




-
近年来,难降解有机污染物的降解问题引起了人们的广泛关注,因为它们对自然环境和人类健康造成了极大的危害[1]。特别是,煤化工生产过程产生的高浓度盐水中含有大量的难降解的有机物(如PAHs、杂环化合物等),这些物质通常具有复杂的芳族分子结构,这使得它们更稳定并且更难以降解。如果不加以处理,这些有害物质会引起某些健康危害和环境污染[2]。虽然传统的处理技术,如物理吸附[3-4],化学氧化[5]和生物方法[6],已经研究从废水中去除难降解有机物,但许多问题仍然是不可避免的,如二次污染[7]和伴随污染物的生物降解延迟[8]。高级氧化过程(AOPs),定义为利用羟基(·OH)进行氧化的那些技术[9-10],在过去几十年中,这些技术在废水处理技术的研究中受到越来越多的关注。这些过程(如光催化氧化,芬顿化学和臭氧催化氧化[11-12])已成功应用于去除或降解顽固污染物,或用作预处理将污染物转化为短链化合物,然后再通过常规或生物方法处理[13]。其中之一的经典方法是非均相臭氧催化氧化工艺,它利用不溶性催化剂对有机污染物进行氧化还原和矿化,臭氧催化氧化因其效率高、操作简单而被认为是一种很有前途的工艺[14-15]。这一过程的关键因素是制备有效的非均相催化剂,因此,须进一步探索研究,以找出廉价、高效和稳定的非均相催化剂,在催化臭氧氧化中高效降解有机物污染物。
因此,本研究采用浸渍-煅烧法制备了负载活性金属氧化物的活性氧化铝型催化剂,以煤化工高浓盐水为目标污染物,探索催化剂的制备工艺和反应操作条件对高盐废水COD去除率的影响,为高效的臭氧催化体系的开发及其在煤化工高盐废水处理领域的应用提供参考。
-
实验水样来自某化工企业煤制天然气废水,共分为3种废水:水样1来自二次反渗透浓盐水;水样2来自二沉池出水;水样3来自一次反渗透浓盐水,基本水质指标如表1所示。由表1可知,水样1中TDS超过了3.5%,属于高盐废水,且COD较高;水样2、水样3中的TDS和COD相对降低,采用3种水样对比实验来考察臭氧催化氧化对高盐废水中的COD去除效果。
-
采用浸渍-煅烧的方法制备催化剂,具体制备过程如下:取一定质量的活性Al2O3或者陶瓷材料球状载体(载体粒径为3~6 mm),浸渍至含有1 mol·L−1的Mn、Fe、Cu、Ni、Co、Ce等过渡金属的硝酸盐溶液中,室温下,以150 r·min−1振荡30 min,使其完全混匀,过滤得到固体,在105 ℃下烘干,然后在500 ℃下煅烧4 h后,制备所得的催化剂。
-
臭氧催化氧化实验装置示意图如图1所示。臭氧催化氧化实验分为以下2步。间歇性实验:在1 L废水中加入一定量的催化剂,反应装置为玻璃柱(内径4 cm,高度1.5 m),废水采用蠕动泵循环以便混合均匀,控制臭氧发生器出口O3气体流量为0.8 L·min−1,O3浓度为3~10 mg·L−1,每隔一段时间检测COD的变化。连续性实验:反应装置连续进出水,出水不返回到进水端,每隔60 min检测COD的变化。
-
采用GB 11914-1989中重铬酸钾滴定法,测定废水中化学需氧量(COD);X射线衍射分析(XRD)由X射线衍射仪(D8 Advance,布鲁克,德国)进行物相分析,测试条件为CuKα辐射,电压为40 kV,电流为40 mA,扫描角度为3°~90°,扫描速度为3(°)·min−1;比表面积和孔径(BET)采用ASAP 2020型比表面积与孔径测定仪(麦克仪器公司,美国)测试,在77 K液氮温度下,进行N2吸附-脱附测定;采用S-4800场发射扫描电子显微镜(SEM)观察样品的整体形貌;采用Noran7型X射线能谱仪(EDS)进行样品微区成分分析;采用原子吸收光谱法(AAS)测定溶液中的金属离子浓度。
-
首先单独使用活性氧化铝和陶粒进行臭氧催化氧化实验,以水样1为实验水样,实验发现活性氧化铝对COD的去除率达到了14.6%,高于陶粒的8.9%,而且活性氧化铝能提高污水的可生化性。相比于陶粒,活性氧化铝臭氧催化氧化污水后,B/C值为0.42,远高于陶粒的0.23。由此可见,氧化铝载体催化性能优于陶粒。
以活性氧化铝作为载体制得金属氧化物负载型催化剂,考虑的活性组分主要有稀土元素(Ce)、过渡金属(Mn、Fe、Cu、Ni、Co)。各种金属元素都以氧化物的形式负载于活性氧化铝上,以水样1作为目标污染物,以反应60 min COD去除率为评价因子,考察不同活性组分对催化剂催化性能的影响,结果如图2所示。实验发现在活性氧化铝表面负载活性组分后的催化活性明显强于未负载活性组分的情况。其中活性氧化铝负载Cu、Mn、Ni、Fe、Co的催化活性较高,反应60 min后,COD去除率分别为35.29%、34.01%、33.73%、33.01%、31.99%,明显强于负载Ce和Zn条件下的去除率,该结果和DHANDAPANI等[16]的研究结果一致。其可能的原因是,催化剂表面的羟基相当于Bronsted酸,金属离子和其中的不饱和氧原子相当于Lewis酸和碱,但由于不同氧化物的存在,使得催化剂表面的酸碱性有所不同,O3的作用方式不同,对产生羟基自由基的促进作用就会有所差别。由于铜、锰、镍氧化物的催化活性高和不饱和氧原子反应较快,且给出质子的倾向较强,因此,在臭氧的结合下对COD的去除效果最好。锰、镍等活性组分存在多种价态,金属氧化物价态变化所转移的电子促进臭氧分子分解产生·OH,有利于有机污染物的降解[17-18]。
此外,前30 min,COD去除较快,之后去除率变化较小。这主要是因为反应开始前,废水pH为8.56,碱性条件有利于臭氧与水溶液中的OH−反应产生·OH,故初始反应速率较快。随着反应的进行,废水中的有机物逐渐被降解为小分子有机酸、醛、酮,致使废水的pH降低至6以下,从而导致表面(S)直接吸附臭氧分子释放出一个氧气分子而不产生·OH,导致催化活性降低,反应如式(1)~式(3)所示。在30 min后,去除率接近稳定[19]。
-
采用BET对活性氧化铝载体催化剂的比表面积、孔容和平均孔径进行检测,结果表明,MnOx-NiOx/γ-Al2O3的比表面积为153.525 m2·g−1,孔容为0.421 cm3·g−1,平均孔径为6.858 nm,γ-Al2O3的比表面积为189.997 m2·g−1,孔容为0.481 cm3·g−1,平均孔径为6.325 nm。相比未负载的活性氧化铝,MnOx-NiOx/γ-Al2O3的比表面积和孔容降低,平均孔径增加,可能的原因是锰、镍等活性金属进入氧化铝的中孔内,起到了一定的孔支撑作用。同样活性金属进入中孔内,阻塞了一些中孔通道,导致催化剂比表面积和孔容降低。而且比表面积越大,越有利于臭氧分子的吸附,增加了臭氧在水中的溶解度,有利于传质反应的进行;同时强化了臭氧分子与催化剂表面活性位点的接触,促进了臭氧分子分解产生·OH[20]。
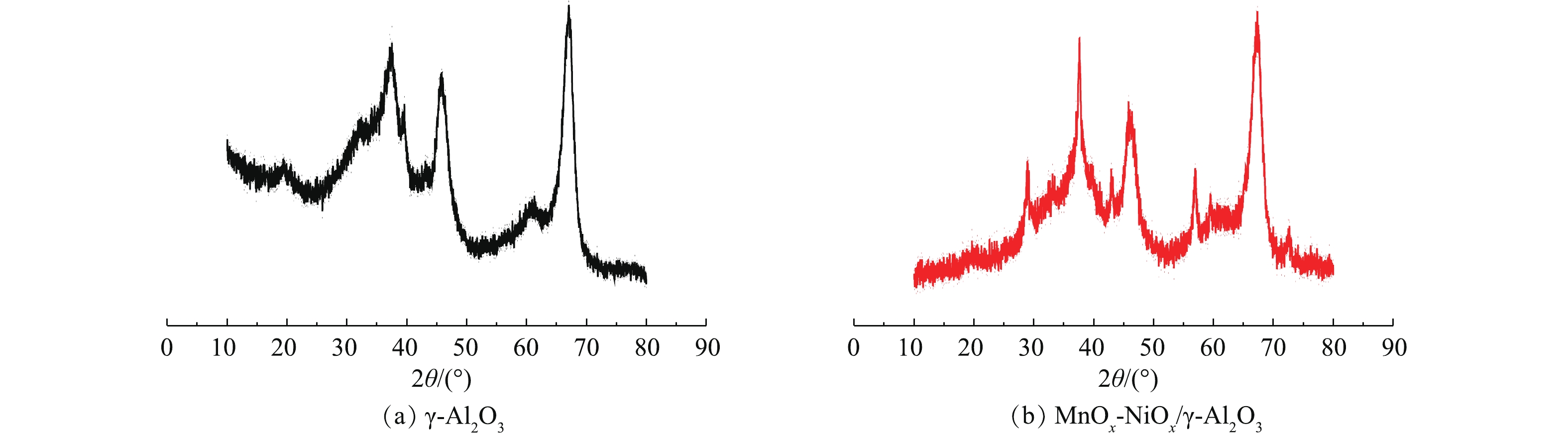
图3为γ-Al2O3、MnOx-NiOx/γ-Al2O3的XRD图谱。由图3(b)可知,对于MnOx-NiOx/γ-Al2O3,在29.1°和56.8°附近出现的衍射峰主要对应于四方α-MnO2(JCPDS 42-1169)。以上结果表明,负载的Mn主要以四方α-MnO2存在。在MnOx-NiOx/γ-Al2O3的XRD图谱中未出现NiOx明显的特征峰,这可能是镍元素含量过低导致的。在2种催化剂的XRD图谱中均出现了γ-Al2O3的特征衍射峰(2θ为67.13°、45.99°、37.56°),由Jade5.0软件根据Sherrer公式计算晶粒尺寸大小,γ-Al2O3和MnOx-NiOx/γ-Al2O3的平均晶粒分别为23.9 nm和22.8 nm,由此可知,负载Mn、Ni后,γ-Al2O3催化剂晶粒略有减小,这与BET结果相吻合。
图4为各催化剂的SEM表征图。由图4(a)和图4(b)可知,500 ℃活化的γ-Al2O3和MnOx-NiOx/γ-Al2O3均具有多孔表面,而后者被紧密堆积的层状颗粒覆盖,如报道的典型MnOx的形态。鉴于XRD图谱中未出现NiOx的特征峰,进一步采用EDS对MnOx-NiOx/γ-Al2O3进行元素含量测试,结果显示,MnOx-NiOx/γ-Al2O3中Mn和Ni的元素含量分别为2.73%和1.22%,2种元素摩尔比接近2∶1,这说明负载效果较好。
-
固定催化剂投加量为50 g·L−1废水,臭氧投加量为180 mg·(L·h)−1,分别比较了臭氧单独氧化与臭氧催化氧化(以MnOx-NiOx/γ-Al2O3为催化剂)对3种实际废水的COD去除效果,结果如图5所示。结果表明,对于不同废水,臭氧催化氧化均比臭氧单独氧化去除COD效果更好。对于水样1,反应60 min后,O3单独氧化对于COD去除率仅为12.5%,这是由于臭氧难以直接矿化污染物,更多的是产生了氧化中间产物,而催化氧化去除率为51.3%,比前者高38.8%;对于水样2和水样3,臭氧催化氧化COD去除率分别比单独臭氧氧化高出10.3%和25.1%,这说明臭氧催化氧化能有效去除煤化工高盐废水的COD。通过添加催化剂能明显改善COD的去除率,这是由于3种组分在一定的配比下具有良好的协同作用,能提高臭氧快速分解产生·OH的能力,而且在非均相O3催化氧化降解有机污染物过程中,金属氧化物在水溶液中由于水合作用,在其表面覆盖羟基,这些羟基形成具有孤对电子或π电子的吸附中心,可使有机物吸附于催化剂表面。同时,金属氧化物表面的羟基也是O3在催化剂表面产生·OH的活性点,从而O3在催化剂表面分解为具有更强氧化作用的·OH活性物种,生成的·OH可以在催化剂表面和溶液中引发自由基链式反应,实现有机污染物降解[21-22]。
-
本研究对比了单独氧化和臭氧催化氧化2种体系中臭氧的利用效率。在臭氧催化氧化过程中,臭氧在气相和液相中存在质量平衡,计算方法[23]见式(4)。
式中:CT、CO、CR和CD分别为初始加入、排出、残余和消耗的臭氧浓度,mg·L−1。CD可按式(5)进行计算。
则臭氧的利用效率(
Ru )计算方法[24]见式(6)。式中:
v 为气体流速,m3·s−1;V为溶液体积,m3。臭氧单独氧化和臭氧催化氧化去除单位质量COD所消耗的臭氧量的计算方法见式(7)。式中:η为臭氧消耗量与COD去除量的比值;
C0 为初始状态废水的COD,mg·L−1,Ct 为反应t时间后废水的COD,mg·L−1。如图6所示,在15~120 min处理过程中,MnOx-NiOx/γ-Al2O3臭氧催化氧化得到较低的η。在不同的臭氧化体系中,易降解物质在初始阶段逐渐被降解。随着时间的推移,废水中的易降解物质逐渐被消耗,难降解物质相对含量增多,η逐渐降低,但MnOx-NiOx/γ-Al2O3臭氧催化氧化的η仍然低于单独的臭氧氧化,这意味着MnOx-NiOx/γ-Al2O3催化剂可以有效地将臭氧分解成活性氧(ROS)而有利于废水中污染物的降解。
-
以水样1为目标污染物,固定臭氧投加量为350 mg·(L·h)−1,催化剂分别投加50 g·L−1废水和100 g·L−1废水,考察催化剂投加量对废水COD去除率的影响,实验结果如图7所示。结果表明,在30 min之前,随着催化剂投加量的增多,COD去除率增加,这可能是由于催化剂投加量的增加,活性位点增多,碰撞和接触概率增大,能吸附更多的有机物和臭氧,生成更多的·OH。但是随着时间到达60 min,COD的去除率随臭氧投加量的增多变化不明显,可能的原因是催化剂的活性位点已不是限制催化反应的主要因素,臭氧的投加量等成为主要的限制因素。另外,过多的催化剂投加量还会降低臭氧的传质效率,从而降低臭氧的利用率,这和图6的实验结论相一致,因此,催化剂最佳投加量应为100 g·L−1。
以水样1为目标污染物,固定催化剂投加量为100 g·L−1废水,考察臭氧投加量对COD去除率的影响,实验结果如图8所示。由图8可知,COD去除率随着臭氧投加量的增加而增加,当臭氧投加量为180、274和350 mg·(L·h)−1时,反应180 min后,COD去除率为55.3%、60.9%和72.3%。这主要是因为随着臭氧投加量的增多,溶解在水中的臭氧量增加,在催化剂的作用下,产生更多的·OH,从而有利于COD去除率的提高,因此,臭氧最佳投加量应为350 mg·(L·h)−1。
-
以水样1为目标污染物,固定催化剂投加量为120 g,废水流量为1.2 L·h−1,连续进水、出水,废水下进上出,反应器底部曝气,O3投加量约为300 mg·L−1,实验结果如图9所示。可以看出,进水COD约为640 mg·L−1时,出水COD稳定在350~380 mg·L−1,其去除率为41%~45%,催化剂催化性能变化较小。采用原子吸收光谱仪每隔60 min测定实验过程中锰、镍活性组分在水中的溶出情况,结果表明,锰镍离子在水中的含量均小于0.5 mg·L−1,这说明该催化剂的稳定性较好,重复利用性较高。
-
1)活性氧化铝载体催化性能优于陶粒,活性氧化铝负载Cu、Mn、Ni的催化活性较高,将活性组分进行组合制得的MnOx-NiOx/γ-Al2O3催化剂,60 min的臭氧催化氧化能有效去除51.3%的COD。
2)利用BET、SEM-EDS、XRD对催化剂进行表征和分析。结果表明,Mn、Ni成功负载到活性氧化铝表面和孔隙内,2种元素负载量摩尔比约为2∶1,且主要以氧化物形式存在。
3)通过计算臭氧利用效率发现,MnOx-NiOx/γ-Al2O3臭氧催化氧化的臭氧利用效率低于单独的臭氧氧化,这意味着通过MnOx-NiOx/γ-Al2O3催化剂可以有效地将臭氧分解成活性氧。
4)通过优化臭氧投加量和催化剂投加量发现,催化剂投加量为100 g·L−1废水,臭氧投加量为350 mg·(L·h)−1时,反应180 min后COD去除率能达到72.3%。
5)连续进行4 h的臭氧催化氧化实验后发现,MnOx-NiOx/γ-Al2O3稳定性和重复利用性较好,COD去除率能维持在42%左右不变,锰、镍离子的溶出量均小于0.5 mg·L−1。
煤化工高盐废水臭氧催化氧化脱除COD
COD removal from high-salt wastewater in coal chemical industry by ozone catalytic oxidation
-
摘要: 针对煤化工高盐废水中有机物难降解问题,采用浸渍-煅烧法制备了负载有活性金属氧化物的活性氧化铝型催化剂,探索催化剂的制备工艺和反应操作条件对废水COD去除率的影响。结果表明:活性氧化铝载体催化性能优于陶粒,活性氧化铝负载Cu、Mn、Ni的催化活性较高,将2种活性组分进行组合制得的MnOx-NiOx/γ-Al2O3催化剂,在经过60 min的臭氧催化氧化后,COD的去除率可达51.3%;利用BET、SEM-EDS、XRD对催化剂进行了表征和分析,Mn、Ni成功负载到活性氧化铝表面和孔隙内,2种元素负载量摩尔比约为2∶1,且主要以氧化物形式存在;通过计算臭氧利用效率,发现MnOx-NiOx/γ-Al2O3臭氧催化氧化的η值低于单独的臭氧氧化,这意味着通过MnOx-NiOx/γ-Al2O3催化剂可以有效地将臭氧分解成活性氧;通过优化臭氧和催化剂投加量后发现,在臭氧为350 mg·(L·h)−1、催化剂投加量为100 g·L−1废水中,反应180 min后,COD去除率可达到72.3%;在连续进行4 h的臭氧催化氧化实验后,MnOx-NiOx/γ-Al2O3稳定性和重复利用性均较好,COD去除率能维持在约42%,锰、镍离子的溶出量均小于0.5 mg·L−1。以上研究结果可为高效的臭氧催化体系在煤化工高盐废水处理领域的应用提供参考。
-
关键词:
- 煤化工高盐废水 /
- MnOx-NiOx/γ-Al2O3催化剂 /
- COD去除率 /
- 臭氧催化氧化
Abstract: Aiming at the problem of refractory degradation of organic matters in high-salt wastewater from coal chemical industry, an active alumina-type catalyst loaded with active metal oxide was prepared by an impregnation-calcination method. The effects of catalyst preparation and reaction conditions on COD removal rate from wastewater were investigated. The experimental results showed that the catalytic performance of activated alumina carrier was better than that of ceramsite, and activated alumina supported with Cu, Mn, Ni had high catalytic activity, of which the MnOx-NiOx/γ-Al2O3 catalyst prepared by the combination of the two active components could catalyze the ozone oxidation reaction with 51.3% COD removal after 60 minutes. The catalyst was characterized and analyzed by BET, SEM-EDS, XRD. The results showed that Mn, Ni elements were successfully loaded on the surface and pores of activated alumina, and their molar ratio was about 2∶1, and their oxide forms mainly appeared. Through calculating the ozone utilization efficiency, the η value of the MnOx-NiOx/γ-Al2O3 catalyst was lower than that of ozone alone. This implied that the MnOx-NiOx/γ-Al2O3 catalyst could effectively decompose ozone into ROS. After optimizing the dosage of ozone and the dosage of the catalyst, the optimum dosages of catalyst and ozone were 100 g·L−1 and 350 mg·(L·h)−1, respectively, at which the COD removal rate could reach 72.3% after 180 min reaction. After 4 hours ozone catalytic oxidation, the stability and reusability of MnOx-NiOx/γ-Al2O3 were good, COD removal rate maintained about 42% and the release amounts of manganese and nickel ions were less than 0.5 mg·L−1. This study can provide a reference for the development of an efficient ozone catalytic system and its application in the field of high-salt wastewater treatment in the coal chemical industry. -
燃料乙醇是我国重点发展的新能源之一,其生产废水的处理和资源化利用很受关注[1]。燃料乙醇的生产以稻谷为原料,经过发酵、精馏后的酒糟通过简单的挤压处理后,所得滤渣经干燥进入干酒糟工艺(distillers dried grains,DDG)处理技术[2]。产生废水主要成分为糖类、蛋白质、纤维素等,其中粗蛋白含量约占27%[3]。该废水经厌氧生物处理后,TN主要以
NH+4 -N形式存在,且浓度较高,一般为300~600 mg·L−1;即使经过好氧硝化反应也仅发生氮形态的变化,而出水TN未降低,这主要是由于好氧进水碳氮比失调导致反硝化反应效果差。为实现TN达标排放,需要在好氧系统段补充进水碳源。影响反硝化效率的因素较多,活性污泥中微生物组成种类也会影响碳源的选择。在调整脱氮系统工艺前,通过在现场开展指导性实验,研究投加不同碳源时的反硝化脱氮效率,以期获得适宜的碳源补充方案,提高A/O反硝化系统脱氮效率。本研究从脱氮反应速率和经济性综合分析以选择适宜的碳源,并优化运行条件,应用于工程实践中,指导DDG废水处理的A/O脱氮系统高效运行。
1. 实施方案
1.1 分析方法
研究采用快速消解法测定水样中的COD;重量法测定活性污泥浓度;玻璃电极测定水样中的pH;碱性过硫酸钾消解-紫外分光光度法测定TN;半自动凯氏定氮仪测定TNK。
1.2 水质概况
某污水站主要有高浓度废水和低浓度废水两股来水。高浓度废水即为DDG废水(俗称清液)。低浓度水中包含实验室废水、净水器排泥水、锅炉除盐水、设备冲洗水、循环水排水和生活区废水等。经过处理后的外排水水质满足《广东省地方标准-水污染物排放限值》(DB 44/26-2001)第二时段一级标准,TN满足《发酵酒精和白酒工业水污染排放标准》(GB 27631-2001)(即50 mg·L−1)排放标准,排至市政污水管网。水质指标如表1所示。
表 1 水质指标汇总Table 1. Quality and flow indicators of wastewater项目(m3·d−1) 水量/(mg·L−1) COD/(mg·L−1) SS/(mg·L−1) TN/(mg·L−1)  -N/
-N/清液 1 800~2 100 45 000~65 000 5 000~8 000 13 00~16 00 10~23 低浓度水 360~400 100~200 20~80 30~50 5~10 出水标准 — 100 60 50 10 | Show Table DownLoad:
CSV
DownLoad:
CSV
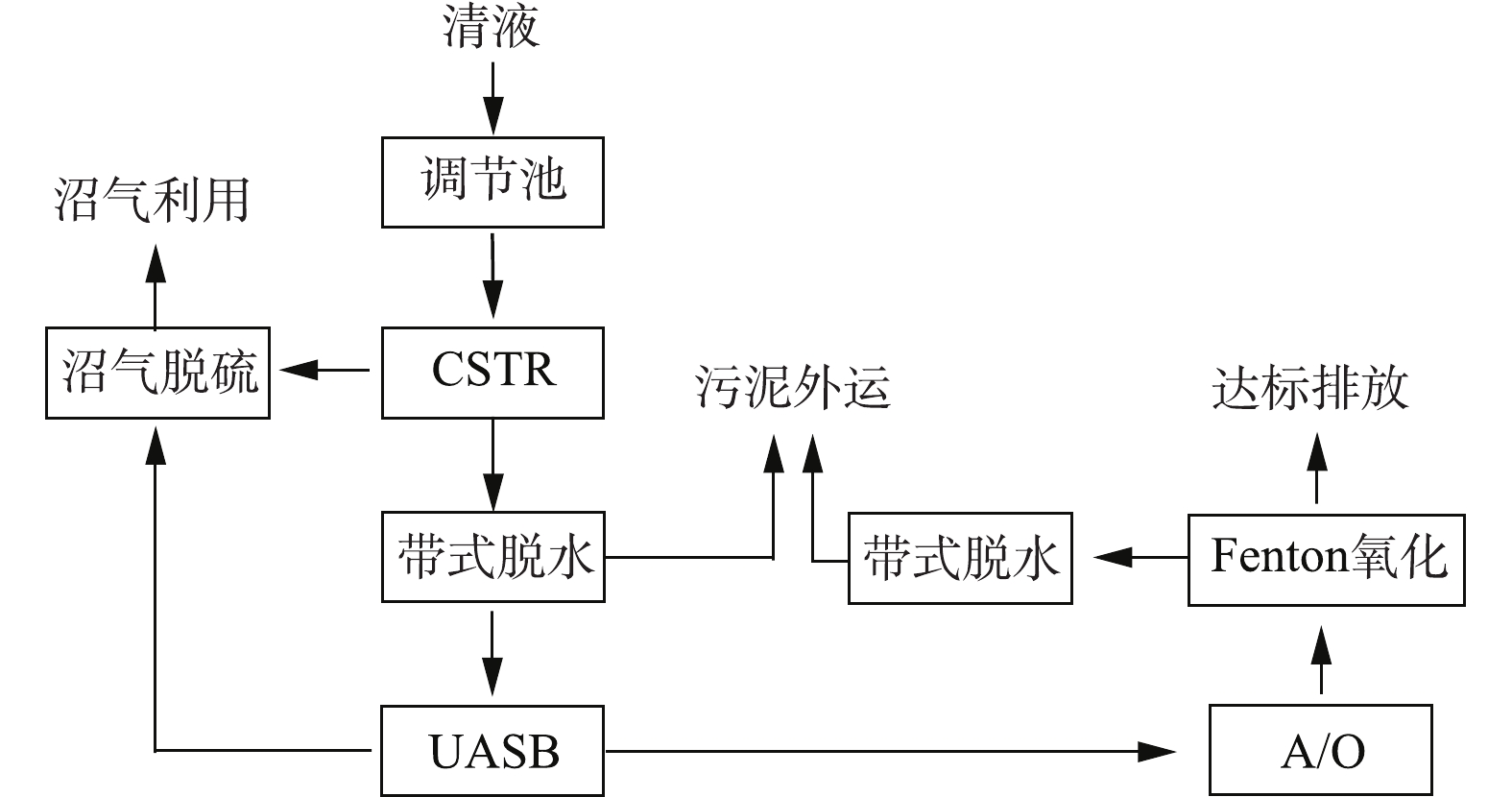
1.3 工艺流程
该污水站采取的主要工艺为CSTR+UASB+A/O+芬顿,废水处理量约2 800~3 400 m3·d−1。工艺流程见图1。
1.4 污水站水质问题分析及碳源种类选择
该污水站运行稳定,除TN外,其他指标均达到排水要求。外排水TN一直为300~360 mg·L−1。为解决这个问题,对该污水站A/O系统的进、出水水质进行了分析,结果如表2所示。
表 2 A/O系统进出水水质Table 2. Water quality index of A/O system工艺段 COD/(mg·L−1) TN/(mg·L−1) TNK/(mg·L−1)  -N/(mg·L−1)
-N/(mg·L−1)A/O进水 (mg·L−1) 480~560 476~556 394~462 A/O出水 450~610 378~410 15~23 未检出 | Show TableDownLoad:
CSV
由表2可知,该污水站A/O系统进水的COD/TN约3.1~4.0。TNK反映了水中
NH+4 -N和有机氮的情况,说明系统进水中的总氮主要以NH4+-N和有机氮为主,故可认为A/O工艺适合该废水的处理。杨建等[4]的研究表明,实验室酒精废水经过两级厌氧消化后系统BOD/COD约为0.5。据此计算,该污水站A/O系统进水BOD/TN约为1.6~2.0,明显低于反硝化过程BOD/TN>3的需求[5-6]。因此,可以判断该污水站TN去除率低归因于A/O生化系统进水的BOD/TN失调。影响TN去除率因素较多,针对上述问题结合工程实践,通过对比研究寻找合适的碳源、确定优化运行条件。根据生产实际,把葡萄糖、乙醇和清液作为主要考察对象,主要原因有:1)葡萄糖作为典型碳源添加剂,广泛应用于市政污水处理厂,操作方便且安全稳定;2)该工厂主要产品为燃料乙醇,有丰富的乙醇可作碳源;3)该污水站来水清液含有大量氨基酸、残糖和纤维素等有机物,而发酵酒精清液的BOD/COD一般为0.4~0.5,可生化性较好,因此,清液可作为碳源加以利用,也可实现污水处理的可持续发展[7-8]。
1.5 实验方法
1)取400 mL A/O系统生化污泥,用去离子水进行清洗以除去原污水中TN对实验的干扰。按照实际进水流量比例,取A/O出水250 mL与上述污泥混合,混合后的污泥浓度约8 500 mg·L−1。
2)取上述比例混合后的污泥300 mL,分别添加葡萄糖、乙醇和清液,每组实验取3组平行样。在500 mL烧杯内搅拌,搅拌强度以水面无漩涡为标准,防止搅拌剧烈充氧。通过前期摸索性实验得知,当反应起始的COD/TN不低于20时,反硝化过程的最大反应速率不受碳源浓度的影响,因此,实验中确保COD/TN不低于20。为避免因添加碳源带来的pH干扰会影响反应初期的反硝化速率,在添加碳源后统一调整pH为6.9。
3)在不同碳源和不同COD/TN的情况下,通过监测反应系统上清液pH和TN的变化来研究反硝化过程。
2. 结果分析与讨论
2.1 葡萄糖为碳源对处理效果的影响
以葡萄糖为碳源的研究结果如图2所示。由图2可知,在反应初始阶段(0~24 h),TN浓度从222.5 mg·L−1下降至215.6 mg·L−1,而反应初始pH由6.90降至6.55。此阶段pH的下降可能是葡萄糖水解产生有机酸导致的[9]。该阶段TN去除量很少,可能是污泥系统中的微生物种群对于基质的变化需要有适应过程,原污水中的葡萄塘较少。加入葡萄糖后,参与反硝化过程的大多数微生物需要时间去适应。反应第2阶段(24~36 h),TN浓度从215.6 mg·L−1下降至130.9 mg·L−1,而pH从6.55下降至6.34。这主要是因为在该系统内主要存在两大类反应:水解酸化和反硝化。在此阶段,葡萄糖水解产酸的速率明显大于反硝化的产碱速率,所以虽然TN已经有了明显下降,但pH仍然下降。反应第3阶段(36~48 h),TN浓度从130.9 mg·L−1下降至52.0 mg·L−1,而pH从6.34上升至8.17,这是典型的反硝化过程,表明在此阶段系统的反硝化产碱速率已明显超过了有机物的水解酸化速率,故系统的pH明显上升。
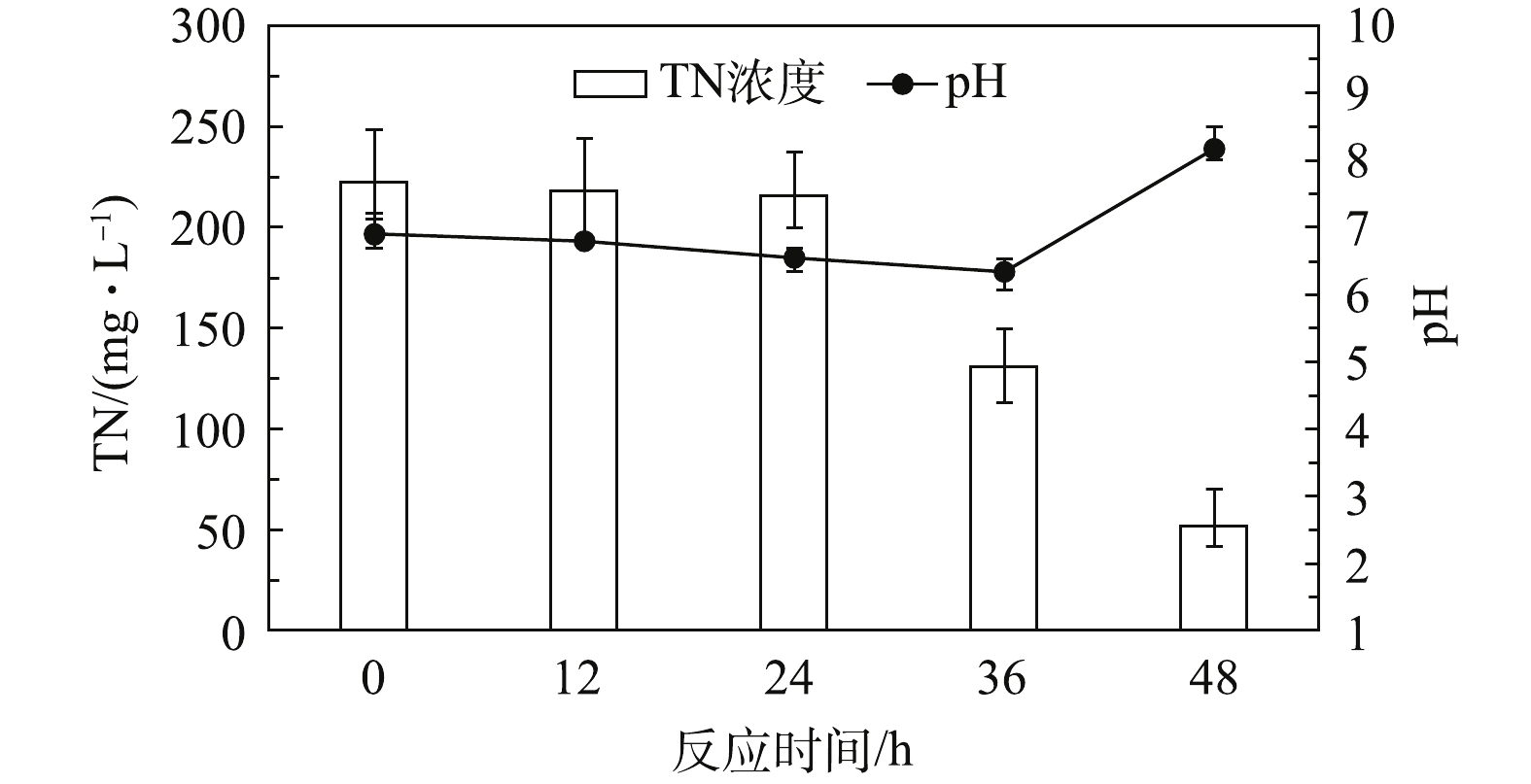 图 2 以葡萄糖为碳源的反硝化系统变化曲线Figure 2. Variation of denitrification system with glucose as carbon source
图 2 以葡萄糖为碳源的反硝化系统变化曲线Figure 2. Variation of denitrification system with glucose as carbon source2.2 乙醇为碳源对处理效果的影响
以乙醇为碳源的实验结果如图3所示。反应初始阶段(0~24 h)的TN由225.6 mg·L−1下降至210 mg·L−1,而反应初始pH由6.97上升至7.17。结果表明:反应初始阶段系统中已经有了微弱的反硝化作用;与葡萄糖作为碳源情况类似,污泥中的微生物菌群在更换反应基质后都需要一定的时间去适应[10]。
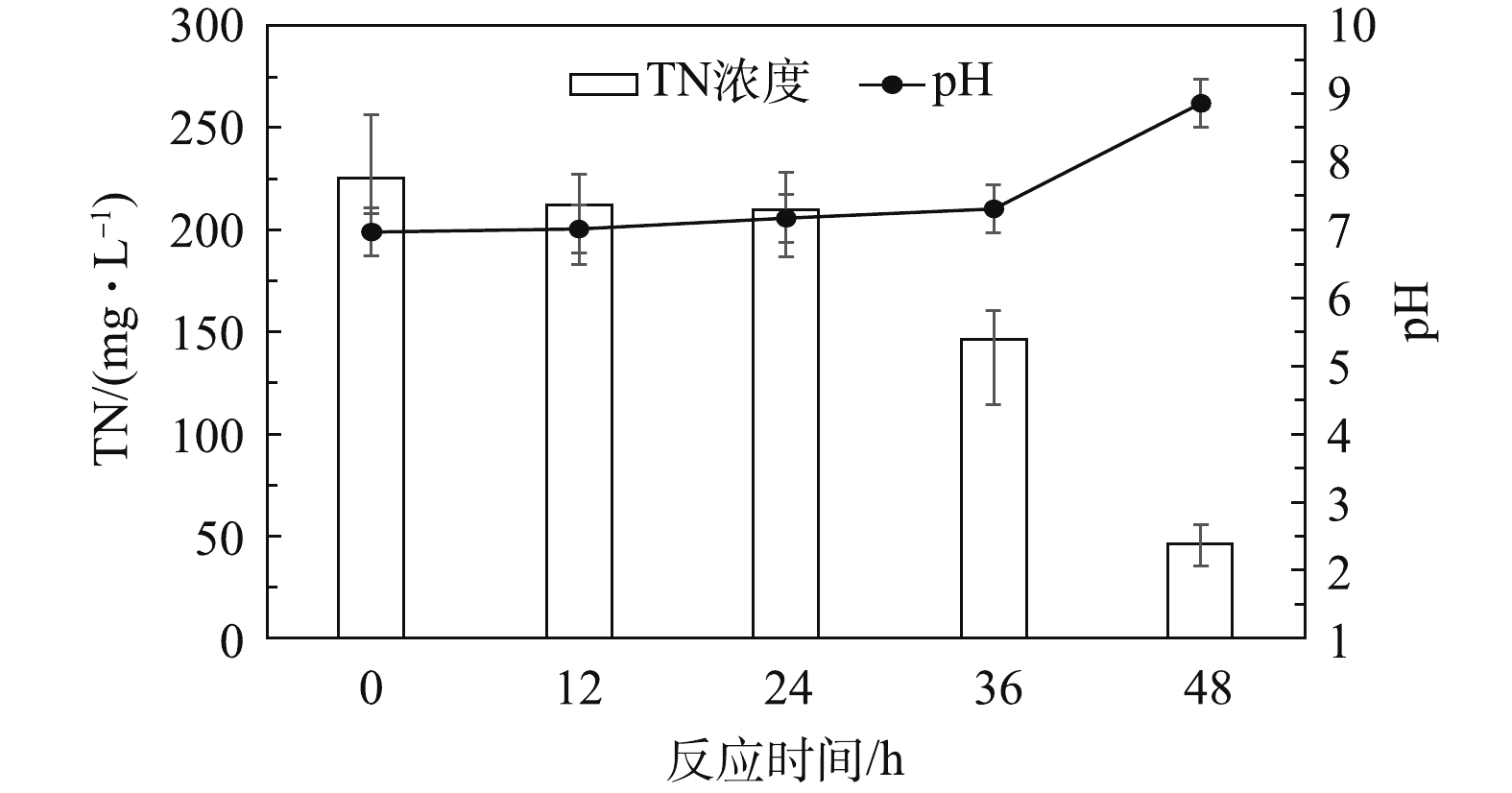 图 3 以乙醇为碳源的反硝化系统中各指标的变化Figure 3. Variation of denitrification system with ethanol as carbon source
图 3 以乙醇为碳源的反硝化系统中各指标的变化Figure 3. Variation of denitrification system with ethanol as carbon source反应第2阶段(24~36 h)的TN由210.0 mg·L−1下降至146.4 mg·L−1,而pH从7.17上升至7.31。说明微生物经一定时间的适应后,反硝化反应速率明显增加。反应第3阶段(36~48 h)的TN由146.4 mg·L−1下降至46.5 mg·L−1,而pH从7.31上升至8.86,表明此阶段反硝化过程反应速率达到了较高水平。
与葡萄糖作为碳源相比,乙醇为碳源的反应体系中pH值提升速率更快,这可能是由于实际生产废水中本来残存乙醇,微生物菌群对于乙醇的适应能力要强于对葡萄糖的适应能力。
2.3 清液为碳源对处理效果的影响
清液实际上是发酵酒精产物,其中所含营养物主要有糖分、蛋白质、纤维素、残糖、短纤维、挥发酸、有机氮、有机磷、钾等,还有大量胶质和菌丝体等[11]。以清液为补充碳源加入后,体系中各参数变化如图4所示。反应初始阶段(0~15 h)的TN由308.1 mg·L−1下降至270.3 mg·L−1,而反应pH基本维持不变。这表明在反应初期,虽然有明显的反硝化作用,但清液中存在的有机酸、氨基酸等物质会消耗反硝化过程产生的碱性物质,故反应体系的pH无明显变化。反应第2阶段(15~24 h)的TN由270.3 mg·L−1下降至179.6 mg·L−1,pH由6.89上升至6.93。在此阶段,TN下降明显,而pH上升速率较慢。这是由于清液中存在的酸性物质继续缓冲了反硝化产生的碱,导致系统的pH变化缓慢。反应第3阶段(24~36 h)的TN由179.6 mg·L−1下降至131.9 mg·L−1,pH由6.93上升至7.18。该阶段TN的反应速率开始下降,但pH上升幅度比第2阶段要快。这可能是由于清液中的酸性物质已消耗殆尽,缓冲作用已明显削弱,故系统pH变化明显。反应第4阶段(36~48 h)的TN在132 mg·L−1左右不再继续下降,但pH从7.15上升至7.65。这可能是由于经过一段时间的反应,清液中的含氮有机物开始了氨化反应,氨化反应产生的游离氨使系统的pH升高[12-13]。
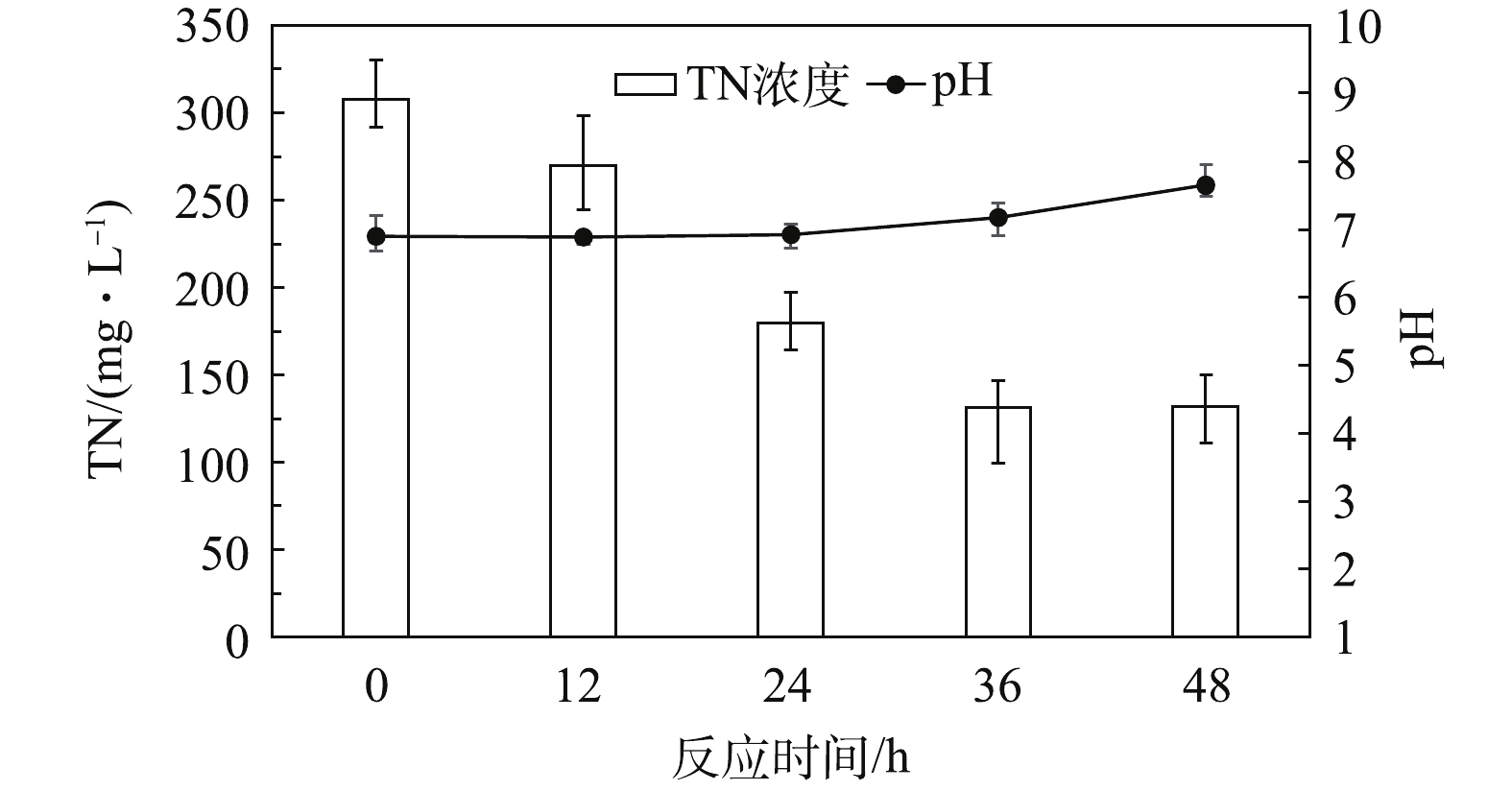 图 4 以清液为碳源的反硝化系统变化曲线Figure 4. Variation of denitrification system with raw wastewater as carbon source
图 4 以清液为碳源的反硝化系统变化曲线Figure 4. Variation of denitrification system with raw wastewater as carbon source相比葡萄糖和乙醇,清液作为碳源时,系统的最终pH最低。这可能是由于清液中含有大量酸性物质所致。这部分酸性物质主要由乳酸、琥珀酸、乙酸和丁酸等发酵副产物组成。而葡萄糖比乙醇更容易发生水解酸化,使得葡萄糖作碳源时反硝化反应的最终pH更低。
2.4 不同碳源反应条件下TN去除速率对比
根据上述实验结果,对比3种碳源在反硝化反应中的TN去除速率,计算公式如式(1)所示,结果如表3所示。
表 3 3种碳源的TN去除速率Table 3. The removal rate of total nitrogen with addition of three carbon sources反应时间/h TN去除速率/(mg·(L·h)−1) 葡萄糖 乙醇 清液 12 0.38 1.10 3.15 24 0.20 0.20 7.56 36 7.06 5.30 3.98 48 6.58 8.33 0.00 平均值 3.55 3.73 3.67 | Show TableDownLoad:
CSV
ΔN=Ns−N0ΔT (1) 式中:ΔN单位为TN反应速率即在单位时间内TN下降浓度,mg·(L·h)−1;
Ns 为在一定反应时间后的TN浓度,mg·L−1;N0为在反应初始期的TN浓度,mg·L−1;ΔT为反应时间,h。由表3可知,在较长反应时间内,以上3种碳源条件下的平均TN去除速率接近,分别为3.55、3.73和3.67 mg·(L·h)−1。这表明在碳源充足的情况下,当反应时间足够长时,碳源种类对反硝化过程的影响较小。但对于某一时段内的反应速率,3种碳源还是有明显区别的。分时段内3种碳源下的最大TN去除速率分别为葡萄糖7.06 mg·(L·h)−1、乙醇8.33 mg·(L·h)−1、清液7.56 mg·(L·h)−1。乙醇去除速率最快主要是由于反硝化细菌优先利用小分子醇类[14],而葡萄糖作为相对复杂的有机物,需要水解酸化后才被吸收。而清液由于能被反硝化细菌快速利用的小分子酸类物质占比较小,该条件下反应速率低于乙醇做碳源的条件。吴代顺等[15]研究表明,当利用乙醇作为碳源进行反硝化反应时,最大反硝化速率超过葡萄糖等有机物。
2.5 经济性比较
假定A/O系统的进水参数为:流量200 m3·h−1;COD 1 800 mg·L−1、TN 450 mg·L−1;投加碳源后的COD/TN=20。以此计算3种碳源投加条件下,生物脱氮过程增加的运行成本,结果见表4,其中葡萄糖和乙醇的单价采用市场采购价格,而清液单价以清液未进行厌氧反应而损失的沼气发电收益来计。
表 4 3种不同碳源运行成本对比Table 4. Comparison of operation costs among three different carbon sources碳源 单位时间碳源投加量/(kg·h−1) 单位TN碳源投加量/(kg·kg−1) 单位体积废水增加的运行成本/(元·t−1) 备注 葡萄糖 1 620 20.3 24.3 葡萄糖市场价格为3 000元·t−1 乙醇 3 348 41.9 100.4 乙醇市场价格为6 000元·t−1 清液 47 787 587.0 9.0 投加清液减少沼气发电收益,该厂清液发电收益为28.6元·m−3清液 | Show TableDownLoad:
CSV
上述计算过程未考虑添加碳源后系统运行成本的增加。这是由于即使添加的碳源全部以曝气方式去除,在该污水站的曝气系统效率及电价基础上,每吨污水的处理成本增加量也不到0.5元。与表4中的成本增加相比,影响较小,所以未作过多考虑。表4计算结果显示,清液成本为仅为乙醇成本的9%,葡萄糖成本的37%。从经济角度出发,应该充分利用废水中的有机物来进行生物脱氮;而充分利用原废水中的碳源也可实现污水资源利用,符合污水处理可持续发展的需要。
3. 工程运行调整及工艺验证
投加不同碳源时,清液的最大反硝化速率是乙醇的90%;从经济成本方面考虑,投加清液的成本仅是乙醇的10%。通过实验研究分析,综合脱氮反应速率和运行成本经济性两方面考虑,以清液作为反硝化碳源具有较明显的优势。
该污水站A/O系统进水流量200 m3·h−1,反硝化停留时间为50 h,内回流比为450%,外回流比100%。未添加清液时,A/O进水COD在1 792~2 023 mg·L−1。外加碳源清液的COD约50 000~62 000 mg·L−1,TN约1 100~1 500 mg·L−1,对不同清液投加量下的A/O系统TN去除率进行了比较,结果如图5所示。未添加清液前,A/O系统进水的COD/TN在5~6,TN去除率约41%;在添加清液后,COD/TN在6.0~9.3,TN去除率上升幅度较快。这主要是由于清液中的有机物被反硝化细菌快速利用,TN去除得以加速。当COD/TN在9.3~12.1时,进水COD在3 058~5 097 mg·L−1,TN在378~422 mg·L−1,出水COD在425~504 mg·L−1,TN在46~48 mg·L−1,TN去除率可以稳定在85%以上,最高达到89%,且水中TNK未检出,说明系统中氨化反应和硝化反应进行彻底,则TN主要以
NO−3 -N存在。而当COD/TN继续升高至12.9时,TN去除率反而下降。这主要是因为清液自身带有一定浓度的有机氮,这些氮的存在影响了原水中TN去除量的计算,从而影响了系统的TN去除率。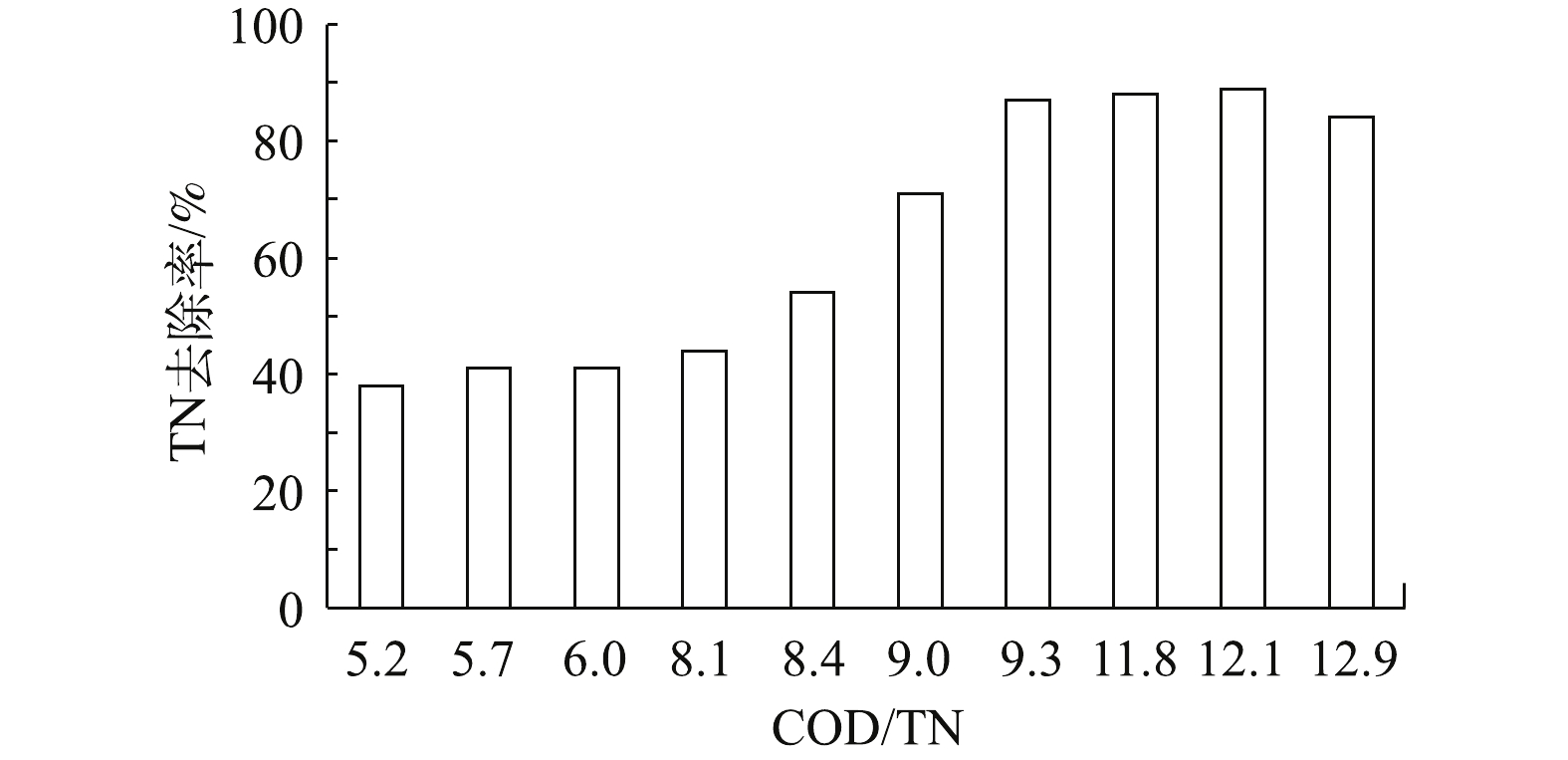 图 5 现场调整中不同COD/TN对应TN的平均去除率Figure 5. Relationship between different COD / TN ratio and TN average removal rate in field process regulation
图 5 现场调整中不同COD/TN对应TN的平均去除率Figure 5. Relationship between different COD / TN ratio and TN average removal rate in field process regulation根据上述实验结果对污水站的运行参数进行了相应调整,当混合液回流比和污泥回流比分别为450%和100%时,采用COD/TN=12.1,即每kg TN投加清液约307 L,该污水站的外排TN可稳定在50 mg·L−1以下。综上所述,DDG项目废水脱氮工艺中,A/O系统进水TN约300~600 mg·L−1时,可通过投加清液作为碳源,以确保A/O系统废水COD/TN在12.1以上,使A/O系统保持较高的TN去除率的同时,对运行成本的影响有限。
4. 结论
1)对于反硝化脱氮碳源不足废水处理,通过投加葡萄糖、乙醇、清液3种不同碳源进行反硝化试验。试验可知采用葡萄糖、乙醇和清液分别做反硝化碳源时,脱氮效率分别为76.6%、79.4%和57.2%;3种不同碳源投加方案下,以葡萄糖做碳源时的TN去除速率最小,清液次之,乙醇做碳源的TN去除速率最大为8.33 mg·(L·h)−1,实验表明反硝化细菌优先利用小分子醇和酸类有机物作为反硝化碳源。
2)对3种碳源条件的经济性比较发现,清液为乙醇成本的9%、为葡萄糖的37%。利用原废水中的有机物经济性最佳,也符合污水处理可持续发展的需要。
3)在DDG项目废水处理脱氮工艺中,A/O系统进水TN约300~600 mg·L−1时,可通过投加清液作为碳源确保A/O系统废水的COD/TN在12.1以上,从而使A/O具有较高TN去除率的同时,对运行成本的影响有限。
4)实际工程应用研究为DDG废水的处理提供了经济合理的碳源补充方案,能为可生化性较好的发酵行业废水处理提供参考。为获得更好的脱氮效率,下一步还需探讨好氧系统内回流比及反硝化停留时间对反硝化效率的影响。
-
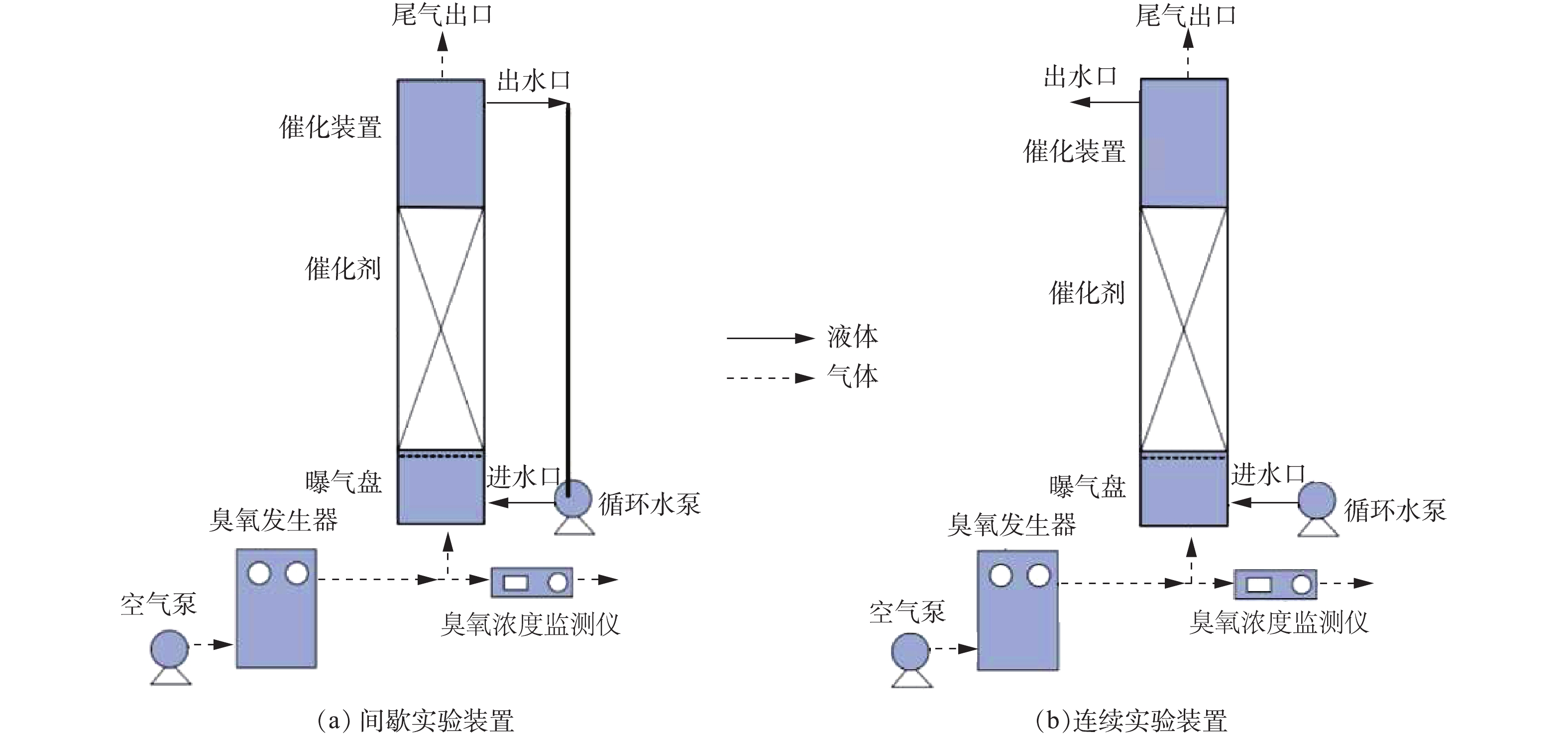
图 1 臭氧催化氧化间歇实验装置和连续实验装置示意图
Figure 1. Ozone catalytic oxidation batch experiment schematic diagram and continuous experiment schematic diagram
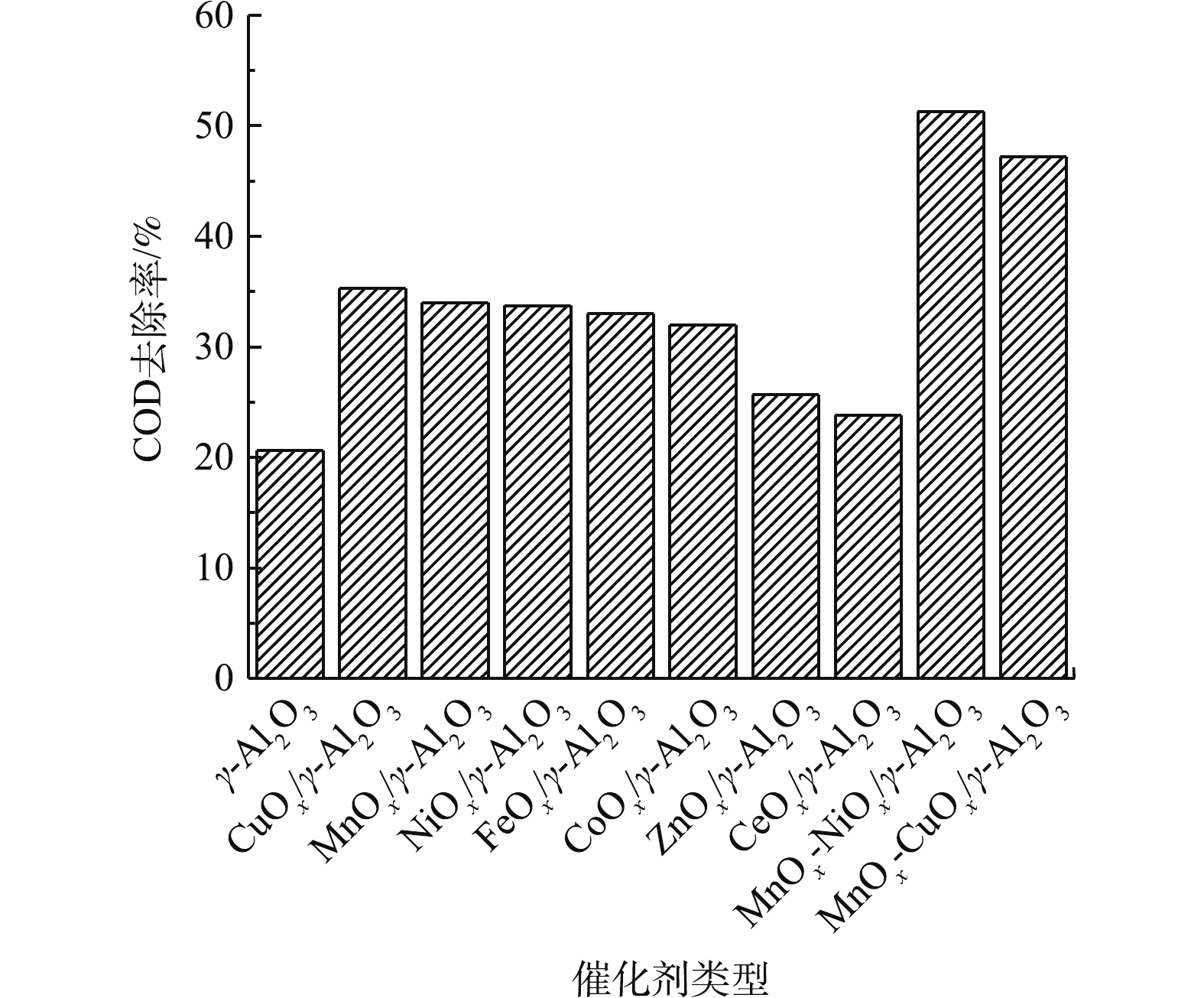
图 2 不同活性组分催化剂的催化效果
Figure 2. Catalytic effects of catalysts with different active components
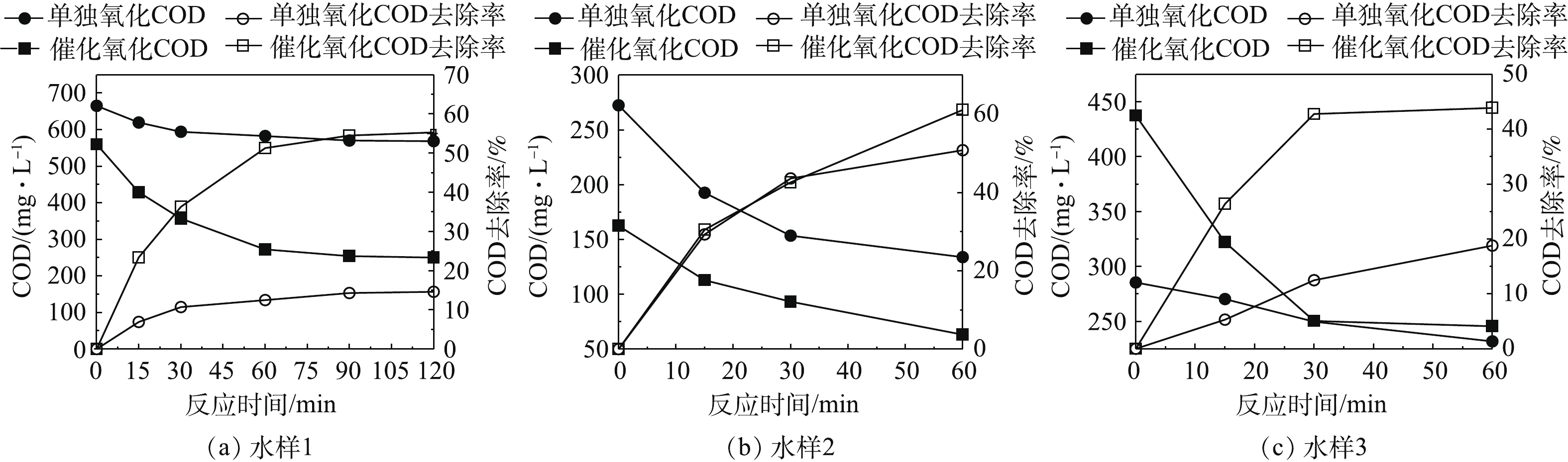
图 5 不同水样臭氧单独氧化与臭氧催化氧化工艺效果比较
Figure 5. Comparison of ozone oxidation and ozone catalytic oxidation process treating different water samples
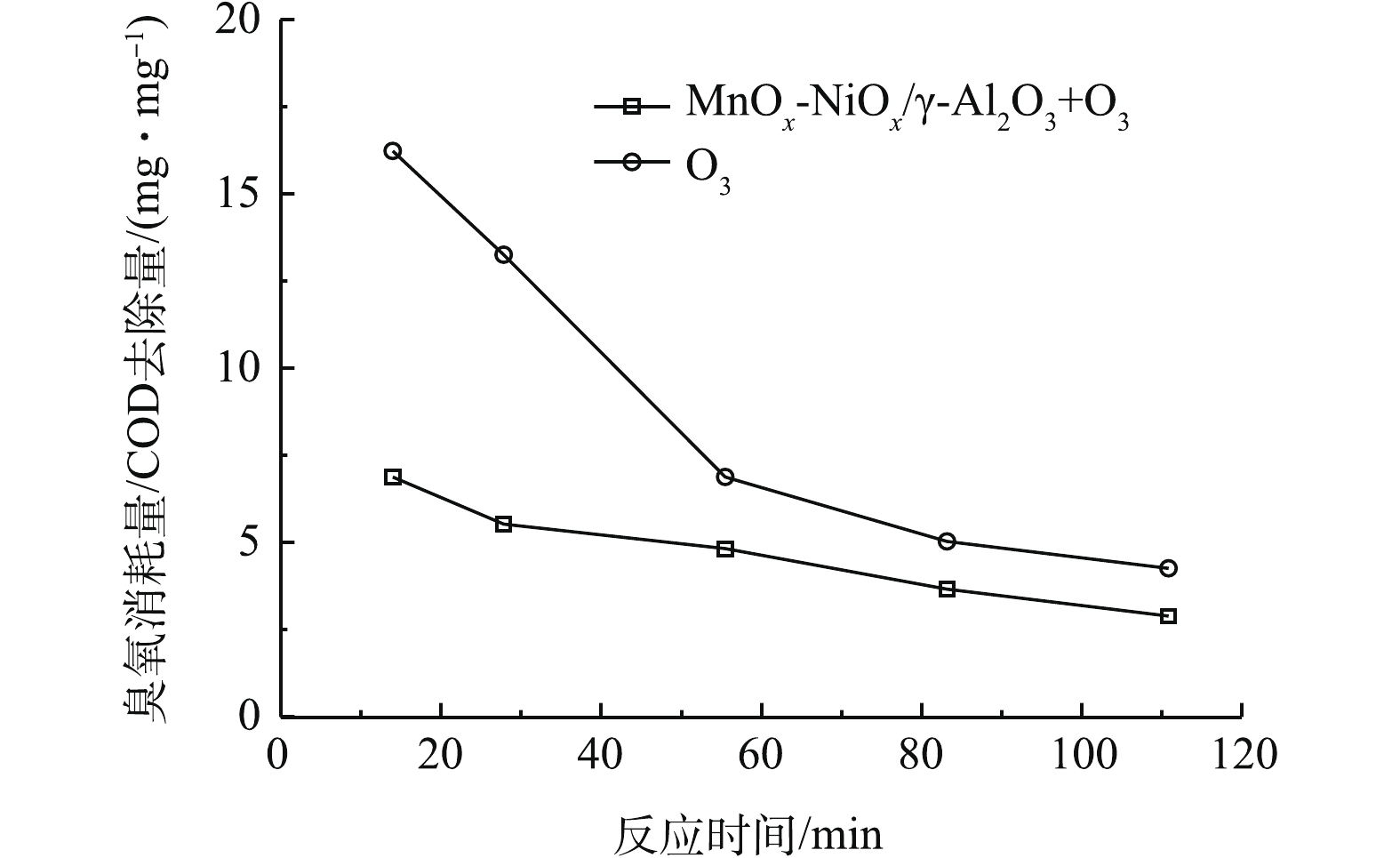
图 6 在单独氧化和臭氧催化氧化体系中臭氧消耗量/COD去除量的变化
Figure 6. Changes of ozone consumption / COD removal in the oxidation alone and ozone catalytic oxidation processes
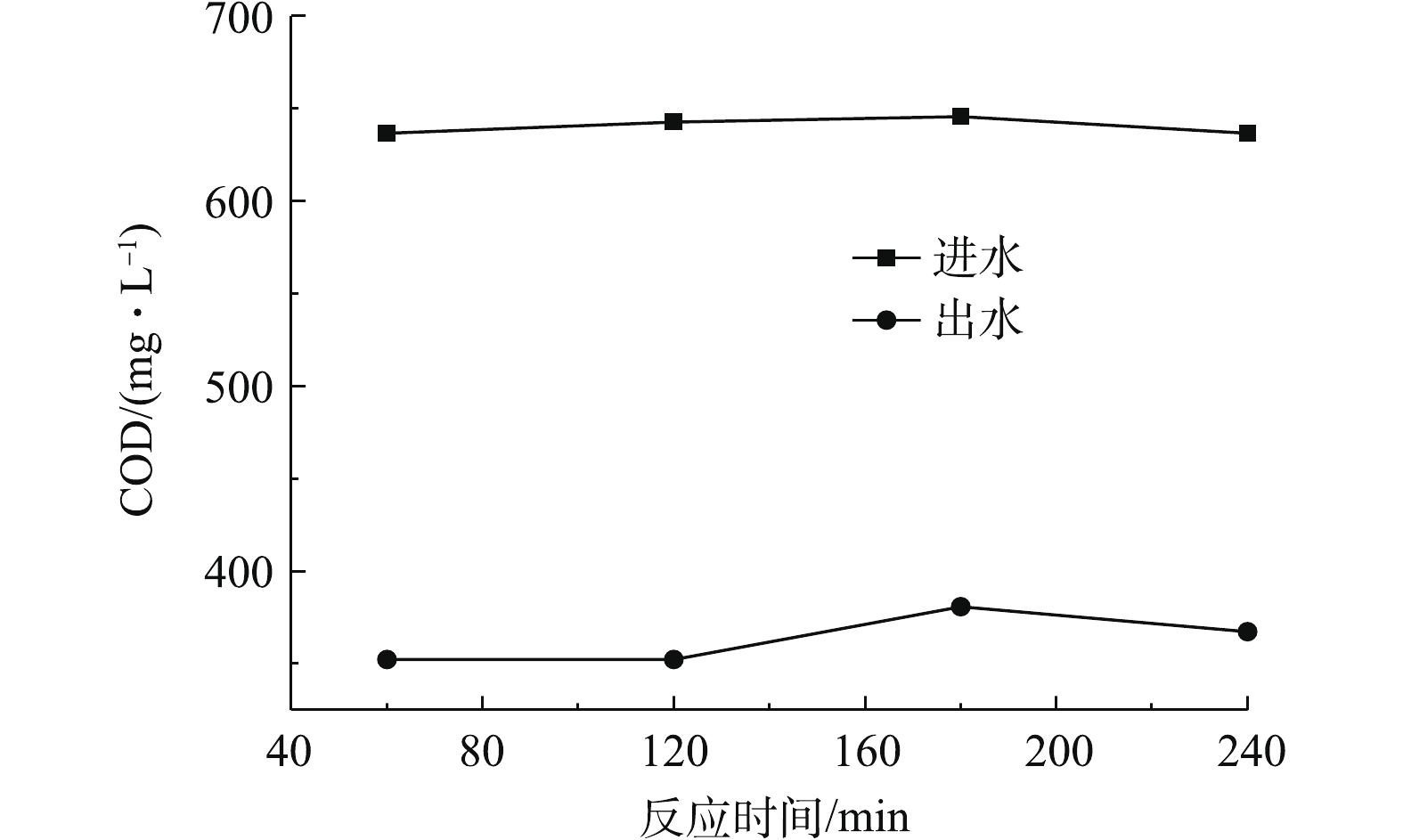
图 9 臭氧催化氧化连续实验中进出水中的COD变化
Figure 9. Changes of COD of influent and effluent water in the continuous experiments of ozone catalytic oxidation
表 1 实验水样基本水质
Table 1. Basic water quality of experimental water samples
水样序号 pH 电导率/(mS·cm−1) TDS/(g·L−1) 氯离子/(mg·L−1) COD/(mg·L−1) 1 8.09 14.10 7.05 3 400 457~665 2 7.55 2.97 1.50 570 126~272 3 8.36 4.57 2.28 1 040 285~437
下载: 导出CSV
-
[1] DENISE M A, ISABELLE M C, DARREN S D, et al. Organic and inorganic composition and microbiology of produced waters from pennsylvania shale gas wells[J]. Applied Geochemistry, 2015, 60(4): 116-125. [2] ZHANG L, LIU X, GUO X, et al. Investigation on the degradation of brilliant green induced oxidation by NiFe2O4 under microwave irradiation[J]. Chemical Engineering Journal, 2011, 173(3): 737-742. doi: 10.1016/j.cej.2011.08.041 [3] GUPTA V K, SUHAS. Application of low-cost adsorbents for dye removal: A review[J]. Journal of Environmental Management, 2009, 90(8): 2313-2342. doi: 10.1016/j.jenvman.2008.11.017 [4] ADAK A, BANDYOPADHYAY M, PAL A. Removal of crystal violet dye from wastewater by surfactant-modified alumina[J]. Separation & Purification Technology, 2005, 44(2): 139-144. [5] SLOKAR Y M, MAJCEN L M A. Methods of decoloration of textile wastewaters[J]. Dyes & Pigments, 1998, 37(4): 335-356. [6] MANAL M A, EL-NAGGAR S, EL-AASAR A, et al. Bioremediation of crystal violet using air bubble bioreactor packed with pseudomonas aeruginosa[J]. Water Research, 2005, 39(20): 5045-5054. doi: 10.1016/j.watres.2004.08.001 [7] LIAO Y, FU M, CHEN L, et al. Catalytic oxidation of toluene over nanorod-structured Mn-Ce mixed oxides[J]. Catalysis Today, 2013, 216(18): 220-228. [8] CHEN C C, LIAO H J, CHENG C Y, et al. Biodegradation of crystal violet by pseudomonas putida[J]. Biotechnology Letters, 2007, 29(3): 391-396. doi: 10.1007/s10529-006-9265-6 [9] MEHRJOUEI M, SIEGFRIED M, DETLEV M. A review on photocatalytic ozonation used for the treatment of water and wastewater[J]. Chemical Engineering Journal, 2015, 263(1): 209-219. [10] ANA M, REY A, FERNANDO J, et al. Solar photo-ozonation: A novel treatment method for the degradation of water pollutants[J]. Journal of Hazardous Materials, 2016, 317(1): 36-43. [11] 杨静, 王建兵, 王亚华, 等. 高级氧化工艺处理煤化工浓盐水[J]. 环境工程学报, 2015, 9(8): 3680-3686. doi: 10.12030/j.cjee.20150815 [12] 王超, 赵旭, 侯子义, 等. 光电催化氧化处理反渗透浓水[J]. 环境工程学报, 2014, 1(8): 3189-3194. [13] WANG J L, XU L J. Advanced oxidation processes for wastewater treatment: formation of hydroxyl radical and application[J]. Critical Reviews in Environmental Science and Technology, 2012, 42(3): 251-325. doi: 10.1080/10643389.2010.507698 [14] 王利平, 沈肖龙, 倪可, 等. 非均相催化臭氧氧化深度处理炼油废水[J]. 环境工程学报, 2015, 9(5): 2297-2302. [15] 杜松, 金文标, 王吉坤, 等. 非均相催化臭氧氧化处理煤化工高含盐废水[J]. 煤炭科学技术, 2018, 46(9): 54-58. [16] DHANDAPANI B, OYAMA S T. Gas phase ozone decomposition catalysts[J]. Applied Catalysis B: Environmental, 1997, 11(2): 129-166. doi: 10.1016/S0926-3373(96)00044-6 [17] CHEN C, LI Y, MA W, et al. Mn-Fe-Mg-Ce loaded Al2O3 catalyzed ozonation for mineralization of refractory organic chemicals in petroleum refinery wastewater[J]. Separation and Purification Technology, 2017, 183(1): 1-10. [18] 亓丽丽. 非均相臭氧催化氧化对氯苯酚机理研究及其工艺应用[D]. 哈尔滨: 哈尔滨工业大学, 2013. [19] 庄海峰. 催化臭氧化-生物组合工艺深度处理煤制气废水效能的研究[D]. 哈尔滨: 哈尔滨工业大学, 2015. [20] 朱秋实, 陈进富, 姜海洋, 等. 臭氧催化氧化机理及其技术研究进展[J]. 化工进展, 2014, 33(4): 1010-1014. [21] IKHLAQ A, BROWN D R, KASPRZYK-HORDERN B. Mechanisms of catalytic ozonation: An investigation into superoxide ion radical and hydrogen peroxide formation during catalytic ozonation on alumina and zeolites in water[J]. Applied Catalysis B: Environmental, 2013, 129(1): 437-449. [22] QI F, XU B, CHEN Z, et al. Catalytic ozonation of 2-iso-propyl-3-methoxypyrazine in water by γ-AlOOH and γ-Al2O3: Comparison of removal efficiency and mechanism[J]. Chemical Engineering Journal, 2013, 219(1): 527-536. [23] DAI Q, WANG J, YU J, et al. Catalytic ozonation for the degradation of acetylsalicylic acid in aqueous solution by magnetic CeO2 nanometer catalyst particles[J]. Applied Catalysis B: Environmental, 2014, 144(1): 686-693. [24] AFZAL S, QUAN X, ZHANG J. High surface area mesoporous nanocast LaMO3(M=Mn, Fe) perovskites for efficient catalytic ozonation and an insight into probable catalytic mechanism[J]. Applied Catalysis B: Environmental, 2017, 206(1): 692-703. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 6191
- HTML全文浏览数: 6191
- PDF下载数: 112
- 施引文献: 0
