




-
近年来,环境问题日益严峻,影响了人类健康和社会的可持续发展。如果说催化技术奠定了现代工业发展的基础,那么其也将是解决人类面临重大生存问题的关键技术。太阳光能量丰富,取之不尽,用之不竭,是理想的能量来源。光催化技术可以利用太阳能消除大气污染物,如氮氧化物、挥发性有机污染物、温室气体CO2等,进而改善我们的生存环境。
20世纪30年代,研究者发现在氧气及紫外光照射下,二氧化钛(TiO2)可以降解染料和纤维,并且反应前后TiO2无耗损[1]。但是由于当时人们对半导体理论理解不深以及测试分析水平不成熟,这种光催化降解有机污染物的现象被忽略,而被理解为紫外光的作用促使氧气在TiO2表面产生高活性物种。20世纪70年代,日本科学家Fujishima 和Honda 在Nature上发表“本多藤岛效应”的光催化现象,即TiO2在紫外光下能够光电催化分解水产生氢气[2],他们提出的太阳光催化分解水制氢的方法受到广泛的关注。1976年美国的Carey等发现光催化可以氧化多氯联苯[3];1983年Pruden和Ollis发现烷烃氯化物这种有机污染物可以被TiO2光催化降解成无污染的H2O和CO2[4];1989年Tanaka等提出在光催化反应中可以产生羟基自由基,这种活性物种将促进有机物污染物的氧化降解[5]。近几十年来,光催化已成为国内外最热门的研究领域之一,各国科研工作者都做出了不懈的努力。
半导体材料是多相光催化的核心,其中,催化剂的光吸收、光生电子-空穴分离、表面反应的性质在光催化反应性能方面起重要作用。锐钛矿TiO2因其较高的光催化性能、较好的稳定性以及低廉的价格等优点,成为应用最广泛的光催化材料。其较高的导带和较低的价带位置,可以增强光生电子、空穴的还原和氧化能力,深度还原和氧化反应物分子,避免二次污染和有毒中间物种生成。但是,TiO2作为直接利用太阳光的催化材料仍然存在一些不足,如:只能吸收紫外光,太阳能的利用率低;光催化反应的还原位点和氧化位点同时在TiO2半导体材料表面,极容易造成光生电子和空穴复合;对反应物吸附性能较差等。因此,寻求高性能的光催化材料已成为研究重点。
近年来,稀土元素以其丰富的能级结构,在光、电、磁等方面得到广泛应用。从电子结构看,5d轨道提供电子转移轨道,可作为光催化反应中光生电子的“转移站”;同时,在形成的氧化物中,正离子外层d和s电子的空态可以形成交叠导带,具有半导体性质。因此,稀土材料,尤其是铈基材料[6-8],在光催化领域具有潜在的应用前景。
-
铈(Ce)作为镧系元素,是稀土中丰度最高、最廉价的元素。由于外层特殊的电子结构(4f1、5d1、6s2),Ce元素具有可变价态。萤石结构的二氧化铈(CeO2)晶胞结构如下:阳离子按面心立方点阵排列,且每个金属阳离子被8个阴离子包围,阴离子填充全部的四面体孔隙,并与4个金属阳离子配位,进而促使氧化物中存在许多八面体空位和可迁移的氧空位[9]。因此,CeO2在较温和的条件下可以发生可逆的氧化-还原反应,具有良好的储存、释放氧性能。比如,在还原性气氛中,CeO2中Ce4+能被还原成Ce3+并释放氧,萤石型的晶胞结构保持不变,形成一种亚稳结构;而在氧化性气氛中,Ce3+又能被氧化成Ce4+储存氧,再次转变为稳定的萤石结构[10]。CeO2是常见的n型半导体,理论能带宽度是6 eV,主要由价带O2p和导带Ce5d构成;但是,由于CeO2-x中少量氧空位的存在,价带和导带分别变成O2p与4f能级,能带宽度会减小到3 eV;当在Ce2O3中,4f能级分裂为4f Empty和4f Full,价带和导带分别变成O2p和4f Full,能带宽度变为2 eV[11]。因此,相比于传统的金属氧化物TiO2光催化剂,CeO2因其特殊的电子结构,从而具有近紫外-可见光的响应,可以增强太阳光的吸收利用。同时,由于具有稳定的晶体结构、优异的氧流动性、较好的光学性质等优点,铈基催化剂被认为最具潜力的光催化材料之一,在大气污染治理领域都具有广泛的应用前景。
然而,目前CeO2光催化材料仍然存在一些不足,比如:禁带宽度为2.79 eV,只能吸收波长范围小于444 nm的光,不能充分利用太阳光;光生电子和空穴易于复合,导致量子效率较低,这严重降低了光催化性能;比表面积较小,不能有效地吸附反应物分子,影响表面反应等。针对上述问题,研究者对CeO2材料进行表面改性和优化,从而通过引入表面缺陷、形成异质结、产生更多的反应位点等方式提高光催化反应中光吸收、光生电子-空穴分离、以及表面反应这三个方面的效率,进而改善光催化反应性能。
-
研究表明通过贵金属沉积、半导体复合、碳材料修饰的方式,可以显著地提高铈基材料的光催化性能。在CeO2表面沉积适量的贵金属可以起到如下作用:
(1)贵金属沉积后,半导体的能带将弯向表面生成的损耗层,在金属-CeO2界面上形成俘获电子的浅势阱能垒,抑制光生电子-空穴的复合;
(2)光生电子快速地从CeO2表面传输到贵金属粒子上,加快电子运输到吸附氧的速率;
(3)Ag、Au负载后会产生等离子体共振效应,增强光催化剂的可见光吸收;
(4)高度分散的贵金属粒子具有特殊的电子结构,表面易吸附反应物分子,从而作为催化反应活性中心。比如:Primo等用生物聚合物模板法合成出CeO2纳米颗粒,将贵金属Au负载于CeO2纳米颗粒表面,发现相比于原来的CeO2,Au-CeO2具有优异的光催化分解水的活性,并且1% wt贵金属Au负载时性能最优[12]。Zhao等合成了CeO2@Ag@CdS复合材料,Ag的加入可以增强复合材料可见光的吸收,以及加快光生电子的迁移[13]。类似地,在Pt[14]、Pd[15]上都有所报道。另外,半导体复合也可以提高铈基催化剂的光催化性能。通过半导体的复合形成Z型、type Ⅱ型异质结,扩展CeO2光谱响应范围,同时加快光生电子-空穴的分离。常见的复合半导体有CeO2-TiO2,CeO2-CuO, CeO2-ZnO, Fe3O4/CeO2,CeO2/CdS,CeO2/g-C3N4等[16-21]。
-
离子掺杂是利用物理或化学方法,将离子引入到CeO2晶格结构内部,从而形成缺陷或改变晶格类型,影响光生电子和空穴的迁移方向、改变CeO2的能带结构,进而提高铈基材料的光催化性能。常用的掺杂离子包括非金属离子、过渡金属离子、稀土金属离子及其它离子。大量的研究表明非金属离子掺杂改性CeO2可以显著提高其光催化反应的活性。Wu等在非金属N元素掺杂过程中,发现少量CeO2的晶格氧被替换,减小CeO2禁带宽度,使得CeO2的激发波长由紫外扩展到了可见光区,提高了可见光下的催化活性,实现了直接利用太阳光能中的可见光来降解有机污染物[22]。除了N的研究[23-24],还有F[25]掺杂,以及S、N[26]共掺杂等。另外,过渡金属离子、稀土金属离子掺杂的相关报道也不少。过渡金属离子主要集中在 Fe3+、Cr3+、V4+等的掺杂[27-30],稀土金属离子主要是Eu3+、Y3+、Pr3+等[31-33]。一般来说,掺杂离子的电位要与CeO2的价带、导带相匹配,掺杂的稀土离子半径与Ce4+相近。利用离子掺杂的手段,形成捕获中心和掺杂能级,并增大载流子扩散长度,有效抑制光生电子与空穴的复合。同时。离子掺杂会引起CeO2的晶格扭曲,有利于更多氧缺陷和Ce3+物种的生成,提高可见光的吸收。
-
通常,催化剂的催化属性很大程度上依赖于其结构,因此,调控催化剂结构(特殊孔道、空心结构)是提升催化活性的重要途径。近年来,新型结构的CeO2光催化材料,如零维的量子点[34],一维的纳米棒[35]、纳米管[36],介孔结构[37],3D结构[38]等,吸引不少研究者的关注。Qi等可控合成了多层CeO2介孔材料,发现特殊的三层空心球结构有利于太阳光的充分利用,且大比表面积、高孔容、低密度的优点促进反应物分子的接触和连续碰撞,加强与活性组分的相互作用,因此相比于单层、双层空心球、纳米颗粒,CeO2三层介孔空心材料具有优异的光催化产氧性能[39]。Wang等通过前驱物MOF的晶体工程调控,得到活性位高分散的铈基材料,如图1所示,将Cu2+嵌入到Ce-MOF骨架中,以此为前驱物焙烧,促进Cu物种在CeO2中的高分散[40]。另外,关于CeO2的晶面效应在光催化反应中的作用机制也有一些报道[41-42]。Li等发现在光催化反应过程中,光生电子倾向于迁移到CeO2{111}晶面,而空穴倾向于迁移到CeO2{100}晶面[41];Zou等比较了CeO2 {110}{100}{111}晶面负载氮化碳的复合催化剂的光解水产氢性能,发现光催化性能与复合材料界面间的电子结构密切相关,CeO2 {110}与氮化碳间的电子转移作用最强,产生的内建电场有利于光生电子-空穴分离,因而具有最优的光催化反应性能[43]。
-
大气中的挥发性有机污染物(VOCs)主要来源于工业生产废气、交通运输中排放尾气以及装修房屋用到的涂料、胶合剂等[44]。在室温下VOCs易挥发,对环境和人体健康产生诸多危害。因此,VOCs的治理技术受到不少研究者的关注。近年来光催化氧化技术因为无二次污染、能耗低等优点,在去除低浓度空气污染物方面受到关注[45]。光催化氧化技术是指在光照下纳米半导体催化剂将室内空气中的挥发性有机污染物转化为无污染的H2O和CO2。其关键是高效的光生电子和空穴对分离的光催化反应,其中空穴的高氧化电位和在光生电荷存在下生成的反应活性中间体是光催化降解VOCs的主要驱动力。
稀土铈基材料由于具有较好的储-释氧能力和丰富的表面氧缺陷含量等优点,应用于甲醛、乙醛、苯、甲苯等VOCs污染物光催化消除反应中[46-53],其反应性能如表1所示。研究表明光催化消除VOCs反应性能与铈基催化剂表面的结构密切相关,特别是表面氧缺陷浓度。Eu掺杂CeO2催化剂,相比于纯CeO2,甲醛光催化降解性能显著提升,这主要由于Eu的掺入,提高了样品氧缺陷浓度,进而加快光生电子-空穴的分离;同时缺陷引入促进更多表面羟基的生成(O 1s XPS结果),样品与甲醛的接触角实验结果进一步说明了Eu掺杂CeO2催化剂有利于甲醛的吸附、活化(图2b)[46]。Muñoz-Batista等比较了CeO2-TiO2和TiO2光催化降解乙醛反应的光吸收效率和量子效率,发现催化反应性能依赖于催化剂的表面性质,在紫外光和可见光下,CeO2-TiO2都具有最优的反应活性和稳定性[49]。另外,有研究报道一些铈基材料还可以将光能转化为热能,进而提高光催化氧化性能。例如:CeMnxOy/TiO2材料可以吸收太阳光中全光谱(200—2400 nm),产生热能,提高催化体系的热量,达到光热协同高效催化消除苯污染物[50]。
对于光催化氧化VOCs多相反应,表面吸附与光催化反应间的关系值得研究。Liu等利用动力学手段,研究了Ce-TiO2光催化降解苯反应的决速步骤,他们认为光催化氧化苯的反应可以分为吸附和光催化两步反应,当吸附速率大于光催化反应速率时,光催化反应是决速步,反之则是吸附是决速步。他们比较苯分子在Ce-TiO2上吸附和光催化反应速率,发现光催化反应是决速步,且这个反应速率依赖于催化剂中Ce3+含量(图3c),反应机理如图2d所示[51]。Wu等利用叔丁醇作为捕获剂和ESR表征技术,测出在Mn-TiO2/CeO2光催化氧化甲苯反应中的活性氧物种,提出•OH氧化机制[52]。另外,研究发现光催化降解VOCs反应机制与光波长也有关系。当紫外光照射CeO2-TiO2复合材料时,光生电荷在TiO2表面激发,TiO2表面的光生空穴将甲苯氧化,而光生电子通过Ce3+/4+氧化还原电对转移到CeO2;当可见光照射时,光生电荷在CeO2表面激发,CeO2表面的光生空穴将甲苯氧化,而光生电子被CeO2中氧空位捕获[53]。
目前对于光催化降解VOCs的技术虽然得到显著地发展,但是该技术效率低、性能不稳定、反应会产生有毒中间产物等瓶颈问题仍然限制了该技术的进一步成功应用。后期研究将进一步关注功能性稀土铈基材料的可控合成:利用载体与活性组分强相互作用提高催化剂的稳定性;形成异质结和核壳结构从空间上促进光生电子-空穴分离,以及通过大比表面积有机材料,增大反应面积,提高其VOC降解能力。最终构建高效、绿色、环保的稀土铈基光催化氧化VOCs的反应体系。
-
氮氧化物(NOx)是造成大气污染的主要污染物之一,不仅能引起酸雨、雾霾、光化学烟雾、温室效应、臭氧层破坏等恶劣的环境现象,而且对人体以及动植物也会产生严重的毒害作用。随着社会经济的迅速发展,机动车辆的不断增加,人类向大气中排放的NOx越来越多。因此,大气治理刻不容缓,而解决问题的关键是控制氮氧化物的排放以及如何有效地消除NOx。如今,选择性NOx催化还原是应用最广泛的脱硝技术,主要其具有效率高、副产物少、操作简单等优点,但是存在能耗大、成本高等缺点,因此需要开发低温下高效的脱硝工艺和脱硝催化剂。
光催化消除氮氧化物因为能耗小、操作简单等优点受到不少研究者的关注。早在1984年,Courbon和Pichat就报道了TiO2在紫外光、常温下可以将少量的NO转化为N2和N2O [64]。Liu等研究了反应条件对光催化脱硝的影响,发现在8% O2,5% H2O的条件下,脱硝效率最高[65]。上述研究表明,利用光催化反应可以实现对少量氮氧化物有效消除。但是,在低温下,硝酸盐物种易形成、黏附于催化剂表面,导致催化剂中毒,削弱光催化反应活性。因此,如何防止副产物的生成是解决问题的关键。
目前,光催化消除少量NOx主要有两种反应机理:1)光催化氧化机理。NO先被光生空穴氧化成NO2,再进一步被羟基自由基、超氧自由基氧化成NO3-[66-67]。2)光催化还原机理。NO在催化剂表面氧空位吸附,进一步解离成N2和表面氧,表面氧释放出O2和电中性的氧空位,电中性的氧空位被光生空穴氧化成氧空位[68]。光催化NO还原成N2因转化率较低而研究较少,文献中主要研究的是光催化氧化NO成NO3-。稀土CeO2材料因丰富的氧空位、表面缺陷、可变的价态等优点,成为光催化氮氧化物消除反应的理想材料[54-56,69]。一方面,CeO2是碱性氧化物,可以有效地吸附、活化NO;另一方面,CeO2催化剂表面存在氧空位,可以通过掺杂、还原等方式增强表面氧空位浓度,进而增加可见光的吸收、减少光生电子-空穴的复合几率,提高光催化效率。Yu等合成了CoOx负载的N掺杂CeO2样品,用于光催化氧化NO反应。结果表明,N的掺入提高了催化剂对NO分子的吸附能力,同时Co与Ce物种间的电子作用有利于催化剂中氧空位的产生,增强O2的活化以及反应性能[69]。Cao等研究了Ce-TiO2光催化氧化NO的反应性能,发现Ce掺入到TiO2的晶胞中,改变其表面电荷密度(图3a—d),增加了激发电子数,进而有利于光催化消除NO反应效率的提高[54]。Li等发现在Au/CeO2−TiO2光催化界面处形成Au−Ce3+物种,可以锚定O2分子,进而与光生电子反应产生更多的活性自由基(图3e)[55]。另外,Ângelo等还研究了光照强度、NO停留时间、浓度、湿度这些反应因素对光催化氧化NO反应性能的影响规律。他们发现增大光照强度、延长停留时间有助于提高NO的转化率和选择性;低浓度NO有利于反应转化率的提升,但不利于选择性;适当的反应湿度可以产生更多的活性自由基,氧化NO,提高反应活性,但是过多的水汽会阻碍NO分子的吸附,不利于NO氧化[70]。因此,选择合适的反应条件对催化反应性能十分关键。
综上所述,目前光催化催化消除NO技术主要利用将NO氧化成硝酸根,此技术只适用于低浓度NO。但是,光催化消除NO方法存在转化率低、选择性差等缺点,大部分还处于实验阶段。如何有效地利用铈基催化剂提高光催化消除NO的性能以及相关的反应机制等科学问题仍需要深入研究。
-
随着工业化进程的不断推进,环境污染和能源枯竭等问题也日益突出。人类的社会活动加快了化石能源的消耗,同时化石能源的燃烧导致大气中温室气体排放量的增加,破坏自然界中碳循环平衡,引发全球气候变暖问题。积极应对能源危机和稳定控制大气中CO2总量,成为各国政府和科学家们的重大研究课题之一。利用光催化技术在太阳能驱动下将 CO2还原为甲醇、甲烷等清洁的碳氢燃料不仅能减缓全球气候变暖,更能实现工业废气的资源化利用,达到缓解石化能源危机的目的,最终实现碳的循环利用和开发清洁能源。
CO2是一种相对稳定的化合物,虽然光催化还原CO2的研究起步较早,但是太阳能转换效率最高只有1‰量级,转化效率较低。因此,光催化还原CO2极具挑战性。其中,将CO2分子还原形成•CO2-自由基的活化过程很难进行,导致光催化还原CO2反应效率至今未达到实际工业应用的要求。基于此,研发高效的绿色催化剂是实现温和条件下CO2再循环利用技术的关键。
研究表明,稀土的存在可以调节催化剂表面酸碱性。其中,CeO2材料作为稀土催化材料中最重要组成,凭借独特的4f电子、丰富的表面氧缺陷、良好的Ce3+/Ce4+切换能力等特点,在能源化工、环境保护和精细化学品生产等新兴领域展现出良好的催化性能和应用前景[57-60]。CeO2具有较强碱性可吸附活化CO2酸性分子,同时表面Ce3+/Ce4+氧化还原电对可以有效地分离光生电子和空穴。Ye等首次可控合成单金属铈层状双氢氧化物(MCe-LDHs),通过调变Ce3+/Ce4+浓度比,控制反应物CO2分子吸附活化能力[57]。Ce3+/Ce4+氧化还原电对可以改变Ce掺杂的TiO2表面电荷密度,进而影响CO2分子吸附活化位点[58]。另外,稀土铈基材料表面吸附的氧物种在光催化还原CO2反应中起到重要作用。研究表明,CeO2表面上的氧空位有助于羟基物种吸附,羟基物种可以提供电子,作为路易斯碱位与CO2分子中C空轨道作用,从而降低CO2吸附能(图4a)[60]。Wang等的研究也进一步证明,Cr掺入介孔CeO2光催化剂中,可以提高表面活性氧物种的含量,最终提升光催化还原CO2的反应性能[29]。
相比于其它传统无机材料,CeO2一个突出特征在于其具有较低的氧空位形成能,如CeO2(101)表面的氧空位形成能在2.0 eV左右,要远低于TiO2(101)表面的氧空位形成能(4.8 eV)。研究表明,表面少量缺陷相比于其体相结构和化学组成具有更重要的作用,微量缺陷的产生往往伴随着化学键的断裂与重组、晶格畸变、电子局域化等一系列的变化,进而对材料的物理化学性能甚至催化性能产生非常重要的影响。CeO2表面的氧空位可以作为路易斯酸性位,接受CO2分子中O原子的p电子,进而促进CO2分子的吸附和活化[61-63,71-73]。Wang等利用草酸和N2预处理CeO2催化剂,发现表面氧空位浓度增加,原位红外技术表明产生的氧空位可以有助于CO2分子还原成CO,但其表面强烈吸附的碳酸盐物种反而会影响催化剂的性能[61]。Rhatigan等利用理论计算也进一步证明确实存在氧空位的催化剂有助于CO2分子吸附活化和H2O分子的解离[71]。另外,Pu等提出了CeO2表面的氧空位作为路易斯酸位和其负载的Cu2O协同促进CO2分子吸附、活化、还原的反应机制,如图4b所示。类似于氧化还原机制(Mars van Krevelen),即Cu2O表面铜原子的d电子与CO2分子中的空轨道作用,同时CO2中氧原子与CeO2表面的氧空位作用,形成羧基中间物种,进一步光电子还原成CO[63]。
随着以CO2为主的温室气体排放量不断增加,寻求新型能源来构建低碳型社会的诉求越来越迫切。通过催化剂将大气中的温室气体CO2光催化还原方法是一项极具挑战性的前沿科技。其目标是合成有效的光催化材料来驱动氧化还原反应,实现其转换效率和选择性超过自然界光合作用。但是目前反应效率不高以及选择性差限制了此技术实际应用。后期可以从以下方面进行改性:(1)选择合适的半导体催剂,提高产物的选择性和产率。(2)选择合适的还原剂。目前水是光催化还原CO2反应中常见的还原剂,但是CO2在水中溶解度不高,且存在产物H2和H2O2竞争反应。因此可以选择一些有机溶剂代替水。(3)设计合适的光催化反应器,包括反应入射光强度、波长、温度、压力等。最终实现光催化还原CO2技术的大规模商业化应用。
-
稀土二氧化铈因其特殊的4f电子结构,具有近紫外-可见光的响应,同时,稳定的晶体结构、优异的氧流动性、较好的光学性质等优点,使其成为最具潜力的光催化材料之一,在光催化消除VOCs、NOx、温室气体CO2等环境领域都具有潜在的应用前景。但是目前对铈基材料的研究还处于实验阶段,主要关注其结构性质、结构与性能间的关系、以及反应机理。后期应重点开发高效铈基光催化材料,提高催化剂的量子效率和催化性能,进而推动稀土铈基光催化剂的工业化应用。
稀土铈基纳米材料在光催化消除环境污染物中的研究进展
Research progress on ceria-based nanomaterials for photocatalytic elimination of environmental pollutants
-
摘要: 当前环境污染问题已经成为影响社会可持续发展的重大问题之一,而光催化技术的迅猛发展为上述问题提供了技术支撑。稀土二氧化铈(CeO2)由于具有稳定的晶体结构、优异的氧流动性、较好的光学性质等优点,近年来在大气污染治理领域受到广泛关注。本综述总结了近年国内外研究者在稀土铈基纳米材料光催化消除环境污染物领域中所取得的主要研究进展,重点讨论了高性能铈基催化剂的设计理念,以及在光催化消除挥发性有机污染物、氮氧化物、温室气体CO2等领域中的基础应用研究,并对稀土铈基纳米材料光催化消除环境污染物的未来发展方向进行了展望。Abstract: Concerns related to air pollution and greenhouse gases emission have created great challenges for sustainable development of the society. Major attention has been given for the advance of photocatalysis to overcome these environmental crises. Recently, cerium dioxide (CeO2) received extensive attention as a promising photocatalyst, owning to its stable crystal structure, excellent oxygen mobility and good optical properties for photocatalytic elimination of environmental pollutants. This work provides a comprehensive review on recent research progress made by domestic and foreign researchers in terms of ceria-based nanomaterials. The main topics include the design of ceria-based catalysts with high-performance and their applications on the photocatalytic elimination of volatile organic compounds (VOCs), nitrogen oxides (NOx), greenhouse gas as CO2, etc. Furthermore, a perspective of future work on the development of ceria-based nanomaterials and their photocatalytic applications on pollutant removal is also presented at the end.
-
湖泊是淡水资源的重要载体,对区域生态环境的维护发挥着重要的作用[1]。随着中国经济的快速发展以及城镇面积的大幅扩张,国内生产与生活用水需求日益增长,由此造成的湖泊环境与生态破坏现象不胜枚举,湖泊水华暴发、水体缺氧等问题频频发生,并由此引发了一系列经济与社会问题[2]。洱海亦出现了水体富营养化的现象[1],水生生态系统逐步退化、湖泊水体富营养化进程加快等问题日益突出[3],同时针对洱海的相关研究[4]发现,其水质呈现变坏的趋势,暗藏蓝藻水华暴发的隐患。
蓝藻水华通常是指在富营养化水体中出现蓝藻大量繁殖的现象,主要表现为水体表面覆盖着一层蓝绿色并伴有恶臭气味的浮沫,水体藻细胞浓度一般都达到并超过1.5×107个·L−1,叶绿素a质量浓度大于10 mg·m−3[5]。叶绿素a(chlorophyll a, Chl-a)是浮游植物(藻类)中最常见的色素,其质量浓度是评价水体富营养化程度的核心参数[6]。传统的湖泊营养状态评价方法主要依赖于对水体的实地取样分析,然而该方法易受局部天气与环境影响,取样与测试过程也比较耗时耗力、成本较高,较难实现对湖泊富营养状态在精细时空尺度上的监测[7]。与之相比,遥感(remote sensing, RS)技术具有覆盖范围广、获取资料快、周期短等优点,可弥补传统水质采样的诸多不足,因此,已被广泛应用于湖泊水环境和水生态等方面的监测。目前,在利用遥感技术的水质参数反演方面,反演方法经历了分析法、经验法、半经验法、机器学习和综合法[8],已建立起具有较高精度和一定普适性的水质参数反演模型,可以用于宏观的水质评价[9-10]。并且形成了遥感反演叶绿素a质量浓度的多种算法,但不同的算法也存在一定的局限性[11],且不同的算法在不同的传感器之间的适应性也存在差异[12]。
已有许多针对不同地区、不同季节、不同水质参数、不同的反演方法和算法、不同的卫星遥感数据源的水质反演方面的研究。潘鑫等[13]利用高分六号卫星影像,采用3种模型对太湖进行叶绿素a质量浓度反演,得出了适合高分六号卫星影像太湖叶绿素a质量浓度反演的模型。郑震[14]基于OLI影像,建立了叶绿素a质量浓度反演的数学回归模型,分析了东张水库叶绿素a质量浓度的时空分布特点。陈命男[15]利用Landsat 8数据,建立了淀山湖的叶绿素a反演的回归模型。但雨生等[16]基于Sentinel-2数据建立了可靠的BP神经网络模型,用以监测平寨水库水质。马丰魁等[17]以密云水库为研究对象,采用BP神经网络算法反演4个水质参数,并且得到了较为可信的研究结果。徐鹏飞等[18]建立了神经网络模型,对千岛湖清洁水体的叶绿素a质量浓度进行反演,并利用该模型对千岛湖的叶绿素a质量浓度进行时空特征分析。
由此可见,已有许多利用遥感数据反演水质参数的研究,分析方法亦较为成熟,这些研究为不同地区湖泊的水质监测提供了可靠的参考依据。但内陆水体光学特征具有较强的区域性和季节性[8],而且针对叶绿素a反演的各种算法仍受到季节和地理位置等的限制[11],致使各地区建立的水质参数反演模型不具有普适性。为此,针对不同地区、不同季节及不同的传感器[11],仍需要根据实际情况有针对性地建立适合当地的相关模型,为水污染防治提供合理的数据支撑[19]。近年来,利用遥感技术监测洱海水质情况的研究主要包括蓝藻水华的空间分布特征[20]、土地利用变化与水质的关系[21]、干季水质的时空变化[22]等。毕顺等[23]利用OLCI数据,采用了三波段模型对洱海2017年4月19日叶绿素a质量浓度的分布进行了估算。但该研究的三波段模型中第三波段的选取需要满足一系列的假设条件,且三波段模型主要适用于中高浓度叶绿素水体,不适用于高度浑浊水体[11];还有研究[24]表明,OLCI数据虽具有较高辐射分辨率,但其空间分辨率(为300 m)较低,在中小型内陆水体的监测上能力有限。由于洱海属中型湖泊,因此,选用空间分辨率较高的多光谱遥感影像能较为准确地获取水质采样点的反射率数据,这也是提高模型水质反演精度较为主要的因素之一[25]。
鉴于以上所述,本研究选取空间分辨率较高、也是近些年最流行的多光谱遥感数据之一的Sentinel-2数据,以较少利用Sentinel-2数据反演叶绿素a质量浓度的洱海作为研究区域,建立2种叶绿素a质量浓度反演模型,反演洱海叶绿素a质量浓度的空间分布,旨在利用不同数据源和方法探索适用于洱海流域的叶绿素a质量浓度反演模型,为相关部门的水质监测和水污染防治提供参考。
1. 研究区域简介和数据获取
1.1 研究区域
洱海位于大理市,湖泊面积约256 km2,是云南省第二大高原淡水湖泊,也是大理市和周边乡村居民的生产生活用水供给源地。其水量补给主要为大气降水和入湖径流,周边主要入湖河流有29条。洱海区域属高原季风气候类型,四季温和、平均温度较小,日照差大,光照充足,干湿季节分明,雨季季节分配不均[1]。作为受人类活动干扰严重的中型湖泊,2010—2019年期间洱海综合营养状态指数为38.8 ~ 43.1,属中营养水平;2014—2019年,整体水质类别为Ⅱ ~ Ⅲ类,水质呈现变坏趋势,污染物主要来源于降水产生的地表径流所携带的禽畜养殖、农村生活和农田污染[3-4]。
1.2 研究数据来源
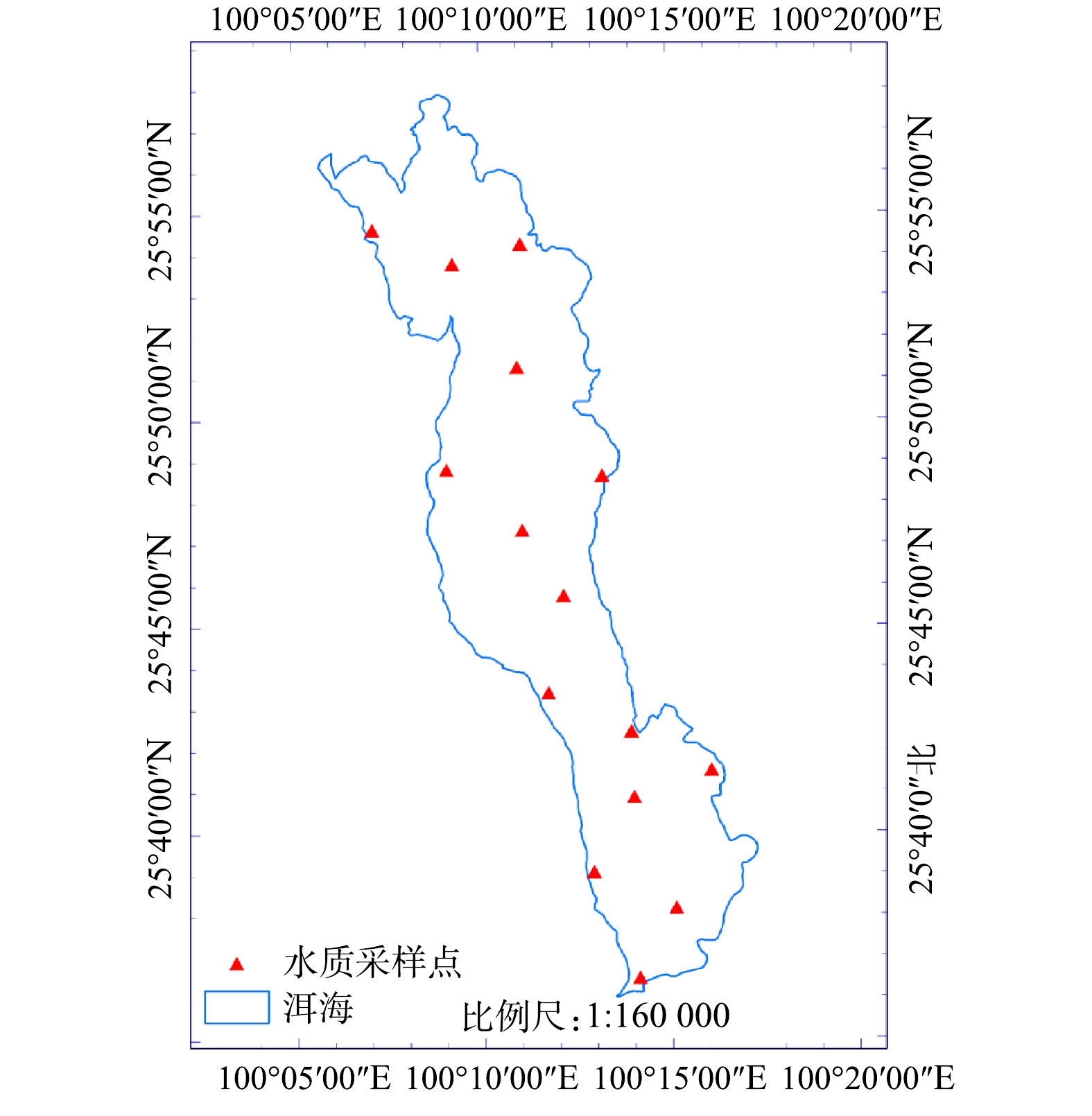
1)叶绿素a质量浓度数据。叶绿素a质量浓度实测数据来源于云南省水环境监测中心大理州分中心,水质监测点共计15个(图1),采用2019年10月9日、2019年11月8日2期监测数据,共计30条,即每月监测1次,每次采集15个水质样品,水质采样在当天完成。
2) Sentinel-2卫星数据及预处理。此次研究采用的是Sentinel-2数据,卫星数据从欧洲空间局(European Space Agency, ESA)网站下载,数据信息见表1。Sentinel-2是欧洲空间局哥白尼计划下的一个地球观测任务,该计划是由2颗相同的卫星Sentinel-2A与Sentinel-2B组成的卫星群,单颗卫星重返周期为10 d,Sentinel-2的卫星2颗互补,重返周期为5 d。Sentinel-2的每颗卫星都搭载相同的多光谱影像仪(multispectral instrument, MSI)。该影像仪可拍摄涵盖可见光、近红外与短波红外的13个波段影像。MSI的拍摄方式是推扫式,影像幅宽达到290 km。通过Sentinel-2获取的各波段信息如表2所示。
表 1 Sentinel-2数据信息Table 1. Sentinel-2 data information序号 日期 卫星 产品级别 1 2019年11月9日 Sentinel-2A Level-2A 2 2019年10月12日 Sentinel-2B Level-2A | Show Table DownLoad:
CSV
表 2 Sentinel-2传感器波段信息Table 2. Spectral bands for the Sentinel-2 sensors
DownLoad:
CSV
表 2 Sentinel-2传感器波段信息Table 2. Spectral bands for the Sentinel-2 sensors波段号 波段 Sentinel-2A Sentinel-2B 空间分辨率/m 中心波长/nm 波宽/nm 中心波长/nm 波宽/nm B1 沿海气溶胶 442.7 21 442.3 21 60 B2 蓝 492.4 66 492.1 66 10 B3 绿 559.8 36 559.0 36 10 B4 红 664.6 31 665.0 31 10 B5 植被红边 704.1 15 703.8 16 20 B6 植被红边 740.5 15 739.1 15 20 B7 植被红边 782.8 20 779.7 20 20 B8 近红外 832.8 106 833.0 106 10 B8a 窄波近红外 864.7 21 864.0 22 20 B9 水蒸气 945.1 20 943.2 21 60 B10 短波红外-卷云 1 373.5 31 1 376.9 30 60 B11 短波红外 1 613.7 91 1 610.4 94 20 B12 短波红外 2 202.4 175 2 185.7 185 20 | Show TableDownLoad:
CSV
在光学数据中,Sentinel-2卫星在红边范围含有3个波段的数据,为快捷反演大区域叶面积指数、叶绿素质量浓度等生物物理量指标提供了可能[26]。本次研究直接获取Sentinel-2卫星的L2A级数据。L2A级数据在L1C级数据的基础上,利用ESA提供的Sen2cor模型进行处理生成。从2018年3月开始,ESA逐渐向全球用户提供L2A级产品,并于2018年12月覆盖到全球。L2A级数据在生成过程中,会对L1C级数据进行大气校正、云雪检测、地形校正、场景分类等一系列处理[27],可直接用于下游产品。
为便于使用ENVI 5.3提取各采样点反射率,利用SNAP软件将2期影像都按照同样的方法进行重采样,将其重采样为10 m,之后,波段由13减至12个(减少了短波红外-卷云B10波段)。分别提取水质采样点2期影像的水体反射率。
2. 叶绿素a质量浓度反演模型的建立与验证
2.1 反演波段的选择
在水质参数的反演工作中,获取与水质参数关系密切的敏感波段,将其作为模型的输入因子,这样建立的模型具有更高的预测精度[28]。本研究通过Pearson相关分析来获取与叶绿素a质量浓度关系密切的敏感波段,Pearson相关系数是一种线性相关系数,通过Pearson相关分析可以得出不同波段或者波段组合与叶绿素a质量浓度的相关性强弱,可进一步剔除弱相关的、可能干扰反演模型建立的波段信息。由于水体的反射特性主要位于可见光和近红外波段,且有研究[29]表明,可见光和近红外的反射率可以成功地用以反演水体的叶绿素a质量浓度,因此,首先选择4个可见光波段和4个红边波段进行相关性分析,然后利用置信度为99.9%(P < 0.001)的波段进行波段组合,进一步分析各波段组合的反射率与叶绿素a质量浓度的相关性。
目前,常用的波段组合方式有单波段比值、双波段比值、波段差值、三波段和四波段模型。对于Sentinel-2数据来说,双波段比值对叶绿素质量浓度更加敏感[30],因此,双波段比值是本研究首选的波段组合方式之一,另外还选择了单波段比值。根据但雨生等[16]基于Sentinel-2卫星数据与平寨水库叶绿素a质量浓度的相关分析研究结果,叶绿素a质量浓度与组合波段B5/B4、(1/B4-1/B5)×B6、(1/B4-1/B5)×B7和(1/B4-1/B5)×B8的Pearson相关系数最大。另外,叶绿素质量浓度与归一化植被指数(normalized difference vegetation index, NDVI)呈线性关系[31],所以本研究选择具有代表性的波段比值、三波段模型以及NDVI筛选与叶绿素a质量浓度显著相关的敏感波段。利用15个采样点提取的2期共30条水体反射率数据,与实测叶绿素a质量浓度数据进行Pearson相关分析,分别在单波段、单波段比值和双波段比值中选取相关系数最大的叶绿素a反演波段。如表3所示,B6、B7/B6和(B6+B8)/(B7+B8a)反射率与叶绿素a质量浓度呈现0.001水平的显著性,且相关系数最大。因此,最终选择B6、B7/B6和(B6+B8)/(B7+B8a)作为叶绿素a质量浓度的反演波段。
表 3 各波段/波段组合反射率与叶绿素a质量浓度的相关性Table 3. Correlation between reflectance of each band/band combination and chlorophyll-a mass concentration波段/波段组合 Pearson相关系数 P 样本数 波段/波段组合 Pearson相关系数 P 样本数 B2 -0.148 0.435 30 B8/B6 0.297 0.110 30 B3 -0.099 0.602 30 B8/B7 0.470 0.009 30 B4 0.233 0.216 30 B7/B6 -0.713 < 0.001 30 B5 0.186 0.324 30 (1/B4-1/B5)×B6 -0.128 0.501 30 B6 0.811 < 0.001 30 (1/B4-1/B5)×B7 -0.086 0.653 30 B7 0.724 < 0.001 30 (1/B4-1/B5)×B8 -0.098 0.606 30 B8 0.788 < 0.001 30 (1/B4-1/B5)×B8a -0.070 0.713 30 B8a 0.694 < 0.001 30 (B6+B7)/(B8+B8a) -0.382 0.037 30 B5/B4 -0.009 0.963 30 (B6+B8)/(B7+B8a) 0.821 < 0.001 30 B8a/B6 0.081 0.669 30 (B8-B6)/(B8+B6) 0.363 0.048 30 B8a/B7 0.264 0.158 30 (B8-B7)/(B8+B7) 0.477 0.008 30 B8a/B8 -0.621 < 0.001 30 (B8-B4)/(B8+B4) 0.489 0.006 30 | Show TableDownLoad:
CSV
需要说明的是,在进行Pearson相关分析之前,需要对各波段、波段组合提取的采样点反射率数据以及实测叶绿素a质量浓度数据进行Shapiro-Wilk检验。在25组数据中(波段或波段组合的采样点反射率数据24组(表3);实测叶绿素a质量浓度数据1组),6组数据符合正态分布,其余数据基本符合正态分布。
2.2 BP神经网络模型的构建
BP (back propagation)神经网络是一种按误差逆向传播算法训练的多层前馈网络,能学习和存储大量的“输入-输出”模式的映射关系,而无需事前揭示描述这种映射关系的数学方程[32]。BUCKTON等[33]和SCHILLER等[34]利用MERIS数据和2层BP神经网络模型反演了叶绿素、悬浮物、黄色物质3个水质参数,得出神经网络模型完全可以用来反演Ⅰ类水质和Ⅱ类水质参数,且反演精度远高于经验模型的结论。BP神经网络具有自适应、自学习、自组织和非线性映射的功能,非常适用于模拟各种错综复杂的非线性关系。已有研究[16]证明,具有1个隐含层的3层网络可以逼近任意非线性函数。本研究所采用的即是具有1个输入层、1个隐含层和1个输出层的3层BP神经网络结构。其中,隐含层的神经元节点数可根据3种经验公式[32]选择,本研究由经验公式(式(1))进行确定。
M=√m+n+a (1) 式中:M为隐含层神经元个数;m和n分别为输入层和输出层神经元个数;a为1 ~ 10的整数。
根据输入输出层神经元个数3和1,由式(1)计算得到M的取值为3 ~ 12的整数。分别从单波段、单波段比值、双波段比值中选取与叶绿素a质量浓度显著相关且相关系数最大的波段和波段组合B6、B7/B6和(B6+B8)/(B7+B8a)作为神经网络的输入数据(波段组合使用软件PIE-Basic 6.3完成),与之相对应的实测叶绿素a质量浓度作为输出数据。30组数据随机分为训练数据23组和检验数据7组,采用feedforwardnet函数建立神经网络。trainglm作为训练函数,双曲正切函数sigmoid为传递函数,线性函数purelin作为输出层函数。训练次数设置为1 000次,学习速率为0.01,训练目标为1×10−6。设置参数后,分别选择不同的神经元节点数对神经网络进行反复训练,最终获得决定系数R2最大、均方根误差(root mean square error, RMSE)最小的神经网络结构,此时隐层神经元节点数为4 (表4)。
表 4 隐含层不同神经元个数的R2和RMSETable 4. R2 and RMSE of different numbers of neurons in the hidden layer神经元个数 决定系数 均方根误差 3 0.770 0.004 5 4 0.925 0.002 8 5 0.564 0.005 1 6 0.335 0.004 7 7 0.587 0.006 1 8 0.361 0.004 9 9 0.791 0.007 4 10 0.851 0.004 5 11 0.661 0.007 1 12 0.418 0.007 9 | Show TableDownLoad:
CSV
2.3 模型精度的检验
根据构建BP神经网络时随机产生的训练样本23组,也就是利用与BP神经网络相同的特征波段B6、B7/B6和(B6+B8)/(B7+B8a)及其数据,建立多元线性回归模型(式(2))。
C=0.0310x1+0.0016x2+0.0334x3−0.0302 (2) 式中:C为Chl-a质量浓度,mg·L−1;x1代表B6波段;x2代表单波段比值B7/B6;x3代表双波段比值(B6+B8)/(B7+B8a)。
采用决定系数R2、均方根误差(RMSE)、平均绝对误差百分比(mean absolute percentage error, MAPE)3个指标评价模型效果。MAPE的计算方法见式(3),7条检验数据对2种模型的检验结果如表5所示。
表 5 检验数据对2种模型误差检验结果Table 5. Error testing of two models using test data样本编号 实测值/(mg·L−1) 线性回归模型估测值 BP神经网络估测值 线性回归模型相对误差/% BP神经网络相对误差/% 1 0.014 0 0.007 0 0.013 1 49.82 6.36 2 0.006 5 0.008 3 0.011 3 27.90 74.01 3 0.015 6 0.008 6 0.013 8 44.72 11.66 4 0.017 0 0.012 0 0.014 7 29.33 13.73 5 0.021 8 0.019 0 0.017 6 12.69 19.48 6 0.011 0 0.009 2 0.012 6 16.63 14.81 7 0.015 4 0.013 2 0.013 9 14.20 9.49 注:线性回归模型的平均绝对误差百分比(MAPE)为27.90%,均方根误差(RMSE)为0.004 5,决定系数(R2)为0.788;BP神经网络的平均绝对误差百分比(MAPE)为21.36%,均方根误差(RMSE)为0.002 8,决定系数(R2)为0.925。 | Show TableDownLoad:
CSV
δ=1m∑mi=1 |yi−ˆyiyi|×100% (3) 式中:δ为所有检验样本的相对误差绝对值的平均值;m为检验样本总数;yi为第i个检验样本的叶绿素a质量浓度的实测值;
ˆyi 3. 结果与讨论
3.1 建模数据的时间匹配
从本研究的叶绿素a质量浓度数据的采集时间和Sentinel-2卫星影像的时间来看,2种数据没有在时间上做到非常好的匹配。10月9日,叶绿素a质量浓度实测数据与10月12日Sentinel-2卫星数据间隔3 d;11月8日,叶绿素a质量浓度实测数据与11月9日Sentinel-2卫星数据间隔1 d。根据来源于美国国家海洋和大气管理局国家环境信息中心(NOAA National Centers for Environmental Information)的气象站点(该气象站点位于洱海湖体西南部,大理市气象局东南部)数据,研究区10月9日—10月10日无降水,10月11日降水量为0.508 mm,10月12日降水量为0.254 mm,10月9日—10月12日4 d内最大平均风速为2.16 m·s−1,日平均温度变化最大为1.2 ℃;11月8日—11月9日无降水量,最大平均风速为1.59 m·s−1,日平均温度升高2.3 ℃。此外,来源于美国国家航空航天局(National Aeronautics and Space Administration, NASA)提供的大气再分析资料MERRA-2数据显示,10月9日—10月12日,大理市地区日照时数的最大值和最小值分别为5.92 h和4.85 h,变化值最大为1.07 h;11月8日—11月9日,日照时数减少0.10 h。因此,时间上的差异对数据产生的影响很小。此外,鉴于本研究存在的数据在时间匹配上的误差,且叶绿素a质量浓度变化受风速、降水、流速、营养元素等因子影响[35],可建立包含风速、降水、流速、营养元素等因子的模型,以减少因为数据不能在时间上完全匹配所产生的叶绿素a质量浓度的估算误差,这可以在一定程度上避免因卫星数据时间分辨率以及卫星数据云量过多造成的与实测数据的时间匹配问题。
3.2 相关分析结果
数据的Shapiro-Wilk检验结果显示,B9、B5/B4、B7/B6、B8a/B8、(1/B4-1/B5)×B6、(1/B4-1/B5)×B7没有呈现显著性(p > 0.05),说明以上6组数据符合正态分布。其余19组数据的峰度绝对值小于10并且偏度绝对值小于3,这些数据的正态检验直方图也都显示为钟形(中间高两端低),说明这些数据基本符合正态分布。根据本研究的实测叶绿素a质量浓度数据与对应水质样品采样点的Sentinel-2影像反射率的相关分析结果,建立模型所采用的对叶绿素a质量浓度敏感的波段都处于0.001的显著水平,分别是在单波段、单波段比值、双波段比值中相关系数最大的波段和波段组合B6、B7/B6和(B6+B8)/(B7+B8a)。根据表3,B5/B4、(1/B4-1/B5)×B6、(1/B4-1/B5)×B7和(1/B4-1/B5)×B8与叶绿素a质量浓度的相关系数很小,分别为−0.009、−0.128、−0.086、−0.098。这说明以上波段组合与叶绿素a质量浓度存在弱相关关系或几乎没有相关关系,与但雨生等[16]的研究存在差异。在但雨生等[16]的研究中,采用与叶绿素a实测数据准同步的Sentinel-2影像,在秋季,采用了同样的相关分析方法,因此,出现这种差异可能由不同研究区的水体光学特征的差别所引起,也就是说可能由于不同研究区水体的固有光学特征和表观光学特征的差异,导致同一卫星传感器的所接收的水体反射信息存在差别。此外,NDVI与叶绿素a质量浓度的相关系数为0.489,P值为0.006,说明NDVI与叶绿素a质量浓度存在0.01水平的显著性。
3.3 BP神经网络与多元线性回归模型
本研究选用多元线性回归模型以及可以模拟各种非线性关系的BP神经网络模型来进行湖泊水叶绿素a质量浓度的反演。从对2种模型的检验结果来看,BP神经网络模型的MAPE为21.36%,小于线性回归模型27.90%;RMSE为0.002 8,小于线性回归模型的0.004 5。从2种模型的决定系数R2来看,本研究的多元线性回归模型的3个自变量(B6、B7/B6和(B6+B8)/(B7+B8a))只能用来解释因变量(叶绿素a质量浓度)方差的78.8%,远低于BP神经网络模型(R2 = 0.925)。这说明BP神经网络模型的预测精度要高于线性回归模型。这与赵玉芹等[36]基于SPOT 5遥感影像建立了多元线性回归模型、BP神经网络模型和径向基神经网络模型,对渭河陕西段水域的4种水质参数进行反演所得出的结论几乎一致,均认为BP神经网络模型的预测精度最高。同样,岳佳佳等[37]以黄石磁湖为研究区,利用IKONOS遥感影像和水质采样数据构建了多元线性回归模型、BP神经网络模型和径向基神经网络模型,对比后,亦得出神经网络模型反演结果优于多元线性回归模型。此外,杨柳等[38]也使用ETM+数据和准同步实测的2种水质数据,建立多个隐含层为1的BP神经网络模型,得出了BP神经网络方法反演水质参数结果好于线性回归方法的结论。
在水质参数反演中,BP神经网络虽然较多元线性回归模型有较高的精度,但是也存在一些缺点。BP神经网络的设计通常需要确定网络的层数、输入层的节点数、隐含层的节点数、输出层神经元个数、网络的初始权值,需要选择不同的传递函数和训练方法。本研究使用feedforwardnet函数建立神经网络,该函数自动对数据进行归一化处理,可以根据train函数自动确定网络的输入层和输出层数。如图2所示,本研究的BP神经网络输入层数为3,分别为Sentinel-2卫星数据的B6、B7/B6和(B6+B8)/(B7+B8a)3个特征数据。将这3个特征数据按照不同的权值wij分别输入到隐含层各个神经元,再和各神经元阈值bj求和,之后激活传递函数(双曲正切函数sigmoid),接着通过一定的权值wjk由隐层进入输出层,与输出层阈值bk求和后激活输出函数(线性函数purelin),随后按减少误差的原则,从输出层经过隐含层,回到输入层,不断调整各连接权值,进行反复训练。虽然这样可以不断地提高正确率,但BP神经网络的一些参数的选择依然没有依据,隐含层的神经元个数也只能依据经验公式进行试凑,网络的权值是通过训练样本和学习率参数得到的,这也导致了BP神经网络的不稳定。除此之外,BP神经网络每次训练的初始权值是随机给定的,导致了其不可重现性;网络还存在样本依赖性,主要依赖于选择的训练样本是否典型,所以,应避免样本集合代表性差、矛盾样本多、存在冗余样本等问题;再者,网络容易陷入局部最优,需要实时检测误差率的变化以确定最佳训练次数。相比之下,径向基网络模型具有结构可靠、不依赖初始权值等优点。吴倩等[39]认为径向基网络模型是值得推广的光谱反演叶绿素模型。同时需要注意的是,吴倩等[39]的研究采用地物光谱仪测定采样点水体反射光谱,波谱区间为325 ~ 1 075 nm,光谱采样间隔为1.6 nm,光谱分辨率为3.5 nm,光谱分辨率较高,且径向基网络模型同时也存在丢失信息、数据不充分时无法工作等缺点。本研究旨在探索适用于秋季洱海流域的叶绿素a质量浓度的遥感反演模型,采用的是Sentinel-2多光谱数据,波段配置不同也会影响叶绿素a的反演[11],因此,依然使用了在各个地区利用遥感反演水质参数比较成熟的BP神经网络模型。
3.4 叶绿素a质量浓度的空间分布
如表5所示,BP神经网络的模型预测误差要比多元线性回归模型小,采用构建好的BP神经网络模型对洱海秋季2019年10月12日、11月9日2 d的叶绿素a质量浓度进行反演,结果见图3和图4。由反演结果可以看出,利用Sentinel-2影像可从宏观上反演叶绿素a质量浓度,并且可以非常直观地展现叶绿素a质量浓度的空间分布,这也是遥感监测的优势体现。利用这一优势,结合地理信息系统(geographic information system, GIS)以及人工智能(artificial intelligence, AI)技术,构建蓝藻水华及富营养化监测预警系统是未来的研究方向之一[40]。如以10 mg·m−3叶绿素a质量浓度作为藻华发生的临界值[41],利用以上技术结合适用于当地的BP神经网络模型,可以构建藻华发生的预警系统,这也是本研究的实际意义之一。
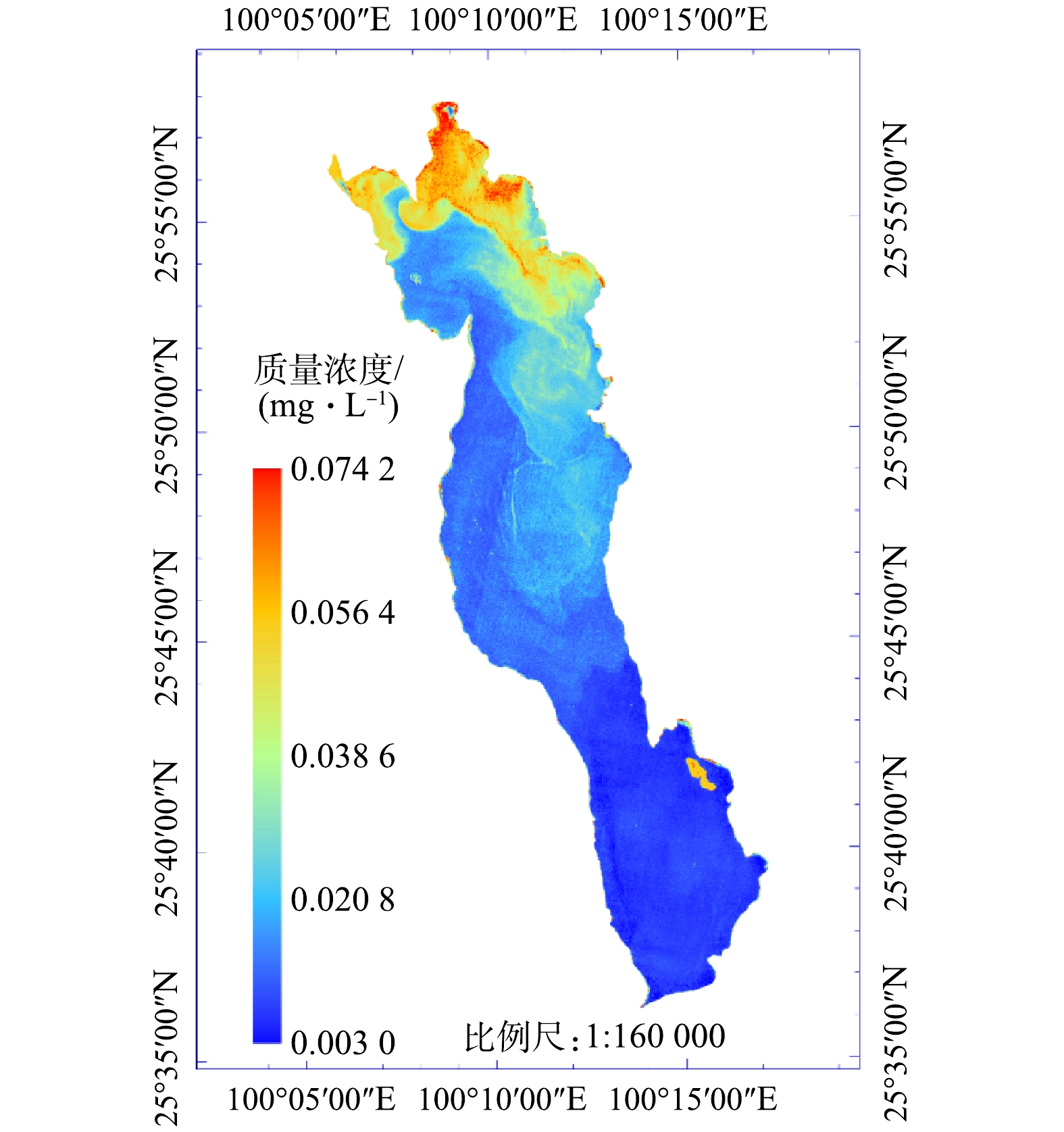 图 3 洱海 2019 年 10 月 12 日叶绿素 a 质量浓度分布图Figure 3. Distribution of chlorophyll a mass concentration in Erhai lake on October 12th, 2019
图 3 洱海 2019 年 10 月 12 日叶绿素 a 质量浓度分布图Figure 3. Distribution of chlorophyll a mass concentration in Erhai lake on October 12th, 2019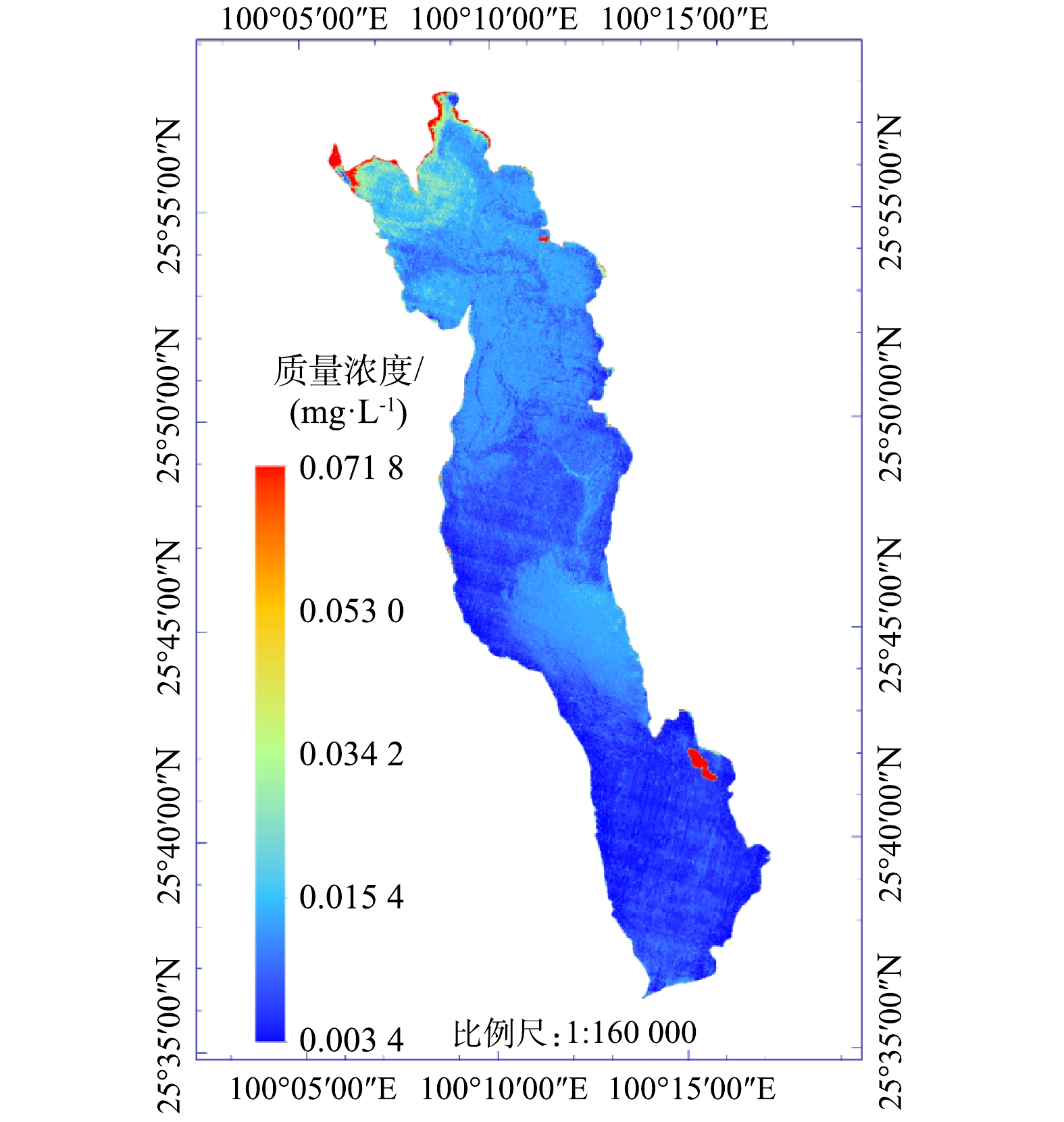 图 4 洱海 2019 年 11 月 9 日叶绿素 a 质量浓度分布图Figure 4. Distribution of chlorophyll a mass concentration in Erhai lake on November 9th, 2019
图 4 洱海 2019 年 11 月 9 日叶绿素 a 质量浓度分布图Figure 4. Distribution of chlorophyll a mass concentration in Erhai lake on November 9th, 2019由洱海2 d的反演结果可以看出,叶绿素a质量浓度均呈现洱海北部高于南部的分布状态,且洱海北部沿岸地区叶绿素a质量浓度值最大。10—11月,叶绿素a质量浓度出现扩散,北部较高质量浓度逐渐向洱海中部扩散,叶绿素a质量浓度最大值降低,分布面积扩大。从叶绿素a质量浓度来看,10月最大值为0.074 2 mg·L−1,最小值为0.003 0 mg·L−1;11月最大值为0.071 8 mg·L−1,最小值为0.003 4 mg·L−1。10月与11月相比较而言,洱海叶绿素a质量浓度区间基本没有发生变化。根据11月的反演结果,洱海叶绿素a质量浓度仍然呈北部大于南部的趋势。这与祁兰兰等[22]利用GF-1卫星数据的研究结果(2014—2019年11月,洱海叶绿素a质量浓度在空间上均呈现南部>中部>北部)相反。
为了进一步探寻造成此结果的原因,首先想到的是将叶绿素a质量浓度与水体富营养化联系起来。这是因为叶绿素a质量浓度是藻类生物量的指标,可以很容易的与水体富营养化联系,而营养化水平与叶绿素a质量浓度又是成正相关的[42],因此,将2019年11月9日Sentinel-2A的真彩色合成影像通过3%的线性拉伸显示(图5),通过目视解译,可以看出洱海北部的水华现象。其次,由于本研究使用的是Sentinel-2数据,而通过查询,没有与祁兰兰等[22]研究中相同日期(2019年11月22日)的Sentinel-2数据,未能与其做对比实验。相应地,在11月9日,也没有查询到高分一号WFV洱海区域的数据,也不能利用祁兰兰等[22]采用的反演模型对本研究结果做对比验证。但是,WANG等[41]的研究显示,洱海叶绿素a质量浓度在2016年11月、2017年11月均为北部高于南部;TAN等[42]的研究同样表明2016年11月洱海北部叶绿素a质量浓度高于南部。这种结果的差异可能是祁兰兰等[22]研究所采用模型的区域性限制,该模型最早是利用环境一号卫星CCD数据以太湖为研究区建立的[43],后来被朱利等[44]证明高分一号WFV数据与环境一号CCD数据在波段设置上具有很强的一致性,并将该模型用于高分一号WFV数据的太湖叶绿素a质量浓度反演。
 图 5 洱海 2019 年 11 月 9 日 Sentinel-2A真彩色合成图Figure 5. Sentinel-2A true-color composite image of Erhai lake on November 9, 2019
图 5 洱海 2019 年 11 月 9 日 Sentinel-2A真彩色合成图Figure 5. Sentinel-2A true-color composite image of Erhai lake on November 9, 20194. 结论
1)在秋季的洱海流域,对于Sentinel-2卫星数据,与叶绿素a质量浓度具有显著的相关关系(P < 0.001),且在单波段、单波段比值和双波段比值中相关系数最大的分别为B6、B7/B6和(B6+B8)/(B7+B8a)。
2)使用BP神经网络,建立了适用于秋季洱海流域的叶绿素a质量浓度反演模型。对比使用同样数据建立的多元线性回归模型,BP神经网络模型的平均绝对误差百分比(MAPE)和均方根误差(RMSE)均小于多元线性回归模型,决定系数R2大于多元线性回归模型。总体来说,BP神经网络模型的精度高于线性回归模型,可使用Sentinel-2数据,利用本研究构建的BP神经网络模型反演叶绿素a质量浓度,能得到较可靠的反演结果。
3)通过构建的具有4个隐含层神经元节点的3层BP神经网络模型,反演洱海2019年10月12日、11月9日叶绿素a质量浓度,结果均显示洱海北部叶绿素a质量浓度明显高于南部。因此,基于Sentinel-2影像和BP神经网络模型可以宏观监测叶绿素a质量浓度的空间分布。
-
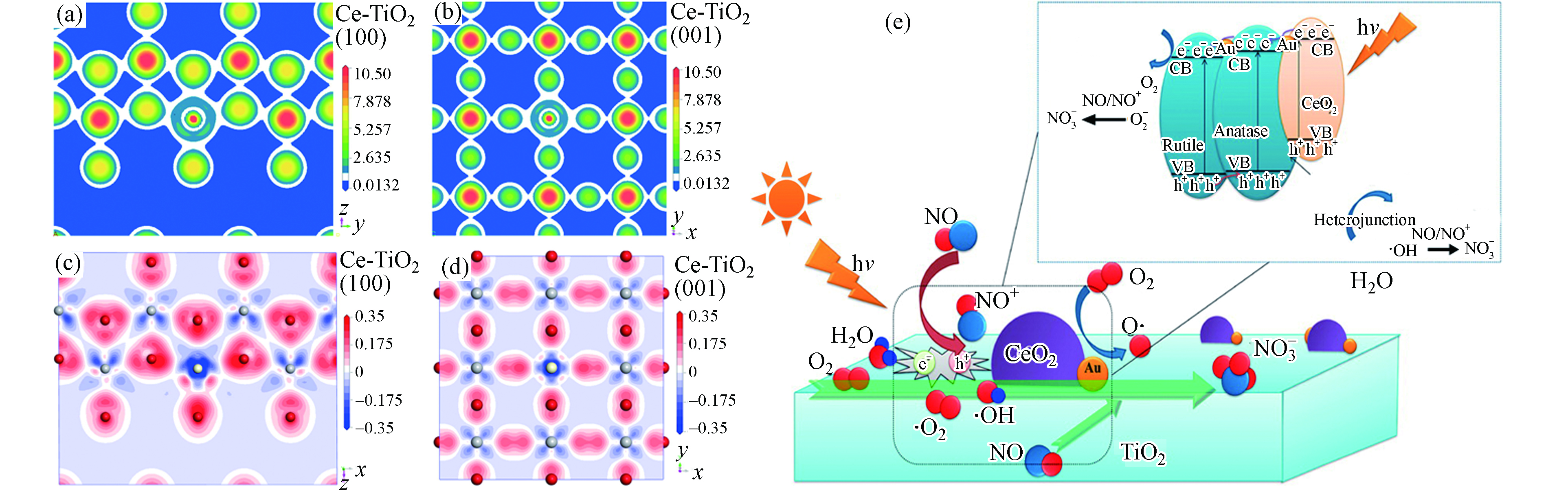

图 2 Eu掺杂CeO2催化剂:(a)O 1s XPS结果,(b)与甲醛的接触角结果[46];Ce-TiO2:(c)光催化降解苯反应速率与Ce3+含量的关系,(d)光照下的反应机制[51]。
Figure 2. The results of Eu doped CeO2 catalysts: (a) XPS spectra of O 1s, (b) contact angles with formaldehyde[46]; Ce-TiO2: (c) the relationship between the degradation rates of benzene and the Ce3+ contents, and (d) reaction mechanism[51]

表 1 铈基材料在光催化消除大气污染物中的研究。
Table 1. Study of ceria-based materials photocatalytic removal of atmospheric pollutants.
催化剂Catalyst 大气污染物Atmospheric pollutant 污染物浓度及反应条件Pollutant concentration reaction conditions 净化效率Elimination efficiency 参考文献Reference Eu/CeO2 HCHO 500 μg·m−3,100 W100 W卤钨灯(λ>420 nm) 80% [46] Ce-GO-TiO2 HCHO 2000 μg m−3,氙灯(全光谱) 85% [47] CeO2-TiO2 乙醛 300 μg·m−3,45%湿度,紫外/可见光 70% (紫外光)5% (可见光) [48] CeO2–TiO2/g-C3N4 甲苯 700 μg·m−3,75%湿度,6 W日光灯 6.37×10−10 mol·s−1·m−1 (紫外光)3.52×10−10 mol·s−1·m−1 (可见光) [49] Pt@CeO2 苯甲醇 0.1 mmol,300 W氙灯(λ<420 nm) 40% [50] Ce3+-TiO2 苯 5.5 μg·m−3, (52%±2%)湿度,8 W汞灯 70% (紫外光)15% (可见光) [51] Mn-TiO2/CeO2 甲苯 30 μg·m−3,50%湿度4 W紫外灯 50% [52] CeO2/TiO2锐钛矿 甲苯 700 μg·m−3,90%湿度6 W灯(λ>290 nm) 1.2×10−9 mol·s−1·m−1 (紫外光)3.5×10−10 mol·s−1·m−1 (太阳光) [53] Ce-TiO2 NO 1.25 μg·m−3,50%湿度卤素灯(λ>400 nm) 27.38% [54] Au/CeO2/TiO2 0.500 μg·m−3150 W卤钨灯/4 W汞灯 80% [55] CeO2/g-C3N4 100 μg·m−3,70%湿度500 W氙灯(λ>420 nm) 55% [56] MCe-LDHs CO2 300 W氙灯,水 CO:13.5 μmol·g−1 [57] Ce-TiO2 8 W汞灯,NaOH溶液 CH4:16 μmol·g−1 [58] Fe-CeO2 300 W氙灯,水 CH4:17.5 μmol·g−1CO:75 μmol·g−1 [59] CeO2 300 W氙灯 CO:0.2 μmol·g−1 [60] CrCeO2 500 W氙灯(λ>420 nm) CH4:10.5 μmol·g−1CO:16 μmol·g−1 [29] CeO2-x 300 W氙灯,水 CO:13×10−6(V/V) [61] Pd/Ce-TiO2 氙灯,CO2/H2:1/4 CH4:225 μmol·g−1CO:28 μmol·g−1 [62] Cu2O-CeO2 300 W氙灯,水 CO:1.1 μmol·g−1 [63]
下载: 导出CSV
-
[1] HASHIMOTO K, IRIE H, FUJISHIMA A. TiO2 photocatalysis: A historical overview and future prospects [J]. Japanese Journal of Applied Physics, 2005, 44(12): 8269-8285. doi: 10.1143/JJAP.44.8269 [2] FUJISHIMA A, HONDA K. Electrochemical photolysis of water at a semiconductor electrode [J]. Nature, 1972, 238(5358): 37-38. doi: 10.1038/238037a0 [3] CAREY J H, LAWRENCE J, TOSINE H M. Photodechlorination of PCB's in the presence of titanium dioxide in aqueous suspensions [J]. Bulletin of Environmental Contamination and Toxicology, 1976, 16(6): 697-701. doi: 10.1007/BF01685575 [4] PRUDEN A L, OLLIS D F. Degradation of chloroform by photoassisted heterogeneous catalysis in dilute aqueous suspensions of titanium dioxide [J]. Environmental Science & Technology, 1983, 17(10): 628-631. [5] HISANAGA T, HARADA K, TANAKA K. Photocatalytic degradation of organochlorine compounds in suspended TiO2 [J]. Journal of Photochemistry and Photobiology A:Chemistry, 1990, 54(1): 113-118. doi: 10.1016/1010-6030(90)87015-4 [6] CHEN X X, ZHAN S J, CHEN D S, et al. Grey Fe-CeO2-σ for boosting photocatalytic ozonation of refractory pollutants: Roles of surface and bulk oxygen vacancies [J]. Applied Catalysis B:Environmental, 2021, 286: 119928. doi: 10.1016/j.apcatb.2021.119928 [7] YANG C, ZHANG G H, MENG Y, et al. Direct Z-scheme CeO2@LDH core–shell heterostructure for photodegradation of Rhodamine B by synergistic persulfate activation [J]. Journal of Hazardous Materials, 2021, 408: 124908. doi: 10.1016/j.jhazmat.2020.124908 [8] HAO C C, TANG Y B, SHI W L, et al. Facile solvothermal synthesis of a Z-Scheme 0D/3D CeO2/ZnIn2S4 heterojunction with enhanced photocatalytic performance under visible light irradiation [J]. Chemical Engineering Journal, 2021, 409: 128168. doi: 10.1016/j.cej.2020.128168 [9] SHOKO E, SMITH M F, MCKENZIE R H. Charge distribution and transport properties in reduced ceria phases: A review [J]. Journal of Physics and Chemistry of Solids, 2011, 72(12): 1482-1494. doi: 10.1016/j.jpcs.2011.09.002 [10] GHOM S A, ZAMANI C, NAZARPOUR S, et al. Oxygen sensing with mesoporous ceria-zirconia solid solution [J]. Sensors and Actuators B:Chemical, 2009, 140(1): 216-221. doi: 10.1016/j.snb.2009.02.078 [11] KULLGREN J, CASTLETON C W M, MULLER C, et al. B3LYP calculations of cerium oxides [J]. The Journal of Chemical Physics, 2010, 132: 1-12. [12] PRIMO A, MARINO T, CORMA A, et al. Efficient visible-light photocatalytic water splitting by minute amounts of gold supported on nanoparticulate CeO2 obtained by a biopolymer templating method [J]. Journal of the American Chemical Society, 2012, 134(3): 1892. doi: 10.1021/ja211206v [13] ZHAO M, LI H H, SHEN X P, et al. Facile electrochemical synthesis of CeO2@Ag@CdS nanotube arrays with enhanced photoelectrochemical water splitting performance [J]. Dalton Transactions, 2015, 44(46): 19935-19941. doi: 10.1039/C5DT03661E [14] LI S X, CAI J B, WU X Q, et al. TiO2@Pt@CeO2 nanocomposite as a bifunctional catalyst for enhancing photo-reduction of Cr (VI) and photo-oxidation of benzyl alcohol [J]. Journal of Hazardous Materials, 2018, 346: 52-61. doi: 10.1016/j.jhazmat.2017.12.001 [15] SARAVANAKUMAR K, KARTHIK R, CHEN S M, et al. Construction of novel Pd/CeO2/g-C3N4 nanocomposites as efficient visible-light photocatalysts for hexavalent chromium detoxification [J]. Journal of Colloid and Interface Science, 2017, 504: 514-526. doi: 10.1016/j.jcis.2017.06.003 [16] JIANG B T, ZHANG S Y, GUO X Z, et al. Preparation and photocatalytic activity of CeO2/TiO2 interface composite film [J]. Applied Surface Science, 2009, 255: 5975-5978. doi: 10.1016/j.apsusc.2009.01.049 [17] WANG N, PAN Y, LU T, et al. A new ribbon-ignition method for fabricating p-CuO/n-CeO2 heterojunction with enhanced photocatalytic activity [J]. Applied Surface Science, 2017, 403: 699-706. doi: 10.1016/j.apsusc.2017.01.232 [18] RAJENDRAN S, KHAN M M, GRACIAL F, et al. Ce3+-ion-induced visible-light photocatalytic degradation and electrochemical activity of ZnO/CeO2 nanocomposite [J]. Scientific Reports, 2016, 6: 31641-31652. doi: 10.1038/srep31641 [19] MOHAMMADIYAN E, GHAFURI H, KAKANEJADIFARD A, Synthesis and characterization of a magnetic Fe3O4@CeO2 nanocomposite decorated with Ag nanoparticle and investigation of synergistic effects of Ag on photocatalytic activity [J]. Optik, 2018, 166: 39-48. [20] YOU D T, PAN B, JIANG F, et al. CdS nanoparticles/CeO2 nanorods composite with high-efficiency visible-light-driven photocatalytic activity [J]. Applied Surface Science, 2016, 363: 154-160. doi: 10.1016/j.apsusc.2015.12.021 [21] LI X Z, ZHU W, LU X W, et al. Integrated nanostructures of CeO2/attapulgite/g-C3N4 as efficient catalyst for photocatalytic desulfurization: Mechanism, kinetics and influencing factors [J]. Chemical Engineering Journal, 2017, 326: 87-98. doi: 10.1016/j.cej.2017.05.131 [22] WU C L, Solvothermal synthesis of N-doped CeO2 microspheres with visible light-driven photocatalytic activity [J]. Materials Letters, 2015, 139: 382-384. [23] CHEN J, SHEN S H, WU P, et al. Nitrogen-doped CeOx nanoparticles modified graphitic carbon nitride for enhanced photocatalytic hydrogen production [J]. Green Chemistry, 2015, 17: 509-517. doi: 10.1039/C4GC01683A [24] JORGE A B, FRAXEDAS J, CANTARERO A, et al. Nitrogen Doping of Ceria [J]. Chemistry of Materials, 2008, 20(5): 1682-1684. doi: 10.1021/cm7028678 [25] AHMAD S, GOPALAIAH K, CHANDRUDU S N, et al. Anion (fluoride)-doped ceria nanocrystals: Synthesis, characterization, and its catalytic application to oxidative coupling of benzylamines [J]. Inorganic Chemistry, 2014, 53: 2030-2039. doi: 10.1021/ic403166q [26] MANSINGH S, PADHI D K, PARID K M. Enhanced visible light harnessing and oxygen vacancy promoted N, S co-doped CeO2 nanoparticle: A challenging photocatalyst for Cr(Ⅵ) reduction [J]. Catalysis Science & Technology, 2017, 7: 2772-2781. [27] WANG Y G, WANG F, CHEN Y T, et al. Enhanced photocatalytic performance of ordered mesoporous Fe-doped CeO2 catalysts for the reduction of CO2 with H2O under simulated solar irradiation [J]. Applied Catalysis B:Environmental, 2014, 147: 602-609. doi: 10.1016/j.apcatb.2013.09.036 [28] PENG D Z, CHEN S Y, CHEN C L, et al. Understanding and Tuning Electronic Structure in Modified Ceria Nanocrystals by Defect Engineering [J]. Langmuir, 2014, 30(34): 10430-10439. doi: 10.1021/la501576c [29] WANG Y G, BAI X, WANG F, et al. Nanocasting synthesis of chromium doped mesoporous CeO2 with enhanced visible-light photocatalytic CO2 reduction performance [J]. Journal of Hazardous Materials, 2019, 372: 69-76. doi: 10.1016/j.jhazmat.2017.10.007 [30] CUI X, LIU Z Y, Li G S, et al. Self-generating CeVO4 as conductive channel within CeO2/CeVO4/V2O5 to induce Z-scheme-charge-transfer driven photocatalytic degradation coupled with hydrogen production [J]. International Journal of Hydrogen Energy, 2019, 44: 23921-23935. doi: 10.1016/j.ijhydene.2019.07.096 [31] XU B, YANG H, ZHANG Q T, et al. Design and synthesis of Sm, Y, La and Nd-doped CeO2 with a broom-like hierarchical structure: a photocatalyst with enhanced oxidation performance [J]. ChemCatChem, 2020, 12: 2638-2646. doi: 10.1002/cctc.201902309 [32] CODURI M, SCAYINI M, ALLIETA M, et al. Defect structure of Y-doped ceria on different length scales [J]. Chemistry of Materials, 2013, 25: 4278-4289. doi: 10.1021/cm402359d [33] HAO S Y, HOU J, APREA P, et al. Mesoporous Ce-Pr-O solid solution with efficient photocatalytic activity under weak daylight irradiation [J]. Applied Catalysis B:Environmental, 2014, 160-161: 566-573. doi: 10.1016/j.apcatb.2014.06.013 [34] SREEKANTH T V M, NAGAJYOTHI P C, DILLIP G R, et al. Determination of Band Alignment in the Synergistic Catalyst of Electronic Structure-Modified Graphitic Carbon Nitride-Integrated Ceria Quantum-Dot Heterojunctions for Rapid Degradation of Organic Pollutants [J]. The Journal of Physical Chemistry C, 2017, 121(45): 25229-25242. doi: 10.1021/acs.jpcc.7b08568 [35] ZHAO K, QI J, YIN H J, et al. Efficient water oxidation under visible light by tuning surface defects on ceria nanorods [J]. Journal of Materials Chemistry A, 2015, 3: 20465-20470. doi: 10.1039/C5TA05817A [36] WU J S, Wang J S, DU Y C, et al. Chemically controlled growth of porous CeO2 nanotubes for Cr(Ⅵ) photoreduction [J]. Applied Catalysis B:Environmental, 2015, 174-175: 435-444. doi: 10.1016/j.apcatb.2015.03.040 [37] MANWAR N R, CHILKALWAR A A, NANDA K K, et al. Ceria Supported Pt/PtO-Nanostructures: Efficient Photocatalyst for Sacrificial Donor Assisted Hydrogen Generation under Visible-NIR Light Irradiation [J]. ACS Sustainable Chemistry & Engineering, 2016, 4(4): 2323-2332. [38] WANG A Q, ZHENG Z K, WANG H, et al. 3D hierarchical H-2-reduced Mn-doped CeO2 microflowers assembled from nanotubes as a high-performance Fenton-like photocatalyst for tetracycline antibiotics degradation [J]. Applied Catalysis B:Environmental, 2020, 277: 119171. doi: 10.1016/j.apcatb.2020.119171 [39] QI J, ZHAO K, LI G D, et al. Multi-shelled CeO2 hollow microspheres as superior photocatalysts for water oxidation [J]. Nanoscale, 2014, 6(8): 4072-4077. doi: 10.1039/C3NR06822F [40] WANG F, TIAN J, LI M, et al. A photoactivated Cu-CeO2 catalyst with Cu--Ce active species designed through MOF crystal engineering [J]. Angewandte Chemie, 2020, 132: 8280-8286. doi: 10.1002/ange.201916049 [41] LI P, ZHOU Y, ZHAO Z Y, et al. Hexahedron prism-anchored octahedronal CeO2: crystal facet-based homojunction promoting efficient solar fuel synthesis [J]. Journal of the American Chemical Society, 2015, 137(30): 9547-9550. doi: 10.1021/jacs.5b05926 [42] JIANG D, WANG W Z, ZHANG L, et al. Insights into the surface-defect dependence of photoreactivity over CeO2 nanocrystals with well-defined crystal facets [J]. ACS Catalysis, 2015, 5(8): 4851-4858. doi: 10.1021/acscatal.5b01128 [43] ZOU W X, DENG B, HU X X, et al. Crystal-plane-dependent metal oxide-support interaction in CeO2/g-C3N4 for photocatalytic hydrogen evolution [J]. Applied Catalysis B:Environmental, 2018, 238: 111-118. doi: 10.1016/j.apcatb.2018.07.022 [44] XU C C, ZHANG Q, XU Q, et al. Research progress of organic-inorganic photocatalysts for degrading VOCs [J]. Environmental Engineering, 2020, 38(1): 28-36. [45] SHEN L J, CHENG R, CHEN Y H, et al. Research progress on the purification technologies of indoor volatile organic compounds [J]. Chemical Industry and Engineering Progress, 2017, 36(10): 3887-3896. [46] HUANG Y C, LONG B, TANG M N, et al. Bifunctional catalytic material: An ultrastable and high-performance surface defect CeO2 nanosheets for formaldehyde thermal oxidation and photocatalytic oxidation [J]. Applied Catalysis B:Environmental, 2016, 181: 779-787. doi: 10.1016/j.apcatb.2015.08.047 [47] LI J, ZHANG Q, LAI A C K, et al. Study on photocatalytic performance of cerium–graphene oxide–titanium dioxide composite film for formaldehyde removal [J]. Physica Status Solidi (a), 2016, 213(12): 3157-3164. doi: 10.1002/pssa.201600261 [48] MUÑOZ-BATISTA M J, DE LOS MILAGROS BALLARI M, KUBACKA A, et al. Acetaldehyde degradation under UV and visible irradiation using CeO2–TiO2 composite systems: Evaluation of the photocatalytic efficiencies [J]. Chemical Engineering Journal, 2014, 255: 297-306. doi: 10.1016/j.cej.2014.06.056 [49] MUÑOZ-BATISTA M J, FERNÁNDEZ-GARCíA M, KUBACKA A. Promotion of CeO2–TiO2 photoactivity by g-C3N4: Ultraviolet and visible light elimination of toluene [J]. Applied Catalysis B:Environmental, 2015, 164: 261-270. doi: 10.1016/j.apcatb.2014.09.037 [50] LIU H H, LI Y Z, YANG Y, et al. Highly efficient UV-Vis-infrared catalytic purification of benzene on CeMnxOy/TiO2 nanocomposite, caused by its high thermocatalytic activity and strong absorption in the full solar spectrum region [J]. Journal of Materials Chemistry A, 2016, 4: 9890-9899. doi: 10.1039/C6TA03181A [51] LIU T X, LI X Z, LI F B. Enhanced photocatalytic activity of Ce3+-TiO2 hydrosols in aqueous and gaseous phases [J]. Chemical Engineering Journal, 2010, 157: 475-482. doi: 10.1016/j.cej.2009.12.010 [52] WU M Y, LEUNG D Y C, ZHANG Y G, et al. Toluene degradation over Mn-TiO2/CeO2 composite catalyst under vacuum ultraviolet (VUV) irradiation [J]. Chemical Engineering Science, 2019, 195: 985-994. doi: 10.1016/j.ces.2018.10.044 [53] MUÑOZ-BATISTA M J, KUBACKA A, FONTELLES-CARCELLER O, et al. Surface CuO, Bi2O3, and CeO2 species supported in TiO2-anatase: study of interface effects in toluene photodegradation quantum efficiency [J]. ACS Applied Materials & Interfaces, 2016, 8(22): 13934-13945. [54] CAO X J, YANG X Y, LI H, et al. Investigation of Ce-TiO2 photocatalyst and its application in asphalt-based specimens for NO degradation [J]. Construction and Building Materials, 2017, 148: 824-832. doi: 10.1016/j.conbuildmat.2017.05.095 [55] ZHU W, XIAO S N, ZHANG D Q, et al. Highly efficient and stable Au/CeO2-TiO2 photocatalyst for nitric oxide abatement: potential application in flue gas treatment [J]. Langmuir, 2015, 31(39): 10822-10830. doi: 10.1021/acs.langmuir.5b02232 [56] TIAN N, HUANG H W, LIU C Y, et al. In situ co-pyrolysis fabrication of CeO2/g-C3N4 n–n type heterojunction for synchronously promoting photo-induced oxidation and reduction properties [J]. Journal of Materials Chemistry A, 2015, 3(33): 17120-17129. doi: 10.1039/C5TA03669K [57] YE T, HUANG W M, ZENG L M, et al. CeO2-x platelet from monometallic cerium layered double hydroxides and its photocatalytic reduction of CO2 [J]. Applied Catalysis B:Environmental, 2017, 210: 141-148. doi: 10.1016/j.apcatb.2017.03.051 [58] MATĔJOVÁ L, KOČÍ K, RELI M, et al. Preparation, characterization and photocatalytic properties of cerium doped TiO2: On the effect of Ce loading on the photocatalytic reduction of carbon dioxide [J]. Applied Catalysis B:Environmental, 2014, 152-153: 172-183. doi: 10.1016/j.apcatb.2014.01.015 [59] ZHAO Y X, CAI W, CHEN M D, et al. Turning the activity of Cr-Ce mixed oxide towards thermocatalytic NO oxidation and photocatalytic CO2 reduction via the formation of yolk shell structure hollow microspheres [J]. Journal of Alloys and Compounds, 2020, 829: 154508-154520. doi: 10.1016/j.jallcom.2020.154508 [60] ZHU C Z, WEI X Q, LI W Q, et al. Crystal-plane effects of CeO2 {110} and CeO2 {100} on photocatalytic CO2 reduction: synergistic interactions of oxygen defects and hydroxyl groups [J]. ACS Sustainable Chemistry & Engineering, 2020, 8(38): 14397-14406. [61] WANG M, SHEN M, JIN X X, et al. Mild generation of surface oxygen vacancies on CeO2 for improved CO2 photoreduction activity [J]. Nanoscale, 2020, 12: 12374-12382. doi: 10.1039/D0NR00717J [62] LI N X, ZOU X Y, LIU M, et al. Enhanced visible light photocatalytic hydrogenation of CO2 into methane over a Pd/Ce-TiO2 Nanocomposition [J]. Journal of Physical Chemistry C 2017, 121(46): 25795-25804. [63] PU Y, LUO Y D, WEI X Q, et al. Synergistic effects of Cu2O-decorated CeO2 on photocatalytic CO2 reduction: Surface Lewis acid/base and oxygen defect [J]. Applied Catalysis B:Environmental, 2019, 254: 580-586. doi: 10.1016/j.apcatb.2019.04.093 [64] COURBON H, PICHAT P. Room-temperature interaction of N18O with ultraviolet-illuminated titanium dioxide [J]. Journal of the Chemical Society, Faraday Transactions 1:Physical Chemistry in Condensed Phases, 1984, 80(11): 3175-3185. doi: 10.1039/f19848003175 [65] LIU H, ZHANG H R, YANG H M. Photocatalytic removal of nitric oxide by multi-walled carbon nanotubes‐supported TiO2 [J]. Chinese Journal of Catalysis, 2014, 35(1): 66-77. doi: 10.1016/S1872-2067(12)60705-0 [66] WANG J Y, HAN F M, RAO Y F, et al. Visible-light-driven nitrogen-doped carbon quantum dots/CaTiO3 composite catalyst with enhanced NO adsorption for NO removal [J]. Industrial & Engineering Chemistry Research, 2018, 57(31): 10226-10233. [67] DUAN Y Y, LUO J M, ZHOU S C, et al. TiO2-supported Ag nanoclusters with enhanced visible light activity for the photocatalytic removal of NO [J]. Applied Catalysis B:Environmental, 2018, 234: 206-212. doi: 10.1016/j.apcatb.2018.04.041 [68] WU Q P, van de KROL R. Selective photoreduction of nitric oxide to nitrogen by nanostructured TiO2 photocatalysts: role of oxygen vacancies and iron dopant [J]. Journal of the American Chemical Society, 2012, 134(22): 9369-9375. doi: 10.1021/ja302246b [69] YU Y, ZHONG Q, CAI W, et al. Promotional effect of N-doped CeO2 supported CoOx catalysts with enhanced catalytic activity on NO oxidation [J]. Journal of Molecular Catalysis A:Chemical, 2015, 398: 344-352. doi: 10.1016/j.molcata.2015.01.002 [70] ÂNGELO J, MAGALHÃES P, ANDRADE L, et al. Optimization of the NO photooxidation and the role of relative humidity [J]. Environmental Pollution, 2018, 240: 541-548. doi: 10.1016/j.envpol.2018.04.051 [71] RHATIGAN S, NOLAN M, CO2 and water activation on ceria nanocluster modified TiO2 rutile (110) [J]. Journal of Materials Chemistry A, 2018, 6: 9139-9152. [72] LI W Q, JIN L, GAO F, et al. Advantageous roles of phosphate decorated octahedral CeO2 {111}/g-C3N4 in boosting photocatalytic CO2 reduction: Charge transfer bridge and Lewis basic site [J]. Appl Catal B:Environ, 2021, 294: 120257. doi: 10.1016/j.apcatb.2021.120257 [73] PU Y, LI W Q, CAI Y D, et al. Effects of different treatment atmospheres on CeO2/g-C3N4 photocatalytic CO2 reduction: good or bad?[J]. Catal. Sci. Technol. , 2021, 11(8): 2827–2833. -

 点击查看大图
点击查看大图

计量
- 文章访问数: 5656
- HTML全文浏览数: 5656
- PDF下载数: 214
- 施引文献: 0
