



=\mathrm{ln}q_e-k_1t $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead>
<tr>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">吸附模型Adsorption model</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">准一级动力学模型Pseudo first order kinetic model</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">准二级动力学模型Pseudo second order kinetic model</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">表达式Expression</td>
<td align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{ln}\left(q_e-q_t\right)=\mathrm{ln}q_e-k_1t $</tex-math><alternatives><img class="graphic" src="2023092902_M1.jpg"><img class="graphic" src="2023092902_M1.png"></alternatives></inline-formula></td>
<td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \dfrac{t}{{q}_{t}}=\dfrac{1}{{k}_{2}{q}_{e}^{2}}+\dfrac{t}{{q}_{e}} $</tex-math><alternatives><img class="graphic" src="2023092902_M2.jpg"><img class="graphic" src="2023092902_M2.png"></alternatives></inline-formula></td>
</tr>
<tr>
<td align="center" valign="middle"><i>R</i><sup>2</sup></td>
<td align="center" valign="middle">0.768 8</td>
<td align="center" valign="middle">0.999 9</td>
</tr>
<tr>
<td align="center" valign="middle">拟合参数Parameters</td>
<td align="center" valign="middle"><i>k</i><sub>1</sub>=0.034 70<i>q</i><sub><i>e</i></sub>=53.515 6</td>
<td align="center" valign="middle"><i>k</i><sub>2</sub>=0.011 56<i>q</i><sub><i>e</i></sub>=121.654 5</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">拟合方程Equation</td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M3">$ \mathrm{ln}\left(53.5156-q_t\right)=1.7285-0.0347t $</tex-math><alternatives><img class="graphic" src="2023092902_M3.jpg"><img class="graphic" src="2023092902_M3.png"></alternatives></inline-formula></td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M4">$ \dfrac{t}{{q}_{t}}=0.005 9+\dfrac{t}{121.654 5} $</tex-math><alternatives><img class="graphic" src="2023092902_M4.jpg"><img class="graphic" src="2023092902_M4.png"></alternatives></inline-formula></td>
</tr>
</tbody>
<tfoot>
<tr>
<td colspan="3" align="left" valign="middle"> 注:<i>q</i><sub><i>t</i></sub>表示时间为<i>t</i>时的吸附量,<i>q</i><sub><i>e</i></sub>为平衡吸附量,<i>k</i><sub>1</sub>为准一级吸附速率常数, <i>k</i><sub>2</sub>为准二级吸附速率常数.</td>
</tr>
</tfoot>
</table></div></foreignObject></svg>"></inline-formula></td>
<td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \dfrac{t}{{q}_{t}}=\dfrac{1}{{k}_{2}{q}_{e}^{2}}+\dfrac{t}{{q}_{e}} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead>
<tr>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">吸附模型Adsorption model</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">准一级动力学模型Pseudo first order kinetic model</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">准二级动力学模型Pseudo second order kinetic model</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">表达式Expression</td>
<td align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{ln}\left(q_e-q_t\right)=\mathrm{ln}q_e-k_1t $</tex-math><alternatives><img class="graphic" src="2023092902_M1.jpg"><img class="graphic" src="2023092902_M1.png"></alternatives></inline-formula></td>
<td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \dfrac{t}{{q}_{t}}=\dfrac{1}{{k}_{2}{q}_{e}^{2}}+\dfrac{t}{{q}_{e}} $</tex-math><alternatives><img class="graphic" src="2023092902_M2.jpg"><img class="graphic" src="2023092902_M2.png"></alternatives></inline-formula></td>
</tr>
<tr>
<td align="center" valign="middle"><i>R</i><sup>2</sup></td>
<td align="center" valign="middle">0.768 8</td>
<td align="center" valign="middle">0.999 9</td>
</tr>
<tr>
<td align="center" valign="middle">拟合参数Parameters</td>
<td align="center" valign="middle"><i>k</i><sub>1</sub>=0.034 70<i>q</i><sub><i>e</i></sub>=53.515 6</td>
<td align="center" valign="middle"><i>k</i><sub>2</sub>=0.011 56<i>q</i><sub><i>e</i></sub>=121.654 5</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">拟合方程Equation</td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M3">$ \mathrm{ln}\left(53.5156-q_t\right)=1.7285-0.0347t $</tex-math><alternatives><img class="graphic" src="2023092902_M3.jpg"><img class="graphic" src="2023092902_M3.png"></alternatives></inline-formula></td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M4">$ \dfrac{t}{{q}_{t}}=0.005 9+\dfrac{t}{121.654 5} $</tex-math><alternatives><img class="graphic" src="2023092902_M4.jpg"><img class="graphic" src="2023092902_M4.png"></alternatives></inline-formula></td>
</tr>
</tbody>
<tfoot>
<tr>
<td colspan="3" align="left" valign="middle"> 注:<i>q</i><sub><i>t</i></sub>表示时间为<i>t</i>时的吸附量,<i>q</i><sub><i>e</i></sub>为平衡吸附量,<i>k</i><sub>1</sub>为准一级吸附速率常数, <i>k</i><sub>2</sub>为准二级吸附速率常数.</td>
</tr>
</tfoot>
</table></div></foreignObject></svg>"></inline-formula></td>
</tr>
<tr>
<td align="center" valign="middle"><i>R</i><sup>2</sup></td>
<td align="center" valign="middle">0.768 8</td>
<td align="center" valign="middle">0.999 9</td>
</tr>
<tr>
<td align="center" valign="middle">拟合参数Parameters</td>
<td align="center" valign="middle"><i>k</i><sub>1</sub>=0.034 70<i>q</i><sub><i>e</i></sub>=53.515 6</td>
<td align="center" valign="middle"><i>k</i><sub>2</sub>=0.011 56<i>q</i><sub><i>e</i></sub>=121.654 5</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">拟合方程Equation</td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M3">$ \mathrm{ln}\left(53.5156-q_t\right)=1.7285-0.0347t $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead>
<tr>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">吸附模型Adsorption model</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">准一级动力学模型Pseudo first order kinetic model</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">准二级动力学模型Pseudo second order kinetic model</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">表达式Expression</td>
<td align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{ln}\left(q_e-q_t\right)=\mathrm{ln}q_e-k_1t $</tex-math><alternatives><img class="graphic" src="2023092902_M1.jpg"><img class="graphic" src="2023092902_M1.png"></alternatives></inline-formula></td>
<td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \dfrac{t}{{q}_{t}}=\dfrac{1}{{k}_{2}{q}_{e}^{2}}+\dfrac{t}{{q}_{e}} $</tex-math><alternatives><img class="graphic" src="2023092902_M2.jpg"><img class="graphic" src="2023092902_M2.png"></alternatives></inline-formula></td>
</tr>
<tr>
<td align="center" valign="middle"><i>R</i><sup>2</sup></td>
<td align="center" valign="middle">0.768 8</td>
<td align="center" valign="middle">0.999 9</td>
</tr>
<tr>
<td align="center" valign="middle">拟合参数Parameters</td>
<td align="center" valign="middle"><i>k</i><sub>1</sub>=0.034 70<i>q</i><sub><i>e</i></sub>=53.515 6</td>
<td align="center" valign="middle"><i>k</i><sub>2</sub>=0.011 56<i>q</i><sub><i>e</i></sub>=121.654 5</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">拟合方程Equation</td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M3">$ \mathrm{ln}\left(53.5156-q_t\right)=1.7285-0.0347t $</tex-math><alternatives><img class="graphic" src="2023092902_M3.jpg"><img class="graphic" src="2023092902_M3.png"></alternatives></inline-formula></td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M4">$ \dfrac{t}{{q}_{t}}=0.005 9+\dfrac{t}{121.654 5} $</tex-math><alternatives><img class="graphic" src="2023092902_M4.jpg"><img class="graphic" src="2023092902_M4.png"></alternatives></inline-formula></td>
</tr>
</tbody>
<tfoot>
<tr>
<td colspan="3" align="left" valign="middle"> 注:<i>q</i><sub><i>t</i></sub>表示时间为<i>t</i>时的吸附量,<i>q</i><sub><i>e</i></sub>为平衡吸附量,<i>k</i><sub>1</sub>为准一级吸附速率常数, <i>k</i><sub>2</sub>为准二级吸附速率常数.</td>
</tr>
</tfoot>
</table></div></foreignObject></svg>"></inline-formula></td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M4">$ \dfrac{t}{{q}_{t}}=0.005 9+\dfrac{t}{121.654 5} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead>
<tr>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">吸附模型Adsorption model</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">准一级动力学模型Pseudo first order kinetic model</td>
<td align="center" valign="middle" class="table_top_border table_bottom_border2">准二级动力学模型Pseudo second order kinetic model</td>
</tr>
</thead>
<tbody>
<tr>
<td align="center" valign="middle">表达式Expression</td>
<td align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{ln}\left(q_e-q_t\right)=\mathrm{ln}q_e-k_1t $</tex-math><alternatives><img class="graphic" src="2023092902_M1.jpg"><img class="graphic" src="2023092902_M1.png"></alternatives></inline-formula></td>
<td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \dfrac{t}{{q}_{t}}=\dfrac{1}{{k}_{2}{q}_{e}^{2}}+\dfrac{t}{{q}_{e}} $</tex-math><alternatives><img class="graphic" src="2023092902_M2.jpg"><img class="graphic" src="2023092902_M2.png"></alternatives></inline-formula></td>
</tr>
<tr>
<td align="center" valign="middle"><i>R</i><sup>2</sup></td>
<td align="center" valign="middle">0.768 8</td>
<td align="center" valign="middle">0.999 9</td>
</tr>
<tr>
<td align="center" valign="middle">拟合参数Parameters</td>
<td align="center" valign="middle"><i>k</i><sub>1</sub>=0.034 70<i>q</i><sub><i>e</i></sub>=53.515 6</td>
<td align="center" valign="middle"><i>k</i><sub>2</sub>=0.011 56<i>q</i><sub><i>e</i></sub>=121.654 5</td>
</tr>
<tr>
<td align="center" valign="middle" class="table_bottom_border">拟合方程Equation</td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M3">$ \mathrm{ln}\left(53.5156-q_t\right)=1.7285-0.0347t $</tex-math><alternatives><img class="graphic" src="2023092902_M3.jpg"><img class="graphic" src="2023092902_M3.png"></alternatives></inline-formula></td>
<td align="center" valign="middle" class="table_bottom_border"><inline-formula><tex-math id="M4">$ \dfrac{t}{{q}_{t}}=0.005 9+\dfrac{t}{121.654 5} $</tex-math><alternatives><img class="graphic" src="2023092902_M4.jpg"><img class="graphic" src="2023092902_M4.png"></alternatives></inline-formula></td>
</tr>
</tbody>
<tfoot>
<tr>
<td colspan="3" align="left" valign="middle"> 注:<i>q</i><sub><i>t</i></sub>表示时间为<i>t</i>时的吸附量,<i>q</i><sub><i>e</i></sub>为平衡吸附量,<i>k</i><sub>1</sub>为准一级吸附速率常数, <i>k</i><sub>2</sub>为准二级吸附速率常数.</td>
</tr>
</tfoot>
</table></div></foreignObject></svg>"></inline-formula></td>
</tr>
</tbody>
<tfoot>
<tr>
<td colspan="3" align="left" valign="middle"> 注:<i>q</i><sub><i>t</i></sub>表示时间为<i>t</i>时的吸附量,<i>q</i><sub><i>e</i></sub>为平衡吸附量,<i>k</i><sub>1</sub>为准一级吸附速率常数, <i>k</i><sub>2</sub>为准二级吸附速率常数.</td>
</tr>
</tfoot>
</table></div></foreignObject></svg>)
-
随着铬化合物在工业、医药、染料及有机合成等领域的广泛应用,导致大量重金属离子Cr(Ⅵ)进入水体中而造成了严重的铬污染,对人类和其他生物会造成持久性毒害[1 − 2]. 在众多Cr(Ⅵ)的去除方法中,吸附法具有简单、经济、高效、易于操作、易回收等优点,而成为大规模处理含铬废水的首选方法[3]. 决定吸附法去除效果的关键因素是吸附剂. 目前,常用的Cr(Ⅵ)的吸附剂,如磺化褐煤、黏土矿物、沸石、有机树脂、工业副产物(如煤、粉煤灰)和壳聚糖等,存在吸附效率低、吸附平衡时间长、再生困难等一系列问题,从而限制了吸附法的大规模应用. 因此开发效率高、易再生的新型吸附剂迫在眉睫[4].
近年来,过渡金属氧化物由于其具有可控的形态、可变的表面化学、独特的结晶性质和可调谐的带边,特别是对金属离子吸附具有高选择性,因此在重金属离子处理领域被广泛研究[5]. 但由于纯金属氧化物的稳定性较差、吸附容量低,因此需要通过复合改性来提高其性能[6]. 例如,Yaacob[6]制备了一种TiO2-ZrO2复合材料,表明少量的ZrO2能有效提高对含油废水的降解能力. 彭宽宽[7]制备了一种介孔Fe2O3-MgO复合氧化物具有良好的Cr(Ⅵ)吸附能力. 尽管纳米复合物在Cr(Ⅵ)吸附领域有一定研究,但是主要还是氧化物之间的复合,且仍然存在吸附容量低、吸附速率慢的问题,因此研发新复合体系对于提高过渡金属氧化物吸附Cr(Ⅵ)的能力至关重要.
相对于氧化物而言,硒化物具有较低电负性,其较弱的金属-硒键更有利于重金属离子的吸附,而氧化物与硒化物复合材料对重金属离子的吸附作用还尚未见报道,且硒化对重金属离子的吸附机理还需要深入研究. 基于此,本文以Cu2O为对象,采用一锅水热法,原位硒化构筑了一种新型Cu2O-Cu2Se纳米复合物,系统研究了各种吸附条件对新型纳米复合物吸附Cr(Ⅵ)性能的影响,同时通过吸附动力学研究探讨了该纳米复合物对Cr(Ⅵ)的吸附机理,表明Cu2O-Cu2Se纳米复合物不仅对Cr(Ⅵ)具有高效的化学吸附,而且具有优异的吸附容量和稳定性,因此其有望在废水处理领域得到广泛的应用.
-
重铬酸钾(K2Cr2O7)、硒粉、五水合硫酸铜(CuSO4·5H2O)、乙二胺四乙酸(EDTA)、氢氧化钠(NaOH)、氨水(NH3·H2O)、盐酸(HCl)、硫酸、磷酸、二苯碳酰二肼、葡萄糖均购买自重庆兴玻化工有限公司.
-
采用一锅法原位硒化制备Cu2O-Cu2Se纳米复合物. 首先将0.01 mol硒粉和0.13 mol NaOH溶解于去离子水中得到A液. 将0.02 mol CuSO4·5H2O和0.1 mmol EDTA溶解于去离子水中得到B液. 然后将A、B液混合后倒入50 mL水热反应釜中,在160 ℃下反应24 h后离心洗涤至中性,最后在60 ℃下真空干燥6 h,即得到Cu2O-Cu2Se纳米复合物. 纯Cu2O采用同样的方法,在未加入硒粉的情况下制得,做为对比.
-
采用X射线衍射仪(XRD:岛津XRD-
7000 )对试样进行了晶体结构分析,使用场发射电子显微镜加能谱(SEM-EDS:日本电子JEM-2100F)对试样的微观形貌和元素含量进行了分析. 此外,使用Zeta电位及纳米粒度分析仪(DLS:Malvern ZEN3690 )对试样的等电点进行分析. -
取30 mL重铬酸钾配制的Cr(Ⅵ)溶液于反应管中,加入一定量吸附剂,用硝酸和氨水调节pH值,持续搅拌30 min到吸附平衡,取1 mL吸附后的溶液进行离心,定容至50 mL,加入显色剂二苯碳酰二肼,用紫外分光光度计(T6新世纪)在540 nm测量其吸光度. 通过标准曲线计算Cr(Ⅵ)的质量浓度,采用公式(1)和公式(2)分别计算Cr(Ⅵ)的吸附率和吸附容量:
式中:Re代表吸附达到平衡时的吸附率;qe代表吸附达到平衡时的吸附量(mg·g−1);C0和Ce分别代表吸附前、吸附后溶液中Cr(Ⅵ)的浓度(mg·L−1);V代表溶液体积(L);m代表吸附剂的质量(g).
-
将离心收集的15 mg吸附后的吸附剂,浸入0.1 mol·L−1的NaOH溶液中搅拌4 h,离心后再用0.1 mol·L−1的H2SO4溶液进行离心洗涤3次,以除去吸附的Cr(Ⅵ),最后用乙醇离心洗涤3次后真空干燥6 h,即得再生的吸附剂. 将其用于吸附过程,再次离心洗涤干燥,进行下一次吸附,重复操作进行5个循环.
-
Cu2O-Cu2Se纳米复合物和Cu2O的晶体结构通过XRD测定. 由图1a所示,两者在29.6°、36.4°、42.2°、61.6°、73.9°和77.3°的特征峰对应于Cu2O晶体(JCPDS No.05-0667)的(110)、(111)、(200)、(220)、(311)和(222)晶面[8]. Cu2O晶体在43.3°、50.4°和74.1°出现了单质铜的特征峰,这可能是由于葡萄糖过度还原导致的[9].
相对于Cu2O,原位硒化的Cu2O-Cu2Se纳米复合物在2θ=27.1°、31.4°、44.9°、53.3°、55.9°、65.5°、72.2°、74.5°具有明显的特征峰,分别对应Cu2Se晶体(JCPDS No.88-2043)的(111)、(200)、(220)、(311)、(222)、(400)、(331)、(420)晶面[10],说明采用一锅法能够成功原位硒化Cu2O得到Cu2O-Cu2Se纳米复合物. 同时用SEM分析了各试样的形貌,Cu2O(图1b)是大小为1 μm左右的不规则颗粒,而Cu2O-Cu2Se纳米复合物(图1c)是由50—100 nm左右的小颗粒组成,且具有多孔珊瑚状形貌,由于氧化亚铜材料的吸附活性位点是Cu+,因此相对于Cu2O,原位硒化后的纳米复合物形成的多孔珊瑚状形貌可以提供更多的吸附活性位点,这将有利于重金属Cr(Ⅵ)的吸附[7]. 进一步采用EDS面扫来分析纳米复合物的元素组成,结果如图1d-g所示,从元素分布可以清晰的看出,纳米复合物中Cu、Se、O元素分布均匀. 此外,从Cu2O-Cu2Se纳米复合物的元素组成情况(表1)可以看到,原位硒化后的纳米复合物是由Cu、O、Se组成,摩尔分数分别55.91%、36.25%、7.84%,进一步表明原位硒化所得试样是以Cu2O为主的Cu2O-Cu2Se纳米复合物,同时说明Cu2O-Cu2Se纳米复合物是Cu2O表面少量硒化的产物,由此构成了多孔珊瑚状的Cu2O-Cu2Se的复合结构,从而能够增强吸附.
-
为了更清楚地研究Cu2O-Cu2Se纳米复合物的吸附性能,首先研究了不同吸附剂加入量下pH、时间及Cr(Ⅵ)的初始浓度对纳米复合物吸附性能的影响. 由图2a可知,随着溶液pH的增加,Cr(Ⅵ)的吸附率从100%急剧降低至0%,这表明pH对纳米复合物去除Cr(Ⅵ)的能力具有显著影响. 当pH值为2时,Cr(Ⅵ)吸附率达到100%,这是由于Cr(Ⅵ)在偏酸性条件下主要以HCrO4−和Cr2O72-形态存在[11]. Cu2O-Cu2Se纳米复合物的等电点pHpzc约为7.2(图2b),当溶液pH值小于7.2时,纳米复合物的表面H+浓度高,使其对HCrO4−的静电引力大,因此在酸性条件下,Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)具有高效吸附能力. 在碱性条件下,Cr(Ⅵ)主要以CrO42-的形态存在,当溶液pH值大于7.2时,Cu2O-Cu2Se纳米复合物更倾向于吸附阳离子,同时水中的OH−也随之增加,也会对Cr(Ⅵ)的吸附产生竞争作用而导致纳米复合物对Cr(Ⅵ)的吸附率降低[12]. 进一步在酸性条件(pH=2),对不同时间下纳米复合物的Cr(Ⅵ)吸附性能进行了测试,结果如图2c所示,当加入纳米复合物10 min后,Cr(Ⅵ)的吸附率达到90%以上,当加入量从5 mg增加至15 mg时,吸附10 min后Cr(Ⅵ)的吸附率可以从91%增加至99%,在30 min时基本达到吸附-脱附的平衡,吸附率可达到100%. 这说明Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)具有超高速吸附. 另外,也测试了Cr(Ⅵ)初始浓度对Cu2O-Cu2Se纳米复合物的吸附性能的影响,如图2d所示,从图中可以看出提高吸附剂的加入量有利于高浓度Cr(Ⅵ)的去除,当加入量为15 mg,Cr(Ⅵ)初始质量浓度从10 mg·L−1到50 mg·L−1时,Cu2O-Cu2Se纳米复合物的吸附率一直保持100%,之后随着初始质量浓度的增加,吸附率逐渐降低. 这是因为当含铬离子溶液浓度较高时,吸附剂的表面更易饱和,导致离子交换减少[13]. 此外,进一步研究了Cr(Ⅵ)初始浓度与吸附量及吸附剂加入量的关系,结果如图2e所示,从结果可知,当Cr(Ⅵ)初始浓度为50 mg·L−1时,吸附量达到最高,且加入5 mg吸附剂的吸附量最佳,在吸附30 min后达到148.8 mg·g−1.
与文献中其他吸附剂进行对比,结果如表2所示,原位硒化构筑的Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)的吸附量和吸附速率优于不少复合材料[11,14 − 16]. 与水凝胶、壳聚糖和金属氧化物等相比,Cu2O-Cu2Se纳米复合物显示了优异的高速吸附效率. 此外,由图2f的Cu2O-Cu2Se纳米复合物的循环性能测试结果可见,经5次再生循环后纳米复合物的吸附量降至89.6 mg·g−1,但其仍可达初始吸附量(110.6 mg·g−1)的80%以上,说明Cu2O-Cu2Se纳米复合物不仅具有高效吸附能力,还具有良好的循环再生稳定性.
为了进一步阐明原位硒化对Cu2O-Cu2Se的吸附性能的影响,取5 mg Cu2O和Cu2O-Cu2Se纳米复合物分别在30 mL 50 mg·L−1 Cr(Ⅵ)溶液中测试其吸附性能,采用溶液中剩余Cr(Ⅵ)占初始浓度的占比(C/C0)作为去除效果,结果如图3a所示,Cu2O-Cu2Se纳米复合物吸附30 min后基本达到吸附平衡,C/C0为0.20,比未硒化Cu2O吸附后的C/C0降低了60%,表明原位硒化能显著提升Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)的去除能力. 进一步用FT-IR测试了吸附前后的Cu2O-Cu2Se纳米复合物,以揭示其吸附机理. 从图3b中可以看到,未吸附与已吸附Cr(Ⅵ)的Cu2O-Cu2Se纳米复合物的FT-IR谱图均在
3415 cm−1处出现一个宽峰,对应于O—H拉伸,另外在1618 cm−1处的附加谱带与羟基的弯曲振动有关. 未吸附Cr(Ⅵ)的Cu2O-Cu2Se纳米复合物在617 cm−1处有1个强的吸收峰,对应于Cu—O振动模式,与已吸附Cr(Ⅵ)试样的谱图对比,已吸附Cr(Ⅵ)的纳米复合物在617 cm−1处的振动峰消失了,说明Cr(Ⅵ)与Cu2O-Cu2Se纳米复合物之间发生了化学反应,因此Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)的吸附属于化学吸附[17]. 而由图3c可知Cu2O吸附前后其在617 cm−1处的吸收峰变化不大,表明Cu2O对Cr(Ⅵ)的吸附化学较弱,这说明通过原位硒化后的纳米复合材料不仅具有多孔珊瑚形貌而能提供更多的吸附活性位点Cu+,同时表面形成的Cu2Se由于具有较弱的Cu—Se键而促进了Cu+对Cr(Ⅵ)的化学吸附,从而显著提高了Cr(Ⅵ)的吸附速率[8]. -
为了明确Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)的吸附机制,对实验数据进行了吸附动力学分析,结果如图4所示. 首先采用准一级动力学方程和准二级动力学方程分别对5 mg Cu2O-Cu2Se纳米复合物吸附Cr(Ⅵ)与时间关系的实验数据(图2c)进行拟合,拟合曲线如图4a-b所示. 从拟合数值可知(表3),Cu2O-Cu2Se纳米复合物吸附Cr(Ⅵ)的准二级动力学方程的R2更接近于1,理论平衡吸附量与相应的实验值121.0 mg·g−1也符合得较好,这表明Cu2O-Cu2Se纳米复合物吸附Cr(Ⅵ)的过程中,化学吸附为主要作用方式,这与FT-IR结果一致,表明纳米复合物对Cr(Ⅵ)的吸附是化学键的作用力发生而引起的一种吸附,因此具有高效的吸附性能. 采用Langmuir和Freundlich等温吸附模型对5 mg Cu2O-Cu2Se纳米复合物吸附Cr(Ⅵ)与时间关系的实验数据(图2c)进行拟合,拟合曲线如图4c-d所示. 从拟合数值可知(表4),Langmuir模型能更好地拟合纳米复合物对Cr(Ⅵ)的吸附过程,说明Cu2O-Cu2Se纳米复合物是单分子吸附,而其理论最大吸附量与实验值148.8 mg·g−1接近,表明纳米复合物的表面具有剩余价键力的活性点,由于只有在表面存在剩余价键力的活性点处才能产生化学吸附,因此Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)的吸附是单分子层化学吸附[18]. 与物理吸附相比,化学吸附具有一定的选择性,其作用力比范德瓦尔引力大得多,所以吸附位阱更深,作用距离更短,因此所制Cu2O-Cu2Se纳米复合物具有高速吸附效率.
-
(1)本文采用一锅法原位硒化制备了Cu2O-Cu2Se纳米复合物.
(2)Cu2O-Cu2Se纳米复合物具有的多孔珊瑚状形貌和表面形成的Cu2Se,不仅增加了材料的吸附活性位点Cu+,还促进了Cu+对Cr(Ⅵ)的化学吸附,从而显著增强了Cr(Ⅵ)的吸附效率,与纯Cu2O的Cr(Ⅵ)吸附率(55%)相比,Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)的吸附率增加至85%.
(3)在酸性条件下10 min内Cu2O-Cu2Se纳米复合物吸附率可达90%以上,30 min达到吸附平衡,吸附量可达100%,表现出对Cr(Ⅵ)的超高速吸附性能. 当吸附剂加入量为5 mg,pH为2,在30 mL 50 mg·L−1的Cr(Ⅵ)溶液中纳米复合物的吸附容量能够达到148.8 mg·g−1,优于大多数吸附材料. 且经5次再生循环后,Cu2O-Cu2Se纳米复合物仍具有较高的Cr(Ⅵ)吸附率.
(4)吸附动力学分析表明,Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)的吸附符合准二级动力学方程,吸附过程和Langmuir方程能更好地拟合,说明纳米复合物以单分子层化学吸附为主,有利于对Cr(Ⅵ)的高速吸附.
原位硒化衍生新型Cu2O-Cu2Se纳米复合物用于高速吸附重金属离子Cr(Ⅵ)
Novel Cu2O-Cu2Se nanocomposite derived from in-situ selenization for high speed adsorption of Cr(Ⅵ) heavy metal ions
-
摘要: 金属氧化物具有高的选择性而在废水中重金属离子Cr(Ⅵ)处理领域受到广泛关注,然而其稳定性较差、吸附容量低限制了其应用。为克服这些问题,本文通过Cu2O原位硒化构筑了新型Cu2O-Cu2Se纳米复合物. 该纳米复合物具有多孔珊瑚状结构,能够提供更多的吸附活性位点Cu+,其对Cr(Ⅵ)的吸附符合准二级动力学方程和Langmuir等温吸附模型,主要为单分子层化学吸附,同时表面形成的较弱的Cu—Se键促进了Cu+对Cr(Ⅵ)的化学吸附,从而使其具有高速吸附Cr(Ⅵ)的能力. 当纳米复合物加入量为5 mg,pH=2,在30 mL 50 mg·L−1 Cr(Ⅵ)溶液中,吸附30 min后吸附量可达148.8 mg·g−1,优于大多数吸附材料.Abstract: The high selectivity of metal oxides in the treatment of Cr(Ⅵ) ions in wastewater has garnered significant attention. However, their limited stability and adsorption capacity impede their practical application. To overcome these issues, we have synthesized a novel Cu2O-Cu2Se nanocomposite through in-situ selenization of bare Cu2O. The porous coral-like structure of the nanocomposite provides ample active sites for Cu+ adsorption. The adsorption behavior of Cu2O-Cu2Se nanocomposite for Cr(Ⅵ) follows the pseudo second order kinetic model and Langmuir isothermal adsorption model, indicating predominantly single-molecular layer chemisorption. The formation of weak Cu—Se bonds on the surface enhances the chemisorption of Cr(Ⅵ) by Cu+, and thus enabling rapid removal of Cr(Ⅵ). With just 5 mg of nanocomposite added at an initial concentration of 50 mg·L−1 and pH = 2, a maximum adsorption capacity of 148.8 mg·g−1 was achieved within only 30 minutes, which is superior to most adsorbents.
-
Key words:
- Cr(Ⅵ) /
- nanocomposite /
- adsorption /
- selenation
-
中国是抗生素生产和使用大国[1]。作为中国最常用的5类抗生素之一,β-内酰胺类抗生素生产量和销售份额在众多抗生素中名列前茅[2],在全球制药行业也具有举足轻重的地位[3-4]。青霉素和头孢菌素是使用最广泛的抗生素。2010年,青霉素和头孢菌素2种抗生素使用量占所有抗生素使用总量的近60%,与2000年相比增长了41%[5]。环境中β-内酰胺类抗生素的残留主要来自制药工业[6-9]、临床[10-12]和畜禽养殖[13-14]。抗生素生产过程产生的制药废水[8-9]及菌渣[15-16]中含有高浓度的抗生素残留[17]。LI等[18]对青霉素G生产废水(废母液)的研究表明,由于溶剂萃取过程的高温酸性环境使大部分青霉素G母体降解(残留浓度为153.0 μg·L−1),降解产物脱羧青霉噻唑酸和青霉醛的浓度可分别达到389.0 mg·L−1和75.3 mg·L−1。与青霉素G生产废水相比,青霉素生产排放菌渣中的青霉素G残留更高,可达5 000 mg·L−1[19],是β-内酰胺类抗生素排放强度最高的排放源。
尽管β-内酰胺类抗生素是生产和应用最广泛的抗生素,但由于其结构中的β-内酰胺环具有易水解和生物降解的特性,通常环境中检出浓度较低,甚至一些类型低于检测限。青霉素生产废水处理后,出水和受纳河流下游青霉素G浓度仅为1.68 μg·L−1和0.35 μg·L−1,但却发现大量对β-内酰胺类和其他各类抗生素具有多抗性的抗药细菌,这表明即使废水中抗生素母体浓度很低,其排放仍然可能导致环境细菌抗药性潜在风险[18]。头孢菌素生产废水中也出现了类似的现象[7]。制药菌渣中的青霉素G[20]、头孢菌素C[16]残留及转化产物导致在好氧堆肥过程中[17, 21]或土壤施用菌渣肥[20]后亦出现抗性基因和抗药菌升高的现象[20, 22]。因此,深入认识环境中β-内酰胺类抗生素的环境行为、抗性的产生和传播、高浓度污染源的抗生素去除等,对于该类物质的环境管理和污染控制至关重要。
本研究在对β-内酰胺类抗生素污染特征、环境行为和控制技术的研究进展进行梳理的基础上,重点关注了β-内酰胺类抗生素中最重要的“母体”抗生素原料药——青霉素G和头孢菌素C的环境行为和控制,并在构筑抗生素和抗性基因控制多级屏障技术体系和危险废弃物制药菌渣无害化处理及资源化利用等方面提出展望,以期为β-内酰胺类抗生素残留效价的削减和抗性的控制提供参考。
1. β-内酰胺类抗生素的性质和主要环境排放源
β-内酰胺类抗生素是指化学结构包含一个β-内酰胺环,具有抗菌活性的天然或经化学手段合成的有机化合物[23-25]。表1和图1列举了主要的β-内酰胺类抗生素及其分子结构。
表 1 主要的β-内酰胺类抗生素Table 1. Major β-lactam antibiotics分类 亚类 化合物名称 青霉素类 天然青霉素 青霉素G 耐酸青霉素 青霉素V、苯氧乙基青霉素 耐酶青霉素 苯唑西林、氯唑西林、双氯西林、氟氯西林 广谱青霉素 氨苄西林、阿莫西林、匹氨西林 抗绿脓杆菌青霉素 羧苄西林、磺苄西林、替卡西林等 头孢菌素类 第1代 头孢噻吩、头孢噻啶、头孢唑啉等 第2代 头孢呋辛、头孢孟多、头孢替安等 第3代 头孢哌酮、头孢噻肟、头孢克肟等 第4代 头孢吡肟、头孢匹罗等 第5代 头孢洛林、头孢托罗、头孢吡普等 非典型类 头霉素类 头孢西丁、头孢美唑、头孢替坦等 拉氧头孢类 拉氧头孢 β-内酰胺酶抑制剂 克拉维酸、舒巴坦等 单环β-内酰胺类 氨曲南、卡芦莫南 碳青霉烯类 亚胺培南,美罗培南,帕尼培南等 | Show Table DownLoad:
CSV
DownLoad:
CSV
青霉素G和头孢菌素C是2种重要的天然抗生素和原料药。青霉素于1928年被发现。1949年,科学家通过X射线晶体学研究揭示了青霉素的完整结构,并于1959年通过全合成证实了该结构[26-27]。1945年,分离得到的头孢子菌可以产生多种成分的抗生素,1955年,分离得到头孢菌素C[28-29]。在不改变核心结构β-内酰胺环基础上,通过人工修饰侧链[30],又发展出了第4代头孢菌素、β-内酰胺抑制剂、碳青霉烯、单环菌素等多种β-内酰胺类抗生素[4, 31]。β-内酰胺类抗生素从20世纪初开始广泛使用,不仅应用于人类疾病的临床治疗,还可用于畜牧水产养殖和植物病虫害的防治[12-14, 32-33]。由于β-内酰胺类抗生素的分子结构与细胞壁黏肽结构中的D-丙氨酰-D-丙氨酸相似,可以竞争性地结合转肽酶,不可逆地抑制肽聚糖的形成,抑制细胞壁的合成,从而起到抗菌作用[34-37]。β-内酰胺环是抗菌活性基团,而侧链基团改变会引起抗菌谱或对β-内酰胺酶的敏感性等药理学特性变化[34, 38]。图2显示了青霉素G和头孢菌素C的分子结构。头孢菌素C-7位基团影响抗菌活性,而C-3和C-4位取代基主要决定药代动力学[30-31]。与青霉素相比,头孢菌素具有较低的致敏性和对β-内酰胺酶的敏感性,因此,被认为是广谱抗生素,可用于治疗革兰氏阴性和革兰氏阳性细菌的感染,并有效地阻止微生物的生长[31]。
环境中β-内酰胺类抗生素主要来自制药工业[6-9]、医用[10-12]和兽用[13-14]。抗生素原料药发酵生产过程产生的制药废水(废母液)[8-9]及菌渣[15-16]含有高浓度的抗生素残留[17],是强度最高的排放源。制药废母液中单一抗生素(如土霉素)的浓度可达1 000 mg·L−1[39]。在印度一个大型药物生产基地的污水处理厂中检出高浓度的氟喹诺酮类抗生素残留[9]。由于β-内酰胺环具有不稳定易水解的特性,与土霉素、氟喹诺酮类等结构相对稳定的抗生素相比,青霉素G残留浓度相对较低,浓度为150 μg·L−1左右。然而,其降解产物脱羧青霉噻唑酸和青霉醛的浓度可以达到389.0 mg·L−1和75.3 mg·L−1[18]。有学者[7]在中国北方一个年产头孢菌素3 000 t的制药园区排放的废水中,发现了700 μg·L−1的头孢呋辛残留。因此,尽管制药行业污水相对生活污水(含医院污水)排放量不大,但其污染物排放强度高,如果不进行有效处理,制药废水中的抗生素残留就可能直接排放到水环境或者通过管网进入城市污水处理厂。现行的污水处理技术对常规有机物处理效果良好,但是对抗生素的去除效率还较低,这也会导致抗生素从污水厂向环境中的排放[40-42]。
临床使用的抗生素可通过粪便、尿液及医疗废弃物排入环境。头孢菌素通过口服和肠胃外途径给药,排泄半衰期为0.25~9 h[43]。大多数头孢菌素以母体的形式排泄,代谢率为5%~65%,使得头孢菌素母体和在体内产生的代谢产物,以及排泄后形成的初级转化产物进入环境中[44]。医院是头孢菌素类抗生素的主要排放源[45],其中第3代和第4代头孢菌素抗生素检出较多。也有研究表明,医院废水不是城市污水中抗生素残留的主要来源。在欧洲,医院抗生素用量仅占总用量的5%~20%,而英国、美国和德国的社区使用量占比为70%~75%[46]。另外,抗生素也广泛用于畜牧水产养殖[47]:美国畜牧商每年将约11 200 t抗生素投加进牛、猪和家禽的饲料中,以促进畜禽的生长[48];兽药的使用也导致残留的抗生素随着动物排泄物进入环境中[13, 49]。排泄物中的抗生素一部分会直接渗入地下水,另一部分则通过粪便进入土壤[50],随着雨水淋洗作用迁移至河流[51]或湖泊[1, 52]。
2. 环境中β-内酰胺类抗生素的污染特征
来自制药、医用和兽用的β-内酰胺类抗生素残留,可通过在污水处理厂和土壤中的迁移进入水体被底泥吸附,或者随水体迁移进入其他水环境中。抗生素残留在医院废水[11, 46]、制药废水[6, 18]、地表水[11]、河流[24, 53]、污水厂废水[11, 54]、污泥[41]和菌渣[15, 17, 22]等不同环境基质中均有检出。β-内酰胺环不稳定,易水解开环[38, 55],同时还可以与β-内酰胺酶发生酶解反应,上述过程均使其丧失抗菌活性。因此,β-内酰胺类抗生素尽管是应用最广泛的抗生素,但通常在环境中浓度较低,甚至低于检测限。
表2列出了不同环境介质中β-内酰胺类抗生素的分布浓度。医院废水中浓度为20.0 ng·L−1~4.1 μg·L−1,与其使用量和预期排放浓度相比,检出浓度较低[56-57];青霉素G、青霉素V和头孢克洛等未检出[11]。在地表水和河水中这类抗生素的残留浓度较低,阿莫西林、头孢克洛、氯唑西林、青霉素G和青霉素V的浓度在0~250 ng·L−1[11];长江、黄河和珠江中β-内酰胺类抗生素残留浓度分别为(123.1±128.2)、(343.5±345.6)及(1 606.3±1 384.3)ng·L−1[24]。头孢菌素在不同基质中浓度在0.30~30 ng·L−1,污水厂废水中浓度最高[31]。城市污水厂中检出氨苄西林、头孢氨苄、头孢噻肟,浓度分别为77.2~383.0、65.7~525.0、38.4~93.0 ng·L−1[54]。制药废水中抗生素浓度比医院废水和污水厂浓度高。青霉素制药废水中青霉素G浓度只有0.153 mg·L−1,而水解产物青霉噻唑酸、脱羧青霉噻唑酸、青霉二酸、青霉异二酸和青霉醛的浓度分别高达8.49、389.00、23.50、1.05和75.30 mg·L−1[18]。相比之下,由于污水处理过程或水循环过程中,抗生素母体被迅速降解,使得活性污泥和河流沉积物中抗生素的浓度较低[58]。在活性污泥、剩余污泥和脱水污泥中,均未检出氨苄西林和阿莫西林[41],但其水解产物可能存在较高的浓度[18]。制药菌渣中,抗生素残留高于制药废水,例如青霉素鲜菌渣中青霉素G残留浓度为5 000 mg·L−1[15-17, 19]。作为β-内酰胺类抗生素强度最高的排放源,菌渣的无害化处理和资源化利用是抗生素残留削减的关键。
表 2 β-内酰胺类抗生素在不同环境介质中的分布浓度Table 2. Distribution concentration of β-lactam antibiotics in different environmental media环境基质 检测方法 抗生素及其转化产物名称 浓度 来源 医院废水 HPLC-MS/MS 阿莫西林、青霉素G、青霉素V、头孢克洛、氯唑西林、头孢氨苄 0~4 100 ng·L−1 [11] 制药废水 HPLC-MS/MS 青霉素G、青霉噻唑酸、脱羧青霉噻唑酸、青霉二酸、青霉异二酸、青霉醛、头孢呋辛、头孢唑啉、头孢噻肟、头孢曲松、阿莫西林、头孢菌素 0.13~703.84 mg·L−1 [7-8, 18, 59] 地表水 HPLC-MS/MS 氨苄西林、哌拉西林、阿莫西林、头孢克洛、氯唑西林、青霉素G、青霉素V 0~250 ng·L−1 [11, 56] 表层海水 HPLC-MS/MS 头孢氨苄 10~180 ng·L−1 [60] 城市污水 HPLC-MS/MS 青霉素G、头孢氨苄、头孢噻肟、阿莫西林、青霉素V、头孢克洛、氨苄西林、氯唑西林、头孢菌素、苯唑西林 0~1 400 ng·L−1 [11, 54, 57, 61-63] 河流底泥 HPLC-MS/MS 青霉素G、青霉噻唑酸、脱羧青霉噻唑酸、青霉二酸、青霉异二酸、青霉醛 0~6.56 mg·kg−1 [18, 53] 活性污泥 HPLC-MS/MS 青霉素G、青霉噻唑酸、脱羧青霉噻唑酸、青霉二酸、青霉异二酸、青霉醛 0.034~470 mg·kg−1 [18, 41] 制药菌渣 HPLC-UVHPLC-MS/MS 青霉素G、头孢菌素C 2 000~5 000 mg·L−1(鲜菌渣)70~420 mg·kg−1(干菌渣) [15-17, 19, 64] | Show TableDownLoad:
CSV
3. β-内酰胺类抗生素的环境行为
β-内酰胺类抗生素的环境行为主要包括吸附、迁移和降解。其中,吸附和迁移是β-内酰胺类抗生素在土壤中的重要环境行为。β-内酰胺类抗生素的生物降解主要由微生物的活动驱动,受到环境条件、微生物种群、化合物结构等因素影响[65]。非生物降解指抗生素不经生物作用而自行降解的过程,通常经过光解、水解等理化作用转化为其他物质。直接光解是去除湖水中头孢曲松钠的主要过程[52],也可能是地表水中头孢菌素类抗生素最重要的消除过程[33],但是在污水处理过程中光解作用贡献较小[40]。β-内酰胺类抗生素中的β-内酰胺环不稳定,在酸、碱、热、氧化剂、紫外线以及极性试剂存在的条件下易水解开环,从而丧失抗菌活性[38, 55, 66]。水解是β-内酰胺类抗生素最主要的降解途径,通过一些强化手段还可能应用于废水中抗生素的去除。因此,下面重点讨论β-内酰胺类抗生素在环境中的吸附迁移和水解过程,并特别关注大量生产的青霉素G和头孢菌素C这2种重要母体抗生素的降解途径和降解产物。
3.1 吸附与迁移
吸附对抗生素在环境中的滞留、迁移及转化有重要影响,也会影响到抗生素在环境基质中的活性和生物转化作用。吸附作用可将抗生素暂时以非生物活性的形态储存在土壤中,降低急性毒性并延长在基质中的停留时间[67]。吸附系数(Kd)用来衡量吸附能力。吸附能力强则容易在环境中累积;而吸附能力较弱则易随雨水发生迁移,并转至其他水体中,严重时还会影响地下水质。土壤的温度、pH、阳离子交换容量(cation-exchange capacity,CEC)、黏土矿物含量和有机碳含量(organic carbon,OC)会影响抗生素的吸附[5]。温度升高会影响活性污泥对不同抗生素的吸附作用。例如,温度升高后,污泥对阿莫西林、头孢噻肟、头孢克洛和头孢唑林的吸附作用增强,对普鲁卡因和头孢曲松的吸附作用降低。与青霉素类抗生素相比,活性污泥对头孢类抗生素的吸附作用受pH的影响更大[68]。
迁移是指抗生素随水体移动到其他环境基质中,或经过土壤渗透进入土壤深层或地下水的过程[69]。青霉素制药废水中,青霉素G及水解产物在处理后会随着废水的排放向收纳河流下游迁移[18]。迁移行为的影响因素包括吸附特性、淋洗速率和降解程度等。由于β-内酰胺类抗生素在土壤中的吸附能力较弱,很可能对地表及地下水造成污染[70]。抗生素在环境中的迁移能力取决于其理化特性、土壤理化特性、有机粪肥的施用和天气条件。酸雨可使抗生素加速从动物粪便向地表土壤迁移,而长时间的降雨还会促进抗生素在土壤中向下迁移[5, 50, 65]。
3.2 水解
水解是有机化合物,特别是酯类和酰胺类化合物在环境中最重要的降解途径。β-内酰胺环不稳定易水解开环。这一特性也是这类抗生素在无大量微生物种群的系统(如河流和地下水)中的主要降解途径[71]。温度和pH是影响水解速率最重要的参数。水解速率通常随着温度的升高而增加[71-72]。氨苄青霉素、头孢洛汀和头孢西丁3种抗生素在通常环境条件(pH=7,25 ℃)下的水解半衰期为5.3~27.0 d。碱性条件下的水解速率明显快于酸性和中性条件下的水解速率。温度升高10 ℃时,水解速率增加2.5~3.9倍[71]。在对阿莫西林的水解研究中也得到了类似结论[55]。
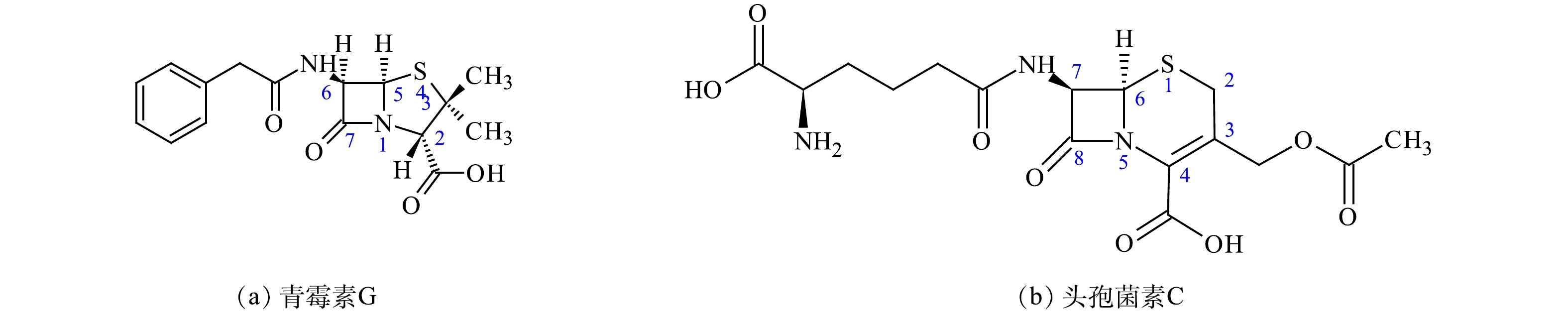
KESSLER等[73]探究了青霉素G在酸性条件下的两种降解途径,确定了青霉胺作为最终降解产物的酸性降解途径。青霉素G在酸性、碱性、中性的水解途径和酶解途径如图3所示[23,74]。在青霉素制药废水中检测到这些产物,并通过定量测定验证了产物之间的转化关系。在制药废水处理过程中,出水的青霉素G浓度为(1.68±0.48)μg·L−1,去除率为96.7%;脱羧青霉噻唑酸是青霉素的主要降解产物,浓度为(44.5±2.5)mg·L−1,摩尔浓度占总摩尔浓度的68.8%。高浓度的青霉素降解产物被排放到河流中,在污水排放的受纳河流下游,脱羧青霉噻唑酸、青霉噻唑酸和青霉二酸都具有相对的环境持久性[18]。
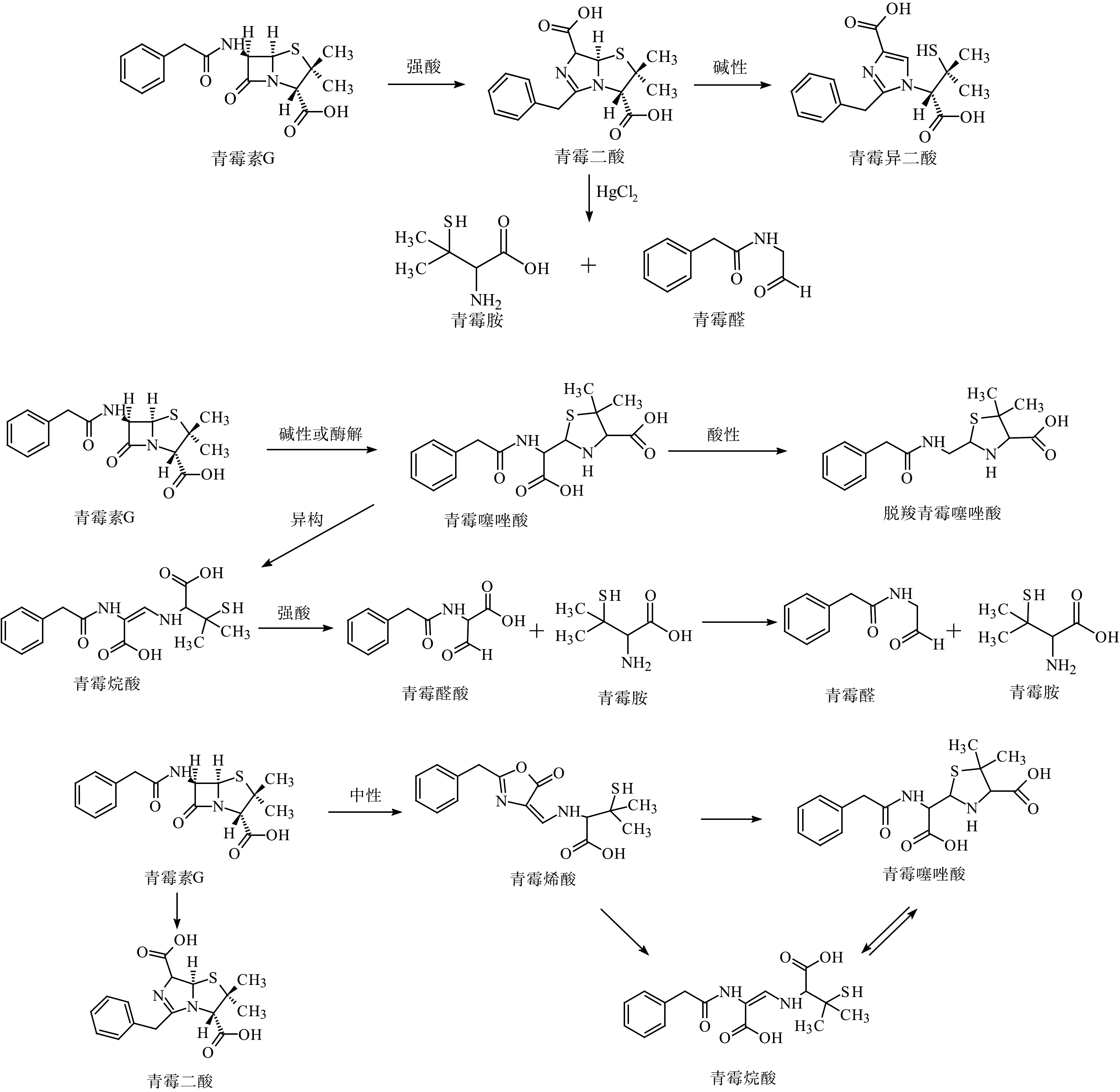 图 3 青霉素G在不同条件下的水解途径Figure 3. Hydrolysis pathway of penicillin G under different conditions
图 3 青霉素G在不同条件下的水解途径Figure 3. Hydrolysis pathway of penicillin G under different conditions与青霉素相比,头孢菌素中四元环和六元环形成的稠合结构对水解反应更稳定,但也会发生各种化学和酶促转化。C-3位取代基、β-内酰胺环、C-4位羧基和C-7位侧链酰胺键都是头孢菌素易发生降解反应的位置(图2(b))[75]。C-3位存在离去基,使得亲核试剂或β-内酰胺酶可以水解β-内酰胺环的C—N键[23]。在酸性、碱性和中性水解中,C-3位侧链分离是头孢菌素的主要降解途径之一。头孢菌素C的水解途径[23]如图4所示。与青霉素类似,β-内酰胺环也是头孢菌素水解反应的关键位点。开环反应根据侧链结构的不同分为:中性条件的一般开环水解、侧链α-氨基参与的分子内氨解及头孢菌素环缩合成噻唑结构。另外,还存在pH依赖型的立体异构,即在中性或酸性环境下C-6位发生立体异构,而碱性环境下C-7位发生立体异构和C-7位侧链的甲氧亚胺键构型发生顺反异构[75-76]。HIRTE等[55]研究阿莫西林在酸性、碱性、中性条件下的降解过程中,识别鉴定出了45个产物,证明了阿莫西林青霉噻唑酸、阿莫西林2',5'-哌嗪二酮和阿莫西林脱羧青霉噻唑酸并非稳定产物,并首次报道了青霉胺氧化成二硫化青霉胺的过程。
 图 4 头孢菌素C在不同条件下的水解途径Figure 4. Hydrolysis pathway of cephalosporin C under different conditions
图 4 头孢菌素C在不同条件下的水解途径Figure 4. Hydrolysis pathway of cephalosporin C under different conditions4. β-内酰胺类抗生素抗性的产生和传播
抗生素的作用目标是细菌。若抗生素在环境中长期稳定存在,则会影响基质中的群落结构,导致环境中微生物菌群失调,诱导微生物抗性的产生,也会对动物、植物产生毒性作用,可能存在潜在的环境和健康风险。环境中抗生素残留对抗生素抗性细菌(ARB)及抗生素抗性基因(ARGs)的形成具有促进作用。
世界卫生组织将抗生素抗性列为一类亟需解决的全球性公共健康问题。各种抗生素第一次观察到抗性的时间序列已被系统总结[38]。19世纪40年代,临床上就出现了青霉素抗性菌。1946—1948年,青霉素抗药菌株比例从14%上升到59%。抗药菌的抗药机制[35, 37-38]主要包括4个方面:抗生素作用靶点突变、酶促反应使抗生素失活、细胞膜特性改变和外排泵机制、改变代谢途径。对于β-内酰胺类抗生素,细菌主要通过分泌β-内酰胺酶,释放到细胞外周胞质中水解药效基团β-内酰胺环,从而使其到达作用靶点前失活。此外,细菌也通过改变抗生素作用对象青霉素结合蛋白(PBPs)的结构产生抗性,但由于PBPs变异比较困难,故分泌β-内酰胺酶是β-内酰胺类抗生素最重要的抗性机制[77]。抗性基因是通常由基因突变产生的并赋予宿主细胞对抗生素有耐受性的一类基因。而抗性基因通过整合到质粒和整合子等水平转移元件上,实现了在不同菌种之间的水平转移[37]。初级抗性在微生物中天然存在,例如铜绿假单胞菌对青霉素G的抗性,相同物种生物通过细胞分裂(垂直转移)“继承”该抗性。在微生物与抗生素接触过程中会产生次级抗性,质粒介导的抗性可在微生物之间转移,染色体外遗传物质通过结合在不同细菌之间转移(水平转移)[78]。
碳青霉烯类抗生素是抗菌谱最广、抗菌活性最强的非典型β-内酰胺类抗菌药物。这类抗生素对超广谱β-内酰胺酶(ESBL)和头孢菌素酶 (AmpC酶)具有高度稳定性,是医学临床治疗中产生超广谱β-内酰胺酶类耐药肠肝菌科细菌等多重耐药革兰氏阴性菌感染的重要药物。随着这些药物广泛应用于临床,碳青霉烯类耐药肠杆菌科细菌等各种广泛耐药细菌出现并流行,碳青霉烯抗菌药物的疗效已经明显降低[79-80]。20世纪80年代,还出现铜绿假单胞菌、沙雷菌等对碳青霉烯类药物耐药的现象。KPC型碳青霉烯酶于1996年在美国发现[81]。目前,常见的碳青霉烯酶类耐药基因主要有KPC、IMP、VIM和NDM等,国内以KPC和IMP为主。碳青霉烯类抗生素抗药性产生的机理主要有3种:1)细菌通过产生碳青霉烯酶产生抗药性;2)产生质粒或染色体介导β-内酰胺酶合并外膜蛋白的缺失或数量的减少;3)药物作用靶位的特异性改变。其中,碳青霉烯酶所造成的危害最为严重[82]。由于碳青霉烯类耐药肠杆菌具有流行范围广、传播速度快、多重耐药以及引起的疾病死亡率高等特点,美国己经将碳青霉烯类耐药肠杆菌列为最为紧急的3大威胁之一[83]。目前,动物养殖、环境和临床细菌的碳青霉烯耐药菌和抗性基因及其关联性研究是一个热点。
不同环境基质中都已检出β-内酰胺类抗生素抗药菌和抗性基因。对自然水体中β-内酰胺酶基因和整合子的发生和分子多样性评估[84]发现,在77.8%的肠杆菌科和10.5%的气单胞菌中检测到β-内酰胺酶基因,最常检出基因是blaTEM。海水中超过90%的细菌菌株对1种以上的抗生素具有抗性,而20%的细菌对至少5种抗生素具有抗性。医院和集约化养殖场的废水可能是环境中抗药菌和抗性基因的主要污染源[85]。污水处理厂则被认为是环境中抗性基因传播的热点地区,因为污水处理过程为抗药菌增殖以及抗性基因在不同微生物之间的水平转移提供了便利条件[38]。目前,已有86种β-内酰胺抗性基因在11个国家的污水处理厂进水、出水和活性污泥中的分布和变化情况被报道[38]。污水处理厂中抗生素残留在抗性菌及抗性基因的维持和传播中起着重要作用,除了选择抗性表型,还可能干扰土著细菌群落。制药废水的抗生素残留进入水体后也能够改变水体的微生物系统[6, 86],通过选择性压力诱导水体中的微生物产生抗性基因,并进行传播扩散[85]。本课题组的前期研究也发现,由于可能受到废水中青霉素G及转化产物的影响,在青霉素生产废水和下游河流中发现了大量抗药菌,对β-内酰胺类抗生素具有极高的抗性,对其他各类抗生素也有广泛的抗性,具有一定的环境风险[86]。此外,携带了各种抗性基因的I型整合子(intI1)在废水和下游河流的菌株中也被检出,进一步说明了抗性基因在细菌间水平转移的可能性。制药菌渣中的青霉素G[20]、头孢菌素C[16]残留及转化产物在堆肥过程[17, 21]或施用菌渣肥[20]后,在土壤中也会诱导产生抗性基因[20, 22]和抗药菌[21]。
当考虑导致抗生素抗药菌和抗性基因出现、扩增和持续存在的因素时,传统的假设是“抗生素的使用是这些过程主要驱动力”[87]。处于亚抑制浓度的抗生素可能会影响细胞功能,并改变毒力因子基因表达或抗生素抗性转移。一些研究表明,医院废水中抗药性的增长与头孢菌素的使用呈正相关[10, 85];但也有证据表明,抗药性并不总与所用药物的量或环境中残留物的浓度有关[48]。肠胃外施用头孢噻呋对胃肠道中头孢噻呋抗药细菌的流行具有有限或短期的影响。虽然这种反应可能足以解释对头孢菌素广泛抗药的模式,但大约2/3的头孢噻呋代谢物会从尿液中排出,这增加了环境选择在扩增和维持抗生素抗药性起重要附加作用的可能性[87]。不同的基因可能具有不同的维持动力学,可能与携带它们的宿主微生物有关[85, 88]。环境中β-内酰胺类抗生素抗药菌和抗性基因发展和传播的机制还需要进一步深入探讨。
5. 废水和菌渣中β-内酰胺类抗生素的控制技术
与临床使用和动物养殖抗生素造成的环境广泛存在的污染相比,制药行业的污染源相对集中,但污染排放强度却是最高,故制药废水和菌渣是β-内酰胺类抗生素排放强度最高的排放源。若不对制药废水和菌渣进行有效控制,抗生素母体及残留效价则可能直接排放到环境中,或通过管网进入城市污水处理厂,从而导致抗生素从污水厂向环境中的排放。因此,必须在源头对抗生素残留效价进行控制,阻断抗生素和抗性基因向环境中排放。
对废水和菌渣中β-内酰胺类抗生素的去除技术,主要有生物处理和物化处理两类。通常采用厌氧、好氧和缺氧等生物处理过程去除制药废水中的COD,并实现氨氮的硝化反应和反硝化脱氮。以山西某制药厂为例,该厂青霉素生产废水生化处理后出水COD为218.0 mg·L−1,无法达到一级排放标准[89]。使用UASB反应器处理制药废水时,COD去除率为39%~85%,效果不稳定;同时6-氨基青霉烷酸和阿莫西林的去除率分别只有19%~33%和13%~47%[90]。当模拟配水中阿莫西林浓度从250 mg·L−1提高至650 mg·L−1时,活性污泥对阿莫西林的去除率从88.79%下降至完全无法去除[91]。因此,尽管β-内酰胺类抗生素容易生物降解,但因制药废水水质成分复杂,残留的抗生素和残留基质、试剂可能会影响生物处理效果[38, 61, 92]。抗生素的浓度、稳定性和对微生物的抑制作用都会造成处理效果的不稳定[31],更为严重的是可能会导致废水生物处理过程中抗性基因的产生和排放[93-95]。解决抗生素废水生物处理技术瓶颈的关键,是在废水进入生物处理之前,利用预处理技术选择性去除废水中的抗生素效价残留,降低对微生物的抑制作用,从而达到稳定的处理效果[96]。
抗生素生产过程除了产生制药废水,还会产生大量生产菌渣。制药菌渣含水率高,主要成分为菌丝体、剩余培养基、代谢中间产物、有机溶媒以及少量残留的抗生素[97]。2008年,抗生素菌渣被列入《国家危险废物名录》,如何实现抗生素菌渣合理有效利用与安全处置已成为制药企业亟待解决的难题[98]。许多研究者通过生物处理技术对菌渣进行无害化处理和资源化利用。例如,厌氧消化可以将头孢菌素C菌渣中低品位的有机质转化为高品位沼气,可实现菌渣资源化利用[99];好氧堆肥可以消除青霉素G菌渣中的抗生素残留,但堆肥过程也会导致抗药菌和抗性基因的产生[100-103],在其他青霉素菌渣好氧堆肥的研究中也得出了相似的结论[17]。最近的研究[104]发现,采用高温(70 ℃)厌氧消化对螺旋霉素菌渣进行预处理具有可行性。高温消化可大幅度降低抗生素的浓度,同时由于相应宿主的减少,抗药基因的丰度也在7 d之内减少。实际上,处理菌渣问题的关键是研发出去除抗生素效价残留的有效技术,以阻断抗性传播为目标,并在此基础上实现菌渣无害化处理及资源化利用。
氧化等一些物理化学技术通常对于水中抗生素的去除具有很好的效果。表3列出了常见的物理化学去除工艺及效果。吸附[105-106]和膜过滤[57]是常规的物理处理技术。这类技术通过分子间作用力或孔径截留作用将抗生素从一个相中分离转移至另一相,但抗生素本身没有发生降解反应。由于头孢菌素具有在相对较宽的波长范围内的光吸收作用,故光催化技术广泛用于头孢菌素类抗生素的去除技术的研究中[33, 52],但目前还难以进行实际工程应用。臭氧、芬顿等高级氧化技术在β-内酰胺类抗生素的去除研究中体现出反应速度快和去除效率高的特点,可以高效去除溶液中抗生素母体[107-108],但在氧化过程产生的降解产物的结构、毒性,特别是在复杂实际废水中的适用性还有待进一步探究[31]。在青霉素制药废水深度处理工艺探究中发现,Fenton氧化法、Cu/Fe催化还原法、臭氧氧化法均可有效降低COD残留。在Fenton氧化最优条件下,出水COD含量可降至22.5 mg·L−1 [89]。采用Fenton氧化和Fe/C微电解2种方法预处理7-氨基头孢烷酸制药废水。结果表明:2种方法均可有效提高COD去除率和废水可生化性;但Fenton氧化法存在药耗量大,处理成本高的问题;而Fe/C微电解发也存在反应时间较长的问题[109]。物理化学处理技术在制药菌渣无害化和资源化过程中也有一些初步研究。例如,青霉素菌渣经过100 ℃热水解作用,98%的抗生素残留被去除,制取的菌渣肥可以作为有机肥料应用于农业生产[15, 20]。也有研究者发现,微波加热温度可显著影响菌渣中残留抗生素的去除率,700 W辐射功率下快速升温至100 ℃条件下持续处理15 min,头孢菌素C的去除率可以达到99.9%[16]。
表 3 常见的物理化学处理工艺及处理效果Table 3. Common physical and chemical treatment process and treatment effect处理工艺 基质 处理条件 抗生素名称 浓度 去除效果 来源 吸附 制药废水 pH 为2~7 温度 30 ℃吸附剂量0.1~3.5 g 阿莫西林 317 mg·L−1 膨润土为吸附剂,去除率88.01%;活性炭为吸附剂,去除率94.67% [59] 膜处理 城市污水 MF微滤2 min,平衡罐5 min,RO反渗透 0.5 min,加氯反冲洗 阿莫西林 90 ng·L−1 头孢氨苄去除率87%~100%,其余抗生素去除率100% [57] 氯化 模拟配水 阿莫西林和头孢拉啶:ClO2和抗生素摩尔比为 0.25~2.0,pH=8.0反应时间1 min;青霉素:ClO2和抗生素摩尔比为 0.25~1.5,pH=3.5反应时间2 h 阿莫西林头孢拉啶青霉素G 1.6 mg·L−11.8 mg·L−125.0 mg·L−1 阿莫西林和头孢拉啶在1 min内全部降解,青霉素可以在2 h内全部降解 [110] 芬顿 模拟配水 pH为3,H2O2浓度25 mmol·L−1Fe3+浓度1.5 mmol·L−1 青霉素G 400 mg·L−1 配水中COD的去除率达56%,TOC的去除率达46% [111] 模拟配水 m(COD): m(H2O2): m(Fe2+)=1:3:0.3pH=3 阿莫西林氨苄青霉素氯唑西林 104 mg·L−1105 mg·L−1103 mg·L−1 2 min抗生素完全降解,10 min生物降解性提高到0.37,60 min COD和DOC降解率为81.4%和54.3% [112] 光催化 模拟配水 pH 为3.0~9.0,太阳光源,催化剂TiO2 0.1~0.7 g·L−1或不同比例铁碳混合物 阿莫西林 100 mg·L−1 最优条件下去除率可以达到85% [113] 模拟配水 pH为 3.0~11.0,紫外光源365 nm,6 W催化剂TiO2 0.5~2.0 g·L−1,H2O2 50~300 mg·L−1 阿莫西林氨苄青霉素氯唑西林 104 mg·L−1105 mg·L−1103 mg·L−1 pH=5,TiO2 1.0 g·L−1,抗生素去除率均达50%,DOC去除率81%;pH=5,TiO2为1.0 g·L−1,H2O2为100 mg·L−1,抗生素30 min内完全降解,24 h矿化率达40% [114] 臭氧 模拟配水 pH为2.5~7.2O3浓度为1.6×10−4 mol·L−1 阿莫西林 210 mg·L−1 4 min阿莫西林去除率90%,20 min阿莫西林矿化率可以达到18.2%,延长时间后,矿化率保持较低水平 [115] 模拟配水 pH为3.0~11.0,O3量为3 g·(h·L)−1H2O2浓度0~200 mmol·L−1 头孢曲松青霉素G − 60 min抗生素去除率95%,TOC去除率45%,提高废水可生化性 [110] 模拟配水 预臭氧pH 为7.0~12.0,O3量1 800 mg·(h·L)−1;H2O2 100 mmol·L−1 青霉素G 600 mg·L−1 pH=7,预臭氧工艺提高废水可生化性,但并不能完全去除生态毒性,还存在严重生物抑制作用;1 h内臭氧COD去除率为37%,而经预臭氧COD去除率达76% [116] 水热 制药菌渣 高压釜内部容积1.0 L,内径70 mm,温度100~220 ℃ 头孢菌素C − 在100~220 ℃和0~60 min内,头孢菌素C的去除率为99.0%~99.9% [66] 热水解 制药菌渣 盐酸2 mol·L−1,投加量10~60 mL,温度50~100 ℃ 青霉素G 9 g·mL−1(效价) 最优条件盐酸投加40 mL浸泡1 h,90 ℃水浴中加热机械搅拌3 h,过滤,效价去除率80.93% [64] 制药菌渣 加水倍数为3,水解温度60 ℃,水解时间30 min 青霉素G 5 000 mg·kg−1 水解后菌渣青霉素残留小于0.5 mg·kg−1,凯式氮削减率大于45%,有效降低青霉素残留及凯氏氮对厌氧消化的影响,为厌氧消化高效、稳定进行创造有利条件 [19] 热水解 制药菌渣 温度60~100 ℃ 青霉素G 2 000 mg·L−1 60 ℃时,20 min去除率为20%;100 ℃时,20 min去除率为98% [15] 微波 制药菌渣 温度100 ℃,微波功率为300、500、700 W 头孢菌素C 420 mg·kg−1 前150 s降解效率较低,此后降解速率明显提升,不同功率条件微波辐射15 min,降解率均超过99.9% [16] | Show TableDownLoad:
CSV
综上所述,尽管生物处理过程能削减部分抗生素,但抗生素残留会导致处理效果不稳定和抗性基因产生、排放问题。高级氧化技术可以在生物处理之后进一步去除残留抗生素,阻断抗生素向环境排放。但该技术主要用于模拟废水的研究,在实际废水处理中还存在操作困难、选择性差、成本高等问题,且可能产生具有较强毒性的未知中间产物[117]。因此,如何从制药废水中高效选择性去除残留抗生素及其相关物质仍然是一个技术挑战。阻断抗性基因向环境中排放的关键,是在生物处理工序之前有效削减和控制抗生素效价残留,并根据抗生素特性,开发针对抗生素抗菌活性基团的源头预处理技术。笔者所在研究团队根据四环素类抗生素药效官能团易水解的特性,探究了四环素类抗生素的水解动力学及其效价变化,通过优化水解条件可以显著加快四环素和土霉素的水解,并有效降低抗生素对生物处理微生物的抑制[118]。在此基础上进行了现场中试实验[119],验证了强化水解预处理工艺,并与生物处理结合进行现场工程应用,实现了常规指标、抗生素及抗性基因的源头控制。该技术思路有望实现β-内酰胺类抗生素废水的无害化处理。同时,抗生素及其代谢产物残留,还有抗性基因均是菌渣资源化利用造成环境风险的重要安全隐患,在以后的研究中需要从这两个方面对菌渣资源化利用进行安全性评估,为菌渣利用安全性评估方法和控制标准的制定提供科学基础。
6. 研究展望
1)由于β-内酰胺类抗生素具有易水解和生物降解的特性,在环境中通常浓度很低或低于检测限。然而,抗生素母体的削减并不意味着环境效应的消除,各种水解、降解产物等相关物质可能会贡献残留效价,导致环境中β-内酰胺抗药菌和抗性基因的产生和传播。因此,需要对β-内酰胺类抗生素的降解产物和降解途径进行深入探究,进一步明确降解产物与细菌抗性产生和传播之间的关系。
2)对β-内酰胺类抗生素残留的去除,除了母体之外,还应关注抗生素残留效价,将具有抑菌活性的中间产物、前驱体或者生产副产物,以及经过物化或者生物降解等过程后保留抗菌活性的中间转化产物纳入β-内酰胺类抗生素污染控制评价体系。为了明确残留抗生素和效价的控制目标和标准,需要对抗生素在促进抗性基因产生和发生水平转移方面的浓度阈值进行研究。
3)针对β-内酰胺类抗生素制药废水等高浓度抗生素残留的废水,需要开发选择性地去除抗生素效价的预处理技术,从源头解决由于抗生素效价残留造成生物处理效果不稳定的问题,阻止抗性的产生;同时采用末端保障深度处理技术,阻断抗生素残留、抗性菌和抗性基因向环境的传播,保障废水的安全排放。为了构筑抗生素和抗性基因控制多级屏障技术体系,应当将源头控制预处理技术和末端保障深度处理技术联合起来。
4)包括β-内酰胺类抗生素在内的发酵类抗生素的制药菌渣已被列入《国家危险废物名录》。针对其产生量大、处理难度大等现实问题,并根据《制药工业污染防治技术政策》(征求意见稿) 中提出的“鼓励开发发酵菌渣在生产工艺中的再利用技术、无害化处理技术、综合利用技术”政策建议,如何实现抗生素菌渣合理有效利用与安全处置已成为制药企业亟待解决的难题[98]。未来的研究将以去除抗生素效价残留、阻断抗性传播为目标,开发和优化菌渣处理技术,同时围绕菌渣资源化利用过程中抗生素残留效价和抗性发展进行系统评价,为菌渣无害化处理及资源化利用提供参考。
-
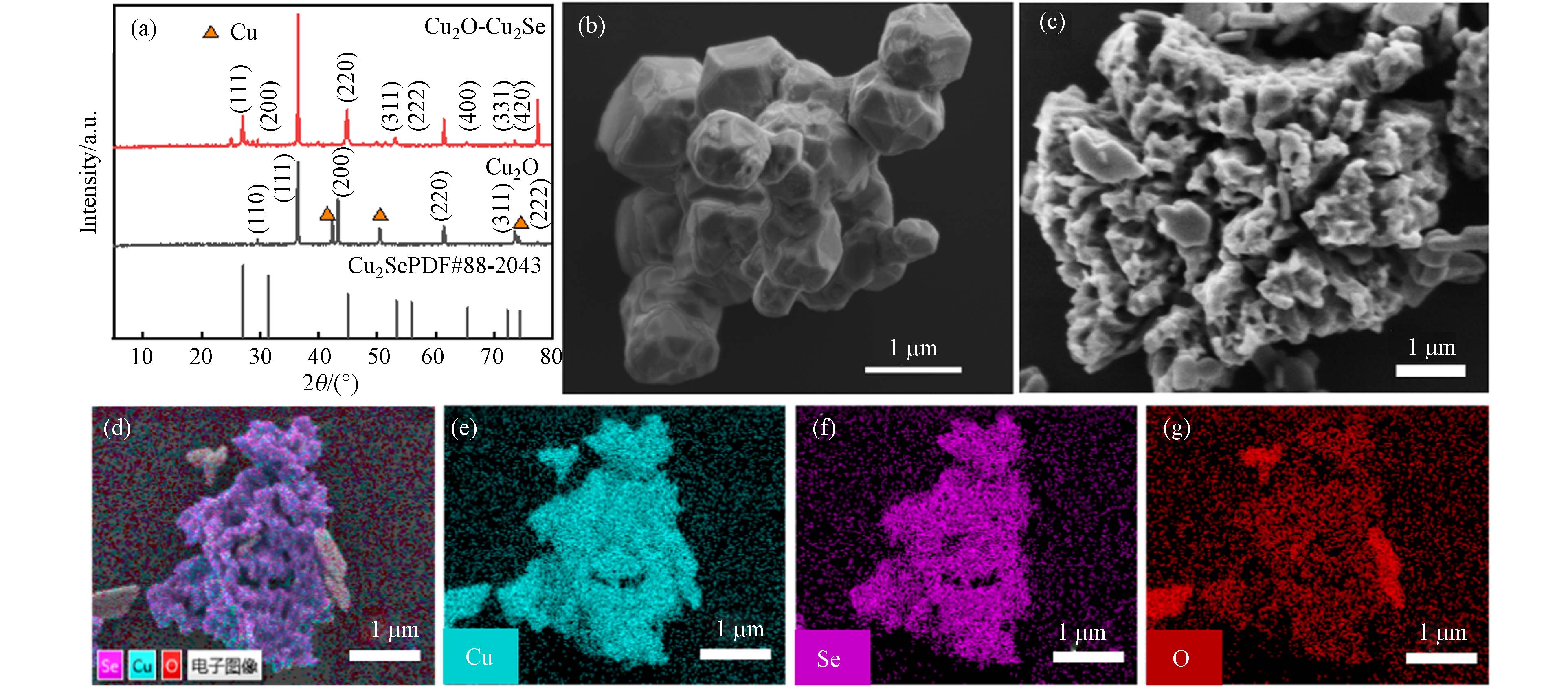
图 1 (a)Cu2O-Cu2Se纳米复合物和纯Cu2O的XRD图谱, (b-c)纯Cu2O和Cu2O-Cu2Se纳米复合物的SEM图,(d-g)Cu2O-Cu2Se纳米复合物的Cu、Se、O元素的SEM图和EDS面扫图
Figure 1. (a)XRD images of Cu2O-Cu2Se nanocomposite and pure Cu2O,(b-c)SEM image of pure Cu2O and Cu2O-Cu2Se nanocomposite,(d-g) SEM images of Cu, Se, O elements and EDS-mappings of Cu2O-Cu2Se nanocomposite
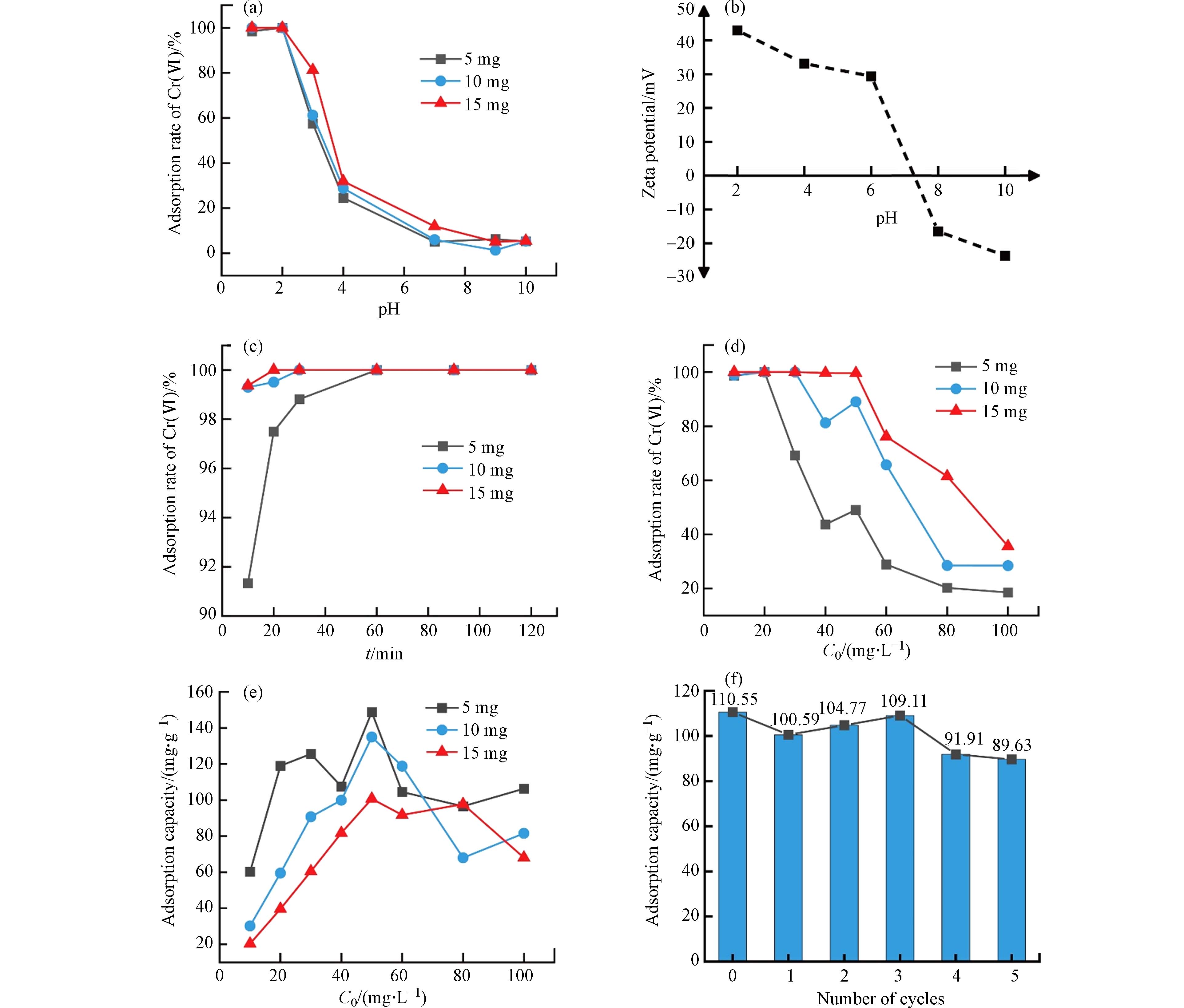
图 2 (a)pH对Cu2O-Cu2Se纳米复合物的吸附性能的影响,(b)Cu2O-Cu2Se纳米复合材料的Zeta电位分布图,(c-d)时间和Cr(Ⅵ)初始浓度对Cu2O-Cu2Se纳米复合物的吸附性能的影响,(e)Cu2O-Cu2Se纳米复合材料吸附量随Cr(Ⅵ)初始浓度的变化,(f)Cu2O-Cu2Se纳米复合物的循环吸附能力
Figure 2. (a)Effect of pH on adsorption properties of Cu2O-Cu2Se nanocomposite,(b)Zeta potential distribution of Cu2O-Cu2Se nanocomposite,(c-d)Effect of time and initial concentration of Cr(Ⅵ) on adsorption properties of Cu2O-Cu2Se nanocomposite,(e)Change of adsorption capacity of Cu2O-Cu2Se nanocomposite with initial concentration of Cr(Ⅵ),(f)Cyclic adsorption capacity of Cu2O-Cu2Se nanocomposite
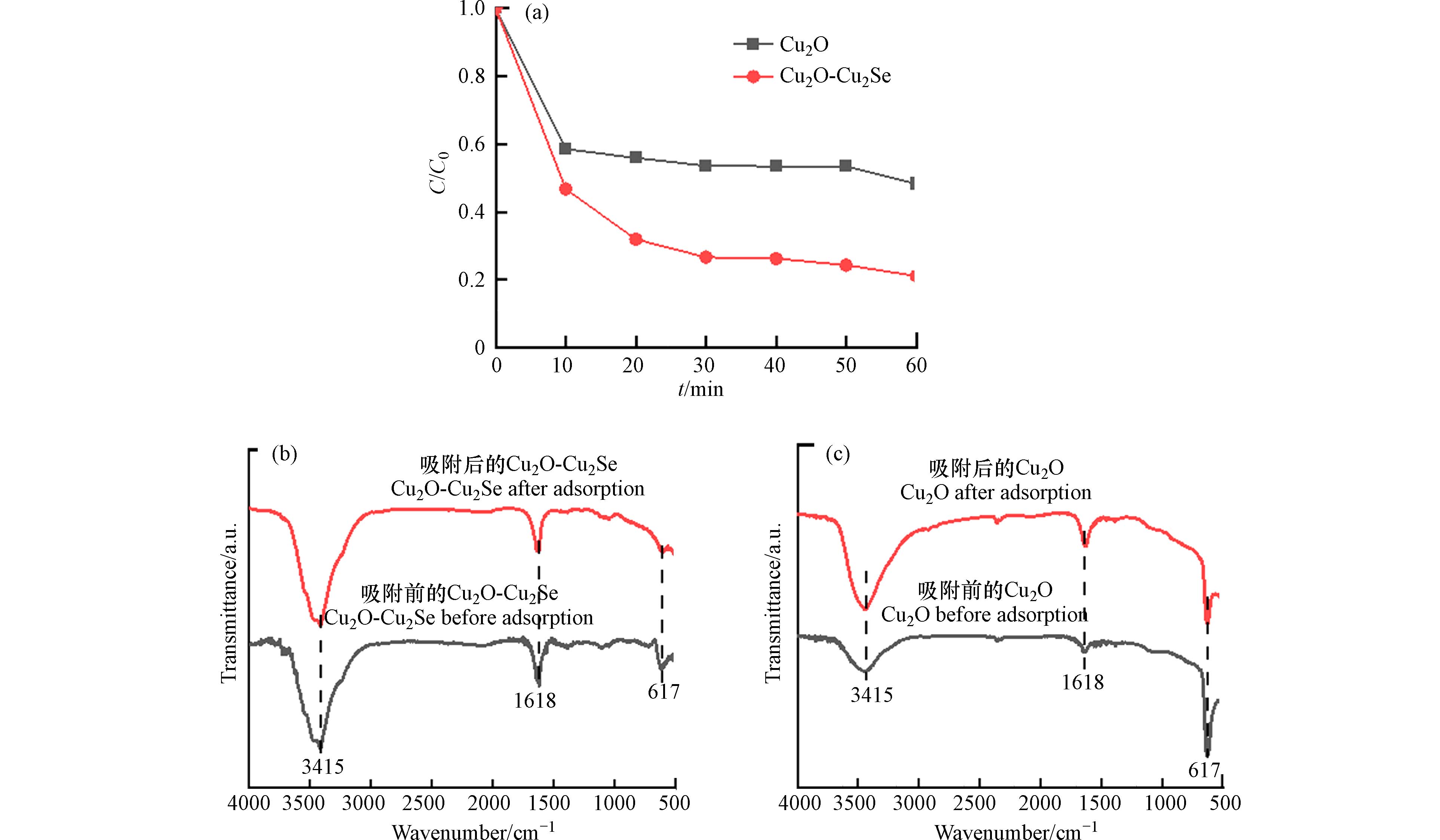
图 3 (a)纯Cu2O和Cu2O-Cu2Se纳米复合物对Cr(Ⅵ)的去除能力对比,(b-c)吸附Cr(Ⅵ)前后的Cu2O-Cu2Se纳米复合物和纯Cu2O的FT-IR谱图
Figure 3. (a)Comparison of Cr(Ⅵ) adsorption rates of pure Cu2O and Cu2O-Cu2Se nanocomposite,(b-c)FT-IR spectra of Cu2O-Cu2Se nanocomposite and pure Cu2O before and after Cr(Ⅵ) adsorption
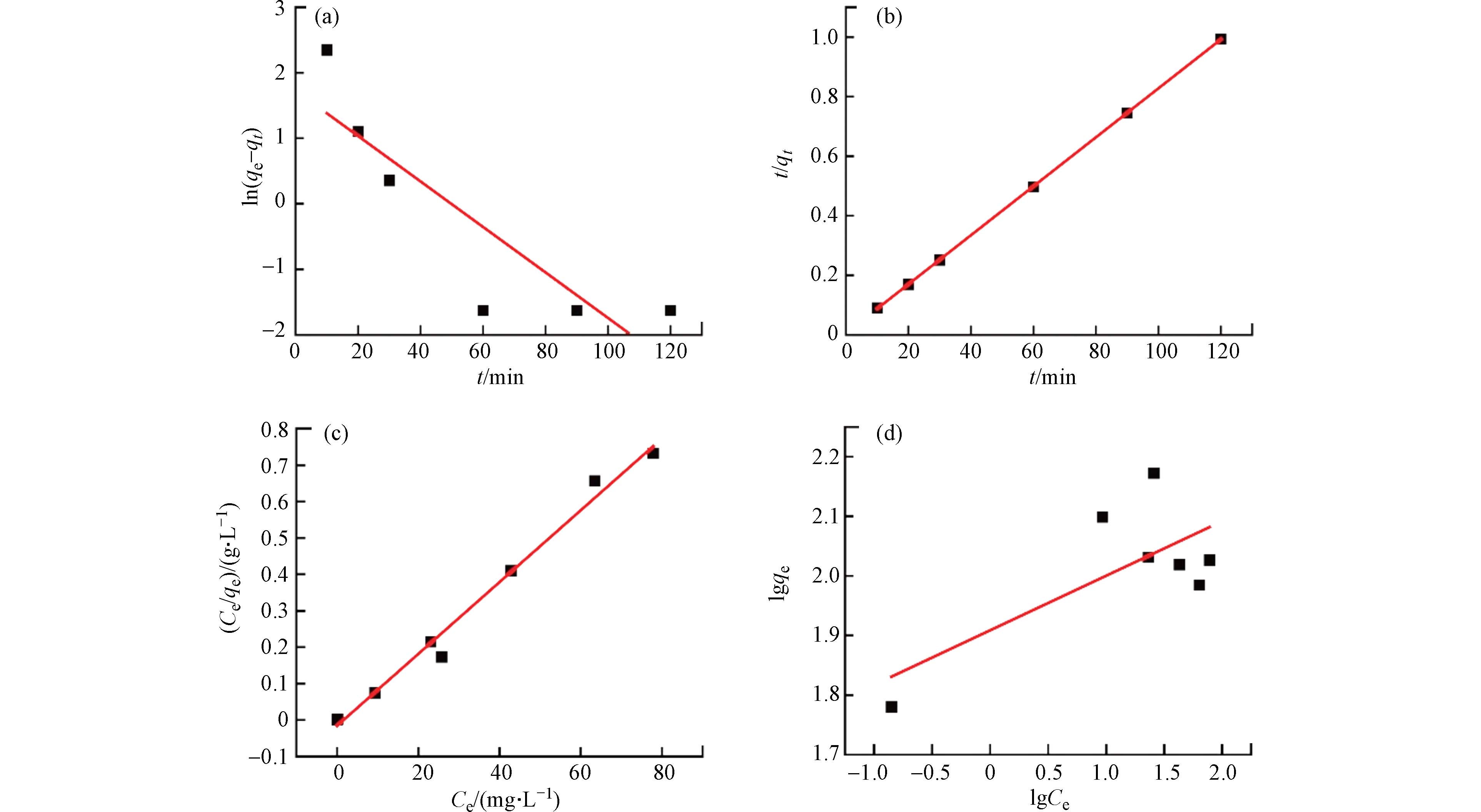
图 4 Cu2O-Cu2Se纳米复合物的(a-b)准一级吸附动力学、准二级吸附动力学模型线性拟合,(c-d)Langmuir 模型、Freundlich 模型线性拟合
Figure 4. (a-b)fitted curves of pseudo first order kinetic model and pseudo second order kinetic model,(c-d)Langmuir model and Freundlich model of Cu2O-Cu2Se nanocomposite
表 1 Cu2O-Cu2Se纳米复合物的元素含量百分比
Table 1. Cu2O-Cu2Se nanocomposite elemental content percentage
元素Element 质量分数/%Mass percentage 原子百分比/%Atomic percent O 12.20 36.25 Cu 74.77 55.91 Se 13.03 7.84
下载: 导出CSV
表 2 各种材料的Cr(Ⅵ)离子吸附性能
Table 2. Several materials used for the removal of Cr(Ⅵ) ions
材料Material pH Cr(Ⅵ)初始浓度/(mg·L−1)Cr(Ⅵ) initial concentration 吸附容量/(mg·g−1)Adsorption capacity 达到吸附平衡时间/minTime to reach adsorption equilibrium 参考文献Reference Cu2O-Cu2Se复合材料 2 50 148.78 30 本文 磁性阳离子水凝胶 3—12 10 285 180 [14] 聚苯胺@磁性壳聚糖 2 100 186.6 420 [15] 纳米磁性MnFe2O4 2 55 34.84 1440 [11] 活性氧化铝 4 60 5.4 90 [16]
下载: 导出CSV
表 3 动力学方程的拟合结果
Table 3. Fitting results of the kinetic equations
吸附模型Adsorption model 准一级动力学模型Pseudo first order kinetic model 准二级动力学模型Pseudo second order kinetic model 表达式Expression ln(qe−qt)=lnqe−k1t tqt=1k2q2e+tqe R2 0.768 8 0.999 9 拟合参数Parameters k1=0.034 70qe=53.515 6 k2=0.011 56qe=121.654 5 拟合方程Equation ln(53.5156−qt)=1.7285−0.0347t tqt=0.0059+t121.6545 注:qt表示时间为t时的吸附量,qe为平衡吸附量,k1为准一级吸附速率常数, k2为准二级吸附速率常数.
下载: 导出CSV
表 4 等温吸附模型的拟合结果
Table 4. Fitting results of the isothermal adsorption model
吸附模型Adsorption model Langmuir Freundlich R2 0.9872 0.5077 拟合参数Parameters KL=0.62qm=101.42 mg·g−1 KF=81.031/n=0.09 拟合方程Equation ce/qe=0.0099ce-0.0159 lgqe=0.0916lgce+1.9087 注:ce为平衡时的浓度,qe为平衡时的吸附量,qm为最大吸附量, KL为Langmuir吸附常数,n和KF为Freundlich吸附常数.
下载: 导出CSV
-
[1] 张春梅, 杨婷婷, 陆桂花, 等. 纳米纤维素/壳聚糖气凝胶对六价铬的吸附性能[J]. 功能材料, 2022, 53(10): 10180-10184. ZHANG C M, YANG T T, LU G H, et al. Adsorption properties of cellulose nanocrystalline/chitosan aerogels for hexavalent chromium[J]. Journal of Functional Materials, 2022, 53(10): 10180-10184 (in Chinese).
[2] REN L L, XU J, ZHANG Y C, et al. Preparation and characterization of porous chitosan microspheres and adsorption performance for hexavalent chromium[J]. International Journal of Biological Macromolecules, 2019, 135: 898-906. doi: 10.1016/j.ijbiomac.2019.06.007 [3] 陶正凯, 荆肇乾, 王郑. 纳米纤维素材料在重金属废水治理中的应用[J]. 材料导报, 2023, 37(6)∶214—211. TAO Z K, JING Z Q, WANG Z. Application of nanocellulose materials for the treatment of wastewater containing heavy metals[J]. Materials Reports, 2023, 37(6): 214-221 (in Chinese).
[4] VAKILI M, DENG S B, LI T, et al. Novel crosslinked chitosan for enhanced adsorption of hexavalent chromium in acidic solution[J]. Chemical Engineering Journal, 2018, 347: 782-790. doi: 10.1016/j.cej.2018.04.181 [5] QUMAR U, HASSAN J Z, AHMAD BHATTI R, et al. Photocatalysis vs adsorption by metal oxide nanoparticles[J]. Journal of Materials Science & Technology, 2022, 131: 122-166. [6] YAACOB N, SEAN G P, NAZRI N A M, et al. Simultaneous oily wastewater adsorption and photodegradation by ZrO2-TiO2 heterojunction photocatalysts[J]. Journal of Water Process Engineering, 2021, 39: 101644. doi: 10.1016/j.jwpe.2020.101644 [7] 彭宽宽, 贾志刚, 诸荣孙, 等. 介孔铁镁复合氧化物对Cr(Ⅵ)的吸附性能[J]. 硅酸盐学报, 2011, 39(10): 1651-1658. PENG K K, JIA Z G, ZHU R S, et al. Adsorption of chromium(Ⅵ) onto mesoporous Fe-Mg composite oxide sorbent[J]. Journal of the Chinese Ceramic Society, 2011, 39(10): 1651-1658 (in Chinese).
[8] 黄凤萍, 邹正波, 李春花, 等. 二维层状RGO/Cu2O材料的光催化性能及机理[J]. 化工新型材料, 2022, 50(10): 197-201,207. HUANG F P, ZOU Z B, LI C H, et al. Photocatalytic performance and mechanism of 2D layered RGO/Cu2O material[J]. New Chemical Materials, 2022, 50(10): 197-201,207 (in Chinese).
[9] 张亚兰, 贾相华, 郑友进, 等. 氧化亚铜微纳米颗粒的制备及光催化性能[J]. 牡丹江师范学院学报(自然科学版), 2017(1): 22-24. ZHANG Y L, JIA X H, ZHENG Y J, et al. The preparation of micro/nano cuprous oxide particles and photocatalytic performance[J]. Journal of Mudanjiang Normal University (Natural Sciences Edition), 2017(1): 22-24 (in Chinese).
[10] 蒋立君, 樊姗, 张新雨, 等. 简易合成Cu2Se/rGO水凝胶用于高性能锌离子混合电容器[J]. 广州化工, 2022, 50(21): 59-61,98. doi: 10.3969/j.issn.1001-9677.2022.21.019 JIANG L J, FAN S, ZHANG X Y, et al. A facile synthesis of Cu2Se/rGO hydrogel for high performance zinc-ion hybrid capacitors[J]. Guangzhou Chemical Industry, 2022, 50(21): 59-61,98 (in Chinese). doi: 10.3969/j.issn.1001-9677.2022.21.019
[11] EYVAZI B, JAMSHIDI-ZANJANI A, DARBAN A K. Synthesis of nano-magnetic MnFe2O4 to remove Cr(III) and Cr(Ⅵ) from aqueous solution: A comprehensive study[J]. Environmental Pollution, 2020, 265(Pt A): 113685. [12] FU Y, YAO Y F, FORSE A C, et al. Solvent-derived defects suppress adsorption in MOF-74[J]. Nature Communications, 2023, 14(1): 2386. doi: 10.1038/s41467-023-38155-8 [13] ZHANG X X, GUO X Z, CHEN S S, et al. A stable microporous framework with multiple accessible adsorption sites for high capacity adsorption and efficient separation of light hydrocarbons[J]. Chemical Engineering Journal, 2023, 466: 143170. doi: 10.1016/j.cej.2023.143170 [14] 饶品华, 张文启, 黄中子, 等. 磁性阳离子水凝胶的合成及其对六价铬的吸附去除[J]. 环境化学, 2011, 30(11): 1858-1863. RAO P H, ZHANG W Q, HUANG Z Z, et al. Synthesis of magnetic cationic hydrogel and its adsorption for chromium(Ⅵ)[J]. Environmental Chemistry, 2011, 30(11): 1858-1863 (in Chinese).
[15] LEI C, WANG C W, CHEN W Q, et al. Polyaniline@magnetic chitosan nanomaterials for highly efficient simultaneous adsorption and in situ chemical reduction of hexavalent chromium: Removal efficacy and mechanisms[J]. The Science of the Total Environment, 2020, 733: 139316. doi: 10.1016/j.scitotenv.2020.139316 [16] MOR S, RAVINDRA K, BISHNOI N R. Adsorption of chromium from aqueous solution by activated alumina and activated charcoal[J]. Bioresource Technology, 2007, 98(4): 954-957. doi: 10.1016/j.biortech.2006.03.018 [17] 赵妍, 陈升宇, 赵隆磊, 等. 绿色合成氧化亚铜/无纺布选择性去除阴离子染料[J]. 环境化学, 2023, 42(10)∶3534-3545. ZHAO Y, CHEN S Y, ZHAO L L, et al. Green synthesis of Cu2O/non-woven fabrics for selective removal of anionic dyes. Environmental Chemistry, 2023, 42(10): 3534-3545 (in Chinese).
[18] 李若男, 张国凤, 陈舜胜. 纤维素纳米纤维基吸附剂的制备及水中重金属的去除[J]. 化工环保, 2022, 42(2): 195-202. doi: 10.3969/j.issn.1006-1878.2022.02.012 LI R N, ZHANG G F, CHEN S S. Preparation of cellulose nanofiber-based adsorbent and removal of heavy metal from water[J]. Environmental Protection of Chemical Industry, 2022, 42(2): 195-202 (in Chinese). doi: 10.3969/j.issn.1006-1878.2022.02.012
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 905
- HTML全文浏览数: 905
- PDF下载数: 12
- 施引文献: 0
