

-
挥发性有机化合物(VOCs)对人体有害[1],且可与大气中的NOx发生光化学反应,形成灰霾和近地表臭氧污染[2-4]。催化氧化法可在250~500 ℃将VOCs彻底分解为小分子,适合应用于不具有回收价值的VOCs废气处理,其中催化剂是核心[5-6]。含氯VOCs(CVOCs)一般作为有机合成原料和溶剂使用,常见于石化、制药等行业VOCs废气中。与普通碳氢VOCs不同,CVOCs容易使催化剂氯中毒失活[7]。因此,开发抗氯中毒能力强、环境友好的CVOCs催化剂是未来研究的重点。
钒基催化剂以V2O5-WO3/TiO2催化剂应用最为广泛,一般用于选择性催化还原(SCR)脱硝[8]。V2O5-WO3/TiO2催化剂之所以能够在成分复杂的工业烟气中成功应用,与催化剂抗硫、氯中毒能力强有关。因此,钒基催化剂对于VOCs的催化性质也受到越来越多的关注[9-10]。钒基催化剂在CVOCs催化氧化中的主要优点为稳定性好、副产物少[11]。与钒基催化剂相比,商用的铂、钯贵金属基催化剂不仅容易氯中毒失活,且在氯苯的催化氧化过程中会生成多氯代副产物,其毒性较初始VOCs更大[12-13]。其他过渡金属组分(如Mn、Ce等)在CVOCs催化氧化过程中均会出现明显的失活[14]。
将钒基催化剂应用于CVOCs催化氧化仍有一些问题有待解决。如V2O5-WO3/TiO2催化剂用于脱硝时,催化剂中V2O5的质量分数一般为0.5%~1.5%,WO3的质量分数通常在5%~10%,其余为TiO2和少量黏结剂。在脱硝反应中,WO3提供了大量Bronsted酸性位点,可以有效吸附NH3,从而增强催化剂活性[15]。有研究[10]认为,WO3也可起到吸附VOCs反应物的作用。然而,VOCs与NH3的性质明显不同,WO3在氯苯催化氧化中的作用是否显著仍须进一步探讨。此外,也有必要明确CVOCs中氯元素对传统V2O5-WO3/TiO2催化剂的毒害作用大小。
本研究通过合成V2O5/TiO2、WO3/TiO2和V2O5-WO3/TiO2催化剂,采用氯苯作为CVOCs的模型化合物,同时开展催化评价和原位红外实验研究,结合宏观催化性能和微观反应过程2个方面的结果,分别明确V2O5和WO3在氯苯催化氧化过程中的作用。本研究将有助于进一步优化CVOCs催化氧化的钒基催化剂,也对V2O5-WO3/TiO2催化剂用于同时脱硝脱CVOCs有借鉴意义。
-
TiO2粉末购自阿拉丁试剂(上海)有限公司,晶型为锐钛矿,纯度≥99.8%;偏钒酸铵、钨酸铵、草酸均购自国药集团化学试剂有限公司,分析纯。
-
采用浸渍法制备V2O5/TiO2、WO3/TiO2和V2O5-WO3/TiO2催化剂。将TiO2粉末在马弗炉中550 ℃焙烧2 h后作为催化剂载体备用。在室温下,将0.025 7 g偏钒酸铵、0.128 6 g偏钒酸铵、1.049 8 g钨酸铵和0.025 7 g偏钒酸铵与1.049 8 g钨酸铵的混合物分别加入去离子水中,并加入过渡金属摩尔量2倍的草酸,使上述活性组分前驱体溶解。取TiO2载体10 g,并分为5份,其中4份分别加入上述前驱体溶液中,搅拌混匀后,在45 ℃旋转蒸发,过夜干燥,之后以5 ℃·min−1的升温速率在500 ℃焙烧4 h,得到粉末催化剂。以V2O5和WO3在催化剂中所占的质量分数计,分别记为1%和5%的V2O5/TiO2催化剂(V1Ti和V5Ti)、4%的WO3/TiO2催化剂(W4Ti)、1%的V2O5与4%的WO3的V2O5-WO3/TiO2催化剂(V1W4Ti)。将部分TiO2载体和上述粉末催化剂压片、筛分为40~60目颗粒,用于活性评价,剩余粉末样品用于原位红外表征。
-
粉末样品的X射线衍射(X-ray diffraction,XRD)谱图通过岛津Rigaku D/max-RA衍射仪得到;比表面积和孔结构在康塔Autosorb iQ自动表面积和孔结构分析仪上通过N2吸附和脱附测定。
-
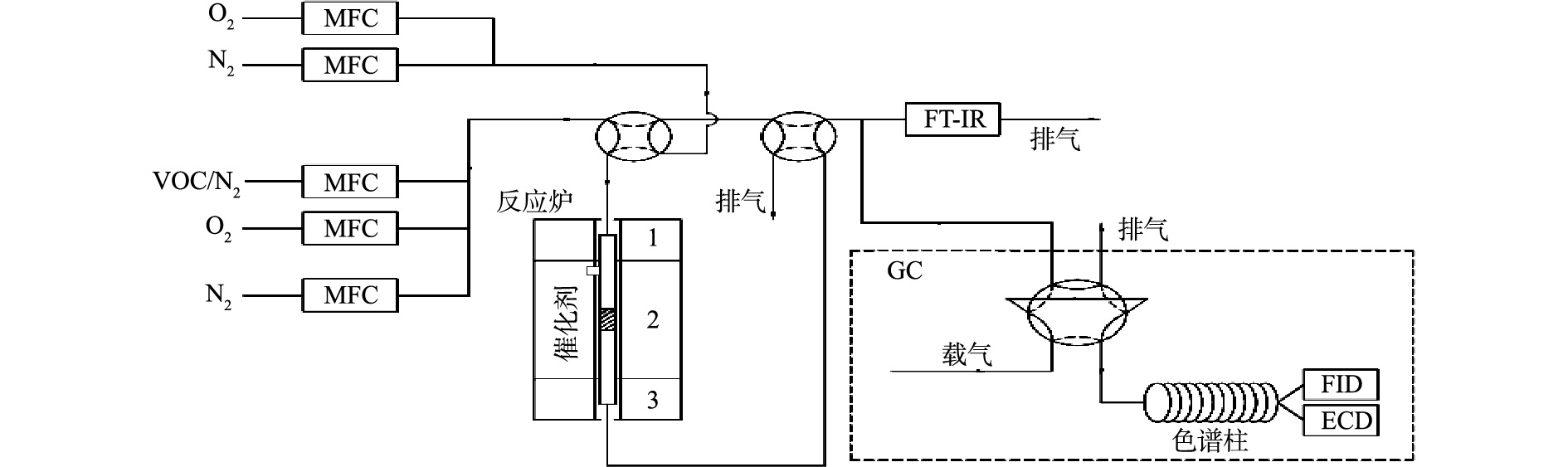
催化剂评价在固定床-气相色谱联用实验平台上进行,实验装置如图1所示。催化反应管为内径4 mm的石英管,设置筛网,用于放置催化剂。催化反应管通过电炉加热,电炉控温热电偶紧贴催化反应管中催化剂放置的位置。氯苯、O2、N2均通过钢瓶产生,通过质量流量控制器控制气体流量,总气量为100 mL(标况),其中氯苯为4.5 mmol·m−3(标况,体积分数为10−4),O2为10%,其余为N2。实验气路设置有旁路和反应路(旁路不通过催化剂床层),均在80~90 ℃保温,基本消除了对氯苯的吸附。通过配备有FID和ECD检测器的安捷伦GC 7890A气相色谱对氯苯进出口浓度进行测试,通过配备有甲烷转化炉和FID的岛津GC 2010气相色谱对CO2和CO出口浓度进行测试。催化剂评价使用100 mg的催化剂,对应的质量空速(WHSV)为60 000 mg·(g·h)−1。活性评价通过程序升温的方式测试,温度为100~400 ℃,每个测试点间隔50 ℃并稳定1 h,升温速率为10 ℃·min−1;稳定性评价在300 ℃进行,时间为6 h。
氯苯转化率通过式(1)计算,CO2和CO选择性通过式(2)计算。
式中:x为氯苯转化率;Cinlet和Coutlet分别为氯苯进出口浓度;S(COx)为CO2或CO选择性(x= 1或2;当表示为COx时,指的是CO2和CO的总和);C(COx)为出口CO2或CO的浓度(x= 1或2;当表示为COx时,指的是CO2和CO的总和)。空白测试表明,在温度为100~400 ℃时,没有催化或气相均相反应发生。
-
原位红外谱图通过配备有MCT检测器的热电Nicolet 6700红外光谱仪采集,原位池采用CaF2作为窗片。实验开始前,使用N2将原位池吹扫干净,将约50 mg催化剂样品放置于原位池中,并于400 ℃在10% O2/N2气氛下预处理1 h,之后分别采集400、300、200 ℃的样品,并在N2气氛下采集100 ℃的背景。为了测试催化剂样品对于氯苯的吸附性能,在100 ℃将4.5 mmol·m−3氯苯/N2气流通入原位池中30 min,之后使用N2吹扫30 min,分别采集红外谱图。为了测试不同温度下催化剂对于反应中间产物的氧化能力,将4.5 mmol·m−3氯苯/10% O2/N2气流通入原位池中,并升温至200 ℃,反应30 min,之后在10% O2/N2气氛下分别升温至300 ℃和400 ℃,分别采集红外谱图。
-
图2为TiO2载体及所制备的催化剂XRD表征结果。可以看出,TiO2粉末在550 ℃焙烧后仍维持了锐钛矿晶体结构,基本观察不到金红石相的形成。在所研究的催化剂负载量下,通过XRD表征,没有发现归属于V2O5或WO3的衍射峰,这说明催化剂表面活性组分没有发生大幅团聚。
对上述样品进行了N2吸附和脱附测试,结果如表1所示。可见,TiO2载体的比表面积最大,催化剂由于负载活性组分,比表面积均小于载体,负载量越大,催化剂的比表面积越小。其中,V5Ti的比表面积不仅明显小于TiO2,同时小于V1W4Ti,这说明WO3在TiO2上的分散性更好。虽然XRD表征结果没有观测到V5Ti催化剂上V2O5出现严重团聚,但V5Ti催化剂表面可能已经形成了V2O5微晶,堵塞了载体孔道。此外,样品比表面积的改变对于孔容和平均孔径的影响很小,所得到的数据几乎在误差允许的范围之内。
TiO2和催化剂上氯苯转化率与温度的函数关系如图3所示。可见,TiO2具有一定的催化能力,400 ℃时氯苯转化率可以达到40%以上。1%的V2O5负载使得氯苯转化率大幅提高,在相同温度下,V1Ti催化剂几乎可以实现氯苯的完全转化。相比较而言,WO3对于氯苯的催化能力较弱,W4Ti催化剂的催化活性甚至低于TiO2载体。且在W4Ti催化剂活性评价后期,随着反应温度的升高,催化剂上氯苯转化率出现了一定程度的下降,说明WO3出现了明显的氯中毒(积碳不会导致高温转化率降低)。与V1Ti催化剂相比,V1W4Ti催化剂活性出现了进一步的提升,在300 ℃时,氯苯转化率提高20%左右。需要指出的是,WO3负载会减少原有TiO2载体的暴露。因此,V1W4Ti催化剂对氯苯的催化活性并非V1Ti和W4Ti催化剂活性的叠加,说明V2O5和WO3之间存在协同作用,如WO3增强了V2O5的氧化能力,或者V2O5可以使WO3脱氯。当V2O5负载量进一步提高至5%时,V5Ti催化剂的活性又明显高于V1W4Ti催化剂,氯苯在约350 ℃即可实现100%转化。
TiO2和催化剂在氯苯催化氧化中的稳定性评价结果如图4所示。可见,TiO2、V1Ti、W4Ti和V1W4Ti催化剂在氯苯催化氧化过程中均出现了活性下降。TiO2活性在前3 h出现明显下降,3 h之后基本保持稳定。V1Ti催化剂活性明显高于TiO2,其活性下降幅度明显降低,但仍然没能达到稳态。W4Ti催化剂活性下降幅度最大,由40%下降至10%左右。与V1Ti和W4Ti催化剂相比,V1W4Ti的稳定性得到了进一步提高。与以上样品明显不同,V5Ti催化剂在稳定性评价过程中活性十分稳定。
选择以上催化剂中表现最好的V5Ti催化剂,研究了其在氯苯催化氧化过程中CO2和CO选择性。如图5所示,氯苯在V5Ti催化剂的作用下,几乎全部转化为COx,其选择性几乎与氯苯转化率相等。结合V5Ti催化剂对氯苯的稳定性评价结果(图4)可知,氯苯在V5Ti催化剂作用下确实发生了催化氧化反应,并非吸附在催化剂表面。然而,产物中出现了大量CO,说明氯苯在V2O5上的转化过程中出现了不完全氧化的现象,且V2O5对CO的氧化能力不强。因此,钒基催化剂用于VOCs的降解,可能须配合CO氧化催化剂。需要特别指出的是,在V5Ti催化剂对氯苯的催化氧化过程中,ECD检测器上没有发现多氯代副产物。
为了研究氯苯在催化剂上的转化过程,采用原位红外研究了氯苯在不同催化剂上的吸附和吹扫过程以及程序升温表面反应过程,结果分别如图6和图7所示,红外谱图中归属于中间产物的红外峰列于表2。
V1Ti、W4Ti、V1W4Ti和V5Ti催化剂上氯苯吸附和吹扫过程的原位红外结果如图6所示,红外谱图中不同波数红外峰的强度变化代表了催化剂表面物种数量的增减。如图6所示,红外谱图中主要包括氯苯的物理吸附峰、中间氧化产物的红外峰以及由于催化剂表面羟基物种变化形成的红外峰。具体来说,波数位于1 581、1 478 cm−1和1 444 cm−1位置的红外峰归属于物理吸附态的氯苯[16];1 596 cm−1和1 635 cm−1位置的红外峰归属于酚类物种[16](这2个位置的红外峰可能被其他红外峰掩盖而难以在1个催化剂上全部被观测到);1 561 cm−1的红外峰归属于醋酸盐物种[17]。以上红外峰在不同催化剂上均有出现。对于V5Ti催化剂,1 680 cm−1和1 661 cm−1位置的红外峰归属于醌类物种[18-19]。此外,在波数1 617 cm−1(V1Ti,图6(a))、1 628 cm−1(W4Ti、V1W4Ti,图6(b)和图6(c))、1 606 cm−1(V5Ti,图6(d))位置的红外峰为催化剂表面羟基物种变化引起的(如—OH物种的改变,或表面吸附有H2O)[16]。
必须指出,除V5Ti催化剂之外,其他催化剂在1 350~1 400 cm−1位置均出现了明显的倒峰,同时在1 250~1 350 cm−1位置出现了正峰,且这些红外峰强度的变化规律与归属于中间物种的红外峰不一致。猜测此现象与TiO2载体相关,因为随着活性组分负载量的增加,上述位置红外峰强度明显降低。TiO2载体可能与氯苯或中间产物之间存在某种作用,使得其对红外光的吸收发生了改变,造成对于某些波数红外光吸收增加,而对于某些波数红外光吸收减少。由于在上述位置中间产物的红外峰强度较低,受到此现象的干扰较大,因而后续对于此波数范围的红外谱图不再进行分析。
对1 400~1 800 cm−1位置的谱图分析可知,氯苯在上述催化剂上均出现了一定的转化,以V5Ti催化剂上的中间产物最多,V1W4Ti其次,V1Ti和W4Ti催化剂上的中间产物最少。这说明相对于WO3,V2O5可以更有效地活化氯苯分子。在吹扫过程中,催化剂上归属于氯苯的红外峰(1 581、1 478和1 444 cm−1)的峰强度均出现了一定的下降,说明氯苯被逐渐移除(或转化为中间产物);V1Ti、W4Ti和V1W4Ti催化剂上中间产物的峰强度均没有出现明显变化,V5Ti催化剂上中间产物的峰强度出现了明显的增加,说明100 ℃时中间物种在催化剂表面较为稳定,且V5Ti催化剂的氧化能力远比其他催化剂强,在较低温度且没有氧气通入的情况下即可使苯环发生大量裂解。
V1Ti、W4Ti、V1W4Ti和V5Ti催化剂在不同温度下的程序升温表面反应的原位红外结果如图7所示。归属于氯苯的红外峰(1 581、1 478和1 444 cm−1)强度很低,仅在200 ℃时可见,说明此温度条件下几乎不存在物理吸附的氯苯。归属于酚类物种和醋酸盐物种的红外峰(1 596 cm−1和1 561 cm−1)仅在V1W4Ti催化剂上200 ℃时氯苯的氧化过程中可见(图7(c)),这可能是由于此位置红外峰在其他催化剂上较弱,被其他物质红外峰覆盖所致。不同于100 ℃下的吸附和吹扫结果(图6),在每种催化剂上均可以观察到归属于醌类物种的红外峰(1 680 cm−1和1 661 cm−1)。除此之外,催化剂表面出现了新的红外峰,须进一步分析。其中1 581、1 551和1 414 cm−1位置的红外峰也可以归属为醋酸盐物种[17](此处1 581 cm−1的红外峰与物理吸附氯苯的红外峰不同),与红外峰位置在1 561 cm−1的醋酸盐物种的不同之处在于甲基含氯数量不同;1 509 cm−1和1 437 cm−1位置的红外峰归属于马来酸盐物种[17, 20],400 ℃时,此位置的红外峰强度相对较强,说明马来酸盐物种难以分解。此外,不同于H2O吸附在V2O5上在1 606 cm−1位置形成的红外峰,W4Ti和V1W4Ti催化剂上在1 614 cm−1位置出现了红外峰。这2种催化剂在苯的催化过程中在1 614 cm−1位置并没有观测到红外峰,说明1 614 cm−1位置的红外峰很可能与氯物种有关,这可能是由醋酸盐等中间产物进一步氯化形成的,如形成酰氯物种[16]。根据活性评价结果(图3),WO3抗氯中毒能力较差,因而氯元素会在WO3上大量累积,更容易形成表面氯化的中间产物。
总体来说,在200 ℃氯苯通入原位池的过程中,即催化剂表面物种积累的阶段,V1W4Ti催化剂表面中间物种的峰强度最大,其次是V1Ti催化剂,之后是V5Ti催化剂,最后为W4Ti催化剂。催化剂表面中间产物的多少与催化剂捕集和氧化氯苯分子的能力有关,捕集能力越强、氧化能力越弱的催化剂,其表面中间产物积累量将越大。根据程序升温过程中峰强度下降幅度来看,V5Ti催化剂的氧化能力最强,随着温度的升高,催化剂表面中间产物的峰强度几乎下降为零,说明中间物种几乎全部分解。而其他3种催化剂上均出现了中间产物峰强度先升高后下降的趋势,可能是催化剂表面吸附的氯苯进一步转化为中间产物或原位池中残留部分氯苯在催化剂表面氧化所致,说明这3种催化剂氧化能力远低于V5Ti催化剂。相比较而言,V1WTi催化剂的氧化能力略高于V1Ti催化剂(400 ℃时2种催化剂表面中间产物的峰强度接近,但200 ℃时V1W4Ti催化剂表面的中间产物峰强度更大)。而200 ℃时W4Ti催化剂表面中间产物的峰强度很弱,且随着温度的提高,中间产物的峰强度下降幅度也很小,说明W4Ti催化剂捕集和氧化氯苯分子的能力较差。
上述催化评价和原位红外实验结果说明,V2O5是关键的氧化活性组分,WO3不能很好地捕集和氧化氯苯分子,且容易发生氯中毒失活。因此,提高钒基催化剂活性的关键在于增加V2O5含量。此外,当V2O5和WO3同时存在时,二者之间具有协同作用,催化剂的活性和稳定性(相对于WO3/TiO2)均得到提高。因此,对于钒基催化剂用于氯苯的催化氧化,提高V2O5含量较提高WO3含量更为有效。
图8反映了氯苯浓度和O2浓度与氯苯在V5Ti催化剂上反应速率的关系(氯苯转化率低于15%)。将反应速率(−r)作为氯苯进口浓度(Cchlorobenzene)和O2进口浓度(Coxygen)的函数,两边取对数并进行线性拟合,可以得到氯苯和O2的反应级数,结果如图8所示。拟合后的数据显示,氯苯的反应级数为0.73,接近一级反应;O2的反应级数为0.24,接近零级反应。这说明氯苯的活化吸附较难进行,提高氯苯浓度,可以有效提高反应速率;而催化剂表面存在大量活性氧物种(晶格氧或表面氧),气氛中的氧仅作为对于活性氧物种的补充,提高气氛中氧浓度对于反应速率影响较小,这是一种典型的Mars-van Krevelen氧化还原反应机理。
保持氯苯进口浓度不变,空速对于V5Ti催化剂上氯苯转化率的影响结果如图9所示。可见,当催化剂质量一定时,提高反应空速,相同温度下氯苯转化率随之下降;反之,氯苯转化率提高。总体来说,当反应温度高于350 ℃时,氯苯在以上空速下均可实现完全转化。
-
1) V2O5在氯苯的催化氧化过程中起到最为重要的作用,在100 ℃时,V2O5即可氧化氯苯形成大量中间产物,随着温度的升高,中间产物可有效地从钒基催化剂表面移除。
2) WO3氧化氯苯的能力较弱,且容易发生中毒失活,催化氧化氯苯的性能甚至低于TiO2载体。
3) V2O5和WO3之间存在协同作用,这可能是由于WO3增强了V2O5的氧化能力,或者V2O5可以使WO3脱氯,提高了WO3的稳定性。
4)氯苯在上述催化剂上的转化过程类似,均为苯环逐渐氧化开环及后续中间产物的氧化过程,酚类物种、醌类物种、马来酸盐和醋酸盐物种是重要的中间产物。
5)传统V2O5-WO3/TiO2脱硝催化剂直接用于CVOCs的催化氧化并不高效,应通过提高V2O5含量或改变助剂、载体等方式进一步优化。
钒基催化剂降解氯苯的原位红外分析
In-situ FT-IR analysis of chlorobenzene degradation by vanadia based catalysts
-
摘要: V2O5-WO3/TiO2催化剂被广泛应用于脱硝,且由于V2O5抗氯中毒能力强,对于氯代挥发性有机物(CVOCs)的催化降解也具有较好效果。通过浸渍法制备了不同V2O5和WO3含量的负载型催化剂,采用氯苯作为CVOCs的模型化合物,对催化剂进行了活性评价和原位红外实验研究,在分子层面明确V2O5和WO3在氯苯催化氧化过程中的作用。结果表明:增加V2O5含量是提高催化剂活性和稳定性的关键;氯苯在不同活性组分上的降解途径类似,均为苯环逐渐氧化开环及后续中间产物的氧化过程;V2O5对氯苯具有较强的氧化能力,在100 ℃即可观察到大量中间产物,且随着温度的升高,中间产物可迅速被氧化分解。相对而言,WO3的氧化性能很差,仅在温度达到300 ℃才可明显观察到中间产物,但V2O5和WO3之间存在协同作用。以上分子层面的反应机制研究,有助于明确催化剂各组分的具体作用,进一步指导开发性能更好的钒基催化剂,用于CVOCs的催化氧化。
-
关键词:
- V2O5-WO3/TiO2 /
- V2O5 /
- 氯苯 /
- 催化氧化 /
- 原位红外
Abstract: V2O5-WO3/TiO2 catalysts were widely used for denitrification in coal-fired power plants, and had good performance on catalytic degradation of chlorinated volatile organic compounds (CVOCs) due to the strong resistance to chlorine poisoning of V2O5. In this study, supported catalysts with different contents of V2O5 and WO3 were synthesized by impregnation method. Chlorobenzene was used as the model compound of CVOCs to conduct the activity evaluations and in-situ FT-IR experiments of above catalysts, and then to clarify the roles of V2O5 and WO3 in the catalytic oxidation process of chlorobenzene at the molecular level. The results showed that the increase of V2O5 content was the key to improve the activity and stability of the catalyst. The degradation pathway of chlorobenzene on different active components was similar: benzene ring opening by gradual oxidation, subsequent oxidation of the intermediates. V2O5 had a strong oxidation of chlorobenzene, and lots of intermediates could be observed at 100 ℃. And with the increase of temperature, intermediates could be rapidly oxidized and decomposed. In contrast, the catalytic performance of WO3 was very poor, intermediates could be clearly observed only when the reaction temperature reached 300 ℃, but there was a synergistic effect between V2O5 and WO3. By studying the reaction mechanism at the molecular level above, it was helpful to further develop V2O5-based catalysts with better performance for the catalytic oxidation of CVOCs.-
Key words:
- V2O5-WO3/TiO2 /
- V2O5 /
- chlorobenzene /
- catalytic oxidation /
- in-situ FTIR
-
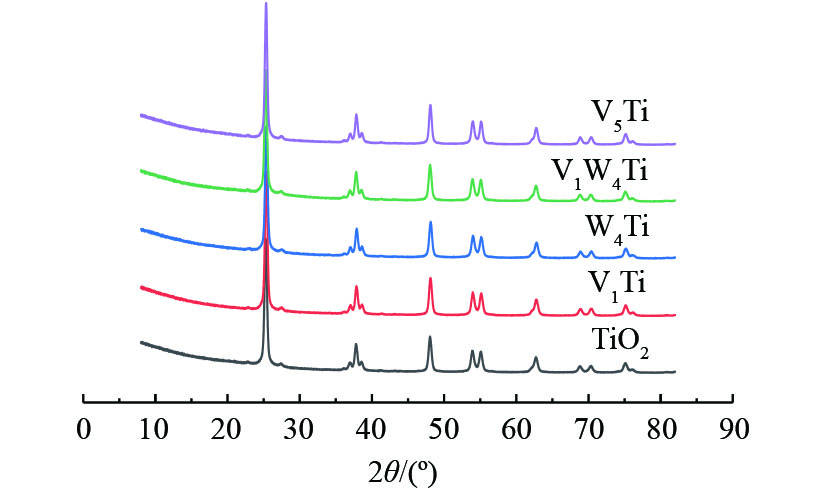
图 2 TiO2和V1Ti,W4Ti,V1W4Ti和V5Ti催化剂的XRD谱图
Figure 2. XRD patterns of TiO2 and the V1Ti, W4Ti,V1W4Ti and V5Ti catalysts
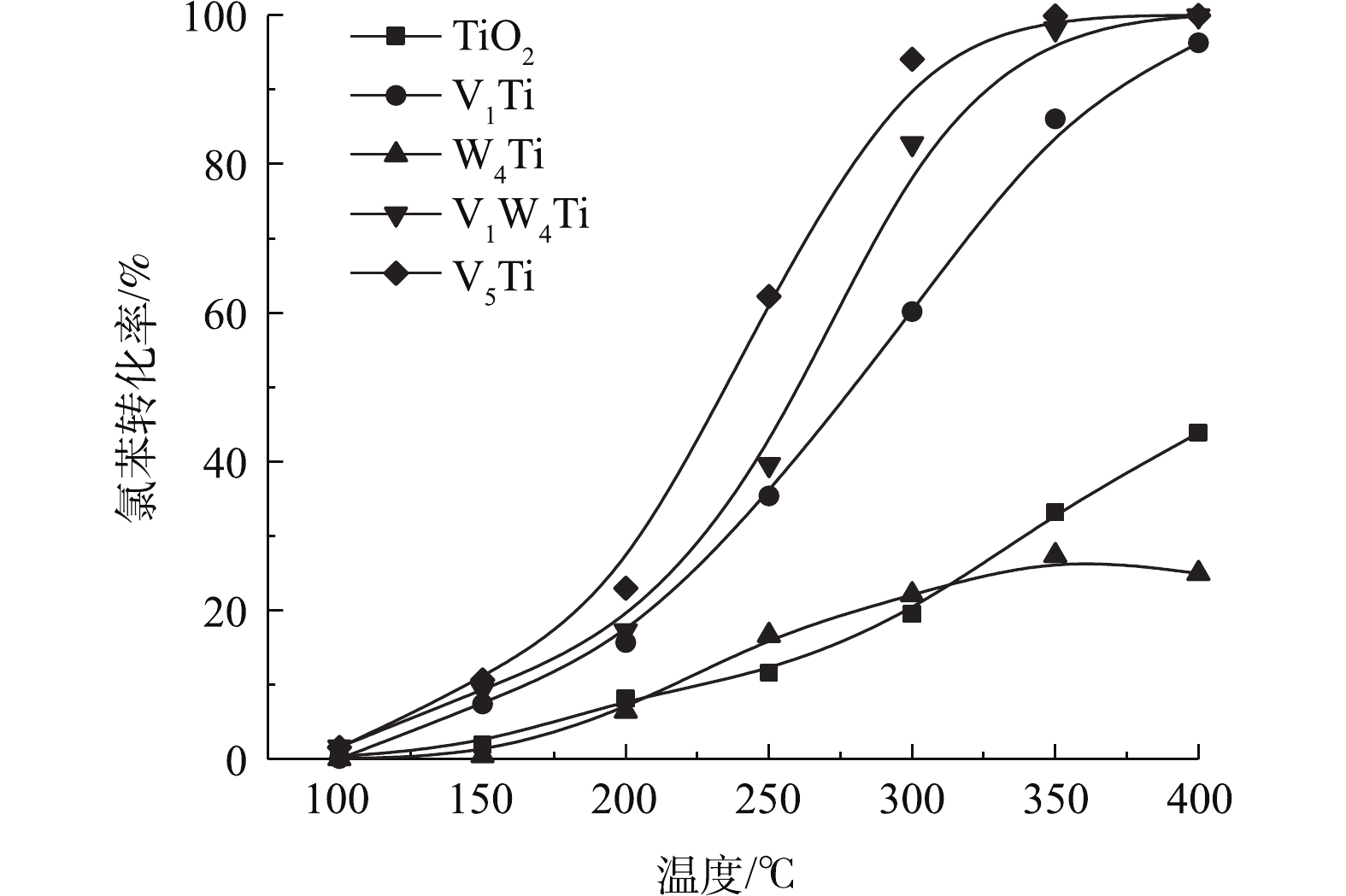
图 3 氯苯在TiO2和V1Ti,W4Ti,V1W4Ti和V5Ti催化剂上的转化率与温度的函数关系
Figure 3. Chlorobenzene conversion as a function of temperature over TiO2 and the V1Ti, W4Ti, V1W4Ti and V5Ti catalysts

图 4 氯苯在TiO2和V1Ti,W4Ti,V1W4Ti和V5Ti催化剂上300 ℃的连续运行6 h的稳定性评价
Figure 4. Stability evaluation of chlorobenzene oxidation over TiO2 and the V1Ti, W4Ti, V1W4Ti and V5Ti catalysts for 6 hours running continuously at 300 ℃
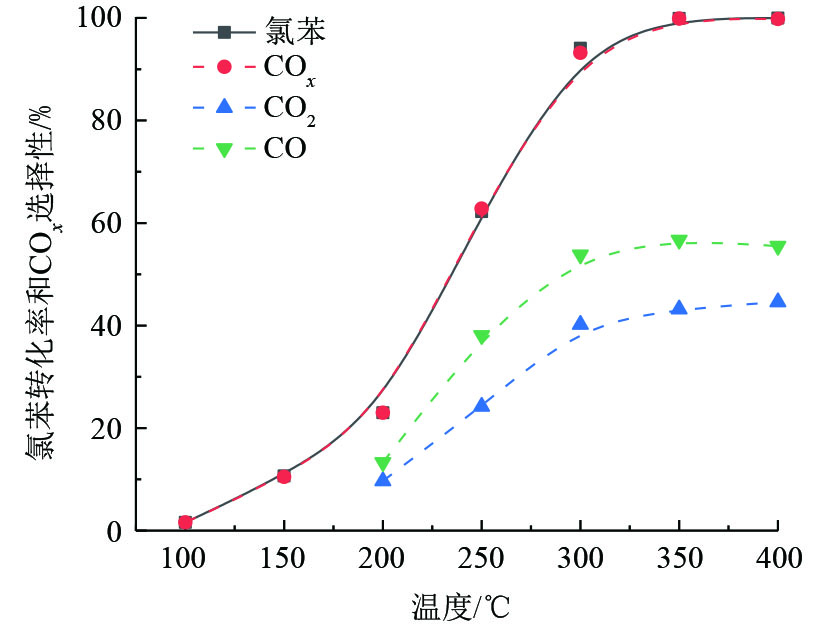
图 5 V5Ti催化剂催化氧化氯苯的转化率和COx选择性
Figure 5. Chlorobenzene conversion and COx selectivity as a function of temperature over the V5Ti catalyst
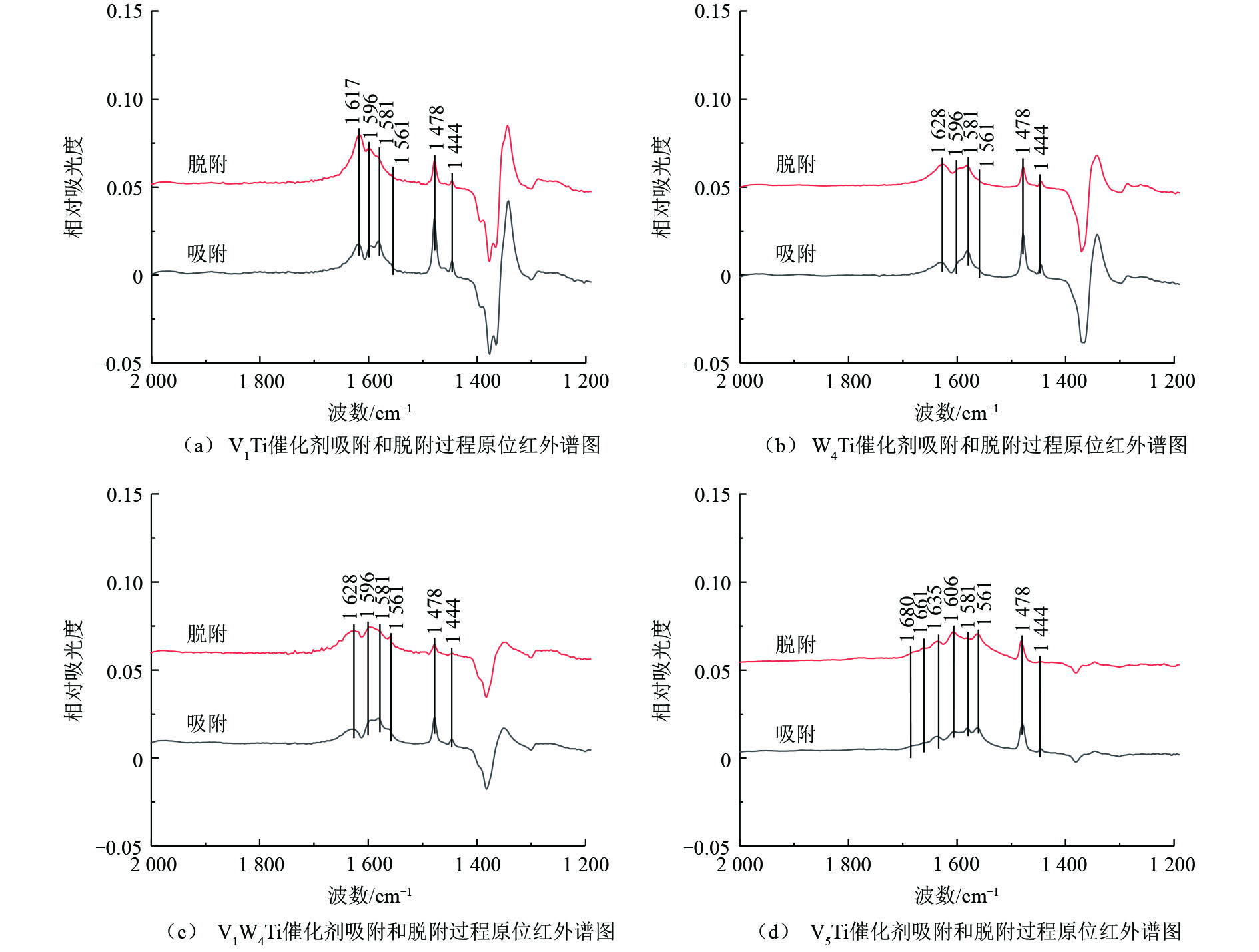
图 6 催化剂吸附和脱附过程的原位红外谱图
Figure 6. In-situ FT-IR spectra of catalysts collected during adsorption and desorption processes
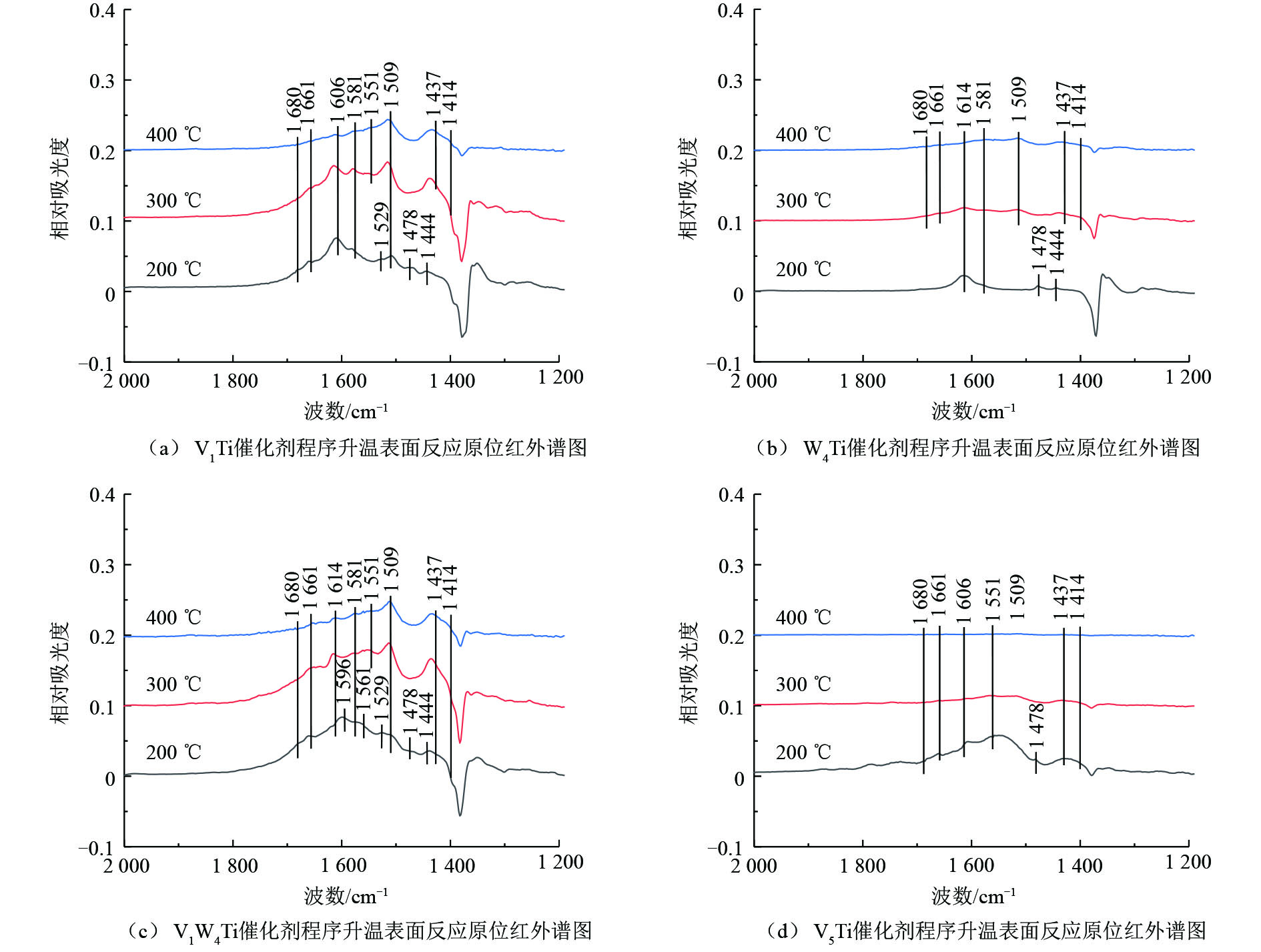
图 7 催化剂程序升温表面反应的原位红外谱图
Figure 7. In-situ FT-IR spectra of catalysts collected during temperature programmed surface reaction processes
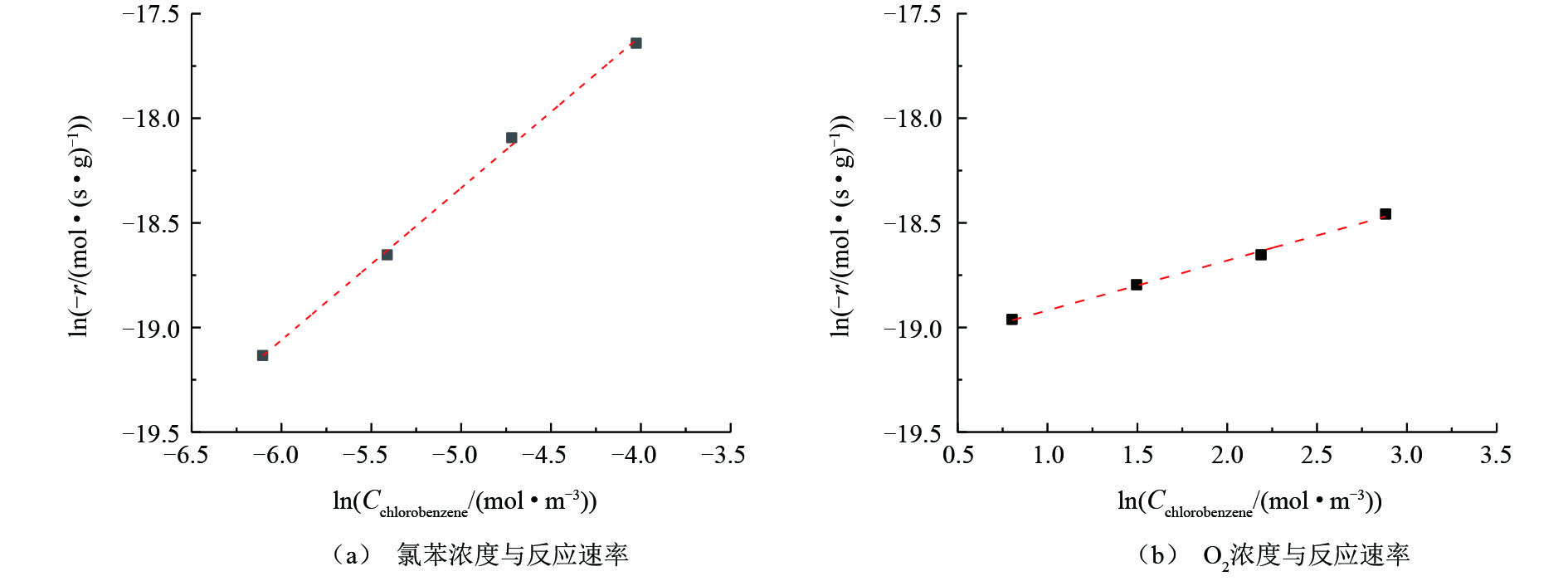
图 8 氯苯浓度和O2浓度与反应速率的影响
Figure 8. Effect of chlorobenzene concentration and O2 concentration on reaction rate

图 9 空速对V5Ti催化剂上氯苯转化率的影响
Figure 9. Effect of WHSV on chlorobenzene conversionover the V5Ti catalyst
表 1 TiO2和V1Ti,W4Ti,V1W4Ti和V5Ti催化剂的比表面积、孔容和平均孔径
Table 1. Specific surface area, pore volume and pore size of TiO2 and V1Ti, W4Ti, V1W4Ti and V5Ti catalysts
样品 比表面积/(m2·g−1) 孔容/(cm3·g−1) 平均孔径/nm TiO2 68.1 0.32 12.4 V1Ti 61.4 0.34 12.3 W4Ti 64.7 0.32 12.4 V1W4Ti 60.6 0.34 12.4 V5Ti 52.1 0.32 12.3 注:比表面积使用Brunauer-Emmett-Teller(BET)方法计算;孔容和平均孔径使用Barrett-Joyner-Halenda(BJH)方法计算。  下载: 导出CSV
下载: 导出CSV
-
[1] KAMPA M, CASTANAS E. Human health effects of air pollution[J]. Environmental Pollution, 2008, 151: 362-367. doi: 10.1016/j.envpol.2007.06.012 [2] SHAO M, ZHANG Y, ZENG L, et al. Ground-level ozone in the Pearl River Delta and the roles of VOC and NOx in its production[J]. Journal of Environmental Management, 2009, 90: 512-518. [3] VOLKAMER R, JIMENEZ J L, SANMARTINI F, et al. Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected[J]. Geophysical Research Letters, 2006, 33(17): 254-269. [4] ZHANG Y H, SU H, ZHONG L J, et al. Regional ozone pollution and observation-based approach for analyzing ozone-precursor relationship during the PRIDE-PRD2004 campaign[J]. Atmospheric Environment, 2008, 42: 6203-6218. doi: 10.1016/j.atmosenv.2008.05.002 [5] LI W B, WANG J X, GONG H. Catalytic combustion of VOCs on non-noble metal catalysts[J]. Catalysis Today, 2009, 148: 81-87. doi: 10.1016/j.cattod.2009.03.007 [6] LIOTTA L F. Catalytic oxidation of volatile organic compounds on supported noble metals[J]. Applied Catalysis B: Environmental, 2010, 100: 403-412. doi: 10.1016/j.apcatb.2010.08.023 [7] LI W J, ZHAO P, LIU S T. SnOx-MnOx-TiO2 catalysts with high resistance to chlorine poisoning for low-temperature chlorobenzene oxidation[J]. Applied Catalysis A: General, 2014, 482: 363-369. doi: 10.1016/j.apcata.2014.06.013 [8] FORZATTI P. Present status and perspectives in de-NOx SCR catalysis[J]. Applied Catalysis A: General, 2001, 222: 221-236. doi: 10.1016/S0926-860X(01)00832-8 [9] BERTINCHAMPS F, GREGOIRE C, GAIGNEAUX E M. Systematic investigation of supported transition metal oxide based formulations for the catalytic oxidative elimination of (chloro)-aromatics: Part I: Identification of the optimal main active phases and supports[J]. Applied Catalysis B: Environmental, 2006, 66: 1-9. doi: 10.1016/j.apcatb.2006.02.011 [10] BERTINCHAMPS F, GREGOIRE C, GAIGNEAUX E M. Systematic investigation of supported transition metal oxide based formulations for the catalytic oxidative elimination of (chloro)-aromatics: Part II: Influence of the nature and addition protocol of secondary phases to VOx/TiO2[J]. Applied Catalysis B: Environmental, 2006, 66: 10-22. doi: 10.1016/j.apcatb.2006.02.012 [11] WEBER R, SAKURAI T, HAGENMAIER H. Low temperature decomposition of PCDD/PCDF, chlorobenzenes and PAHs by TiO2-based V2O5-WO3 catalysts[J]. Applied Catalysis B: Environmental, 1999, 20: 249-256. doi: 10.1016/S0926-3373(98)00115-5 [12] VANDEN BRINK R W, LOUW R, MULDER P. Formation of polychlorinated benzenes during the catalytic combustion of chlorobenzene using a Pt/gamma-Al2O3 catalyst[J]. Applied Catalysis B: Environmental, 1998, 16: 219-226. doi: 10.1016/S0926-3373(97)00076-3 [13] GIRAUDON J M, ELHACHIMI A, LECLERCQ G. Catalytic oxidation of chlorobenzene over Pd/perovskites[J]. Applied Catalysis B: Environmental, 2008, 84: 251-261. doi: 10.1016/j.apcatb.2008.04.023 [14] WANG X Y, KANG Q, LI D. Catalytic combustion of chlorobenzene over MnOx-CeO2 mixed oxide catalysts[J]. Applied Catalysis B: Environmental, 2009, 86: 166-175. doi: 10.1016/j.apcatb.2008.08.009 [15] CHEN J P, YANG R T. Role of WO3 in mixed V2O5-WO3/TiO2 catalysts for selective catalytic reduction of nitric-oxide with ammonia[J]. Applied Catalysis A: General, 1992, 80: 135-148. doi: 10.1016/0926-860X(92)85113-P [16] WANG J, WANG X, LIU X L, et al. Catalytic oxidation of chlorinated benzenes over V2O5/TiO2 catalysts: The effects of chlorine substituents[J]. Catalysis Today, 2015, 241: 92-99. doi: 10.1016/j.cattod.2014.04.002 [17] LICHTENBERGER J, AMIRIDIS M D. Catalytic oxidation of chlorinated benzenes over V2O5/TiO2 catalysts[J]. Journal of Catalysis, 2004, 223: 296-308. doi: 10.1016/j.jcat.2004.01.032 [18] BUSCA G, RAMIS G, LORENZELLI V. FT-IR study of the surface-properties of polycrystalline vanadia[J]. Journal of Molecular Catalysis, 1989, 50: 231-240. doi: 10.1016/0304-5102(89)85066-7 [19] LOMNICKI S, LICHTENBERGER J, XU Z T, et al. Catalytic oxidation of 2, 4, 6-trichlorophenol over vanadia/titania-based catalysts[J]. Applied Catalysis B: Environmental, 2003, 46: 105-119. doi: 10.1016/S0926-3373(03)00215-7 [20] RAMSTETTER A, BAERNS M. Infrared spectroscopic investigation of the adsorption states of 1-butene, 1,3-butadiene, furan, 2,5h-furanone, and maleic-anhydride on alumina-supported V2O5-P2O5 catalyst. 1. Adsorption under nonreactive conditions[J]. Journal of Catalysis, 1988, 109: 303-313. doi: 10.1016/0021-9517(88)90213-8 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4050
- HTML全文浏览数: 4050
- PDF下载数: 68
- 施引文献: 0
