



-
石油烃污染地块主要是石油在开采、运输、使用和储存过程中泄露而造成的[1],据相关资料显示,我国采油区点位超标率已达23.6%[2],随着交通基础设施发展,加油站的泄露成为主要污染源之一[3]。石油类污染成分复杂,通常有几十、上百种成分,按结构可分为烷烃、环烷烃、芳香烃和烯烃[4]。这些物质进入到土壤、地下水、地表水和大气中都会给人体健康及生态环境安全造成威胁。
建设用地在开展修复活动之前,需要评估其可能会对环境和人体造成的危害程度。依据风险程度不同制定相应的管理方法[5]。石油烃由于成分复杂,各组分毒理性质不同,如何科学、准确的开展评估工作成为了国内外有关学者的关注问题。1995年美国材料与试验协会(ASTM)出台了《石油泄漏场地基于风险的纠正行动标准导则》等相关技术导则,美国GSI公司基于该系列准则开发了RBCA商用模型,目前已成为广泛应用的风险评估模型之一[6-7]。我国的风险评估工作起步较晚,目前出台的国家导则规定了石油烃(C10~C40)的相关限值,但没有规定具体的风险评估模型和方法[8-9]。依据上海市的相关技术文件[10],本研究使用分段评估的方法,以某石油烃污染地块为例,利用《建设用地土壤污染风险评估技术导则:HJ25.3—2019》(简称“RAG-C”模型)和RBCA模型分别进行人体健康风险评估,分析了两者的异同,以期为石油烃的风险评估技术提供借鉴。
-
该石油烃污染地块位于我国某工业百强市,由于曾作为油品加工企业生产用地,企业在环保整治专项行动中关停,经若干年的生产活动所产生的石油类物质进入环境,存在污染风险。
-
依据本地块地质探勘资料,除表层杂填土外,主要地层结构由黏土组成。潜水含水层埋深约1.5 m,土壤相关理化参数,见表1。
-
初步调查阶段采用专业判断法在原厂的生产区域内进行布点采样,详细调查阶段在初步调查的超标点位附近按照20 m×20 m的网格进行加密布点,在厂区其余区域按照40 m×40 m的网格布设采样点位,最大采样深度达12 m。在S1~S25点位采集土壤样品、SW1~SW8采集土壤及地下水样品,W2-1~W3-8采集地下水样品,共计采集土壤样品216个,地下水样品20个,地块中石油烃污染分析数据,见表2。
-
RBCA模型的评价方法是将石油烃分成若干馏分,依据分段的毒性参数开展风险评估。石油烃属于非致癌物质,模型计算其危害商,判定标准为1[11]。非致癌物质的危害商(HQ)计算,见式(1):
式中:IR为摄入比例;EF为暴露频率;ED为暴露持续时间;BW为受体质量;AT为平均暴露时间;RfD为参考剂量。下标oral、dermal、inh分别为经口摄入、皮肤接触和呼吸吸入。
-
我国在借鉴美国等发达国家经验的基础上编制了《污染场地风险评估技术导则:HJ 25.3—2014》,并于2019年更新为《建设用地土壤污染风险评估技术导则: HJ 25.3—2019》[9],其核心理念是基于风险地块管理。计算非致癌风险危害商时,参考上海市相关技术文件中规定的石油烃的毒性参数,对石油烃进行分段评估,见式(2):
式中:Ernc为非致癌暴露量;OIS、DCS、PIS、IOV、IIV分别为经口、皮肤接触和吸入土壤颗粒,室外吸入蒸汽,室内吸入蒸汽;c为污染物浓度;下标sur、sub分别表示表层土、下层土;RfD为参考剂量;SAF为参考计量分配系数。
-
RBCA模型与RAG-C相似处居多,尤其在评估程序、多层次评估架构和风险计算方法等方面[12]。但由于两国国情不同,在计算时仍有诸多差异。如RBCA不考虑室内土壤灰尘的暴露途径,RAG-C不考虑土壤淋溶到地下水中对地表水环境质量的影响。RAG-C的敏感人群包含儿童与成人,RBCA另外考虑了对建筑工人及青年的影响。
-
模型参数包括:1)污染区参数、土壤参数、建筑物参数和暴露参数,依据本地块的土壤类型、实测水文地质参数和导则相关规定参数输入;2)石油烃分段比例及毒理参数,见表3,取自上海市相关技术文件附件2~附件4;3)敏感受体及暴露参数,本地块后续作为第二类用地,不考虑对儿童作为敏感受体。暴露参数,见表4。
-
在计算风险时,以稳定地下水埋深作为划分表层与深层土壤的依据。当各层存在多个样品时取较大值进行风险计算。当石油烃进行分段计算时,各馏段的风险总和为该物质的风险值[13]。
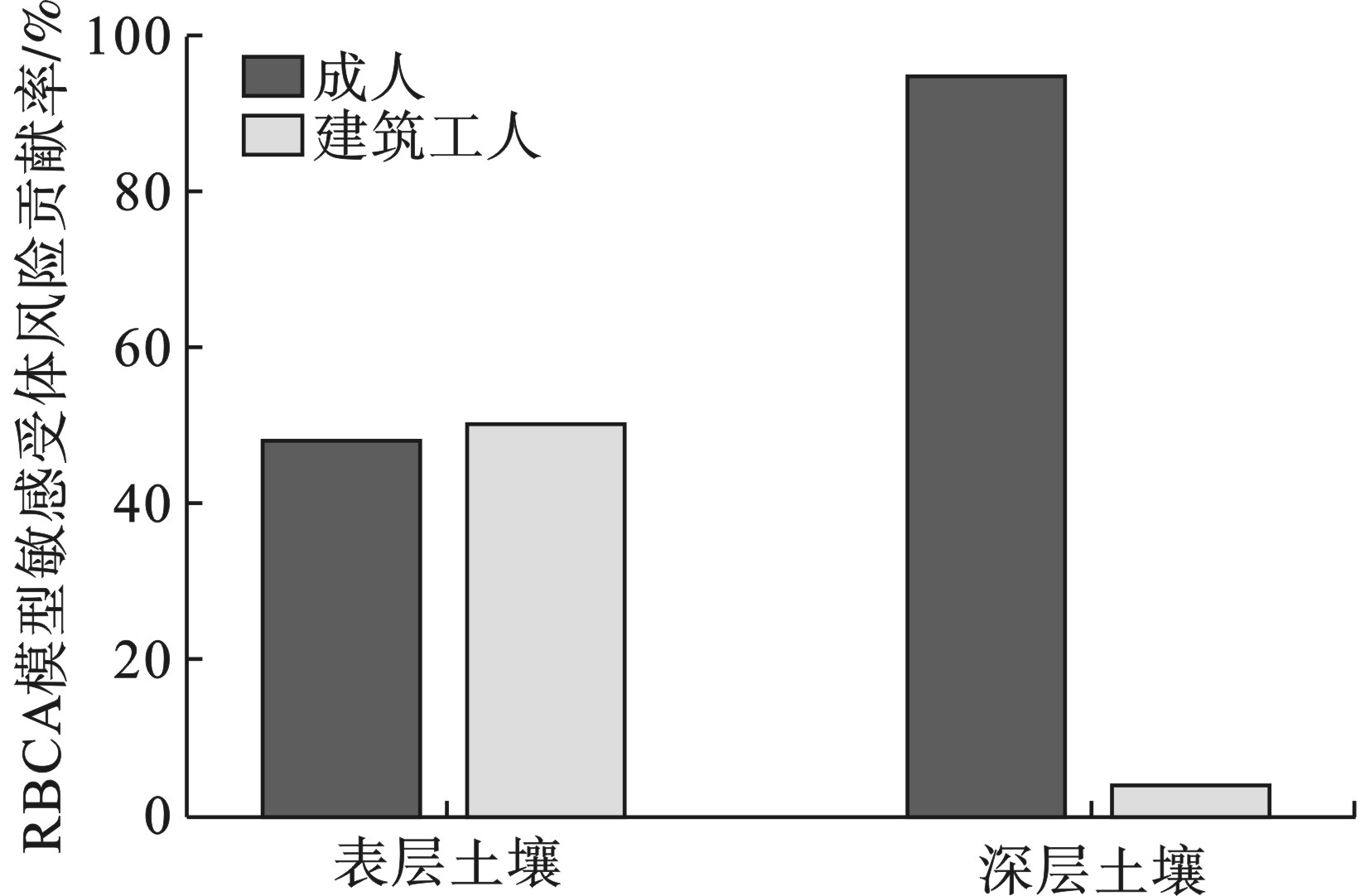
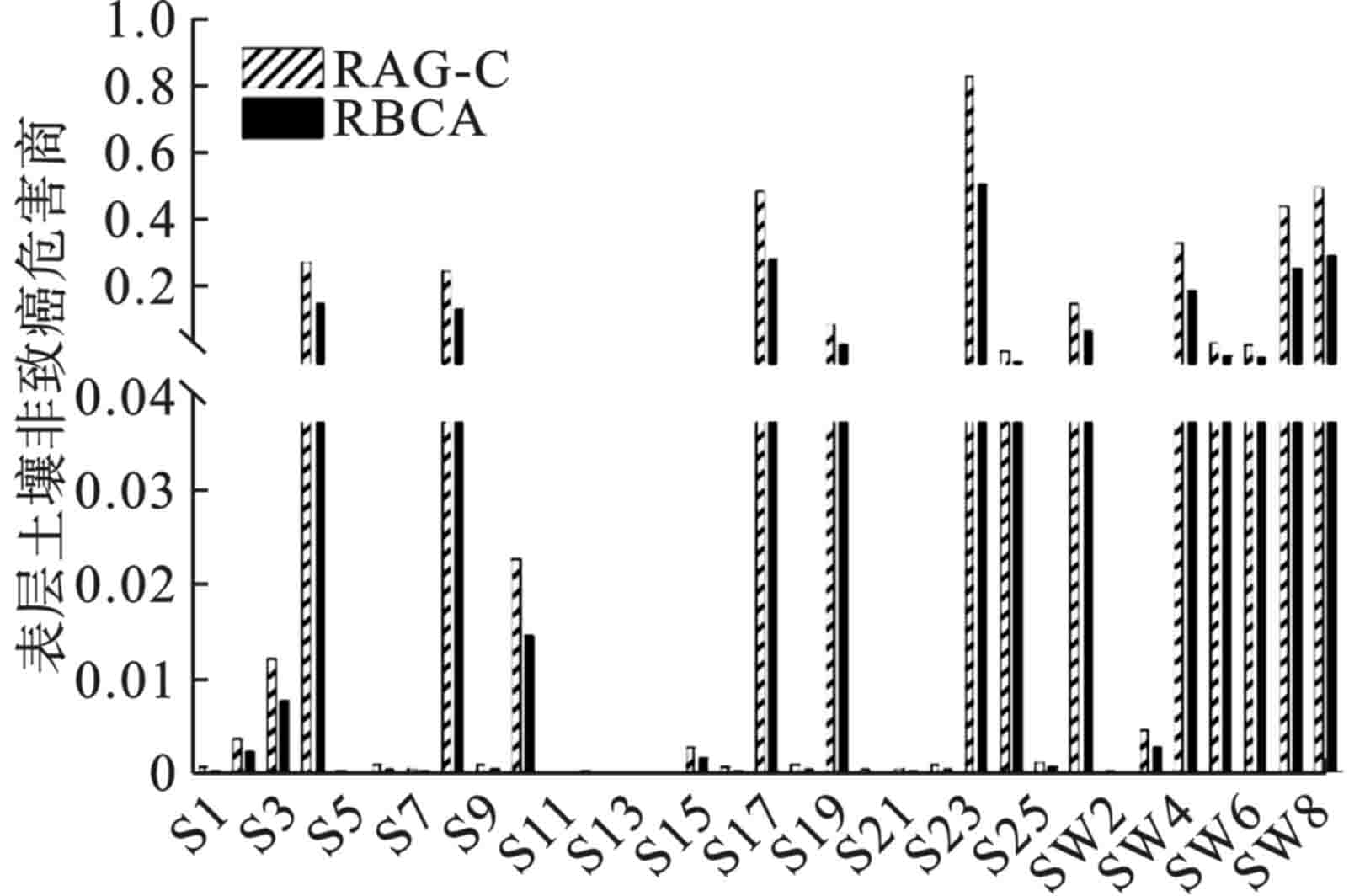
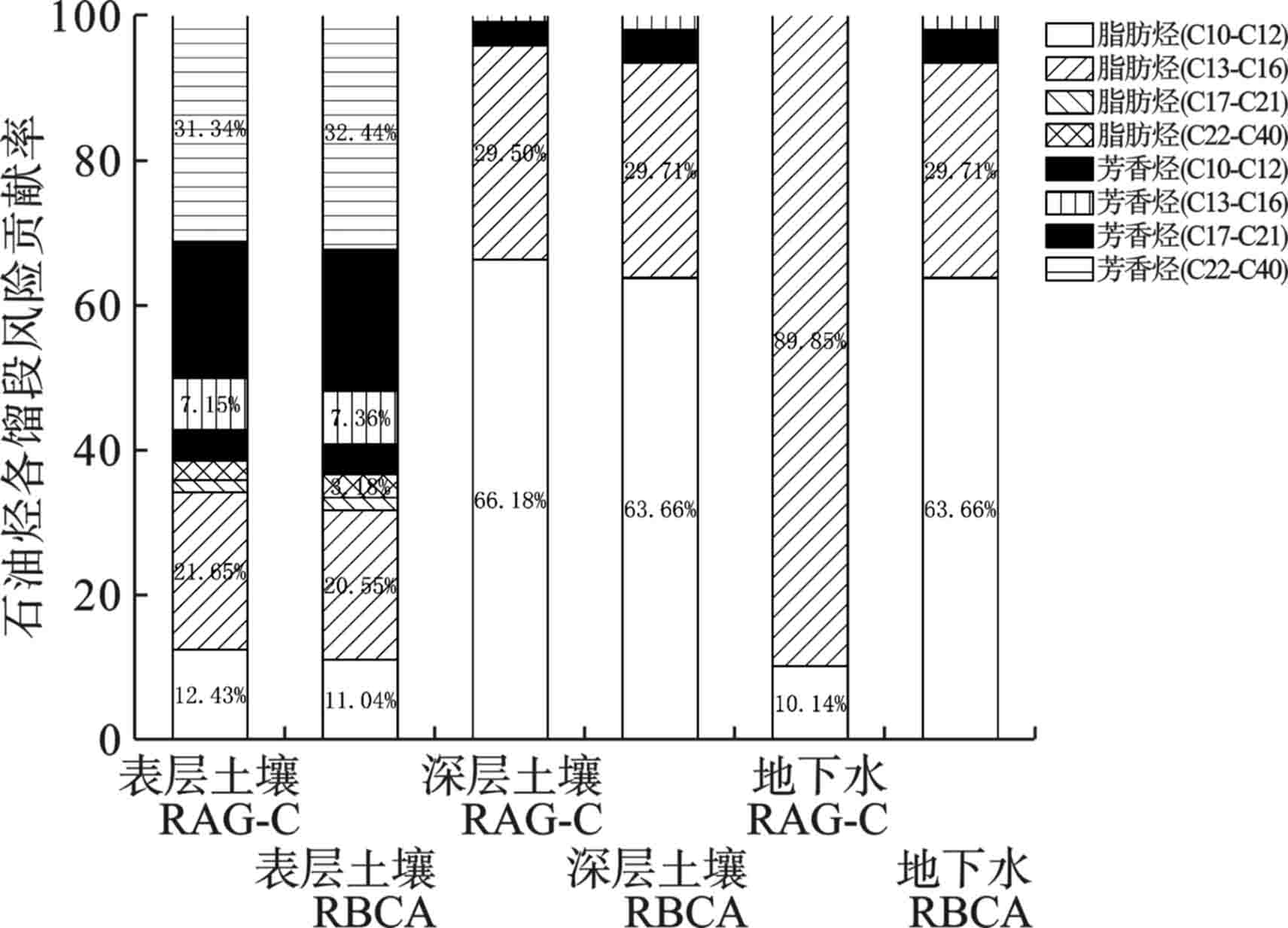
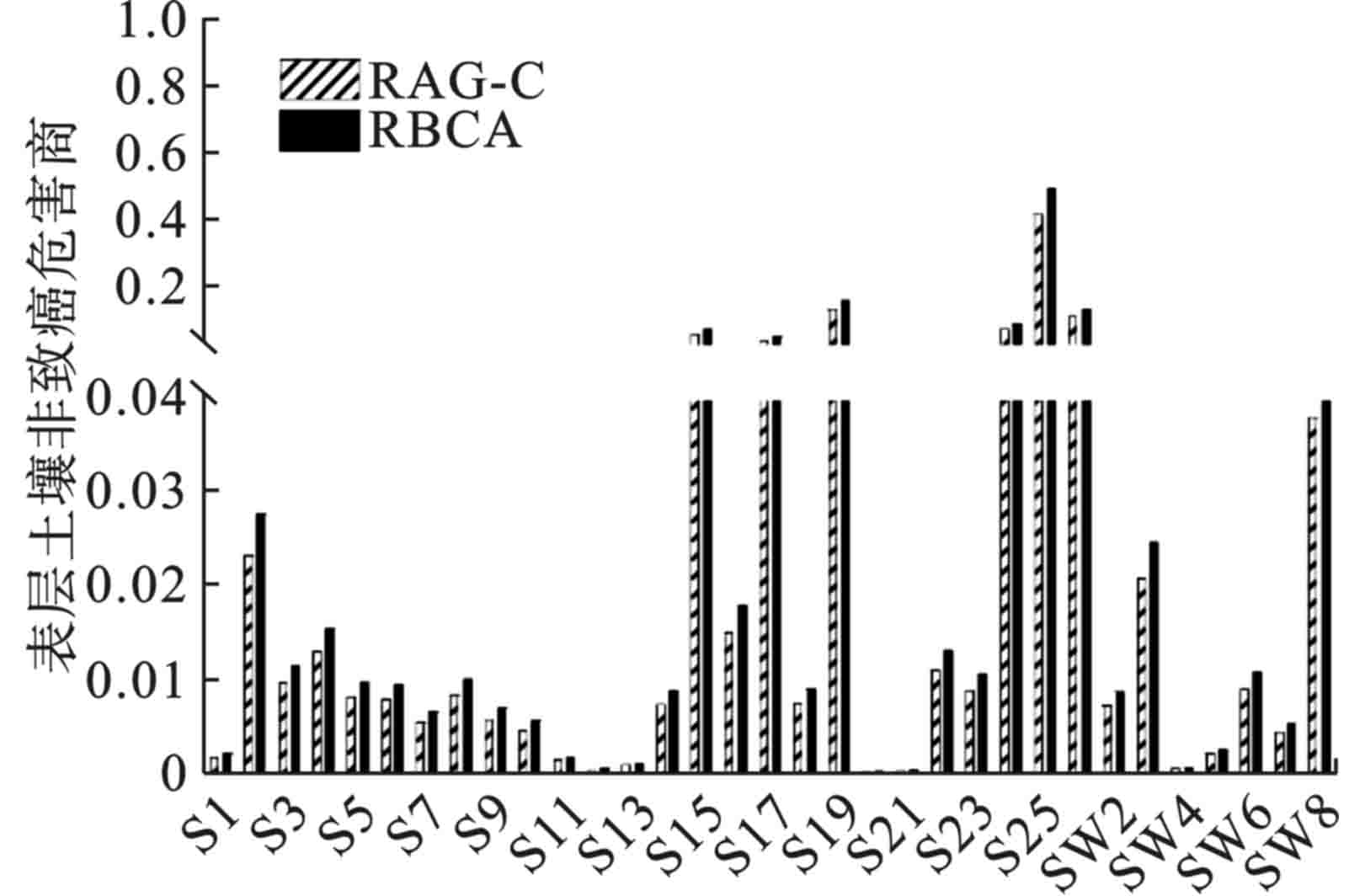
由表层土壤的非致癌危害商,见图1。所有点位的非致癌危害商均低可接受水平1,81.8%点位的风险水平低于0.04。2个模型的计算结果有所差异,但处于同一数量级之内。RBCA的风险值相对较高,原因在于RBCA在计算时考虑了建筑工人的暴露风险,建筑工人对非致癌危害商的贡献率占比达到51.15%,见图2,而RAG-C则仅考虑了成人作为敏感受体时的情形。
66.7%点位的风险水平低于0.04,2个模型计算结果显示深层土壤的人体健康风险在可接受水平之内。虽然仍然考虑了对建筑工人的影响,但其贡献率占比缩减至4.6%,见图2,导致RBCA计算的整体风险低于RAG-C,其计算结果占后者的比例为64.72%。
深层土壤的非致癌危害商大于表层土壤,见图3。
由地下水的非致癌危害商,见图4。
2个模型中超风险点位率均为65%。SW4点位的非致癌风险均为最高,在RBCA和RAG-C模型的计算结果分别超过可接受水平的129、229倍。RBCA模型由于在地下水风险表征中未考虑对建筑工人的影响,因此计算结果低于RAG-C模型,其计算风险占后者比例约56.65%。
-
暴露途径是依据实际情况选取污染物迁移和暴露于人体的方式,2种模型的暴露途径相近,但不同途径的风险贡献率差别较大。表层土壤中的风险贡献率在2个模型中的排序为皮肤接触土壤>经口摄入土壤>吸入室外空气中来自表层土壤的污染物>吸入土壤颗粒物。在吸入土壤颗粒物这一暴露途径上存在数量级的差别,原因在于2个模型的评估所采用的特征参数不同,RAG-C模型以实测依据环境空气中颗粒物含量来评估呼吸吸入颗粒物风险,RBCA模型则是以颗粒物释放因子为关键参数[14]。
深层土壤的暴露途径相对表层较少,吸入室内空气来自下层土壤的污染物大于吸入室外空气中来自下层土壤的污染物所带来的风险。原因在于敏感受体在室内的暴露频率大于室外,更易受到室内污染物的影响。在具体到不同碳链时,芳香烃(C13~C16)风险贡献率在2个模型中的差异最大,RBCA模型是RAG-C的3.26倍。
地下水中暴露途径与深层土壤类似,吸入室内空气中地下水气态污染物大于吸入室外空气中地下水气态污染物所造成的风险。2个模型在关键参数的选取上存在差异,RAG-C模型采取默认亨利常数,RBCA模型采取有效亨利常数用于计算气态污染物的扩散因子[15],见表5。
-
2个模型对各馏段在土壤中的风险贡献率基本一致,在表层土壤中芳香烃(C22~C40)>脂肪烃(C13~C16)>芳香烃(C17~C21)>脂肪烃(C10~C12)>芳香烃(C13~C16)>芳香烃(C10~C12)>脂肪烃(C22~C40)>脂肪烃(C17~C21)。深层土壤中脂肪烃(C10~C12)>脂肪烃(C13~C16)>芳香烃(C10~C12)>芳香烃(C13~C16)。
RAG-C模型在地下水风险计算中的主要风险来源于脂肪烃(C10~C12)及脂肪烃(C13~C16),分别占比10.14%和89.85%。RBCA模型则与之相反,分别占比63.66%和29.71%。芳香烃(C10~C12)和芳香烃(C13~C16)的占比约0.01%和0.004%,远小于RBCA模型中的4.46%和2.17%。石油烃各馏段的非致癌风险贡献率,见图5。
-
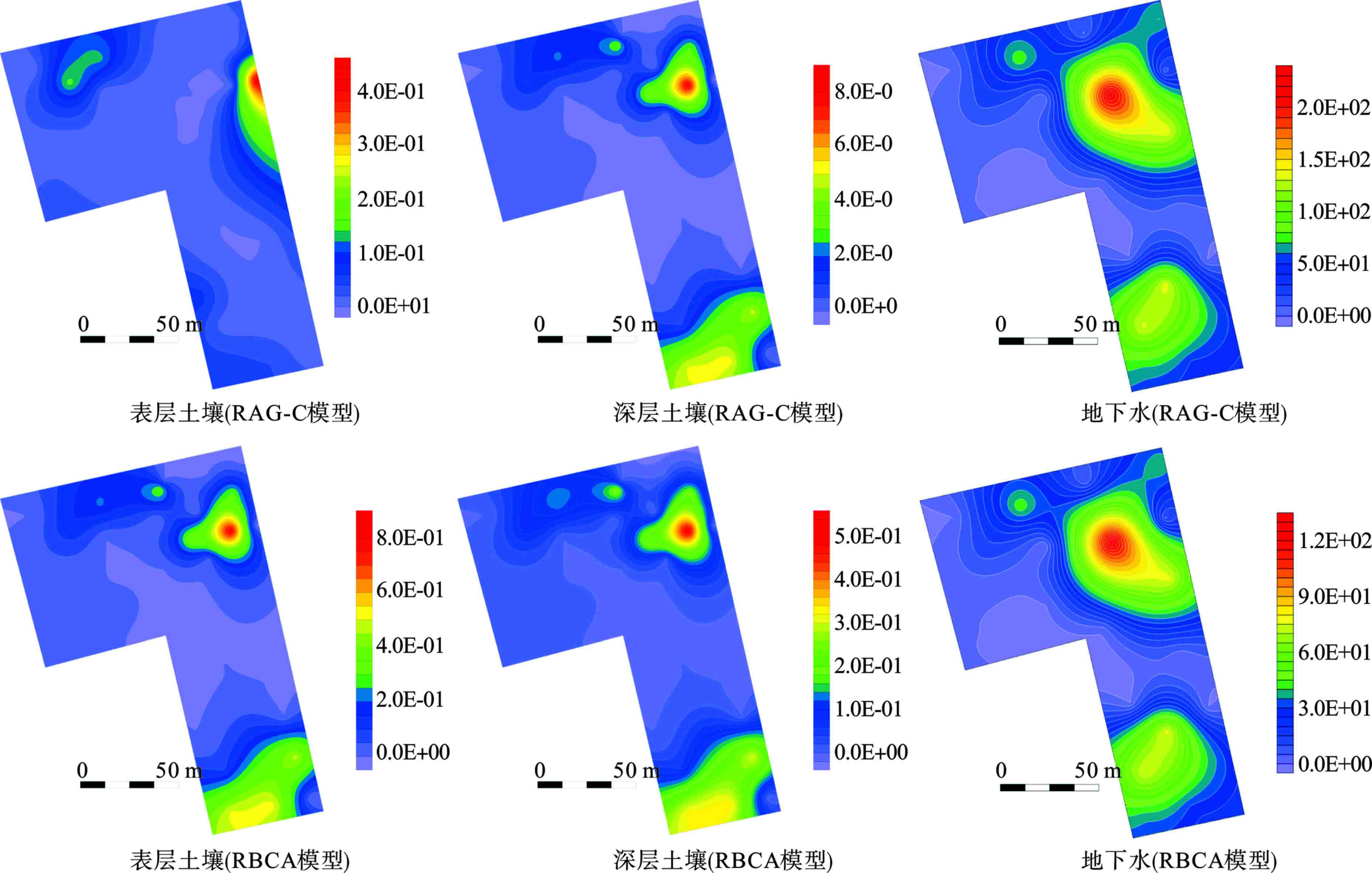
利用Surfer 15.3中的克里金(Kriging)插值法对石油烃的风险空间分布情况进行了模拟,见图6。
图6可知,虽然最大风险值有所不同,但2个模型所表征的风险分布范围并无明显差异。在表层土壤中,污染指数在地块东北侧最大,西北侧次之,南侧最小。在深层土壤中,东北侧最大,南侧次之,西北侧最小。地下水中的风险指数分布与深层土壤相近,污染物存在由土壤向地下水中迁移的可能性,进而导致地下水的污染。其风险指数亦为东北侧大于南侧,西北侧最小。
从地下水中的超风险的范围来看,RAG-C模型计算出的结果比RBCA模型大1.98%,见表6。
同时,由于的非致癌危害商存在明显差异,RAG-C模型计算出的污染程度更大。以上2个因素导致通过RAG-C模型进行模拟计算的地块在后期地下水修复中成本会相对更高。
-
(1)研究地块的关注污染物为石油烃,采用分段的方法开展评估工作,各馏段含量占比依据污染类型分配。计算结果可知石油烃在土壤中的非致癌风险未超过可接受水平,在地下水中的非致癌危害商存在超标现象,需开展后续修复及管控工作。
(2)从风险计算的角度分析,2种模型在风险计算结果上存在差异,但没有数量级区别。RBCA模型考虑到了对建筑工人的影响,在表层土风险计算时风险大于RAG-C模型。在深层和地下水风险计算时RAG-C模型相对保守,计算所得风险值更大。
(3)不同暴露途径在2个模型中的贡献序列基本一致,但部分途径如吸入土壤颗粒物在2个模型中差异很大,部分馏段如芳香烃(C13~C16)在2个模型中亦差异明显。
(4)2种模型内不同馏段的风险贡献率在土壤中基本一致。但在地下水中则相反,芳香烃(C10~C12)及芳香烃(C13~C16)的贡献率存在数量级差别。
(5)从风险的空间分布来看,深层土壤与地下水较为接近。2种模型所得超风险范围差异较小,由于RAG-C模型在地下水的模拟中更加保守,导致后期地下水修复工作量及修复成本均大于RBCA。
RAG-C及RBCA模型在污染地块风险评估中的应用比较
——以石油烃(C10~C40)的分段评估为例Comparison of application of RAG-C and RBCA models for risk assessment in a contaminated site
-
摘要: 通过RAG-C及RBCA 2个模型比较分析,采用分段评估的方法对石油烃污染地块展开风险评估工作。不同模型的风险计算结果有所不同,但仍处于同一数量级之内,仅针对成人受体而言,RAG-C计算结果是RBCA的1.7倍左右。从暴露途径来看,吸入土壤颗粒物途径贡献率相对较小,但在2个模型中存在数量级差异。石油烃不同馏段在土壤中的风险贡献率基本一致,在地下水中的最大贡献率不同,脂肪烃(C13~C16)在RAG-C模型中占比89.85%,脂肪烃(C10~C12)在RBCA模型中占比63.66%。从空间分布来看RAG-C模拟计算的超风险范围比RBCA大1.98%左右。Abstract: Risk assessments of petroleum hydrocarbon contaminated site were performed by the fractionation assessment method, based on the comparative analysis of RAG-C and RBCA models. The risk calculation results of two models were different. However, it still existed within the same order of magnitude. The RAG-C calculation results were about 1.7 times than those of RBCA for adults. From the perspective of exposure pathways, the contribution rate of inhalation of soil particulates was relatively small. There was a difference between the two models for the order of magnitude. The risk contribution rates of different fractions of petroleum hydrocarbons in soil were basically comparable, and the maximum contribution rates in groundwater were different. Aliphatic hydrocarbons(C13~C16) accounted for 89.85% in the RAG-C model, and aliphatic hydrocarbons(C10~C12) accounted for 63.66% in the RBCA model. In terms of spatial distribution, unacceptable risk range of RAG-C was about 1.98% larger than that of RBCA.
-
Key words:
- petroleum fractionation /
- risk assessment /
- RAG-C model /
- RBCA model
-
近年来,我国城镇居民用水量不断增大,给水厂自来水产量也随之提高[1]。在自来水生产的同时,会产生大量的污泥,这部分污泥源于生产工艺中的沉淀池排泥水和滤池反冲洗水,主要为原水中的无机颗粒、有机物以及净水过程中投加的混凝剂[3]。其产量约占自来水总生产水量的4%~7%,为避免资源浪费,应设法将其回收利用[2-4]。
化学调理脱水是污泥脱水常用的方法[5]。常用的化学调理剂有PAM等有机高分子调理剂和铁系、铝系等无机调理剂[6]。PAM在较少的投加量下便可达到较理想的效果,如给水厂中常用阳离子PAM来对污泥进行调理,但其具有轻微毒性,泥水分离后的水不能回用,会对给水厂造成较大的损失。铁系混凝剂絮体形成快,但本身不稳定,且投加到水体中存在色度问题[7-8]。对于铝系混凝剂而言,由于铝形态的不同,其絮凝效果和机理也相应不同[9]。羟基铝盐絮凝剂的形态可分为Ala、Alb、Alc。Ala为低聚态铝,如单体铝AlCl3;Alb为中聚态铝,如Al13(分子式为[AlO4Al12(OH)24(H2O)12]7+);Alc为高聚态铝,如Al30。相对于单体铝来讲,聚合铝具有投加量少、污泥产生量少、电中和能力高、对pH和温度有更高的适应能力等优点[10]。Al13是铝离子水解过程的中间产物,是一种高电荷的纳米粒子。与其他混凝剂相比,Al13电中和能力更强,其在混凝、除氟和污泥调理等方面均有应用[11]。刘沛[8]和CAO等[9]分别将AlCl3和Al13应用于市政污泥脱水,发现经2者调理后的污泥脱水性能均有提高,而且Al13的效果更为显著。
本研究采用无机盐调理剂AlCl3与自制Al13溶液对给水厂污泥进行调理,探究不同的药剂投加量对毛细脱水时间和污泥比阻的影响,并分析污泥调理过程中絮体粒径及形态的变化,从而得出2种调理剂形成的絮体特征;最后,结,2种药剂对有机物的去除率、调理后污泥上清液的余铝含量,比较其实际应用潜力。
1. 材料与方法
1.1 供试污泥和试剂
本研究所用污泥取自北京市某给水厂污泥浓缩池,该水厂以南水北调南干渠原水为水源,日产水量50×104 t。污泥基本性质见表1。
表 1 污泥基本性质Table 1. Basic properties of sludge含水率/% 有机质/% pH CST/s d(0.5)/μm SRF×1013/(m·kg−1) 98.5 30.9 7 77.5 51.13 1.63 注:d(0.5)表示污泥中体积累积百分比为50%时颗粒的最大直径。 | Show Table DownLoad:
CSV
DownLoad:
CSV
六水合氯化铝(AlCl3·6H2O)、氢氧化钠(NaOH)、硫酸钠(Na2SO4)、氯化钡(BaCl2)均为分析纯;铝十三溶液(Al13,Al含量9.6 g·L−1)于实验室采用慢速滴碱法自制[12]。经Ferron比色法[9]检测,AlCl3和Al13溶液的Ala、Alb、Alc含量分别为95.32%、3.91%、0.77%和3.44%、93.96%、2.60%。
1.2 实验及检测方法
调理方法。烧杯搅拌实验在六联搅拌器上进行,6个烧杯中分别倒入500 mL污泥,AlCl3的投加量(以Al含量计)为0.300、0.600、0.800、0.900、1.000、1.200 g·L−1;Al13投加量(以Al含量计)分别为0.024、0.096、0.192、0.288、0.336、0.360 g·L−1。搅拌器程序设定为:250 r·min−1快搅30 s,加药后200 r·min−1搅拌1 min,再以40 r·min−1慢搅10 min。
毛细吸水时间(CST)由CST测定仪(RTC-304B,英国Triton公司)直接测定;污泥比阻(SRF)在压力为0.6 MPa下将一定量污泥倒入布氏漏斗中抽滤测定[14];含水率和有机质使用电热恒温干燥箱(DHG-9146A,北京天林恒泰科技有限公司)和箱式电阻炉(SX2-2 5-10TP,上海一恒科学仪器有限公司)测定[13]。
污泥上清液有机物的测定。参考NIU等[14]分离EPS的方法分步分离有机物,用荧光分光光度计(F-7000,日立公司)测定三维荧光谱图,采用150 W氙灯为激发光源,激发波长(Ex)200~400 nm,发射波长(Em)220~550 nm,狭缝宽度5 nm。DOC由总有机碳测定仪(岛津,岛津公司)测定。
污泥上清液余铝的测定。调理后的污泥上清液过0.45 μm膜,稀释一定倍数用电感耦合等离子光谱仪(9800,岛津公司)测定。
絮体粒径和分形维数的动态变化用马尔文激光粒度分析仪(Mastersizer 2000,马尔文公司)测定。混凝程序:A段快搅阶段,250 r·min−1快搅30 s,加AlCl3或Al13后200 r·min−1搅拌1 min;B段慢搅阶段,40 r·min−1慢搅15 min;C段破碎阶段,250 r·min−1快搅1 min破碎絮体;D段恢复阶段,40 r·min−1慢搅15 min使絮体再生成,参考SUN等[15]的方法计算分形维数。
污泥絮体微观结构用场发射扫描电子显微镜(SU-8020,日立公司)观察。
2. 结果与讨论
2.1 AlCl3和Al13调理对污泥脱水性能的影响
图1为经不同浓度Al13和AlCl3调理后污泥的CST和SRF。由图1(a)可知,随着Al13投加量的逐渐增加,污泥CST以较快的速度下降,在投加量为0.336 g·L−1时,CST从79.1 s减小到33.1 s,再继续增加投加量,CST反而上升;相比之下,随着AlCl3投加量的逐渐增加,CST下降速度较为缓慢,当其降至最小值34.8 s时,所对应的最佳投加量为0.800 g·L−1,继续增加投加量,CST缓慢上升。由图1(b)可知,Al13投加量为0.024和0.096 g·L−1时,SRF改善效果不明显,继续增加到0.336 g·L−1时,SRF改善效果最佳,为4.98
× 1012 m·kg−1,继续增加投加量,SRF增大,污泥脱水效果变差;AlCl3投加量为0.300 g·L−1时,SRF达到6.53× 1012 m·kg−1,继续增加投加量至0.600、0.800、0.900 g·L−1,SRF维持在5.50× 1012 m·kg−1左右,最小值为5.45× 1012 m·kg−1,再继续增加投加量,污泥脱水效果变差。在最佳投加量的基础上继续增加投加量,CST和SRF效果都变差,这是因为调理剂投加过多,会引起颗粒复稳,脱水性能将变差[16]。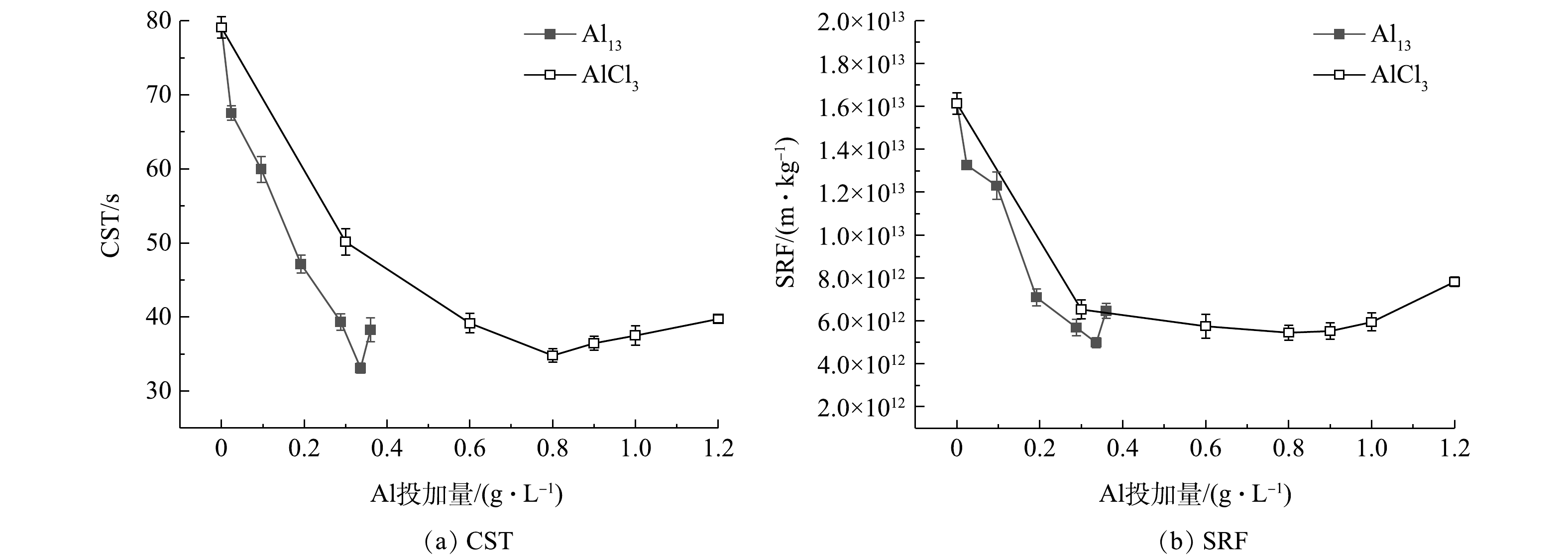 图 1 不同浓度Al13和AlCl3调理后的污泥的CST和SRFFigure 1. CST and SRF of sludge treated with different concentrations of Al13 and AlCl3
图 1 不同浓度Al13和AlCl3调理后的污泥的CST和SRFFigure 1. CST and SRF of sludge treated with different concentrations of Al13 and AlCl32.2 AlCl3和Al13调理对污泥中有机物的影响
给水厂污泥主要成分为原水中的无机颗粒、有机物以及净水工艺中投加的混凝剂[17]。污泥中水分分为自由水、毛细结合水、表面吸附水和内部结合水[18]。与污水厂污泥不同,给水厂污泥微生物含量较少,内部结合水的含量不足以影响污泥脱水效能,因此在本研究中可忽略。有机物相对于无机颗粒更易于与水分结合,其存在状态在给水厂污泥中有3种:有机物单独存在、有机物吸附在无机颗粒上、有机物被净水过程中投加的混凝剂包裹。经过Al13和AlCl3调理后,有机物与调理剂结合,结合在有机物上的水分子脱出。
图2是原泥及AlCl3、Al13最佳投加量调理后的污泥上清液有机物三维荧光图,原泥游离的有机物中主要呈现2个峰:Ex/Em 265-290 nm/320-375 nm、252-272 nm/430-440 nm,分别为微生物代谢产物峰和腐殖酸峰[9]。原泥和Al13、AlCl3调理过的污泥上清液各区域的荧光强度按照游离的有机物、吸附在无机颗粒的有机物、被混凝剂包裹的有机物的顺序依次减小,这说明有机物含量逐渐降低。Al13调理过的污泥游离有机物的荧光强度低于AlCl3,而混凝剂包裹的有机物荧光强度略高于AlCl3,因此Al13对游离有机物去除率相对较高,对混凝剂包裹的有机物去除率相对较低。有研究表明,污泥脱水性能与蛋白质含量相关[19]。蛋白质是污泥絮体的主要组成部分[20],其水化层会包围水相中的水分,使这部分水不易排出[21]。三维荧光图谱中Ex/Em 200-250 nm/280-380 nm为蛋白质区域[9],由图2(a)、图2(d)、图2(g)可知,Al13调理后的污泥上清液在蛋白质区域的荧光强度相对原泥明显减少,而AlCl3变化较小,因此Al13对蛋白质的去除效果优于AlCl3。该结果验证了污泥比阻实验中,Al13调理后的污泥易脱水的结论。
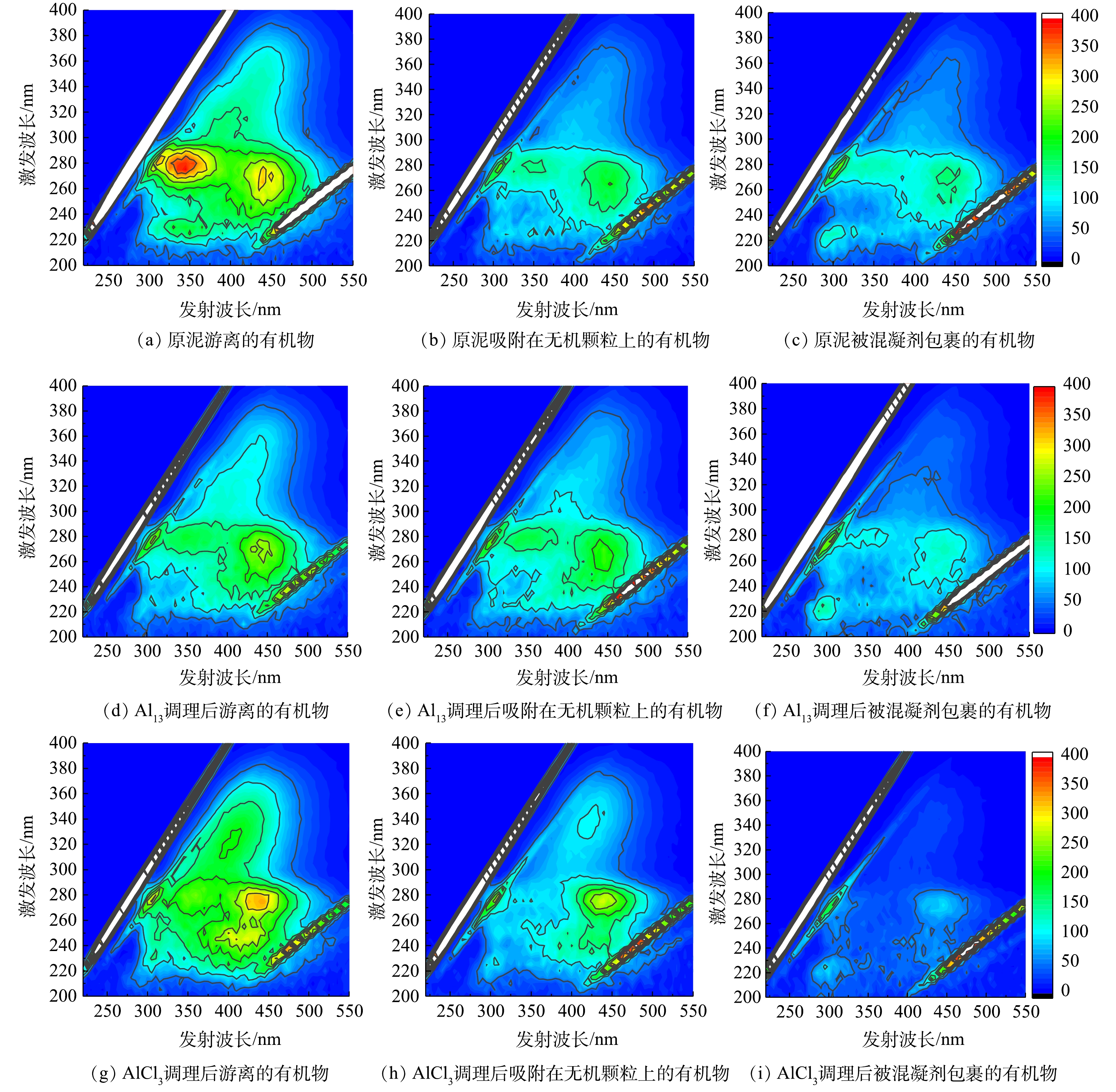 图 2 原泥以及Al13和AlCl3最佳投加量调理后污泥上清液的三维荧光图谱Figure 2. 3D-EEM spectra of sludge supernatant about raw sludge and conditioned sludge using Al13、AlCl3
图 2 原泥以及Al13和AlCl3最佳投加量调理后污泥上清液的三维荧光图谱Figure 2. 3D-EEM spectra of sludge supernatant about raw sludge and conditioned sludge using Al13、AlCl3图3为原泥和Al13、AlCl3调理过的污泥上清液不同层次有机物的DOC浓度(以总悬浮物(TSS)计)。图中3种污泥上清液的浓度按照游离的有机物、吸附在无机颗粒上的有机物、被混凝剂包裹的有机物的顺序减少,与三维荧光谱图中的趋势相同。经Al13和AlCl3调理过后的污泥各组分DOC浓度均有不同程度降低。原泥和Al13、AlCl3调理后的污泥游离有机物DOC浓度分别为2.16和1.03、2.15 mg·g−1,Al13调理过后的污泥游离有机物DOC浓度比原泥降低了1.13 mg·g−1,而AlCl3调理过后的污泥游离有机物DOC浓度仅比原泥降低了0.01 mg·g−1,因此Al13调理过后的污泥更易脱水。
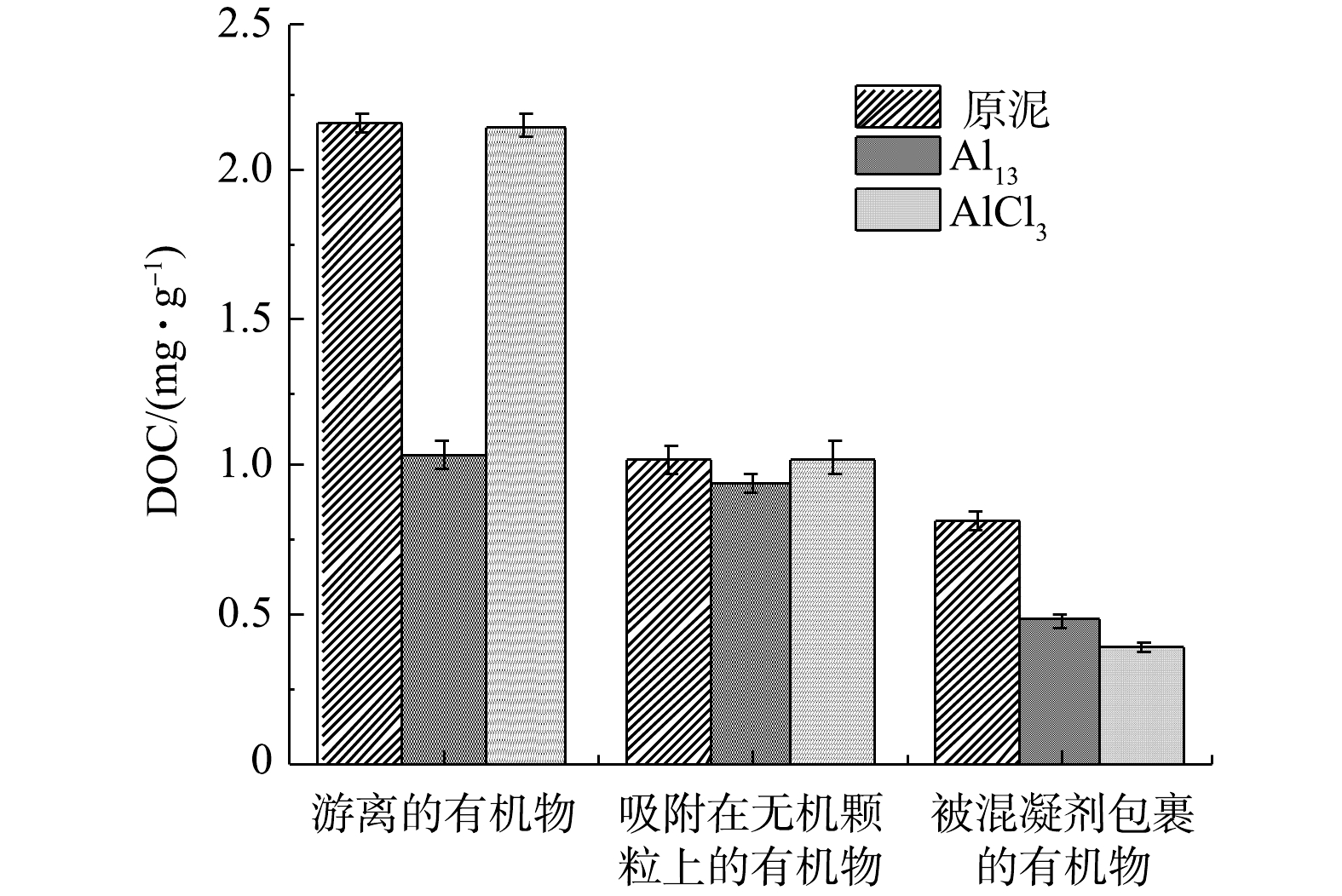 图 3 原泥和Al13、AlCl3调理后的污泥不同上清液的DOC值Figure 3. DOC content of raw sludge and conditioned sludge using Al13、AlCl3
图 3 原泥和Al13、AlCl3调理后的污泥不同上清液的DOC值Figure 3. DOC content of raw sludge and conditioned sludge using Al13、AlCl32.3 AlCl3和Al13调理对污泥絮体性质的影响
图4是在AlCl3和Al13最佳投加量时,污泥随时间变化的絮体粒径(d(0.5))和分形维数。原泥粒径为52.42 μm,快搅30 s后加入Al13和AlCl3,絮体粒径均有所增长,在快搅阶段投加Al13的污泥形成的絮体粒径平均为57.29 μm,大于加入AlCl3的54.42 μm;慢搅阶段加入AlCl3的絮体粒径增长较快,最大能够增长到126.83 μm,稳定后平均粒径为110.47 μm,而加入Al13的污泥絮体大小则较稳定,一直保持在82.00 μm左右,稳定后平均粒径为81.70 μm;加入AlCl3和Al13的污泥絮体破碎阶段的粒径分别为88.83和69.96 μm;AlCl3在破碎后再生成的絮体稳定后平均粒径为137.39 μm,大于破碎前的粒径,Al13为78.36 μm。AlCl3和Al13的强度因子分别为80.41%和85.63%,恢复因子分别为224.40%和71.79%。强度因子越高,絮体抗剪切能力越强,恢复因子越高,絮体破坏后越容易重新团聚成絮体,因此Al13形成的污泥絮体抗剪切能力更强,AlCl3形成的絮体受到破坏后更容易恢复[22]。
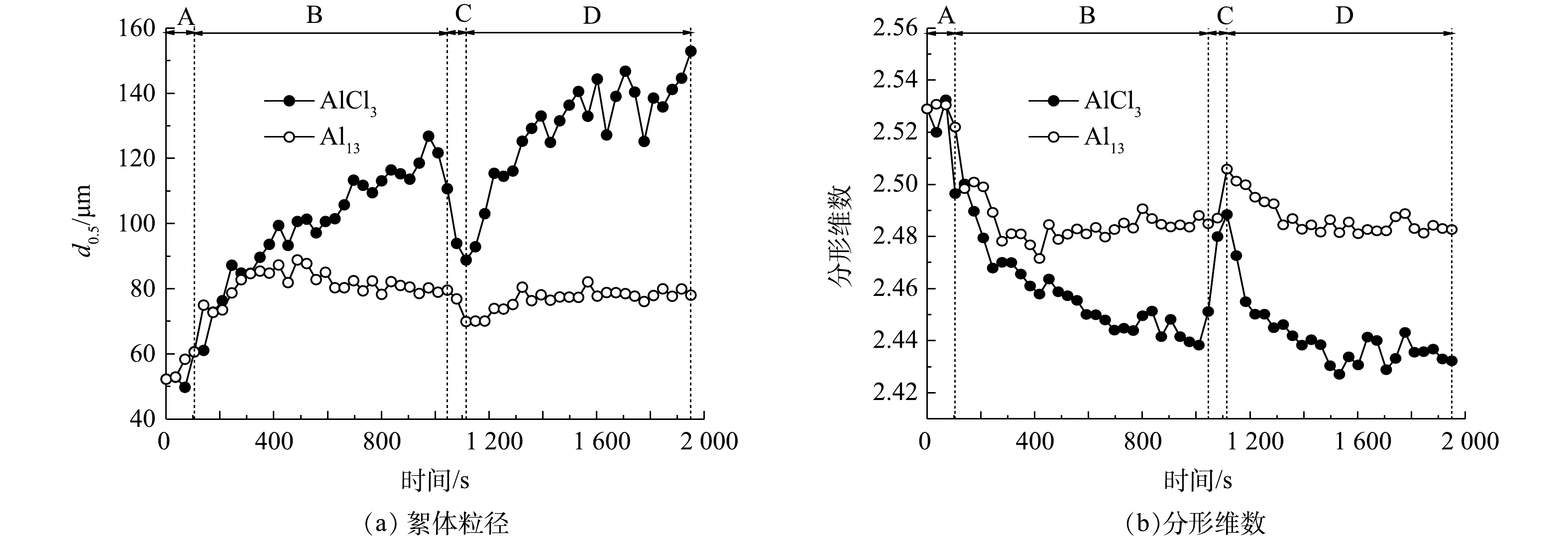 图 4 Al13、AlCl3最佳投加量时污泥的絮体粒径和分形维数变化Figure 4. Particle size and fractal dimension of flocs under the optimal dosage of Al13 and AlCl3
图 4 Al13、AlCl3最佳投加量时污泥的絮体粒径和分形维数变化Figure 4. Particle size and fractal dimension of flocs under the optimal dosage of Al13 and AlCl3分形维数是表示分形体不规则的量度[23],分形维数越高,絮体越紧实[24]。从图4中可看出,Al13和AlCl3的分形维数稳定时分别在2.48和2.44左右,相差不大,2者絮体密实程度无明显差别。从图5扫描电镜图中可以看出,原泥表面光滑,絮体规则而紧密,经AlCl3和Al13调理过后的污泥絮体松散。
 图 5 原泥和AlCl3、Al13最佳投加量时污泥絮体的扫描电镜Figure 5. SEM of raw sludge and conditioned sludge under the optimal dosage of Al13 and AlCl3
图 5 原泥和AlCl3、Al13最佳投加量时污泥絮体的扫描电镜Figure 5. SEM of raw sludge and conditioned sludge under the optimal dosage of Al13 and AlCl32.4 AlCl3和Al13调理对污泥上清液性质的影响
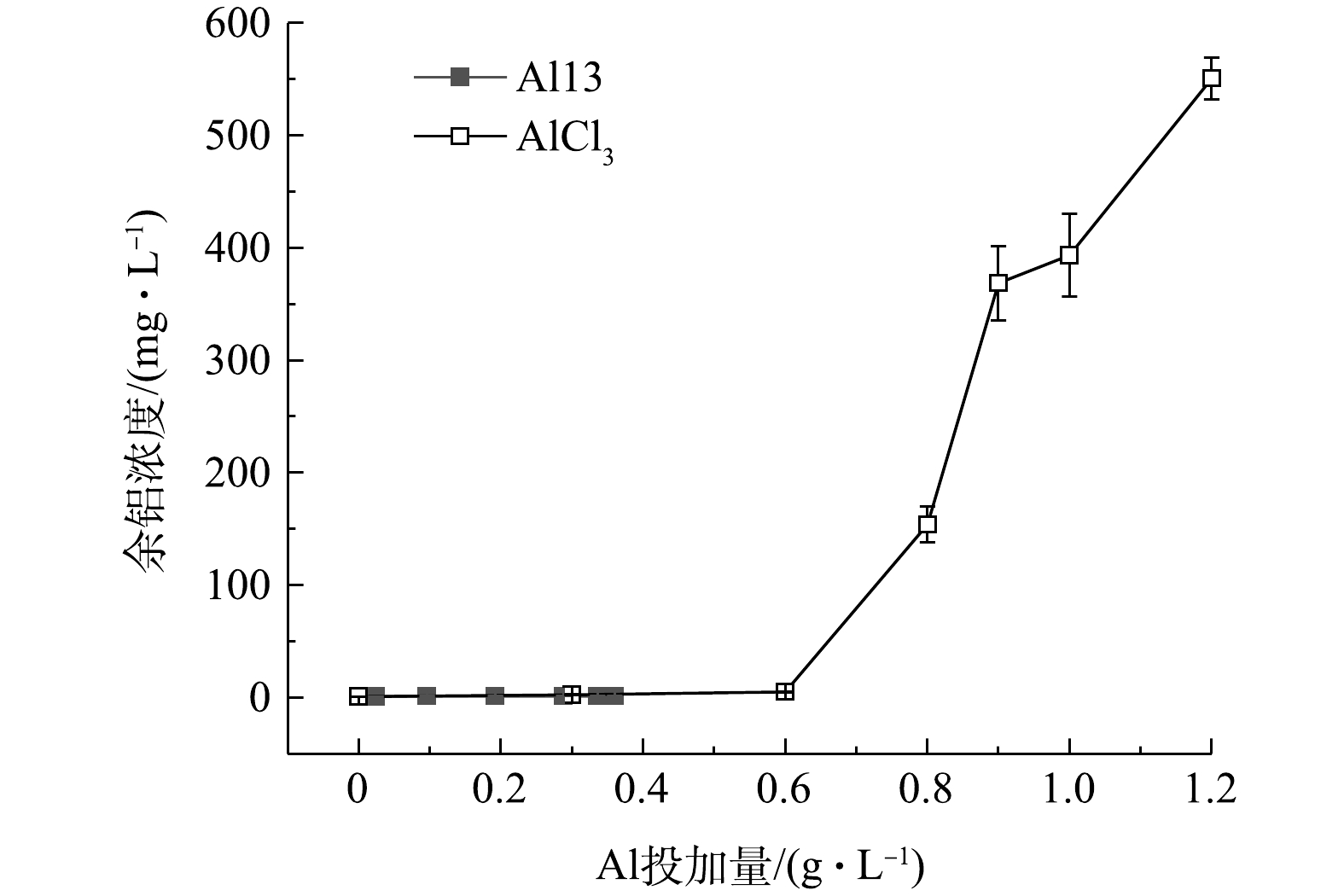
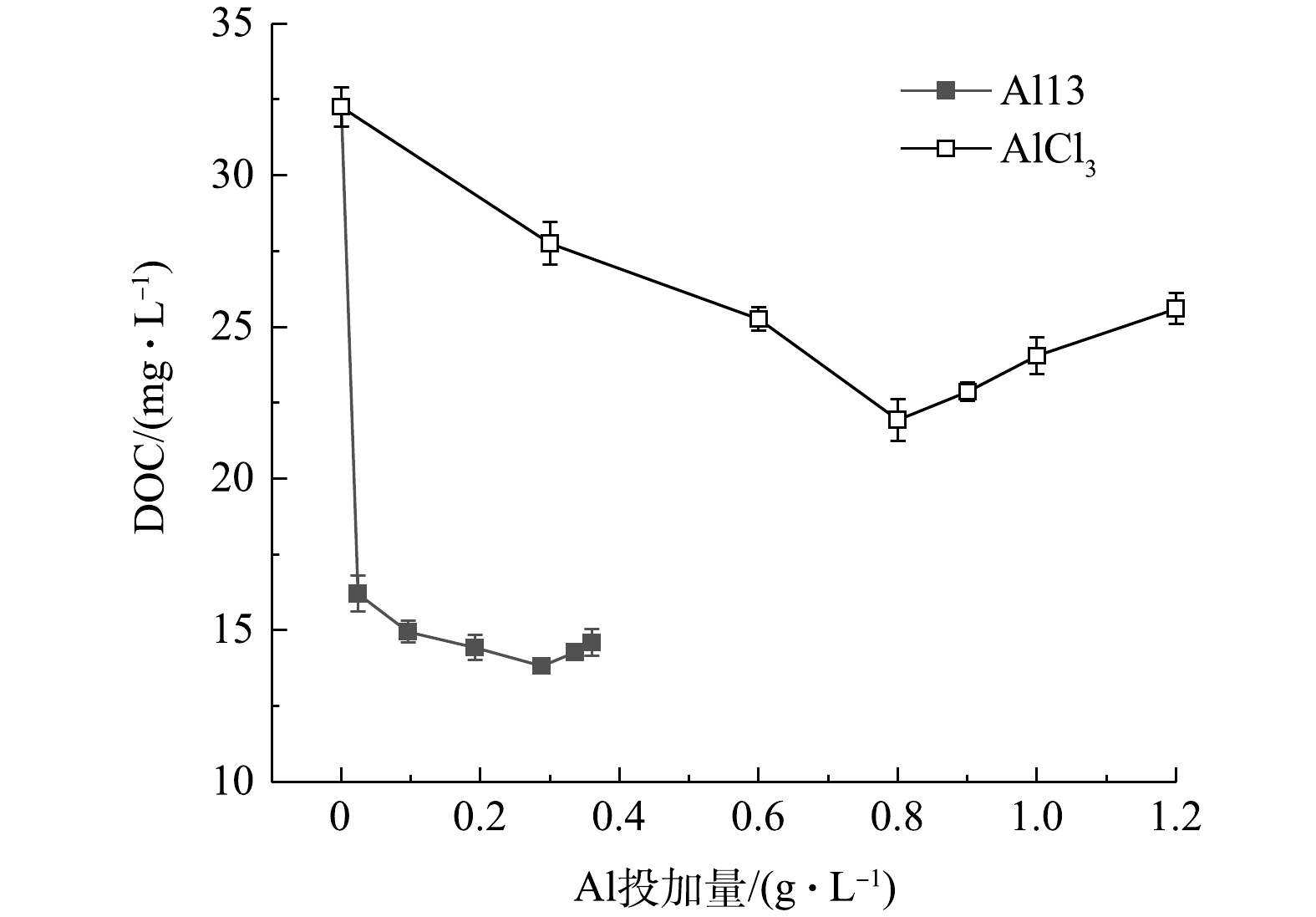
图6是Al13和AlCl3不同投加量下的污泥上清液余铝浓度,Al13调理后的污泥上清液余铝浓度最高为1.35 mg·L−1,远远低于AlCl3调理后的污泥上清液余铝浓度。未投加铝盐混凝剂的原泥上清液中余铝浓度为0.83 mg·L−1。这是因为,制水工艺中投加的铝盐混凝剂沉降下来,富集了这部分混凝剂的沉后水污泥排入污泥池中,使得污泥中铝离子含量偏高。因此,如何在保证调理效果的基础上降低余铝浓度,是下一步要解决的重要问题。图7为投加2种调理剂后溶液DOC含量的变化,原泥DOC浓度为32.25 mg·L−1,Al13的加入使得DOC含量迅速下降到16.21 mg·L−1,继续投加,DOC变化幅度较小,最佳投加量时为14.27 mg·L−1;随着投加量的增加,AlCl3调理后的污泥DOC变化较为平缓,最佳投加量时的浓度为21.93 mg·L−1,再继续增加投加量,DOC浓度变高,这是因为投加量过多,体系pH下降,引起了颗粒的复稳。考虑到污泥脱水后脱出水的回用,如果投加Al13的污泥上清液余铝和溶解性有机物含量更低,则回用后续处理将会更简单。
2.5 AlCl3和Al13污泥调理机制
综合前4节的分析可知,Al13和AlCl3加入给水厂污泥后,与污泥中的有机质相互作用,降低了污泥体系中有机物的含量,使得污泥颗粒聚集成尺寸更大、结构更疏松的絮体,最终造成水分更容易脱出。同时发现,有机物的含量与污泥脱水性能联系密切,在加入Al13的污泥中,有机物去除量更高,因此该调理剂的脱水性能优于AlCl3。图8为Al13、AlCl3与有机物的相互作用模型,AlCl3(Ala)可通过络合作用与给水污泥中的有机物结合,并形成Ala-有机物络合物,由于给水污泥中的有机物来源于天然水体,其中的大部分有机物分子量较小,不足以给Ala形成的絮凝体提供凝核,因此部分Ala-有机物络合物未能沉淀;相比之下,带有更多正电荷的Al13(Alb)不仅能够有效中和有机物的负电荷,还可作为核团与可溶性有机物形成絮体[25]。因此,Al13调理后的污泥相对AlCl3来讲,上清液中的有机物浓度更低。
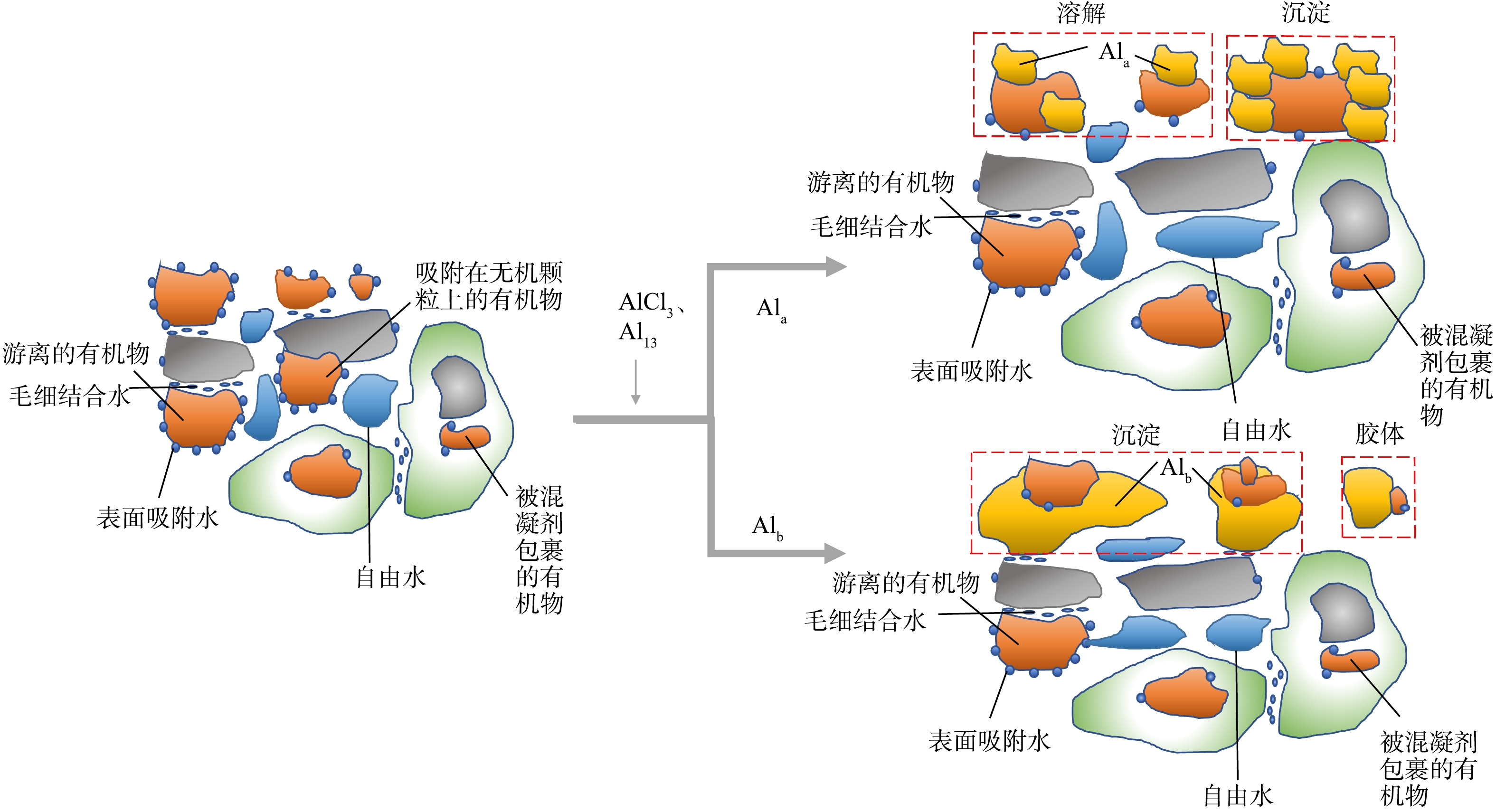 图 8 Al13、AlCl3与有机物相互作用模型Figure 8. Model of interaction of Al13、AlCl3 and organic matters
图 8 Al13、AlCl3与有机物相互作用模型Figure 8. Model of interaction of Al13、AlCl3 and organic matters3. 结论
1)以Al含量计,Al13和AlCl3的最佳投加量分别为0.336和0.800 g·L−1,此时Al13的CST和SRF均小于AlCl3,Al13在投加量较少的情况下便可取得良好的脱水效果。调理后的污泥絮体相对原泥更松散,有利于水分的脱出。
2)相比于AlCl3,Al13更能有效地与有机物形成沉淀,调理后的污泥有机物含量更低,在蛋白质含量降低的体现上尤为突出。Al13调理后的污泥上清液余铝浓度也远远低于AlCl3,更利于给水厂的回用。
-
表 1 土壤理化性质参数汇总表
类型 含水率/% 比重 重度/KN·m−3 孔隙比 Kv/cm·s−1 Kh/cm·s−1 有机质/g·kg−1 黏土 28.5 2.75 19.0 0.826 3.29E-07 4.82E-07 15.8 注:Kv为垂直渗透系数,Kh为水平渗透系数。
下载: 导出CSV
表 2 关注污染物分析结果
污染物 介质 样品数量/个 检出率/% 浓度范围 超标率/% 最大值 平均值 石油烃(C10~C40) 土壤/ mg·kg−1 216 98.6 4.3~43 500 7.4 43 500 1 566 地下水/ mg·L−1 20 100 0.42~950 90.9 950 190.9 注:土壤标准参考《土壤环境质量建设用地土壤污染风险管控标准(试行):GB36600—2018》中第二类筛选值,地下水评价标准《上海市建设用地地下水污染风险管控筛选值补充指标》(沪环土〔2020〕62号)。
下载: 导出CSV
表 3 石油烃分段毒性参数
污染物 碳段分配比例/% RfDo/mg·(kg·d)−1 RfC/mg·m−3 SAF 无量纲 ABSgi无量纲 ABSd无量纲 脂肪烃(C10~C12) 6.6 1.00E-01 5.00E-01 5.00E-01 5.00E-01 1.00E-01 脂肪烃(C13~C16) 13.5 1.00E-01 5.00E-01 5.00E-01 5.00E-01 1.00E-01 脂肪烃(C17~C21) 25.4 2.00E+00 / 5.00E-01 5.00E-01 1.00E-01 脂肪烃(C22~C40) 40.2 2.00E+00 / 5.00E-01 5.00E-01 1.00E-01 芳香烃(C10~C12) 1.1 4.00E-02 2.00E-01 5.00E-01 5.00E-01 1.00E-01 芳香烃(C13~C16) 2.0 4.00E-02 2.00E-01 5.00E-01 5.00E-01 1.00E-01 芳香烃(C17~C21) 4.2 3.00E-02 / 5.00E-01 5.00E-01 1.00E-01 芳香烃(C22~C40) 7.0 3.00E-02 / 5.00E-01 5.00E-01 1.00E-01 注:RfDo为经口摄入参考剂量; RfC为呼吸吸入参考浓度; SAF为参考剂量分配比例; ABSgi为消化道吸收因子; ABSd为皮肤吸收效率因子。
下载: 导出CSV
表 4 第二类用地类型下暴露参数
暴露参数 非致癌效应平均时间ATnc/a 成人平均体重BWa/kg 成人暴露期EDa/a 气态污染物入侵持续时间t/a 成人暴露频率EFa/d•a−1 皮肤暴露频率EFd/d•a−1 皮肤接触面积SAEa/cm2 成人皮肤表面土壤粘附系数SSARa/mg•cm−2 成人每日摄入土壤量OSIRc/mg•d−1 第二类用地 成人 25 61.8 25 25 250 250 3 023 0.2 100 施工人员(RBCA) 1 61.8 1 25 180 180 3 023 0.5 100
下载: 导出CSV
表 5 不同暴露途径的非致癌风险贡献率分析
% 模型 馏段 表层土壤 深层土壤 地下水 经口摄入 皮肤接触 吸入颗粒物 室外表层土壤 室外下层 室内下层 室外蒸汽 室内蒸汽 RAG-C模型 1 32.29 39.04 0.20 28.47 8.54 91.46 2.65 97.35 2 37.91 45.84 0.23 16.03 8.54 91.46 2.64 97.36 3 45.27 54.73 - - - - - - 4 45.27 54.73 - - - - - - 5 39.60 47.88 0.24 12.28 8.62 91.38 3.40 96.60 6 42.52 51.41 0.26 5.81 8.75 91.25 3.93 96.07 7 45.27 54.73 - - - - - - 8 45.27 54.73 - - - - - - RBCA模型 1 37.61 45.48 0.000036 16.90 4.42 95.58 0.92 99.08 2 41.35 50.00 0.000040 8.66 9.03 90.97 0.92 99.08 3 45.27 54.73 - - - - - - 4 39.82 60.18 - - - - - - 5 41.16 49.77 0.000039 9.08 12.92 87.08 1.12 98.88 6 42.73 51.67 0.000041 5.59 28.56 71.44 1.43 98.57 7 45.27 54.73 - - - - - - 8 45.27 54.73 - - - - - - 注:馏段中1、2、3、4、5、6、7和8分别代表脂肪烃(C10~C12)、脂肪烃(C13~C16)、脂肪烃(C17~C21)、脂肪烃(C22~C40)、芳香烃(C10~C12)、芳香烃(C13~C16)、芳香烃(C17~C21)和芳香烃(C22~C40)。
下载: 导出CSV
-
[1] 李辈辈. 石油污染土壤的生物修复[J]. 上海国土资源, 2018, 39(4): 55 − 57. doi: 10.3969/j.issn.2095-1329.2018.04.012 [2] 陈能场, 郑煜基, 何晓峰, 等. 全国土壤污染状况调查公报[J]. 中国环保产业, 2014(5): 10 − 11. [3] 王博. 加油站石油污染修复技术研究[D]. 北京: 清华大学, 2010. [4] 环境保护部自然生态保护司. 土壤修复技术方法与应用[M]. 北京: 中国环境科学出版社, 2011. [5] 周启星, 宋玉芳. 污染土壤修复原理与方法[M]. 北京: 科学出版社, 2004. [6] UK Environment Agency. Fact Sheet for the RBCA Tool Kit for Chemical Releases[EB/OL]. (2009-03-11)[2018-08-10]. http://www.environment-agency.gov.uk. [7] CHANG S H, KUO C Y, WANG J W, et al. Comparison of RBCA and Caltox for setting risk-based cleanup levels based on inhalation exposure[J]. Chemosphere, 2004, 56(4): 359 − 367. doi: 10.1016/j.chemosphere.2004.01.006 [8] 中华人民共和国生态环境部. 建设用地土壤污染状况调查技术导则: HJ 25.1—2019[S]. 北京: 中国环境出版集团, 2020. [9] 中华人民共和国生态环境部. 建设用地土壤污染风险评估技术导则: HJ 25.3—2019[S]. 北京: 中国环境出版集团, 2020. [10] 上海市生态环境局. 上海市建设用地土壤污染状况调查、风险评估、风险管控与修复方案编制、风险管控与修复效果评估工作的补充规定(试行) [EB/OL]. (2021-05-15)[2020-06-10]. https://sthj.sh.gov.cn/hbzhywpt6061/20200610/ebf9b3dc47c541a690e7bc813b19fea5.html. [11] US Environmental Protection Agency. Technical background document for the supplemental report to congress on remaining fossil fuel combustion wastes groundwater pathway human health risk assessment[EB/OL]. [2007-06-10]. http://www.epa.gov/epaoswer/other/fossil/ffc2_395.pdf. [12] 陈梦舫, 骆永明, 宋静, 等. 中、英、美污染场地风险评估导则异同与启示[J]. 环境监测管理与技术, 2011, 23(3): 14 − 18. doi: 10.3969/j.issn.1006-2009.2011.03.004 [13] US Environmental Protection Agency. Guidelines for the Health Risk Assessment of Chemical Mixtures[EB/OL]. [2007-06-10]. http://www.epa.gov/NCEA/raf/pdfs/chemmix/chemmix1986.pdf. [14] CONNOR J A, BOWERS R, MCHUGH T, et al. Software guidance manual RBCA tool kit for chemical releases[M]. Houston, Texas, USA: GSI Environmental Inc, 2007. [15] 张斌, 邹卉, 肖杰, 等. RAG-C和RBCA模型中场地特征参数的差异及其启示[J]. 环境工程, 2015, 33(9): 130 − 133. doi: 10.13205/j.hjgc.201509029 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2510
- HTML全文浏览数: 2510
- PDF下载数: 29
- 施引文献: 0
