


-
近年来,土壤污染问题已引起广泛关注。据《全国土壤污染状况调查公报》[1]显示,全国土壤点位超标率为16.1%,主要存在重金属和有机物污染。石油生产、有机肥料和农药的生产及使用、电子垃圾的处理和火力发电厂燃料的燃烧等工农业生产活动造成有机污染物和重金属大量进入环境,造成土壤污染[2-3],且呈现多类型污染物复合存在的态势[4-5]。根据针对土壤与地下水修复行业的调查可知,2018年,复合污染修复项目占工业污染场地项目的37.5%、重金属污染占32.8%、有机污染占29.7%[6]。
土壤修复技术逐渐趋于多样化,其中,热处理技术由于具有良好的修复效果和较短的修复周期而被广泛应用于有机污染修复[7]。2007—2017年,热处理修复技术的应用比例为38.7%,2018年的应用比例为43.4%[6]。常用热处理技术包括工业炉窑协同处置、热解吸和原位热脱附等。早期的热处理修复研究主要集中在单一污染物。魏萌[8]的研究表明,热处理对焦化场地土壤多环芳烃具有良好的去除效果,而对土壤中的重金属全量则无明显影响。BONNARD等[9]发现,热脱附后,土壤中多环芳烃的去除率可达到94%,而重金属全量未发生改变,但对蚯蚓的生物毒性增大了,该处理通过改变重金属形态提高了重金属的生物有效性,进而增加了其遗传毒性。焦文涛等[10]发现,在高温条件下,土壤中稳定金属的存留率会增加,导致其生物毒性增大。焦化厂、电子垃圾拆解厂、火力发电厂以及污水灌溉等在生产使用过程中可能造成重金属和有机物的复合污染[5,11-14]。在热处理修复土壤有机污染时,往往忽略了土壤中重金属的赋存形态以及健康风险的变化。工业窑炉高温协同处置重金属污染土壤时,也应关注重金属风险的变化。
与大气环境、水环境管理思路不同,我国污染地块的管理思路主要是基于用地功能、环境和健康风险考虑。依据《建设用地土壤污染风险评估技术导则》(HJ 25.3-2019)[15] (以下简称《导则》),土壤重金属的健康风险采用全量进行评估,但土壤重金属的赋存形态是决定其在土壤中的迁移性、生物可利用性以及毒性的重要因素[16]。环境中的重金属不能完全被人体所吸收,因此,仅以重金属全量为评价依据可能会造成高估其环境风险[17]。XU等[18]发现,添加生物炭可降低土壤中重金属的可提取态,增加其残渣态,从而可降低白菜对重金属的可吸收性。XIA等[19]发现,添加羟基磷灰石会降低重金属的酸可提取态,增加其残渣态,从而降低了其浸出毒性。重金属可通过多种途径引起暴露及生物吸收,对生态环境和人体健康造成一定的威胁[20]。而热处理对土壤重金属赋存形态和生物可给性的研究较少。基于生物可给性的重金属风险评估更能反映土壤的实际风险,体外模拟实验能够反映土壤重金属在消化系统中的生物可利用性[21],其中基于生理学的提取实验(PBET)模型等被广泛认可和应用[22-23]。
本研究通过研究电镀企业场地土壤热处理对重金属赋存形态和生物可给性的变化,探究热处理对重金属人体健康风险的影响,以期为污染土壤热处理修复工程的健康风险评估和生态效应评估提供参考。
-
供试土壤样品取自于浙江台州某电镀退役场地,共4个采样点,编号为S1、S2、S3和S4(图1)。其中,S1、S3取自原电镀车间,分别为0~1 m杂填土和2~3 m粉土;S2、S4取自原污水站,深度分别为1~2 m粉土和3~4 m粉土。样品采集后,去除石块等杂质风干、过2 mm筛网备用。
-
将土壤样品置于马弗炉中进行热处理,取30 g 土样置于100 mL陶瓷坩埚中,将马弗炉升温至相应温度(200、400、600 ℃)后,将坩埚放入马弗炉保持15 min。处理结束后,取出坩埚置于干燥器中进行冷却,待测。
-
土壤pH使用pH计(SevenCompact-S210)测定,水土比为2.5∶1。4种重金属(Cu、Pb、Ni和Cd)全量使用盐酸-硝酸-氢氟酸-高氯酸进行消解,使用火焰原子吸收分光光度法进行测定。采用改进的BCR连续提取法进行重金属形态分析,用0.11 mg·L−1醋酸提取土壤中酸可提取态(F1);用0.5 mg·L−1(pH=1.5)盐酸羟胺提取土壤中可还原态(F2);加 8.8 mg·L−1过氧化氢在85 ℃下水浴 1 h(重复2次),再用1 mg·L−1 (pH=2)醋酸铵提取可氧化态(F3);用全量消解方法测定残渣态(F4)。重金属生物可给性采用PBET体外模拟方法,健康风险基于生物可给性进行评估。
-
1) PBET。模拟胃液由1.25 g 胃蛋白酶、0.50 g苹果酸钠、0.50 g柠檬酸钠、420 μL乳酸及500 μL醋酸配合而成,溶解后定容到1 L,并用1∶1 HCl溶液调节pH至2.5。称0.4 g土壤样品加入40 mL模拟胃液在37 ℃、100 r·min−1下振荡1 h,之后,抽取20 mL模拟胃液过0.22 μm滤膜待测。胃液提取结束后,用NaHCO3粉末调节消化液pH至7.0,加入52.5 mg胆汁盐和15 mg胰液素(模拟肠液)然后继续振荡2 h,结束后过0.22 μm滤膜待测。
2)生物可给性计算。胃肠阶段的生物可给性(BA)[24]由公式(1)计算。
式中:BA为重金属在胃或肠阶段的生物可给性; CIV为胃或肠阶段反应液中重金属含量,mg·L−1;VIV为反应液的体积,L;TS为土壤样品中重金属总量,mg·kg−1;MS为反应器中土样样品的质量,kg。
3)健康风险评估。在暴露评估和毒性评估的基础上,利用风险评估模型计算土壤中污染物经暴露的致癌风险和危害商。当危害商的值大于1时,风险为“不可接受”。人体对土壤重金属暴露可通过3个途径:经口摄入、呼吸作用和皮肤接触。其中,经口摄入为主要暴露途径[25-26],因此,本研究对电镀退役场地土壤健康风险评估条件为“一类用地经口暴露途径”,健康风险评估基于溶解在肠胃中的生物可利用部分进行评估。本研究采用US EPA的风险评价模型,其中涉及的Cu、Pb、Ni和Cd均具有非致癌风险,危害商计算方式及其涉及的参数见参考文献[27-29]。
-
表1为供试土壤pH、重金属全量和重金属形态比例。由表1可知,S1点位重金属全量较高,其中Pb和Ni污染较重;同一点位下,不同深度下重金属质量分数存在差异,4种重金属形态主要以可还原态和残渣态的形式存在。S1点位取自镀镍车间0~1 m,长期直接与污染源接触,造成Pb和Ni污染严重;污水站S2、S4点位土壤重金属质量分数则较低。重金属进入土壤后,能够被土壤中的矿物质、微生物、有机物等物质通过吸附-解吸、氧化-还原、络合等作用固定在表层[30],但是在某些环境条件下(例如酸雨淋滤和水流渗透等),会跟随水分迁移到土壤深层[31],因此造成不同深度的重金属质量分数存在差异。
-
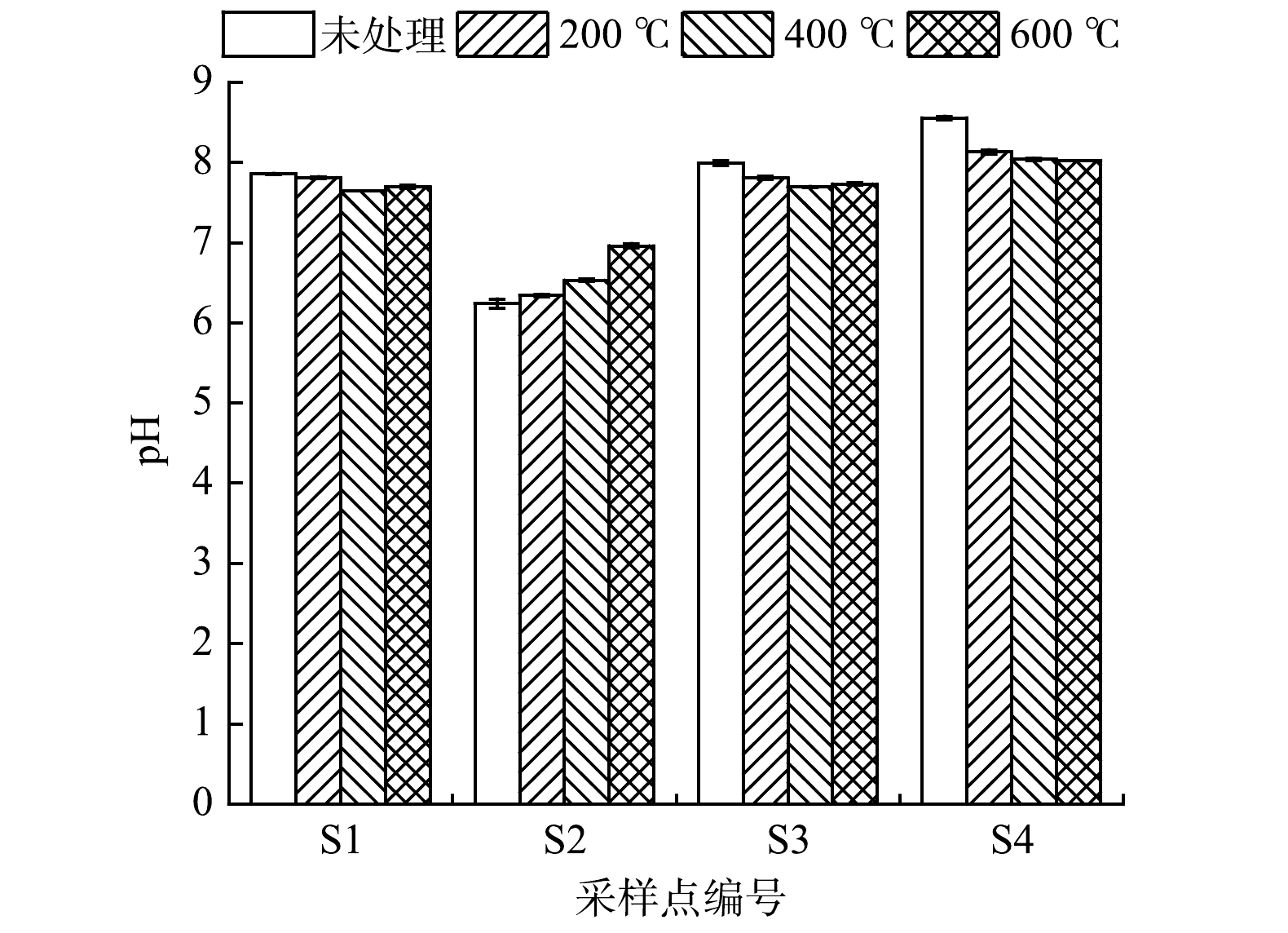
热处理前,原始土壤S2呈弱酸性,pH为6.24,S1、S3和S4的pH分别为7.86、8.00、8.57,呈弱碱性。热处理前后土壤pH变化见图2。由图2可看出,各点位土壤经热处理后,均未改变其弱酸性或弱碱性的性质。随着温度的升高,S1点位的土壤pH无显著变化(7.86~7.64),这可能是由于土壤中含有缓冲物质CaCO3[32]。S2点位的土壤经热处理后,pH有略微升高,600 ℃处理时最大pH为6.96。这可能是由于随着温度的升高,土壤中水和铁或铁的氧化物形成了脱水赤铁矿[33-34]。S3和S4点位土壤经热处理后较处理前略微降低。这可能是由于有机质释放CO2,矿化作用释放质子[35]。
-
热处理前后土壤中重金属全量变化见图3。由图3可以看出,经热处理后,土壤中重金属全量未发生明显变化。重金属及其化合物的熔点和沸点是决定其在热处理中全量变化的重要因素[36]。Cu和Ni的熔点分别为1083和1453 ℃,其氧化物的最低熔点为1326和1 980 ℃[37],均远高于本实验中的最高温600 ℃,因此,Cu和Ni全量无明显变化。Pb和Cd单质的熔点327和321 ℃[37],在实验设置温度范围内,相对易挥发,但在热处理过程中,当土壤处于氧化性气氛下,重金属主要以其氧化物形式存在,Pb和Cd氧化物的熔点分别888和1426 ℃[37]且热处理时间短,导致重金属并未有明显挥发,这与王昕晔[38]研究结果一致。
-
图4反映了热处理前后重金属赋存形态的变化。由图4可看出,热处理后,S1土壤中Cu、Pb和Ni酸可提取态呈现出增加的趋势,而Cd的趋势相反。Cu可还原态所占比例为32%~37%,经热处理后,可还原态减少,残渣态与可氧化态2种形态之和明显增加。热处理后,S1土样中Cu酸可提取态仍然存在,且有升高的趋势,这与李进平等[39]的研究结果相一致。这可能是由于:高温下有机质分解,附着其上的酸可提取态被释放出来。S1中Pb在600 ℃处理下,酸可提取态增加了17%,Pb可还原态从79%减少至40%。S2和S4土样在400 ℃和600 ℃处理下,酸可提取态未检出,S4残渣态有所增加,这与张怡斐[40]的研究结果一致。随着热处理温度的增加,S1和S2中Ni的酸可提取态也随之增加,其中S1从未处理的2%增加到17%,S2由10%增加到23%。这与LENG等[41]报道的变化规律相同,在高温处理下,产生了不利于Ni固定的尖晶石。Cd的赋存形态以残渣态为主,热处理后,S1酸可提取态占比降低了12%,残渣态占比增加了19%。S4中酸可提取态Cd也呈现出降低的趋势,400 ℃处理下有异常,残渣态呈现增加的趋势。这可能是由于土壤中的铁锰氧化物与其反应络合转化成残渣态[42],说明热处理有利于土壤中Cd的稳定。
-
1)土壤重金属的生物可给性。热处理前后基于PBET的生物可给性见表2。由表2可知,经热处理后,S1中Cu、Pb和Ni 3种元素的胃肠阶段生物可给性都随着温度的增加而增加,600 ℃处理下的生物可给性增加较为明显。而Cd的生物可给性随温度的增加而降低,400 ℃处理下有异常,这与图4酸可提取态的变化趋势相同。酸可提取态增加,则相应的生物可给性亦增加。S2、S3和S4样品在热处理情况下,Cu的胃肠生物可给性总和变化与酸可提取态变化相同,其中,在600 ℃下S3增加程度最大达到5.24%。
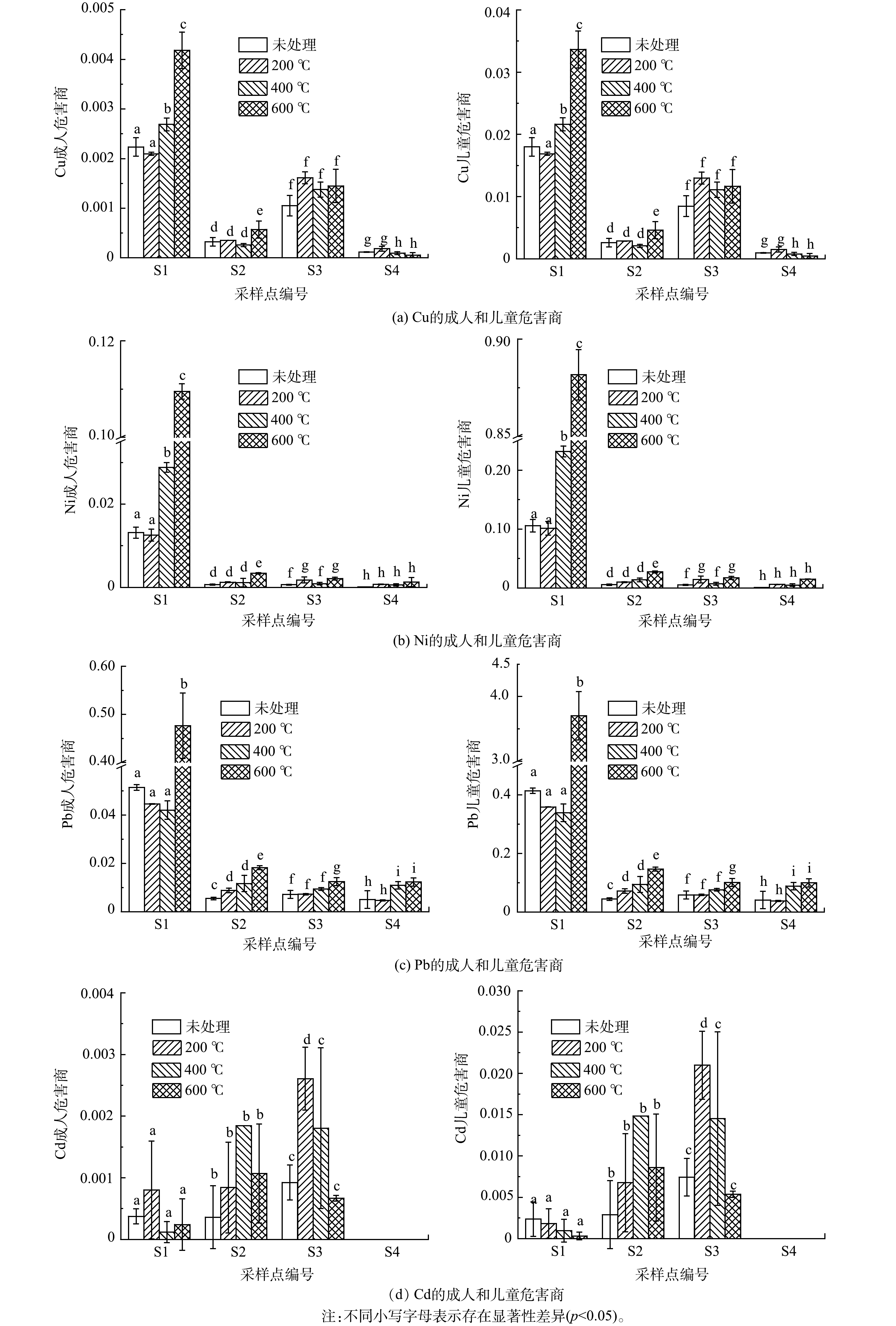
2)基于PBET的热处理前后土壤重金属风险评估。热处理前后基于PBET成人和儿童的危害商如图5所示。热处理后,S1点位Cu、Ni和Pb成人和儿童风险均增加。其中,Pb在600 ℃处理下,对儿童的危害商从0.41增加至3.70,其他点位金属在热处理后的风险仍小于1,热处理后Cd对成人和儿童的风险无显著性差异。4种金属对儿童的风险均大于成人,未处理原始土样重金属的危害商均小于1。分别对成人和儿童重金属危害商进行显著性差异分析,S1为高浓度污染的点位,Cu、Ni和Pb在600 ℃处理下呈现出显著性差异(p<0.05),健康风险明显增加。这是由于热处理后生物可给性增大(表2)以及酸可提取态的变化(图4)造成的,酸可提取态重金属易被人体肠胃消化吸收,从而造成健康风险增加[43]。S1中,Cd热处理后健康风险无显著性差异(p>0.05)。这可能是由于表2中生物可给性较小,变化不明显。
-
1) 在600 ℃处理下,重污染土壤中Cu、Pb和Ni的酸可提取态增加,而Cd的酸可提取态减少。
2) 重金属污染土壤中,Cu、Pb和Ni胃肠阶段生物可给性随热处理温度的升高而增加,而Cd的生物可给性则随热处理温度的变化呈现相反的规律,与其酸可提取态的变化趋势相同。
3) 体外模拟实验和人体健康风险研究结果表明,热处理会增加土壤Cu、Pb、Ni的成人和儿童健康风险,但Cd的健康风险无显著性差异。热处理引起的土壤重金属可给性变化与酸可提取态的变化密切相关。
热处理对土壤重金属形态的影响及健康风险
Effects of thermal treatment on soil heavy metals speciation and health risks
-
摘要: 为探究土壤热修复后的土壤重金属形态以及健康风险的变化,以退役电镀企业地块的污染土壤为研究目标,分别在200、400和600 ℃下处理土壤15 min,以分析热处理对土壤重金属Cu、Pb、Ni和Cd赋存形态的影响、生物可给性变化以及重金属人体健康风险的差异。结果表明,经热处理后,土壤Cu、Pb和Ni的酸可提取态增加,增加了胃肠阶段的生物可给性,而Cd酸可提取态减少,生物可给性降低。在基于生物可给性的风险评估中,热处理会增加Cu、Pb和Ni的健康风险,在600 ℃处理下,Pb对儿童的危害商从0.41增加至3.70,Cd的健康风险无显著性差异。本研究结果可为场地土壤热处理后的重金属健康风险及生态效应评估提供参考。Abstract: In order to study the variation of chemical speciation of soil heavy metals and health risks after thermal treatment, the temperature was set at 200, 400 and 600 ℃ respectively for 15 minutes to analyze chemical speciation of Cu, Pb, Ni and Cd, the variation of its bioavailability and the differences in human health risks in the contaminated soils of the decommissioning electroplating site. The results showed the acid soluble fraction of Cu, Pb and Ni and its bioavailability in the gastrointestinal stage were increased after thermal treatment, while the acid soluble fraction of Cd and its bioavailability were decreased. In the risk assessment based on bioavailability, thermal treatment increased the health risk of Cu, Pb and Ni. the hazard quotient of Pb for children increased from 0.41 to 3.70 at 600 °C, but there was no significant difference in hazard quotient of Cd. This work could provide a reference for the evaluation of the health risk and ecological effects of heavy metals in contaminated sites after thermal treatment.
-
Key words:
- thermal treatment /
- soil heavy metals /
- speciation /
- health risk
-
石油类污染物的意外泄漏事故频繁发生,导致一定数量的非水相液体 (NAPL) 通过包气带进入地下造成污染[1]。在包气带内挥发性有机物 (VOCs) 会通过挥发作用进入土壤气体,VOCs气体在迁移过程中发生生物降解,源区轻非水相液体 (LNAPL) 由生物降解作用导致的衰减占LNAPL总质量损失的90%~99% [2-3]。因此,挥发性石油烃在包气带中的气相自然衰减和生物降解动力学规律的研究对VOCs蒸气入侵风险评估和石油场地监测自然衰减应用都具有重要意义[4-5],气相生物降解速率是蒸气入侵建模和定量风险评估的关键输入参数[6-7]。
当VOCs在污染源挥发成为气态在包气带扩散迁移过程中,吸附、生物降解等机制会导致显著的质量衰减,这被称为气相自然衰减[8-9]。在某汽油污染场地,从地下水源挥发的总碳氢化合物质量的68%在毛细管层被生物降解,石油烃蒸气潜水面上1 m内包气带内被降解殆尽[10]。HÖHENER等[11]通过微宇宙实验、柱实验及现场研究测定了包气带中13种VOCs的气相生物降解规律。VOCs气相自然衰减受到土壤类型、温度、含水率、营养水平等多种因素影响。土壤类型,特别是土壤孔隙度、含水量、土壤渗透性、有效扩散性和有机碳吸附能力是影响VOCs气相迁移归趋的重要因素[12]。YAO等[13]报道,土壤质地对VOCs浓度衰减的影响,其中土壤粒径的对底板下土壤气衰减因子的影响平均约为0.4个数量级。BEKELE等[14]研究了三氯乙烯 (TCE) 在5种不同的土壤中的气相吸附,粘土质量分数增加1倍会导致TCE蒸气吸附量增加11倍。但是,目前关于包气带VOCs气相自然衰减的研究仍然很不充分,对于不同种类石油烃蒸气在不同土壤中的吸附与生物降解规律以及我国不同区域土壤的气相自然衰减潜力尚不清楚。
本研究采用微宇宙实验,选取4种正构烷烃 (正戊烷、正己烷、正庚烷、正辛烷) 、4种环烷烃 (环戊烷、环己烷、环庚烷、环辛烷) 及4种苯系物 (苯、甲苯、乙苯、对二甲苯) ,对其在黑土、黄土、红土及石英砂等4种土壤中的气相自然衰减规律和生物降解速率进行了系统研究。
1. 材料与方法
1.1 实验材料
实验试剂采用色谱纯的正戊烷、正己烷、正庚烷、正辛烷、环戊烷、环己烷、环庚烷、环辛烷、苯、甲苯、乙苯、对二甲苯。本研究所用的黄土、黑土、红土和石英砂分别取自河北省、黑龙江省、云南省和江西省,土壤经自然风干后过2 mm标准方孔筛备用,其基本理化性质如表1所示。
表 1 实验土壤的基本理化性质Table 1. Basic physical-chemical characteristics of experimental soils土壤类型 pH 孔隙度 含水率 有效扩散系数 (20 ℃)/ (正戊烷,m2·d−1) 密度/ (g·cm−1) 有机质质量分数/% 土壤质地 机械组成/% 砂粒 粉粒 黏粒 黄土 7.8 0.5 0.02 0.28 1.3 0.66 壤土 49.6 39.6 10.7 黑土 6.1 0.8 0.09 0.80 0.6 36.70 壤质砂土 74.6 19.6 5.8 红土 5.7 0.6 0.02 0.36 1.0 0.33 粘土 11.2 5.4 83.4 石英砂 7.6 0.4 0.01 0.19 1.5 0.32 砂土 97.4 1.8 0.9 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 实验方法
微宇宙实验在50 mL的玻璃旋盖小瓶中进行,分别取20 g土壤及20 μL混合污染物于瓶中,使用特氟龙微型阀瓶盖密封。为了研究非生物作用对气相自然衰减的贡献,同时做灭菌对照组,使用120 ℃高压蒸汽灭菌30 min。每隔一段时间进行气体取样,采用250 μL气密性微量取样针取100 μL气体,用气相色谱仪测定污染物蒸气的质量浓度 (g·m−3) 。
1.3 分析方法
污染物气体质量浓度采用安捷伦7890B 气相色谱仪,检测器为火焰离子化检测器(FID),温度250 ℃,HP-5毛细管柱( 30 m×0. 32 mm×0. 25 μm),进样口温度设置为200 ℃,分流比10∶1。测试正构烷烃时,柱箱升温程序为初始温度55 ℃保持时间2.5 min,升温至140 ℃,保持时间0.2 min,升温速率为30 ℃·min−1。测试环烷烃时,柱箱温度为50 ℃保持时间2 min,升温至75 ℃,保持时间0.2 min,升温速率为10 ℃·min−1。测试苯系物时色谱柱箱恒温120 ℃。
自然衰减通常用一级动力学模型描述。气态烃的表观一级衰减速率是通过质量浓度数据明显下降阶段的ln (Ct/C0) 与时间 (t) 线性回归来确定,其中C0是污染蒸气的初始质量浓度,Ct是t时刻污染蒸气质量浓度[15]。非灭菌土壤中的表观一级衰减速率是生物降解和非生物自然衰减速率的总和,而灭菌土壤中的污染物质量浓度下降仅由非生物机制引起,因此标记为“非生物衰减速率”。对于同一种气态烃,非灭菌土壤中的“表观一级衰减速率”减去灭菌土壤中的“非生物衰减速率”是生物降解的真正贡献,标记为“生物降解速率”[16]。
2. 结果与讨论
2.1 正构烷烃在4种土壤中的气相自然衰减
4种正构烷烃 (正戊烷、正己烷、正庚烷、正辛烷) 在灭菌组和非灭菌组的衰减去除率均表现为黑土>黄土>红土>石英砂 (表2) 。4种正构烷烃的生物降解速率表现为黑土>黄土>红土,石英砂未发生明显的生物降解,因此无法拟合石英砂的降解速率 (图1、表3) 。灭菌组中气态烃的自然衰减机制主要是吸附,而土壤有机质质量分数是4种土壤气相吸附能力差异大的主要原因。4种土壤的有机质的质量分数顺序:黑土 (36.70%) >黄土 (0.66%) >红土 (0.33%) >石英砂 (0.32%) 。这与灭菌组中的气态烃衰减去除率一致。其他研究[14,17]也发现,土壤有机质质量分数是土壤对气态VOCs吸附的主要因素,高有机质质量分数会增加土壤对VOCs的吸附。BEKELE等[14]报道,土壤有机质质量分数增加1倍导致总三氯乙烯蒸气吸附量增加7倍。UGWOHA和ANDRESEN [17]发现,土壤有机质质量分数从0增加至5%后气态烷烃的吸附质量增加了2倍,土壤有机质量分数的增加会增强汽油烃蒸气的吸附速度和吸附量。气态VOCs需要首先被土壤吸附固定,然后才能被土壤微生物代谢降解[18],较高的有机质质量分数会提高土壤吸附固定VOCs的能力,从而会提高其生物降解能力。因此,土壤有机质质量分数是4种土壤气相吸附能力和生物降解能力差异大的主要原因。除有机质质量分数外,土壤的生物降解能力还与土壤土著微生物数量密切相关,经测试发现,本研究所用的黑土 (5.2×106 cfu·g−1) 和黄土 (2.1×106 cfu·g−1) 中的微生物数量远大于红土 (1.2×105 cfu·g−1) 和石英砂 (2.0×103 cfu·g−1) 。综上所述,较高有机质质量分数和微生物数量使得黑土对于气态烃具有较强的自然衰减能力。
表 2 石油烃在不同土壤中自然衰减去除率Table 2. The remove rates of petroleum hydrocarbons in different soils% 化合物 黄土 黑土 红土 石英砂 灭菌组 非灭菌组 生物降解的贡献* 灭菌组 非灭菌组 生物降解的贡献* 灭菌组 非灭菌组 生物降解的贡献* 灭菌组 非灭菌组 生物降解的贡献* 正戊烷 24.61 27.82 3.21 42.91 68.18 25.27 21.33 23.06 1.73 3.56 5.08 1.52 正己烷 35.35 45.64 10.29 64.67 87.24 22.57 32.89 36.05 3.16 2.18 3.03 0.86 正庚烷 51.17 74.77 23.61 74.62 93.20 18.58 40.25 45.29 5.04 0.61 1.39 0.78 正辛烷 63.14 78.43 15.28 78.12 96.55 18.44 49.37 46.77 2.60 5.63 7.87 2.24 环戊烷 13.30 33.68 20.38 20.90 43.04 22.14 5.29 6.29 1.00 5.58 9.62 4.05 环己烷 12.96 28.24 15.28 46.68 70.96 24.28 1.75 7.09 5.34 4.29 9.91 5.62 环庚烷 8.23 45.91 37.68 61.22 84.85 23.63 9.88 12.89 3.01 12.67 14.29 1.62 环辛烷 57.60 66.28 8.67 71.90 85.35 13.45 9.14 17.37 8.23 12.56 21.14 8.58 苯 69.62 81.55 11.93 77.66 92.32 14.66 64.88 75.69 10.81 27.15 27.51 0.36 甲苯 72.54 80.57 8.03 82.64 91.98 9.34 67.44 75.63 8.19 26.43 27.00 0.57 乙苯 72.31 79.12 6.81 80.98 85.64 4.66 53.64 56.86 3.23 19.38 19.88 0.50 对二甲苯 75.80 82.81 7.01 77.57 83.36 5.80 51.45 55.65 4.20 19.46 20.00 0.54 注:“*”表示生物降解贡献的去除率为非灭菌组与灭菌组去除率之差。 | Show TableDownLoad:
CSV
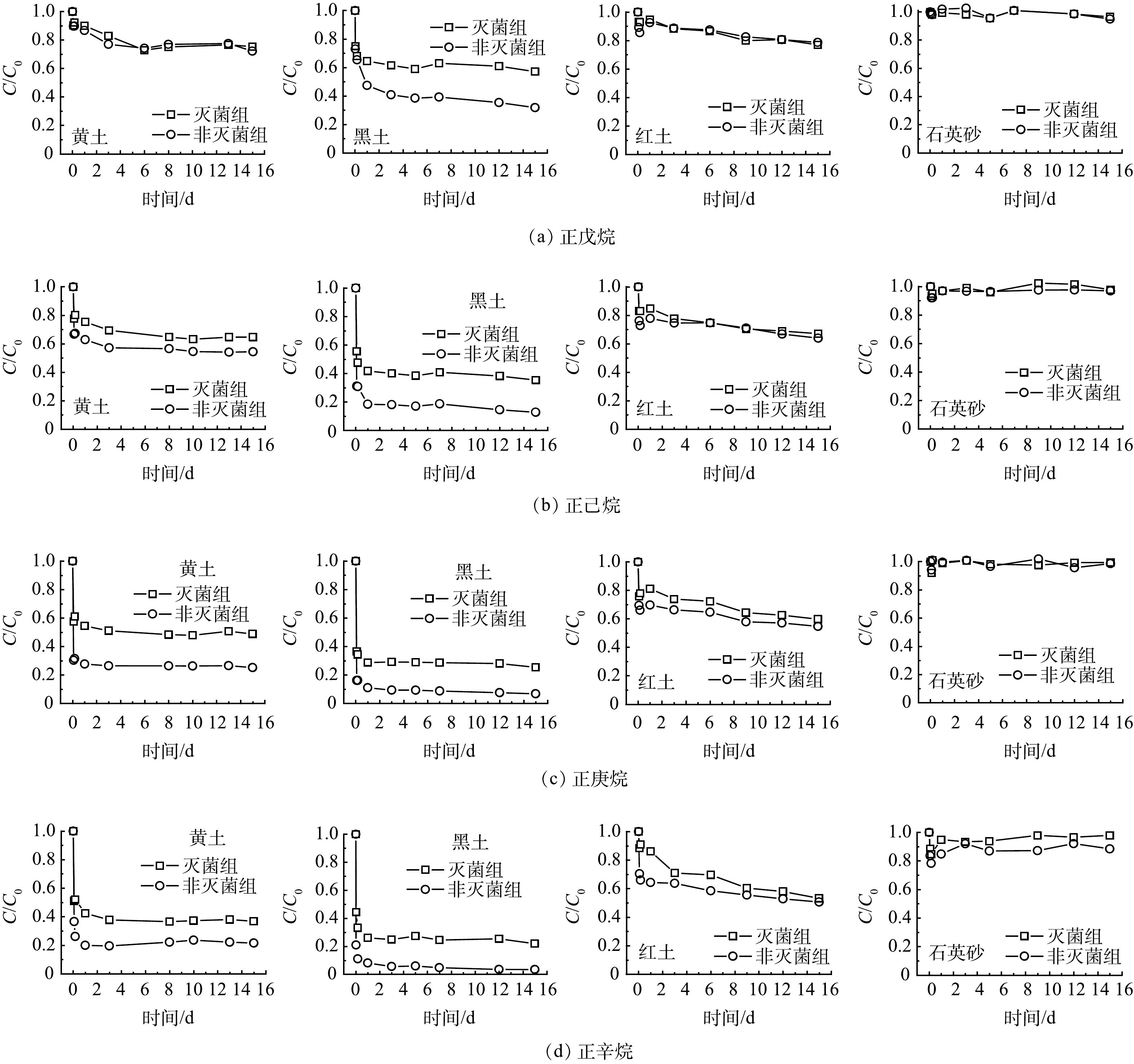 图 1 正构烷烃在4种土壤中的质量浓度变化曲线Figure 1. N-alkanes concentration attenuation curve in four soils表 3 石油烃在不同土壤中的一级衰减速率常数Table 3. The first order attenuation rate constants of petroleum hydrocarbons in different soils
图 1 正构烷烃在4种土壤中的质量浓度变化曲线Figure 1. N-alkanes concentration attenuation curve in four soils表 3 石油烃在不同土壤中的一级衰减速率常数Table 3. The first order attenuation rate constants of petroleum hydrocarbons in different soilsd−1 化合物 黄土 黑土 红土 石英砂 灭菌组 非灭菌组 生物降解 灭菌组 非灭菌组 生物降解 灭菌组 非灭菌组 生物降解 灭菌组 非灭菌组 生物降解 正戊烷 0.03 0.04 0.01 0.03 0.06 0.03 0.02 0.02 — — — — 正己烷 0.03 0.06 0.03 0.05 0.18 0.13 0.016 0.02 0.004 — — — 正庚烷 0.05 0.18 0.13 0.26 0.54 0.28 0.02 0.05 0.03 — — — 正辛烷 0.06 0.15 0.09 0.02 0.12 0.10 0.03 0.05 0.02 — — — 环戊烷 0.01 0.02 0.01 0.04 0.07 0.03 0.01 0.01 — — — — 环己烷 0.01 0.03 0.02 0.20 0.26 0.06 0.01 0.01 — — — — 环庚烷 0.02 0.05 0.03 0.28 0.32 0.04 0.01 0.01 — — — — 环辛烷 0.07 0.09 0.02 0.33 0.39 0.06 0.01 0.01 — — — — 苯 25.68 27.84 2.16 28.24 235.48 7.24 24.24 25.78 1.54 — — — 甲苯 30.72 38.96 8.24 29.36 39.12 9.76 25.68 28.44 2.76 — — — 乙苯 15.6 17.08 1.48 324.98 30.82 5.84 11.76 12.72 0.96 — — — 对二甲苯 14.4 15.58 1.18 23.76 27.66 3.90 11.04 11.94 0.90 — — — | Show TableDownLoad:
CSV
2.2 环烷烃在4种土壤中的气相自然衰减
4种环烷烃 (环戊烷、环己烷、环庚烷、环辛烷) 在不同土壤中的衰减规律与正构烷烃相似,在灭菌组和非灭菌组的衰减去除率整体上符合黑土>黄土>红土>石英砂的规律 (表2、图2) 。环烷烃在黑土、黄土和红土中的生物降解速率均很小 (<0.06 d−1) ,而石英砂中几乎不存在生物降解。这说明环烷烃蒸气的生物降解性极低 (表3) 。HöHENER等[11]也发现,气相环烷烃的自然衰减速率很低,实验和场地等不同方式获得的气相一级衰减速率常数均低于0.31 d−1。这可能由于环烷烃的溶解度较低,导致土壤气体和水之间的界面传质速率较低[19]。而且短链烷烃 (<C9) 对许多微生物都有毒性,中间链长的正构烷烃 (C10~C24) 降解速度最快,短链烷烃通过其溶剂作用来抑制微生物活性,它们在作为溶剂存在时会破坏脂质膜[20-21]。大量有机地球化学研究证实环烷烃的生物降解性低于正构烷烃[22-23],且生物降解随着环数的增加而降低[24]。环烷烃通过氧化酶的作用降解为环醇,进一步脱氢为酮,环烷烃代谢的主要产物是环酮和环烷烃-羧酸[20,24]。
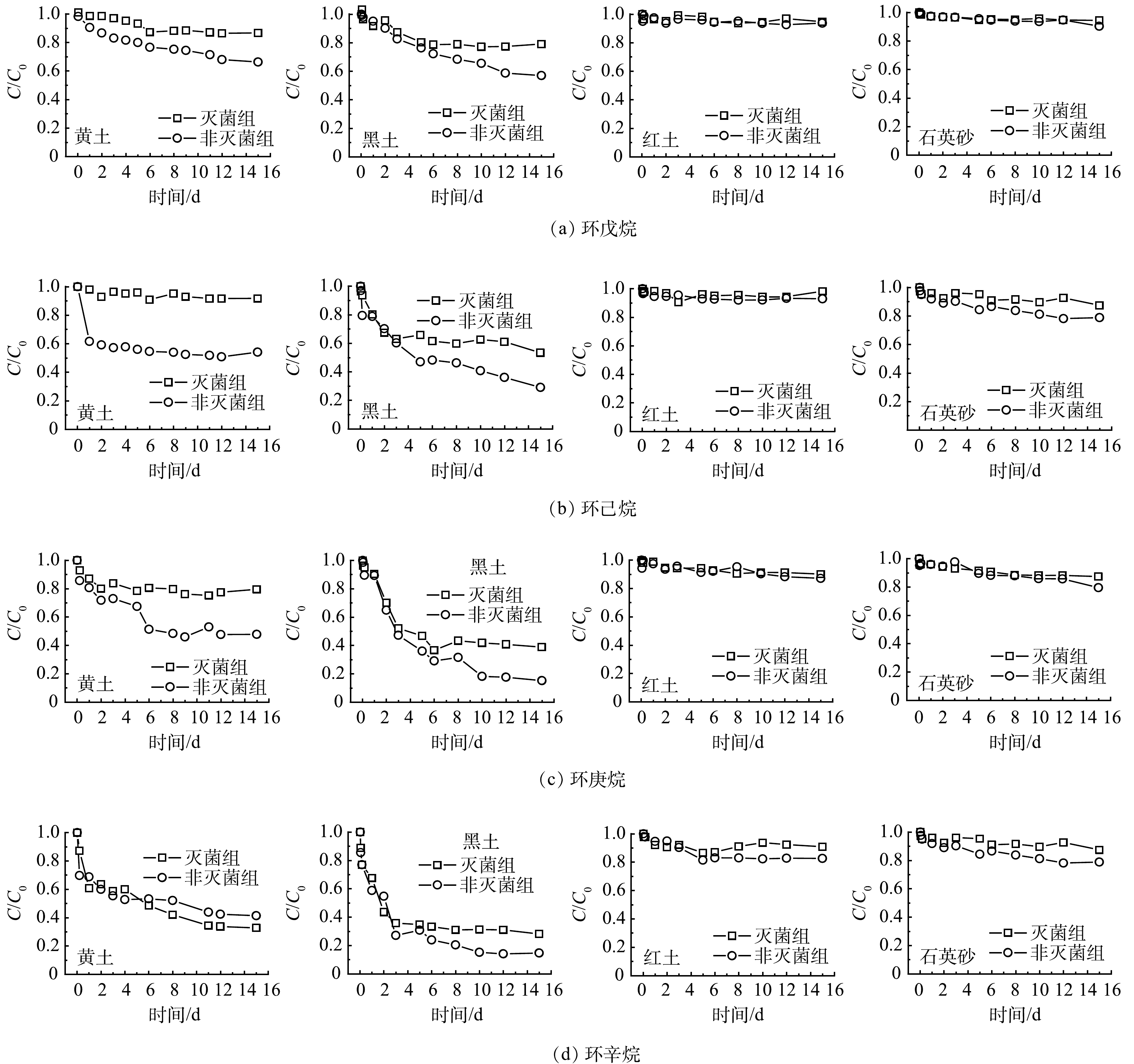 图 2 环烷烃在4种土壤中的质量浓度变化曲线Figure 2. Cyclic alkanes concentration attenuation curve in four soils
图 2 环烷烃在4种土壤中的质量浓度变化曲线Figure 2. Cyclic alkanes concentration attenuation curve in four soils2.3 苯系物在4种土壤中的气相自然衰减
4种苯系物 (苯、甲苯、乙苯、对二甲苯) 在4种土壤在灭菌组和非灭菌组中的自然衰减去除率均表现为黑土>黄土>红土>石英砂 (表2、图3) 。4种苯系物的生物降解速率均符合黑土 (3.90~9.76 d−1) >黄土 (1.18~8.24 d−1) >红土 (0.90~2.76 d−1) 的规律,而石英砂几乎不存在生物降解 (表3) 。因为黑土中的高有机质质量分数会促进苯系物蒸气的吸附,进而促进生物降解。UGWOHA和ANDRESEN[17]发现土壤有机质的质量分数增加5%导致E20乙醇汽油中苯的吸附量增加76%。在不同的土壤中苯系物的一级生物降解速率均为甲苯>苯>乙苯>对二甲苯。苯系物的生物降解速率依赖于分子化学结构,甲苯通常认为是苯系物中最容易被生物降解的。因为环上存在取代基提供了攻击侧链或氧化芳香环的替代途径[25]。苯氧化的第一步是由双加氧酶催化的羟基化,苯环上取代基的存在允许两种可能的机制:攻击侧链或氧化芳环,所有这些途径汇聚形成邻苯二酚中间体。苯主要中间产物是邻苯二酚,甲苯和乙苯在单独的途径上降解,产生各自的主要中间体3甲基邻苯二酚和3-乙基邻苯二酚,二甲苯均代谢为单甲基化儿茶酚[25]。BTEX化合物之间存在复杂的底物相互作用以及其生物降解过程中的竞争性抑制效应[26];有研究报道[27],在混合物中苯和甲苯根据竞争性抑制动力学被去除,而对二甲苯在苯和甲苯的存在下通过共代谢过程部分去除,甲苯、苯或乙苯的存在对二甲苯的降解速率有负面影响。尽管芳环上没有官能团可能使苯难以被生物转化,但苯在水中的溶解度 (1 791.00 mg·L−1) 在4种苯系物中最高(甲苯、乙苯及对二甲苯溶解度分别为535.00、161.00、156.00 mg·L−1),也促进了苯的生物降解[25]。
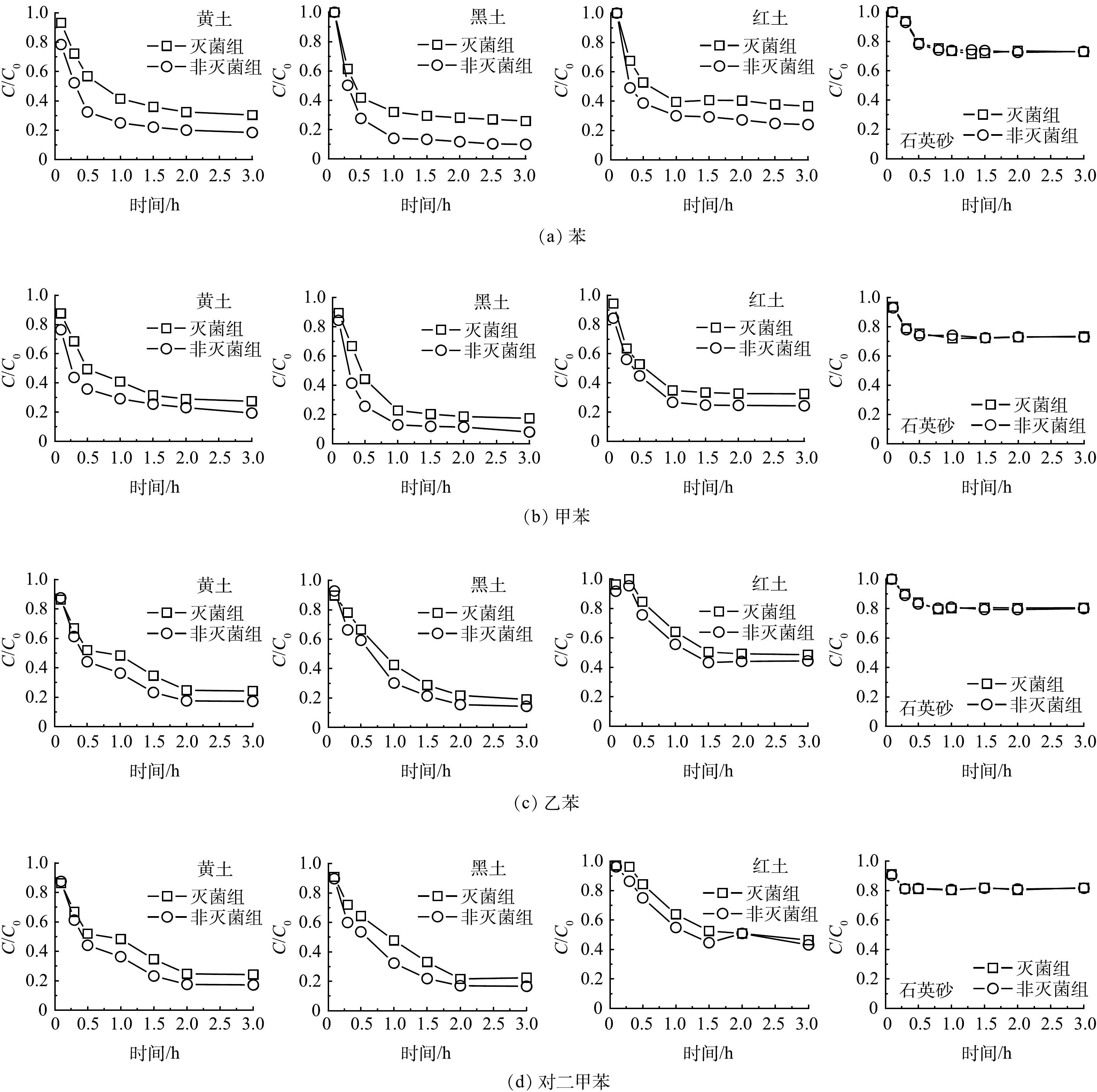 图 3 苯系物在4种土壤中的质量浓度变化曲线Figure 3. Monoaromatic hydrocarbons concentration attenuation curve in four soils
图 3 苯系物在4种土壤中的质量浓度变化曲线Figure 3. Monoaromatic hydrocarbons concentration attenuation curve in four soils2.4 不同种类石油烃的气相自然衰减潜力对比
苯系物的自然衰减去除率和生物降解速率远高于正构烷烃和环烷烃,环烷烃最难被生物降解。4种苯系物中一级生物降解速率均为甲苯>苯>乙苯>对二甲苯,甲苯最容易被生物降解。苯、甲苯、乙苯、对二甲苯生物降解速率0.90~9.76 d−1,正戊烷、正己烷、正庚烷、正辛烷生物降解速率小于0.28 d−1,环戊烷、环己烷、环庚烷、环辛烷生物降解速率小于0.06 d−1。苯系物的表观一级衰减速率和生物降解速率都远高于正构烷烃和环烷烃,可能因为苯系物的溶解度远高于正构烷烃和环烷烃[22]。PASTERIS等[21]也报道过,柱实验数据拟合的甲苯的一级生物降解速率常数为3.2 d−1,而短链烷烃如戊烷和己烷、环烷烃和的生物降解速率较慢,估计为0.1~1.2 d−1。脂肪族烃缺乏官能团且水溶性极低,微生物对脂肪烃都表现出较低的化学反应性和生物利用度,烷烃的微生物降解的敏感性:正构烷烃>异构烷烃>环烷烃[20]。
3. 结论
1) 正构烷烃、环烷烃和苯系物蒸气在4种土壤中的自然衰减,整体自然衰减去除率黑土>黄土>红土>石英砂,黑土生物降解速率高于黄土,红土和石英砂的生物降解能力很低。
2) 苯系物的气相自然衰减速率和生物降解速率远高于正构烷烃和环烷烃。包气带中的气相生物降解是石油污染场地一种重要但被忽视的自然衰减机制,该过程对于挥发性石油烃蒸气入侵暴露以及污染物自然衰减均具有重要的影响。
3) 气相自然衰减和气相生物降解动力学数据可以为蒸气入侵定量风险评估和石油污染场地自然衰减速率定量评估模型提供关键输入参数。
-

图 3 重金属全量随热处理温度的变化
Figure 3. Total concentration of heavy metals before and after thermal treatment
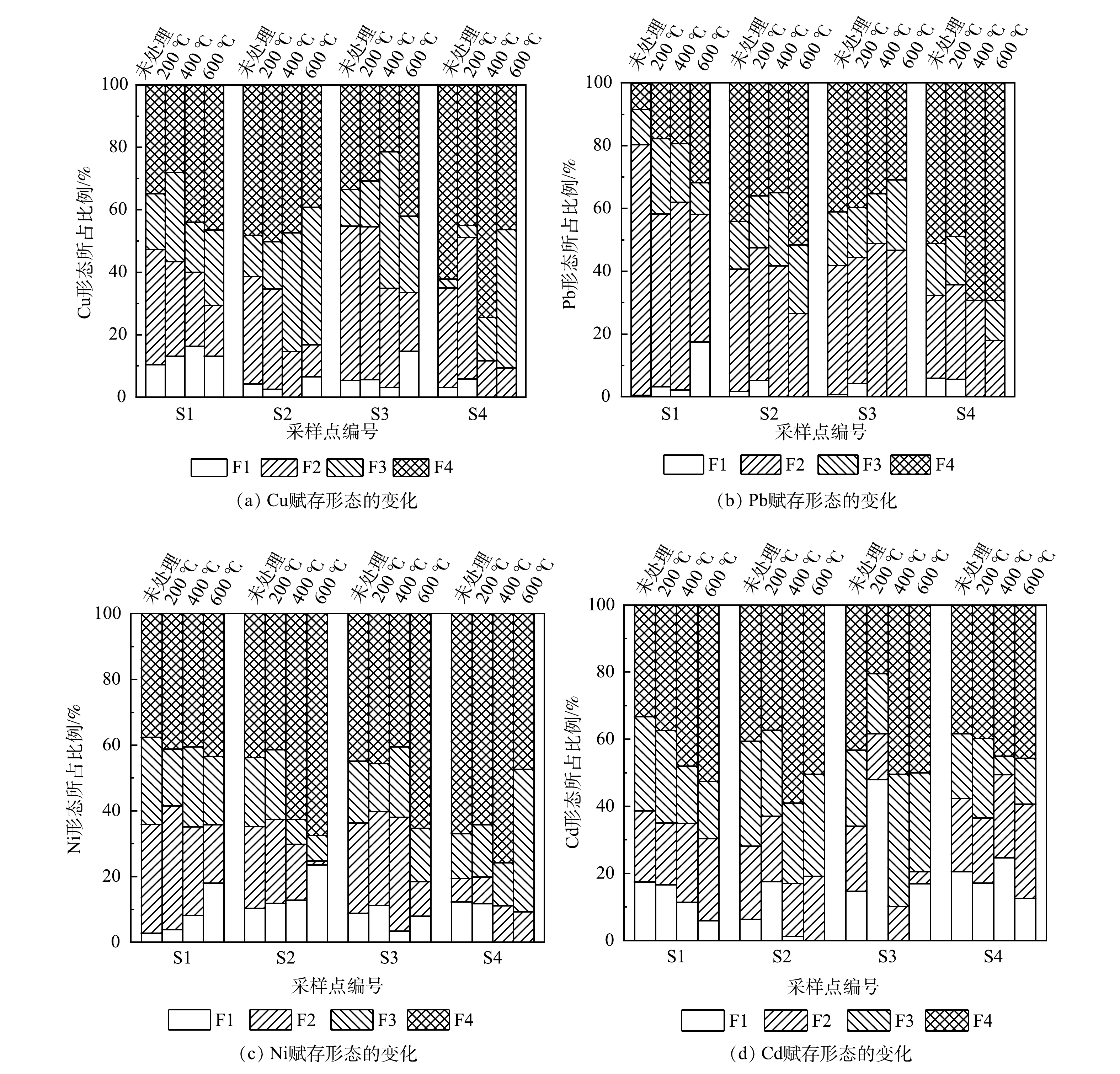
图 4 重金属赋存形态的变化(F1为酸可提取态;F2为可还原态;F3为可氧化态;F4为残渣态)
Figure 4. Speciation of heavy metals before and after thermal treatment (F1 acid soluble fraction; F2 reducible fraction; F3 oxidizable fraction; F4 residual fraction)
表 1 土壤重金属全量及形态分布特征
Table 1. Total concentration and speciation of heavy metals in soil
采样点位编号 pH 重金属 重金属全量/(mg·kg−1) 重金属形态占比% 酸可提取态 可还原态 可氧化态 残渣态 S1 7.86 Cu 3.49×102 10.3 36.91 17.84 34.88 Pb 5.62×103 0.47 79.85 11.17 8.36 Ni 8.32×103 2.71 33.13 26.55 37.61 Cd 6.06 17.48 21.11 28.08 33.36 S2 6.24 Cu 1.24×102 4.22 34.38 13.26 48.13 Pb 6.59×10 1.68 31.44 12.13 35.60 Ni 1.17×102 10.28 24.89 21.02 43.81 Cd 6.10 6.37 21.75 31.26 40.56 S3 8.00 Cu 2.54×102 5.42 49.39 11.71 33.49 Pb 6.44×10 0.68 41.11 17.11 47.12 Ni 8.57×10 8.84 27.44 18.88 44.84 Cd 6.26 14.67 19.41 22.67 43.18 S4 8.57 Cu 6.59×10 3.11 31.83 2.89 62.16 Pb 3.99×10 5.97 26.39 16.46 51.19 Ni 7.12×10 12.22 7.14 13.67 66.97 Cd 5.41 20.54 21.77 19.25 38.49
下载: 导出CSV
表 2 热处理前后基于PBET生物可给性
Table 2. Bioavailability of in PBET before and after thermal treatment
采样点位编号 处理温度/℃ Cu生物可给性/% Pb生物可给性/% Ni生物可给性/% Cd生物可给性/% GP IP GP IP GP IP GP IP S1 未处理 8.28 7.88 1.70 0.33 1.22 0.77 0.38 0.04 200 8.88 6.23 1.42 0.36 0.99 0.83 0.29 0.06 400 12.32 6.67 1.20 0.47 1.35 3.02 0.43 0.03 600 17.64 11.74 17.71 1.63 8.26 8.21 0.24 0.03 S2 未处理 0.00 6.61 3.21 12.21 3.15 5.18 0.57 0.07 200 1.56 5.96 11.11 13.39 8.56 4.83 0.92 0.09 400 2.18 3.25 2.30 28.93 9.51 5.35 1.08 0.13 600 2.59 7.53 19.48 45.14 11.84 26.55 0.47 0.12 S3 未处理 1.86 8.55 8.41 16.75 0.00 9.46 0.61 0.09 200 7.21 7.30 9.50 16.02 16.77 15.71 0.94 0.26 400 7.16 6.67 1.88 29.91 11.05 4.21 1.83 0.29 600 5.89 9.76 8.33 34.12 4.12 28.87 0.12 0.07 S4 未处理 0.00 4.54 11.27 30.51 0.00 1.75 0.73 0.00 200 1.83 5.05 0.00 24.79 11.04 1.96 1.02 0.00 400 0.65 2.82 11.34 39.77 7.42 9.37 0.96 0.00 600 0.00 3.23 14.81 50.96 1.32 32.05 0.06 0.00 注:GP为胃阶段,IP为肠阶段。
下载: 导出CSV
-
[1] 生态环境部, 国土资源部. 全国土壤污染状况调查公报[J]. 中国环保产业, 2014, 36(5): 1689-1692. [2] TOUZOUT N, MEHALLAH H, MORALENT R, et al. Phytotoxic evaluation of neonicotinoid imidacloprid and cadmium alone and in combination on tomato (Solanum lycopersicum L.)[J]. Ecotoxicology, 2021, 30(6): 1126-1137. [3] ABDEL-SHAFY H I, MANSOUR M S M. A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation[J]. Egyptian Journal of Petroleum, 2016, 25(1): 107-123. doi: 10.1016/j.ejpe.2015.03.011 [4] 汤超, 王潇. 农用地土壤污染治理与修复技术研究[J]. 低碳世界, 2020, 10(4): 15-17. doi: 10.3969/j.issn.2095-2066.2020.04.009 [5] 杨悦锁, 陈煜, 李盼盼, 等. 土壤、地下水中重金属和多环芳烃复合污染及修复研究进展[J]. 化工学报, 2017, 68(6): 2219-2232. [6] 李书鹏. 土壤与地下水修复行业2018年发展报告[R]. 中国环境保护产业协会. 北京, 2019. [7] 赵涛, 马刚平, 周宇, 等. 多环芳烃类污染土壤热脱附修复技术应用研究[J]. 环境工程, 2017, 35(11): 178-181. [8] 魏萌. 焦化污染场地土壤中PAHs的赋存特征及热脱附处置研究[D]. 北京: 首都师范大学, 2013. [9] BONNARD M, DEVIN S, LEYVAL C, et al. The influence of thermal desorption on genotoxicity of multipolluted soil[J]. Ecotoxicology and Environmental Safety, 2010, 73(5): 955-960. doi: 10.1016/j.ecoenv.2010.02.023 [10] 焦文涛, 韩自玉, 吕正勇, 等. 土壤电阻加热技术原位修复有机污染土壤的关键问题与展望[J]. 环境工程学报, 2019, 13(9): 2027-2036. doi: 10.12030/j.cjee.201905138 [11] 朱岗辉, 孙璐, 廖晓勇, 等. 郴州工业场地重金属和PAHs复合污染特征及风险评价[J]. 地理研究, 2012, 31(5): 831-839. [12] WU Q, LEUNG J Y S, GENG X, et al. Heavy metal contamination of soil and water in the vicinity of an abandoned e-waste recycling site: Implications for dissemination of heavy metals[J]. Science of the Total Environment, 2015, 506: 216-217. [13] DONAHUE W F, ALLEN E W, SCHINDLER D W. Impacts of coal-fired power plants on trace metals and polycyclic aromatic hydrocarbons (PAHs) in lake sediments in Central Alberta, Canada[J]. Journal of Paleolimnology, 2006, 35(1): 111-128. doi: 10.1007/s10933-005-7878-8 [14] SONG Y F, WILKE B M, SONG X Y, et al. Polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs) and heavy metals (HMs) as well as their genotoxicity in soil after long-term wastewater irrigation[J]. Chemosphere, 2006, 65(10): 1859-1868. doi: 10.1016/j.chemosphere.2006.03.076 [15] 生态环境部. 建设用地土壤污染风险评估技术导则:HJ 25.3-2019[S]. 北京: 中国环境科学出版社, 2019. [16] 李非里, 刘丛强, 宋照亮. 土壤中重金属形态的化学分析综述[J]. 中国环境监测, 2005(4): 21-27. doi: 10.3969/j.issn.1002-6002.2005.04.007 [17] RUBY M V, DAVIS A, SCHOOF R, et al. Estimation of lead and arsenic bioavailability using a physiologically based extraction test[J]. Environmental Science and Technology, 1996, 30(2): 422-430. doi: 10.1021/es950057z [18] XU W, HOU S, LI Y, et al. Bioavailability and speciation of heavy metals in polluted soil as alleviated by different types of biochars[J]. Bulletin of Environmental Contamination and Toxicology, 2020, 104(19): 484-488. [19] XIA W Y, FENG Y S, JIN F, et al. Stabilization and solidification of a heavy metal contaminated site soil using a hydroxyapatite based binder[J]. Construction & Building Materials, 2017, 156(15): 199-207. [20] 王璇, 于宏旭, 熊惠磊, 等. 南方某典型矿冶污染场地健康风险评价及修复建议[J]. 环境工程学报, 2017, 11(6): 3823-3831. doi: 10.12030/j.cjee.201603219 [21] JUHASZ A L, SMITH E, WEBER J, et al. Comparison of in vivo and in vitro methodologies for the assessment of arsenic bioavailability in contaminated soils[J]. Chemosphere, 2007, 69(6): 961-966. doi: 10.1016/j.chemosphere.2007.05.018 [22] GONZÁLEZ-GRIJALVA B, MEZA-FIGUEROA D, ROMERO F M, et al. The role of soil mineralogy on oral bioaccessibility of lead: Implications for land use and risk assessment[J]. Science of the Total Environment, 2019, 657: 1468-1479. doi: 10.1016/j.scitotenv.2018.12.148 [23] 尹乃毅, 都慧丽, 张震南, 等. 应用SHIME模型研究肠道微生物对土壤中镉、铬、镍生物可给性的影响[J]. 环境科学, 2016, 37(6): 2353-2358. [24] LU Y, YIN W, HUANG L, et al. Assessment of bioaccessibility and exposure risk of arsenic and lead in urban soils of Guangzhou City, China[J]. Environmental Geochemistry and Health, 2011, 33(2): 93-102. doi: 10.1007/s10653-010-9324-8 [25] 孙立强, 孙崇玉, 刘飞, 等. 淮北煤矿周边土壤重金属生物可给性及人体健康风险[J]. 环境化学, 2019, 38(7): 1453-1460. doi: 10.7524/j.issn.0254-6108.2018092801 [26] TAO X Q, SHEN D S, SHENTU J L, et al. Bioaccessibility and health risk of heavy metals in ash from the incineration of different e-waste residues[J]. Environmental Science and Pollution Research, 2015, 22(5): 3558-3569. doi: 10.1007/s11356-014-3562-8 [27] Environmental Protection Agency. Office of emergency and remedial response. Risk assessment guidance for superfund (RAGS) part A[J]. Saúde Pública, 1989, 804(7): 636-640. [28] Environmental Protection Agency. Exposure factors handbook[S]. National Center for Environmental Assessment. EPA/600. National Technical Information Service, Washington, DC: Environmental Protection Agency, 2011. [29] LI H H, CHEN L J, YU L, et al. Pollution characteristics and risk assessment of human exposure to oral bioaccessibility of heavy metals via urban street dusts from different functional areas in Chengdu, China[J]. Science of the Total Environment, 2017, 586(15): 1076-1084. [30] 王学锋, 杨艳琴. 土壤-植物系统重金属形态分析和生物有效性研究进展[J]. 化工环保, 2004, 24(1): 24-28. doi: 10.3969/j.issn.1006-1878.2004.01.008 [31] CHEN G, ZENG R, DU R, et al. Transfer of heavy metals from compost to red soil and groundwater under simulated rainfall conditions[J]. Journal of Hazardous Materials, 2010, 181(1/2/3): 211-216. doi: 10.1016/j.jhazmat.2010.04.118 [32] 蔡荣欣. 土壤的热修复[J]. 上海化工, 2018, 43(8): 41-44. doi: 10.3969/j.issn.1004-017X.2018.08.014 [33] HUANG Y T, HSEU Z Y, HSI H C. Influences of thermal decontamination on mercury removal, soil properties, and repartitioning of coexisting heavy metals[J]. Chemosphere, 2011, 84(9): 1244-1249. doi: 10.1016/j.chemosphere.2011.05.015 [34] VIDONISH J E, ZYGOURAKIS K, MASIELLO C A, et al. Pyrolytic treatment and fertility enhancement of soils contaminated with heavy hydrocarbons[J]. Environmental Science & Technology, 2016, 50(5): 2498-2506. [35] MA F, ZHANG Q, XU D, et al. Mercury removal from contaminated soil by thermal treatment with FeCl3 at reduced temperature[J]. Chemosphere, 2014, 117(1): 388-393. [36] 裘知, 孙福成. 重金属热处理过程中的挥发及其抑制[C]//环境污染防治应用技术交流会, 2010:246-247. [37] 赵天从. 重金属冶金学[M].北京: 冶金工业出版社, 1981. [38] 王昕晔. 垃圾焚烧过程中铅和镉的挥发特性及其排放控制研究[D]. 南京: 东南大学, 2016. [39] 李进平, 胡云娇, 陈思奇. 污泥热干化过程中重金属Pb、Cu、Zn的形态转化及稳定特性[J]. 环境工程学报, 2015, 9(12): 6041-6044. doi: 10.12030/j.cjee.20151262 [40] 张怡斐. 市政污泥热处理过程中主要污染物的迁移转化[D]上海:上海交通大学, 2011. [41] LENG L, YUAN X, HUANG H, et al. The migration and transformation behavior of heavy metals during the liquefaction process of sewage sludge[J]. Bioresource Technology, 2014, 167: 144-150. doi: 10.1016/j.biortech.2014.05.119 [42] 王涵. 污水污泥热处理后重金属的赋存形态与浸出特性研究[D]. 哈尔滨:黑龙江大学, 2015. [43] GARAU M, GARAU G, DIQUATTRO S, et al. Mobility, bioaccessibility and toxicity of potentially toxic elements in a contaminated soil treated with municipal solid waste compost[J]. Ecotoxicology and Environmental Safety, 2019, 186: 109766. doi: 10.1016/j.ecoenv.2019.109766 -

 点击查看大图
点击查看大图

计量
- 文章访问数: 5092
- HTML全文浏览数: 5092
- PDF下载数: 58
- 施引文献: 0
