




-
醛酮类化合物被广泛应用于有机合成、化工、合成纤维、染料、农药、木材加工及制漆等行业。一些醛酮类化合物有毒或为致癌物,会刺激皮肤与粘膜及毒害中枢神经系统,具有遗传毒性等。国内外对醛酮类分析检测方法报道较多,但多数针对水中[1-4]、环境空气[5-11]、车间空气[12-13]、车内空气[14-15]和汽车尾气[16-17]等方面的研究,分别采用不同的衍生化试剂及检测手段,本文主要研究了固定源废气中醛酮类污染物的测定,用2,4-二硝基苯肼(DNPH)作为衍生化试剂,在酸性条件下,与醛酮反应生成2,4-二硝基苯腙类化合物,对采样、样品稳定性、腙类化合物的萃取等进行了系统研究。
-
Agilent 1100型液相色谱仪,二极管阵列检测器,配有自动进样器。Agilent ODS-C18色谱柱:250 mm×4.6 mm。重蒸蒸馏水;乙腈、二氯甲烷、正己烷:色谱纯。
2,4-二硝基苯肼(国药沪试)吸收液:称取4.0 g 2,4-二硝基苯肼固体于棕色试剂瓶中,加入180 mL盐酸,再加入820 mL水,超声30 min。形成饱和溶液,先后用二氯甲烷和正己烷萃取纯化。吸收液应在采样前48 h内制备和纯化。
醛、酮类-DNPH衍生物-乙腈标准溶液:浓度200 µg/mL(美国AccuStandard公司):包括甲醛-DNPH、乙醛-DNPH、丙烯醛-DNPH、丙酮-DNPH、丙醛-DNPH、丁烯醛-DNPH、丁醛-DNPH、苯甲醛-DNPH、异戊醛-DNPH、正戊醛-DNPH、邻甲基苯甲醛-DNPH、间甲基苯甲醛-DNPH、对甲基苯甲醛-DNPH、正己醛-DNPH、2,5-二甲基苯甲醛-DNPH,2-丁酮-DNPH。
醛、酮类化合物-乙腈标准溶液:浓度1 000 µg/mL(美国AccuStandard公司)。
-
(1)等速采样研究
美国EPA 0011方法[18]采用等速采样采集固定源废气中的醛酮类化合物。本实验参考该方法,进行模拟实验。
模拟实验1:在玻璃纤维滤筒上加入醛酮混合标准溶液(加标量为20.0 µg),将3支装有100 mL DNPH饱和溶液的气泡吸收瓶和一支空吸收瓶串联到烟尘采样器,在采样管不加热的情况下,以10 L/min模拟采样60 min,分别测定玻璃纤维滤筒和吸收瓶中DNPH饱和吸收液中醛、酮类化合物的含量。
模拟实验2:采样方式同模拟实验1,采样结束后,用二氯甲烷清洗采样时接触到的所有表面(包括探头喷嘴、探针配件、探针衬垫、第一吸收瓶、吸收瓶连接器),将清洗液与吸收瓶中DNPH饱和吸收液合并,用二氯甲烷萃取,按照废气样品的分析步骤分析测定。
模拟实验3:在已采集颗粒物的玻璃纤维滤筒上加入醛酮混合标准溶液(加标量为20.0 µg),将3支装有100 mL DNPH饱和溶液的气泡吸收瓶和一支空吸收瓶串联到烟尘采样器,采样管温度大于120 ℃,以10 L/min模拟采样60 min,分别测定玻璃纤维滤筒和吸收瓶中DNPH饱和吸收液的采样效率(测定方法同模拟实验1)。
(2) 恒流采样研究
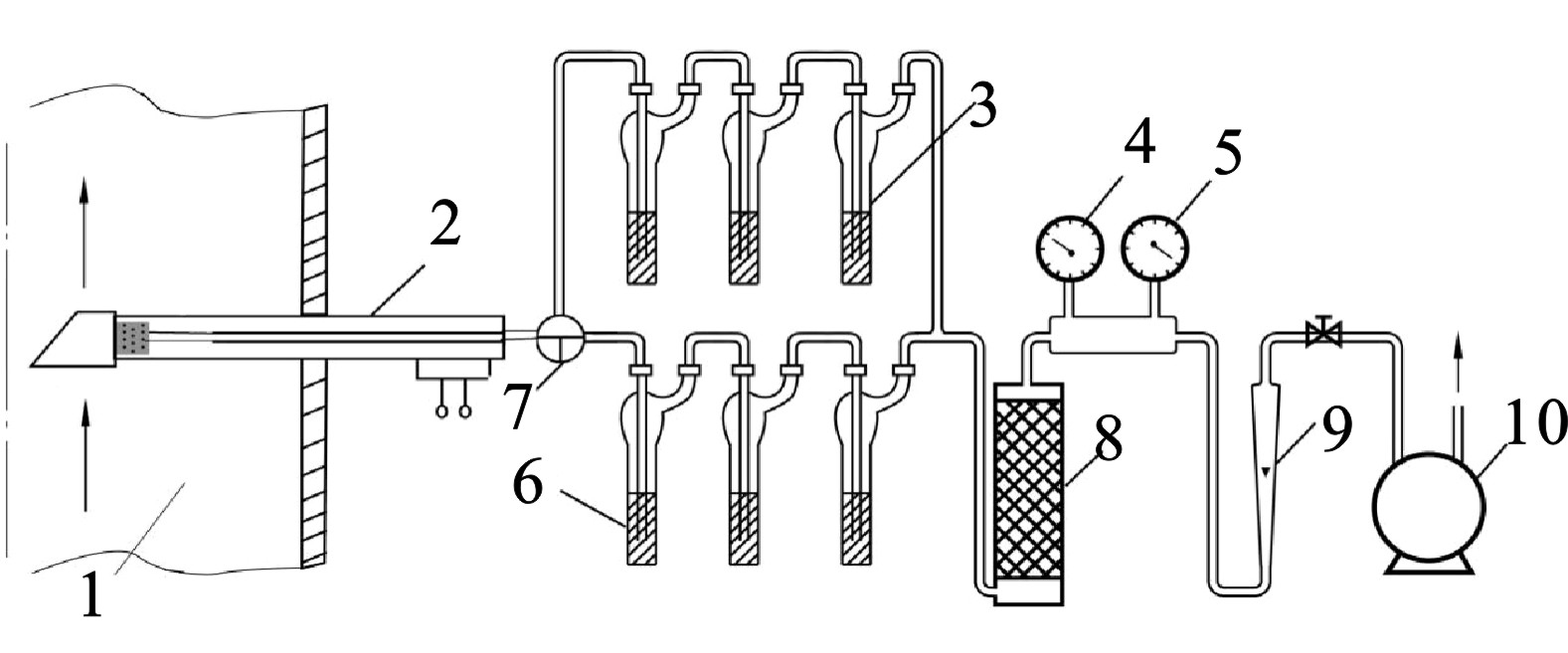
固定污染源废气的布点、采样及参数测定应符合GB/T 16157和HJ/T 397中的相关规定,采样装置,见图1。
串联3支各装有50 mL DNPH饱和吸收液的棕色气泡吸收瓶,与烟气采样器连接,按照气态污染物采集方法,以0.2 ~0.5 L/min的流量,连续采样1 h,或在1 h内以等时间间隔采集3~4个样品,采样期间流量波动应≤±10%。采样过程中,应保持采样管保温夹套温度不低于120 ℃,以避免采集气体中的水汽于吸收瓶之前凝结。
采样结束后,切断采样泵和吸收瓶之间的气路,抽出采样管,取下吸收瓶,用密封帽密封避光保存。
-
样品应于4 ℃以下密封避光冷藏保存,样品采集后3 d之内完成试样制备,制备好的试样在3 d内完成分析。
-
将吸收瓶中的样品转移至250 mL分液漏斗中,用10 mL二氯甲烷-正己烷混合溶液或二氯甲烷萃取、萃取3次,收集有机相于150 mL三角瓶中,加入无水硫酸钠至硫酸钠颗粒可自由流动。浓缩至近干,更换溶剂为乙腈,并用乙腈定容至10.0 mL。
-
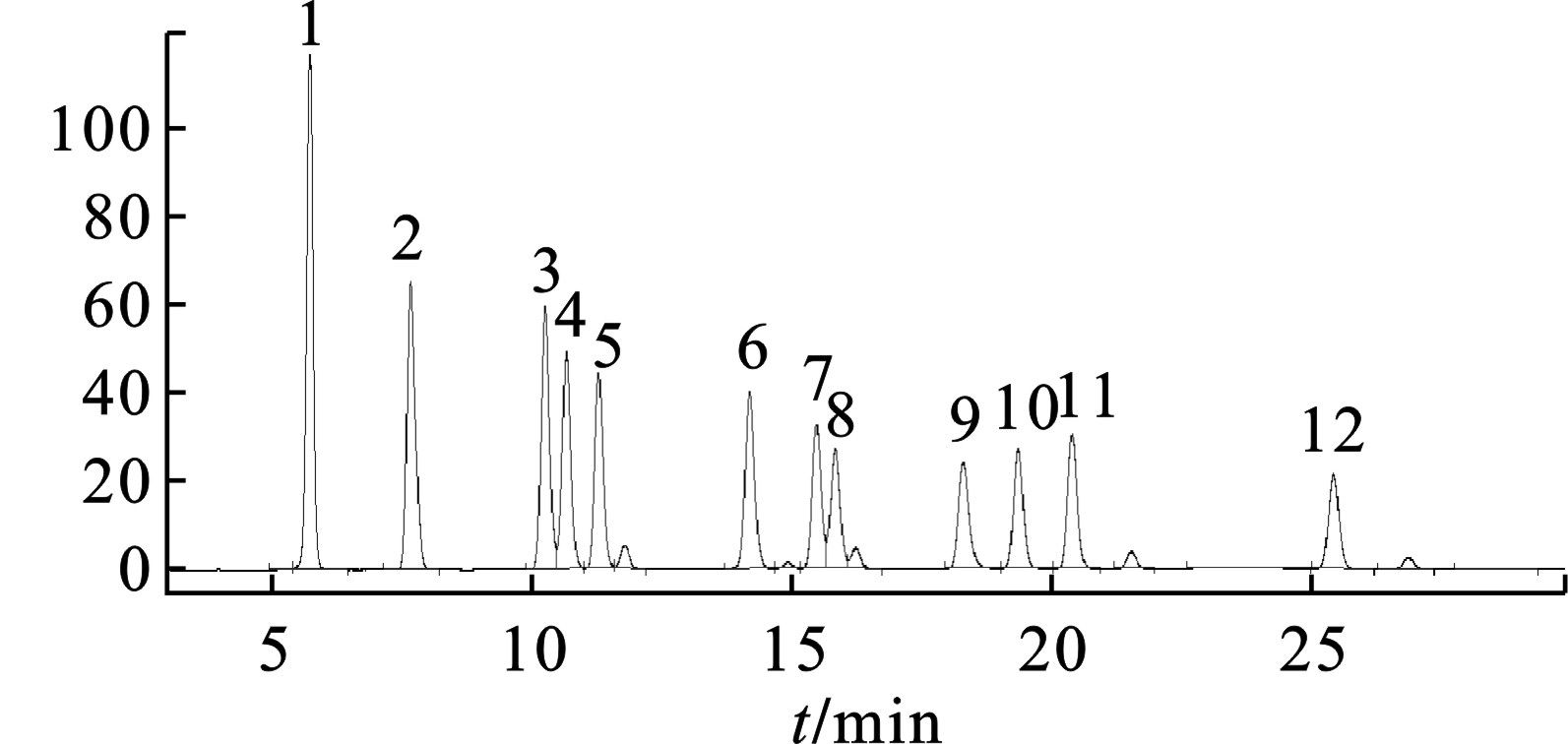
色谱条件:柱温箱温度:35 ℃;进样体积:10 μL;紫外检测器波长:360 nm。流动相A:乙腈,流动相B:水,流动相C:甲醇。梯度洗脱程序,见表1。
定性定量方法:根据保留时间、样品的紫外光谱和标准溶液的紫外谱图比较进行定性,外标法定量。
-
本实验室分别以二氯甲烷、正己烷、正己烷/二氯甲烷(7+3,V/V)和二氯甲烷/正己烷(1+1,V/V)为萃取剂,对加标量为2.0 μg醛酮衍生物的2,4-二硝基苯肼吸收液进行萃取,结果表明正己烷/二氯甲烷(7+3,V/V)和二氯甲烷对醛酮衍生物的萃取效率高于其他溶剂,但二氯甲烷在下层,方便萃取操作,见表2。
-
按照1.2.1.1连接采样系统,分别按模拟实验1~3操作步骤,以10 L/min流量采气60 min后,将吸收瓶中吸收液转移至1 000 mL分液漏斗中,用二氯甲烷萃取吸收液,按照废气样品的分析步骤分析;玻璃纤维滤筒放入棕色样品瓶中,加入2 mL DNPH乙腈溶液,10 μL盐酸,再加入适量乙腈,放置30 min,超声15 min,然后将提取液过滤后转移至浓缩瓶中,用乙腈第二次冲洗滤筒,冰水浴超声15 min,将2次洗脱液混合后浓缩分析,见表3。
表3可知,模拟实验1实验结果表明,当采样管不加热时,玻璃纤维滤筒中未检出醛酮类化合物,但吸收液中苯甲醛、甲基苯甲醛和2,5-二甲基苯甲醛回收率较低;模拟实验2实验结果表明,当采样管不加热时,高沸点醛酮类化合物(苯甲醛、甲基苯甲醛和二甲基苯甲醛)会附着在采样时接触到的采样系统表面(尤其是排气筒是高湿的情况)。模拟实验3实验结果表明,当采样管加热时,玻璃纤维滤筒中也未检出醛、酮类化合物,吸收液中醛、酮类化合物的采样效率在50%以上(2,5-二甲基苯甲醛除外),因此,模拟固定污染源废气实验中醛、酮类化合物各组分主要分布在气相中。因为无法模拟真正的颗粒物,也无法找到合适的污染源,去验证高沸点醛酮类是否存在于颗粒物,完全采用等速采样采集固定源样品,操作十分复杂,不易推广。另外实验结果显示,高温高湿条件下,在加热的采样管壁和玻璃纤维滤膜中均未检出醛酮类化合物,即醛、酮类化合物各组分主要分布在气相中,因此本方法采样方式确定为恒流采样。
-
用液体吸收法采集空气样品时,通常使用两个采样瓶串联采样,但本方法在样品采集的同时需要进行衍生化,因此,我们试验了以串联四支各装有50 mL吸收液的气泡式吸收瓶,在第一支吸收瓶口加入醛酮混合标准溶液(加标量为40.0 µg),按照气态污染物采集方法,采气流量分别为0.2、0.5、0.8 L/min,模拟采集有组织排放废气中醛、酮类化合物样品连续采样1 h,分别测定每一吸收瓶中醛酮类化合物的浓度,计算每一吸收瓶的吸收效率(每一吸收瓶的采样量与总采样量之比),见表4~6。
表4~6可知,在0.2~0.5 L/min采样流量条件下,除2-丁酮外,其他化合物采样效率都能稳定达到70%以上。对于大多数化合物第一和第二吸收瓶合并吸收效率都在90%以上,但丙酮和2-丁酮在0.8 L/min采气流速下,第三支吸收瓶中的吸收效率仍在10%以上,因此,在采集有组织排放废气样品时采样流量选择0.2~0.5 L/min,必须串联3支装有50 mL DNPH饱和吸收液的气泡吸收瓶。
-
在ODS-C18和乙腈/水二元混合溶剂组成的色谱体系中,12种醛酮腙类化合物中有2组难分离物质对,分别是丙烯醛/丙酮,2-丁酮/正丁醛。在乙腈-水二元梯度体系中,当提高乙腈的比例时,有利于丙烯醛和丙酮的分离,但另外一组难分离物质对的分离度又会降低,在甲醇-水二元梯度体系中2组难分离物质对都能较好分离,但初始柱压较高,醛类-DNPH有同分异构体的峰出现,基线有漂移;在甲醇-乙腈-水-四氢呋喃四元梯度体系中丙烯醛/丙酮和2-丁酮/正丁醛都可以得到较好分离,但梯度洗脱程序复杂,另外,四氢呋喃的引入,使得醛类-DNPH都有同分异构体的峰出现。经过多次实验,根据谱图中醛酮腙类化合物各组分的分离情况和出峰时间的长短,综合比较分离效果、基线漂移,以及待测组分与样品基质中干扰物质的分离等情况,最后采用梯度洗脱和甲醇-乙腈-水作为流动相以达到最佳分离,见图2。
-
对样品稳定性进行了测试,将醛酮类化合物标准溶液加到DNPH饱和吸收液中,在4 ℃以下密闭、避光保存一定时间后,按照样品分析步骤进行测定,见表7。
表7可知,多数醛酮类化合物在实验条件下存放7 d都比较稳定,但2-丁酮样品的测定结果随存放时间变化较大,而且采用不同萃取溶剂的变化趋势相反,当采用二氯甲烷萃取样品时,样品测定结果随存放时间变长而逐渐降低;而当采用正己烷/二氯甲烷(7+3,V/V)萃取样品时,样品测定结果随存放时间变长而逐渐增大,因此,在样品采集后放置时间较长时,推荐采用二氯甲烷-正己烷混合溶液萃取样品。
-
串联四支各装有50 mL吸收液的气泡式吸收管,按照气态污染物采集方法,以0.5 L/min的流量,连续采样20 L,测定后3支吸收管中各醛酮类化合物的空白值;其他组分是将1.0 µg标准溶液加于第二支装有50 mL吸收液的吸收管中,采用同样方法采样,进行7次平行测定。方法的检出限为0.005~0.010 mg/m3。
-
分别采集东北制药总厂、中远船务、大连船舶重工和大连机车厂等企业有组织排放样品,见表8。
将采集后的有组织排放样品混合均匀作为实际样品。其中一个为实样样品本底,另外6个再加入5.0 µg醛酮类化合物标准溶液,重复测定六次计算回收率和相对标准偏差,加标回收率在64.6%~109%之间,变异系数在3.9~10.1%之间,见表9。
-
本方法适用于固定污染源废气中12种醛、酮类污染物的检测。醛、酮类化合物各组分主要分布在气相中,在采集有组织排放废气样品时采样流量选择0.2~0.5 L/min,必须串联3支装有50 mL DNPH饱和吸收液的气泡吸收瓶。采集后的样品用二氯甲烷-正己烷混合溶液或二氯甲烷萃取,加标回收率在64.6%~109%之间,变异系数在3.9%~10.1%之间。当采集有组织排放废气20 L,定容体积10.0 mL时,方法的检出限为0.005~0.010 mg/m3。
固定污染源废气中醛酮类化合物测定方法研究
Determination of aldehyde and ketone compounds from an emission of stationary source
-
摘要: 建立了固定污染源排放废气中的醛、酮类化合物的测定方法。用酸性2,4-二硝基苯肼(DNPH)吸收液采集废气样品,并发生衍生化反应,生成2,4-二硝基苯腙类化合物,用溶剂萃取后,经高效液相色谱分离检测。加标回收率在64.6%~109%之间,当采样体积20 L时,方法的检出限为0.005~0.010 mg/m3。可用于固定污染源废气中12种醛、酮类污染物的检测。Abstract: A determination method for aldehyde and ketone compounds in the waste gas from an emission of a stationary source was established. The waste gas was collected by aqueous acidic 2,4-dinitrophenylhydrazine, which can introduce the derivative reaction to produce the 2,4-dinitrophenylhydrazones. Its derivative was extracted, and then analyzed by using high performance liquid chromatography. The recovery ranged from 64.6% to 109%. The detection limit for 20 L sample was 0.005 mg/m3 to 0.010 mg/m3. This method could determine 12 kinds of aldehydes and ketones in the waste gas from an emission of a stationary source.
-
煤矸石是煤炭开采过程中的大宗工业固废,目前处置方式以堆存为主,存在占用土地、生态破坏、地下水污染等问题。煤矿瓦斯在经燃烧利用所产中低温烟气余热未得到有效利用,造成能源的大量浪费。
煤矸石在矿物组成上与土壤具有高度的相似性,但因其粒径粗大、保水性能差等原因,煤矸石大规模生态化利用受限。魏忠义和王秋兵[1]研究认为微粒含量上升即持水率提高是煤矸石复垦的先决条件,胡振琪等[2]认为矿区复垦重点在于土壤性能综合提升。煤矸石颗粒粉化可提升基质持水能力,实现基质成土的初期演替。自然环境中,煤矸石等岩石颗粒的风化粉化主要由温度和水分驱动,但自然扰动强度小、频率低,煤矸石风化进程缓慢。蔡毅等[3]对淮南矿区煤矸石进行采样分析,结果表明风化2~12年的煤矸石持水率才具有较为明显的提升。张清峰等[4]研究表明煤矸石经36个月风化后含水率趋于稳定并超过10%。针对煤矸石风化粉化,梁冰等[5]利用木霉菌对煤矸石进行分解改良,作用30 d后,0.5 mm以下微粒上升了5%左右,时间成本较高;尚志等[6]、ZHANG等[7]采用冻融循环提高煤矸石黏粒细砂含量,研究表明冻融对土壤矿物化学性能改变不大,但对其粒径粉化效果显著,但冻融循环受环境制约难以大规模推广。张素等[8]认为干湿/冷热扰动有利于基质微粒含量的快速提升。通过瓦斯烟气余热利用对煤矸石进行强化风化处理,增强环境扰动的强度和频率,有望实现煤矸石的快速粉化及矿物转化,均衡其土壤学要素,达到综合性能的提升,开辟煤矸石原位风化成土的新途径。所以将煤矸石与瓦斯烟气余热进行协同处理处置,实现资源与能源的高效利用,是煤炭企业绿色可持续发展的重要课题。
针对煤矸石粒径粗大、持水能力薄弱的问题,本研究利用瓦斯烟气余热,强化煤矸石干湿/冷热的交替循环,加速煤矸石结构风化粉化,促进煤矸石的矿物分解与重构,提升风化产物的透气保墒性能,并通过植物生长考核其风化产物的生态化应用效果,以实现煤矸石与瓦斯烟气余热的资源能源的高效利用。
1. 材料与方法
1.1 实验原料
实验所用煤矸石来自云南恩洪煤矿,岩性以页岩、砂岩、泥岩为主,矿物成分主要为钙长石、伊利石、方解石等,以及少量黄铁矿。实验设置3组样品,初始持水率为6.3%±0.5%,每组样品重量为1 kg,样品粒径分布如表1所示。
表 1 煤矸石样品初始粒径分布Table 1. The initial particle size distribution of coal gangue samples5~10 mm 2~5 mm 1~2 mm 0~1 mm 68%±5% 6%±2% 12%±2% 14%±2% | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 实验方法
1) 干湿交替实验。对煤矸石进行高频干湿/冷热交替的扰动模拟,将煤矸石用滤布包裹后放入直径15 cm的PVC反应器,向容器中注入1 000 mL超纯水对煤矸石浸泡30 min并记录容器总重;提起滤布取出样品,沥干30 min,然后在45~50 ℃下烘干8 h,此为1个循环。样品分别经历10、20、30次循环,样品命名为CG1、CG2、CG3,干湿交替过程中堆浸液循环使用,每次浸泡时补充超纯水至初始记录总重,堆浸液命名为R1、R2、R3。反应完成后底部煤矸石微粒采用真空抽滤进行分离。
2) 紫花苜蓿生长试验。处理后的样品各取80 g,混匀后放入种植盆,每盆播种20颗紫花苜蓿种子,观察紫花苜蓿发芽及生长状况。每组样品设计3组平行样。
1.3 分析方法
1) 堆浸液。堆浸液静置沉淀30 min,样品过0.45 μm滤膜,使用电导率仪 (上海雷磁,DDS-11A) 检测其电导率,为减小实验误差,相邻2次电导率取平均值作图。样品分别循环处理完成后,30次循环的堆浸液上清液过0.45 μm滤膜烘干得到堆浸液盐分结晶,测定浸出液盐分的元素种类。样品粒径检测采用干筛法,筛分时间t (
m样品50g×20 2) 风化产物。利用XRD (日本 Rigaku Ultima IV) 检测颗粒矿物种类;将干湿交替循环结束的样品经105 ℃干燥,利用体式显微镜 (江西凤凰 XTL-165-VB) 表征循环处理风化产物表面形貌,用XRF (美国 Thermo Scientific™ Niton™ XL3t 600) 表征结晶盐元素含量,使用SEM-EDS (捷克 TESCAN MIRA LMS) 表征20次循环颗粒表面结晶盐形貌及元素种类;紫花苜蓿发芽后记录发芽时间,待其继续生长7 d后测定其发芽率、用直尺测量茎长。
2. 结果与讨论
2.1 煤矸石干湿/冷热交替盐分溶出规律
如图1所示,干湿/冷热交替过程中,R1、R2、R3堆浸液电导率呈现上升、下降、再次上升的趋势,将0~6次循环划分为干湿交替初期。将7~22次循环划分为干湿交替中期,将22次以后的循环划分为后期。溶液电导率与其离子质量浓度呈正比,说明干湿交替初期煤矸石盐分快速溶出,造成堆浸液电导率上升,中期时堆浸液内盐分下降,而后煤矸石颗粒再次发生溶出。
 图 1 循环过程堆浸液电导率变化情况Figure 1. Changes in conductivity of heap leaching solution during circulation
图 1 循环过程堆浸液电导率变化情况Figure 1. Changes in conductivity of heap leaching solution during circulation干湿/冷热交替初期盐分大量溶出,电导率持续上升说明除颗粒表面游离盐分外,煤矸石可溶性组分也不断溶解。干湿交替过程提高了水分的迁移速率,增加了与可溶性组分的接触面积,对其进行溶解,而后在干燥的条件下水分受热蒸发,向颗粒表面移动,盐分随着水流一起向外转移,并进入溶液。这与SHEILA[9]、ZHANG等[10]研究结果相符,干湿交替促进水分不断侵入新区域,还萌生、连接了微裂隙,提高了岩石颗粒孔隙度。可溶组分的持续溶解和溶出通道的形成加速了干湿交替初期煤矸石所含盐分向溶液转移的过程,导致盐分质量浓度的显著上升。
干湿/冷热交替中期堆浸液电导率下降可能的原因是煤矸石颗粒经历了初期的快速溶出,颗粒表面活性组分含量下降,盐分溶出作用受限。而溶液中盐分在干燥作用下发生重结晶,不但堵塞盐分溶出通道还粘附在容器上,从溶液中脱除;除重结晶作用,盐分还被煤矸石中微粒吸附、固定,当盐分脱除速度高于盐分的溶出速度时,溶液盐分质量浓度下降。ZHAO等[11]对砂岩的研究结果表明,随着干湿交替进行,颗粒中低溶性组分相对含量增加,盐分溶出速率下降。HAYNES等[12]、曹勇等[13]认为非晶铝硅酸盐、页硅酸盐以及采矿伴生的黏土对溶液离子有较强吸附作用。本试验初期的盐分溶出使矿物向非晶格形式或次生黏土矿物转变,溶液中离子的吸附固定作用增强,总体而言,溶液盐分含量变化受溶出作用与多方面的固定作用影响,盐分的固定作用强于溶出作用时,溶液盐分质量浓度下降。
干湿/冷热交替后期溶液电导率上升说明颗粒表面可溶组分再次增加,其可能的原因是煤矸石颗粒形成了大量的活性界面,水的溶出作用会降低表面活性,所以这部分活性界面的形成应来源于干湿交替过程中颗粒的物理性损伤。ZHU等[14]研究煤矸石粉改良膨润土裂隙发育的机理,结果表明干湿交替过程中颗粒内部会形成不与外界相连的裂隙,随着循环次数的增加,裂隙不断发育、连接,最终形成宏观裂隙或断面,与外界联系。HUANG等[15]的研究结果也表示风化导致煤矸石中粗孔、大裂隙发育明显,提升了煤矸石元素的溶出速率。MA等[16]研究了干湿交替对固废再生絮凝膏体的破坏作用,发现样品宏观裂隙是逐渐发育的过程,颗粒受到周期性损伤,在中途的4~5个循环时变质程度显著加大。随着干湿/冷热交替的不断进行,煤矸石内微裂隙逐渐发育,形成宏观断面,暴露出新鲜界面,使后期溶液盐分质量浓度再次上升。
2.2 干湿/冷热交替作用下煤矸石矿物溶出与重构过程分析
如图2所示,煤矸石颗粒表面出现盐分重结晶,能谱表征显示其为硫酸盐,结果表明干湿/冷热交替过程中煤矸石发生了涉硫组分的溶出与重构。煤矸石中涉硫组分以黄铁矿为主,占总硫组分90%以上,所以认为颗粒表面硫酸盐的来源是黄铁矿氧化溶解。硫酸盐随水分向颗粒表面转移,而后水分蒸发,盐分再结晶并附在颗粒表面。黄铁矿氧化过程如式(1)所示。黄铁矿在氧气和水分作用下氧化,干湿交替增大了煤矸石的孔隙度,从而加速氧气与水分的扩散,提高了黄铁矿的氧化速度。
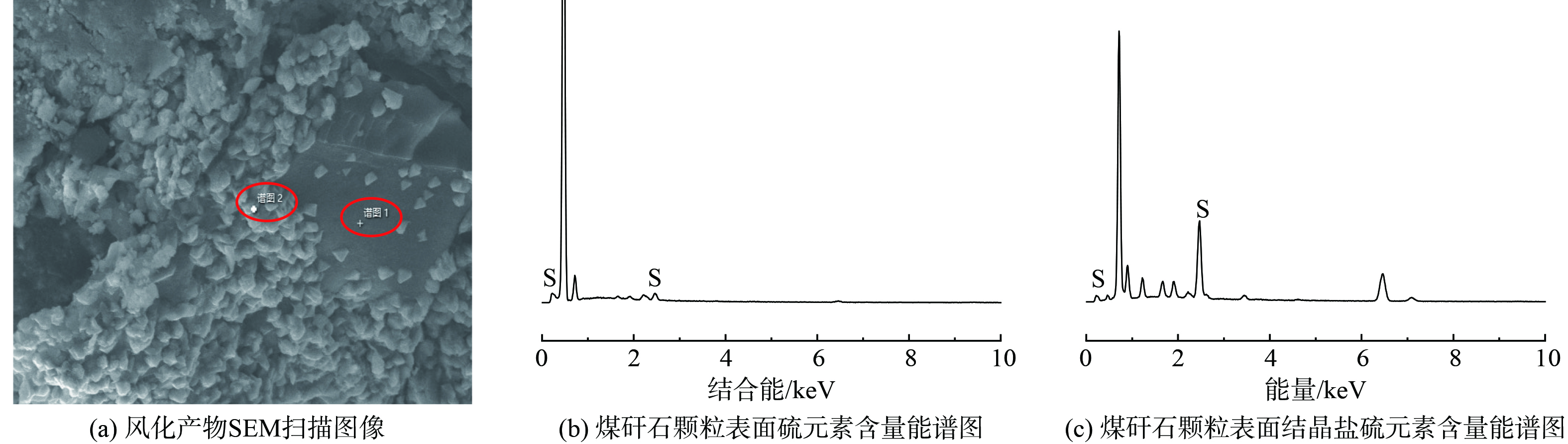 图 2 煤矸石颗粒表面结晶盐显微及能谱分析Figure 2. Microscopic and energy spectrum analysis of crystallized salt on the surface of coal gangue particles
图 2 煤矸石颗粒表面结晶盐显微及能谱分析Figure 2. Microscopic and energy spectrum analysis of crystallized salt on the surface of coal gangue particlesstringUtils.convertMath(!{formula.content}) (1) 干湿/冷热交替处理30次的风化产物进行XRD表征,其结果与原煤矸石结果对比如图3所示。处理后煤矸石所含黄铁矿、钙长石、方解石等衍射强度显著降低,黄铁矿峰趋于消失。但煤矸石为复杂矿物混合物,杂峰较多,且本试验未外加溶蚀药剂对矸石浸出,所以图3显示的前后样品结果变化幅度较小,难以直接说明煤矸石矿物溶出重构过程。CHETTY[17]认为矿渣脉石矿物所含长石、伊利石在表面溶蚀作用下向高岭石转变,YUAN等[18]认为中性条件下,长石向高岭石转变并生成正硅酸。基于图3显示的煤矸石黄铁矿、钙长石、方解石发生溶出,在H+存在及中性条件下的条件下,对含钾含钙矿物进行热力学分析,反应如式(2)~(7)所示。
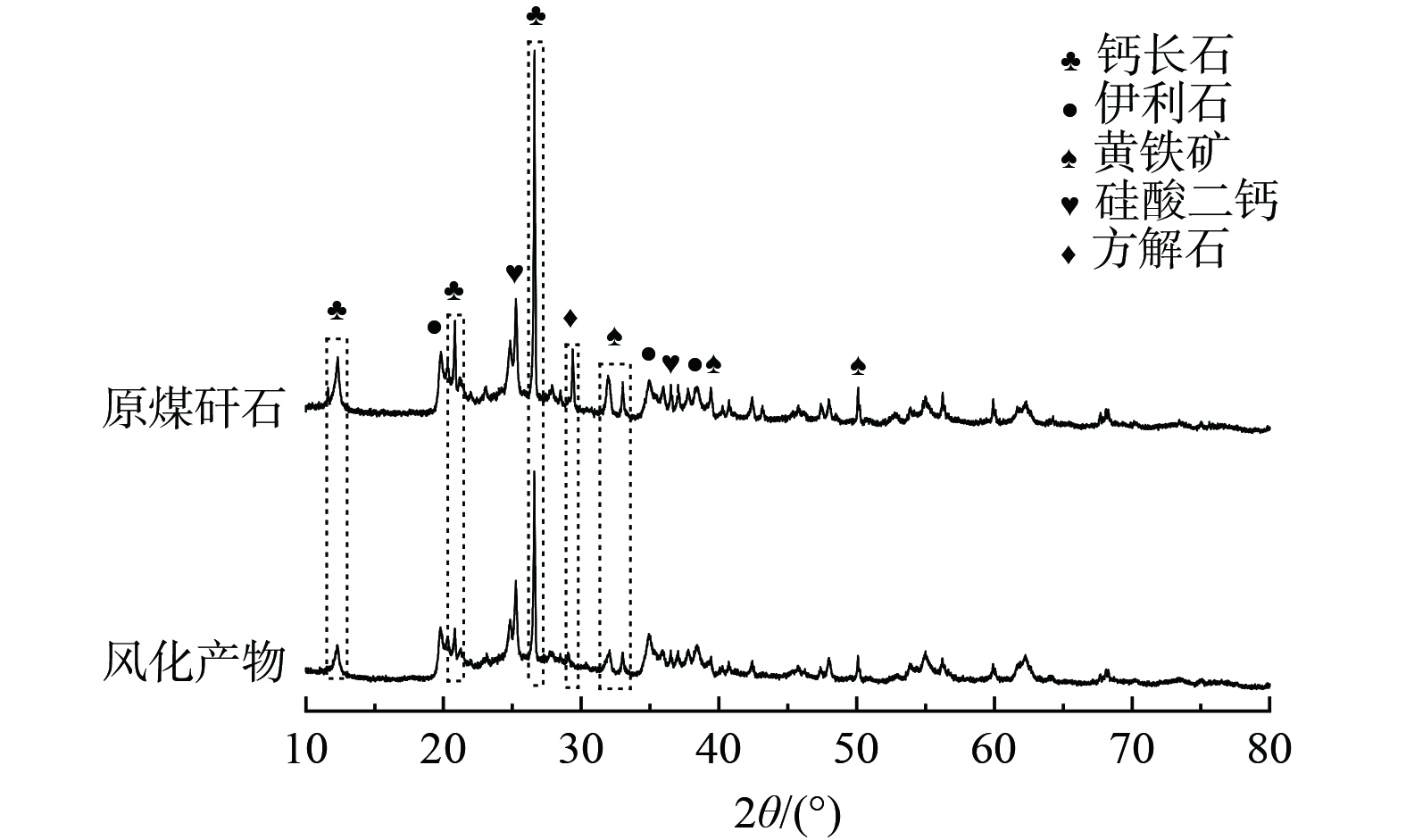 图 3 30次循环处理产物与原煤矸石XRD结果对比图Figure 3. Comparison chart of XRD results between 30 cycles of processing products and raw coal gangue
图 3 30次循环处理产物与原煤矸石XRD结果对比图Figure 3. Comparison chart of XRD results between 30 cycles of processing products and raw coal ganguestringUtils.convertMath(!{formula.content}) (2) stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) stringUtils.convertMath(!{formula.content}) (5) stringUtils.convertMath(!{formula.content}) (6) stringUtils.convertMath(!{formula.content}) (7) 公式(2)~(7)说明钙长石、伊利石等含钾含钙矿物在H+存在时具有自发反应趋势,而在无H+时反应难度较大。煤矸石中含有的黄铁矿在干湿/冷热交替下氧化溶解速率提升,H+产率上升,从而加快长石等硅酸盐矿物溶解速率。NORDSTROM等[19]研究认为硅酸盐矿物、碳酸盐矿物在高浓度酸性矿山废水中起到显著的中和作用,说明黄铁矿的大量氧化形成的H+可与伴生矿物快速反应并导致钾、钙组分的溶出。
样品处理完成后,将30次处理的浸出液烘干得到盐结晶,进行XRD表征,结果如图4所示。堆浸液结晶盐主要成分为硫酸钙、硫酸钠、硫酸钾,说明溶出的盐分中SO42−、K+、Ca2+、Na+含量最高。其中SO42−、K+、Ca2+来源于矿物的溶出,Na+在原煤矸石XRD表征中没有对应的晶型含钠矿物,所以认为钠元素为煤矸石自身所含的可溶性组分,在处理过程中直接溶解在水中,在水分蒸发后与溶液中硫酸根结合,形成硫酸钠。
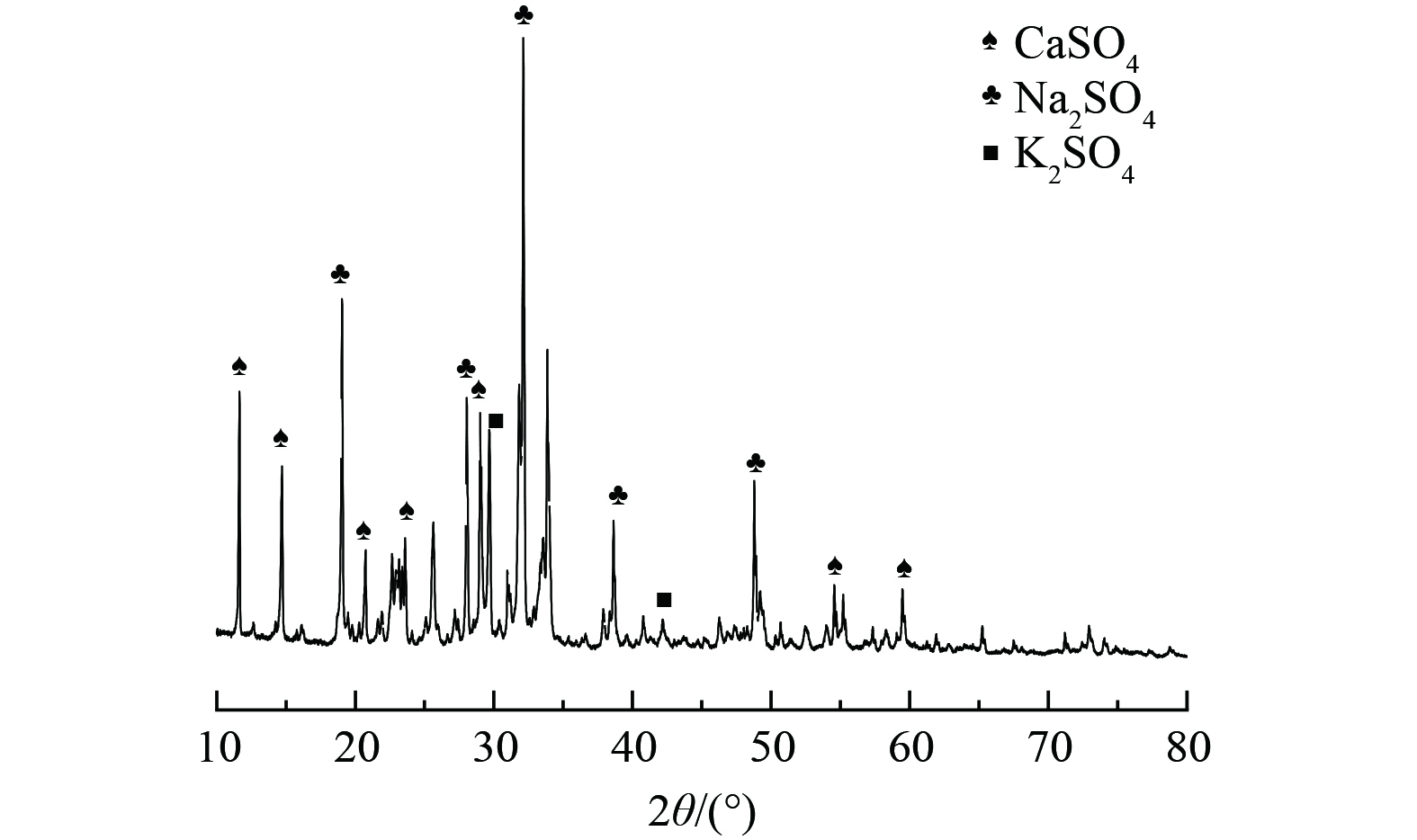 图 4 30次循环浸出液结晶盐XRD结果Figure 4. XRD results of crystallized salt in leaching solution after 30 cycles
图 4 30次循环浸出液结晶盐XRD结果Figure 4. XRD results of crystallized salt in leaching solution after 30 cycles使用XRF测定堆浸液烘干后的盐结晶,结果如表2所示。除硅酸盐矿物组分外,Fe含量也较高,根据前文结果,认为这部分Fe来自黄铁矿的氧化溶出。干湿/冷热交替过程导致岩石矿物碎裂,进而生成大量高反应活性界面,热力学不平衡也会导致矿物化学键的减弱,所以也存在含钾含钙矿物直接与表面水膜发生物质交换的可能。
表 2 堆浸液结晶盐XRF检测结果Table 2. XRF detection results of heap leach liquid crystallization saltmg·kg−1 样品编号 K Ca S Fe R1 2 371 9 557 160.4×103 1 734 R2 4 203 7 396 142.3×103 1 458 R3 820 18.1·103 96.5×103 1 487 | Show TableDownLoad:
CSV
2.3 煤矸石粉化过程分析
如图5所示,干湿/冷热交替处理完成后观察20次处理的颗粒表面形貌,图(a)、(b)、(c)、(d)表明10~30次处理后产物颗粒都出现盐分重结晶现象。所以煤矸石颗粒的粉化除2.2所述可溶矿物的溶解外,还受盐分结晶的挤压作用。高润东等[20]研究认为干湿交替使含硫混凝土盐分溶出,随着蒸发进行,盐分发生重结晶,而结晶作用可对颗粒产生挤压应力,促使裂隙的进一步发育。CELIK和SERT[21]对安山岩进行冻融、热冲击和盐结晶处理,试验表明空隙的重结晶作用有助于裂隙和孔洞的扩大。AN等[22]对长石砂岩进行干湿交替处理,试验发现盐的结晶作用对砂岩的劣化效果最为显著。干湿/冷热交替加速颗粒组分的结晶频率,从而促使颗粒裂隙发育,侵蚀离子、水分子进入颗粒内部,与新鲜活化界面反应、溶出,使煤矸石粉化反应不断进行。
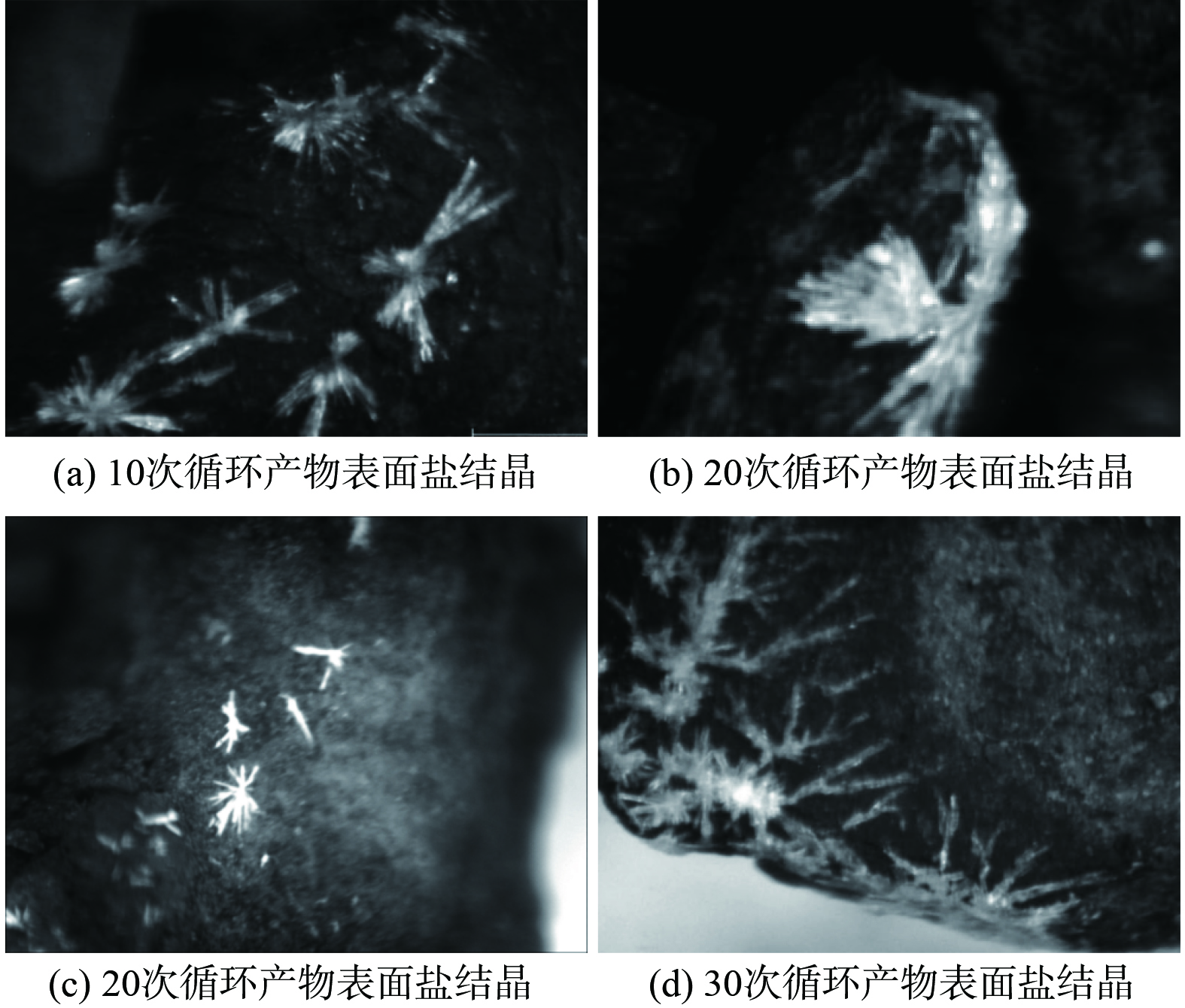 图 5 20次循环处理产物表面盐结晶Figure 5. Salt crystallization on the surface of the product after 20 cycles
图 5 20次循环处理产物表面盐结晶Figure 5. Salt crystallization on the surface of the product after 20 cycles如图6所示,图(a)、(b)、(c)、(d)表明10~30次处理后煤矸石颗粒都受到损伤,出现裂隙,根据煤矸石颗粒岩性,认为这部分裂隙除盐结晶作用外,还可能是黏土矿物不均匀性膨胀、收缩造成的。煤矸石中伊利石、长石等具有不同的膨胀性能,梁冰等[23]对弱崩解泥岩开展干湿循环试验,结果表明含伊利石、高岭石较多的泥岩在干湿交替过程中,界面结合处发生力学破坏能,加速促进裂隙、微裂隙的扩展。YANG等 [24]对泥岩进行干湿交替处理,发现水的侵入导致黏土矿物膨胀,并产生拉伸应力,失水导致的收缩使裂隙不断发展,进而有利于下一次水分的侵入,展示了混合矿物颗粒裂隙发育原理。因此,煤矸石在干湿交替过程中会发生物理性损伤而粉化。
煤矸石颗粒在干湿/冷热交替过程中发生盐分溶解作用、盐结晶挤压作用、黏土矿物崩解作用,3类作用促使煤矸石颗粒不断粉化,形成微粒,过程如图7所示。
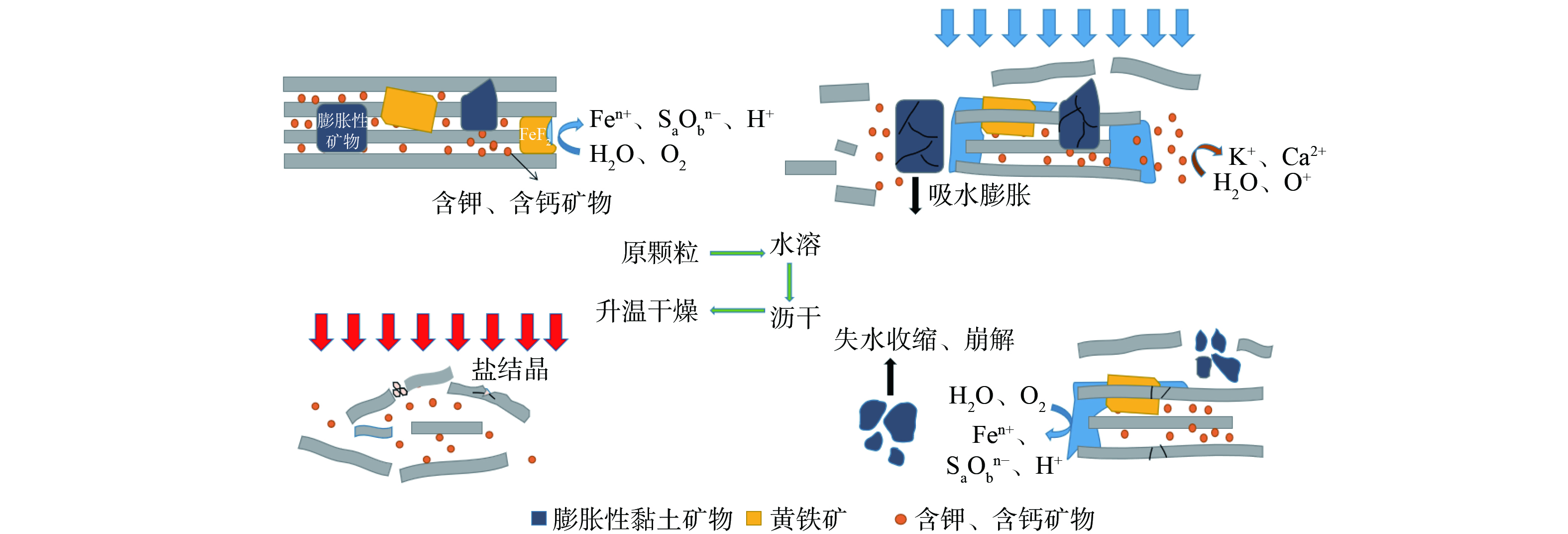 图 7 干湿交替作用下煤矸石风化粉化机理图Figure 7. Mechanism of coal gangue weathering and pulverization under the alternating action of dry and wet
图 7 干湿交替作用下煤矸石风化粉化机理图Figure 7. Mechanism of coal gangue weathering and pulverization under the alternating action of dry and wet2.4 煤矸石风化产物土壤性能
干湿/冷热交替处理后检测煤矸石粉化效果,测定各粒径范围内煤矸石颗粒的含量变化,结果如表3、图8所示。干湿交替对煤矸石颗粒粉化具有显著效果,3组样品中,5~10 mm颗粒显著下降,1 mm以下微粒显著上升,20和30次循环后1 mm以下细砂含量上升30%以上,可达35.4%,较低扰动强度 (10次循环) 对1 mm以下微粒提升也有较好的效果。ZHANG等[25]实验表明干湿交替处理过程中,10次循环时,岩石颗粒微观结构已发生较大变化,20~30个循环后,颗粒劣化速度趋于稳定。1~5 mm颗粒含量略微上升,说明在此粒径范围内集中的主要是结构较为稳定的矿物或岩石颗粒,粉化作用较5~10 mm颗粒弱。
表 3 干湿/冷热交替处理前后煤矸石样品粉化粒径分布Table 3. Pulverized particle size distribution of coal gangue samples before and after dry-wet/cold-heat alternate treatment粒径分布/mm CG3 CG2 CG1 处理前/% 处理后/% 处理前/% 处理后/% 处理前/% 处理后/% 5~10 71.5 28 66.1 24.5 63.1 34.2 2~5 4.8 7.2 9.5 15 7.1 9.3 1~2 12.2 18 12.2 15.6 15 18.5 0~1 11.5 46.9 12.4 44.9 15.2 38 | Show TableDownLoad:
CSV
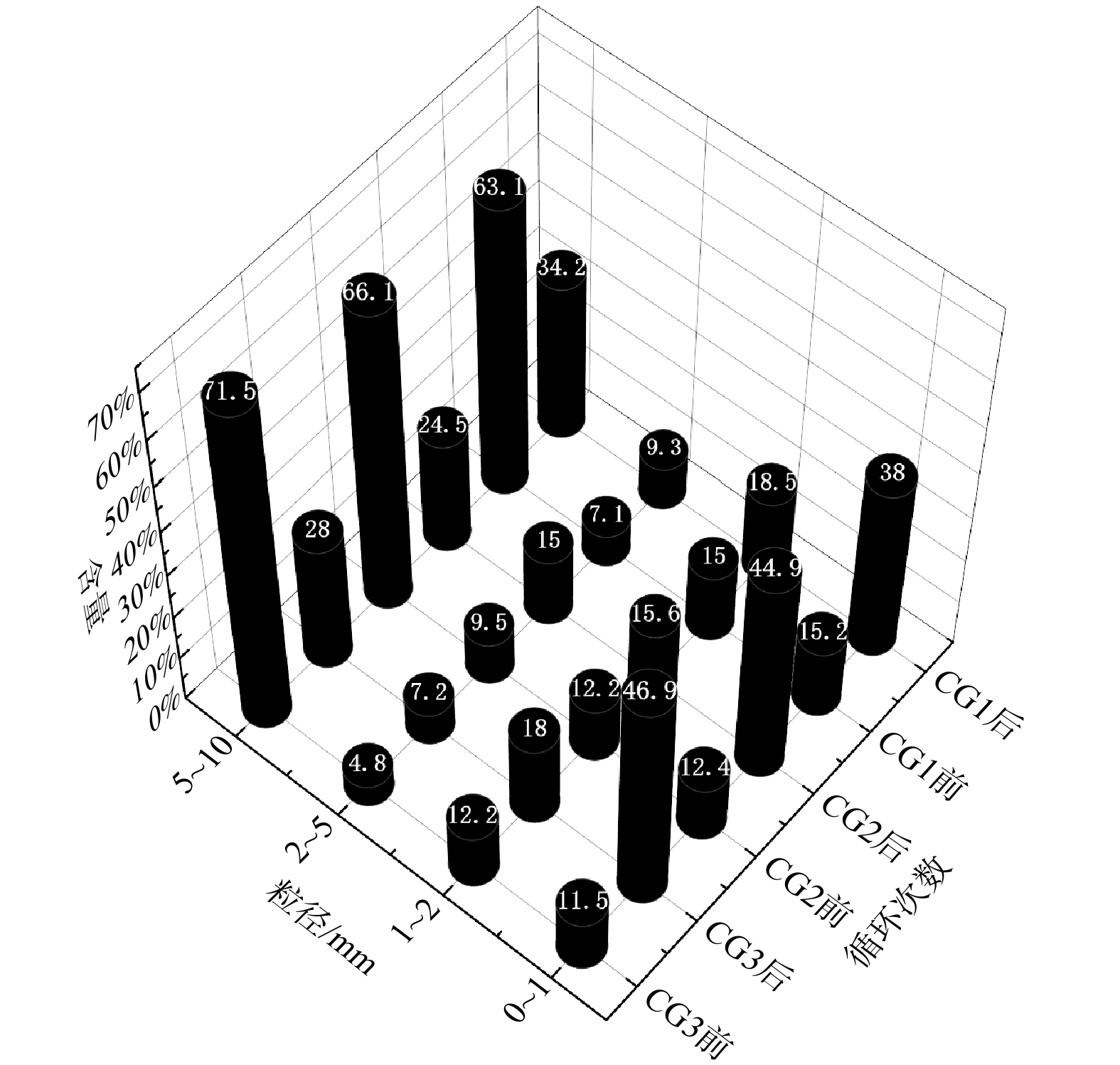 图 8 干湿交替前后煤矸石粒径分布Figure 8. Particle size distribution of coal gangue before and after dry-wet alternation
图 8 干湿交替前后煤矸石粒径分布Figure 8. Particle size distribution of coal gangue before and after dry-wet alternation煤矸石经历干湿/冷热交替后,其粒度显著下降,风化产物的持水率与孔隙度上升,与循环次数呈正比,如图9所示。持水率和空隙度是土壤物理性质的重要指标,两者的提高有利于风化基质的生态化利用。风化产物持水率在30次循环后最大,达到12.82%,该指标满足砂土持水标准。毛管空隙度上升可提高风化产物透气性能,未经处理的煤矸石粒径粗大,颗粒间缝隙大,因此宏观上较为透气,但颗粒本身致密,无法满足土壤基质缓冲、透气的要求。本试验中煤矸石毛管孔隙度随循环次数增加而不断上升,与DEHESTANI等[26]研究结果一致,即岩石颗粒的有效孔隙度与干湿循环次数呈正比。
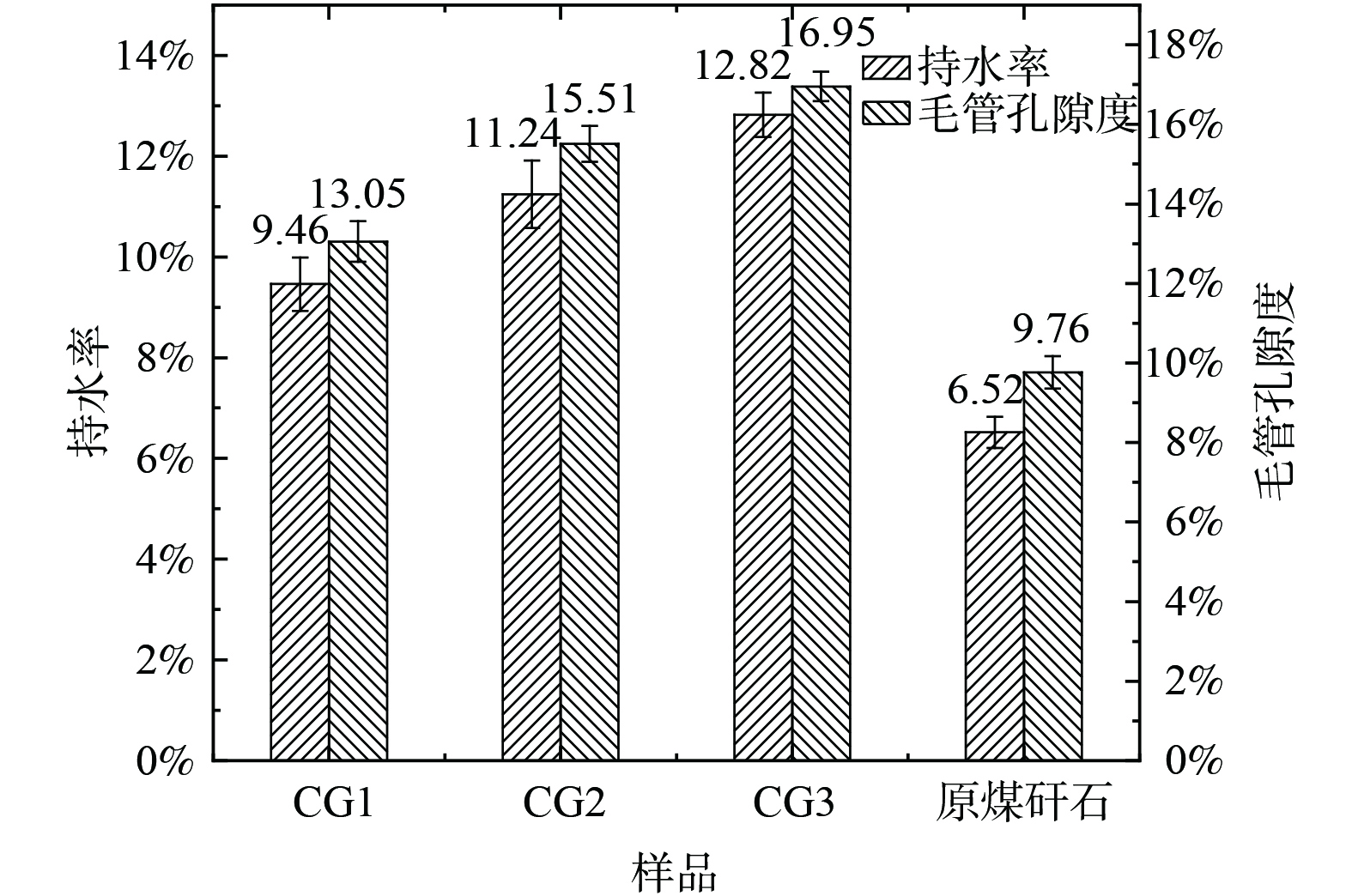 图 9 干湿交替前后煤矸石持水率及孔隙度Figure 9. Water holding capacity and porosity of coal gangue before and after dry-wet alternation
图 9 干湿交替前后煤矸石持水率及孔隙度Figure 9. Water holding capacity and porosity of coal gangue before and after dry-wet alternation20与30次循环后,煤矸石1 mm以下微粒增长量相似,但30次循环后风化基质毛管孔隙度提升较多,说明30次循环后样品含有较多的微裂隙或小孔,此结果与前文所述煤矸石电导率变化情况相符合,证明煤矸石颗粒的崩解存在一个缓慢发育的周期,由微裂隙/微孔逐渐扩大,最终形成断面,微粒含量提高。
紫花苜蓿是常见的牧草,是牧业种植的重要经济作物。风化产物进行紫花苜蓿种植,其考核结果如表4所示,经过干湿交替处理的煤矸石基质发芽率、同生长时间茎长等均优于未处理的对照组,图10为3组样品经干湿交替后种植效果展示。
表 4 紫花苜蓿生长指标Table 4. Growth index of alfalfa样品 发芽率/% 茎长/cm 发芽时间/d 原煤矸石 20 0.97 20 10次循环产物 40 1.76 20 20次循环产物 60 2.21 21 30次处理产物 85 2.64 18 | Show TableDownLoad:
CSV
3. 结论
1) 干湿/冷热交替可显著降低煤矸石的结构强度,加速煤矸石的风化粉化,风化产物中0~1 mm微粒含量上升30%以上,风化产物粒度级配得到优化,显著提升了煤矸石风化产物的透气保墒性能。
2) 干湿/冷热交替促进煤矸石发生矿物分解与重构,加速盐分的溶出与重结晶,产生挤压应力加速煤矸石碎裂及矿物稳定化;硫化矿物发生氧化溶出,并轰击硅酸盐矿物,导致K、Ca、S、Si等元素的溶出,提升煤矸石风化产物的肥力性能。
3) 紫花苜蓿种植结果表明,基于余热利用的干湿循环可显著加速煤矸石风化成土过程,显著提升风化产物的土壤学性能,有效促进苜蓿等绿肥植物的生长,进而为煤矸石的大规模生态化利用提供可靠路径。
-
表 1 梯度洗脱程序
t/min 流动相流速/mL·min−1 乙腈/% 水/% 甲醇/% 0 1.0 20 35 45 6 1.0 0 30 70 20 1.0 0 20 80 30 1.0 35 20 45 33 1.0 20 35 45
下载: 导出CSV
表 2 不同萃取溶剂萃取效率比较
目标化合物 加标量/μg 加标回收率测定结果/% 正己烷 二氯甲烷 正己烷/二氯甲烷(7+3,V/V) 正己烷/二氯甲烷(1+1,V/V) 甲醛 2.0 94.9 105.0 110.0 111.0 乙醛 2.0 104.0 106.0 103.0 105.0 丙烯醛 2.0 51.0 78.1 87.0 54.0 丙酮 2.0 77.8 77.5 86.6 77.0 丙醛 2.0 95.2 96.0 102.0 95.0 丁烯醛 2.0 83.1 84.9 91.5 81.5 2-丁酮 2.0 55.7 69.5 55.6 51.9 丁醛 2.0 88.0 99.7 95.0 95.5 苯甲醛 2.0 93.6 96.0 98.0 104.0 异戊醛 2.0 82.7 93.4 97.5 95.0 正戊醛 2.0 82.9 92.2 90.0 89.0 正己醛 2.0 98.9 89.8 96.0 96.0
下载: 导出CSV
表 3 醛酮类化合物在采样体系中的分布
μg 序号 化合物名称 加标量 模拟实验1 模拟实验2 模拟实验3 玻璃纤维滤筒 吸收液 吸收液和二氯甲烷清洗液 玻璃纤维滤筒 吸收液 1 甲醛 20.0 0 19.4 20.3 0 16.6 2 乙醛 20.0 0 18.6 18.0 0 15.3 3 丙烯醛 20.0 0 19.4 15.1 0 16.5 4 丙酮 20.0 0 15.4 16.3 0 15.8 5 丙醛 20.0 0 15.4 16.1 0 16.1 6 丁烯醛 20.0 0 18.7 17.6 0 17.9 7 2-丁酮 20.0 0 11.4 12.8 0 12.5 8 正丁醛 20.0 0 13.8 14.8 0 14.2 9 苯甲醛 20.0 0 7.1 12.9 0 14.7 10 异戊醛 20.0 0 16.5 16.4 0 15.9 11 正戊醛 20.0 0 15.7 16.9 0 15.8 12 邻-甲基苯甲醛 20.0 0 3.0 13.5 0 12.3 13 间-甲基苯甲醛 20.0 0 0 11.4 0 10.1 14 对-甲基苯甲醛 20.0 0 0 9.5 0 10.6 15 正己醛 20.0 0 14.1 16.9 0 15.7 16 2,5-二甲基苯甲醛 20.0 0 0 8.7 0 6.7
下载: 导出CSV
表 4 采样流量0.2 L/min实验结果
化合物名称 第1吸收瓶 第2吸收瓶 第3吸收瓶 采样效率/% 采样量/µg 效率/% 采样量/µg 效率/% 采样量/µg 效率/% 甲醛 34.27 85.7 0.14 0.4 0.02 0.1 86.1 乙醛 38.27 95.7 1.03 2.6 0.10 0.3 98.5 丙烯醛 36.05 90.1 0.25 0.6 0 0 90.8 丙酮 28.42 71.1 5.77 14.4 0.35 0.9 86.4 丙醛 34.89 87.2 1.65 4.1 0 0 91.4 丁烯醛 37.36 93.4 0 0 0 0 93.4 2-丁酮 19.69 49.2 5.03 12.6 0.42 1.1 62.9 正丁醛 27.25 68.1 0.81 2.0 0 0 70.2 苯甲醛 39.81 99.5 0 0 0 0 99.5 异戊醛 34.71 86.8 1.30 3.3 0 0 90.0 正戊醛 33.19 83.0 0.82 2.1 0 0 85.0 正己醛 34.59 86.5 0.95 2.4 0 0 88.9
下载: 导出CSV
表 5 采样流量0.5 L/min实验结果
化合物名称 第1吸收瓶 第2吸收瓶 第3吸收瓶 采样效率/% 采样量/µg 效率/% 采样量/µg 效率/% 采样量/µg 效率/% 甲醛 36.47 91.2 1.97 4.9 0.02 0.1 96.2 乙醛 35.42 88.6 3.84 9.6 0 0 98.2 丙烯醛 35.27 88.2 0.06 0.2 0 0 88.3 丙酮 27.57 68.9 6.48 16.2 0.66 1.7 86.8 丙醛 34.67 86.7 3.12 7.8 0.12 0.3 94.8 丁烯醛 36.93 92.3 0 0 0 0 92.3 2-丁酮 18.23 45.6 5.78 14.5 0.74 1.9 61.9 正丁醛 27.85 69.6 1.98 5.0 0.15 0.4 75.0 苯甲醛 38.03 95.1 0 0 0 0 95.1 异戊醛 36.11 90.3 4.75 11.9 0 0 102 正戊醛 33.04 82.6 1.74 4.4 0 0 87.0 正己醛 34.59 86.5 1.67 4.2 0 0 90.7
下载: 导出CSV
表 6 采样流量0.8 L/min实验结果
化合物名称 第1吸收瓶 第2吸收瓶 第3吸收瓶 第4吸收瓶 采样效率/% 采样量/µg 效率/% 采样量/µg 效率/% 采样量/µg 效率/% 采样量/µg 效率/% 甲醛 35.70 89.3 0.85 2.1 0 0 0 0 91.4 乙醛 32.72 81.8 4.48 11.2 0.47 1.2 0 0 94.2 丙烯醛 33.27 83.2 1.03 2.6 0 0 0 0 85.8 丙酮 20.16 50.4 10.87 27.2 3.59 9.0 1.11 2.8 89.3 丙醛 31.02 77.6 5.28 13.2 0.90 2.3 0 0 93.0 丁烯醛 36.04 90.1 0.19 0.5 0 0 0 0 90.6 2-丁酮 14.18 35.5 7.70 19.3 3.07 7.7 1.35 3.4 65.8 正丁醛 29.02 72.6 2.48 6.2 0.23 0.6 0 0 79.3 苯甲醛 38.94 97.4 0.23 0.6 0 0 0 0 97.9 异戊醛 33.74 84.4 5.75 14.4 0 0 0 0 98.7 正戊醛 35.19 88.0 1.94 4.9 0.24 0.6 0 0 93.4 正己醛 34.36 85.9 2.08 5.2 0 0 0 0 91.1
下载: 导出CSV
表 7 醛酮类化合物加标样品的稳定性(以回收率表示)
% 化合物名称 当天 第二天 第三天 第七天 二氯甲烷 二氯甲烷/正己烷 二氯甲烷 二氯甲烷/正己烷 二氯甲烷 二氯甲烷/正己烷 二氯甲烷 二氯甲烷/正己烷 甲醛 85.3 90.1 88.2 84.5 84.2 85.4 83.8 82.1 乙醛 86.7 80.2 89.1 84.2 90.1 82.6 85.0 80.7 丙烯醛 77.2 73.4 75.1 72.9 70.1 70.1 68.4 68.7 丙酮 79.0 78.6 75.5 71.1 73.9 85.5 69.8 78.9 丙醛 78.3 71.6 83.4 71.4 86.6 76.2 70.5 70.8 丁烯醛 84.9 96.5 90.1 93.6 93.7 90.8 89.6 75.1 2-丁酮 62.2 67.1 58.0 63.2 54.1 71.8 31.9 82.9 正丁醛 68.4 70.1 65.0 66.6 65.9 69.9 69.9 68.9 苯甲醛 89.3 83.5 88.3 87.2 92.3 90.5 88.6 93.0 异戊醛 86.7 77.6 89.2 83.6 86.0 83.2 81.8 84.4 正戊醛 71.9 77.8 74.8 76.9 73.5 76.6 76.1 75.7 正己醛 77.5 72.8 79.7 71.9 81.5 70.5 74.2 72.2
下载: 导出CSV
表 8 有组织排放废气实际样品分析结果
mg·m-3 化合物名称 东北制药总厂 中远船务 大连机车厂 大连船舶重工 甲醛 0.071 0.163 0.164 0.059 乙醛 0.024 0.085 0 0 丙酮 0.110 0.218 0.935 0.198 正丁醛 0 0.014 0 0 注:表中分别采集的丙烯醛、丙醛、丁烯醛、2-丁酮、苯甲醛、异戊醛、正戊醛、正己醛等8个化合物检测值均为0。
下载: 导出CSV
表 9 有组织排放废气实际样品加标回收率
化合物名称 样品含量/µg 加标回收率/% 平均回收量/µg 平均回收率/% 相对标准偏差RSD/% 甲醛 3.90 79.8 91.6 80.2 97.6 87.8 93.6 4.42 88.5 7.5 乙醛 3.14 90.6 86.4 94.0 95.8 80.4 89.0 4.47 89.4 5.7 丙烯醛 0 93.0 83.0 89.8 85.6 80.8 92.0 4.37 87.4 5.7 丙酮 3.31 80.8 78.2 84.4 76.8 82.2 90.6 4.11 82.2 5.5 丙醛 1.17 78.3 81.3 80.1 76.1 84.7 74.3 3.96 79.2 4.3 丁烯醛 0 101.0 104.0 102.0 98.3 107.0 91.8 5.04 101.0 5.3 2-丁酮 0 77.9 67.9 68.6 69.1 69.4 68.9 3.52 70.3 5.3 正丁醛 1.34 88.8 87.0 90.2 70.4 72.0 89.4 4.15 83.0 10.1 苯甲醛 0 77.8 83.0 84.4 88.4 86.2 84.8 4.21 84.1 3.9 异戊醛 3.48 97.7 101.0 93.5 103.0 99.5 109.0 5.03 101.0 4.8 正戊醛 0 79.2 97.4 83.6 84.2 80.4 82.0 4.22 84.5 7.1 正己醛 0 68.4 64.6 72.2 68.4 73.0 74.2 3.51 70.1 4.7
下载: 导出CSV
-
[1] 印楠. 废水中甲醛的测度[J]. 山东环境, 1999, 91(3): 10 − 11. [2] 徐晓力, 徐晓虹, 王宣. 气相色谱法测定废水中的 7 种低分子量醛、酮和醇[J]. 甘肃环境研究与监测, 2000, 13(4): 193 − 194. [3] 胡冠九. HPLC法测定水和废水中的醛酮类化合物[J]. 环境监测管理与技术, 2004, 16(2): 25 − 27. doi: 10.3969/j.issn.1006-2009.2004.02.009 [4] KOBAYASHI K, TANAKA M, KAWAI S. Gas chromatographic determination of lowmolecular-weight carbonyl compounds in aqueous solution as their O-(2, 3, 4, 5, 6-pentafluorobenzyl) oximes[J]. Chromatogr, 1980, 187: 413 − 417. doi: 10.1016/S0021-9673(00)80474-4 [5] LEHMPUHL D W, BIRKS J W. New GC/ECD method for the determination of atmospheric aldehydes and ketones based on cartridge sampling and derivatization with 2, 4, 6-Trichlorophenylhydrazine[J]. Chromatogr A, 1996, 740: 71 − 81. doi: 10.1016/0021-9673(96)00109-4 [6] BÜLDT A, KARST U. N-Methyl-4-hydrazino-7-nitrobenzofurazan as a reagent for air monitoring of aldehydes and ketones[J]. Analytical Chemistry, 1999, 71: 1893 − 1898. doi: 10.1021/ac980946f [7] BINDING N, KLÄNING H, KARST U,et al. Analytical reliability of carbonyl compound determination using 1, 5- Dansylhydrazine derivatization[J]. Analytical Chemistry, 1998, 362: 270 − 273. doi: 10.1007/s002160051072 [8] KÖLLIKER S, OEHME M. Structure elucidation of 2, 4-dinitrophenylhydrazone derivatives of carbonyl compounds in ambient air by HPLC-MS and multiple MS/MS using atmospheric chemical ionization in the negative ion mode[J]. Analytical Chemistry, l998, 70: 1979-1985. [9] 谭培功, 于彦彬, 蒋海威, 等. 大气中醛酮类羰基化合物的研究进展[J]. 环境科学进展, 1999, 7(4): 19 − 22. [10] 于彦彬, 谭培功, 刘赞, 等. 高效液相色谱三元梯度分离法测定大气中11种醛酮类化合物的研究[J]. 分析测试学报, 2000, 19(5): 43 − 46. doi: 10.3969/j.issn.1004-4957.2000.03.013 [11] 祝惠英, 郭素荣, 石磊. 毛细管气相色谱法测定空气中低分子醛酮化合物[J]. 青岛大学学报, 2002, 17(1): 90 − 92. [12] 戴天有, 魏复盛, 彭清涛, 等. 空气和废气中10种醛酮污染物的高效液相色谱测定[J]. 环境科学研究, 1996, 9(6): 29 − 33. doi: 10.3321/j.issn:1001-6929.1996.06.010 [13] 戴天有, 魏复盛, 谭培功, 等. 空气和废气中醛酮污染物的气相色谱测定[J]. 环境化学, 1998, 17(3): 293 − 298. [14] 陆豪等. 列车车厢内醛酮化合物的污染状况[J]. 环境科学, 2005, 26(2): 74 − 77. doi: 10.3321/j.issn:0250-3301.2005.02.015 [15] 邹钱秀, 张卫东, 赵琦, 等. 不同类型新车内醛酮类化合物的污染研究[J]. 中国环境监测, 2012, 28(2): 97 − 100. doi: 10.3969/j.issn.1002-6002.2012.02.023 [16] SWARIN S J, LIPARI F. Determination of formaldehyde and other aldehydes by high performance liquid chro-matography with fluorescence detection[J]. Journal of Liquid Chromatography, 1983, 6: 425 − 444. doi: 10.1080/01483918308076059 [17] GENG A C, CHEN Z L, SIU G G. Determination of low-molecular-weight aldehydes in stack gas and automobile exhaust gas by liquid chromatography[J]. Analytica Chimica Acta, 1992, 257(1): 99 − 104. doi: 10.1016/0003-2670(92)80155-Z [18] U. S. Environmental Protection Agency. Sampling for selected aldehyde and ketone emissions from stationary sources. method 0011[S/OL]. (2018-08-01). https://www.epa.gov/sites/default/files/2020-04/documents/method_0011_0.pdf, 1996. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3018
- HTML全文浏览数: 3018
- PDF下载数: 4
- 施引文献: 0
