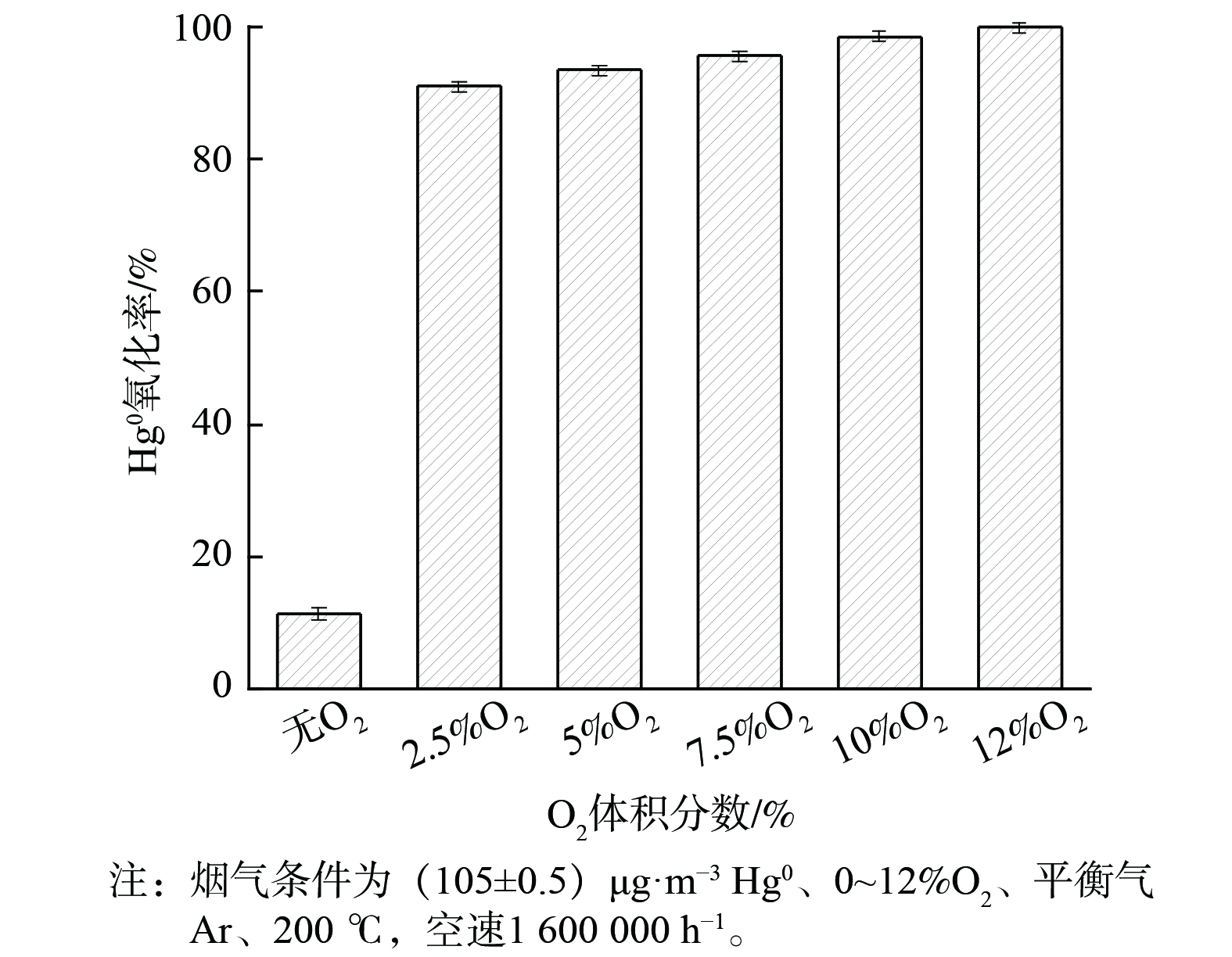
图 3 O2体积分数对2CuVMT催化剂Hg0氧化率的影响
Figure 3. Effect of O2 volume fraction on Hg0 oxidation rate over 2CuVMT catalyst
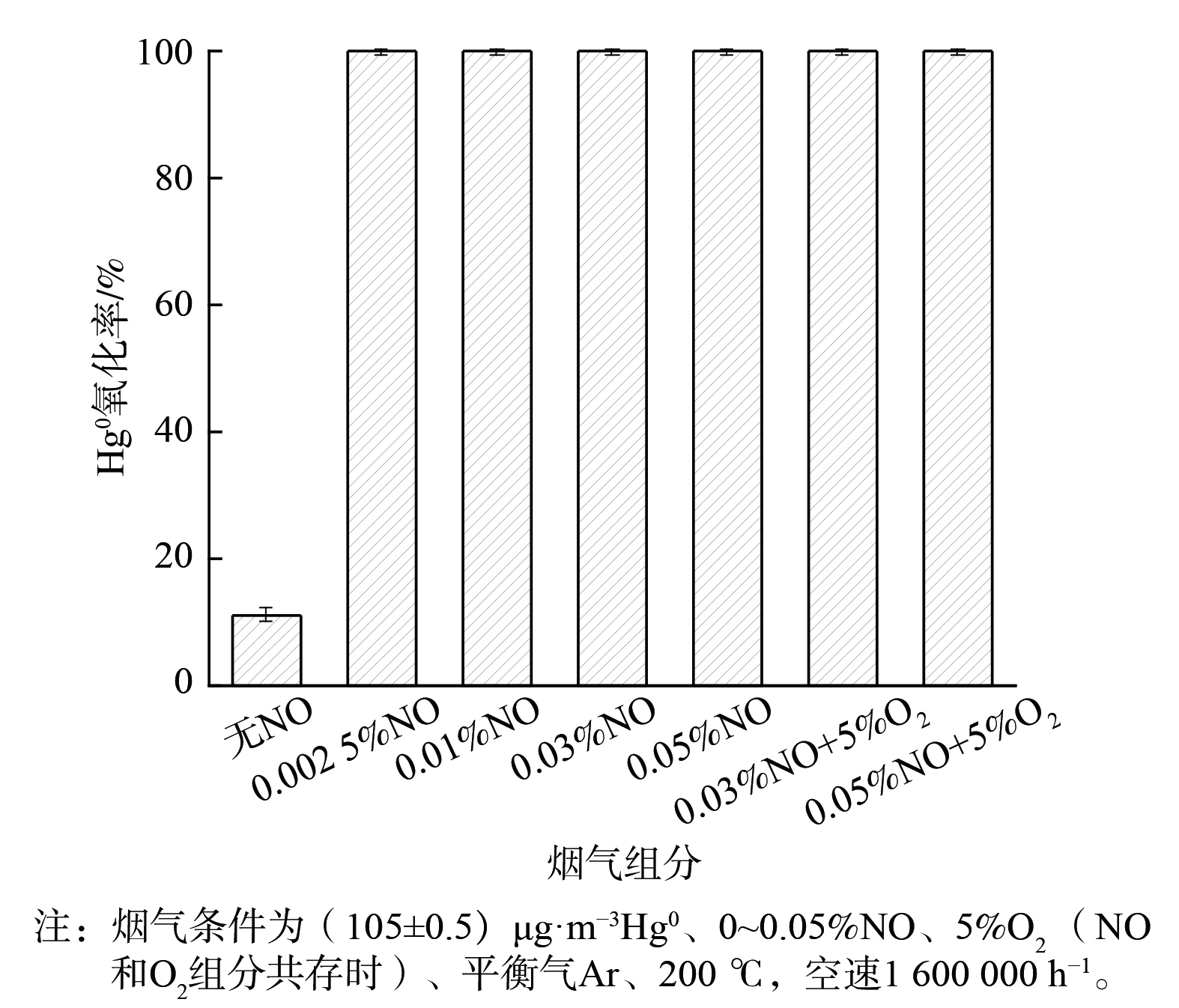
图 4 NO体积分数对2CuVMT催化剂Hg0氧化率的影响
Figure 4. Effect of NO volume fratction on Hg0 oxidation rate over 2CuVMT catalyst

图 5 NH3的体积分数对2CuVMT催化剂Hg0氧化率的影响
Figure 5. Effect of NH3 volume fraction on Hg0 oxidation rate over 2CuVMT catalyst
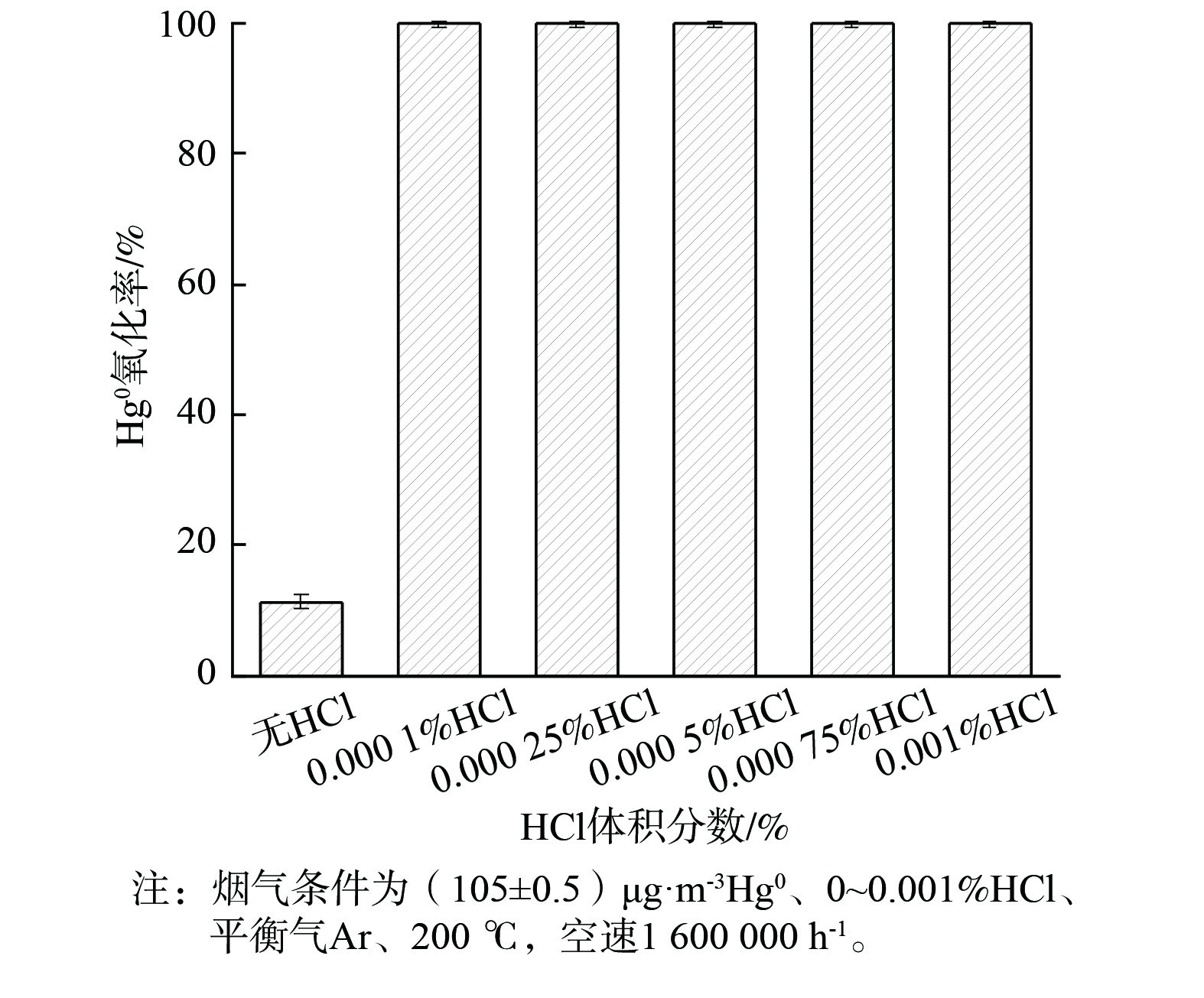
图 7 HCl体积分数对2CuVMT催化剂Hg0氧化率的影响
Figure 7. Effect of HCl volume fraction on Hg0 oxidation rate over 2CuVMT catalyst

图 8 SO2和H2O对2CuVMT催化剂Hg0氧化率的影响
Figure 8. Effect of SO2 and H2O on Hg0 oxidation rate over 2CuVMT catalyst

图 9 反应温度对2CuVMT催化剂Hg0氧化率的影响
Figure 9. Effect of reaction temperature rate on Hg0 oxidation over 2CuVMT catalyst
表 1 CuVMT催化剂的比表面积及孔道结构
Table 1. Specific surface area and pore structure of CuVMT catalysts
催化剂 比表面积/ (m2·g−1) 孔容/ (cm3·g−1) 介孔平均孔径/nm 0CuVMT 68.4 0.36 19.0 2CuVMT 57.3 0.33 21.3  下载: 导出CSV
下载: 导出CSV
-
[1] HU Y A, CHENG H F. Control of mercury emissions from stationary coal combustion sources in China: current status and recommendations[J]. Environmental Pollution, 2016, 218: 1209-1221. doi: 10.1016/j.envpol.2016.08.077 [2] GOLDING G R, KELLY C A, SPARLING R, et al. Evaluation of mercury toxicity as a predictor of mercury bioavailability[J]. Environmental Science & Technology, 2007, 41(16): 5685-5692. [3] CHIU C H, KUO T H, CHANG T C, et al. Multipollutant removal of Hg0/SO2/NO from simulated coal-combustion flue gases using metal oxide/mesoporous SiO2 composites[J]. International Journal of Coal Geology, 2017, 170: 60-68. doi: 10.1016/j.coal.2016.08.014 [4] 左朋莱, 王晨龙, 佟莉, 等. 小型燃煤机组烟气重金属排放特征研究[J]. 环境科学研究, 2020, 33(11): 2599-2604. doi: 10.13198/j.issn.1001-6929.2020.06.07 [5] 魏忠秋, 刁永发, 姚跃辉. 活性焦配方型吸附剂脱除模拟烟气中Hg0[J]. 环境工程学报, 2017, 11(2): 1003-1008. doi: 10.12030/j.cjee.201509220 [6] STREETS D G, HAO J M, WU Y, et al. Anthropogenic mercury emissions in China[J]. Atmospheric Environment, 2005, 39(40): 7789-7806. doi: 10.1016/j.atmosenv.2005.08.029 [7] 国家质量监督检验检疫总局, 中国国家标准化管理委员会. 火电厂大气污染物排放标准: GB 13223—2011[S]. 北京: 中国环境科学出版社, 2012. [8] 鹿存房, 刘清才, 全学军. 利用污泥脱除燃煤电厂烟气中的汞[J]. 环境工程学报, 2017, 11(10): 5559-5564. doi: 10.12030/j.cjee.201611166 [9] 黄永健. 大气气溶胶汞污染研究[D]. 成都: 成都理工大学, 2002. [10] ESWARAN S, STENGER H G. Understanding mercury conversion in selective catalytic reduction (SCR) catalysts[J]. Energy & Fuels, 2005, 19(6): 2328-2334. [11] SCHWÄMMLE T, BERTSCHE F, HARTUNG A, et al. Influence of geometrical parameters of honeycomb commercial SCR-DeNOx-catalysts on DeNOx-activity, mercury oxidation and SO2/SO3-conversion[J]. Chemical Engineering Journal, 2013, 222: 274-281. doi: 10.1016/j.cej.2013.02.057 [12] 陈进生. 火电厂烟气脱硝技术: 选择性催化还原法[M]. 北京: 中国电力出版社, 2008. [13] WAN Q, YAO Q, DUAN L, et al. Comparison of elemental mercury oxidation across vanadium and cerium based catalysts in coal combustion flue gas: catalytic performances and particulate matter effects[J]. Environmental Science & Technology, 2018, 52(5): 2981-2987. [14] ZHAO L K, LI C T, ZHANG J, et al. Promotional effect of CeO2 modified support on V2O5-WO3/TiO2 catalyst for elemental mercury oxidation in simulated coal-fired flue gas[J]. Fuel, 2015, 153: 361-369. doi: 10.1016/j.fuel.2015.03.001 [15] MEI J, SUN P X, XIAO X, et al. Influence mechanism of the compositions in coal-fired flue gas on Hg0 oxidation over commercial SCR catalyst[J]. Journal of Industrial and Engineering Chemistry, 2019, 75: 130-137. doi: 10.1016/j.jiec.2019.03.013 [16] 陶莉, 张旭楠, 李彩亭, 等. 选择性催化还原催化剂氧化脱除烟气中单质汞[J]. 环境工程学报, 2015, 9(6): 2925-2932. doi: 10.12030/j.cjee.20150664 [17] STOLLE R, KOESER H, GUTBERLET H. Oxidation and reduction of mercury by SCR DeNOx catalysts under flue gas conditions in coal fired power plants[J]. Applied Catalysis B:Environmental, 2014, 144: 486-497. doi: 10.1016/j.apcatb.2013.07.040 [18] LI Y, MURPHY P D, WU C Y, et al. Development of silica/vanadia/titania catalysts for removal of elemental mercury from coal-combustion flue gas[J]. Environmental Science & Technology, 2008, 42(14): 5304-5309. [19] YANG J, YANG Q, SUN J, et al. Effects of mercury oxidation on V2O5-WO3/TiO2 catalyst properties in NH3-SCR process[J]. Catalysis Communications, 2015, 59: 78-82. doi: 10.1016/j.catcom.2014.09.049 [20] BERETTA A, USBERTI N, LIETTI L, et al. Modeling of the SCR reactor for coal-fired power plants: impact of NH3 inhibition on Hg0 oxidation[J]. Chemical Engineering Journal, 2014, 257: 170-183. doi: 10.1016/j.cej.2014.06.114 [21] YANG B, LI Z, HUANG Q, et al. Synergetic removal of elemental mercury and NO over TiCe0.25Sn0.25Ox catalysts from flue gas: performance and mechanism study[J]. Chemical Engineering Journal, 2019, 360: 990-1002. doi: 10.1016/j.cej.2018.09.193 [22] CHIU C H, HSI H C, LIN H P, et al. Effects of properties of manganese oxide-impregnated catalysts and flue gas condition on multipollutant control of Hg0 and NO[J]. Journal of Hazardous Materials, 2015, 291: 1-8. doi: 10.1016/j.jhazmat.2015.02.076 [23] SUN X M, WU J, TIAN F G, et al. Synergistic effect of surface defect and interface heterostructure on TiO2/BiOIO3 photocatalytic oxide gas-phase mercury[J]. Materials Research Bulletin, 2018, 103: 247-258. doi: 10.1016/j.materresbull.2018.03.040 [24] WANG T, YANG Y H, WANG J W, et al. Preadsorbed SO3 inhibits oxygen atom activity for mercury adsorption on Cu/Mn doped CeO2(110) surface[J]. Energy & Fuels, 2020, 34(4): 4734-4744. [25] 宗晨曦, 纪蕾朋, 陈奎续, 等. Cu/SAPO-34对模拟烟气中零价汞的脱除性能[J]. 环境工程学报, 2018, 12(6): 1691-1701. doi: 10.12030/j.cjee.201710163 [26] 范红兵, 刁永发, 李攀, 等. 烟气成分对负载V2O5-WO3/TiO2聚苯硫醚纤维脱除烟气中Hg0的影响[J]. 环境工程学报, 2014, 8(7): 2957-2962. [27] YAMAGUCHI A, AKIHO H, ITO S. Mercury oxidation by copper oxides in combustion flue gases[J]. Powder Technology, 2008, 180(1/2): 222-226. [28] LIU Y, WANG Y J, WANG H Q, et al. Catalytic oxidation of gas-phase mercury over Co/TiO2 catalysts prepared by Sol-gel method[J]. Catalysis Communications, 2011, 12(14): 1291-1294. doi: 10.1016/j.catcom.2011.04.017 [29] ZHAO L K, LI C T, WANG Y, et al. Simultaneous removal of elemental mercury and NO from simulated flue gas using a CeO2 modified V2O5-WO3/TiO2 catalyst[J]. Catalysis Science & Technology, 2016, 6(15): 6076-6086. [30] ZHANG X N, LI C T, ZHAO L K, et al. Simultaneous removal of elemental mercury and NO from flue gas by V2O5-CeO2/TiO2 catalysts[J]. Applied Surface Science, 2015, 347: 392-400. doi: 10.1016/j.apsusc.2015.04.039 [31] 李海龙. 新型SCR催化剂对汞的催化氧化机制研究[D]. 武汉: 华中科技大学, 2011. [32] SHEN M Q, LI C X, WANG J Q, et al. New insight into the promotion effect of Cu doped V2O5/WO3-TiO2 for low temperature NH3-SCR performance[J]. RSC Advances, 2015, 5(44): 35155-35165. doi: 10.1039/C5RA04940G [33] LI H L, ZHAO J X, ZHANG W L, et al. NH3 inhibits mercury oxidation over low-temperature MnOx/TiO2 SCR catalyst[J]. Fuel Processing Technology, 2018, 176: 124-130. doi: 10.1016/j.fuproc.2018.03.022 [34] LI H L, WU S K, WU C Y, et al. SCR atmosphere induced reduction of oxidized mercury over CuO-CeO2/TiO2 catalyst[J]. Environmental Science & Technology, 2015, 49(12): 7373-7379. [35] SENIOR C L, SAROFIM A F, ZENG T F, et al. Gas-phase transformations of mercury in coal-fired power plants[J]. Fuel Processing Technology, 2000, 63(2/3): 197-213. [36] ZHAO L K, LI C T, ZHANG X N, et al. A review on oxidation of elemental mercury from coal-fired flue gas with selective catalytic reduction catalysts[J]. Catalysis Science & Technology, 2015, 5(7): 3459-3472. [37] 胡鹏, 段钰锋, 陈亚南, 等. Mo-Mn/TiO2催化剂的协同脱硝脱汞特性[J]. 中国环境科学, 2018, 38(2): 523-531. doi: 10.3969/j.issn.1000-6923.2018.02.014 [38] XU W Q, WANG H R, ZHOU X, et al. CuO/TiO2 catalysts for gas-phase Hg0 catalytic oxidation[J]. Chemical Engineering Journal, 2014, 243: 380-385. doi: 10.1016/j.cej.2013.12.014 [39] LI H L, WU C Y, LI Y, et al. CeO2-TiO2 catalysts for catalytic oxidation of elemental mercury in low-rank coal combustion flue gas[J]. Environmental Science & Technology, 2011, 45(17): 7394-7400. [40] HUANG W J, XU H M, QU Z, et al. Significance of Fe2O3 modified SCR catalyst for gas-phase elemental mercury oxidation in coal-fired flue gas[J]. Fuel Processing Technology, 2016, 149: 23-28. doi: 10.1016/j.fuproc.2016.04.007 [41] CHI G L, SHEN B X, YU R R, et al. Simultaneous removal of NO and Hg0 over Ce-Cu modified V2O5/TiO2 based commercial SCR catalysts[J]. Journal of Hazardous Materials, 2017, 330: 83-92. doi: 10.1016/j.jhazmat.2017.02.013 [42] HE S, ZHOU J S, ZHU Y Q, et al. Mercury oxidation over a vanadia-based selective catalytic reduction catalyst[J]. Energy & Fuels, 2009, 23(1): 253-259. [43] ZHAO L K, LI C T, LI S H, et al. Simultaneous removal of elemental mercury and NO in simulated flue gas over V2O5/ZrO2-CeO2 catalyst[J]. Applied Catalysis B:Environmental, 2016, 198: 420-430. doi: 10.1016/j.apcatb.2016.05.079 [44] ZHANG Q L, XU L S, NING P, et al. Surface characterization studies of CuO-CeO2-ZrO2 catalysts for selective catalytic reduction of NO with NH3[J]. Applied Surface Science, 2014, 317: 955-961. doi: 10.1016/j.apsusc.2014.09.017 [45] LEE S. M, HONG S. C. Promotional effect of vanadium on the selective catalytic oxidation of NH3 to N2 over Ce/V/TiO2 catalyst[J]. Applied Catalysis B:Environmental, 2015, 163: 30-39. doi: 10.1016/j.apcatb.2014.07.043 [46] ZHAO X, HUANG L, LI H, et al. Highly dispersed V2O5/TiO2 modified with transition metals (Cu, Fe, Mn, Co) as efficient catalysts for the selective reduction of NO with NH3[J]. Chinese Journal of Catalysis, 2015, 36(11): 1886-1899. doi: 10.1016/S1872-2067(15)60958-5 [47] DONG L, ZHANG L, SUN C, et al. Study of the properties of CuO/VOx/Ti0.5Sn0.5O2 catalysts and their activities in NO+CO reaction[J]. Acs Catalysis, 2011, 1(5): 468-480. doi: 10.1021/cs200045f [48] CHEN B, XU R, ZHANG R, et al. Economical way to synthesize SSZ-13 with abundant ion-exchanged Cu+ for an extraordinary performance in selective catalytic reduction (SCR) of NOx by ammonia[J]. Environmental Science & Technology, 2014, 48(23): 13909-13916. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3368
- HTML全文浏览数: 3368
- PDF下载数: 78
- 施引文献: 0
