




-
粒径小于5 mm的微塑料已引起专家的广泛关注[1],成为水环境管理的新问题。微塑料可分为原生微塑料和次生微塑料两种。原生微塑料是指化妆品中的微珠或工业树脂颗粒[2]。次生微塑料是由大块塑料经过太阳光照、风、水浸等过程的作用下分解成的塑料颗粒或纤维[3]。大量微塑料长期存在于水体中,容易被浮游生物食入腹中,导致生物行为异常或死亡[4]。此外,微塑料还可经过食物链进行传递,并在生物群落中累积,最终可能危害人类健康[5-6]。微塑料具有粒径小、比表面积大、疏水性强等特性,易成为水环境中有机污染物的运输载体,导致污染扩散[7-8]。
微塑料对有机污染物的吸附性能不仅与微塑料和污染物的性质有关,还与外界环境条件有关[9]。Rochman等[10]指出,有机污染物在橡胶塑料上比玻璃塑料具有更高的吸附亲和力。Liu等[11]发现,PET表面的羰基与氯酚表面的羟基之间会产生氢键。随着pH值从4降到2,大量的H3O+会与氯苯酚争夺吸附位点,降低聚对苯二甲酸乙二醇酯对氯酚的吸附量。Zhang等[12]研究发现,腐植酸可作为风化后的聚苯乙烯表面和土霉素分子之间的桥梁,促进聚苯乙烯对土霉素的吸附。Zhang等[13]研究发现,阳离子Na+和Ca2+会与9-硝基蒽竞争 PP和PS表面的吸附位点。Yu等[14]研究了微塑料与四环素在水溶液中的吸附作用,当溶液中存在Cu2+时聚乙烯对四环素的吸附量会减小,这可能与聚乙烯和四环素形成的胶层表面电位有关。微塑料与有机污染物之间的交互作用是一个有研究意义的课题。
随着纺织业和染色工业的飞速发展,染料废水污染在许多发展中国家仍然是显著的。亚甲基蓝(MB)作为最常见的染料污染物,其具有高毒性,致癌性和诱变效应,对水生生物已构成严重威胁[15]。水体中的微塑料容易积累这些染料污染物,携带有机染料的微塑料可能对水生生物产生更大的毒害作用。现今关于微塑料富集MB的研究还非常有限。因此,需要进一步探究废旧微塑料与MB的作用机理,为评估其潜在的环境风险提供理论依据。
本文以MB为染料污染物的典型代表,以聚苯乙烯(PS)、聚氯乙烯(PVC)和聚甲基丙烯酸甲酯(PMMA)的3种具有不同官能团的聚合物为微塑料的典型代表,研究不同微塑料对MB的吸附行为及溶液pH、盐度、腐殖酸浓度、微塑料粒径、温度和自然水样对微塑料吸附MB的影响。采用扫描电镜、zeta电位、比表面积测试、红外光谱对微塑料的性能进行表征,旨在研究微塑料性能对MB吸附的影响,进一步研究有机染料与微塑料之间的交互作用机理,为科学评价微塑料复杂的环境行为以及作为载体协同迁移污染物的能力提供依据。
-
分析纯的MB标准品购自天津市科密欧化学试剂有限公司。分析纯的氢氧化钠(NaOH)和氯化钠(NaCl)、纯度 > 90%的 腐殖酸(HA)和浓度为37%的盐酸(HCl)购自国药控股化学试剂有限公司。废旧PS、PVC和PMMA塑料来源于湖南省汨罗市废旧塑料回收市场。将废旧塑料放入高速粉碎机中,粉碎的塑料颗粒依次通过10目、18目、40目、120目和200目的筛网,收集粒径范围为 0.074—0.125 mm、0.125—0.425 mm、0.425—1 mm和1—2 mm的颗粒。将制得的废旧微塑料用去离子水和乙醇洗净3次,烘干得到清洁的微塑料粉末。实验中使用的微塑料粒径为0.074—0.125 mm,其他粒径仅用于研究微塑料粒径对吸附的影响。
-
通过扫描电镜(SM-74190UEC,日本)分析PS、PVC和PMMA微塑料的表面形态;通过X射线衍射仪(XRD-7000S/L,日本)检测微塑料的结晶度;通过比表面积测试仪(ASAP 2460,美国)分析微塑料的比表面积;通过Zeta电位分析仪(JS94,中国上海)检测不同pH下微塑料的表面电位;通过傅里叶变换红外光谱仪(FTIR-650S,中国天津)分析3种微塑料吸附MB前后的官能团变化。
-
称取PS、PVC和PMMA微塑料25 mg放入25 mL的棕色玻璃瓶中,加入配制好的5 mg·L−1 的MB溶液20 mL,将所有样品置于25 ℃、180 r·min−1的恒温水浴振荡器中,在相应时间取样,将样品经过0.22 μm的膜过滤后,用紫外分光光度计测量滤液浓度。每组实验重复3次,并设置空白对照组。
-
称取PS、PVC和PMMA微塑料25 mg放入25 mL的棕色玻璃瓶中,分别加入配制好的6个MB溶液浓度梯度(1、3、5、10、15、 20 mg·L−1)。将所有样品置于25 ℃、180 r·min−1的恒温水浴振荡器中。当吸附达到平衡时,将样品取出,经过0.22 μm的膜过滤后,用紫外分光光度计测量滤液浓度。每组实验重复3次,并设置空白对照组。
-
研究环境因素对吸附的影响时仍将MB的浓度设置为5 mg·L−1,PS、PVC和PMMA微塑料的质量为25 mg。除温度实验外,其余实验样品仍置于25 ℃、180 r·min−1的水浴振荡,当吸附达到平衡时取出。为研究pH对吸附的影响,只通过HCl和NaOH将MB溶液的pH调为6个梯度(3.0、4.0、5.0、7.0、8.0、9.0);为研究盐度对吸附的影响,只往MB溶液中加入NaCl将盐度调为 0.5%、1%、2%、3%、3.5%;为研究腐殖酸对吸附的影响,只往MB溶液中加入1、5、10、15、20 mg·L−1的腐殖酸;为研究温度对吸附的影响,分别在15、25、35、45 ℃的条件下进行吸附实验;为了研究自然环境水样中微塑料的实际吸附量,取用湘江水(pH为7.62、总有机碳 (TOC) 为2.203 mg·L−1)配制5 mg·L−1的MB溶液。每组实验重复3次,并设置空白对照组。
-
PS,PVC和PMMA微塑料对MB的吸附动力学实验分别用用准一级动力学模型(1)、准二级动力学模型(2)进行拟合;等温吸附实验数据分别通过Freundlich模型(3)和Langmuir模型(4)进行拟合。
式中,
t 为吸附时间,h;qt 为t 时间的吸附量,mg·g−1;qe 为吸附平衡时的吸附量,mg·g−1;Kd1 为准一级动力学常数,h−1 ;Kd2 为准二级动力学常数,g·(mg·h)−1;Ce 为吸附达到平衡时溶液浓度,mg·L−1;n 为吸附相关常数;KF 为Freundlich模型经验常数,(mg·g−1) (L·g−1) 1/n;KL 为Langmuir模型吸附常数,L·mg−1;qmax 为饱和吸附量,mg·g−1。 -
从电镜图1中可见,PS、PVC和PMMA具有不同的形状和表面形貌。3种微塑料都呈不规则几何形状。PS微塑料的表面孔隙较少,PVC表面褶皱较多,PMMA表面较PS和PVC更粗糙。为了进一步了解微塑料表面性质,对3种微塑料的比表面积进行了检测。PMMA的比表面积为0.183 m2·g−1,PVC的比表面积为0.153 m2·g−1,PS的比表面积为0.109 m2·g−1。这与3种微塑料的形态总体保持一致。通过X射线衍射仪对PS、PVC和PMMA微塑料的结晶度进行表征。从图2a中可知,3种微塑料的结晶度都相对较低。zeta电位是微塑料重要的表面电化学性质,其对有机污染物吸附有显著影响。图2b显示了微塑料表面电位随pH的变化趋势。PS、PVC和PMMA微塑料的zeta电位随pH的增加而降低,且只在pH = 3时微塑料表面带正电,其他实验条件下带负电。
-
PS、PVC和PMMA微塑料对MB的吸附动力学实验结果如图3a所示。3种微塑料对MB都表现出一定的吸附性能。实验开始后,MB的吸附量迅速增加,然后随着时间推移,吸附量缓慢增加,并在24 h达到平衡。
微塑料对MB的吸附能力顺序分别为PMMA > PVC > PS,最大吸附量分别为1.988、1.594、1.419 mg·g−1。为了进一步明确吸附过程,分别用准一级、二级动力学模型对实验结果进行拟合,拟合结果及相关参数见图3b、3c和表1。准一级模型是指主要受物理扩散控制的吸附过程。拟合后的一级方程的线性回归系数R2在0.987—0.989范围内,由方程得到的MB在PS、PVC和PMMA的平衡吸附量低于实际吸附量。可见,MB在PS、PVC和PMMA上的吸附动力学不符合准一级动力学模型,物理扩散不是主要的吸附机理。二级模型是指以化学吸附为主的吸附过程[16]。拟合后的二级方程线性回归系数R2均大于0.999,且基于二级模型计算得到的吸附量与实验平衡吸附量更相符。这表明MB在这3种微塑料上的吸附过程可以用二级模型来描述。因此这3种微塑料对MB的吸附主要受化学吸附控制。
-
PS、PVC和PMMA微塑料对MB的吸附等温线如图4a所示。随着平衡浓度的增大,3种微塑料对MB的吸附量均增加,且相应曲线斜率渐渐变小。微塑料对MB的吸附是MB从水溶液中不断向微塑料固体表面迁移的过程,当微塑料表面的吸附位点饱和后,吸附会趋于平衡。为了进一步了解吸附机理,分别用Langmuir和Freundlich模型对MB在微塑料上的吸附数据进行拟合,拟合结果和等温参数如图4b、4c和表2所示。Freundlich模型的相关系数R2值范围为0.920—0.936。Langmuir模型中PS、PVC和PMMA的相关系数R2分别为0.993、0.993和0.996。因此微塑料对MB的吸附可以很好的符合Langmuir模型。Freundlich 等温吸附是不局限于单分子层的非均匀性吸附,Langmuir等温吸附是指对所有位点对吸附剂具有相同亲和力的单分子层吸附[17]。由此可知,微塑料对MB的吸附为单分子吸附。
-
图5a为pH值对3种微塑料吸附行为的影响。如图5所示,微塑料对MB的吸附量顺序仍然为PMMA > PVC > PS。随着pH(3—9)的增加,3种微塑料对MB的吸附量逐渐增加。因此,这3种微塑料在偏碱性条件下更易富集水中的MB。Li等[18]研究发现碱性水环境条件有利于聚乙烯微塑料对吡虫啉、噻虫嗪、二甲诺康唑的吸附。Bakir等[19]证实低pH值会增加微塑料表面污染物的解吸,从而减少微塑料对有机污染物的吸附。
pH是水环境中的一个基本变量,它会影响微塑料的表面电位,进而影响微塑料对MB的吸附行为。由图2b可知PS的零电荷点为3.09,PVC的零电荷点为3.93,PMMA的零电荷点为3.06。当溶液pH低于零电荷点时,微塑料带正电,微塑料与溶液中的MB会产生静电斥力,抑制微塑料对MB的吸附[20]。当溶液pH高于零电荷点时,微塑料表面带负电荷。随着pH的增大,3种微塑料表面负电荷增加。带负电荷的微塑料会与阳离子染料MB之间会产生静电引力,促进微塑料对MB的吸附。因此,静电相互作用在微塑料对MB的吸附中起着重要作用。
-
NaCl在河流、湖泊和海洋等水环境中广泛存在,对微塑料对有机污染的富集具有重要的影响[13]。图5b显示了盐度对MB在3种微塑料上吸附的影响。盐度从0增加到3.5%时,微塑料对MB的吸附量逐渐减少。Qiu等[21]研究了5种多卤咔唑对PE、PP、PVC的吸附行为,也发现多卤咔唑的吸附量随着盐度的增加而降低。NaCl的存在可能会中和微塑料表面吸附部位的负电荷,从而减少微塑料与MB之间的静电相互作用。此外,微塑料附近的Na+可能会与游离的MB竞争微塑料表面的吸附位点,从而抑制MB的吸附。NaCl的加入还可能提高水溶液的密度和粘度,从而阻碍了MB从水相向微塑料固相的转移[22]。上述讨论说明,PS、PVC和PMMA微塑料在淡水环境中比在海水中更易富集某些有机污染物。
-
腐殖酸中含有丰富的官能团,它们可与天然颗粒或有机污染物相互作用,从而影响微塑料对有机污染物的富集。图5c显示了腐殖酸浓度对MB在3种微塑料上吸附的影响。随着腐殖酸浓度从0增加到20 mg·L−1, PS、PVC和PMMA微塑料对MB的吸附量分别从1.419 mg·g−1降至0.238 mg·g−1,1.594 mg·g−1降至0.615 mg·g−1,1.988 mg·g−1降至0.717 mg·g−1。即使只添加1 mg·L−1的HA,也能显著降低微塑料的吸附能力,说明腐植酸对MB在微塑料上的吸附有显著的抑制作用。Abdurahman等[23]研究发现,腐殖酸会通过疏水作用和π–π相互作用富集在聚苯乙烯微塑料表面。因此溶液中的腐殖酸可能会与MB竞争微塑料表面的吸附位点,从而抑制微塑料对MB的吸附。此外,含氧官能团丰富的腐殖酸还可能通过络合作用和氢键作用与MB分子结合[24]。一般自然水环境中腐殖酸浓度约为4—10 mg·L−1,这些腐殖酸易与有机污染物竞争微塑料表面的吸附位点。
-
不同粒径的PS、PVC和PMMA微塑料对MB的吸附性能如图5d所示。PS、PVC和PMMA微塑料的粒径对MB的吸附有显著影响,吸附量随粒径的增大而减小。这可能是因为粒径较小的PS、PVC和PMMA微塑料具有较大的比表面积,从而增强了它们对MB的吸附能力。Yu等[14]研究PE、PS和 PVC微塑料对四环素的吸附时也发现,小粒径的微塑料对有机污染物具有更好的亲和力。针对微塑料吸附的研究多使用粒径比较大(> 74 μm),但现实环境中的微塑料经过自然老化、风化作用下,可能会以更小的粒径形式存在于自然环境中,这些微小的聚合物,能富集更多的有机污染物。
-
温度是影响MPs吸附有机污染物的关键因素之一。从图5e可以看出,温度对不同微塑料吸附MB的影响不同。PVC和PMMA对MB的吸附量随着温度的升高逐渐增大。当温度从15 ℃升高到25 ℃时,PS对MB的吸附量随着温度的升高而增加。当温度高于25 ℃时,PS的吸附能力随温度的升高而减小。一方面随着温度的升高,微塑料内部链之间的距离会增大,有利于有机污染物在聚合物上的扩散[25]。另一方面微塑料与MB之间的范德华力会随着温度的升高而减小,在高温下MB与微塑料表面之间的氢键可能被破坏[26]。温度对PS、PVC和PMMA微塑料吸附MB的影响不同可能是不同类型微塑料对MB的吸附机理不同所致。PS微塑料对MB的吸附可能受氢键影响较大,而对于PVC和PMMA对MB的吸附可能受氢键影响较小。上述结果说明在水温稍高的环境下PVC和PMMA微塑料表面更易富集MB,而室温环境相对于高温环境更有利于PS对MB的吸附。
-
为研究自然水环境中微塑料对MB的实际吸附效果,以湘江水样为溶剂,研究PS、PVC和PMMA 微塑料对MB的吸附行为。由图5d可知,相比于纯水体系,PS、PVC和PMMA微塑料在自然水环境下对MB的吸附能力有所下降,分别从1.419 mg·g−1降至0.988 mg·g−1,1.594 mg·g−1降至1.170 mg·g−1,1.988 mg·g−1降至1.322 mg·g−1。刘鹏等[27]研究PS对环丙沙星的吸附时也发现,相对于超纯水体系,在天然水环境下微塑料的吸附性能明显降低,与本实验结果相符。湘江水的TOC为2.203 mg·L−1,水中的有机物可能会与MB竞争微塑料表面的吸附位点从而降低微塑料对MB的吸附量[23]。此外,湘江水样中也存在一些金属离子可能会与MB竞争微塑料上的吸附位点。虽然在自然水环境下微塑料对MB的吸附量有所降低,但微塑料表面仍然能富集不少MB,从而增加其环境风险。
-
微塑料作为载体可与水中的染料污染物结合而形成复合污染,增加污染物的环境危害性。因此,研究微塑料对有机污染物的吸附行为,对于评价微塑料复合污染在环境中的风险具有重要意义。为了进一步探究微塑料的吸附机理,通过红外光谱检测分析了3种微塑料吸附MB前后官能团的变化。
图6显示了PS、PVC和PMMA微塑料吸附MB前后的红外光谱。吸附前PS在3446 cm−1处的峰值为O—H伸缩振动;3078、3024、2922、2848 cm−1处为CH2的振动;1558、1492、1447、1024、751 cm−1处的峰属于芳香族环振动。吸附前PVC微塑料3442 cm−1处的强峰对应O—H的振动;2964 cm−1和2920 cm−1为CH的拉伸;在1332 cm−1的峰为CH2的振动,在1124 cm−1处对应C—C的振动。在688 cm−1附近出现了一个强峰,这是由C—Cl的伸缩振动引起的;吸附前PMMA在3432、2850、1640、1462、1070 cm−1处有吸收峰,分别对应于O—H伸缩振动、C—H振动、C=O弯曲振动、O—CH3振动和C—O—C摇摆振动。吸附后3种微塑料都有出现一处新峰。在1628 cm−1处的增强的峰可能对应于MB杂环的C=N和C=C振动[28]。此外,PS微塑料的CH2特征峰、PVC微塑料的CH2和C—Cl特征峰、PMMA微塑料的O—CH3和C—O—C特征峰在吸附后明显减弱,这可能是由于微塑料表面覆盖着一层MB,导致微塑料特征峰减弱。
微塑料的化学成分、粒径、比表面积和官能团都对其吸附有机污染物的性能有显著影响。PMMA是由甲基丙烯酸甲酯聚合而成,PVC由氯乙烯聚合而成,PS由苯乙烯聚合而成。3种微塑料的结晶度都比较低,结晶度低的聚合物链之间的距离有利于有机污染物在聚合物上的扩散[25]。PS、PVC、PMMA塑料在环境中都易富集MB。3种微塑料对MB的吸附能力顺序为PMMA > PVC > PS。微塑料的粒径越小比表面越大,大的比表面积意味着微塑料表面有更多的吸附位点。微塑料的比表面积大小为PMMA(0.183 m2·g−1)> PVC(0.153 m2·g−1)> PS(0.109 m2·g−1)。PMMA微塑料的表面相对粗糙,比表面积更大,因此MB更容易在PMMA的表面扩散。PMMA支链上的极性的酯基官能团,也可能是其对MB吸附能力最强的原因[29]。赵楚云等[30]研究不同微塑料对污染物吸附实验时也发现,PMMA微塑料具有较强的吸附性能。相比于PVC和PMMA,PS微塑料对MB的吸附量最低可能是因为PS塑料中苯环的存在对键的旋转有很强的空间位阻作用,降低了链间的自由体积。
除了不同类型的微塑料对MB的吸附性能不同之外,不同类型微塑料对MB的吸附机理也存在一定的差异。当pH = 3时,3种微塑料的表面带正电荷。微塑料与阳离子染料MB之间存在静电斥力。随着pH增大,微塑料表面所带的负电荷增多,微塑料与MB之间会产生静电吸引力。除了静电相互作用力之外,微塑料与MB之间也存在其他作用力。烷基与芳香环之间会产生CH/π相互作用的氢键[31]。3种微塑料与MB之间可能也存在这种CH/π相互作用。随着温度从25 ℃升高到45 ℃,这种氢键容易断裂,导致PS微塑料吸附效率的下降。而PMMA和PVC的吸附性能则随着温度的升高而增大,说明这种氢键在PMMA和PVC对MB吸附中不起主导作用,PS对MB的吸附作用可能受氢键影响较大。微塑料的特殊官能团也可能对其吸附机理有影响。PMMA和PVC的极性相对较强,PMMA和PVC与MB之间存在极性相互作用。PVC微塑料上的氯原子与苯环上的π电子之间会形成卤素键[32]。PS微塑料可以与含有苯环的MB产生π–π共轭相互作用。总的来说PMMA对MB的主要吸附机制可能是静电相互作用、CH/π相互作用和极性效应;PVC对MB的主要吸附机制可能是静电相互作用、CH/π相互作用、卤素键和极性效应;PS对MB的主要吸附机制可能是静电相互作用、CH/π相互作用和π–π相互作用。
-
(1)3种微塑料对MB的吸附量为:PMMA > PVC > PS。微塑料的单体结构、比表面积和官能团的不同是导致其对MB的吸附能力不同的主要因素。
(2)准二级吸附动力学模型和Langmuir等温吸附模型较好的拟合了3种微塑料对MB的吸附过程,说明微塑料对MB的吸附为单分子层化学吸附。
(3)3种微塑料对MB的吸附都受到静电相互作用力和CH/π相互作用的影响。PMMA和PVC与MB之间存在极性作用。PVC对MB的吸附还受卤素键的影响,PS和MB之间存在π–π相互作用。
(4)溶液pH通过影响微塑料表面所带电荷进而影响吸附性能。NaCl和腐殖酸会与MB竞争微塑料表面的吸附位点。微塑料粒径越小,吸附性能越好。升高温度会促进PMMA和PVC对MB的吸附,却抑制PS对MB的吸附。相对于纯水体系,微塑料在湘江水环境下对MB的吸附能力明显下降。
不同微塑料对亚甲基蓝的吸附行为
Adsorption behavior of methylene blue on diverse microplastics
-
摘要: 微塑料易成为水中染料污染物的载体而形成复合污染,增加污染物的环境危害性。目前,关于微塑料对染料污染物的吸附研究十分有限。本文以亚甲基蓝(MB)为染料污染物的典型代表,系统的研究了聚苯乙烯(PS)、聚氯乙烯(PVC)和聚甲基丙烯酸甲酯(PMMA)的3种微塑料对MB的吸附行为和吸附机理,分析了溶液pH、盐度、腐殖酸、粒径、温度和自然水样对吸附的影响。结果表明,不同微塑料对MB的吸附能力顺序为:PMMA > PVC > PS。微塑料对MB的吸附过程用准二级动力学模型和Langmuir等温模型拟合较好,表明微塑料对MB的吸附为单分子层化学吸附。静电相互作用力和CH/π相互作用会促进微塑料对MB的吸附。PMMA和PVC与MB之间存在极性作用。PVC对MB的吸附还受卤素键的影响,PS和MB之间存在π–π相互作用。 不同微塑料对MB的吸附性能存在差异,主要与微塑料的比表面积和官能团有关。溶液pH通过影响微塑料表面所带电荷进而影响吸附性能;NaCl和腐殖酸会与MB竞争微塑料表面的吸附位点;升高温度能促进PMMA和PVC对MB的吸附,却抑制PS对MB的吸附。相对于纯水体系,微塑料在自然水环境下对MB的吸附能力明显下降。Abstract: Microplastics might be prone to accumulate dye pollutants in the aquatic environment, increasing their potential environmental risks. However, there were rare studies on the interaction between microplastics and dye pollutants. In this study, the interaction between methylene blue (MB) and microplastics (polystyrene (PS), polyvinyl chloride (PVC), and polymethyl methacrylate (PMMA)) were thoroughly investigated, and the effects of pH, salinity, humic acid, particle size, temperature, and natural aquatic environment were considered. The adsorption capacity of MB on microplastics followed the order PMMA > PVC > PS. The sorption process for microplastics to MB could be well described by the pseudo-second-order model and Langmuir model, revealing that the adsorption was monolayer chemisorption. Electrostatic interaction and CH/π interaction could promote the adsorption of MB on microplastics. PMMA and PVC could combine with MB through polar interaction. The adsorption of PVC to MB was affected by halogen bonding, and there was a π-π interaction between PS and MB. The specific surface area and functional groups of microplastics were the main factors affecting the adsorption performance of MB on microplastics. The sorption process exhibited a pronounced pH dependency due to the effect of pH on the surface charge of the microplastics. The presence of NaCl and humic acid could compete with MB for adsorption sites on the surface of microplastics. High temperature promoted the adsorption of MB on PVC and PMMA but inhibited that of PS. Moreover, the adsorption capacities of MB on microplastics were significantly reduced in the natural aquatic environment (Xiang River).
-
Key words:
- microplastics /
- adsorption /
- methylene blue
-
好氧颗粒污泥是微生物在好氧条件下自凝聚形成的一种结构紧密的颗粒状活性污泥[1-2]。与传统活性污泥相比,好氧颗粒污泥具有沉降性能优良、微生物种类多样、生物量高、单级同步脱氮除磷等优点[3-5]。自1991年首次在连续流反应器中培养得到好氧颗粒污泥报道以来[6],有研究[7-8]发现,以葡萄糖或乙酸钠为单一碳培养好氧颗粒污泥存在污泥易膨胀和颗粒污泥结构松散等问题。王芳等[9]采用以葡萄糖和乙酸钠为混合碳源培养的好氧颗粒污泥表面光滑、结构紧密。培养过程中SBR调控方法的不同会导致培养结果也有差异。赵霞等[10]采用调控表面上升气速的方法培养出结构密实且表面有大量丝状菌存在的颗粒污泥;但是通过增大耗氧有机污染物浓度和表面上升气速的方式培养出的好氧颗粒污泥表面光滑且无丝状菌存在[9]。郭承元等[11]以容积负荷和沉降时间为调控参数培养出了表面凹凸不平、内部出现空洞且表面以杆菌为主的好氧颗粒污泥。以上研究在好氧颗粒污泥的培养过程中均采用调控单一条件,同时调控多因素的好氧颗粒污泥培养研究鲜有报道。
因此,本实验在SBR中,采用以葡萄糖和乙酸钠为混合碳源,通过提高进水COD值、表面上升气速,缩短污泥沉降时间的方法培养好氧颗粒污泥,研究污泥颗粒化过程中污泥特性以及污染物去除效果的变化,以期为好氧颗粒污泥培养技术提供理论依据。
1. 材料与方法
1.1 接种污泥和实验用水
1)接种污泥。实验所用接种污泥取自广州市猎德污水处理厂四期二沉池的回流污泥,接种前曝气24 h,接种污泥体积为3.7 L。接种污泥为絮状,灰褐色,含水率为99.14%,沉降速度为12.36 m·h−1、SVI30为110.7 mL·g−1、MLSS为7 588 mg·L−1、MLVSS/MLSS为0.41。
2)实验用水。进水水质采用人工配制的模拟废水,以葡萄糖和乙酸钠为混合碳源,用NaHCO3 调节进水pH为7.5左右,并向进水添加1 mL·L−1的微量元素溶液。模拟废水组成:葡萄糖为500~1 200 mg·L−1、乙酸钠为300~600 mg·L−1、NH4Cl为40~90 mg·L−1、KH2PO4为8~18 mg·L−1、CaCl2为100 mg·L−1、MgSO4·7H2O为30 mg·L−1、FeSO4·7H2O为30 mg·L−1、EDTA为20 mg·L−1。微量元素溶液组成:FeCl3·6H2O为500 mg·L−1、CuCl2·2H2O为30 mg·L−1、MnCl2·4H2O为120 mg·L−1、ZnCl2·6H2O为120 mg·L−1、H3BO3为150 mg·L−1、KI为30 mg·L−1、Na2MoO4·2H2O为60 mg·L−1、CoCl2·6H2O为150 mg·L−1。
1.2 实验装置和运行方式
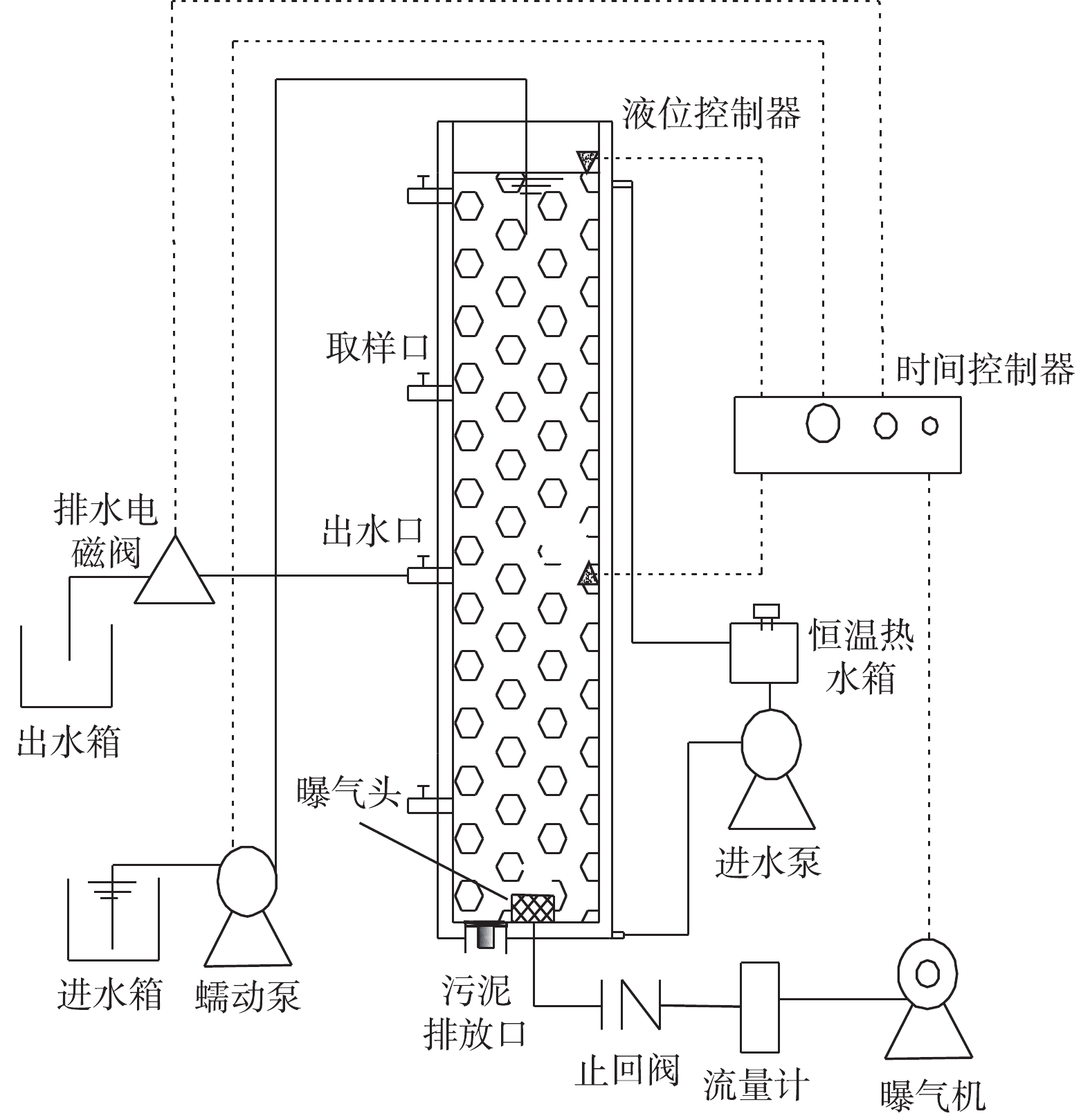
1)实验装置。好氧颗粒污泥培养采用SBR,反应器主体是由有机玻璃制成的圆柱体,其内径D为9.8 cm,总高度为 100 cm,有效高度H为98 cm,有效高径比(H∶D)为 10,反应器有效容积为 7.39 L,实验装置如图1所示。SBR运行过程包括进水、曝气、沉淀、排水和闲置5个阶段,各阶段均通过时间控制器进行全自动控制。
2)运行方式。在污泥颗粒化过程中,以有机污染物浓度,表面上升气速和污泥沉降时间为主要调控参数,各阶段进水碳、氮和磷的质量浓度比控制在130∶10∶1~100∶5∶1,SBR运行周期为6 h,其中包括进水4 min、曝气338~351 min、沉淀2~15 min、排水1 min、闲置2 min,SBR排出比为0.5。采用水浴加热的方法使反应器温度控制在25 ℃。SBR运行的具体参数如表1所示。
表 1 SBR运行参数Table 1. Operating parameters of SBR运行时间/d 进水/min 曝气/min 沉淀/min 排水/min 闲置/min 表面上升气速/(cm·s−1) 混合碳源比例 COD/(mg·L−1) TN/(mg·L−1) TP/(mg·L−1) 1~13 4 338 15 1 2 0.86 5∶3 800 50 6 14~28 4 341 12 1 2 1.25 3∶2 1 000 60 9 29~43 4 343 10 1 2 1.86 5∶3 1 200 70 12 44~57 4 345 8 1 2 2.65 11∶5 1 600 90 16 58~75 4 348 5 1 2 3.87 11∶5 1 600 90 16 76~110 4 351 2 1 2 4.64 2∶1 1 800 100 20 注:混合碳源比例是指葡萄糖和乙酸钠的质量浓度比。 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.3 分析指标和分析方法
MLSS、MLVSS、SVI30、含水率、COD、TN、TP等均采用标准方法[12]测定,粒径分布采用湿式筛分法[13]确定,污泥沉降速度采用重力沉降法[14]测定,利用生物显微镜(BK 500 奥特光学)观测好氧颗粒污泥形态变化。好氧颗粒污泥微观形貌和组成元素采用FIB-SEM双束电镜(LYRA 3 XMU)观察和分析。EPS采用热提取法[15],EPS含量由PN和PS含量之和表示,PS含量采用苯酚-硫酸法[16]测定;PN含量采用BCA分光光度法[17]测定。
2. 结果和讨论
2.1 好氧颗粒污泥形成过程
由图2可知,接种污泥外观为灰褐色、絮状,结构松散,SVI30为110.7 mL·g−1,沉降速度为12.36 m·h−1,MLVSS/MLSS仅为0.41。当SBR运行30 d后,出现少量细小的污泥颗粒,但絮状污泥占主体。第50天,初期好氧颗粒污泥形成,粒径较小,主要呈现椭球状或棒状,SVI30下降为41.66 mL·g−1,沉降速度增大到35.74 m·h−1,MLVSS/MLSS为0.57。随着SBR内选择压不断增强,沉降性能较差的颗粒污泥被筛选出反应器,第70天,好氧颗粒污泥外观呈淡黄色,表面有一层绒毛,颗粒粒径主要集中在1~1.5 mm。在第110天,好氧颗粒污泥为橙黄色,外形较为光滑,整体呈球状或椭球状的立体结构,大多数颗粒粒径分布在1.43~2.26 mm,SVI30为28 mL·g−1,沉降速度为94 m·h−1,MLSS达到17 400 mg·L−1,MLVSS/MLSS为0.72,以上测定结果标志着好氧颗粒污泥培养成功。与其他研究结果[18]相比,该实验培养得到的好氧颗粒污泥沉降性能、微生物量以及微生物活性均较高。
2.2 好氧颗粒污泥微观形貌和元素组成分析
好氧颗粒污泥微观形貌如图3所示。由图3(a)可知:好氧颗粒污泥呈球状结构,外表面具有褶皱和空隙。由于表面褶皱的存在,好氧颗粒污泥比活性污泥具有更大的比表面积,因此更有利于与水体中有机物充分接触,从而增强了对有机污染物的去除效果。此外,好氧颗粒污泥表面的空隙是溶解氧和营养物质渗透到颗粒污泥内部的通道[19]。颗粒表面的微生物以球菌和丝状菌为主,同时还存在少量杆菌和钟虫(图3(b)~图3(e))。球菌呈密集的生长状态(图3(b)),丝状菌相互交联,形成类似网状结构(图3(c))。有研究[20]发现,丝状菌不仅能通过缠绕、连接等作用强化颗粒结构,而且可以消除细菌代谢产物在颗粒内部的积累从而可提高好氧颗粒污泥的结构稳定性。
图4为好氧颗粒污泥局部扫描电镜图像及其面能谱图,扫描结果见图4(a)和图4(b)。其对应的组成元素分析结果如表2所示。由能谱分析结果发现,好氧颗粒污泥中碳元素的质量分数和原子个数比分别达到50%和60%以上,氧元素的质量分数和原子个数比分别达到39%和34%以上,说明颗粒污泥中的元素主要是碳和氧,同时好氧颗粒污泥还含有少量其他元素,如N、Na、Ca、P、S等。
表 2 好氧颗粒污泥组成元素Table 2. Components of aerobic granular sludge元素 质量分数/% 原子个数比/% 元素 质量分数/% 原子个数比/% C 53.3 61.6 S 0.43 0.19 O 39.85 34.58 Mg 0.19 0.11 N 1.36 1.34 Fe 0.38 0.09 Na 1.75 1.06 K 0.16 0.06 Ca 1.75 0.61 Cu 0.02 0 P 0.82 0.37 | Show TableDownLoad:
CSV
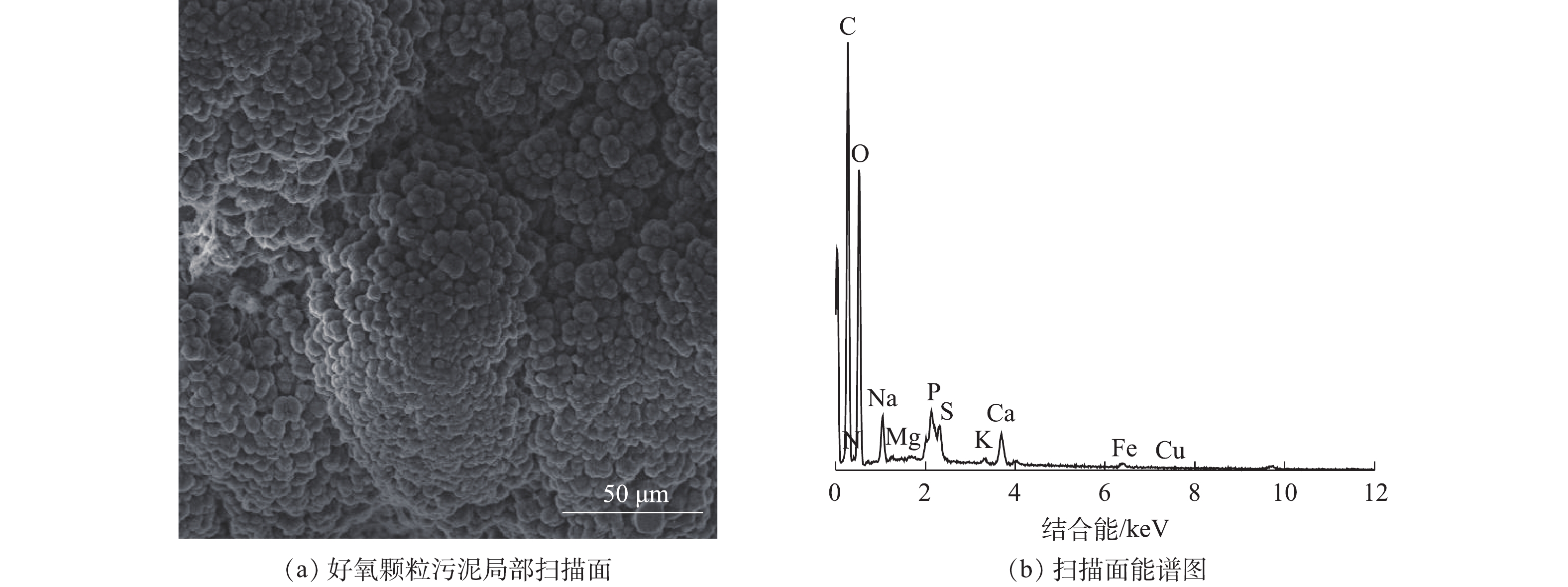 图 4 好氧颗粒污泥局部扫描面及其能谱图Figure 4. Local scanning surface and energy spectrum of aerobic granular sludge
图 4 好氧颗粒污泥局部扫描面及其能谱图Figure 4. Local scanning surface and energy spectrum of aerobic granular sludge2.3 污泥颗粒化过程中污泥理化特性变化
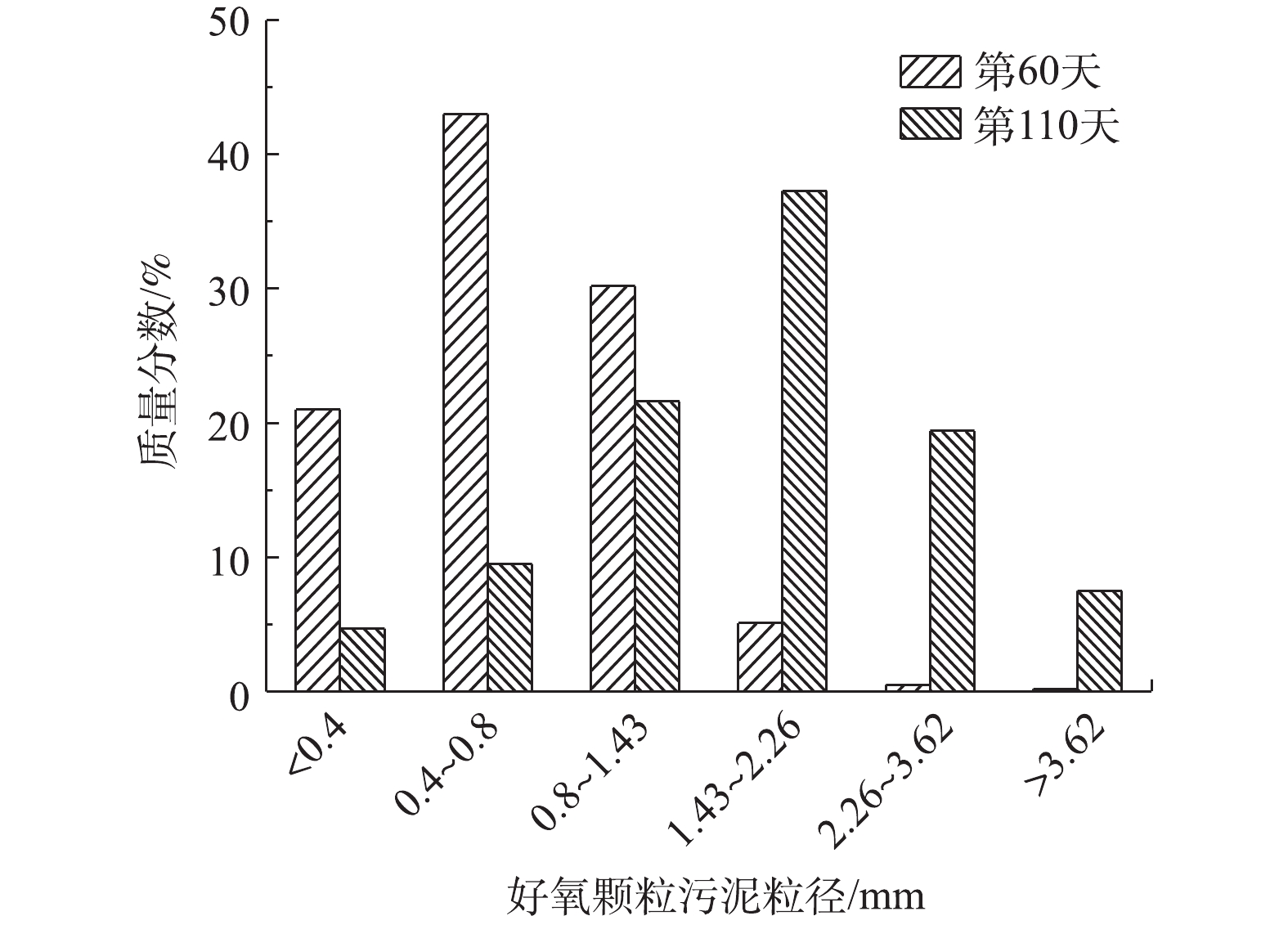
1)好氧颗粒污泥粒径分布变化。如图5所示,第60天,初期好氧颗粒污泥已经形成,其颗粒粒径主要集中在0.4~1.43 mm。随着实验不断进行,第110天,SBR中的好氧颗粒污泥粒径主要分布在1.43~2.26 mm,其质量分数为37%。粒径分布在0.8~3.62 mm的好氧颗粒污泥质量分数为88%,而粒径<0.4 mm和粒径>3.62 mm的好氧颗粒污泥质量分数均不足8%,结合图3(a)可知,SBR中的污泥呈球状立体结构,基本无絮状污泥存在,说明好氧颗粒污泥培养成功且可实现泥水分离。
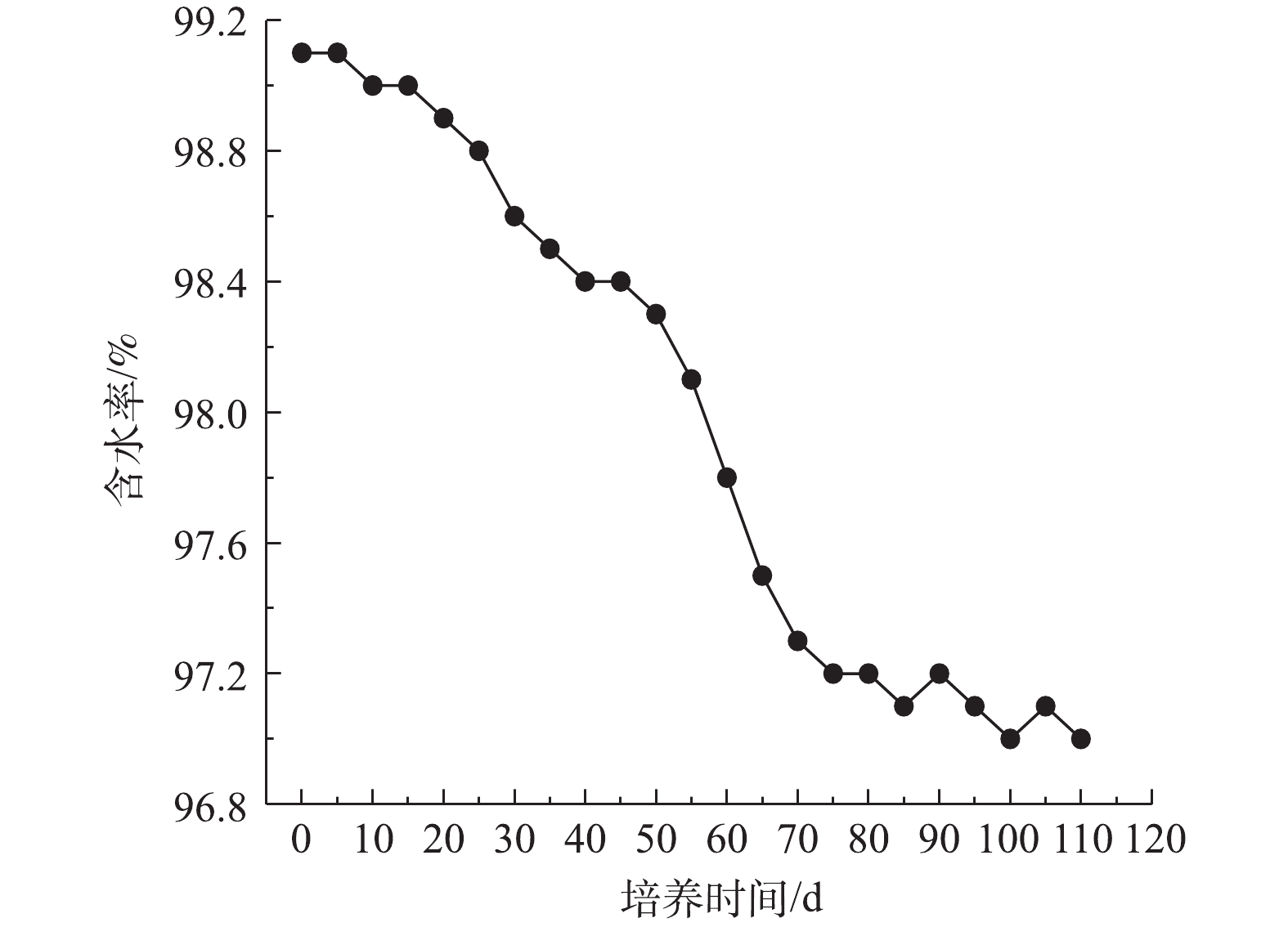
2)好氧颗粒污泥含水率变化。污泥含水率是表征污泥物理性状的一个重要参数。污泥颗粒化过程中污泥含水率随时间变化情况如图6所示。接种污泥含水率为99.14%,在SBR启动初期,因为在曝气状态下接种污泥受到剪切力的作用,污泥原始状态受破坏;此外,污泥沉降时间受到限制(沉降时间为15 min),所以大量沉降性较差的污泥被排出反应器。随着颗粒化逐渐推进,微生物细胞逐渐生长促进污泥间的凝聚,使污泥中的间隙水减少,导致含水率逐渐下降。在第50天由于初期好氧颗粒污泥形成,大量微生物菌群聚集成团,颗粒污泥密实性增加,因此颗粒污泥含水率明显下降。好氧颗粒污泥逐渐趋于成熟状态,其含水率基本维持在较稳定水平。最后培养得到的好氧颗粒污泥含水率为97%。
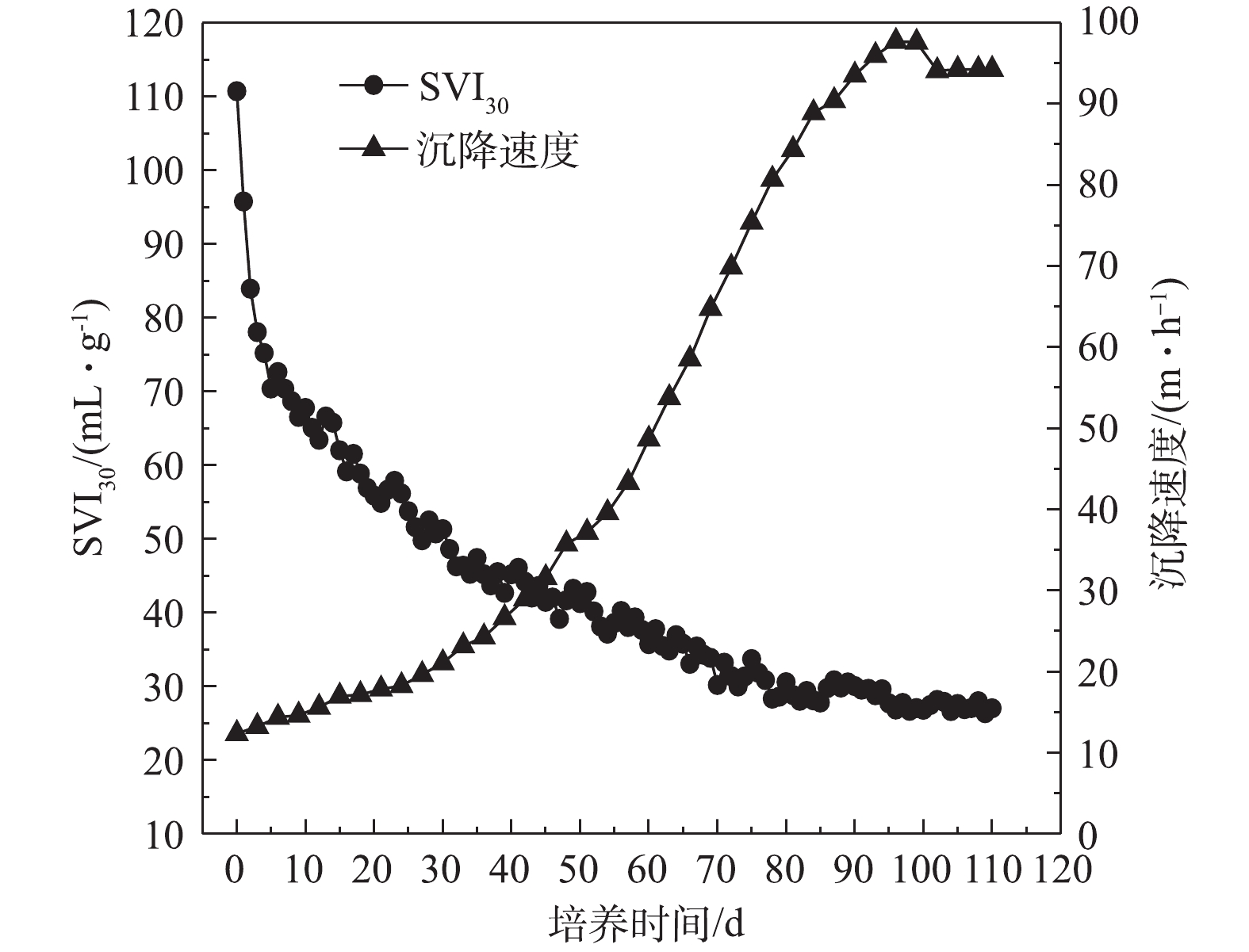
3)好氧颗粒污泥沉降性能和沉降速度变化。由图7可知,接种污泥SVI30为110.7 mL·g−1,沉降性能较差。随着反应器的运行,颗粒化程度不断增加,污泥沉降性能也在不断提升,SVI30不断下降。随着污泥颗粒化的完成,SVI30为28 mL·g−1,远低于普通絮状污泥。接种污泥沉降速度为12.36 m·h−1,好氧颗粒污泥培养初期,污泥沉降速度呈缓慢增大的趋势。第40天,好氧颗粒污泥沉降速度显著增加,最后沉降速度为94 m·h−1。以上表明培养出的好氧颗粒污泥结构密实,沉降性能良好。
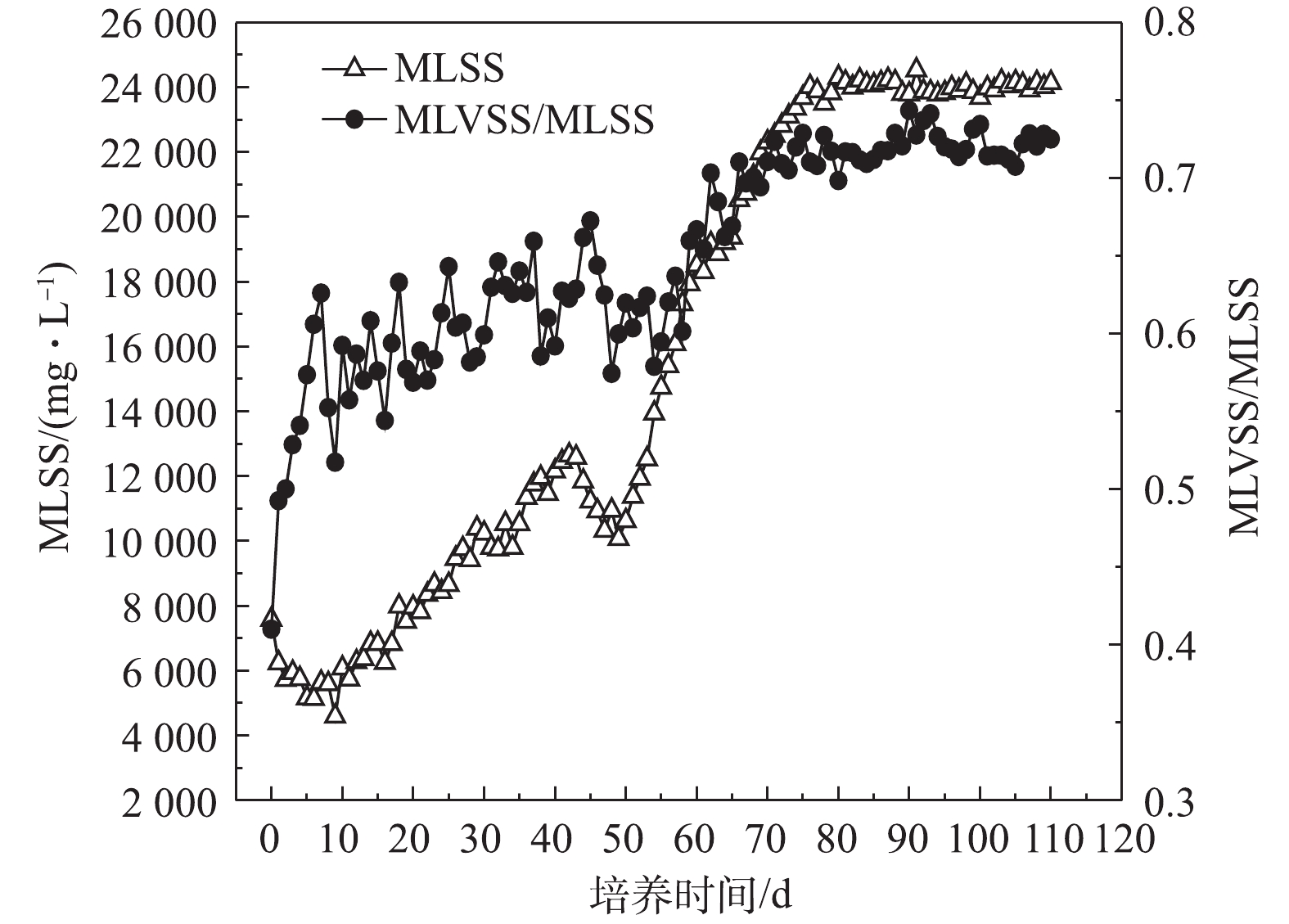
4) MLSS和MLVSS/MLSS变化。MLVSS/MLSS代表污泥中有机固体的比例,表示污泥内部活性微生物量,MLVSS/MLSS越大说明微生物量越高。污泥颗粒化过程中的MLSS和MLVSS/MLSS变化情况如图8所示,接种MLSS为7 588 mg·L−1,在SBR启动初期,污泥沉降时间突然缩短,大量松散絮状污泥被排出反应器,MLSS显著下降。随着接种污泥对进水水质和SBR运行方式的适应,污泥开始逐渐生长,在第20天达到7 990 mg·L−1,超过接种时水平。第44 天,可能是因为颗粒污泥尚未形成,表面上升气速的增加促使粘附在污泥微团表面的污泥进一步被剥落,所以MLSS呈下降趋势。随着SBR的连续运行,第50天,初期好氧颗粒污泥形成,污泥不断颗粒化,好氧颗粒污泥量开始增加,MLSS明显上升,在第87天达到24 232 mg·L−1。当好氧颗粒污泥逐渐趋向成熟,MLSS基本保持在稳定水平。经SBR运行110 d后,培养出的好氧颗粒污泥MLSS为24 140 mg·L−1,约为接种时的3.2倍,说明好氧颗粒污泥具有较高的微生物浓度。接种污泥的MLVSS/MLSS仅为0.41,反映出其内部活性微生物量很少。在整个污泥颗粒化过程中,MLVSS/MLSS整体处于缓慢上升趋势。随着好氧颗粒污泥趋向成熟,SBR运行状态稳定,MLVSS/MLSS值变化不大,基本维持在0.72,说明好氧颗粒污泥微生物量较高。
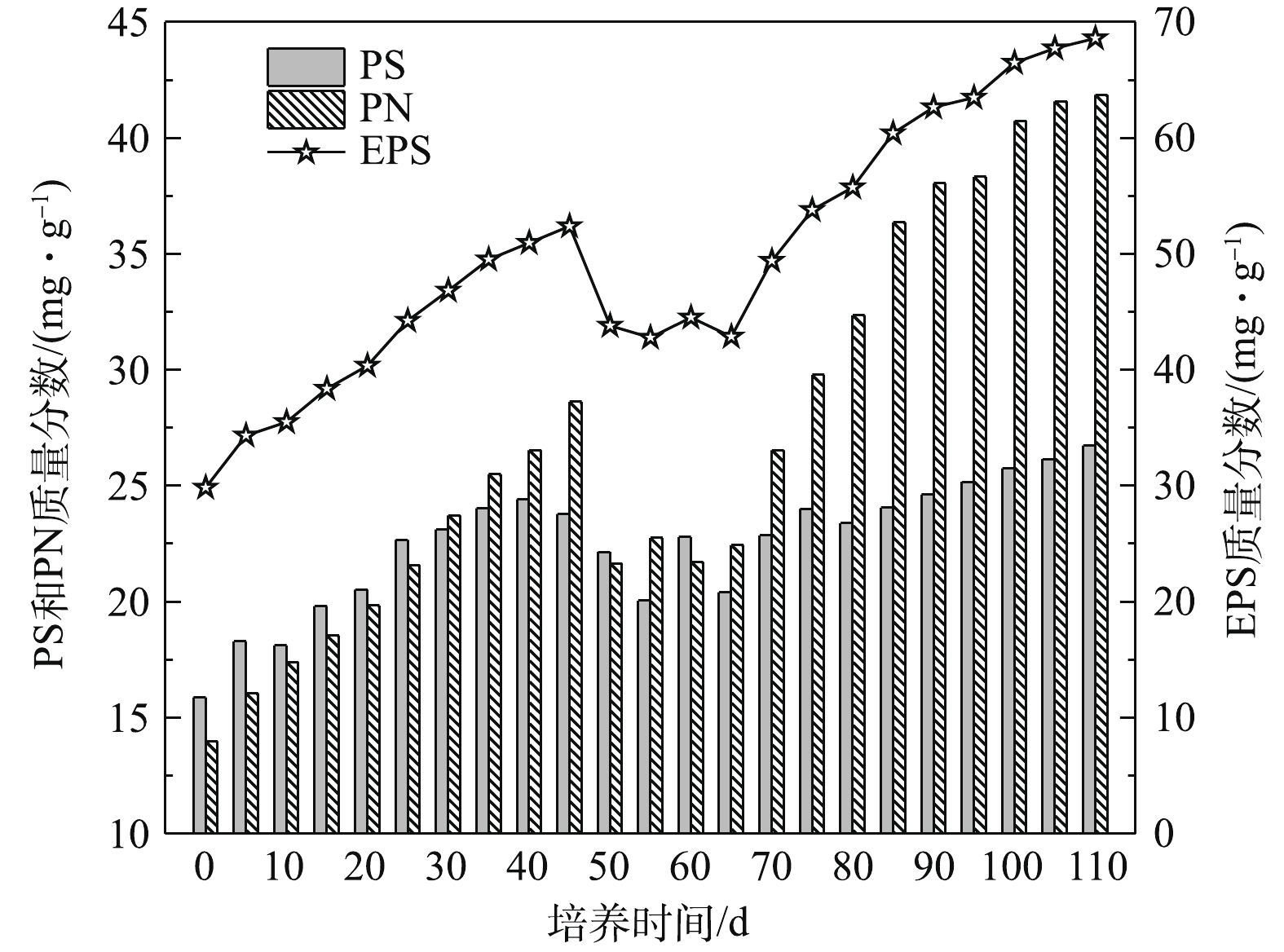
5)污泥颗粒化过程中EPS、PN和PS质量分数变化。如图9所示,接种污泥EPS质量分数为29.86 mg·g−1,其中PN和PS质量分数分别为13.98 mg·g−1和15.88 mg·g−1。在SBR运行初期,EPS、PN和PS含量均缓慢增加,但PS含量略高于PN含量,这是因为污泥内微生物在新环境刺激下需要分泌大量胞外多糖维持正常的生命活动[21]。随后由于沉降时间缩短,表面上升气速和有机污染物浓度提高,SBR内的选择压增强,打破了污泥中微生物细胞间信息交流的动态平衡,使微生物的新陈代谢活动受到抑制,因此EPS、PN和PS含量开始出现短时间的下降趋势。第70天SBR中的污泥以颗粒污泥为主,微生物量提高,EPS和PN含量开始显著增加,而PS含量没有明显波动。随着污泥颗粒化的完成,EPS、PN和PS含量逐渐趋于稳定。第110天的好氧颗粒污泥EPS和PN质量分数分别为68.6 mg·g−1和41.86 mg·g−1,PS质量分数为26.74 mg·g−1,这表明污泥颗粒化与PN含量变化密切相关,而PS含量变化对污泥颗粒化影响不大。在整个污泥颗粒化过程中,EPS含量变化趋势与PN含量变化相一致,这与CHEN等[22]的研究结果相符合。
2.4 污泥颗粒化过程中污染物去除效果
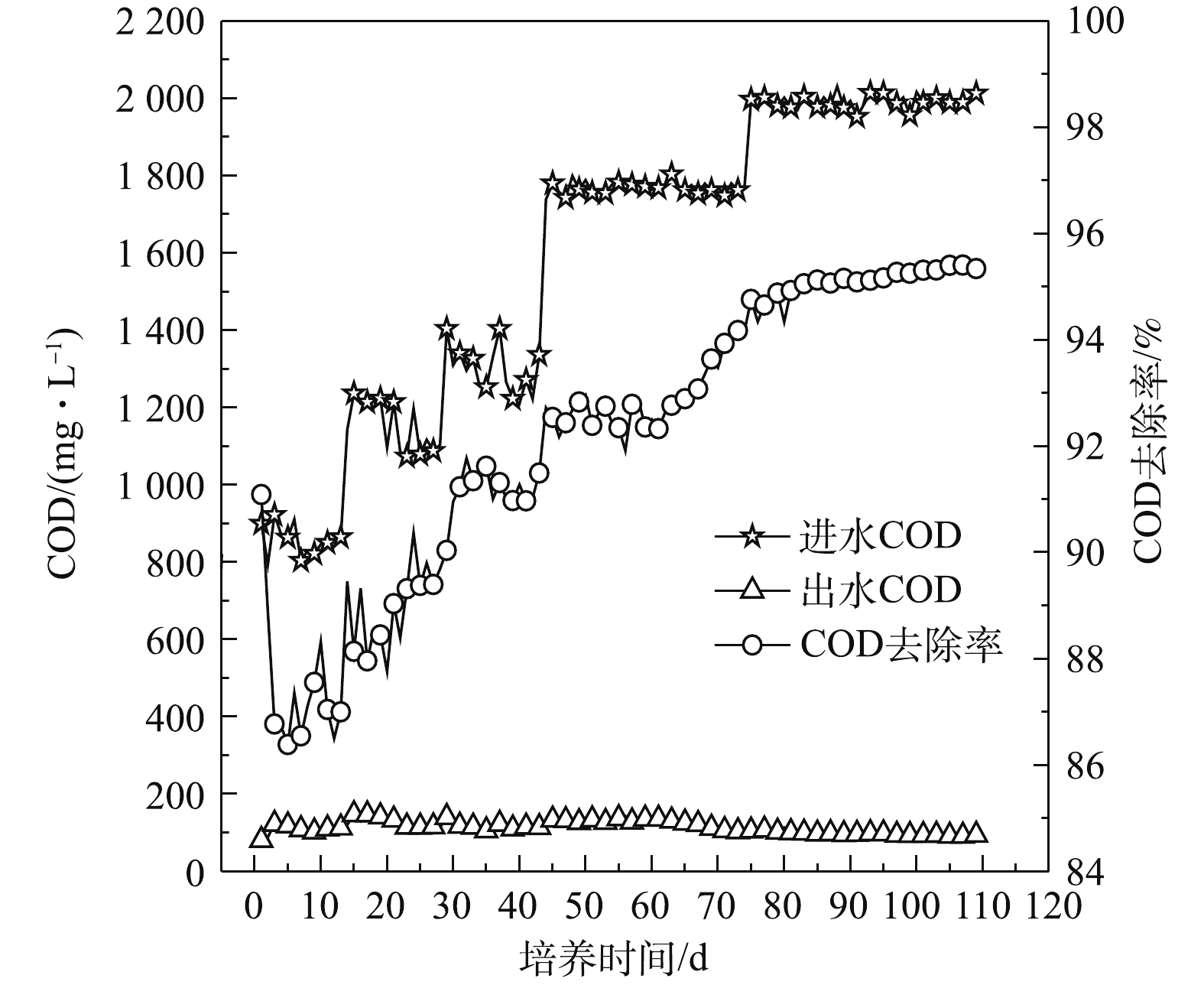
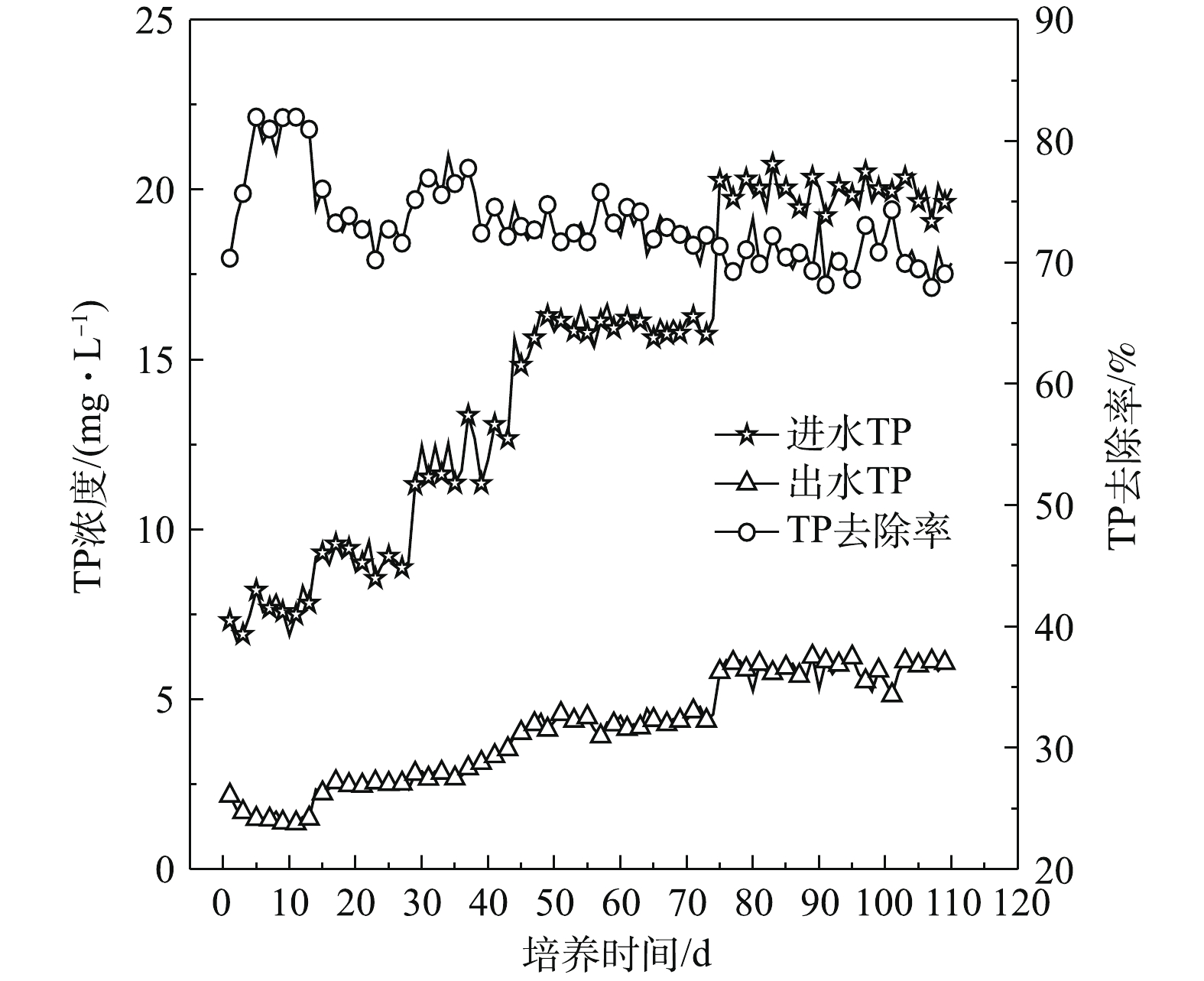
1)污泥颗粒化过程中COD去除效果。接种污泥曝气24 h 后污泥活性较大,对有机物的去除效果较好,如图10所示,第1天,反应器中的污泥对COD去除率为91.09%。随后因为接种污泥受到剪切力的作用,在沉降时间缩短的情况下,大量沉降性较差的絮状污泥被排出反应器,使MLSS和微生物量快速下降,所以在实验初期COD去除率明显下降。随着污泥内部微生物对反应器运行条件的逐渐适应,微生物量不断积累,MLSS开始增加,SBR的污泥颗粒化程度不断提高,因此,COD去除率有一定范围的波动,但整体随着进水COD值的提高在不断增大。在第50天,初期好氧颗粒污泥形成,进水COD为1 764 mg·L−1,出水COD为126 mg·L−1,COD去除率达到92.83%。在好氧颗粒污泥培养后期,COD去除率逐渐趋于稳定,第110天,好氧颗粒污泥培养成功,反应器进水COD为1 989 mg·L−1,出水COD为90 mg·L−1,COD去除率为95%。
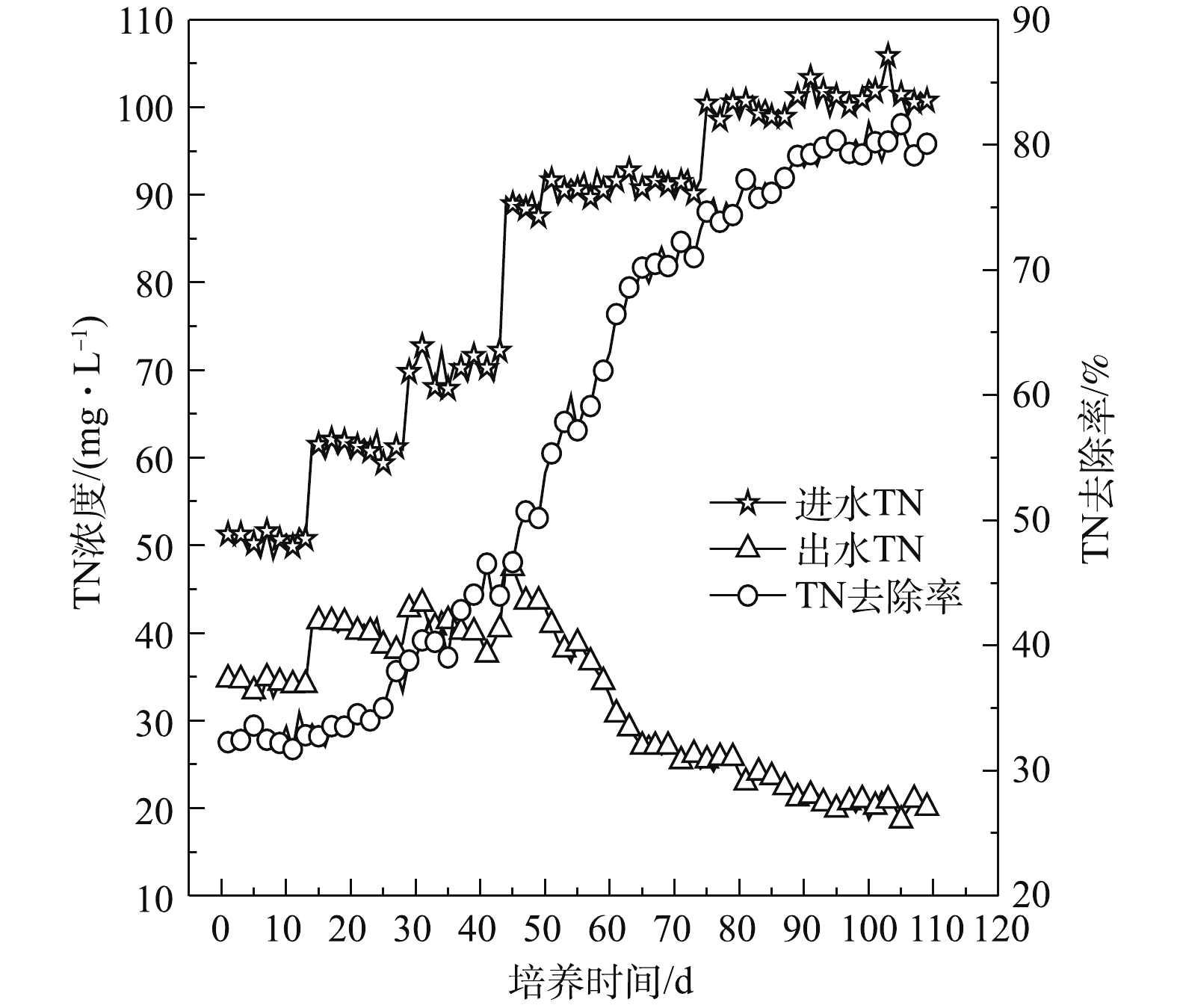
2)污泥颗粒化过程中TN去除效果。污泥颗粒化过程中TN去除效果如图11所示,SBR运行初期,进水TN浓度由最初的50 mg·L−1上升到60 mg·L−1,出水TN浓度随进水TN浓度而增加,TN去除率较低,仅为32%。一方面,因为刚接种污泥内微生物活性较低,对生存环境正处于适应阶段,仅靠微生物的同化作用将TN转化为自身细胞原生质成分;另一方面,又因为初期反应器以絮状污泥为主体,污泥颗粒化现象不明显,尚未形成同步硝化反硝化所需要的微环境[23-24],因此,TN去除率总体较低。之后TN去除率明显增加,出水TN浓度逐渐降低。随着反应器中颗粒污泥粒径不断增大,在第80天TN去除率为75%左右,出水TN浓度为25 mg·L−1。研究发现TN去除效果受颗粒污泥粒径的影响[25],这主要是因为随着颗粒污泥粒径不断增大,其内部因氧气传递的限制产生分层,分别在颗粒污泥表面和内部形成好氧环境和缺氧环境[26];硝化菌在颗粒表面发生硝化作用将
NH+4 -N氧化为硝态氮和亚硝态氮;反硝化菌在颗粒内部发生反硝化作用将进入的硝态氮和亚硝态氮还原成氮气,从而实现对N的去除[27]。当好氧颗粒污泥趋于成熟,硝化和反硝化作用逐渐稳定,出水TN浓度基本维持在20 mg·L−1,TN去除率稳定在80% 左右。有研究者[28]使用相同的混合碳源,通过逐步提高有机负荷和缩短沉降时间的方法培养得到的好氧颗粒污泥对TN的去除率为60%,出水TN浓度为40 mg·L−1,说明本实验的颗粒污泥对TN具有较好的去除效果。结合TN去除率整体变化规律可知,TN去除效果与污泥颗粒化程度密切相关,因此,实现更好的脱氮效果需要加快推进污泥颗粒化程度。3)污泥颗粒化过程中TP去除效果。图12为污泥颗粒化过程中TP去除效果。好氧颗粒污泥去除TP的实质是生物除磷,主要是利用污泥中聚磷菌一类的微生物从外部环境中磷的过剩摄取,并将磷以聚合的形态贮藏在微生物体内,形成高磷污泥,排出SBR,从而实现对污水中TP的去除。由于污泥接种前进行了曝气处理,聚磷菌可吸附污水中大量的磷,而且大量絮状污泥外排,所以TP去除率由最初的70.36%上升到81.99%,随后TP去除率开始下降。当反应器运行一段时间后,絮状污泥凝聚成团或细小颗粒,TP去除率又出现缓慢上升趋势。由于污泥颗粒化程度不断加大,大量沉降性能良好的颗粒污泥被截留在反应器内,污泥排出量越来越少。此阶段,由于污泥中的聚磷菌在好氧条件下,可以通过主动运输的方式将外部环境中的磷摄入体内,用于合成ATP,从而获得能量。因此在SBR实验阶段基本不排泥的情况下,由于好氧颗粒污泥量逐渐增大,对TP仍具有一定的去除效果。随着好氧污泥颗粒化的完成,TP出水浓度为5 mg·L−1, 去除率为70%。
3. 结论
1)在SBR中,经过110 d后培养出致密的好氧颗粒污泥,颗粒粒径主要分布在1.43~2.26 mm,大量球菌和丝状菌附着在好氧颗粒污泥表面。好氧颗粒污泥主要是由碳和氧元素构成,此外还含有少量的金属元素。
2)好氧颗粒污泥SVI30为28 mL·g−1,沉降速度为94 m·h−1,含水率为97%,MLSS为17 400 mg·L−1,MLVSS/MLSS基本维持在0.72,具有较高的微生物量和微生物活性。
3)在污泥颗粒化过程中,PS含量没有明显波动,PN含量却明显增加,由13.98 mg·g−1增加到41.86 mg·g−1,且EPS含量的变化与PN含量变化趋势相一致,表明EPS中的PN含量在增加可促进对好氧颗粒污泥的形成。
4)好氧颗粒污泥具有较好的除污效果,对COD、TN、TP的去除率分别约为95%、80%、70%。
-

图 1 PS、PVC和PMMA微塑料在1 μm和10 μm下的电镜图。
Figure 1. SEM images of 1 μm and 10 μm PS,PVC, and PMMA microplastics.
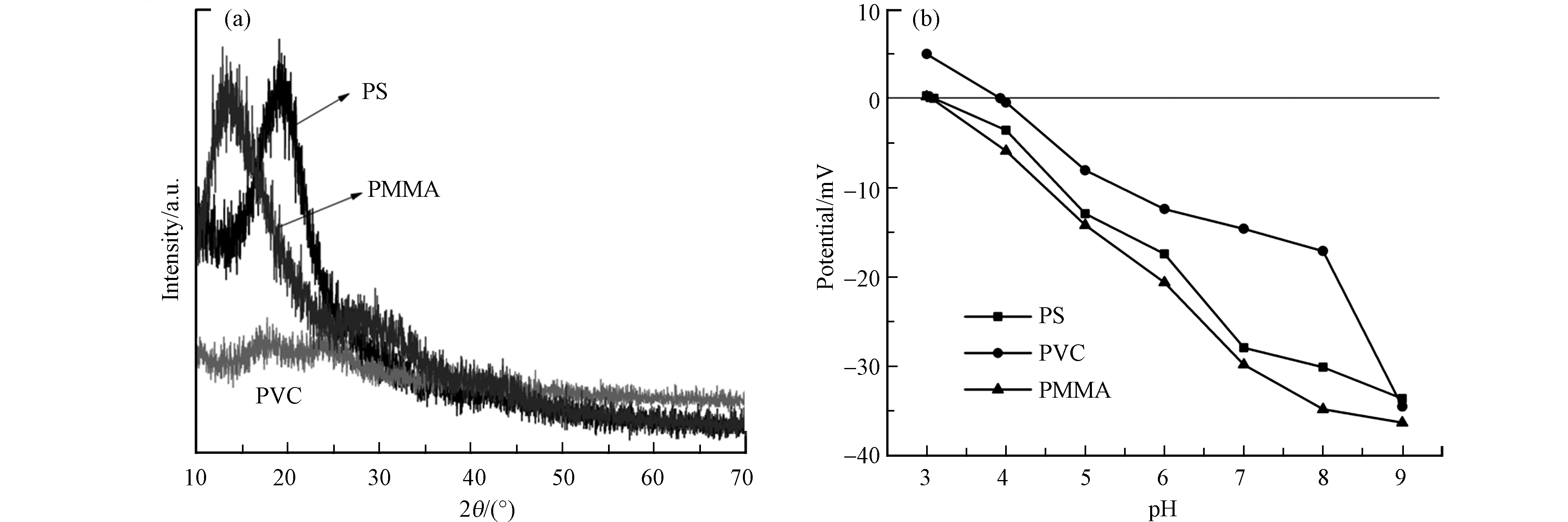
图 2 PS、PVC和PMMA的XRD图 (a),PS、PVC和PMMA的zeta电位图(b)。
Figure 2. The XRD pattern of PS, PVC, and PMMA (a), zeta potential pattern of PS, PVC, and PMMA (b).
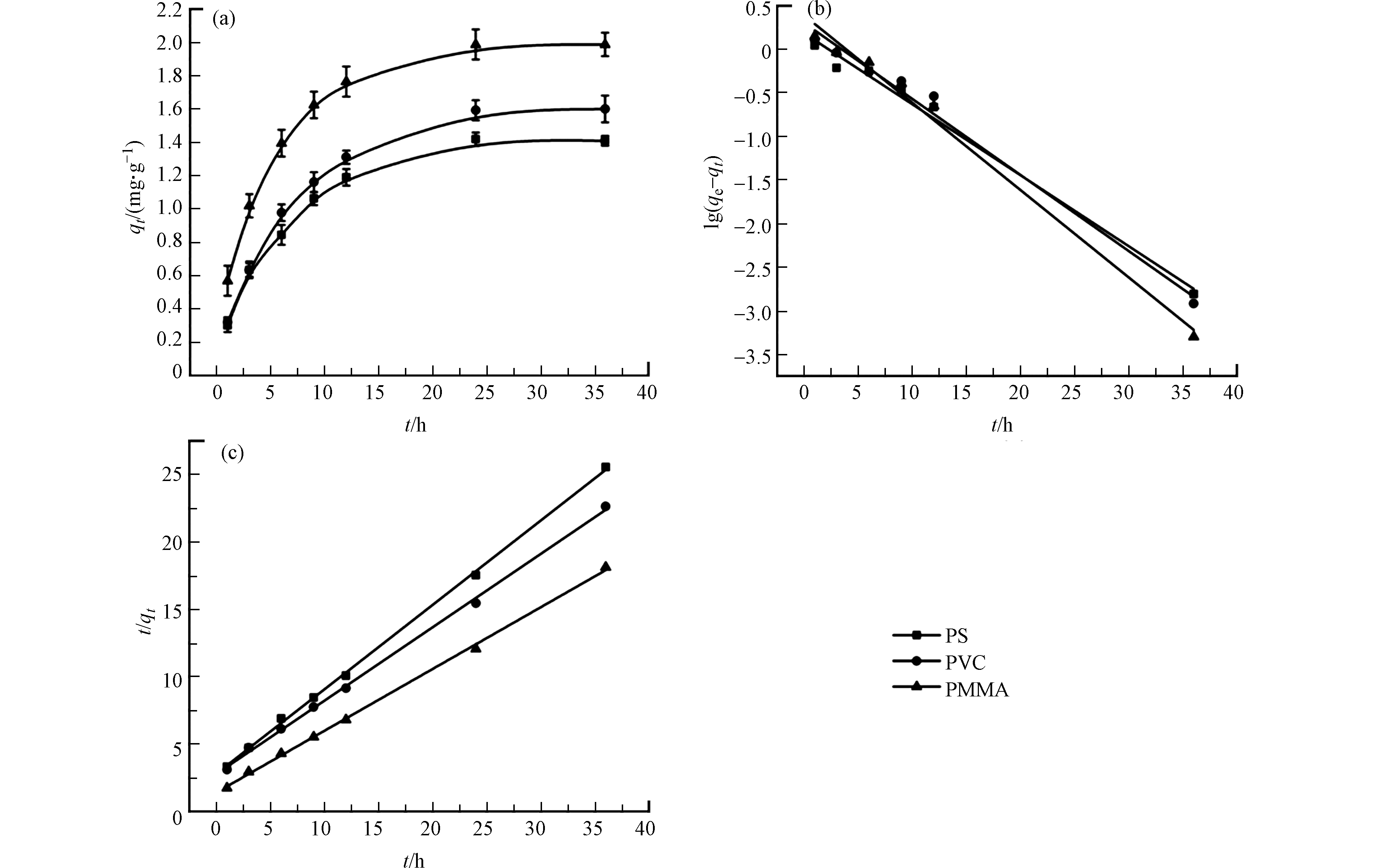
图 3 微塑料对MB的吸附动力学实验数据(a)、一级线性模型(b)和二级线性模型(c)。
Figure 3. Adsorption kinetic experimental data of MB sorption onto microplastics (a), linear plots on pseudo-first-order model (b), and pseudo-second-order model (c).
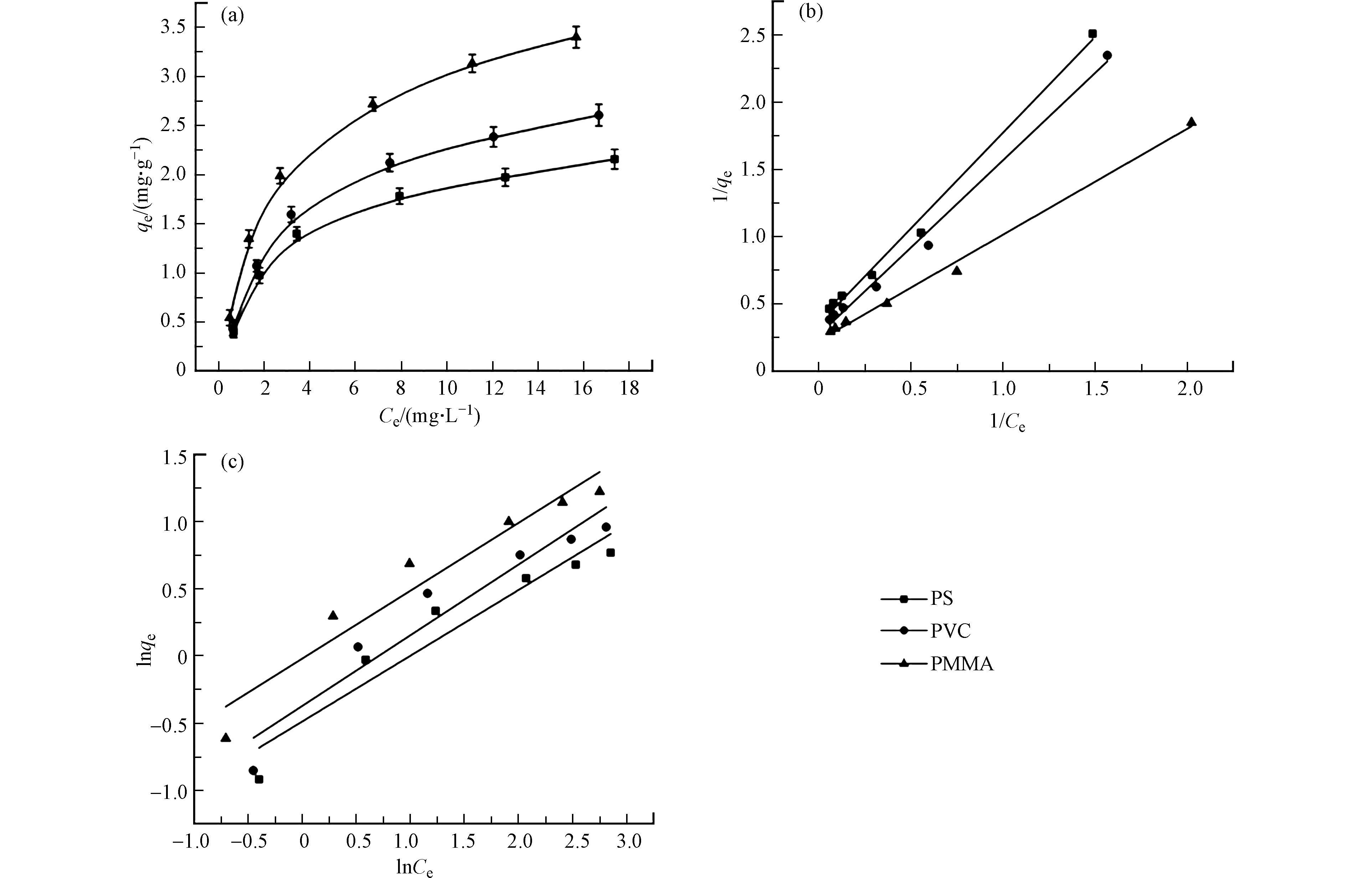
图 4 微塑料对MB的吸附等温实验数据(a)、Langmuir模型(b)和Freundlich模型(c)。
Figure 4. Adsorption isotherm experimental data of MB sorption onto microplastics (a), linear plots on Langmuir model (b), and Freundlich model (c).
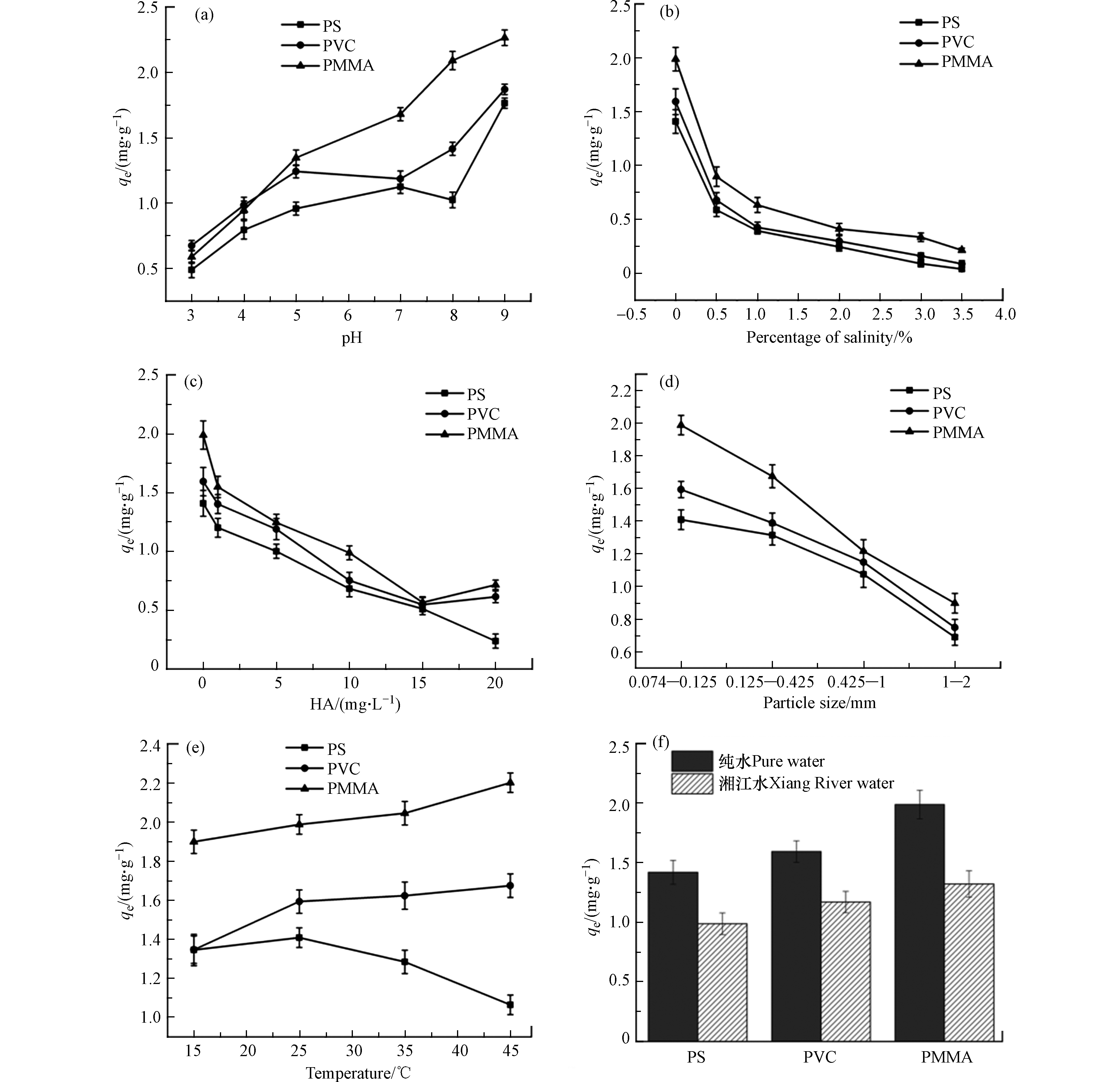
图 5 pH (a) 、盐度(b) 、HA浓度(c) 、微塑料粒径(d) 、温度(e)和湘江水样(f)对微塑料吸附MB的影响。
Figure 5. The effect of pH (a), salinity (b), the concentration of HA (c), particle size (d), temperature (e), and Xiang River water (f) of MB adsorbed on microplastics.
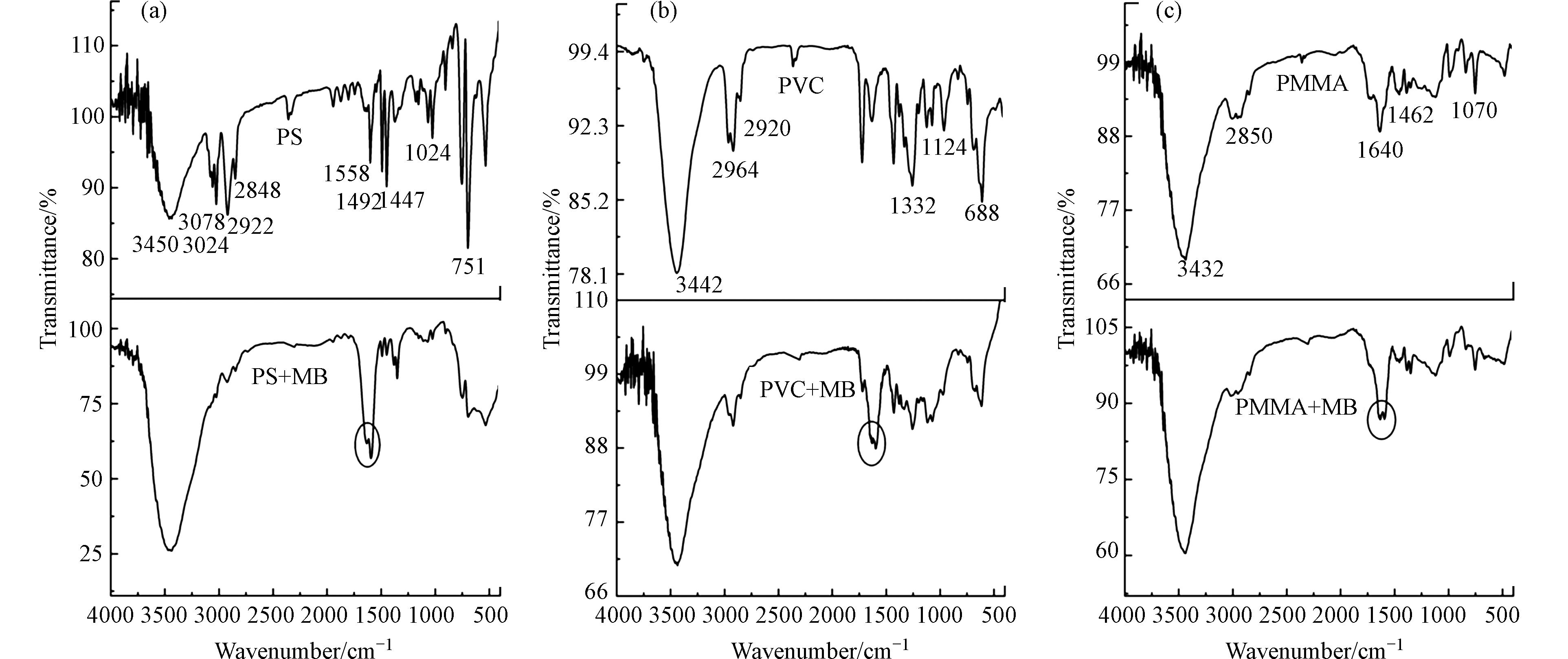
图 6 吸附前后PS (a)、PVC (b)和PMMA (c)的红外光谱图。
Figure 6. FTIR spectra of PS (a), PVC (b), and PMMA (c) before and after adsorption.
表 1 MB在PS、PVC和PMMA微塑料上的吸附动力学参数。
Table 1. The adsorption kinetic parameters of MB on PS, PVC, and PMMA.
微塑料Microplastics 准一级动力学Pseudo-first-order 准二级动力学Pseudo-second-order qe,exp/(mg·g−1) qe,cal/(mg·g−1) Kd1/h−1 R2 qe,cal/(mg·g−1) Kd2/(g·(mg·h)−1) R2 PS 1.419 1.204 0.082 0.989 1.597 1.096 0.999 PVC 1.594 1.357 0.087 0.987 1.836 0.822 0.999 PMMA 1.988 1.478 0.100 0.989 2.182 0.295 0.999
下载: 导出CSV
表 2 MB在PS、PVC和PMMA微塑料上的等温吸附实验参数。
Table 2. The adsorption isotherm parameters of MB on PS, PVC, and PMMA.
微塑料Microplastics Langmuir 模型Langmuir model Freundlich 模型Freundlich model qmax/(mg·g−1) KL/(L·mg−1) R2 n KF/(mg·g−1)(L·g−1)1/n R2 PS 2.872 0.245 0.993 0.489 0.613 0.920 PVC 3.721 0.207 0.993 0.526 0.688 0.927 PMMA 4.407 0.288 0.996 0.505 0.979 0.936
下载: 导出CSV
-
[1] THOMPSON R C, OLSEN Y, MITCHELL R P, et al. Lost at sea: Where is all the plastic? [J]. Science, 2004, 304(5672): 838. doi: 10.1126/science.1094559 [2] FENDALL L S, SEWELL M A. Contributing to marine pollution by washing your face: Microplastics in facial cleansers [J]. Marine Pollution Bulletin, 2009, 58(8): 1225-1228. doi: 10.1016/j.marpolbul.2009.04.025 [3] JEONG C B, KANG H M, LEE Y H, et al. Nanoplastic ingestion enhances toxicity of persistent organic pollutants (POPs) in the monogonont rotifer Brachionus koreanus via multixenobiotic resistance (MXR) disruption [J]. Environmental Science & Technology, 2018, 52(19): 11411-11418. [4] TAYLOR M L, GWINNETT C, ROBINSON L F, et al. Plastic microfibre ingestion by deep-sea organisms [J]. Scientific Reports, 2016, 6: 33997. doi: 10.1038/srep33997 [5] 张凯娜, 李嘉, 李晓强, 等. 微塑料表面土霉素的吸附-解吸机制与动力学过程 [J]. 环境化学, 2017, 36(12): 2531-2540. doi: 10.7524/j.issn.0254-6108.2017032703 ZHANG K N, LI J, LI X Q, et al. Mechanisms and kinetics of oxytetracycline adsorption-desorption onto microplastics [J]. Environmental Chemistry, 2017, 36(12): 2531-2540(in Chinese). doi: 10.7524/j.issn.0254-6108.2017032703
[6] CARBERY M, O'CONNOR W, PALANISAMI T. Trophic transfer of microplastics and mixed contaminants in the marine food web and implications for human health [J]. Environment International, 2018, 115: 400-409. doi: 10.1016/j.envint.2018.03.007 [7] 张瑞昌, 李泽林, 魏学锋, 等. 模拟环境老化对PE微塑料吸附Zn(Ⅱ)的影响 [J]. 中国环境科学, 2020, 40(7): 3135-3142. doi: 10.3969/j.issn.1000-6923.2020.07.040 ZHANG R C, LI Z L, WEI X F, et al. Effects of simulated environmental aging on the adsorption of Zn(Ⅱ) onto PE microplastics [J]. China Environmental Science, 2020, 40(7): 3135-3142(in Chinese). doi: 10.3969/j.issn.1000-6923.2020.07.040
[8] TANG Y Q, LIU Y G, CHEN Y, et al. A review: Research progress on microplastic pollutants in aquatic environments [J]. Science of the Total Environment, 2021, 766: 142572. doi: 10.1016/j.scitotenv.2020.142572 [9] WANG F, WONG C S, CHEN D, et al. Interaction of toxic chemicals with microplastics: A critical review [J]. Water Research, 2018, 139: 208-219. doi: 10.1016/j.watres.2018.04.003 [10] ROCHMAN C M, HOH E, HENTSCHEL B T, et al. Long-term field measurement of sorption of organic contaminants to five types of plastic pellets: Implications for plastic marine debris [J]. Environmental Science & Technology, 2013, 47(3): 1646-1654. [11] LIU Z M, QIN Q D, HU Z X, et al. Adsorption of chlorophenols on polyethylene terephthalate microplastics from aqueous environments: Kinetics, mechanisms and influencing factors [J]. Environmental Pollution, 2020, 265: 114926. doi: 10.1016/j.envpol.2020.114926 [12] ZHANG H B, WANG J Q, ZHOU B Y, et al. Enhanced adsorption of oxytetracycline to weathered microplastic polystyrene: Kinetics, isotherms and influencing factors [J]. Environmental Pollution, 2018, 243: 1550-1557. doi: 10.1016/j.envpol.2018.09.122 [13] ZHANG J H, CHEN H B, HE H, et al. Adsorption behavior and mechanism of 9-Nitroanthracene on typical microplastics in aqueous solutions [J]. Chemosphere, 2020, 245: 125628. doi: 10.1016/j.chemosphere.2019.125628 [14] YU F, YANG C F, HUANG G Q, et al. Interfacial interaction between diverse microplastics and tetracycline by adsorption in an aqueous solution [J]. Science of the Total Environment, 2020, 721: 137729. doi: 10.1016/j.scitotenv.2020.137729 [15] SEERA S D K, KUNDU D, GAMI P, et al. Synthesis and characterization of xylan-gelatin cross-linked reusable hydrogel for the adsorption of methylene blue [J]. Carbohydrate Polymers, 2021, 256: 117520. doi: 10.1016/j.carbpol.2020.117520 [16] SIMONIN J P. On the comparison of pseudo-first order and pseudo-second order rate laws in the modeling of adsorption kinetics [J]. Chemical Engineering Journal, 2016, 300: 254-263. doi: 10.1016/j.cej.2016.04.079 [17] FOO K Y, HAMEED B H. Insights into the modeling of adsorption isotherm systems [J]. Chemical Engineering Journal, 2010, 156(1): 2-10. doi: 10.1016/j.cej.2009.09.013 [18] LI H, WANG F H, LI J N, et al. Adsorption of three pesticides on polyethylene microplastics in aqueous solutions: Kinetics, isotherms, thermodynamics, and molecular dynamics simulation [J]. Chemosphere, 2021, 264: 128556. doi: 10.1016/j.chemosphere.2020.128556 [19] BAKIR A, ROWLAND S J, THOMPSON R C. Enhanced desorption of persistent organic pollutants from microplastics under simulated physiological conditions [J]. Environmental Pollution, 2014, 185: 16-23. doi: 10.1016/j.envpol.2013.10.007 [20] TORRES F G, DIOSES-SALINAS D C, PIZARRO-ORTEGA C I, et al. Sorption of chemical contaminants on degradable and non-degradable microplastics: Recent progress and research trends [J]. Science of the Total Environment, 2021, 757: 143875. doi: 10.1016/j.scitotenv.2020.143875 [21] QIU Y, ZHENG M G, WANG L, et al. Sorption of polyhalogenated carbazoles (PHCs) to microplastics [J]. Marine Pollution Bulletin, 2019, 146: 718-728. doi: 10.1016/j.marpolbul.2019.07.034 [22] WU G G, MA J P, LI S, et al. Magnetic copper-based metal organic framework as an effective and recyclable adsorbent for removal of two fluoroquinolone antibiotics from aqueous solutions [J]. Journal of Colloid and Interface Science, 2018, 528: 360-371. doi: 10.1016/j.jcis.2018.05.105 [23] ABDURAHMAN A, CUI K Y, WU J, et al. Adsorption of dissolved organic matter (DOM) on polystyrene microplastics in aquatic environments: Kinetic, isotherm and site energy distribution analysis [J]. Ecotoxicology and Environmental Safety, 2020, 198: 110658. doi: 10.1016/j.ecoenv.2020.110658 [24] XU B L, LIU F, BROOKES P C, et al. Microplastics play a minor role in tetracycline sorption in the presence of dissolved organic matter [J]. Environmental Pollution, 2018, 240: 87-94. doi: 10.1016/j.envpol.2018.04.113 [25] LEE H, SHIM W J, KWON J H. Sorption capacity of plastic debris for hydrophobic organic chemicals [J]. Science of the Total Environment, 2014, 470/471: 1545-1552. doi: 10.1016/j.scitotenv.2013.08.023 [26] LI H Q, HUANG G H, AN C J, et al. Removal of tannin from aqueous solution by adsorption onto treated coal fly ash: Kinetic, equilibrium, and thermodynamic studies [J]. Industrial & Engineering Chemistry Research, 2013, 52(45): 15923-15931. [27] 刘鹏, 王焓钰, 吴小伟, 等. 粒径对聚苯乙烯微塑料吸附环丙沙星的影响 [J]. 环境化学, 2020, 39(11): 3153-3160. doi: 10.7524/j.issn.0254-6108.2019082802 LIU P, WANG H Y, WU X W, et al. Effects of particle size on the adsorption of ciprofloxacin on polystyrene microplastics [J]. Environmental Chemistry, 2020, 39(11): 3153-3160(in Chinese). doi: 10.7524/j.issn.0254-6108.2019082802
[28] OVCHINNIKOV O V, EVTUKHOVA A V, KONDRATENKO T S, et al. Manifestation of intermolecular interactions in FTIR spectra of methylene blue molecules [J]. Vibrational Spectroscopy, 2016, 86: 181-189. doi: 10.1016/j.vibspec.2016.06.016 [29] RODRIGUES J P, DUARTE A C, SANTOS-ECHEANDÍA J, et al. Significance of interactions between microplastics and POPs in the marine environment: A critical overview [J]. TrAC Trends in Analytical Chemistry, 2019, 111: 252-260. doi: 10.1016/j.trac.2018.11.038 [30] 赵楚云, 李小伟, 张鸿元, 等. 化学预处理对微塑料Pb吸附潜力的影响及机理研究 [J]. 环境科学学报, 2019, 39(10): 3387-3394. ZHAO C Y, LI X W, ZHANG H Y, et al. Effect of chemical pretreatment on adsorption of microplastics to Pb [J]. Acta Scientiae Circumstantiae, 2019, 39(10): 3387-3394(in Chinese).
[31] YAMATE T, KUMAZAWA K, SUZUKI H, et al. CH/π interactions for macroscopic interfacial adhesion design [J]. ACS Macro Letters, 2016, 5(7): 858-861. doi: 10.1021/acsmacrolett.6b00265 [32] MANI D, ARUNAN E. The X–c···π (X = F, cl, br, CN) carbon bond [J]. The Journal of Physical Chemistry A, 2014, 118(43): 10081-10089. doi: 10.1021/jp507849g -

 点击查看大图
点击查看大图
计量
- 文章访问数: 6258
- HTML全文浏览数: 6258
- PDF下载数: 208
- 施引文献: 0











