


-
剩余污泥(waste activated sludge,WAS)是在城市污水处理过程中形成的主要副产物。据报道,2019年我国剩余污泥(80%含水率)的产生量超过6 000×104 t[1-2]。剩余污泥中通常含有有毒有害有机物、重金属、病原菌和寄生虫卵等,具有较大的二次污染风险[3]。目前,污泥的处理以堆埋、焚烧、农业堆肥和自然干化为主,所需费用较高(占污水处理厂总运行费用的50%~60%) [4-5]。厌氧发酵是一种重要的环境生物技术,能够利用剩余污泥生产甲烷和短链脂肪酸(VFA)等多种化学品[3, 6-7]。而且,短链脂肪酸可以作为污水处理厂反硝化的碳源,从而进一步降低污水处理厂的运行成本[8]。因此,将混菌厌氧发酵技术应用于市政污泥处置,是实现其资源化的重要手段。
市政污泥的主要有机成分复杂,包括细胞、胞外聚合物(EPS)和少量纤维素等[9-10],导致了厌氧发酵技术面临生物水解速率慢等诸多问题。例如,厌氧反应器需要较长的水力停留时间(20~30 d),但其有机物去除率仍然不高(30%~50%)[9]。剩余污泥中的EPS组分占污泥有机质干重的50%~80%,具有维持微生物聚集体结构和保持其功能完整性的作用[10]。因此,一般认为EPS组分是导致市政污泥水解困难的主要因素。目前,主要采用预处理过程(超声波处理、水热处理、酸碱处理和高级氧化处理等)来破坏EPS和细胞壁的结构,以降低污泥生物处置的阻力和提高污泥中有机物的可利用性[9, 11]。例如,ZHANG等[12]发现,通过外源投加钢渣和碱处理,污泥中有机物水解程度随pH的增加而增加,20 d后可溶性有机碳质量浓度比空白组增加了1.0 g·L−1。ARENAS等[13]报道,碱性条件下电氧化预处理后可溶性有机物增量最大,总有机碳和可溶性COD(SCOD)的质量浓度分别为2.8和7.8 g·L−1,而空白组仅为0.4和1.1 g·L−1。然而,上述预处理方法选择性不高,并且增加了装置的额外运行成本。
EPS中的酸性多糖(藻酸盐和半乳糖醛酸等)能够与水中阳离子形成凝胶类物质[14-17],可维持污泥结构并阻碍微生物的水解作用。其中,藻酸盐是由β-D-甘露糖醛酸和α-L-古洛糖醛酸按(1→4)糖苷键连接而成。LIN等[18]通过鉴定发现,污泥絮体中藻酸盐类似物的质量分数达到7%。然而,目前有关藻酸盐降解及其对污泥发酵影响的报道仍然较少。因此,本研究首先构建以藻酸盐为底物的恒化器,培养稳定的藻酸盐降解菌群(alginate-degrading consortium,ADC),并通过高通量测序分析菌群结构;其次,利用EPS中存在的典型物质(聚半乳糖醛酸,酪蛋白,纤维素和葡聚糖)作为底物,解析ADC促进EPS水解酸化的功能;最后,将ADC应用到实际剩余污泥体系中,解析3种典型pH(5.0,6.0和7.0)条件下ADC对污泥水解和酸化效率的促进能力,以期为强化污泥产酸提供新的思路。
-
本实验所用的剩余污泥取自福建省福州市金山污水处理厂的二沉池,实验前存放于4 ℃冰箱中,污泥基本特性如表1所示。
-
1)恒化器长期实验。构建恒化器,总体积为3.2 L,工作体积为2.5 L。接种微生物(50 mL)源于以藻酸盐为底物培养的厌氧菌群[19]。恒化器进水藻酸盐的质量浓度为10 g·L−1,无机培养基的成分与ZHANG等[19]报道的相同,并添加10 mmol·L−1 2-溴乙基磺酸钠(BES)以抑制甲烷生成。利用99%纯度的N2将恒化器曝气20 min。恒化器运行时间为60 d,并监测代谢产物的变化。
2)模拟物质厌氧酸化实验。为了解析ADC以EPS作为底物进行水解和厌氧酸化的功能,以聚半乳糖醛酸、纤维素、葡聚糖和酪蛋白作为EPS的典型代表物质,接种ADC作为底物进行为期14 d的厌氧酸化。每1种底物设置3个重复。在120 mL血清瓶中添加60 mL无机培养基,并加入10 mmol·L−1 BES,底物的质量浓度为5 g·L−1、pH为5.5、接种量为10 mL(转速10 000 r·min−1、离心5 min,弃上清液)。利用纯度为99%的N2曝气10 min,密封后放入37 ℃的振荡培养箱进行培养并分析代谢产物。
3)不同pH剩余污泥厌氧酸化实验。将储存在4 ℃冰箱的污泥取出,利用1 mol·L−1 NaOH和HCl将其pH分别调至5.0、6.0和7.0,每个水平分为对照组和实验组2个处理。其中,对照组为原污泥,实验组中接种20 mL ADC菌群。每个处理分装于120 mL血清瓶中(n = 3),液相为60 mL,最后曝气密封并放入恒温箱培养。发酵时间持续12 d,分析污泥发酵过程中的产气(H2和CH4)、水解(SCOD)和酸化(VFA)等参数,并计算水解和酸化效率。
4)水解酸化效率的计算。为了量化在经过不同处理之后,对照组和实验组在不同pH下污泥水解和酸化的情况,利用式(1)和公式(2)分别计算厌氧发酵过程中的水解和酸化效率[20]。
式中:Eh为水解效率;SCODi为对应各个时间点的溶解性COD,g·L−1;TCOD为污泥总COD,g·L−1。
式中:Ea为酸化效率;TVFAi为对应各个时间点总挥发性有机物的质量浓度,g·L−1。
5)分析方法。CH4和H2的体积分数采用气相色谱仪(SP7890,山东鲁南瑞虹化工仪器有限公司)测定。短链脂肪酸样品用0.45 μm 滤膜过滤,之后保存于4℃冰箱中。短链脂肪酸的质量浓度由气相色谱仪(7890,安捷伦科技有限公司)测定。恒化器中藻酸盐的质量浓度采用硫酸-咔唑法测定[21]。TCOD和SCOD用重铬酸钾法测定[22]。污泥中多糖的质量浓度采用硫酸-蒽酮法测定,蛋白的质量浓度则采用Lowry 法分析[23]。pH采用pH计(PHS-3C,上海精密科学仪器有限公司)测定。
6)DNA提取和Illumina Miseq高通量测序。2个DNA样本分别提取自剩余污泥和恒化器中培养60 d的菌群,命名为WAS和ADC。DNA序列扩增(引物341F-806R[19])由ABI GeneAmp® 9700进行,之后使用Illumina Miseq PE 300测序仪进行测序。基于上述测序结果分析菌群的多样性。
-
恒化器运行时间为60 d,运行期间水力停留时间(HRT)通过调控稳定在(4.4 ± 0.2) d,pH维持在酸性(5.5 ± 0.2)条件。如图1(a)所示,恒化器出水藻酸盐的质量浓度远小于进水(10 g·L−1),仅为(0.10 ± 0.06)g·L−1。这说明,在长期培养中,ADC菌群具有良好的藻酸盐降解功能。TSS和VSS分别为(1.4 ± 0.2)和(0.80 ± 0.04)g·L−1。恒化器运行期间,每日产气量小于50 mL(产气速率为(17.2~48.6) mL·d−1),其中,H2在气相中的体积分数为1.6% ± 0.8%。H2的消耗归因于同型产乙酸菌群的作用[8, 24]。为了保证厌氧酸化阶段运行的稳定,期间持续添加BES以抑制产甲烷菌群活性,因此仅监测到痕量的CH4(体积分数于0.1%)。图1(b)显示了恒化器中ADC菌群厌氧产酸情况,VFA的组成成分主要为乙酸、丙酸和丁酸,质量浓度分别为(2.1 ± 0.2)、(0.8 ± 0.1)和(0.6 ± 0.2)g·L−1。根据COD平衡的计算,恒化器的COD转化率为80.0% ± 9.6%。上述结果说明,在长期培养中,ADC具有高效的藻酸盐降解能力。
-
剩余污泥EPS中主要的可生物降解有机组分含有多糖(包括中性糖和酸性糖)和蛋白质[11]。因此,以聚半乳糖醛酸、酪蛋白、纤维素和葡聚糖4种典型有机底物,探究ADC菌群利用不同有机底物产气和产酸的情况。在整个反应期间,4组实验的H2体积分数最高为7.9% ± 0.1%,这与恒化器运行(图1)的结果相似。由于添加了BES,故4组实验均未检测出CH4。
ADC菌群利用4种模拟底物厌氧产酸情况如图2所示,其中分别采用聚半乳糖醛酸(图2(a))代表酸性糖,以葡聚糖(图2(b))和纤维素(图2(c))代表中性糖,以酪蛋白(图2(d))代表蛋白质。结果表明,ADC菌群降解模拟物质中的代谢产物主要以乙酸、丙酸和丁酸为主。其中,代谢聚半乳糖醛酸和葡聚糖生产乙酸的质量浓度最终分别为(0.6 ± 0.03)和(0.9 ± 0.05)g·L−1,而利用酪蛋白和纤维素生产乙酸的质量浓度仅为(0.2 ± 0.01)和(0.4 ± 0.04)g·L−1。以葡聚糖作为底物时,丙酸的最大质量浓度为(0.6 ± 0.04)g·L−1;其余底物中生成丙酸的质量浓度相对稳定。ADC以酪蛋白和葡聚糖为底物时的主要产物是丁酸,质量浓度分别为0.5和0.7 g·L−1。以聚半乳糖醛酸和纤维素为底物时产生了较少的丁酸,质量浓度分别为(0.3 ± 0.05)和(0.3 ± 0.001)g·L−1。上述结果表明,ADC菌群具有降解EPS中各类典型有机质生产短链脂肪酸的能力。
-
在整个反应期间,3组实验的H2体积分数始终低于0.4% ± 0.1%,这与恒化器运行(图1)的结果相似。由于添加了产甲烷抑制剂BES,故4组实验中均未检测出CH4。图3显示了在不同pH处理中空白组和添加ADC组的厌氧产酸情况。在3组pH条件下,WAS的代谢产物以乙酸、丙酸和丁酸为主,并且添加ADC组的产酸质量浓度均高于空白组。在pH=7时,ADC组最终的乙酸、丙酸和丁酸的质量浓度分别累积到(0.9 ± 0.02)、(0.4 ± 0.02)和(0.5 ± 0.03)g·L−1,而对照组仅为(0.4 ± 0.08)、(0.2 ± 0.04)和(0.1 ± 0.01)g·L−1。在pH=6和pH=5的条件下也出现了类似的结果,在厌氧酸化第11 d时测得对照组中乙酸质量浓度分别为(0.4 ± 0.05)和(0.4 ± 0.01)g·L−1,丙酸质量浓度均为(0.2 ± 0.01)g·L−1;而在实验组中的VFA产量得到了明显的提升,其中,乙酸质量浓度为(1.1 ± 0.01)和(0.9 ± 0.02)g·L−1,丙酸质量浓度为(0.4 ± 0.03)和(0.4 ± 0.02)g·L−1。因此,结合第2.2节的实验结果可知,ADC可以通过破坏EPS结构以加速WAS的水解和厌氧产酸。
-
WAS絮体结构的破坏可导致胞外和胞内有机物的释放和溶解[25-26]。因此,通过测定厌氧酸化过程中的SCOD可以量化ADC菌群对WAS水解和酸化阶段的促进作用。图4(a)~图4(c)显示了在ADC组和空白组中不同pH条件下SCOD的变化情况,在pH分别为7.0、6.0和5.0时,空白组中厌氧酸化最终SCOD分别为(3.1±0.12)、(4.4±0.07)和(3.2±0.03)g·L−1;而在ADC组中,相应pH条件下的SCOD均有所提升。图4(d)~图4(f)显示了利用式(1)和式(2)计算得出的WAS水解和厌氧酸化的效率。结果表明,在不同pH条件下,ADC组中的水解和酸化效率均高于空白组。其中,ADC组的水解效率在pH为7.0、6.0、5.0时分别提升了25.4%、13.2%和12.1%,酸化效率分别提升了138.5%、184.0%和103.4%。例如,pH=5.0时,添加ADC后,污泥的水解效率由30.5%±0.3%增加至41.8%±1.6%,酸化效率由34.8%±7.0%增加至70.8%±4.4%。因此,ADC菌群可通过提高EPS中典型大分子有机物的水解和酸化得效率,实现强化WAS厌氧发酵生产VFA。
-
图5显示了经过60 d培养的ADC菌群和WAS菌群的二代测序结果。其中,OTU指数和Shannon指数(图5(a)和图5(b))表明测序结果具有较高的覆盖度,能够体现菌群的多样性。经过恒化器的长期培养,ADC的菌群多样性明显低于WAS菌群。在门水平上(图5(c)),WAS菌群包含绿弯菌门(Chloroflexi,丰度为11.8%)、放线菌门(Actinobacteriota,丰度为17.7%)、拟杆菌门(Bacteroidota,丰度为20.5%)、变形菌门(Proteobacteria,丰度为24.0%)等。而ADC菌群(图5(c))则主要以拟杆菌门(Bacteroidota,丰度为52.7%)和厚壁菌门(Firmicutes,丰度为36.7%)为主。同样,在属水平上(图5(d))也表现出了同样的结果。WAS中属水平下菌群种类更多,而ADC中主要以拟杆菌属(Bacteroides,丰度为37.3%)、颤杆菌克属(Oscillibacter,丰度为18.6%)和厌氧棍状菌属(Anaerotruncus,丰度为10.5%)为主要菌属。ZHANG等[19]发现,拟杆菌属具有藻酸盐降解的功能,但其百分比低于1%。而在本研究中,通过恒化器的长期培养,可以得到拟杆菌属(丰度为37.3%)相对丰度较高的ADC菌群。
有研究表明,EPS的主要组分是蛋白和多糖类物质,并且已有较多采用蛋白酶和多糖水解酶促进污泥水解的研究案例[27-28]。以藻酸盐为代表的酸性多糖是EPS中新分离的组分[16, 29]。本研究中所富集的ADC菌群,具有降解多种典型WAS有机质的能力(图2),能够明显地促进WAS水解和酸化(图3和图4)。综合分析可知,采用ADC菌群与蛋白酶、多糖水解酶联合处理可以促进WAS的资源化,不过仍需要进一步研究。综上所述,ADC菌群是对生物法加速污泥水解和酸化的补充,可为促进污泥资源化提供了新的思路。
-
1)经恒化器培养出的高活性ADC菌群具有较高的藻酸盐转化能力,COD的转化率达到80.0% ± 9.6%。
2) ADC菌群对聚半乳糖醛酸、葡聚糖和酪蛋白等WAS的典型成分均具有较好的厌氧降解能力。
3)在不同的pH条件下,ADC对WAS的水解和酸化过程均存在促进作用。ADC组的水解效率在pH为7.0、6.0、5.0时分别提升了25.4%、13.2%和12.1%,酸化效率分别提升了138.5%、184.0%和103.4%。pH为6.0是ADC菌群促进剩余污泥酸化的最佳工艺条件。
4)经过恒化器的长期富集,ADC菌群以拟杆菌属(Bacteroides,丰度为37.3%)为主。
藻酸盐降解菌群强化剩余污泥厌氧发酵产酸
Enhanced acidogenesis of waste activated sludge fermentation by an alginate-degrading consortium
-
摘要: 厌氧发酵是实现剩余污泥(WAS)资源化的重要技术,而其中的水解阶段是剩余污泥(WAS)厌氧资源化的限速步骤。WAS中的酸性多糖(藻酸盐和半乳糖醛酸等)能够与水中阳离子形成凝胶类物质,从而维持污泥结构并阻碍微生物的水解。利用藻酸盐为底物,经过恒化器培养得到了高效的藻酸盐降解菌群(ADC)。该菌群对WAS的典型有机成分(聚半乳糖醛酸、葡聚糖和酪蛋白等)均具有较好的厌氧降解能力,其代谢产物以乙酸等短链脂肪酸为主。而且,ADC菌群对WAS的水解和酸化过程均存在促进作用;在pH为7.0、6.0、5.0的条件下,水解效率分别提升了25.4%、13.2%和12.1%,酸化效率则分别提升了138.5%、184.0%和103.4%。Illumina Miseq高通量测序结果表明,该菌群以拟杆菌属(Bacteroides,37.3%)为主。本研究结果可为剩余污泥厌氧资源化提供参考。Abstract: Anaerobic fermentation is an important biotechnology to convert the waste activated sludge to valuable biochemicals. But, hydrolysis is known as the rate-limiting step of WAS fermentation. The uronic acids (such as alginate and polygalacturonic acid) in WAS can form hydrogels with cationic ions in wastewater, which maintain sludge structure and retard the microbial hydrolysis. An alginate-degrading consortium (ADC) with high activity was enriched in a mesophilic chemostat using alginate as the substrate. The results showed that the typical organic components of WAS, including polygalacturonic acid, dextran, and casein, could be utilized by the enriched ADC, and the metabolites were volatile fatty acids, like acetate. Moreover, hydrolysis and acidification of WAS were also enhanced by dosing ADC, of which, the hydrolytic efficiency at pH 7.0, 6.0, and 5.0 increased by 25.4%, 13.2%, and 12.1%, respectively, and the acidification efficiency increased by 138.5%, 184.0%, and 103.4%, respectively. The genus Bacteroides (37.3%) was identified as the dominant bacteria in ADC by an Illumina Miseq high-throughput sequencing. The results of this study can provide references for anaerobic resource utilization of WAS.
-
挥发性有机化合物(VOCs)作为PM2.5和臭氧的重要前驱体,已经对大气环境质量和人体健康造成直接和间接的危害[1-2],VOCs废气治理成为了近年来的环境热点话题之一。在VOCs废气的众多处理技术中,催化燃烧技术因处理效率高、能耗低、二次污染小而在工业上应用广泛[3]。为达到VOCs催化燃烧所需温度,工业上多采用电加热使VOCs废气达到起燃温度。由于电加热是采用热传导的加热方式,因而对大气量的VOCs废气加热时能耗巨大。另外,VOCs催化燃烧时,持续的高温环境会使催化剂活性组分烧结而影响VOCs降解效果[4]。微波加热应用于VOCs催化燃烧是一种新技术,它利用电磁波的选择性仅对催化剂进行加热,因而能耗低且催化剂受热均匀、快速[5]。而且,微波对催化剂活性组分的热点效应有利于引发VOCs的催化燃烧,同时微波的偶极极化作用还可降低VOCs的反应阈能而促进其氧化降解[6]。卜龙利等[7]、姚泽等[8]利用吸波型催化剂证实了微波催化燃烧VOCs效果优于电加热催化燃烧。
微波催化燃烧技术的关键是高效而稳定的催化剂。过渡金属氧化物催化剂种类丰富、经济性好、不易中毒,但其对于某些难降解有机物的催化活性差、低温活性以及热稳定性有待提高[9]。贵金属催化剂具有催化活性高、高温下稳定性好和适用范围广的优点,但也存在资源稀少和价格高昂的问题[10]。目前,市面上较为常见的是铜锰铈三元金属氧化物催化剂。胡旭睿[11]研究证实,铜锰铈氧化物催化剂对芳香烃类、醇类、酮类等有机物具有良好的氧化性能,但其矿化效果不佳且对芳香烃的降解效果低于醇、酮类。贵金属Pt、Pd等在低温时对芳烃类和3个碳以上的直链烷烃活化能力强[12]。已有研究证实,将贵金属与过渡金属复合可以增强催化剂活性。SHI等[13]通过还原法和离子交换法合成了Pt /Ce-USY催化剂,其对1,2-二氯乙烷的催化活性高于Pt/USY催化剂,Pt与CeO2之间的相互作用抑制了碳物质的沉积,从而增强了催化剂的耐久性。LEE等[14]将贵金属金、钯沉积在CeO2表面,证实适量Au的加入使得Pd/CeO2催化剂活性增强。CHEN等[15]合成了Pd/Fe3O4催化剂用于去除CO,当沉积适量Fe3O4时,CO氧化的起燃温度明显降低。因此,本研究在铜锰铈三元催化剂基础上复合微量贵金属Pt,以期提高催化剂对芳烃类VOCs的活化能力,进而提高总VOCs的去除效果。
本研究选取油墨印刷VOCs废气中含量最多的2种物质甲苯和乙酸乙酯作为目标污染物,以蜂窝状堇青石为载体制备Pt复合铜锰铈(CMC)负载型催化剂(Pt-CMC/堇青石),重点考察Pt复合前后催化剂对模拟VOCs废气催化燃烧效果的差异,以探究低含量贵金属添加对铜锰铈氧化物催化剂催化活性的影响程度。
1. 材料与方法
1.1 实验原料与仪器
蜂窝状正方体堇青石(7目,边长150 mm);硝酸铜、硝酸锰(50 wt%)、硝酸铈、氯铂酸,分析纯;硅溶胶,分析纯。超声波清洗器(KQ3200,昆山市超声仪器有限公司);电热鼓风干燥箱(101-3AB,天津泰斯特仪器有限公司); 箱式电阻炉(SX-4-10,天津泰斯特仪器有限公司);单模腔微波装置(ZDM-2,南京汇研微波系统工程有限公司);气相色谱仪(GC/FID,6890 N,美国安捷伦科技公司);泵吸式VOC检测仪(XS-2000-VOC,希思智能科技);扫描电子显微镜(JSM-6510LV型,日本电子);比表面积及孔径分析仪(V-sorb2800P型,北京金埃谱公司);X射线衍射仪(X’Pert型,荷兰帕纳科)。
1.2 催化剂制备与表征
本研究采用等体积浸渍法进行负载型催化剂的制备,即载体吸水率测试基础上配置适量体积的浸渍液、然后浸渍液被载体完全吸收的一种催化剂制备方法。首先,将正方体堇青石切割成圆柱型载体(D×L=27×150 mm),称重并测其吸水率,根据吸水率确定助剂硅溶胶的用量;其次,按3∶3∶1的质量负载比例分别称取铜(2.5%(质量分数))、锰、铈的金属盐,称取一定量的硅溶胶以及少量氯铂酸(Pt的质量分数0.01%),加入去离子水超声震荡30 min配制完全溶解的浸渍液;最后,将堇青石载体置于浸渍液中将浸渍液完全吸收,样品静置风干后放入烘箱中80 ℃过夜烘干,电阻炉内500 ℃煅烧4 h,自然冷却后即可制得Pt-CMC/堇青石催化剂。不加入氯铂酸、仅加入氯铂酸和其余步骤均相同情况下,可分别制得CMC/堇青石、Pt /堇青石催化剂。
利用扫描电子显微镜观察催化剂表面形貌及活性组分分布状况,借助BET测试仪分析催化剂的比表面积及孔径孔体积,使用X射线衍射仪测试活性组分的晶体结构及晶粒大小。
1.3 实验装置与方法
实验装置如图1所示,整个装置分为配气、催化燃烧和尾气净化3部分。在配气系统中,空气依次通过变色硅胶柱和活性炭柱去除水分和有机物,其流量通过气体流量计控制;实验时采用微量注射泵将液态甲苯和乙酸乙酯(按一定比例混合)以恒定速度连续均匀地注入三口烧瓶中,三口烧瓶被电加热套加热而将有机物气化,气化的甲苯和乙酸乙酯被空气带出并在混合瓶内混合均匀。实验开始前催化剂置于石英管内,石英管填装催化剂的长度为360 mm,锥形收口的长度是20 mm,上下两端开口外径分别为32 mm和9 mm,壁厚2 mm;石英管固定在单模腔微波装置上形成固定床反应器,实验开始后微波通过单模腔持续辐照在固定床上,催化剂吸波升温达到VOCs催化燃烧温度;石英管下端连接混合瓶,通入的VOCs气体在固定床上发生催化燃烧反应,床中插入的热电偶探针与温度显示仪连接以实时监测床层温度变化,石英管上下分别设进、出气采样口。催化燃烧后的VOCs尾气分别通过缓冲瓶和装有乙醇和碱液的尾气吸收瓶,净化后的废气经通风橱排空。
 图 1 微波催化燃烧VOCs实验装置示意图Figure 1. Schematic diagram of the experimental device for microwave catalytic combustion of VOCs
图 1 微波催化燃烧VOCs实验装置示意图Figure 1. Schematic diagram of the experimental device for microwave catalytic combustion of VOCs本研究利用CMC/堇青石、Pt-CMC/堇青石和Pt /堇青石催化剂在处理气量0.12 m3·h−1、气时空速(gas hourly space velocity,GHSV)1 500 h−1条件下,考察了不同微波功率(30、40、50、60、70 W)和不同VOCs浓度(1 000、1 500、2 000 mg·m−3)下甲苯和乙酸乙酯的催化燃烧效率,分析微波催化燃烧甲苯反应动力学及Pt复合影响,比较3种催化剂的催化活性及Pt复合对总VOCs去除效率的提升作用,探究Pt-CMC/堇青石催化剂对甲苯、乙酸乙酯和总VOCs催化活性的稳定性。
1.4 分析方法
本研究主要利用气相色谱仪对甲苯和乙酸乙酯的进气和出气质量浓度进行定量分析,气相色谱检测条件为[16]:柱箱初始温度100 ℃,以20 ℃·min−1的速率升温至180 ℃,保持3 min,加热器温度为190 ℃,检测器温度300 ℃;设定分流比为50∶1;尾气吹脱用氮气,流量30 mL·min−1。
使用手持泵吸式VOC检测仪对反应器进气和出气的总VOCs浓度进行检测,从而分析反应过程中总VOCs的催化燃烧效率。文中数据为2次平行实验的平均值,以此消除实验偶然性误差的影响。
2. 结果与讨论
2.1 催化剂表征
1) BET。根据表1数据可知,与堇青石载体相比,CMC/堇青石与Pt-CMC/堇青石2种催化剂的比表面积有不同程度的增加,这表明催化剂的吸附能力也相应增强。与堇青石载体相比,CMC/堇青石催化剂总孔容相对减小,平均孔径有所增大。推测其原因是,活性组分负载后,载体表面的部分孔隙被活性组分填充和覆盖,载体结构的原有孔道被拓宽。Pt-CMC/堇青石催化剂活性晶粒间相互积聚生成了包括微孔和介孔在内的新孔道,催化剂的比表面积及孔体积都明显增大,这有利于污染物分子在催化剂表面的吸附与活化。因此,Pt的复合在一定程度上增强了催化剂的吸附活化性能。
表 1 堇青石载体和2种催化剂的比表面积、孔体积及孔径数据表Table 1. Data table of specific surface area, pore volume and pore diameter of Cordierite and two catalysts供试样品 比表面积/(m2·g−1) 微孔面积/(m2·g−1) 孔体积/(cm3·g−1) 总孔容/(cm3·g−1) 平均孔径/nm 堇青石 0.11 ND ND 0.30 36.79 CMC/堇青石 0.66 ND ND 0.09 41.23 Pt-CMC/堇青石 2.95 1.11 0.000 47 0.38 36.85 备注:ND为未检出。 | Show Table DownLoad:
CSV
DownLoad:
CSV
2) XRD。如图2所示,2 θ在10°~40°之间存在着较为密集的堇青石特征衍射峰。对比XRD谱图可以发现,活性组分的负载和制备时的高温煅烧不会改变堇青石自身的晶体结构;与堇青石载体相比,CMC/堇青石和Pt-CMC/堇青石催化剂的特征衍射峰强度减弱,活性晶粒的平均尺寸有不同程度的减小。活性组分负载后堇青石特征峰峰强减弱的原因可能是活性组分一定程度上覆盖和屏蔽了堇青石表面的特征峰[17];贵金属Pt复合后,金属氧化物等活性组分晶体特征峰的峰高有微弱增大,这可能是不同尖晶石活性组分共存所致。6个处于17.5°、21.9°、28.7°、34.1°、36.7°、38.3°位置较为明显的特征峰,均归属于铜锰铈及其金属氧化物,这说明在贵金属Pt复合前后,铜锰铈金属氧化物都是微波催化燃烧VOCs的主要活性组分。在堇青石载体、CMC/堇青石催化剂和Pt-CMC/堇青石催化剂上,3种催化剂的晶粒平均尺寸分别为50.7、13.9和14.5 nm。Pt复合后,催化剂晶粒尺寸变化较小,而比表面积却从0.66 m2·g−1增大到2.95 m2·g−1,由此可以说明贵金属Pt的复合使活性颗粒的分散性增大[18]。
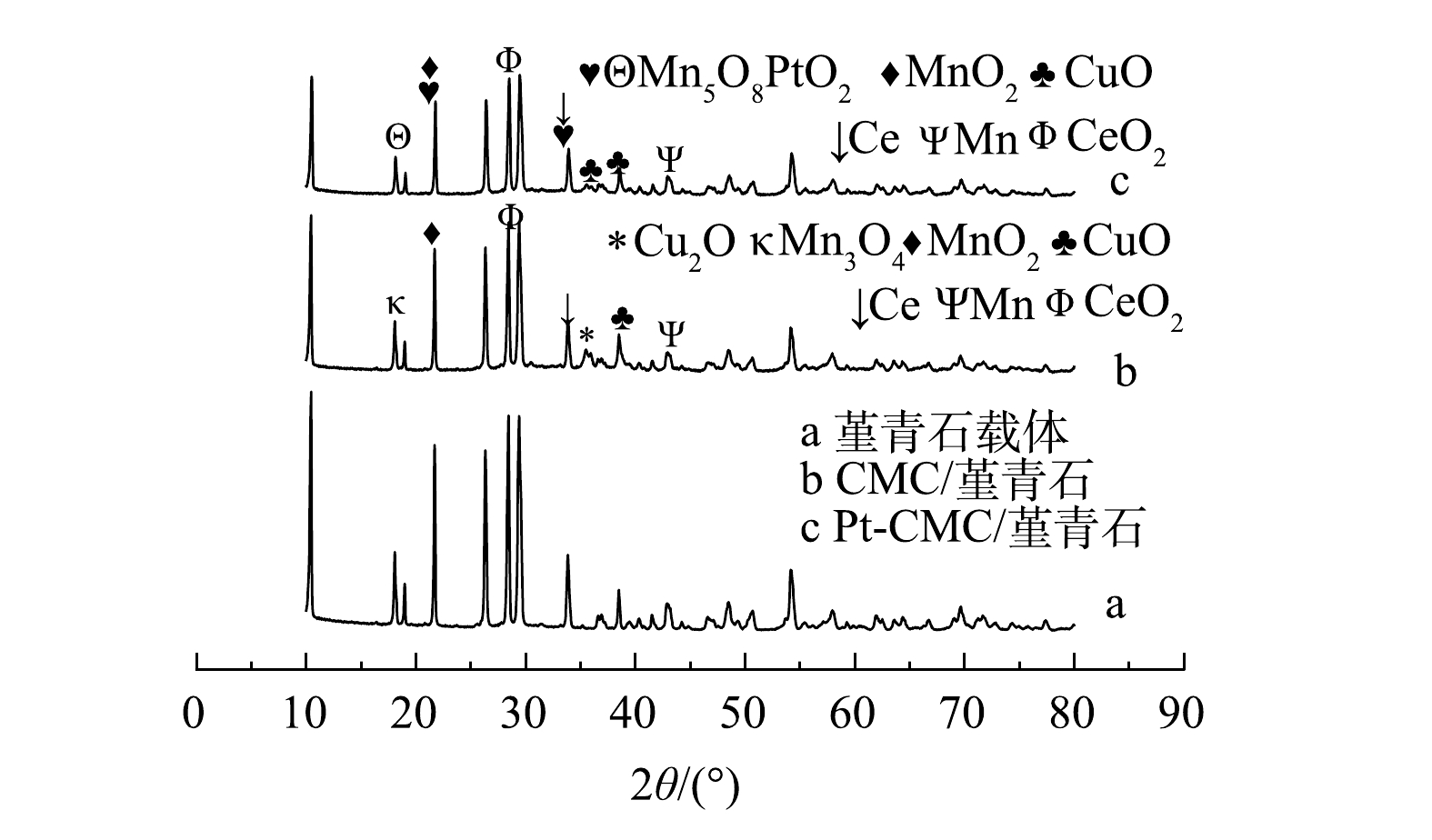 图 2 堇青石载体及CMC/堇青石、Pt-CMC/堇青石催化剂的XRD谱图Figure 2. XRD spectra of Cordierite support and CMC/Cordierite and Pt-CMC/Cordierite catalysts
图 2 堇青石载体及CMC/堇青石、Pt-CMC/堇青石催化剂的XRD谱图Figure 2. XRD spectra of Cordierite support and CMC/Cordierite and Pt-CMC/Cordierite catalysts结合特征峰组分分析可知,少量贵金属Pt的添加改变了催化剂上铜和锰的晶相。贵金属Pt复合前,锰以+4价的MnO2和中间价态的Mn3O4形式存在;贵金属Pt复合后,部分金属锰的价态升高,以Mn5O8形式存在,且Cu2O的晶体在掺杂后未检测到,检测出以+4价存在的PtO2。有文献报道,Cu、Mn金属离子价态升高,可以增强催化剂的活性[19]。
3) SEM。图3给出堇青石载体和CMC/堇青石、Pt-CMC/堇青石催化剂的表面形貌。可以看出,堇青石载体呈明显的层状结构,表面孔隙较多且分布均匀,有利于活性组分的负载。CMC/堇青石催化剂表面存在着较多微米尺寸的活性颗粒,不均匀地分布在载体表面;部分活性组分经高温煅烧后团聚成大块与堇青石结构连结在一起,填充载体原有孔隙的同时也产生了新的孔隙。Pt-CMC/堇青石催化剂表面的活性颗粒在载体表面上分布更均匀、分散度更高。对比Pt复合前后催化剂的微观形貌,可以看出,Pt的复合改变了活性颗粒的分散性,活性物质的再分散可能导致催化剂的吸附性改变。
 图 3 堇青石载体、CMC/堇青石与Pt-CMC/堇青石催化剂的微观表面形貌Figure 3. Microscopic surface morphology of Cordierite, CMC/Cordierite and Pt-CMC/Cordierite catalyst
图 3 堇青石载体、CMC/堇青石与Pt-CMC/堇青石催化剂的微观表面形貌Figure 3. Microscopic surface morphology of Cordierite, CMC/Cordierite and Pt-CMC/Cordierite catalyst2.2 催化剂性能实验
1)吸波升温曲线。图4为堇青石载体、CMC/堇青石、Pt-CMC/堇青石和Pt/堇青石在200 W微波功率辐照下的吸波升温曲线。由图4可见,堇青石载体温度几乎不变,这表明其吸波性能很差;而Pt/堇青石催化剂在15 min内温度也仅升高了3 ℃,其原因是贵金属具有高热稳定性,且吸波性能差。CMC/堇青石和Pt-CMC/堇青石催化剂升温效果明显好于前2种,微波辐照15 min时分别升温至300和355 ℃,说明铜锰铈及其金属氧化物具有良好的吸波性能,是2种催化剂吸波升温的主要原因;Pt复合催化剂升温速率更快,这可能是Pt的复合提高了催化剂上活性晶粒的分散性,降低了金属对微波的反射,以及颗粒尺寸变大增强了微波辐照时的局部热点效应,从而使得催化剂的吸波升温性能提高[20-21]。催化剂的快速升温有助于微波热点效应和高温活性区的出现,从而有助于甲苯和乙酸乙酯的快速氧化与彻底降解。
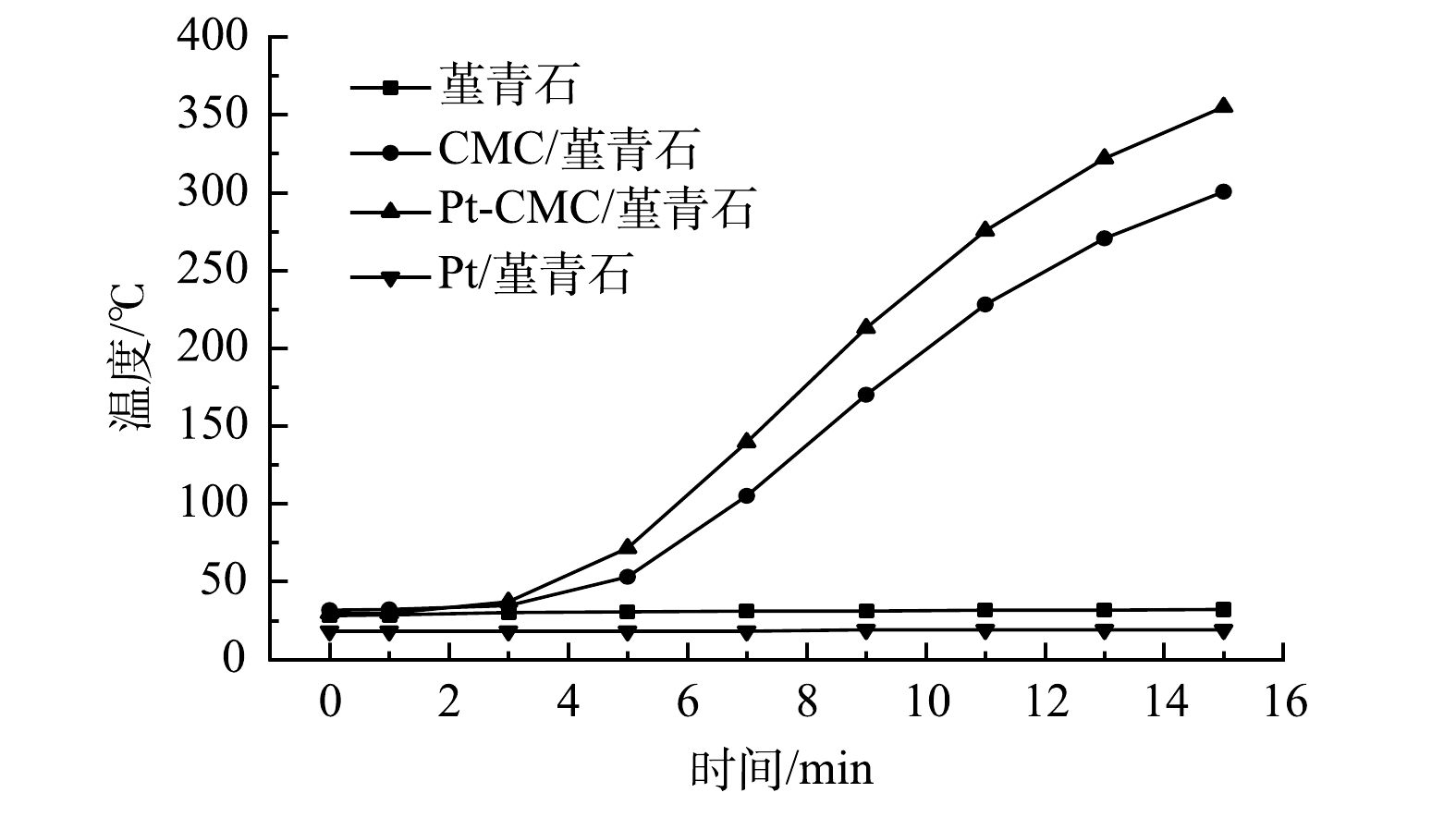 图 4 堇青石载体及3种催化剂的吸波升温曲线Figure 4. Wave absorption heating curve of Cordierite and three catalysts
图 4 堇青石载体及3种催化剂的吸波升温曲线Figure 4. Wave absorption heating curve of Cordierite and three catalysts2)催化剂活性。在甲苯和乙酸乙酯的进气浓度分别为1 000和500 mg·m−3,进气量为0.12 m3·h−1,微波功率分别选取30、40、50、60、70 W的条件下,开展CMC/堇青石和Pt-CMC/堇青石催化剂对甲苯和乙酸乙酯双组分VOCs废气的催化燃烧性能试验,结果如图5所示。图5中纵坐标C/C0为反应实时浓度与进气浓度比值(图6同),可直观表示降解效率并使数据间具有可比性。
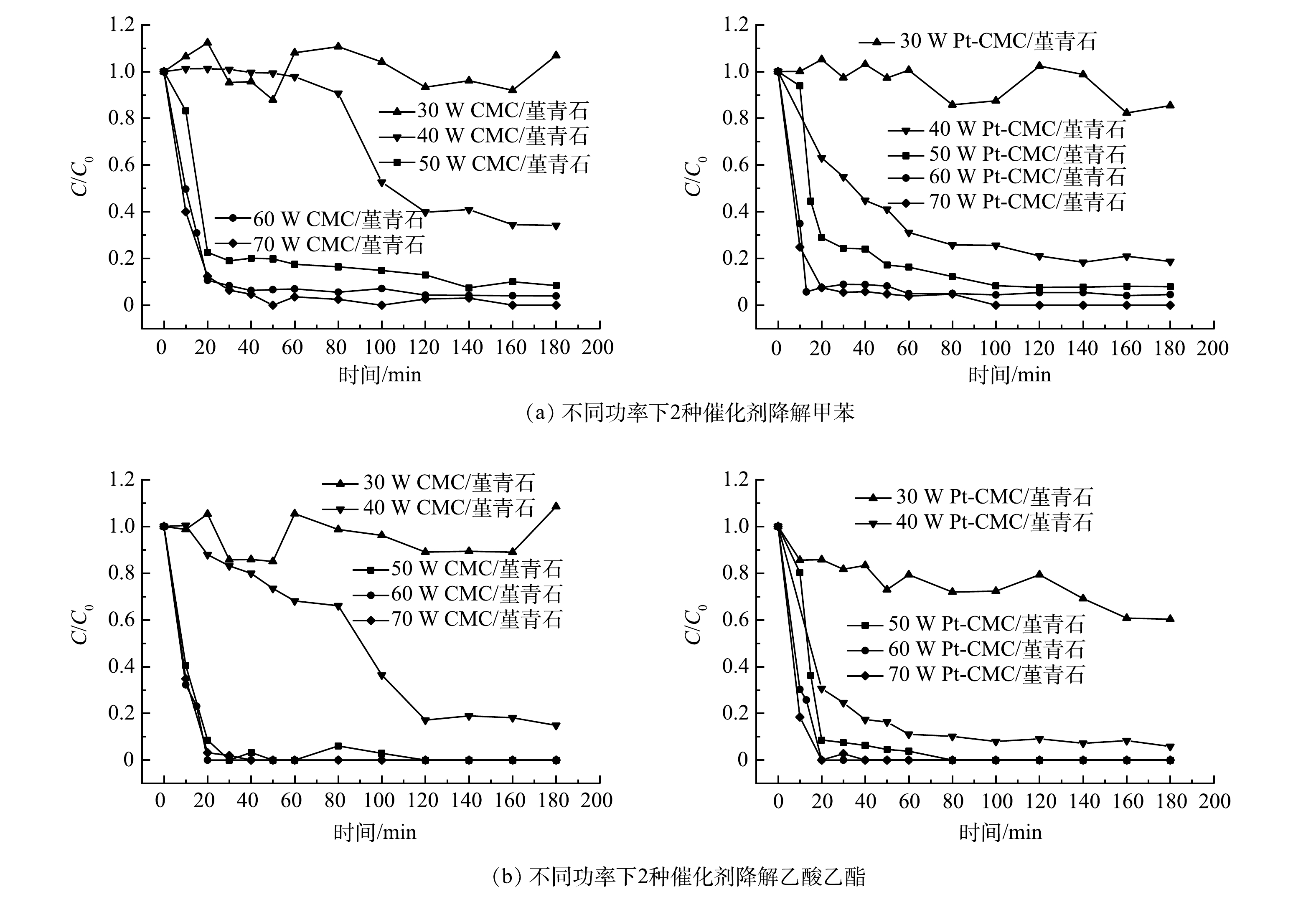 图 5 不同微波功率下CMC/堇青石和Pt-CMC/堇青石催化燃烧甲苯和乙酸乙酯效率曲线Figure 5. CMC/Cordierite and Pt-CMC/Cordierite catalytic combustion efficiency curves of toluene and ethyl acetate under different microwave power
图 5 不同微波功率下CMC/堇青石和Pt-CMC/堇青石催化燃烧甲苯和乙酸乙酯效率曲线Figure 5. CMC/Cordierite and Pt-CMC/Cordierite catalytic combustion efficiency curves of toluene and ethyl acetate under different microwave power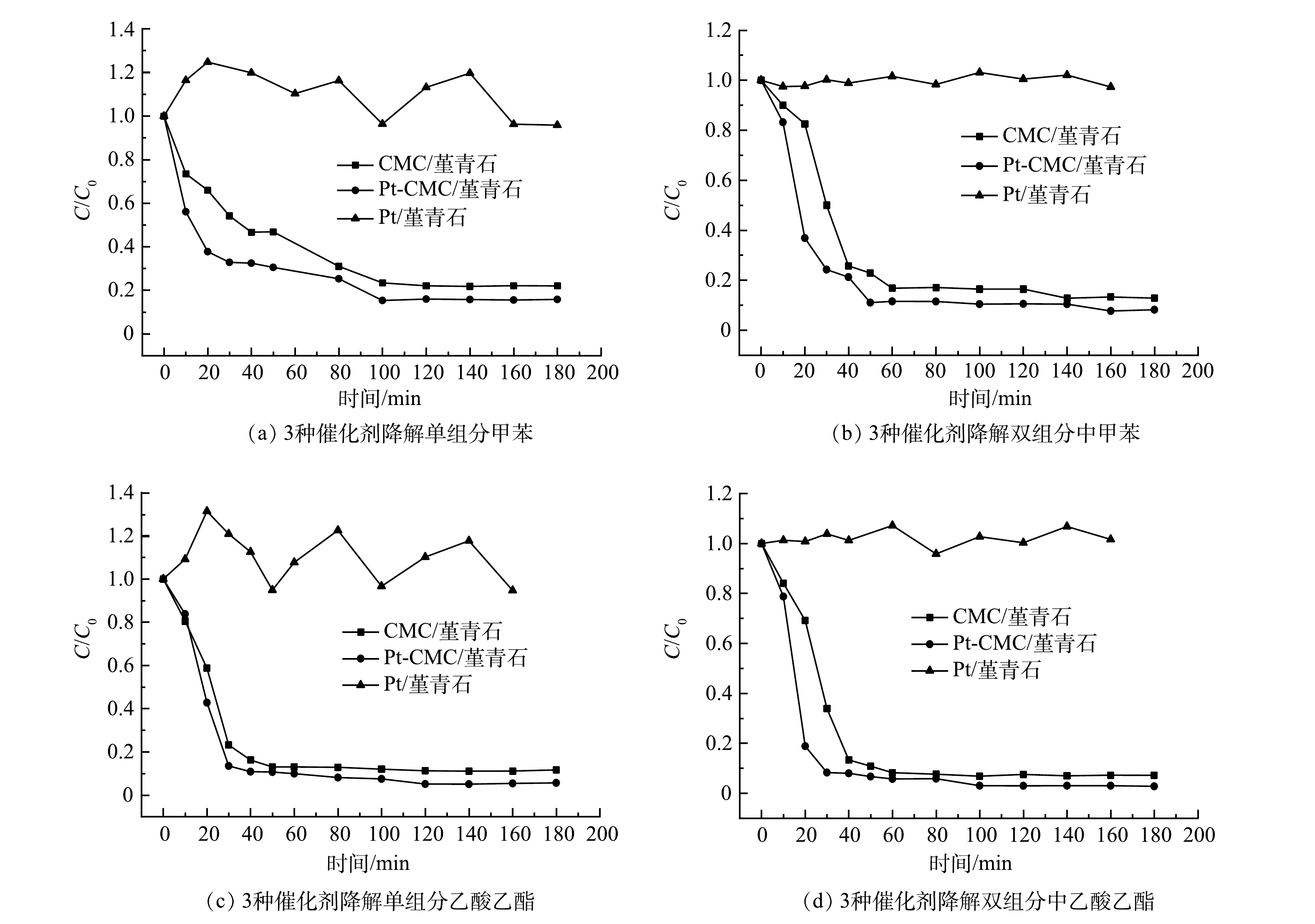 图 6 不同催化剂对甲苯和乙酸乙酯的催化活性比较Figure 6. Comparison of catalytic activity of three catalysts to toluene and ethyl acetate
图 6 不同催化剂对甲苯和乙酸乙酯的催化活性比较Figure 6. Comparison of catalytic activity of three catalysts to toluene and ethyl acetate由图5可见,微波功率30 w时,2种催化剂对甲苯和乙酸乙酯的降解呈不稳定态势,其原因是床层温度偏低(CMC/堇青石床层温度低于66 ℃),催化剂对污染物的去除以吸附-脱附为主。随着微波功率增大,甲苯和乙酸乙酯的降解效率也随之升高,但2种催化剂对甲苯和乙酸乙酯降解效率的差异却由大变小:40 W时,催化剂对甲苯和乙酸乙酯降解效率的差异最大;70 W时,这种差异最小,此时固定床温度高,污染物在催化剂表面完全燃烧,温度的作用掩盖了Pt的复合作用。
同一微波功率下,降解甲苯与乙酸乙酯时, Pt-CMC/堇青石催化剂表现出更高的VOCs催化活性。40 W微波功率下,Pt-CMC/堇青石对甲苯和乙酸乙酯的降解效率分别为82%和93%,而CMC/堇青石降解甲苯和乙酸乙酯的效率分别为66%和82%。推测其原因是,Pt-CMC/堇青石的比表面积相比CMC/堇青石大幅增加,从而有利于甲苯与乙酸乙酯的吸附与活化;同时,活性组分更好的分散性使得Pt-CMC/堇青石升温更快,而高温则有利于VOCs的催化燃烧。如果要使CMC/堇青石催化剂对甲苯和乙酸乙酯的降解效率与Pt-CMC/堇青石相同,则须提高微波功率至47 W。由此可见,Pt的复合提高了催化剂的低温催化活性,有效降低了VOCs的催化燃烧成本。
贺丽娜等[16]证明了甲苯的微波催化燃烧为假一级反应。因此,依据一级反应动力学方程ln(C0/C)=kt,对CMC/堇青石和Pt-CMC/堇青石3种催化剂在微波功率分别为50、60、70 W时降解双组分VOCs废气中甲苯的反应动力学进行线性拟合,相应方程与参数如表2所示。结果表明,CMC/堇青石和Pt-CMC/堇青石微波催化燃烧甲苯的反应符合假一级反应动力学,同时R2值随微波功率的增加而增大,此时一级动力学拟合程度越高,证明高温下的催化燃烧反应为一级反应。反应速度常数k值对比发现,微波功率越大,k值越大,催化反应速度越快;微波功率60 W时,Pt-CMC/堇青石降解甲苯的k值为0.129,明显高于CMC/堇青石的0.111 9。可见,Pt的复合增强了甲苯的吸附活化作用而加快了反应速率,进而提高CMC活性组分对甲苯的微波催化燃烧效率。
表 2 微波催化燃烧甲苯反应动力学Table 2. Reaction kinetics of microwave catalytic combustion of toluene功率条件 催化剂 ln(C0/C)=kt k R2 50 W Pt-CMC/堇青石 y=0.0594x−0.0204 0.0594 0.9828 60 W CMC/堇青石 y=0.1119x−0.1398 0.1119 0.9553 60 W Pt-CMC/堇青石 y=0.129x−0.0797 0.129 0.9887 70 W CMC/堇青石 y=0.1045x−0.0428 0.1045 0.995 70 W CMC/堇青石 y=0.1299x+0.031 0.1299 0.9983 | Show TableDownLoad:
CSV
3)催化剂比较。图6为微波功率40 W、进气量0.12 m3·h−1,甲苯和乙酸乙酯浓度各为1 000 mg·m−3条件下,CMC/堇青石、Pt-CMC/堇青石和Pt/堇青石催化剂降解单组分甲苯、乙酸乙酯及二者混合的效率曲线。无论是单、双组分甲苯和乙酸乙酯的催化燃烧降解,均是Pt-CMC/堇青石的活性最高,CMC/堇青石次之,而Pt/堇青石则未表现出催化活性。对于Pt/堇青石而言,其在微波辐照下几乎不升温,从而无法为VOCs的催化燃烧提供所需的温度条件,因此甲苯和乙酸乙酯几乎不降解。无论是甲苯或乙酸乙酯的单组分降解,还是双组分废气降解,乙酸乙酯的降解效率均高于甲苯。以Pt-CMC/堇青石为例,单组分甲苯和乙酸乙酯的降解效率分别为84%和95%(此时床层温度相近)。乙酸乙酯比甲苯更易于降解的原因可能为:一是乙酸乙酯等含氧有机物比甲苯更易被催化剂吸附活化;二是乙酸乙酯上的C-O键比甲苯的C-H键键能低而更易断裂[22-23]。
比较分析图6中甲苯和乙酸乙酯的降解效率发现:对于单组分甲苯和乙酸乙酯的降解,Pt-CMC/堇青石较CMC/堇青石有约7%和6%的提升;对于甲苯与乙酸乙酯的双组分降解,Pt-CMC/堇青石较CMC/堇青石有约5%和4%的提升。由此可见,Pt复合对甲苯降解效率的提升大于乙酸乙酯。分析认为,CMC/堇青石对于一些C-H键较弱、吸附性好的含氧有机物如乙酸乙酯、丙酮等具有良好的活化性能,但催化氧化C-H键较强的芳香类物质的能力则稍弱,而贵金属Pt活化C-H键的能力很强,弥补了铜锰铈金属氧化物活化苯环能力的不足[24]。此外,CMC/堇青石和Pt-CMC/堇青石对甲苯和乙酸乙酯双组分废气的降解效率明显高于各自单组分,其原因是双组分VOCs废气的初始浓度更高,催化燃烧时放出更多热量使得床层温度更高,从而有利于VOCs的氧化降解。
4)总VOCs去除。微波功率40 W时,对催化剂催化燃烧1 000 mg·m−3甲苯的总VOCs去除率进行测试,其结果和床层温度变化如图7所示。由图7(a)可见,Pt/堇青石的总VOCs去除率为负值且波动幅度大,CMC/堇青石和Pt-CMC/堇青石的总VOCs去除率分别为25%和40%,Pt的复合将总VOCs的去除效率提高了15%。如图7(b)所示,Pt/堇青石的不吸波使其床层温度为常温,催化燃烧反应未发生,甲苯在催化剂表面进行吸附-脱附动态平衡过程,因而其总VOCs去除率波动大且多为负值;Pt-CMC/堇青石床层升温速率快于CMC/堇青石,稳定后的床层温度高出CMC/堇青石床层30 ℃,这是总VOCs去除效率提高的主要原因。分析认为,Pt的复合在提升催化剂吸波升温能力的同时,其对甲苯C-H键的活化能力也有助于提高总VOCs的去除效率。随着微波功率的增加,床层温度急剧升高,总VOCs去除率也随之升高,催化剂对总VOCs去除效率的差别也随之减小,Pt复合的影响被床层温度影响掩盖而体现不出。因此,Pt的复合提高了催化剂的低温催化活性,有助于低温下VOCs的催化氧化。
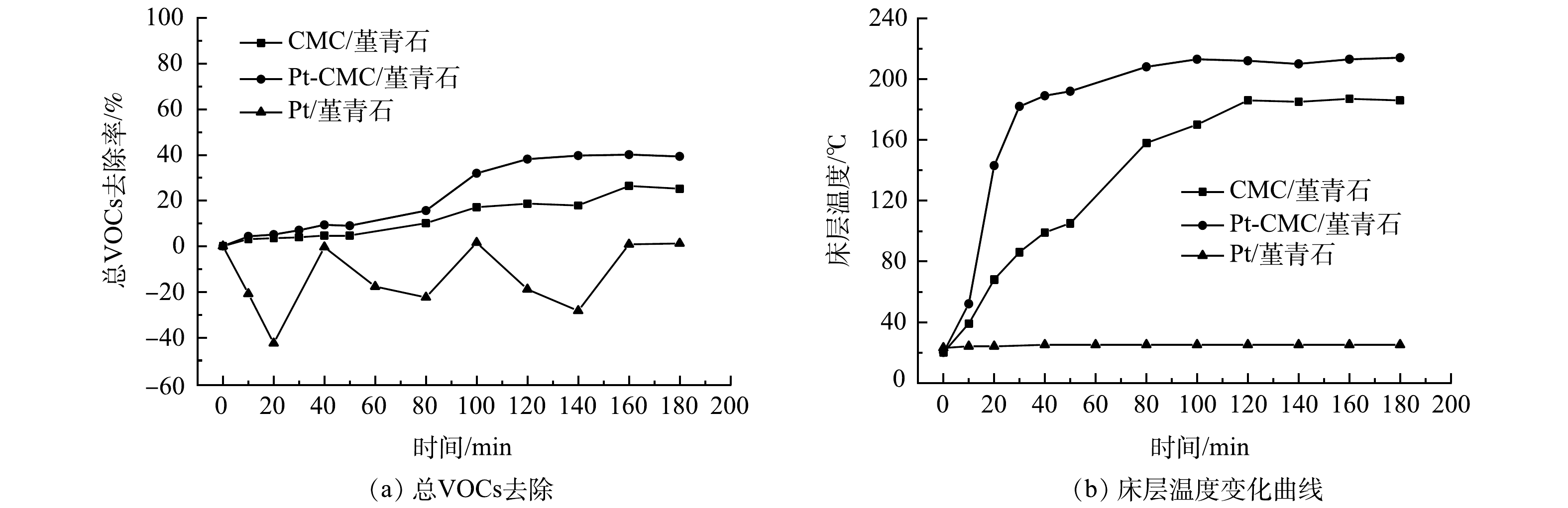 图 7 不同催化剂的总VOCs去除效率及床层温度曲线Figure 7. The total VOCs removal efficiency and bed temperature curves of the three catalysts
图 7 不同催化剂的总VOCs去除效率及床层温度曲线Figure 7. The total VOCs removal efficiency and bed temperature curves of the three catalysts5)稳定性实验。根据图5的实验结果可知,微波功率70 W时Pt-CMC/堇青石催化剂对甲苯和乙酸乙酯的催化效果最好,同时总VOCs的去除率也最高。因此,在微波功率70 W、进气量0.12 m3·h−1、双组分气体中甲苯和乙酸乙酯初始浓度各为1 000 mg·m−3和500 mg·m−3条件下对Pt-CMC/堇青石催化剂进行了连续7次(每次实验3 h)的稳定性测试,结果如图8所示。
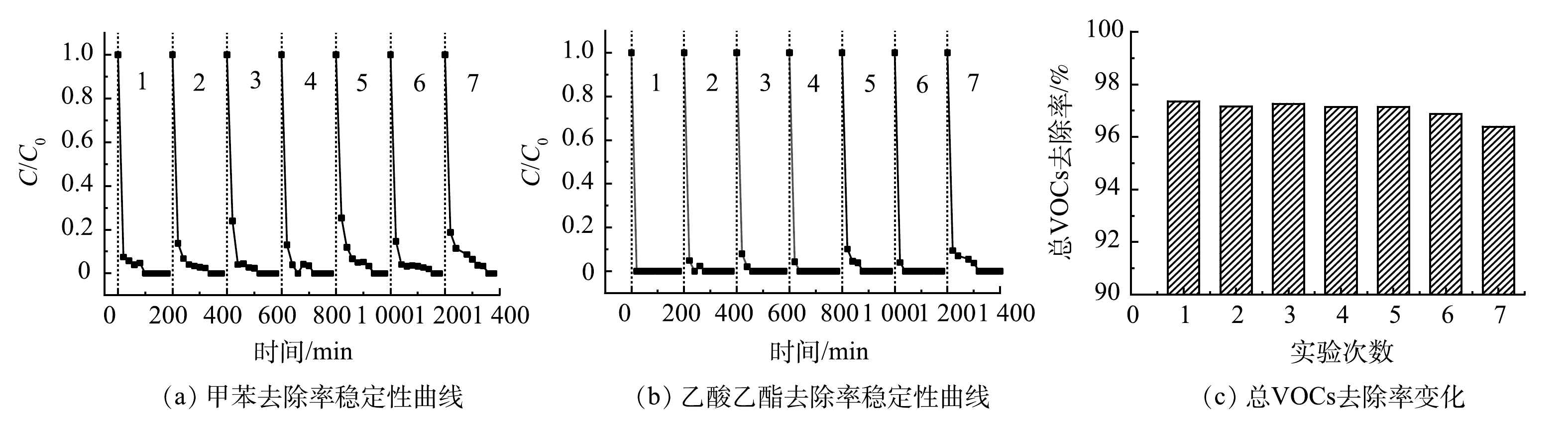 图 8 Pt-CMC/堇青石催化燃烧甲苯和乙酸乙酯稳定性测试结果Figure 8. Pt-CMC/Cordierite catalytic combustion toluene and ethyl acetate stability test results
图 8 Pt-CMC/堇青石催化燃烧甲苯和乙酸乙酯稳定性测试结果Figure 8. Pt-CMC/Cordierite catalytic combustion toluene and ethyl acetate stability test results如图8(a)、8(b)所示,连续稳定性试验中,甲苯和乙酸乙酯被完全去除,乙酸乙酯的去除效率较甲苯更稳定,催化剂展示出良好的催化活性和稳定性。图8(c)的总VOCs去除率从97.5%略微降至96.5%,表明Pt-CMC/堇青石对双组分VOCs废气具有高的矿化效率与稳定性。微波功率70 W的稳定性实验中,催化剂床层温度320 ℃,高于40 W时的210 ℃,可见,床层温度的升高是提升总VOCs去除率的有效手段之一。连续的高温稳定性实验,会引起催化剂孔隙结构的微变和活性颗粒的团聚,进而引起双组分VOCs(特别是甲苯)去除效率的波动;然而,Pt复合增强了催化剂表面活性颗粒的分散性,增大了催化剂的比表面积与孔容,从而有效提高了催化剂活性,保证了催化剂性能的稳定。
3. 结论
1)等体积浸渍法制备的Pt-CMC/堇青石催化剂比CMC/堇青石催化剂具有更大的活性颗粒尺寸,Pt的再分散作用使得催化剂表面活性组分分布更加均匀;Pt的复合提高了催化剂的孔隙率,进而有利于VOCs分子在催化剂表面的吸附与活化;Pt复合后的尖晶石活性组分峰有微弱增大,Cu、Mn价态升高而使催化剂活性增强。
2)复合Pt后,催化剂的吸波升温能力有一定的增强,这与吸波活性组分分布均匀、散热能力增强有关。反应动力学分析表明,Pt的复合加快了催化燃烧反应速率,高温下甲苯的催化燃烧反应为一级反应;复合Pt后的催化剂其低温催化活性明显提高,Pt-CMC/堇青石催化剂表面总VOCs去除效率比CMC/堇青石提高15%。
3)对于Pt-CMC/堇青石和CMC/堇青石催化剂而言,乙酸乙酯的降解效率高于甲苯,可认为含氧的乙酸乙酯比甲苯更易被催化剂吸附活化,C-O键比甲苯的C-H键键能低而更易断裂;Pt复合对甲苯降解效率的提高大于乙酸乙酯,其原因是Pt对苯环中C-H键的活化能力强而有助于苯环的氧化与开环。
-

图 1 以藻酸盐为底物的恒化器厌氧运行状况
Figure 1. Performance of a mesophilic chemostat with alginate as substrate

图 2 ADC菌群利用模拟物质厌氧酸化的情况
Figure 2. Distribution of produced VFAs from typical organics by dosing ADC

图 3 不同pH条件下ADC菌群强化剩余污泥厌氧酸化
Figure 3. Enhanced VFAs production by dosing ADC bacteria under different pH conditions

图 4 不同pH条件下剩余污泥的SCOD值及水解和酸化的效率
Figure 4. Released SCOD and hydrolysis and acidification efficiency of WAS under different pH conditions
表 1 剩余污泥基本特性
Table 1. Basic characteristics of WAS
pH TSS/(g·L−1) VSS/(g·L−1) TCOD/(g·L−1) SCOD/(g·L−1) PS/(mg·L−1) PN/(mg·L−1) 7.2 ± 0.1 21.7 ± 2.2 10.2 ± 0.1 10.4 ± 1.6 0.10 ± 0.01 2.7 ± 0.1 237.3 ± 4.9
下载: 导出CSV
-
[1] 卢怡清, 许颖, 董滨, 等. 去除城市生活污泥中有机络合态金属强化其厌氧生物制气[J]. 环境科学, 2018, 39(1): 284-291. [2] 施正华, 李秀芬, 宋小莉, 等. 采用等电点沉淀法回收市政污泥水解液中的蛋白质[J]. 环境工程学报, 2016, 10(10): 5919-5923. doi: 10.12030/j.cjee.201603072 [3] 董滨, 高君, 陈思思, 等. 我国剩余污泥厌氧消化的主要影响因素及强化[J]. 环境科学, 2020, 41(7): 3384-3391. [4] BAELE D J, KARPE A V, MCLEOD J D, et al. An ‘omics’ approach towards the characterisation of laboratory scale anaerobic digesters treating municipal sewage sludge[J]. Water Research, 2016, 88: 346-357. [5] SMITH K, LIU S, LIU Y, et al. Can China reduce energy for water? A review of energy for urban water supply and wastewater treatment and suggestions for change[J]. Renewable & Sustainable Energy Reviews. 2018, 91: 41-58. [6] LATIF M A, MEHTA C M, BATSTONE D J. Influence of low pH on continuous anaerobic digestion of waste activated sludge[J]. Water Research, 2017, 113: 42-49. doi: 10.1016/j.watres.2017.02.002 [7] 王凯军, 王婧瑶, 左剑恶, 等. 我国餐厨垃圾厌氧处理技术现状分析及建议[J]. 环境工程学报, 2020, 14(7): 1735-1742. doi: 10.12030/j.cjee.201911085 [8] DAI K, ZHANG W, ZENG R J, et al. Production of chemicals in thermophilic mixed culture fermentation: mechanism and strategy[J]. Critical Reviews in Environmental Science and Technology, 2020, 50(1): 1-30. doi: 10.1080/10643389.2019.1616487 [9] ZHEN G, LU X, KATO H, et al. Overview of pretreatment strategies for enhancing sewage sludge disintegration and subsequent anaerobic digestion: current advances, full-scale application and future perspectives[J]. Renewable and Sustainable Energy Reviews, 2017, 69: 559-577. doi: 10.1016/j.rser.2016.11.187 [10] 林治岐, 张信, 邵尉, 等. 废水生物处理过程中微生物胞外聚合物与污染物的分子间相互作用[J]. 中国科学:化学, 2018, 48(9): 1102-1108. [11] XIE G J, LiU B F, WANG Q, et al. Ultrasonic waste activated sludge disintegration for recovering multiple nutrients for biofuel production[J]. Water Research, 2016, 93: 56-64. doi: 10.1016/j.watres.2016.02.012 [12] ZHANG Y, ZHANG C, ZHANG X, et al. Waste activated sludge hydrolysis and acidification: A comparison between sodium hydroxide and steel slag addition[J]. Journal of Environmental Sciences, 2016, 48: 200-208. doi: 10.1016/j.jes.2016.02.010 [13] ARENAS C B, GONZALEZ R, GONZALEZ J, et al. Assessment of electrooxidation as pre- and post-treatments for improving anaerobic digestion and stabilisation of waste activated sludge[J]. Journal of Environmental Management, 2021, 288: 112365. doi: 10.1016/j.jenvman.2021.112365 [14] LIN Y M, DE KREUK M, VAN LOOSDRECHT M C, et al. Characterization of alginate-like exopolysaccharides isolated from aerobic granular sludge in pilot-plant[J]. Water Research, 2010, 44(11): 3355-3364. doi: 10.1016/j.watres.2012.09.017 [15] FELZ S, KLEIKAMP H, ZLOPASA J, et al. Impact of metal ions on structural EPS hydrogels from aerobic granular sludge[J]. Biofilm, 2020, 2: 100011. doi: 10.1016/j.bioflm.2019.100011 [16] 曹达啟, 王振, 郝晓地, 等. 藻酸盐污水处理合成研究现状与应用前景[J]. 中国给水排水, 2017, 33(4): 1-6. [17] 李佳琦, 彭党聪, 董征, 马保卫. 藻酸盐对污泥性能的影响及提取方法的研究[J]. 中国给水排水, 2018, 34(11): 15-19. [18] LIN Y M, SHARMA P K, VAN LOOSDRECHT M C. The chemical and mechanical differences between alginate-like exopolysaccharides isolated from aerobic flocculent sludge and aerobic granular sludge[J]. Water Research, 2013, 47(1): 57-65. [19] ZHANG F, ZHANG W, QIAN D K, et al. Synergetic alginate conversion by a microbial consortium of hydrolytic bacteria and methanogens[J]. Water Research, 2019, 163: 114892. [20] LI J, HAO X, VAN LOOSDRECHT M C M, et al. Effect of humic acids on batch anaerobic digestion of excess sludge[J]. Water Research, 2019, 155: 431-443. doi: 10.1016/j.watres.2018.12.009 [21] FELZ S, VERMEULEN P, VAN LOOSDRECHT M C M, et al. Chemical characterization methods for the analysis of structural extracellular polymeric substances (EPS)[J]. Water Research, 2019, 157: 201-208. doi: 10.1016/j.watres.2019.03.068 [22] GILCREAS F W. Standard methods for the examination of water and waste water[J]. American Journal of Public Health and the Nation's Health, 1966, 56(3): 387-388. doi: 10.2105/AJPH.56.3.387 [23] ZHANG F, QIAN D K, GENG Z Q, et al. Enhanced methane recovery from waste-activated sludge by alginate-degrading consortia: The overlooked role of alginate in extracellular polymeric substances[J]. Environmental Science & Technology Letters, 2021, 8(1): 86-91. [24] ZHANG F, DING J, ZHANG Y, et al. Fatty acids production from hydrogen and carbon dioxide by mixed culture in the membrane biofilm reactor[J]. Water Research, 2013, 47(16): 6122-6129. doi: 10.1016/j.watres.2013.07.033 [25] DOGAN I, SANIN F D. Alkaline solubilization and microwave irradiation as a combined sludge disintegration and minimization method[J]. Water Research, 2009, 43(8): 2139-2148. doi: 10.1016/j.watres.2009.02.023 [26] FANG W, ZHANG P, ZHANG G, et al. Effect of alkaline addition on anaerobic sludge digestion with combined pretreatment of alkaline and high pressure homogenization[J]. Bioresource Technology, 2014, 168: 167-172. doi: 10.1016/j.biortech.2014.03.050 [27] XIN X, HE J, FENG J, LI L, et al. Solubilization augmentation and bacterial community responses triggered by co-digestion of a hydrolytic enzymes blend for facilitating waste activated sludge hydrolysis process[J]. Chemical Engineering Journal, 2016, 284(15): 979-988. [28] TEO CW, WONG PCY. Enzyme augmentation of an anaerobic membrane bioreactor treating sewage containing organic particulates[J]. Water Research, 2014, 48: 335-344. doi: 10.1016/j.watres.2013.09.041 [29] LIN Y, DE K M, VAN LOOSDRECHT M C M, et al. Characterization of alginate-like exopolysaccharides isolated from aerobic granular sludge in pilot-plant[J]. Water Research, 2010, 44(11): 3355-3364. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5209
- HTML全文浏览数: 5209
- PDF下载数: 92
- 施引文献: 0
