






-
水资源紧缺是全球面临的挑战,污水回用是缓解水资源紧缺的重要手段,而其中的关键问题是保障回用水质安全。污水回用主要包括生物处理工艺和深度处理工艺。生物处理主要去除氮、磷和有机物,常用的生物处理工艺包括厌氧好氧工艺(A/O),厌氧缺氧好氧工艺(A/A/O),膜生物反应器(MBR),UNITANK序批式活性污泥工艺和生物滤池等。深度处理工艺主要去除水中的微量有机污染物和杀灭水中的致病微生物,包括病原细菌、病毒及寄生虫卵等。目前污水回用中采用的深度处理工艺包括:氯、氯胺、二氧化氯、臭氧、紫外光照及紫外/过氧化氢等高级氧化(advanced oxidation process,AOP)工艺以及高铁酸盐、高锰酸盐等。
污水消毒是确保回用水水质安全的重要保障[1−2],在水消毒过程中,污水中含有的有机物,如腐殖酸、富里酸和微生物代谢物等与消毒剂发生氧化、加成、取代等反应生成消毒副产物,即DBPs[3−4]。研究表明,大部分DBPs具有潜在的致癌、致畸、致突变性[5−6],其中含氮DBPs具有比常规管制DBPs更高的毒性,目前新型含氮DBPs主要包括卤代乙腈、卤代硝基甲烷以及亚硝胺等[7-11]。
控制污水消毒过程中产生的DBPs是提高回用水水质安全的重要课题,目前对污水DBPs的控制手段主要包括前体物去除、消毒剂种类与投量控制,以及DBPs去除,本研究主要关注DBPs的前体物去除。污水中的有机物(effluent organic matters,EfOM)是DBPs的主要前体物,而因污水中的氨氮和EfOM含有的溶解性有机氮远远高于饮用水源,导致污水的消毒过程会产生大量的含氮的消毒副产物(N-DBPs)[12−13]。此外,污水中的无机离子,如溴离子在臭氧氧化过程中也会催化二甲基亚硝胺(N-Nitrosodimethylamine,NDMA)的生成[14]。
本研究旨在从污水回用角度出发,探究污水深度处理工艺对污水中消毒副产物及其前体物的去除效果。主要研究内容包括:(1)不同生物处理工艺(A/O,A/A/O,MBR,UNITANK,循环式活性污泥法(CAST), 氧化沟和生物滤池)对典型N-DBPs (包括亚硝胺类物质、卤乙腈和卤代硝基甲烷)的去除;(2)不同深度处理工艺(混凝沉淀,PAC,臭氧,二氧化氯,高铁酸盐氧化)对典型N-DBPs及其前体物的去除。
-
4种卤乙腈混合标样(haloacetonitrile,HANs)、三氯硝基甲烷(trichloronitromethane,TCNM)、1,2-二溴丙烷、二甲基亚硝胺(NDMA)、NDMA-d6内标、甲基叔丁基醚(MTBE)、甲醇(Methanol)、浓硫酸(98%H2SO4)、冰醋酸(CH3COOH)、次氯酸钠溶液(NaClO)、亚氯酸钠(NaClO2)、氯化铵(NH4Cl)、无水硫酸钠(Na2SO4)、抗坏血酸(Ascorbic acid)、N,N-二乙基-对苯二胺硫酸盐(DPD)、六水合硫酸亚铁铵(FAS)、碘单质(I2)、碘化钾(KI)、高铁酸钾(K2FeO4)、椰壳炭粉末(PAC)、无水磷酸氢二钠(Na2HPO4)、无水磷酸二氢钠(NaH2PO4)、氢氧化钾(KOH)、明矾。
二氧化氯储备液(ClO2)由亚氯酸钠与浓硫酸反应制备而成,其浓度用DPD/FAS比色法标定。一氯胺(NH2Cl)储备液(10000 mg·L−1,以氯(Cl2)计,全文中含氯浓度均以Cl2计)由氯化铵与次氯酸钠溶液依比例反应制备而成,现配现用。臭氧溶液(O3)使用臭氧发生装置产生臭氧,并溶解在4°C的 pH5磷酸盐缓冲液中,其浓度用靛蓝脱色比色法标定[15],现配现用。高铁酸钾溶液由高纯度(>90%)高铁酸钾固体溶于纯水中配制成,现配现用,由于购买的高铁酸钾固体纯度仅有约50%,需进一步采用有机溶剂洗涤与重结晶法纯化[16],简而言之,将低纯度的高铁酸钾固体用1 mol·L−1的KOH溶液洗涤抽滤,所得滤液与饱和KOH溶液混合置于冰水浴中析出晶体并过滤,所得晶体再依次用正己烷、戊烷、甲醇和乙醚冲洗以脱水脱碱,随后在烘箱中60 ℃干燥,真空干燥器中密封保存。
气相色谱仪(Agilent 7890A,美国安捷伦公司)、气相色谱质谱联用(Trace1300 ISQ,美国赛默飞公司)、氮氧化物分析仪(CLD-88,瑞士ECO Medics公司)、离子色谱仪(ICS-600,美国赛默飞公司)、紫外-可见分光光度计(UV-2700,日本岛津公司)、总有机碳测定仪(TOC-LCPH,日本岛津公司)、哈希水质检测仪(DR2800,美国哈希公司)pH计(S210,瑞士梅勒多-托利多公司)。
-
本研究选择8间污水处理厂的初沉池出水和生化反应池出水作为实验水样探究生物处理工艺对亚硝胺及N-DBPs前体物的影响,样品编号及水厂工艺见表1。由于A/A/O为国内污水处理厂常用的生物处理工艺,因此在探究深度处理工艺对亚硝胺及N-DBPs前体物的影响中,实验所用水样取自B污水厂的二级沉淀池出水,其出水特征更具有代表性。水样基本参数包括DOC、氨氮、UV254和无机离子浓度等均采用国标方法进行。
表2中汇总了各水样的基本参数,其中编号为A1—H1的水样为生物处理池进水,编号为A2—H2的水样为生物处理池出水,水样的DOC、氨氮和亚硝酸盐的浓度反映了含氮消毒副产物的部分前体物水平,这些参数对含氮消毒副产物的生成潜能有影响[17−20]。
-
混凝处理工艺:采用六位组合搅拌器进行混凝试验。在硅烷化玻璃烧杯中装2.1 L污水水样,开始快速搅拌的同时向水样中添加明矾(10 mg明矾·mg−1 TOC),搅拌条件为:100 r·min−1 快速搅拌1 min,30 r·min−1絮凝20 min,沉降时间60 min。沉淀后,收集和过滤上清液样品以进行亚硝胺和DBPs检测和模拟实际处理(uniform formation condition,UFC)试验。
PAC吸附工艺:用0.45 μm玻璃纤维滤膜过滤2.1 L污水样品,粉末活性炭使用前在超纯水中混合24 h。PAC投加量为10 mg·L−1和20 mg·L−1,反应器与混凝实验一致。恒速(100 r·min−1)搅拌4 h后沉降1 h,收集和过滤上清液样品以进行亚硝胺和DBPs检测和UFC试验。
氧化工艺所处理的污水水样均经0.45 µm玻璃纤维滤膜过滤,反应器均采用棕色玻璃瓶,可容纳 >2.1 L水。其中:①二氧化氯氧化工艺:向水样中投加 2 mg·L−1二氧化氯(按Cl2计算),顶空反应2、5、10、20、30 min后测定二氧化氯浓度,并用氮气吹脱剩余二氧化氯以终止反应,选取二氧化氯氧化30 min的水样进行亚硝胺和DBPs检测,所有投量的水样均进行UFC实验。②高铁酸盐氧化工艺:向水样中投加2.5、5、7.5、10 mg·L−1 (Fe)高铁酸钾,混合均匀后静置3 h,期间分别在1、2、3、5、7、10、15、30、60、90、120 min取样并用ABTS法测定高铁酸盐浓度[21],反应3 h后水样经0.45 µm玻璃纤维滤膜过滤后,投量为10 mg Fe·L−1的样品进行亚硝胺和DBPs检测,所有投量的样品均进行UFC实验。③臭氧氧化工艺:向水样中投加0.5、1、2、3 mg·L−1臭氧溶液并迅速摇匀,在15、30、40、60、90、120 s时取样进行臭氧浓度检测,以获得臭氧在水样中的衰减曲线。反应至水样中臭氧浓度低于检出限后,臭氧投量为3 mg ·L−1的样品亚硝胺和DBPs检测,所有投量的样品均进行UFC试验。所有实验均设置2组平行对照。
-
污水后氯胺化实验:采用一氯胺(pH 8.5)作为消毒剂,现配现用,向需要进行UFC实验的2.1 L样品中加入5 mg·L−1的一氯胺溶液,并在黑暗室温条件下维持3 d,保证反应后水样中的总氯含量大于1 mg·L−1,3 d后加入33 mg·L−1 抗坏血酸终止反应,并分装为500 mL、500 mL与30 mL分别进行NDMA、总亚硝胺(TONO)和卤代DBPs萃取检测。所有实验均设置2组平行对照。
-
水样的pH、UVA254、DOC分别采用pH计、UV-vis分光光度计、TOC测定仪。Cl−、Br−、NO3−和NO2−等阴离子采用配有Dionex IonPacTM AS19分析柱(4 mm× 250 mm)和Dionex IonPacTM AG保护柱(4 mm× 50 mm)的离子色谱仪定量,KOH淋洗液梯度程序为:初始KOH淋洗液浓度为10 mmol·L−1,保持10 min,然后以2 mmol·L−1·min−1升至20 mmol·L−1·min−1,保持5 min,淋洗液的流速为1 mL·min−1。氨氮采用纳氏试剂分光光度法测定。
-
样品经含有内标(1,2-二溴丙烷)的MTBE萃取后,依照美国环保署标准方法USEPA Method 551.1[22],采用气相色谱仪测定卤乙腈(HANs)和TCNM。采用Agilent DB-5 0.25 mm × 30 m分离柱,进样口温度为120 ℃,检测器温度为290 ℃。测定时进样体积均为2 μL,进样模式为不分流,氮气流速为1.0 mL·min–1。样品浓度定量采用内标法校正,回收率保证在80%–120%,每次检测前使用现配的标准溶液进行仪器状态检查校正。
-
样品的萃取参考Dai等研究中的固相萃取法[23]。NDMA浓度用GC-MS检测,采用Agilent DB-1701 0.25 mm × 60 m分离柱,载气为高纯氦气,流速为1.0 mL·min–1,升温程序为:37 ˚C维持4 min,以4 ˚C·min−1升至130 ˚C,10 ˚C·min−1升至160 ˚C,40 ˚C·min−1升至250 ˚C并维持2 min。MS端为PCI模式,扫描时间0.7 s,放射电流50 µA,溶剂延迟9.5 min,传输线温度230 ˚C,离子源温度150 ˚C,反应气为甲烷,样品浓度定量采用内标法校正,回收率保证在80%–120%,每次检测前使用现配的标准溶液进行仪器状态检查校正。TONO浓度用氮氧化物分析仪进行检测,样品中的亚硝胺在80 ℃下与I2-KI-醋酸溶液反应后生成一氧化氮气体(NO)进入氮氧化物分析仪,气体流速为200 mL·min−1。
-
图1为A–H生化池进水与出水中总亚硝胺的存在水平,卤乙腈、三氯硝基甲烷未检出。进水中TONO的浓度范围为0.9—2.5 nmol·L−1,出水中的范围为1.4—5.3 nmol·L−1,TONO的浓度在经生物处理后均有不同程度的升高(5.0%—254.5%)。在生物处理的厌氧或缺氧阶段,污水中NO2−浓度在反硝化作用下几乎被完全去除[24],该阶段极少亚硝胺的生成,因此亚硝胺主要在生物处理的好氧阶段生成。在水厂G的进水中具有高浓度NO2−,且其出水中的NO2−浓度更高,在此过程中大量NO2−参与了亚硝胺的生成,导致TONO浓度大幅升高。污水生物处理过程中亚硝胺的降解途径主要是生物降解,生物降解效率与亚硝胺的官能团有直接关系,如亚硝基吗啉(N-nitrosomorpholine,NMOR)等不具有强供电子官能团的亚硝胺难以被生物降解[25−26]。亚硝胺也可能在生物处理过程中生成,主要依靠微生物作用,微生物细胞表面或细胞质膜上吸附的胺类有机物比游离态的胺的亚硝化作用强,且亚硝胺的生成速率由其前体物(EfOM与NO2−)浓度决定[27]。
-
图2a为各深度处理工艺对污水中亚硝胺类物质的去除。原水B(图中标注为effluent)中含有微量的NDMA与TONO,浓度分别为0.14 nmol·L−1和1.35 nmol·L−1,实验结果表明二氧化氯与高铁酸盐对NDMA和TONO的浓度并没有显著影响,但PAC和臭氧处理均会导致NDMA和TONO的增加,PAC处理使NDMA和TONO分别增加了192%和61.5%,臭氧氧化则使NDMA和TONO分别增加了185%和138%。
二氧化氯可以与丁酰肼反应生成NDMA[28],其中一种中间产物为偏二甲肼(UDMH),此外与其他二级胺反应生成的NDMA量极低,对于本实验而言,30 min的二氧化氯氧化对污水中NDMA和TONO的浓度并无明显影响,推测污水中丁酰肼或UDMH的含量很低。
在经20 mg·L−1 PAC吸附处理后,NDMA的浓度增长至0.41 nmol·L−1,同时TONO的浓度增至2.14 nmol·L−1,活性炭在氧气存在的情况下可以催化水中的二级胺生成痕量的亚硝胺类物质,PAC表面的活性位点与氧分子反应生成活性氧(reactive oxygen species,ROS),进而促进PAC表面对氮分子的吸附,同时生成活性氮物质(reactive nitrogen species,RNS),如氧化亚氮和羟胺,进一步与吸附在PAC表面的二级胺反应生成亚硝胺[29−30]。本实验中采用的PAC为400目粉末活性炭,具有较高的比表面积,因此更容易产生亚硝胺。
臭氧(3 mg·L−1)处理的污水中NDMA与TONO浓度也显著提升,分别为0.41 nmol·L−1和3.16 nmol·L−1。臭氧氧化是水处理过程中NDMA生成的一种重要方式,臭氧与二甲胺(DMA)或N,N-二甲基磺胺反应会直接生成NDMA,同时臭氧与一些含氮类药物、染料以及二甲胺反应也会生成NDMA[31−33]。
经各种深度处理工艺后卤代消毒副产物的生成不明显,这是由于采用的深度处理中没有引入氯和氯胺,因此几乎没有卤代消毒副产物的生成。
-
为探究所选深度处理工艺对消毒副产物的前体物的去除效果,将经深度处理的污水再进行后氯胺化处理,并检测其中的各类消毒副产物的生成情况。结果表明,各深度处理工艺对亚硝胺类物质的前体物均有不同程度的去除(图2b),未经深度处理的原水经氯胺化后,NDMA浓度为0.69 nmol·L−1,而混凝、PAC吸附、二氧化氯、高铁酸钾和臭氧处理的污水经氯胺化后NDMA浓度分别为0.38、0.18、0.19、0.33、0.42 nmol·L−1,即NDMA前体物去除率顺序为:PAC(74%)≈二氧化氯(72%)>高铁酸钾(52%)>混凝(45%)>臭氧(39%)。同样的,混凝、PAC吸附、二氧化氯、高铁酸钾和臭氧处理的污水经氯胺化后TONO的浓度分别为6.06、2.67、2.55、2.65、4.50 nmol·L−1,均低于未经深度处理的污水后氯胺化样品(8.51 nmol·L−1),即TONO前体物去除率顺序为:二氧化氯(70%)≈高铁酸钾(69%)≈PAC(69%)>臭氧(47%)>混凝(29%)。
本实验中,采用10 mg·L−1明矾作为絮凝剂对NDMA的前体物去除达到40%,而TONO的前体物去除也低于30%,这可能是由于NDMA前体物的分子量较小(<1kDa),难以通过简单的混凝沉淀达到有效去除。由于亚硝胺类物质的亲水性强(如lg KowNDMA=-0.57)[34],PAC吸附无法对亚硝胺类物质本身起到良好去除效果,甚至可能催化NDMA的生成,但PAC对亚硝胺类的前体物具有吸附作用,能降低亚硝胺在后氯胺化过程中的生成。本实验采用10 mg·L−1的PAC处理污水厂二级出水4 h,对NDMA和TONO的前体物去除率分别达74%与69%。当投量为7—10 mg·L−1时,在饮用水厂采用PAC对进水进行吸附处理能去除约20%的NDMA前体物,且PAC对受轻度污染的水源的NDMA控制效果优于地表水[35],可推测从控制NDMA生成的角度来看,PAC更适用于污水回用。在前人对预氧化处理NDMA的前体物的研究中,采用氯、二氧化氯、臭氧、高铁酸盐和太阳光等对NDMA的前体物均有去除效果[36−37]。
为了比较二氧化氯、高铁酸盐和臭氧在不同的暴露剂量下对污水中含氮消毒副产物前体物的去除效果,将氧化工艺的暴露剂量与对应水样在后氯胺化时的消毒副产物生成量结合分析,见图3。
图3a表明,卤代N-DBPs的生成量会随着二氧化氯的暴露剂量增大而迅速增大,其中HANs的生成量增大了269%—346%,但当二氧化氯的暴露剂量超过9 mg·min−1·L−1时,卤代DBPs的浓度达到平衡,其中二氯乙腈(DCAN)从对照组的6.4 nmol·L−1迅速增至约27.2 nmol·L−1,增加了超过4倍,TCNM从1.4 nmol·L−1增至约5.3 nmol·L−1,BCAN从0.5 nmol·L−1增至约6.8 nmol·L−1。图3b表明,二氧化氯氧化对NDMA的前体物去除效果显著,随着二氧化氯暴露剂量从0 mg·min−1·L−1增至超过10 mg·min−1·L−1,氯胺化后的污水水样中NDMA浓度从0.68 nmol·L−1迅速减至约0.23 nmol·L−1,随后趋于稳定;而二氧化氯对TONO的前体物去除随二氧化氯暴露剂量的增大而逐渐增强,需达到30 mg·min−1·L−1的二氧化氯暴露剂量才趋于稳定(2.56 nmol·L−1)。可能的原因是NDMA的前体物分子量较小,更快被二氧化氯氧化,而二氧化氯氧化其他亚硝胺类的前体物则需更长的暴露时间。
HANs的前体物主要是水中的含氮有机物如氨基酸以及醛类物质,在之前的研究中发现,二氧化氯预氧化通常可降低HANs在后续消毒过程中的生成,而当后氯胺化时投加氯胺的量较低时(NH2Cl:precursor ≤ 10)时,采用二氧化氯预氧化游离酪氨酸会使DCAN的生成增加,这可能是由于游离酪氨酸中的α-氨基被二氧化氯氧化,从而增加了DCAN产率,而当采用高剂量氯胺处理时,二氧化氯的预氧化才能氧化HANs的前体物从而降低其在后消毒过程中的生成[38],推测本实验中的现象是由于实验采用模拟实际水处理的UFC过程,氯胺的浓度较低导致。经二氧化氯预氧化后,丁酰肼在氯胺化过程中的生成的NDMA随二氧化氯投量的增大而升高[28],在本实验中则呈现相反趋势,进一步说明污水中丁酰肼的含量极低。同时,二氧化氯能使一些NDMA的前体物如雷尼替丁、DMAI等快速氧化成DMA,但增加二氧化氯的暴露剂量并不能提高DMA的生成率,因此NDMA的生成控制在较低二氧化氯暴露水平下即达到平衡。而对于总亚硝胺而言,本实验首次发现其前体物的去除随二氧化氯的暴露增大而增大,在较高暴露水平(30 mg·min−1·L−1)才能达到较好去除。
图4a表明,在模拟实际水处理的后氯胺化实验中,DCAN生成量会随着高铁酸盐的引入而迅速减少,从对照组的6.4 nmol·L−1迅速降至0.7 nmol·L−1,已接近检出限浓度(0.5 nmol·L−1),而BCAN和TCNM的浓度随高铁酸盐暴露剂量增大变化不明显,均接近检出限浓度(分别为0.9 nmol·L−1和0.3 nmol·L−1)。高铁酸盐能氧化氨基酸类和胺类物质,破坏C—N键,使含有—NH2官能团的物质如被破坏,而这类物质正是HANs的重要前体物[39−40]。图4b表明,氯胺化后的污水水样中NDMA浓度随高铁酸盐的暴露剂量从0 mg·min−1·L−1增至332 mg·min−1·L−1而从0.68 nmol·L−1逐步减至0.33 nmol·L−1,且尚未到达平衡;而高铁酸盐对TONO的前体物去除随高铁酸盐暴露剂量的增大而迅速增强,在143 mg·min−1·L−1的高铁酸盐暴露下TONO的浓度降至2.79 nmol·L−1,随后趋于稳定。
图5a表明,在模拟实际水处理的后氯胺化实验中,TCNM生成量会随臭氧浓度增大而迅速升高,从对照组的1.37 nmol·L−1以接近线性的趋势逐渐增至47.89 nmol·L−1,而DCAN和BCAN的浓度随臭氧暴露剂量增大变化不明显。图5b表明,氯胺化后的污水水样中,随臭氧的暴露剂量从0 mg·min−1·L−1增至0.19 mg·min−1·L−1,NDMA与TONO的浓度逐渐减至0.44 nmol·L−1和5.17 nmol·L−1,且到达平衡。臭氧氧化的污水会产生大量硝基甲烷,而硝基甲烷正是卤代硝基甲烷(以TCNM为主)的重要前体物[41],因此在本实验中TCNM在氯胺化过程中随前端臭氧暴露剂量增大而急剧增大的现象主要是由于臭氧氧化了污水中的有机物生成硝基甲烷,而N-烷基胺和一些含N-甲胺官能团的抗抑郁类药物在臭氧氧化后会普遍生成一级硝基烷烃,且转化率通常高于50%,说明本实验污水样品中可能含有大量含N-甲胺官能团的有机物。Lee等的研究表明[37],低剂量的臭氧暴露(0.2 mg·min−1·L−1)就能达到约78%的NDMA前体物去除,将臭氧暴露剂量增至1.0 mg·min−1·L−1对NDMA的生成控制并无明显影响,这与本实验中观测到的现象一致。
-
(1)在污水回用过程中,生物处理会促进生成亚硝胺(5.0%—254.5%)。本研究选用A/A/O生物处理后的污水出水进行深度处理,以探究生物处理与深度处理结合的工艺流程对含氮消毒副产物的控制。深度处理工艺中,粉末活性炭会促进亚硝胺的生成(61.5%),臭氧氧化则会直接生成亚硝胺(138%)。因此仅从亚硝胺的控制考虑,污水回用中采用二氧化氯或高铁酸盐氧化更能保障水质安全。
(2)选择的深度处理工艺对亚硝胺前体物均有去除,其中粉末活性炭和二氧化氯对NDMA和TONO的控制效果较好(约70.0%)。但氧化工艺对HANs和TCNM的前体物影响不一,高铁酸盐对两者的前体物均有显著去除,二氧化氯则增加了HANs的前体物(269%–346%),臭氧氧化则会增加TCNM的前体物。仅从典型含氮消毒副产物及其前体物的控制效果考虑,高铁酸盐氧化更适合在污水回用中作为深度处理工艺被应用,但高铁酸盐的大剂量制备工序繁琐复杂,在污水处理的实际应用中需考虑其便利性与实用性。
污水生物与深度处理技术对新型含氮消毒副产物及前体物的控制
Study on the control of emerging nitrogenous disinfection by-products and precursors by biological and advanced treatment for wastewater reclamation
-
摘要: 典型含氮消毒副产物(N-DBPs, 包括亚硝胺、卤乙腈和三氯硝基甲烷)的控制是污水回用过程中需关注的重点。本研究对采用不同生物处理工艺的污水厂进行生化池进出水中N-DBPs的存在水平分析,并选取其中一个污水厂的二级出水进行不同深度处理(粉末活性炭吸附、臭氧氧化、高铁酸盐氧化、二氧化氯氧化),研究其对N-DBPs及其前体物的去除。结果表明,生物处理过程会导致亚硝胺的生成。深度处理工艺中,粉末活性炭会催化亚硝胺的生成,臭氧氧化则会直接生成亚硝胺。选择的深度处理工艺对亚硝胺前体物均有去除,其中粉末活性炭和二氧化氯对二甲基亚硝胺和总亚硝胺的控制效果较好。但氧化工艺对卤乙腈(HANs)和三氯硝基甲烷(TCNM)的前体物影响不一,高铁酸盐对两者的前体物均有显著去除,二氧化氯会增加污水在低浓度氯胺处理时HANs的生成,臭氧氧化则会增加TCNM在后氯胺化过程中的生成。研究结果推动了污水深度处理技术的发展并为相关研究提供了理论指导。Abstract: The control of typical nitrogenous disinfection by-products (N-DBPs, including nitrosamines, haloacetonitriles and trichloronitromethane) is a major concern in wastewater reuse. In this study, the presence levels of N-DBPs in the influent and effluent of biochemical treatment in wastewater treatment plants (WWTPs) with different biological treatment processes were analyzed. The secondary effluent of one of the WWTPs was selected to investigate the removal of N-DBPs and their precursors in different advanced treatments (powdered activated carbon adsorption, ozone oxidation, ferrate oxidation, and chlorine dioxide oxidation). The results showed that the biological treatment process resulted in the generation of nitrosamines. For the advanced treatment processes, powdered activated carbon (PAC) catalyzed the generation of nitrosamines, while ozone oxidation formed nitrosamines directly. The selected advanced treatment processes showed different degrees of removal of nitrosamine precursors, with PAC and chlorine dioxide providing better control of N-nitrosodimethylamine and total nitrosamines. However, the oxidation processes had mixed effects on the precursors of haloacetonitriles (HANs) and trichloronitromethane (TCNM). Ferrate (FeVI) showed significant removal of precursors of both HANs and TCNM, while chlorine dioxide increased the formation of HANs in the effluent treated with low concentration of chloramine. Ozonation increased the generation of TCNM during post-chloramination. The results promote the development of advanced wastewater treatment technologies and provide theoretical guidance for related research.
-
砷是煤中普遍存在、毒性较强且易挥发的痕量元素之一,在煤燃烧过程中,砷可随烟气被释放进入环境导致污染,是重要的人为大气砷排放源[1-4]。烟气中的砷除一部分以气态形式存在,大部分可在灰尘等颗粒物中富集[5-10]。无论是气态还是颗粒态砷最终均进入环境并发生迁移转化[11-12],对人体构成健康风险[13-17]。
将烟气砷污染控制技术按实施位置分为燃烧前、燃烧中和燃烧后三类控制技术[18]。燃烧前控制技术的关键是采用物理或化学前处理方法减少煤中的砷含量。煤中砷酸盐和有机砷较少,黄铁矿形态砷居多[19]。连续化学浸提法分析表明,砷以硫化物结合态为主,其次为有机物结合态,其它形态砷含量与煤种相关[20]。燃烧中砷污染控制技术的核心是通过混煤燃烧、添加化学药剂将挥发性强的气态砷转化为不易挥发的砷酸盐等,然后与粉煤灰发生凝并或被吸附而被除尘器捕集[21-22],以达到炉膛出口烟气砷含量降低的目标。燃烧后脱除主要针对末端烟气中的砷,通过吸附剂对其进行物理、化学吸附,实现气态砷在吸附剂表面的固化和稳定化,从而减少烟气中的气态砷含量。
本文综述了近年来烟气砷污染控制领域的主要研究进展,按燃烧前、燃烧中和燃烧后三类控制技术展开论述[18],对比分析了不同控制技术的原理、效果和优缺点。
1. 烟气砷排放现状(The situation of arsenic emission from flue gas)
燃煤烟气中的砷来源于煤炭,煤中砷含量与地质形成过程密切相关。因此,不同国家和地区燃煤中砷含量差异明显,不同煤种砷含量也不相同。表1至表2分别列出了不同国家、地区和煤种中砷的含量[8, 15, 23-26]。世界上煤中砷平均含量为8.3 mg·kg−1。我国煤中砷平均含量为3.18 mg·kg−1,略低于世界平均水平。我国燃煤发电仍然是最主要的能源利用方式,2019年我国燃煤机组发电量为50465 GWh[27],占总发电量约69%。据统计,2020年我国煤炭产量为39亿t[28-29],其中45%以上的煤炭被用于燃煤发电[8].
表 1 不同国家和地区煤砷含量Table 1. The average contents of arsenic in coal from different countries and areas国家和地区Country and Areas 砷丰度/(mg·kg−1)As 样本数Number of samples 参考文献Ref. 中国 3.18 737 [8] 印度 0.1 4 [26] 美国 1.5 — [30] 俄国 7.8 59 [31] 世界 8.3 119 [32] 中国贵州 3.9 140 [15] Donets Basin 9.4 — [23] Danville 12.7 33 [24] Springfield 9.4 64 [24] 注: —表示文献中未给出, 下同. Note:— Indicates that it is not given in the literature, the same below. | Show Table DownLoad:
CSV
表 2 不同煤种砷的含量Table 2. The average contents of arsenic in different ranks of coal| Show TableDownLoad:
CSV
DownLoad:
CSV
表 2 不同煤种砷的含量Table 2. The average contents of arsenic in different ranks of coal| Show TableDownLoad:
CSV
煤中砷以不同结合形态存在。绝大部分有机结合态砷可被燃烧释放并发生形态转化,粉煤灰中几乎检测不到有机结合态砷;燃烧温度越高,煤中残渣态砷越趋于向可交换态与酸溶态转化,粉煤灰中残渣态比例降低,气态砷所占比例升高[34-35]。热力学平衡计算表明,在900—1200 K氧化气氛条件下,砷主要以As2O3(g)存在[34];煤燃烧后,气态砷占总砷含量的24.5%,大多数砷吸附在颗粒物上[36]。部分不能被脱除的颗粒态、气态砷仍通过末端烟气排放进入大气[8, 37-40]。2003—2006年,我国工业砷排放逐年增加,2006年达到了902.95 t,其中燃煤电厂砷排放量达到了522.13 t,占总排放量半数以上[8]。平均每发电1 MWh将产生大气砷排放1.36—9.07 mg[36]。2000—2006年,燃煤电厂大气砷排放量由354.01 t逐步上升至615.70 t[38]。2020年中国燃煤电厂的大气砷排放量为216—257 t[38]。据统计,我国部分地区大气砷平均浓度超过了《国家环境空气质量标准》和世界卫生组织规定的参考值6.0 ng·m−3和6.6 ng·m−3[41]。燃煤电厂持续产生的大气砷排放不容忽视。此外,烟气砷污染控制还可以减缓SCR催化剂中毒[42-43],降低电厂的运行成本。
2. 燃烧前控制(Pre-combustion removal)
较早用于燃烧前砷控制的技术是通过煤洗选去除矸石进而达到降低砷含量的目的[44-45]。根据水溶性强弱,煤中砷可分为硫化物态(36%)、有机结合态(26%)、砷酸盐(17%)、硅酸盐(16%)、水溶解态与可交换态(5%)[46]。根据砷元素在煤中的主要赋存状态与浸出特征,黄铁矿形态的砷含量较高[19, 47]。煤矸石中含有大量的黄铁矿[48-51],洗选煤技术通过去除煤矸石实现有毒元素协同脱除[52-53]。洗选煤技术可分为湿法洗选煤技术与干法洗选煤技术[45]。干法选煤技术在空气中进行选煤,利用煤与矸石的物理性质差别,通过外力,如气流、振动、摇动与它们组合作用进行筛选,比如风选、跳汰选、电磁选和空气重介质流化床选煤等[44]。湿法选煤技术用液态流体作为分选介质,煤矸石密度比煤大,经过流体时,煤矸石被截留后与煤分离[45],同时一些有毒元素被协同脱除。湿法选煤包括重介质法与浮选法等[54]。湿法洗选是我国现行的主流技术。王文峰等[52]的研究表明,基于重介质的物理洗选煤的砷平均脱除率为62.1%,最低为42.5%,最高为84.3%。表3中总结了一些文献中洗选煤技术的砷脱除效果,部分文献研究发现,洗选煤技术不同程度上降低了煤中的砷含量。
表 3 煤洗选砷脱除效果Table 3. The arsenic removal efficiency of coal washing分类Category 方法Method 最低效率/%Min 平均效率/%Avg 最大效率/%Max 样本数N 文献Ref. 干式 重介质法 42.5 62.1 84.3 6 [52] 干式与湿式 重介质和浮选法 45.3 63.4 84.2 6 [58] 湿式 浮选法 12.5 52.4 89.6 10 [59] 干式与湿式 重介质、跳汰选和浮选法 — 16.5 — 47 [60] | Show TableDownLoad:
CSV
脱除率主要与砷赋存状态密切相关。但也受煤级、粒度以及洗选工艺影响,因此不同样品在不同的洗选工艺中差异较大。褐煤与有机砷含量较高的煤无法通过洗选脱砷,甚至发生严重的砷富集现象。王明仕等[55]的研究中有4个煤样洗选后砷发生富集,它们的平均富集率为82.4%。这是因为煤中的硫与砷具有很强的相关性,但部分煤中硫可能以有机硫或细分散的矿物存在[46, 56],导致砷与硫的脱洗率为负。洗选煤的砷脱除效率还与煤的总砷含量与煤粉粒度有关。研究发现,煤中砷含量为0—0.55 mg·kg−1时,砷主要以有机结合态为主,溶解性差、密度小,洗选过程可表现出一定程度的富集;含量为5.55—8.00 mg·kg−1时,煤中的硫化物态砷含量最高,平均脱除率为67.30%;含量大于8.00 mg·kg−1时平均脱除率为49.82%[46]。此外,有研究发现精末、精小粒、精中粒、精大粒洗选后砷脱除率表现为先变高后降低的趋势,表明它们的脱除程度在精中粒中相对较高[57]。
洗选煤技术降低了煤炭中所含的灰份和硫份,减少很多因燃煤产生的环境问题。通过洗选煤筛分出煤矸石同时将砷含量降低,但不同研究中砷洗选脱除效率相差较大,存在不确定性,脱除效率受到多种条件的影响。另外,洗选煤会产生大量高砷煤泥等废物[46, 52, 57-62],存在较高二次污染风险。
3. 燃烧中控制(Removal during combustion)
燃烧中砷污染控制技术是在煤燃烧过程中通过混煤、添加化学添加剂等方式,使其不易转化为气态砷或富集在细微飞灰颗粒上,而是以固态砷酸盐存在或吸附在粗飞灰颗粒上,从而与底灰飞灰等共同被APCDs捕集。研究证明,炉膛烟气中的As2O3(g)与CaO和CaCO3反应,生成以砷酸钙(Ca3(AsO4)2)、砷酸铁(FeAsO4)为代表的易团聚、热稳定性强、挥发性弱的化合物,减少气态砷释放[15, 63-65]。
3.1 混煤
混煤燃烧是一种洁净煤燃烧技术,混煤在一定程度上解决锅炉结渣问题,还能降低氮、硫氧化物与重金属的排放[66-67]。研究表明,Heshan烟煤与Huolinhe褐煤1:1混燃后,细颗粒物中的砷向粗颗粒物中转移,同时与Heshan烟煤燃烧后相比,PM10中砷降低了33%[66]。经分析,Huolinhe褐煤中含有较多矿物质,混煤燃烧降低了灰熔融温度,促进了铁、钙与铝硅酸盐的反应,抑制细颗粒物产生,而粗颗粒物中的砷更易被脱除。此外,煤中矿物质与砷相互反应产生砷酸盐,抑制砷的挥发。但有研究发现不同配比下,混煤中砷的挥发率均高于两种原煤砷挥发率的加权平均值,导致砷挥发率增加[66-69]。主要原因是褐煤中的高挥发分促进了混煤中的焦炭燃烧,进而加速了硫化物结合态砷分解生成气态砷,因此混煤的砷挥发特性更接近于单一褐煤[69]。目前混煤法进行砷脱除具有一定局限性,煤种、煤质以及矿物含量对脱除效果影响显著,应用范围较窄。
3.2 化学添加剂
郭胜利等[65]通过改性碳酸钙混燃技术,实现了燃煤有毒元素有效控制。硫酸铝(Al2(SO4)3)改性碳酸钙与无烟煤混合燃烧,砷脱除效率达到46.15%。经分析,Al3+半径小于Ca2+,它与碳酸根中氧成键的能量更低。Al3+取代Ca2+的位置,形成点缺陷[70],碳酸钙晶体内部重新排列,Ca2+被活化后利用率增加。Zhao等[15]研究了氧化钙混燃技术,向煤粉中掺入0.3% wt. CaO,燃烧后,PM1中砷降低了56%,PM10中砷降低了6.8%。在该控制技术应用过程中,在900 ℃时主要发生化学吸附且产物为热稳定性很强的砷酸钙。研究发现,气态砷富集在PM1中,氧化钙加入后,大部分砷被吸附剂表面的活化阳离子捕获,气态砷含量明显降低,但炉内仍有部分氧化钙团聚成大于PM1的颗粒,并与气态砷发生反应,导致PM10中砷含量下降不明显。燃烧中砷污染控制不需要对现有APCDs进行改造也无需增加新设备,砷脱除成本低。
4. 燃烧后控制(Post-combustion removal)
燃烧后烟气砷污染控制技术,是利用原有的空气污染控制设备与吸附剂将烟气砷固定并脱除,降低砷排放量。研究发现,APCDs对砷具有协同控制作用[71-75],包括选择性催化还原脱硝(SCR)、静电除尘器(ESP)、布袋除尘器(FF)与湿法烟气脱硫(WFGD),它们的砷脱除效果见表4。
表 4 APCDs的砷脱除效果总结Table 4. Summary of performance of APCDs for arsenic removal from flue gas控制设备Control device 平均脱除效率/% Average removal efficiency 样本数N 文献Ref. SCR 6.4 5 [73] ESP 83.0 4 [71] ESP 96.1 5 [73] ESP 98.3 1 [74] ESP+FF 99.8 1 [75] WFGD 61.0 4 [71] WFGD 48.2 1 [75] WFGD 75.0 1 [74] ESP+WFGD 97.3 — [72] ESP+WFGD 99.6 1 [74] | Show TableDownLoad:
CSV
2020年,燃煤电厂全部完成超低排放改造(ULE)[76]。某电厂改造后,空气污染控制设备(APCDs)对砷的协同脱除效率由约95%提高至约97%[77]。即使完成了ULE改造,2020年中国燃煤电厂的大气砷排放量仍达到216—257 t[38]。,SCR对砷的脱除效果差,ESP最好,WFGD次之。APCDs的砷平均脱除效率已经很高,但现有APCDs无法有效脱除气态砷,尤其是水溶性很低的As2O3(g),而它的毒性极大,加上电厂烟气排放量巨大,这部分砷对生态环境的影响不容忽视。因此燃烧后的烟气还需要添加额外的吸附剂将气态砷固定并脱除。
除尘器可有效捕集到Ca/Fe/Al的砷酸盐[3],其热稳定性强,沸点高不易挥发,环境化学性质较为稳定。因此,将As2O3(g)转化为稳定性强的砷酸盐可以实现气态砷的固化[15, 63-64, 76,78]。CaO、CaSiO3、Fe2O3、γ-Al2O3及其改性材料可以被用于吸附脱除烟气砷。按照文献中的吸附剂组成特点,将其区分为碳基、钙基、铁基、铝基吸附剂、复合金属吸附剂以及其他吸附剂。
4.1 碳基吸附剂
常见的碳基吸附剂包括各种活性炭[36, 79-81]、石墨烯[82]、C60[83]和纳米碳管[84]等。碳基吸附剂具有比表面积大、孔容积大、孔隙率高等特点,因此具有很强的吸附能力[79-81]。Wu等[85]通过密度泛函理论(DFT)模拟了以六元碳环椅形、之字形吸附剂在分子水平上吸附As2O3(g)的过程,认为As2O3分子可以被水平或垂直形式吸附在碳原子表面,6种可能的吸附形式中吸附能最大为− 45.47 kJ·mol−1,最小为− 497.74 kJ·mol−1,这证明活性炭吸附剂与气态砷可发生化学吸附。在模拟烟气条件下,低浓度SO2降低了相邻碳吸附活性位点的静电势,促进了其对As2O3(g)的吸附。由于竞争吸附效应,SO2浓度较高时可抑制其对As2O3(g)的吸附。Charpenteau等[79]研究了商品活性炭、废旧轮胎热解活性炭和黑炭对气态砷的脱除作用,200 ℃时3种吸附剂的砷脱除效率分别为69%、50%和72%;400 ℃时分别为72%、51%和38%。可见不同碳基吸附剂在不同温度时的吸附效果存在明显差异。Marczak等[36]采用商品活性炭将烟煤燃烧烟气的砷浓度由146.2 μg·m−2降低至17.3 μg·m−2,脱除效率达到88.2%。López-Antón等[80]模拟了煤燃烧过程中砷和硒的挥发过程,并采用3种活性炭进行吸附实验,砷吸附容量分别为0.30 mg·g−1、0.35 mg·g−1和0.56 mg·g−1。提高活性炭的用量可以改善气态砷的吸附效果。Player等[81]通过提高活性炭的用量并优化吸附条件,最终实现99.94%的As2O3脱除效率。碳基吸附剂对烟气中气态砷具有较强的吸附效果,但选择性较差,易受烟气组分影响,应用成本相对较高,在实际应用中存在一定难度。另外,碳基吸附剂的热稳定性差,吸附效果受温度影响明显。一般地,超过400 ℃时,砷吸附效果会显著下降[81]。
4.2 钙基吸附剂
钙基吸附剂主要包括氧化钙、碳酸钙、硫酸钙和硅酸钙等。钙基吸附剂对As2O3(g)有较好的脱除效果,可与As2O3(g)发生如下(1)—(7)反应[86-92]:
CaO(s)+As2O3(g)→CaO⋅As2O[88]3 (1) CaO(s)+1/3As2O3+1/3O2→1/3Ca3(AsO4)[86,88−92]2 (2) CaCO3+1/3As2O3+1/3O2→1/3Ca3(AsO4)2+CO[91]2 (3) CaSO4+1/3As2O3→1/3Ca3(AsO4)2+SO2+1/6O[65,88]2 (4) CaSO4+1/2As2O3→1/2Ca2As2O7+SO[87]2 (5) CaSiO3+1/3As2O3+1/3O2→1/3Ca3(AsO4)2+SiO[89]2 (6) 2CaSiO3+As2O3+O2→Ca2As2O7⋅2SiO[87]2 (7) 3Ca2As2O7→2Ca3(AsO4)2+As2O3+O[72]2 (8) 温度较低时钙基吸附剂与As2O3(g)发生物理吸附,类似反应(1),高温时则几乎为化学吸附。研究发现,450 ℃以下,CaO与As2O3以物理吸附为主,750 ℃以上发生化学吸附(反应2)[88]。As2O3(g)与CaO和CaCO3可发生反应(2)、(3)后生成砷酸钙Ca3(AsO4)2[86, 88-92],与CaSO4和CaSiO3反应产物为Ca2As2O7和Ca3(AsO4)2(反应4—7),600 ℃主要产物为单斜晶型的Ca3(AsO4)2,800—1000 ℃时逐步转化为菱面晶型[91],1000 ℃以上时生成的Ca2As2O7则再次分解为Ca3(AsO4)2(反应8),因此CaSO4或CaSiO3与As2O3(g)的最终产物同样也为Ca3(AsO4)2[72]。
从反应(2)—(7)可以看出,除CaSO4外,CaO、CaCO3和CaSiO3与As2O3反应均有O2参与,因此O2可以促进砷的化学吸附[86-92]。As2O3吸附转化过程中CaO晶体表面的晶格氧与表面吸附O2同时对As2O3起到氧化作用[88],促进砷的脱除。此外,研究表明CO2也可促进CaO对砷的脱除效果,但具体机理还需要进一步研究[91]。研究显示,在1300 ℃以下CaO和CaSiO3对砷的吸附容量随温度升高而升高[87, 89-90]。然而,CaSO4的吸附容量却随温度升高而降低。高浓度SO2(5.721 g·m−3)显著抑制了CaO对As2O3的吸附作用,但900 ℃以上时,CaO与SO2反应生成了CaSO4,同样对As2O3(g)具有一定吸附能力,减弱了SO2的抑制效果[88],见化学反应(4)和(5)。SO2浓度较低时(2.002 g·m−3)CaO的吸附容量几乎不受影响[91]。NO对CaO脱除As2O3(g)的影响较小[87, 90]。表5中列出了不同温度与时间下的钙基吸附剂的吸附效果。钙基吸附剂适于高温烟气或混燃过程中气态砷的产生抑制和吸附脱除,在价格成本方面表现出优势。
表 5 钙基吸附剂的砷脱除效果总结Table 5. Summary of performance of calcium-based sorbents for arsenic removal from flue gas吸附剂Sorbent 模拟烟气Simulated flue gas 温度/℃Temp. 吸附时间/minTime 吸附容量/(mg·g−1)Absorption capacity 文献Ref. CaSiO3 N2/O2 1200 10 4.98 [87] CaSiO3 N2/O2/NO/SO2 1200 10 3.50 [87] CaO N2/O2/H2O 750 5 3.75 [88] CaO N2/O2/H2O/SO2 750 5 8.69 [88] CaO N2/O2 800 30 11.82 [91] CaO N2/O2/SO2 800 30 11.51 [91] CaO N2/O2/NO/SO2 1000 10 1.65 [90] CaO N2/O2/NO/SO2 1300 10 1.94 [90] CaSO4 N2/O2 1000 10 3.79 [90] | Show TableDownLoad:
CSV
4.3 铁基吸附剂
Fu等[93]研究发现富砷烟煤燃烧过程中,含铁矿物在砷捕获和形态转化过程中发挥着关键作用。鉴于此,将Fe2O3、Fe3O4或含铁矿物及氧化物称为铁基吸附剂。研究表明,Fe2O3的表面晶格氧将As2O3(g)氧化为As2O5(s),晶格氧可以通过化学吸附O2再生,且As2O5(s)与Fe2O3进一步反应生成砷酸铁(FeAsO4)[80, 92, 94]。DFT研究结果也表明,As2O3(g)可在Fe2O3的(001)面形成8种稳定的吸附结构,吸附能最低为− 275.52 kJ·mol−1[95]。此外,在Fe3O4的(111)面形成四种稳定的吸附结构,均为化学吸附,其中吸附能最低为− 197.96 kJ·mol−1[93]。研究证明,FeAsO4通过下列化学反应(9)生成,该反应的分步反应为(10)—(12)[80]。此外,Fe3O4、铁磁珠与As2O3发生反应(13),可以生成FeAsO4与砷酸亚铁(Fe3(AsO4)2)[96]。
As2O3(g)+Fe2O3+O2(g)→FeAsO4 (9) As2O3(g)↔As2O3(ads) (10) As2O3(ads)+FexOy↔As2O5(ads)+FexOy−2 (11) As2O5(ads)+Fe2O3→2FeAsO4 (12) 4As2O3(g)+3Fe3O4+4O2(g)→6FeAsO4+Fe3(AsO4)2 (13) 表6中列出了不同温度与时间下的铁基吸附剂的吸附效果。在600—900 ℃的温度区间,Fe2O3的砷脱除效率随温度升高由57.02%降低至43.38%[92];在150—900 ℃的温度区间内,Fe2O3/γ-Al2O3的砷脱除效率先升高后降低,600 ℃时效率最高为68%[94]。O2可促进铁基吸附剂对砷的吸附脱除。As2O3(g)转化为FeAsO4会消耗Fe2O3的晶格氧,而O2可以有效补充表面晶格氧[80, 92, 94]。在一定浓度范围内,SO2有利于砷的脱除,可在吸附剂表面与O2、H2O在Fe(Ⅲ)催化作用下生成
HSO−4 SO2−4 NO−3 NO−3 表 6 烟气组分和温度对铁基吸附剂的砷脱除效果Table 6. The arsenic removal efficiencies of iron-based adsorbents in different gas components and reaction temperatures吸附剂Sorbent 模拟烟气Simulated flue gas 温度/℃Temp. 吸附时间/minTime 脱除效率/%Removal efficiency 吸附容量/(mg·g−1)Absorption capacity 文献Ref. Fe2O3 N2/O2 600 90 57.02 1.38 [92] Fe2O3 N2/O2 900 90 43.38 1.08 [92] 铁磁珠 N2/O2/SO2 600 60 42.75 0.53 [96] Fe2O3/γ-Al2O3 N2/O2/SO2/NO 600 — 68.00 — [94] Fe2O3/γ-Al2O3 N2/O2/SO2/NO 900 — 46.00 — [94] | Show TableDownLoad:
CSV
4.4 铝基吸附剂
Hu等[97]通过DFT理论计算,研究了γ-Al2O3(001)表面吸附As2O3(g)的吸附结构和过渡态。结果表明,吸附能最低达到了− 409.13 kJ·mol−1,研究认为As2O3(g)在γ-Al2O3表面存在如图1所示的4种吸附方式[98]。不同烟气组分和温度下铝基吸附剂吸附效果见表7。
 图 1 (a)As3+物理吸附;(b)As3+弱化学吸附;(c)As3+强化学吸附;(d)As5+强化学吸附Figure 1. (a) As3+ physical adsorption; (b) As3+ weak chemical adsorption; (c) As3+ strong chemical adsorption; (d) As5+ strong chemical adsorption(图片借鉴自文献[98])表 7 不同烟气组分和温度下铝基吸附剂的砷脱除效果Table 7. The arsenic removal efficiencies of aluminum-based adsorbents in different gas components and reaction temperatures
图 1 (a)As3+物理吸附;(b)As3+弱化学吸附;(c)As3+强化学吸附;(d)As5+强化学吸附Figure 1. (a) As3+ physical adsorption; (b) As3+ weak chemical adsorption; (c) As3+ strong chemical adsorption; (d) As5+ strong chemical adsorption(图片借鉴自文献[98])表 7 不同烟气组分和温度下铝基吸附剂的砷脱除效果Table 7. The arsenic removal efficiencies of aluminum-based adsorbents in different gas components and reaction temperatures吸附剂Sorbent 模拟烟气Simulated flue gas 温度/℃Temp 吸附时间/minTime 吸附容量/(mg·g−1)Absorption capacity 文献Ref. γ-Al2O3 N2/O2/H2O 300 60 66.62 [99] γ-Al2O3 N2/O2/H2O 400 60 52.30 [99] γ-Al2O3 N2/O2/H2O/SO2 300 60 56.92 [99] γ-Al2O3 N2/O2/H2O/SO2 400 60 46.49 [99] γ-Al2O3 N2/O2/H2O 750 90 9.27 [98] γ-Al2O3 N2/O2/H2O/HCl/SO2 750 90 8.44 [98] γ-Al2O3 N2 400 60 0.99 [100] γ-Al2O3 N2 600 60 1.48 [100] γ-Al2O3 N2/O2 400 60 1.55 [100] γ-Al2O3 N2/O2 600 60 2.17 [100] | Show TableDownLoad:
CSV
模拟烟气条件下,砷脱除效果随温度升高而降低[99]。在较高温度条件下,γ-Al2O3比表面积显著下降并开始向θ-Al2O3转变,导致脱除效果变差[98]。由于竞争吸附效应,SO2一定程度上抑制了γ-Al2O3对As2O3(g)的吸附。NO显著抑制铝基吸附剂对砷的脱除效果,NO与As2O3(g)在γ-Al2O3表面铝原子上发生竞争吸附,同时竞争晶格氧,NO则被氧化为NO2,而As2O3(g)也被转化为氧化形态。尽管晶格氧可以通过O2进行不断补充,NO仍然会表现出抑制效果。SO2与NO同时存在时,它们可在γ-Al2O3上的Al-OH位生成(Al-SO3NO)中间体,随后在O2存在条件下,转化为(Al-SO4)和(NO2),在一定程度上避免了晶格氧耗竭从而减弱了NO对As2O3(g)的吸附抑制效应[99]。铝基吸附剂的适宜脱除温度低于钙基吸附剂,价格相对低廉,脱除效果较好。
4.5 复合金属吸附剂
单金属氧化物吸附剂对砷吸附虽然在As2O3(g)浓度较低时具有较高的脱除效率,但吸附容量有限,较难满足高浓度As2O3(g)或复杂烟气条件下的需求,且γ-Al2O3与CaO高温时会出现比表面积下降[88]或晶型转化[98]等问题。为了提高吸附剂的吸附容量和稳定性,改善吸附效果,研究提出了多种复合吸附剂用于烟气砷污染控制。表8列出了不同复合吸附剂对砷的脱除效果,其中复合吸附剂主要包括α-Al2O3、γ-Al2O3负载Pt吸附剂[101-103]、Fe-Mn双元氧化物(FMBO)[104]、氧化锰改性凹凸棒土(Mn(Ⅳ)/ATP)[105]等。
表 8 不同烟气组分和温度下复合吸附剂的砷脱除效果Table 8. The arsenic removal efficiencies of aluminum-based adsorbents in different gas components and reaction temperatures吸附剂Sorbent 模拟烟气Simulated flue gas 温度/℃Temp. 吸附时间/minTime 吸附容量/(mg·g−1)Absorption capacity 文献Ref. Pd/α-Al2O3 N2/H2/CO/CO2/H2S 204 150 4.74 [101] Pd/γ-Al2O3 N2/H2/CO2 200 300 70.00 [102] FMBO N2/O2/CO2/H2O/NO/SO2 300 30 17.98 [104] FMBO N2/O2/CO2/H2O/NO/SO2 600 30 21.65 [104] FMBO N2/O2/CO2/H2O/NO/SO2 700 30 8.22 [104] Mn(Ⅳ)/ATP N2/O2/CO2/H2O/NO/SO2 600 30 6.66 [105] Mn(Ⅳ)/ATP N2/O2/CO2/H2O/NO/SO2 600 60 10.98 [105] Mn(Ⅳ)/ATP N2/O2/CO2/H2O/NO/SO2 600 180 25.01 [105] | Show TableDownLoad:
CSV
As2O3(g)与Pd/γ-Al2O3发生化学吸附,砷吸附容量达到70 mg·g−1[102],吸附效率达到63.81%[103],吸附容量明显优于单金属氧化物吸附剂。吸附产物为As3Pd8与AsPd2[101-102]。当As/Pd原子比提高时,As3Pd8可向AsPd2转化[101]。在初始较短时间内,反应产物为As3Pd8[101],随着反应时间的延长,产物逐渐转化为AsPd2[102]。
CaO、Fe2O3和γ-Al2O3脱除As2O3(g)时需要晶格氧或吸附剂表面的化学吸附氧将As2O3(g)氧化为As2O5(s),再形成稳定的产物。MnO2可将As2O3(g)氧化为As2O5(s),但无法与As2O5(s)形成稳定的产物,MnO2与Fe2O3制成复合吸附剂(FMBO)后,MnO2将As2O3(g)氧化为As2O5(s),而Fe2O3不再需要消耗晶格氧,便可将As2O5(s)固化在FMBO表面生成FeAsO4。最佳条件下,FMBO的砷吸附容量达到21.65 mg·g−1。凹凸棒土(ATP)是一种含有Fe2O3的硅铝酸盐,具有高比表面积与较大的吸附容量,且天然矿物的成本低、性质稳定。ATP可作为复合材料载体制备复合吸附剂[104-106]。He等[105]制作了氧化锰改性凹凸棒土(Mn(Ⅳ)/ATP)用于脱除烟气砷,脱除机理与FMBO类似,最佳条件时的砷吸附容量为25.01 mg·g−1。
FMBO吸附容量受温度影响明显。在600 ℃以下,温度越高FMBO与Mn(Ⅳ)/ATP的砷吸附容量越高,600 ℃时吸附容量最大,但温度达到700 ℃以上时,由于MnO2发生团聚或分解,比表面积下降,吸附容量下降明显[104-105]。CO2会产生明显的吸附抑制作用,CO2通过占据FMBO与Mn(Ⅳ)/ATP表面的活性位点,浓度高时,表面的铁被还原为二价[107],导致吸附效果下降。FMBO与Mn(Ⅳ)/ATP的吸附机理与实验结论都证实了O2对吸附效果产生促进作用,此处不再赘述。随着NO浓度增大,FMBO与Mn(Ⅳ)/ATP的砷吸附容量先升高后降低,NO可以在它们的表面形成NO+、NO2+和NO2,这些官能团可以将As2O3(g)氧化为As2O5(s),但NO浓度过高会导致晶格氧与化学吸附氧过度消耗,使砷脱除效率下降。SO2对FMBO与Mn(Ⅳ)/ATP的影响规律不同,SO2为1.144—5.721 g·m−3时,浓度越高Mn(Ⅳ)/ATP的砷吸附容量越高。SO2可在H2O和O2存在下生成HSO4−或SO42-。Mn氧化物与ATP结合后更容易吸附H2O并生成羟基,SO2生成双齿表面配合物并与表面羟基结合后稳定存在,而锰氧化物还可加速SO2氧化为硫酸盐的过程。SO2达到5.721 g·m−3时FMBO的吸附容量略有降低,而Mn(Ⅳ)/ATP的吸附容量未受抑制。
Mn(Ⅳ)/ATP与铁锰双元氧化物吸附剂的最佳砷脱除温度为600 ℃,高于SCR的最佳工作温度,但将砷脱除设备置于SCR前,吸附剂可耐受较高SO2、NO与颗粒物的环境。Pd/γ-Al2O3在204 ℃时的砷吸附容量很大,可放置在SCR后或除尘器之后,此时烟气中NOx与颗粒物含量较低。
4.6 其他脱除技术
除了4.2—4.5节中提到的砷脱除技术,还有一些脱除方法包括粉煤灰吸附法、异相凝并技术与液相氧化脱除技术。粉煤灰中含有大量的Ca、Si、Al与Fe的氧化物[108-110],因此利用粉煤灰也可以对烟气砷实现控制。Li等[65]研究了3种粉煤灰回注技术在进行砷原位固定的可行性,在900 ℃,模拟烟气环境中,3种粉煤灰砷吸附容量分别为5.97、8.33、5.54 mg·g−1。Wang等[66]在某电厂实际工况下研究了改性粉煤灰在SCR出口回注技术协同脱除砷等重金属,烟气中总砷浓度降低了78.1%。SO2与NO会抑制粉煤灰的砷脱除效果[65-66]。
异相凝并是一种新兴的烟气砷脱除技术,它向SCR与ESP的烟道间喷入羧甲基纤维素钠、聚丙烯酰胺、磺胺树脂与羟甲基纤维素等凝并剂,使颗粒态砷通过电荷中和、桥架等方式互相团聚成更大的颗粒,且更易以镶嵌的形式与液滴发生吸附,从而形成更大的团聚体。异相凝并吸附剂喷入烟道后,其汽化降低了烟道温度,促进了气态砷的非均相冷凝、成核作用,从而更易被固定[111-112]。凝并剂能够促使PM1长大至1—10 μm;在脱硫石膏中,凝并后砷含量降低67.6%。与未凝并工况对比,气态与颗粒态砷向10 μm以上颗粒转移,最终排放至大气的砷降低69.3%[111]。研究发现,凝并粉煤灰的砷批淋滤浸出浓度降低,在纯净水中的浸出量降低了50%,吸附在凝并飞灰上的砷迁移转化能力减弱[112]。
除了固体吸附材料,也有研究探索了液相氧化剂对砷的脱除效果。As2O5比As2O3的毒性低50倍,且As2O5的溶解性好,将As(Ⅲ)氧化为As(Ⅴ)后不仅毒性降低,也利于溶解吸收。一些氧化剂溶液可以在实验室条件下实现对气态砷的吸收,包括KMnO4、Na2S2O8/H2O2、芬顿试剂、NaClO、NaClO/NaClO2和CH3COOOH/H2O2[113-118]。最佳条件下,吸收效率可接近100%。其中KMnO4的砷脱除效率受SO2影响最大,SO2超过4.290 g·m−3时,砷脱除效率便降低至不到60%;其他氧化剂在SO2超过11.441 g·m−3时仍有超过50%的效率。NO明显抑制砷脱除效率[113-116, 118],这是由于NO持续消耗氧化剂,NO被氧化为硝酸根与亚硝酸根。此外,NO与NaClO2反应产生ClNO和ClNO2中间体,这可能导致NaClO2与NaClO的持续消耗。但Na2S2O8/H2O2氧化剂却非如此[117],随着NO浓度增加,砷脱除效率出现先升高后降低的趋势,这可能是因为NO较低时与As2O3竞争氧化剂分子,当NO较高时,NO的氧化产物NO2对As2O3仍有氧化作用。除NaClO/NaClO2外[118],CO2均对砷脱除起到了抑制作用,而CO2对NaClO/NaClO2氧化剂几乎无影响。除Na2S2O8/H2O2/Ca(OH)2外[117],O2对以上氧化剂也可以产生抑制作用。砷液相氧化脱除技术的最佳温度为50—60 ℃,已经接近电厂烟气出口温度,但在实际条件下需要增加新设备,成本较高,且存在二次污染风险。
4.7 小结
活性炭等碳基吸附剂的优点是吸附容量较大,当气体成分复杂时,吸附剂特异性差,再加上成本高等原因,少有工业应用。单金属氧化物吸附剂的研究众多,包括氧化钙、氧化铝、氧化铁,这些吸附剂具有一定的抵抗酸性气体能力,具有一定的吸附特异性。这类吸附剂能适应更宽的温度,钙基吸附剂能适应超过1000 ℃的高温,而氧化铝、氧化铁的吸附温度范围略低,但SCR的最佳工作温度仅为400 ℃左右,未达到其最佳吸附温度。在实际工况下,单金属氧化物吸附剂的吸附容量仍有待提升,可以通过金属氧化物吸附剂进行修饰、负载、多元复合等方式实现吸附容量的提升,如Pd/γ-Al2O3、FMBO和Mn(Ⅳ)/ATP。Pd/α-Al2O3与Pd/γ-Al2O3吸附容量更大,吸附选择性强,最适应吸附温度与电厂烟气更接近,但缺点是吸附剂的成本极高,工业应用价值低。FMBO与Mn(Ⅳ)/ATP的吸附选择性与抗酸性气体能力更强,成本略高于单金属氧化物吸附剂,具有很好的工业应用潜力。目前已知的燃烧后砷污染控制技术中,粉煤灰、改性粉煤灰与异相凝并技术已经有实际的工业应用,其中粉煤灰吸附剂兼顾了固体废弃物资源化,应用成本较低,但粉煤灰的组成往往随煤种与电厂的工况发生变化,不同粉煤灰的脱除效果差距大。目前异相凝并技术的工业应用效果最好,成本适中,可以进行大规模工业应用。氧化脱除技术在很宽的NO与SO2浓度范围内具有良好的适用性,且脱除效率高。烟气中的砷浓度已经很低,但量大流速快并需要进行长时间吸收。在上述这些研究中,最长反应时间皆未超过30 min,但在实际生产中则是长时间反应;另外,由于烟气中其他还原性物质消耗氧化剂,使氧化剂频繁添加,所产生的还原产物可能随烟气带出,导致二次污染。
5. 总结与展望(Summary and prospect)
考虑到我国能源结构的特点,在工业生产过程中,燃煤产生的砷排放不容忽视。燃煤烟气砷污染控制不仅可以减少大气砷污染,同时也可以最大限度避免SCR催化剂失活,降低烟气治理成本。该部分从燃烧前、燃烧中和燃烧后控制技术进行了总结与展望。
燃烧前控制技术主要是采用洗选方式将燃煤中砷含量很高的煤矸石去除,使原煤中砷含量降低。降低洗选煤的耗水量与二次污染风险,提高其效果的稳定性是该技术重点研究之处。
燃烧中控制技术主要是在煤燃烧过程中通过化学添加剂或混煤等方式,将砷转化为化学性质稳定不易挥发的砷酸盐,主要以颗粒物为载体固定在底灰、飞灰中。受限于燃煤电厂实际情况与现实条件,目前通过吸附剂与煤粉混燃脱砷的相关研究较少,且研究处于有限的工况下,很难代表持续运行情况。例如以下几个方面需要进行深入研究:(1)实际燃煤烟气中吸附剂脱砷的关键影响因素有哪些?如何进行性能、空燃比,过量空气系数等参数的调控;(2)吸附剂在炉膛中的喷射方式,流动与分布情况是否影响其脱除效果,如改变喷射角度、喷射位置、混和与喷入的先后顺序,利用模型与实际工况实验进行详细研究。
燃烧后控制技术在烟气燃烧区域后用吸附剂进行烟气砷脱除。研究表明,金属氧化物进行修饰、负载制备多元复合物后,吸附性能和稳定性得到提升。目前,燃烧后控制技术几乎都在固定床脱砷实验台进行,这些实验装置与燃煤电厂的实际工况差距较大,有必要开展中试规模实验。未来燃烧后砷污染控制的方向和趋势应着眼以下几点:(1)提高废弃吸附剂中砷的热稳定性并降低其生物有效性,降低废旧吸附剂的二次污染风险。(2)研究可循环利用的吸附剂,减少资源浪费;(3)拓展吸附剂的工作温度宽度,使其适应于多APCDs的工况,便于升级改造。将吸附剂进行化学改性,若得到化学性质更稳定的吸附剂,其生物有效性便可能降低;若稳定性适中,可再通过一些方法脱附,便可能进行吸附剂再生后循环利用;若改性后,不同温度时的吸附效果变化,其工作温度便得到一定程度的拓展。
-
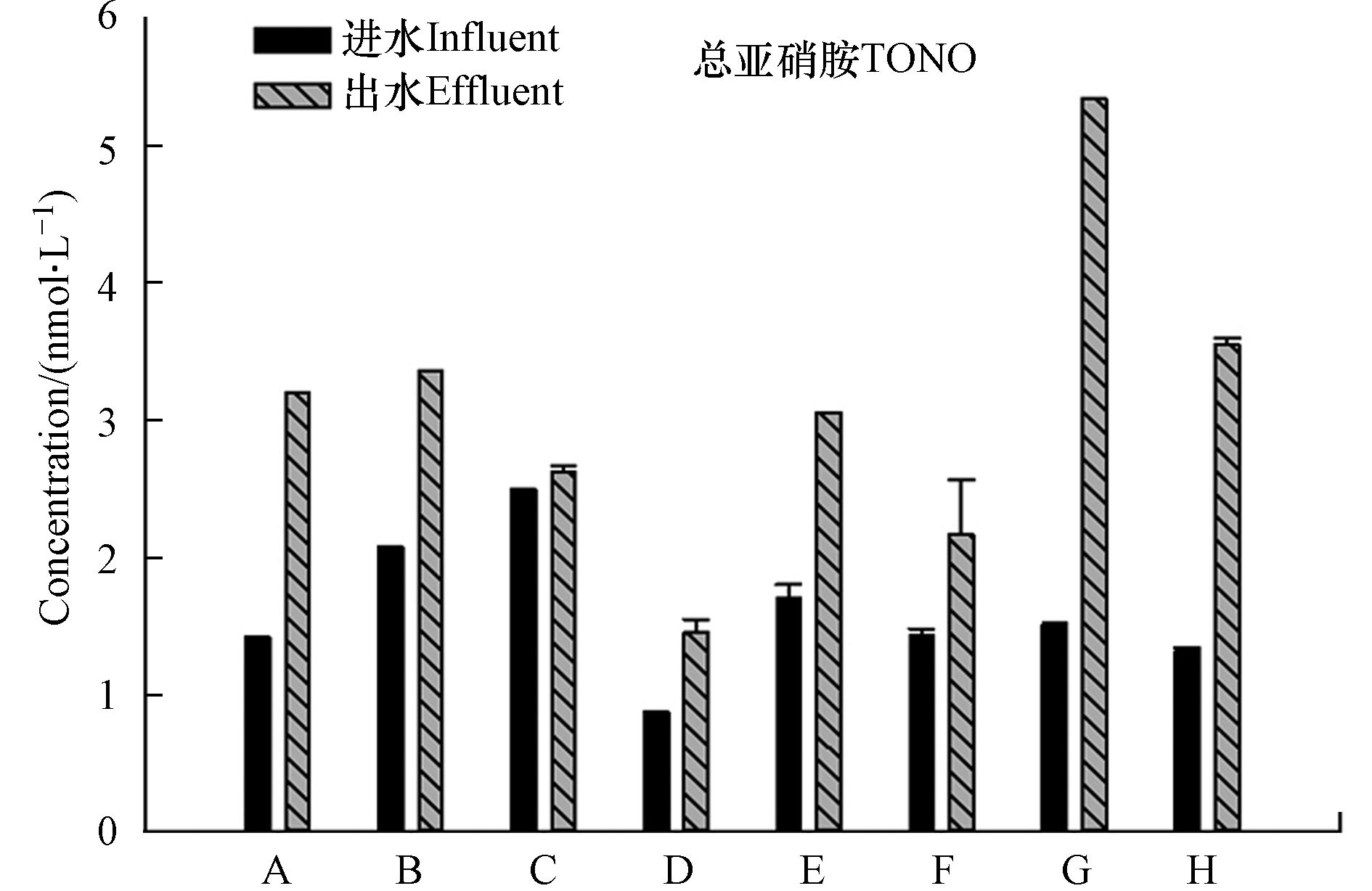
图 1 各水厂生物处理池的进出水中总亚硝胺(TONO)的浓度
Figure 1. Concentrations of TONO in influent and effluent of biological treatment in selected wastewater treatment plants
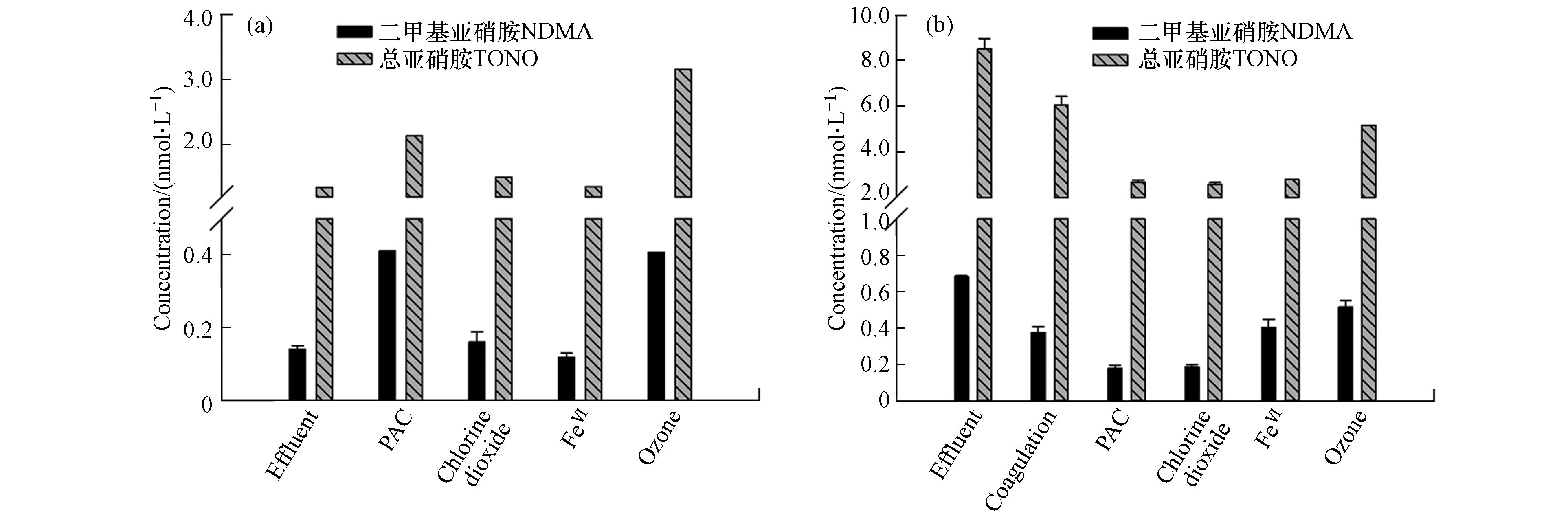
图 2 水样经各深度处理(a)与进一步UFC氯胺消毒后(b)NDMA与TONO的浓度。
Figure 2. Concentrations of NDMA and TONO in effluent after advanced treatment processes (a) and after post-chloramination (b).
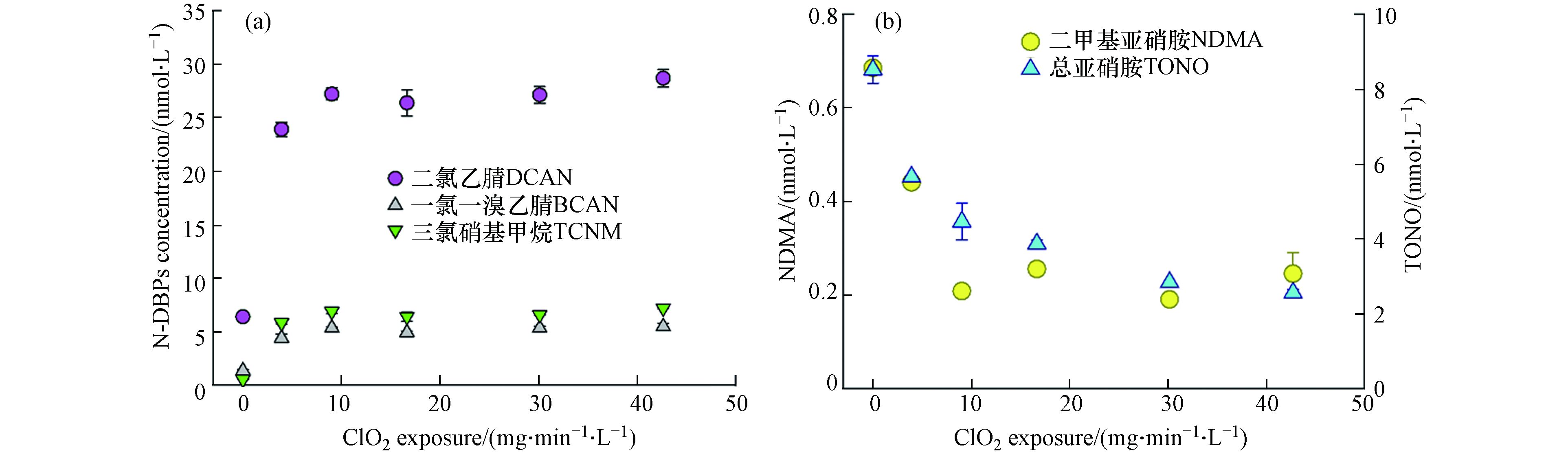
图 3 不同暴露剂量下二氧化氯对卤代N-DBPs(a)及亚硝胺(b)前体物的去除
Figure 3. Removal of the precursors of halogenated N-DBPs (a) and nitrosamines (b) under different exposures of ClO2
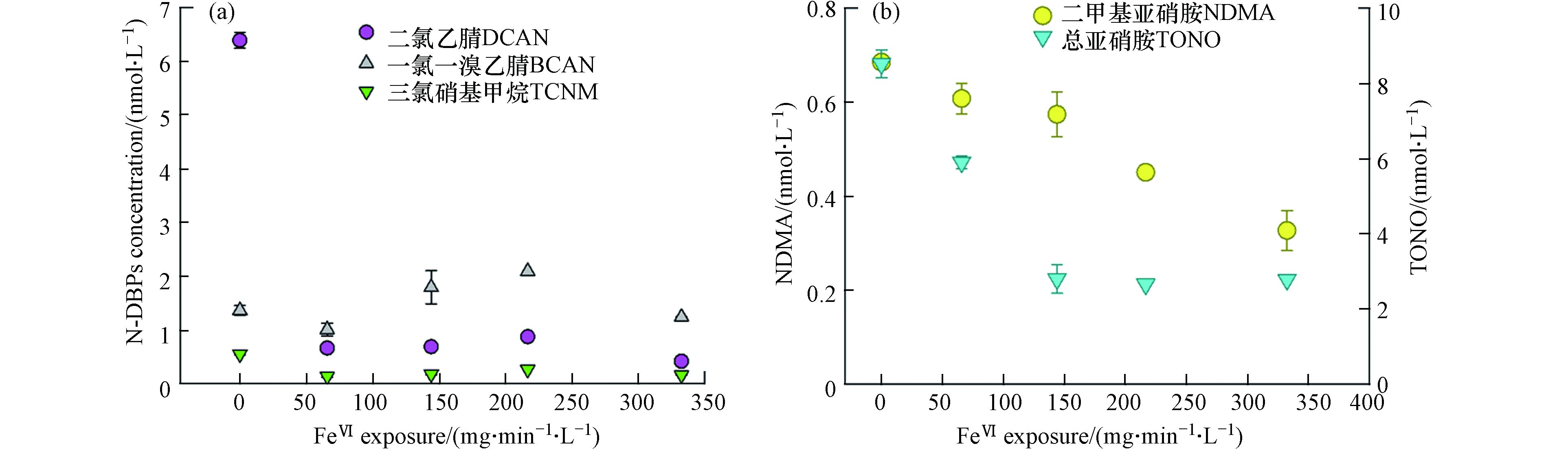
图 4 不同暴露剂量下高铁酸盐对卤代N-DBPs(a)及亚硝胺(b)前体物的去除
Figure 4. Removal of the precursors of halogenated N-DBPs (a) and nitrosamines (b) under different exposures of ferrateVI
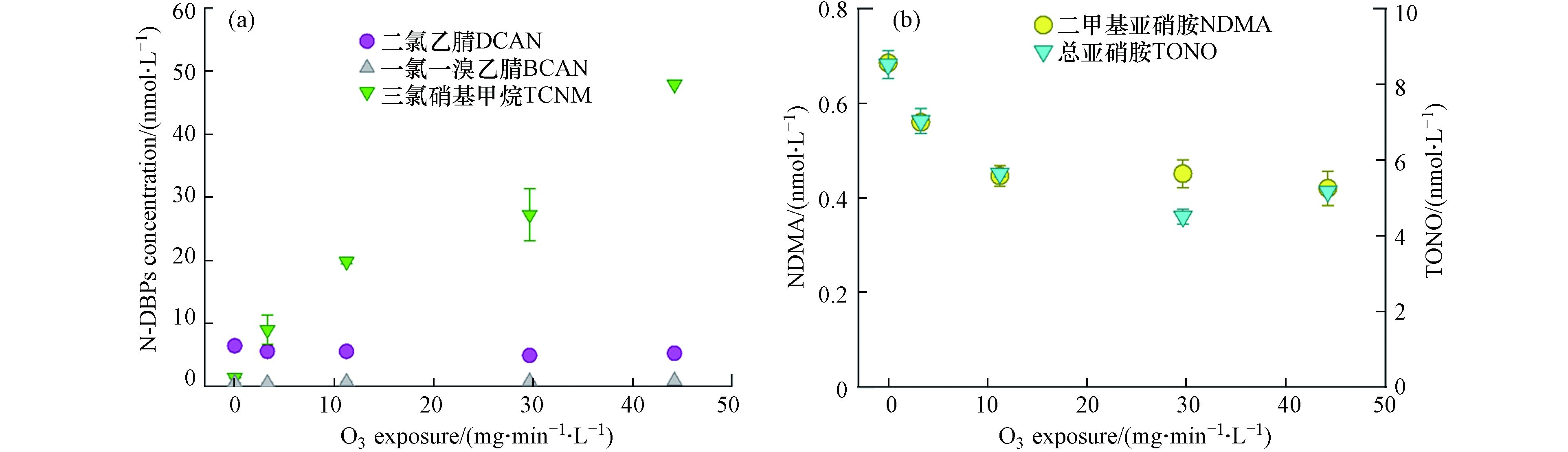
图 5 不同暴露剂量下臭氧对卤代N-DBPs(a)及亚硝胺(b)前体物的去除
Figure 5. Removal of the precursors of halogenated N-DBPs (a) and nitrosamines (b) under different exposures of O3
表 1 样点污水处理厂基本信息
Table 1. Basic information of wastewater treatment plants in the research
污水厂编号Number of wastewater treatment plants 污水处理能力(×104)/(m3·L−1)Wastewater treatment plant’s capacity 采用的生物处理工艺Biological treatment processes 生物处理类型Type of biological treatments A 10 A/O 完全硝化 B 10 A/A/O 完全硝化 C 20 Unitank 部分反硝化 D 10 A/A/O 弱硝化 E 10 CAST 完全硝化 F 5 Carrousel 2000氧化沟 完全反硝化 G 3 生物滤池 完全硝化 H 10 A/A/O 部分硝化 MBR 完全硝化 注Note:完全硝化Good nitrification:NH3 < 2 mg·L−1(按N算,下同), NO2− < 1 mg·L−1, NO3− > 10 mg·L−1。部分硝化Partial nitrification:NH3: 2—9 mg·L−1, NO3−: 2—10 mg·L−1。弱硝化Poor nitrification:NH3 > 9 mg·L−1, NO2− < 1 mg·L−1, NO3− < 2 mg·L−1。完全反硝化:NH3 < 2 mg·L−1, NO2− < 1 mg·L−1, NO3− < 5 mg·L−1。部分反硝化Partial denitrification:NH3 < 2 mg·L−1, NO3−: 5—10 mg·L−1.
下载: 导出CSV
表 2 8间水厂生物池进水与出水的基本水质参数
Table 2. Water parameters of influent and effluent of biological treatment processes in 8 WWTPs
编号No. 工艺Process pH DOC/ (mg·L−1) NH3-N/(mg·L−1) Cl−/(µg·L−1) Br−/(µg·L−1) NO2−/(mg·L−1) NO3−/(mg·L−1) A1 A/O 7.3 14.5 19.8 21.0 196 0.0 4.4×10-4 A2 7.6 11.9 0.10 43.5 51.0 4.1×10-3 10 B1 A/A/O 7.2 28.0 15.7 11.8 61.6 0.0 5.2×10-4 B2 7.3 6.50 0.10 49.8 57.3 5.4×10-3 11 C1 UNITANK 7.4 11.2 10.3 27.9 48.0 7.5×10-2 6.4×10-3 C2 7.2 5.50 1.50 30.7 30.5 0.3 7.3 D1 A/A/O 7.3 16.8 21.7 63.0 50.7 3.1×10-3 0.0 D2 7.1 9.70 9.20 10.9 37.5 0.1 0.1 E1 CAST 7.3 23.5 14.5 28.0 45.6 3.8×10-3 0.0 E2 7.1 6.80 0.20 39.1 22.0 4.6×10-3 13 F1 Carrousel 2000 7.2 23.8 11.6 53.0 53.6 3.4×10-3 0.0 F2 7.4 8.40 0.20 44.3 30.1 1.9×10-3 3.0 G1 生物膜 7.5 9.50 6.20 34.0 56.1 0.3 6.6 G2 7.6 10.7 0.30 36.0 40.7 0.6 10 H1 MBR+A/A/O 7.8 22.7 23.6 47.1 60.9 8.3×10-3 5.3×10-4 H2 7.6 7.80 0.30 42.9 41.9 4.0×10-2 7.7
下载: 导出CSV
-
[1] LI Z G, LIU X Y, HUANG Z J, et al. Occurrence and ecological risk assessment of disinfection byproducts from chlorination of wastewater effluents in East China [J]. Water Research, 2019, 157: 247-257. doi: 10.1016/j.watres.2019.03.072 [2] LAZAROVA V, SAVOYE P, JANEX M L, et al. Advanced wastewater disinfection technologies: State of the art and perspectives [J]. Water Science and Technology, 1999, 40(4/5): 203-213. [3] MUNSON A E, SAIN L E, SANDERS V M, et al. Toxicology of organic drinking water contaminants: Trichloromethane, bromodichloromethane, dibromochloromethane and tribromomethane [J]. Environmental Health Perspectives, 1982, 46: 117-126. doi: 10.1289/ehp.8246117 [4] RICHARDSON S D. Disinfection by-products and other emerging contaminants in drinking water [J]. TrAC Trends in Analytical Chemistry, 2003, 22(10): 666-684. doi: 10.1016/S0165-9936(03)01003-3 [5] MILLS C J, BULL R J, CANTOR K P, et al. Workshop report. Health risks of drinking water chlorination by-products: Report of an expert working group [J]. Chronic Diseases in Canada, 1998, 19(3): 91-102. [6] SEDLAK D L, von GUNTEN U. Chemistry. The chlorine dilemma [J]. Science, 2011, 331(6013): 42-43. doi: 10.1126/science.1196397 [7] KIMURA S Y, KOMAKI Y, PLEWA M J, et al. Chloroacetonitrile and n, 2-dichloroacetamide formation from the reaction of chloroacetaldehyde and monochloramine in water [J]. Environmental Science & Technology, 2013, 47(21): 12382-12390. [8] CHUANG Y H, LIN A Y C, WANG X H, et al. The contribution of dissolved organic nitrogen and chloramines to nitrogenous disinfection byproduct formation from natural organic matter [J]. Water Research, 2013, 47(3): 1308-1316. doi: 10.1016/j.watres.2012.11.046 [9] PLEWA M J, WAGNER E D, MUELLNER M G, et al. Comparative mammalian cell toxicity of N-DBPs and C-DBPs[M]//ACS Symposium Series. Washington, DC: American Chemical Society, 2008: 36-50. [10] CHOI J, VALENTINE R L. Formation of N-nitrosodimethylamine (NDMA) from reaction of monochloramine: A new disinfection by-product [J]. Water Research, 2002, 36(4): 817-824. doi: 10.1016/S0043-1354(01)00303-7 [11] KRASNER S W, MCGUIRE M J, JACANGELO J G, et al. The occurrence of disinfection by-products in US drinking water [J]. Journal - American Water Works Association, 1989, 81(8): 41-53. doi: 10.1002/j.1551-8833.1989.tb03258.x [12] BARKER D J, STUCKEY D C. A review of soluble microbial products (SMP) in wastewater treatment systems [J]. Water Research, 1999, 33(14): 3063-3082. doi: 10.1016/S0043-1354(99)00022-6 [13] KRASNER S W, WESTERHOFF P, CHEN B Y, et al. Impact of wastewater treatment processes on organic carbon, organic nitrogen, and DBP precursors in effluent organic matter [J]. Environmental Science & Technology, 2009, 43(8): 2911-2918. [14] TROGOLO D, MISHRA B K, HEEB M B, et al. Molecular mechanism of NDMA formation from N, N-dimethylsulfamide during ozonation: Quantum chemical insights into a bromide-catalyzed pathway [J]. Environmental Science & Technology, 2015, 49(7): 4163-4175. [15] BADER H, HOIGNÉ J. Determination of ozone in water by the indigo method [J]. Water Research, 1981, 15(4): 449-456. doi: 10.1016/0043-1354(81)90054-3 [16] THOMPSON G W, OCKERMAN L T, SCHREYER J M. Preparation and purification of potassium ferrate. VI [J]. Journal of the American Chemical Society, 1951, 73(3): 1379-1381. [17] LIU P, FARRÉ M J, KELLER J, et al. Reducing natural organic matter and disinfection by-product precursors by alternating oxic and anoxic conditions during engineered short residence time riverbank filtration: A laboratory-scale column study [J]. Science of the Total Environment, 2016, 565: 616-625. doi: 10.1016/j.scitotenv.2016.05.061 [18] WU M R, LIANG Y M, PENG H L, et al. Bioavailability of soluble microbial products as the autochthonous precursors of disinfection by-products in aerobic and anoxic surface water [J]. Science of the Total Environment, 2019, 649: 960-968. doi: 10.1016/j.scitotenv.2018.08.354 [19] ZENG T, MITCH W A. Impact of nitrification on the formation of N-nitrosamines and halogenated disinfection byproducts within distribution system storage facilities [J]. Environmental Science & Technology, 2016, 50(6): 2964-2973. [20] SHAH A D, MITCH W A. Halonitroalkanes, halonitriles, haloamides, and N-nitrosamines: A critical review of nitrogenous disinfection byproduct formation pathways [J]. Environmental Science & Technology, 2012, 46(1): 119-131. [21] SHAO B B, DONG H Y, SUN B, et al. Role of ferrate(IV) and ferrate(V) in activating ferrate(VI) by calcium sulfite for enhanced oxidation of organic contaminants [J]. Environmental Science & Technology, 2019, 53(2): 894-902. [22] EPA U. National primary drinking water standards[S]. 2002 [23] DAI N, ZENG T, MITCH W A. Predicting N-nitrosamines: N-nitrosodiethanolamine as a significant component of total N-nitrosamines in recycled wastewater [J]. Environmental Science & Technology Letters, 2015, 2(3): 54-58. [24] PADHYE L, TEZEL U, MITCH W A, et al. Occurrence and fate of nitrosamines and their precursors in municipal sludge and anaerobic digestion systems [J]. Environmental Science & Technology, 2009, 43(9): 3087-3093. [25] WIJEKOON K C, FUJIOKA T, MCDONALD J A, et al. Removal of N-nitrosamines by an aerobic membrane bioreactor [J]. Bioresource Technology, 2013, 141: 41-45. doi: 10.1016/j.biortech.2013.01.057 [26] TADKAEW N, HAI F I, MCDONALD J A, et al. Removal of trace organics by MBR treatment: The role of molecular properties [J]. Water Research, 2011, 45(8): 2439-2451. doi: 10.1016/j.watres.2011.01.023 [27] RALT D, TANNENBAUM S R. The role of bacteria in nitrosamine formation[M]//ACS Symposium Series. WASHINGTON D C: AMERICAN Chemical Society, 1981: 157-164. [28] GAN W H, BOND T, YANG X, et al. Role of chlorine dioxide in N-nitrosodimethylamine formation from oxidation of model amines [J]. Environmental Science & Technology, 2015, 49(19): 11429-11437. [29] PADHYE L, WANG P, KARANFIL T, et al. Unexpected role of activated carbon in promoting transformation of secondary amines to N-nitrosamines [J]. Environmental Science & Technology, 2010, 44(11): 4161-4168. [30] PADHYE L P, HERTZBERG B, YUSHIN G, et al. N-nitrosamines formation from secondary amines by nitrogen fixation on the surface of activated carbon [J]. Environmental Science & Technology, 2011, 45(19): 8368-8376. [31] OYA M, KOSAKA K, ASAMI M, et al. Formation of N-nitrosodimethylamine (NDMA) by ozonation of dyes and related compounds [J]. Chemosphere, 2008, 73(11): 1724-1730. doi: 10.1016/j.chemosphere.2008.09.026 [32] SHEN R Q, ANDREWS S A. Demonstration of 20 pharmaceuticals and personal care products (PPCPs) as nitrosamine precursors during chloramine disinfection [J]. Water Research, 2011, 45(2): 944-952. doi: 10.1016/j.watres.2010.09.036 [33] SCHMIDT C K, BRAUCH H J. N, N-dimethylsulfamide as precursor for N-nitrosodimethylamine (NDMA) formation upon ozonation and its fate during drinking water treatment [J]. Environmental Science & Technology, 2008, 42(17): 6340-6346. [34] XU B, YE T, LI D P, et al. Measurement of dissolved organic nitrogen in a drinking water treatment plant: Size fraction, fate, and relation to water quality parameters [J]. Science of the Total Environment, 2011, 409(6): 1116-1122. doi: 10.1016/j.scitotenv.2010.12.016 [35] UZUN H, KIM D, KARANFIL T. The removal of N-nitrosodimethylamine formation potential in drinking water treatment plants [J]. Journal - American Water Works Association, 2017, 109(6): 15-28. [36] SELBES M, KIM D, KARANFIL T. The effect of pre-oxidation on NDMA formation and the influence of pH [J]. Water Research, 2014, 66: 169-179. doi: 10.1016/j.watres.2014.08.015 [37] LEE Y, von GUNTEN U. Oxidative transformation of micropollutants during municipal wastewater treatment: Comparison of kinetic aspects of selective (chlorine, chlorine dioxide, ferrateVI, and ozone) and non-selective oxidants (hydroxyl radical) [J]. Water Research, 2010, 44(2): 555-566. doi: 10.1016/j.watres.2009.11.045 [38] YAO D C, CHU W H, BOND T, et al. Impact of ClO2 pre-oxidation on the formation of CX3R-type DBPs from tyrosine-based amino acid precursors during chlorination and chloramination [J]. Chemosphere, 2018, 196: 25-34. doi: 10.1016/j.chemosphere.2017.12.143 [39] YANG X, GUO W H, ZHANG X, et al. Formation of disinfection by-products after pre-oxidation with chlorine dioxide or ferrate [J]. Water Research, 2013, 47(15): 5856-5864. doi: 10.1016/j.watres.2013.07.010 [40] WANG A, LIN C S, SHEN Z, et al. Effects of pre-oxidation on haloacetonitrile and trichloronitromethane formation during subsequent chlorination of nitrogenous organic compounds [J]. International Journal of Environmental Research and Public Health, 2020, 17(3): 1046. doi: 10.3390/ijerph17031046 [41] SHI J L, MCCURRY D L. Transformation of N-methylamine drugs during wastewater ozonation: Formation of nitromethane, an efficient precursor to halonitromethanes [J]. Environmental Science & Technology, 2020, 54(4): 2182-2191. 期刊类型引用(2)
1. 薛彦茵. 废水氮磷生物处理新技术研究. 广东化工. 2022(16): 115-116+129 .  百度学术
百度学术
2. 丁红,王艺洁,武福平. 废铁屑-Ti(RuO_2)三维电极高效处理高氮磷低C/N污水. 环境工程学报. 2021(03): 847-856 . 本站查看
其他类型引用(2)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 4226
- HTML全文浏览数: 4226
- PDF下载数: 144
- 施引文献: 4
