

























-
氮氧化物(NOx)主要来源于化石燃料的燃烧,会形成酸雨,导致光化学烟雾,产生温室效应等[1 − 3],极大的威胁了人体健康和生态环境. 目前应用最为广泛的脱硝技术之一是选择性催化还原法(SCR),V2O5-WO3/TiO2催化剂应用较广泛,活性较高,活性温度窗口窄(300—400 °C),催化剂安装在电厂锅炉除尘和脱硫设备之前,烟道气中存在大量的飞灰和SO2,容易导致催化剂失活,降低催化剂的使用寿命,增加运行成本[4 − 5]. 而且,钒基催化剂毒性大,高温条件下易挥发性及其产生的会造成环境的二次污染. 因此,开发新型的可以置于脱硫和除尘设备之后的低温SCR催化剂已经成为烟气脱硝研究的热点.
近年来,锰基氧化物因其在低温下去除NO的高活性和高选择性得到了广泛的研究[6 − 8]. 然而MnOx比表面积较小,催化剂结构不稳定,并且易被SO2和H2O等中毒,因而在实际应用中不适宜单独使用. Baidya等[9 − 10]研究表明,Ce-ZrO2载体是三效催化剂中的常用载体,铈具有优异的储氧能力和较好的氧化还原性质,铈的掺杂增大了催化剂表面活性氧浓度,从而增强催化剂对NH3的吸附能力,并且能减少催化剂在煅烧过程中比表面积的损失,提高催化剂的低温催化活性. Shen等[11]的研究成果指出铈可以改善催化剂抗SO2的性能. Kambur等[12]认为锆不但氧离子传导能力强,而且能改善催化剂的热稳定性和机械性能,因此广泛应用于催化行业. 另外,Liu等 [13]研究表明,锆铈的共掺杂显著提高了晶格氧的传导能力,进而提高对NO的催化能力. 相关文献[14 − 15]表明Ce-ZrO2载体在三效催化剂系统中取得了良好的效果. 因此,本文将Mn负载在Ce-ZrO2载体上,研究其低温SCR催化活性.
低温SCR催化剂可布置在脱硫设备之后,但脱硫后烟气中存在的碱土金属(Mg、Ca)仍会造成催化剂的中毒. Yang等[16]研究成果表明,碱土金属易沉积在催化剂表面,堵塞孔道,从而影响SCR的性能. 南开大学张晓鹏[17]对烟气中H2O和SO2对低温催化剂的毒化作用、机理、影响程度进行了研究,Mn/Ce-ZrO2低温催化剂具有很好的抗H2O和SO2性能. 杨志琴[18]对以纳米ZrO2为载体的低温SCR催化剂的制备及性能进行了研究,研究结果表明,8%Fe-10%MnOx-CeO2-/ZrO2催化剂具有良好的低温活性和热稳定性,可用于SO2含量较低的烟气的低温脱硝处理,研究结果也为以纳米ZrO2为载体的SCR催化剂的工业化应用奠定了实验研究基础和理论依据. 目前关于碱土金属对低温SCR催化剂中毒特性的研究报道还较少,因此开展此方面的研究具有重要的现实意义[19].
因此,本文利用模拟中毒法使MnOx/Ce-ZrO2中毒,通过活性测试和N2-BET,XRD, H2-TPR,NH3-TPD,XPS等表征测试研究了Mg对催化剂性能的影响,同时考察了再生实验,探究其再生机理,旨在为催化剂中毒和再生的应用提供理论基础.
-
Ce-ZrO2载体的制备:室温条件下,将定量的Ce(NO3)2和Zr(NO3)2(摩尔比Ce:Zr=9:1)溶解于去离子水中,磁力搅拌条件下,缓慢的滴入 NH3·H2O溶液(3 mol·L−1)至pH 为11,继续搅拌2 h,静置1h,随后将得到沉淀物用去离子水反复过滤和洗涤直至pH值接近7,并在120°C下干燥12 h,最后将其在500 °C下焙烧5 h,得到Ce-ZrO2载体.
MnOx/Ce-ZrO2催化剂的制备:将计算好的Mn(NO3)2溶液浸渍在已制备的Ce-ZrO2载体上(Mn/(Ce+Zr)的摩尔比为0.4:1),静置4 h后,将其在120 °C下干燥12 h,最后在500°C下焙烧5 h,即得到MnOx/Ce-ZrO2催化剂(记为MCZ).
-
中毒催化剂制备:将MCZ平均分成四份,将不同量的Mg(NO3)2溶液分别浸渍于其中3份(Mg与Mn的摩尔比分别设为0.1、0.2、0.5),静置4 h后,将其在120°C下干燥12 h,最后在500 °C下焙烧5 h,即得到不同Mg负载比例的中毒催化剂,分别记为Mg0.1-MCZ、 Mg0.2-MCZ、Mg0.5-MCZ.
-
选取中毒催化剂(Mg0.2-MCZ)进行再生研究,将Mg0.2-MCZ置于0.5 mol·L−1的H2SO4溶液中浸泡3 h,去离子水洗涤pH至中性,过滤后于100 °C烘箱内干燥12 h,得到的材料记为ZSMg0.2-MCZ.
-
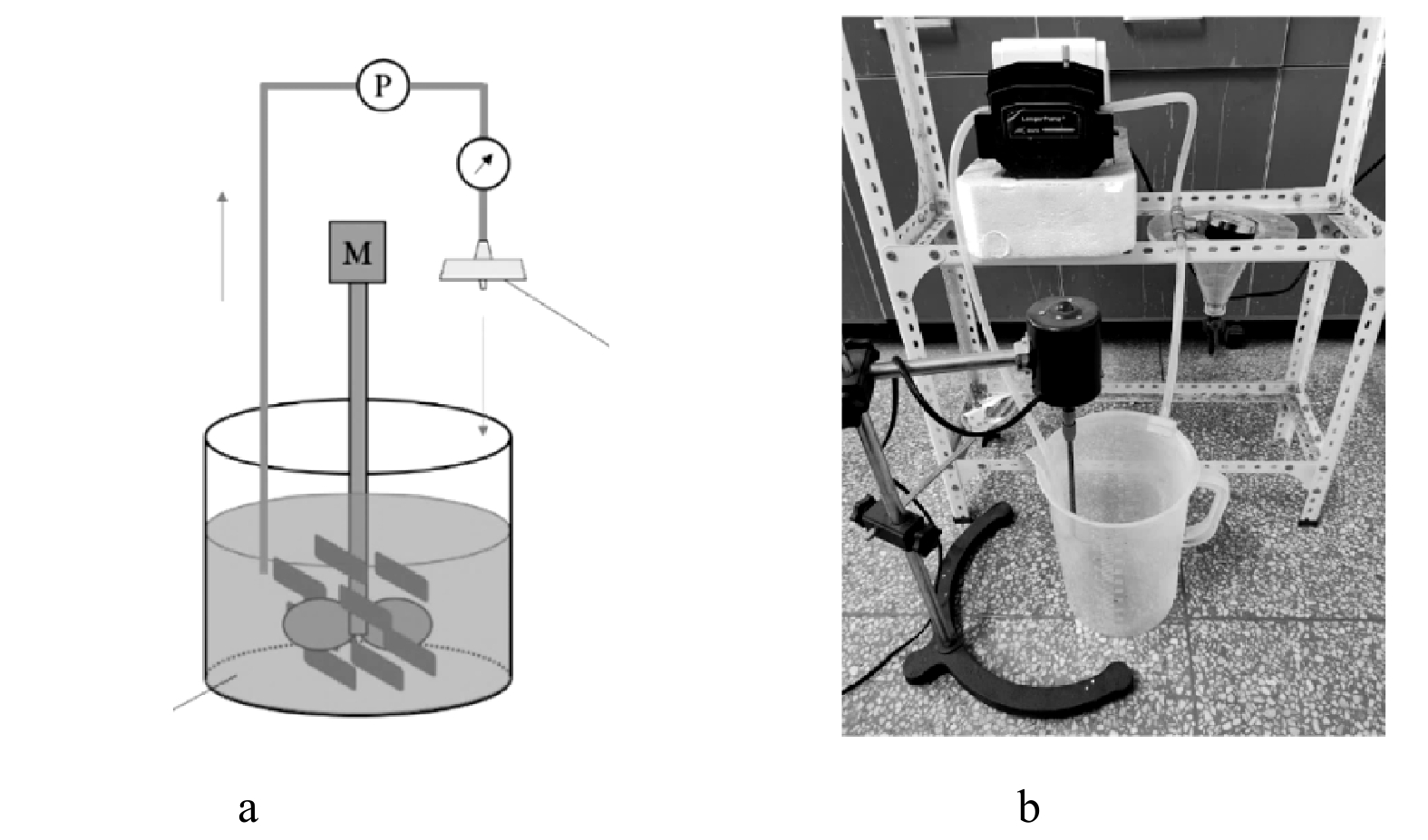
SCR脱硝实验反应在固定床微反应器中进行(图1),选取催化剂粒径60目至100目以防止气路的堵塞。实验的工况条件:反应温度范围为100—220°C;模拟烟气组成为NO为600 cm3·m−3, NH3为600 cm3·m−3,O2为5%,背景气、平衡气均为氩气;烟气流速为1 L·min−1,空速比为
108000 h−1. 为进一步排除催化剂吸附的影响,实验开始前,将催化剂放入反应器中,通入NO气体,确保反应体系中NO浓度均匀不变.采用烟气分析仪(Model 60i,德国赛默飞世尔)测定反应过程前后NO的含量来计算NO转化率,其公式为:
式中,
ηNO 表示NO转化率,Cin 表示进口NO浓度,Cout 表示出口NO浓度. -
催化剂的比表面积、孔体积和平均孔径测定采用美国康塔公司Autosorb-1型物理吸附仪;催化剂晶相结构采用日本理学D/max 2550PC型衍射仪测定;H2-TPR,NH3-TPD测试采用美国康塔公司ChemBET TPR/TPD型化学吸附仪;XPS分析采用英国KRATOS公司XSAM800型电子能谱仪,以Al-Kα作为激发源.
-
表1为4种催化剂的比表面积和粒径分布,由表1可知,随着Mg负载量的提高,催化剂的比表面积明显减小,总孔体积也有轻微下降. 研究认为[20],由于碱土金属的沉积,能堵塞催化剂的孔道和降低其比表面积,影响催化反应的传质过程和催化剂对反应气体的吸附和活化能力,从而降低NOx-SCR脱硝转化率. 此外,添加Mg会导致催化剂平均孔径微小波动,但无明显的规律.
-
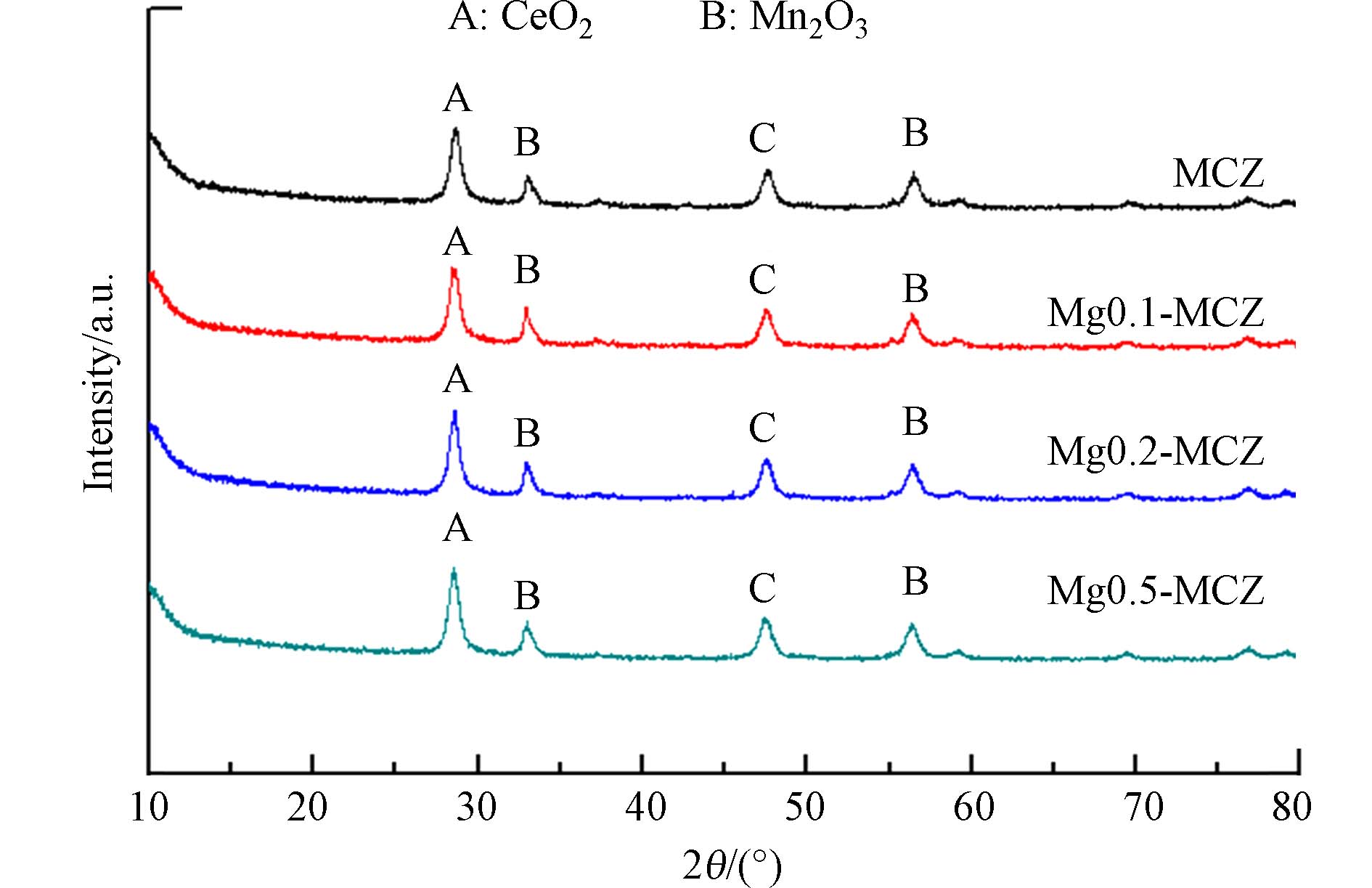
催化剂的晶型结构在某种程度上决定了其催化性能,图2为4种催化剂的XRD衍射图. 4个样品中均只发现CeO2和Mn2O3两种衍射峰,但未检测出ZrO2的衍射峰,可能的原因是ZrO2均匀分散在催化剂中,且分散度较高,另一个可能的原因是在催化剂合成中Zr插入了CeO2晶格中. 3种催化剂中Mn2O3的衍射峰均较弱,说明锰在3种催化剂中的结晶度较低,分散度较高. 研究表明,高分散的锰能提高氧空穴的数量,能促进催化活性[21]. 另外,图2中未检测到Mg及其相关物质的衍射峰,表明Mg的添加不影响催化剂的晶型,也进一步说明了低温SCR催化剂的碱金属中毒并不是催化剂的晶型变化导致的[22].
-
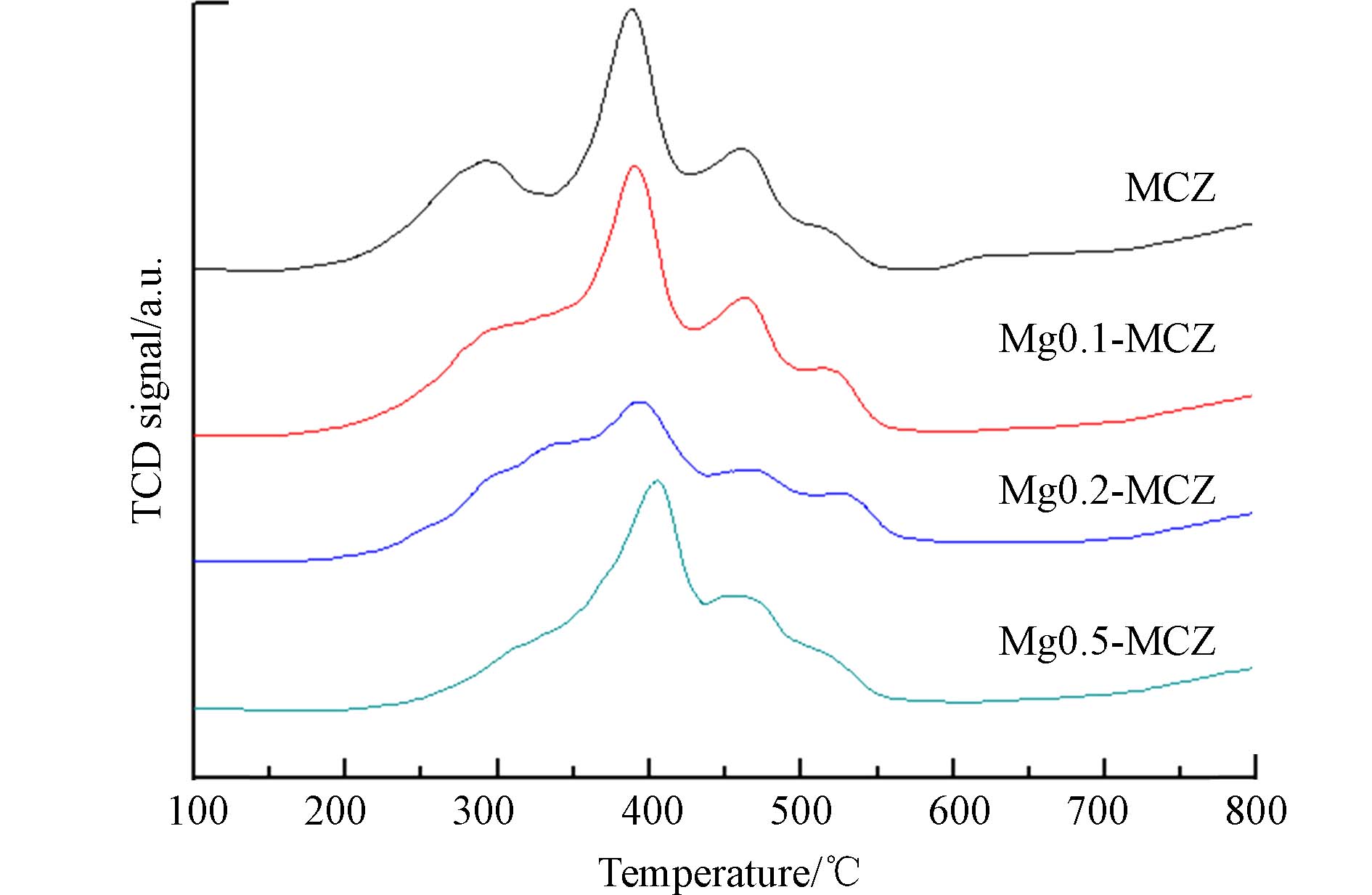
采用H2-TPR技术考察Mg对催化剂表面氧化还原行为的影响. 图3为4种催化剂的H2-TPR谱图. 可以看出MCZ出现了4个还原峰,280 °C左右的还原峰可归属于MnO2到Mn2O3的转化,385 °C和460 °C左右的还原峰可分别归属于Mn2O3到Mn3O4和Mn3O4到MnO的转化. 此外,510 °C左右的还原峰可认为是体相吸附氧的还原[23]. 一般认为,催化剂还原温度越低,催化剂在SCR反应中的还原能力越强. 而随着Mg负载量的提高,催化剂的还原峰逐渐向高温方向移动,说明Mg的添加能够提高MnOx的稳定性而使其还原能力减弱. 并且当Mg负载量为0.5时,第一个和最后一个还原峰已基本消失,表明处于这两个温度的氧化还原反应也已基本停止,进一步验证了碱土金属对催化剂表面活性成分的氧化还原性能的影响.
-
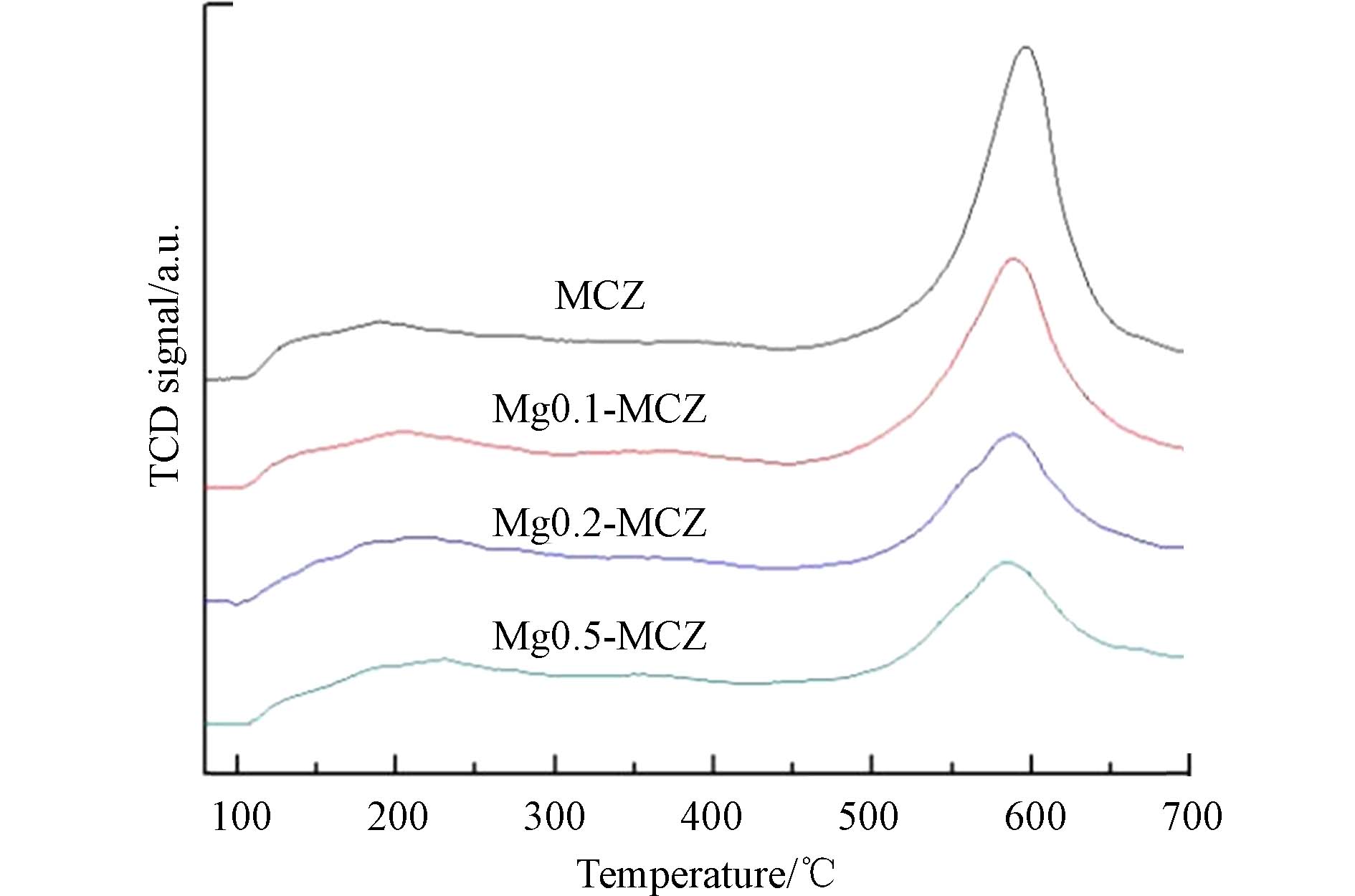
采用NH3-TPD分析了表面酸位酸量结果. 如图4所示,4种催化剂均存在两个明显的脱附峰,位于100—400 °C的峰,反映了催化剂表面弱酸的分布,与物理吸附在催化剂表面的NH3和与表面Brönsted酸结合形成的NH4+的脱附行为相关;位于500—650 °C的峰,反映了催化剂表面强酸的分布,与吸附在催化剂Lewis酸酸位上NH3的脱附行为相关[24]. 随着Mg负载比例的增加,低温段的Brönsted酸位没有发生明显的变化,而高温段的Lewis酸位酸量则越来越低,从而降低了其对NH3的吸附能力,造成催化剂活性的下降. 另外,3种中毒催化剂的吸附峰形状相似,表明Mg的添加没有形成新的酸位点.
-
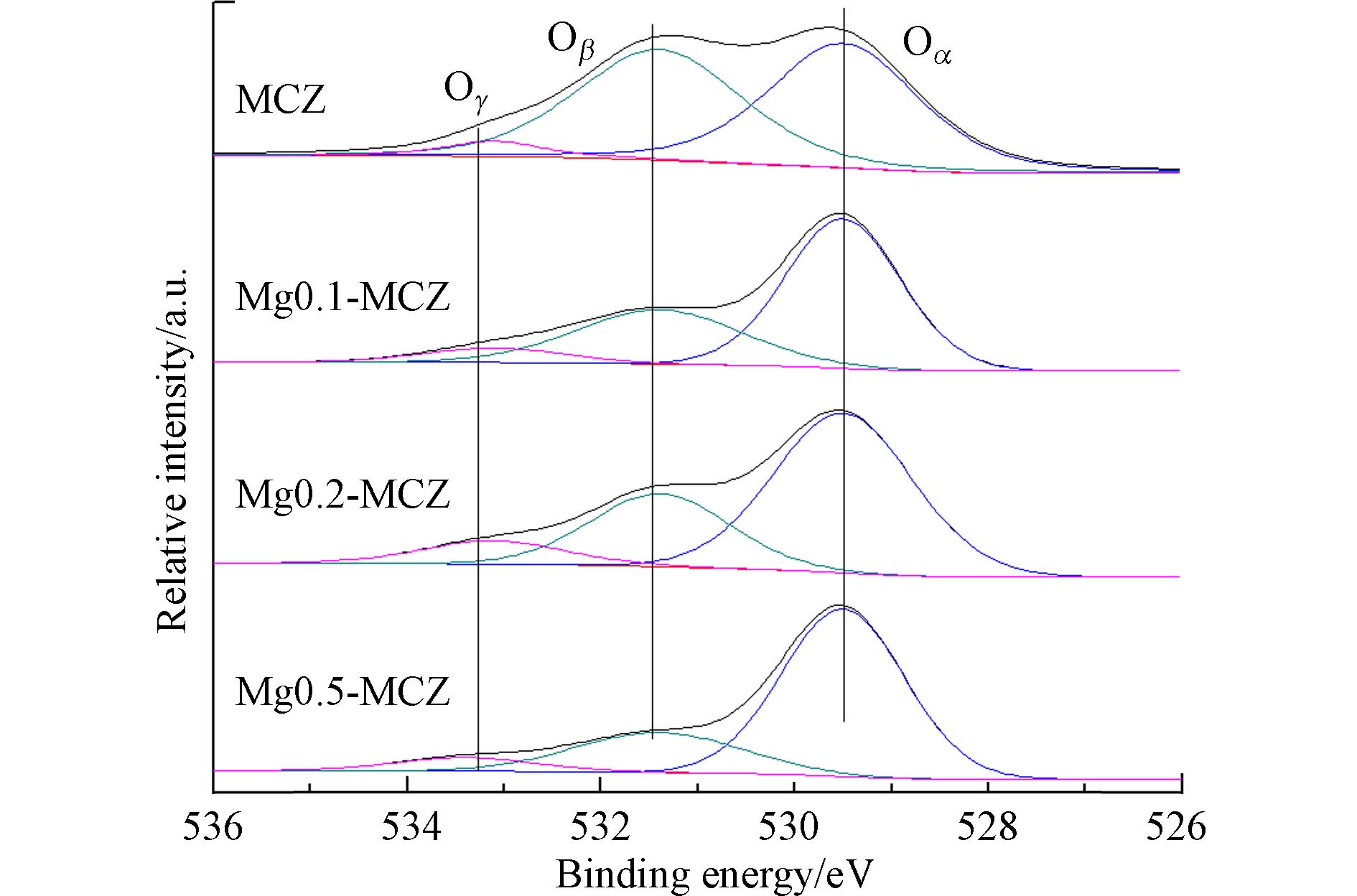
采用XPS分析了Mg中毒对催化剂表面状态和元素价态. 如图5所示,对于O1s,结合能在529.0—530.0 eV(记为Oα)处的晶格氧,结合能在531.3—531.9 eV(记为Oβ)处的化学吸附氧以及结合能在532.7—533.5 eV(记为Oγ)处的羟基氧[25]. 催化剂表面上化学吸附氧能够形成强氧化性的自由基,可以置换空气中的氧和吸附在催化剂上的分子,强化NH3等物质在催化剂表面的吸附和氧化[25 − 26]. 因此化学吸附氧的增加积极影响了SCR反应. 表2为分峰拟合结果得出的两种氧原子的比值,可以看出,随着Mg负载量的增加,表面化学吸附氧含量逐渐降低,主要是由于MgO与氧形成对催化剂表面氧空位的竞争,在催化剂表面的氧空位形成强吸附,使化学吸附氧峰降低,因此催化剂的氧化还原能力逐渐减弱,导致催化剂活性逐渐降低.
-
图6为新鲜催化剂MCZ及负载不同Mg含量后的催化剂的脱硝效率比较. 由图6可知,MCZ的脱硝效率随温度升高而升高,且在160—220 °C之间保持90%以上的脱硝效率,在200 °C时,效率最高达到了97%. 碱土金属Mg的添加使脱硝效率明显降低,在Mg负载量为0.1时,对催化剂的中毒作用较小,在200 °C时仍能达到95.4%的脱除效率. 而随着Mg负载量的继续增加,效率降低则越来越明显,当Mg负载量为0.5时,催化剂的最高效率也只有47.6%,说明Mg对催化剂产生了严重的中毒作用.
-
Mg负载量为0.2的催化剂酸洗前后比表面积和粒径分布的差异如表3所示. 由表3可知,经过酸洗后催化剂比表面积上升明显,达到了50.835 m2·g−1,已经接近于新鲜催化剂水平,这表明酸洗使中毒催化剂微孔及表面的碱土金属和其它微小的颗粒物被清洗下来,有效的恢复了催化剂的比表面积,利于催化反应的进行. 另外,总孔体积和平均孔径也均有小幅度的增长,说明酸洗后中毒催化剂表层上的颗粒变得更小且分散均匀,甚至较新鲜催化剂表层的颗粒更小且分散更均匀,颗粒的均匀也使得微孔变的更多,从而降低了催化反应过程中产生的传质阻力,有利于催化活性的恢复.
-
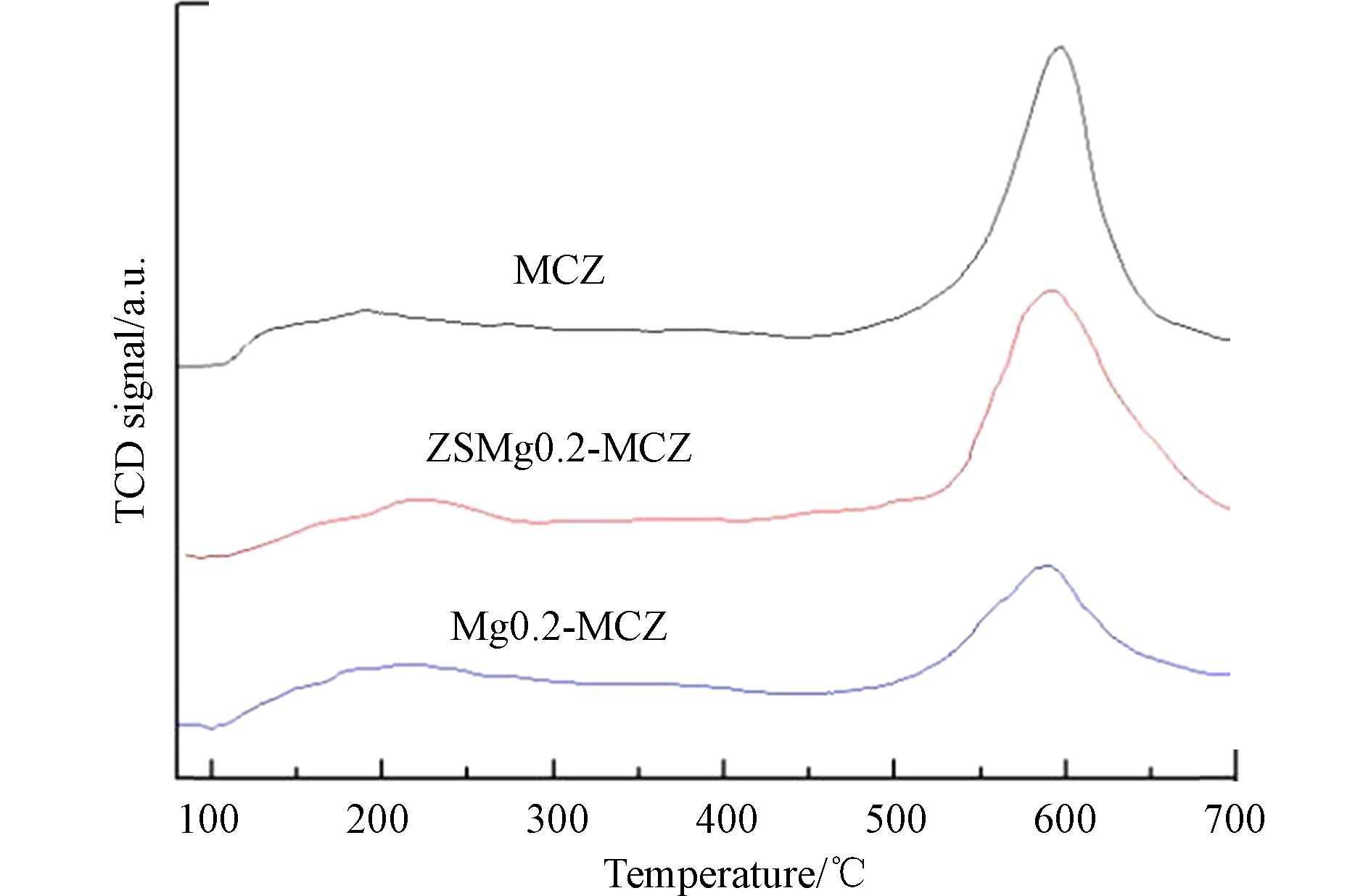
中毒催化剂酸洗再生后的NH3-TPD对比谱图如图7所示,可以看出酸洗后中毒催化剂没有形成新的酸位点,NH3脱附峰面积明显增大,具体表现在低温段的Brönsted酸位没有发生明显的变化,而高温段的Lewis酸位酸量明显增加,进一步结合表4计算出的催化剂表面酸度分析,再生催化剂的表面酸度已经接近于新鲜催化剂. 对于硫酸再生处理催化剂,一般认为经酸洗再生后催化剂表面形成 SO42-基团,有利于对NH3的吸附,根据NOx-SCR反应机理,NH3在催化剂表面的吸附是NOx-SCR反映进行的决定步骤,因此催化剂表面酸位点的恢复对脱硝活性的提高有促进作用.
-
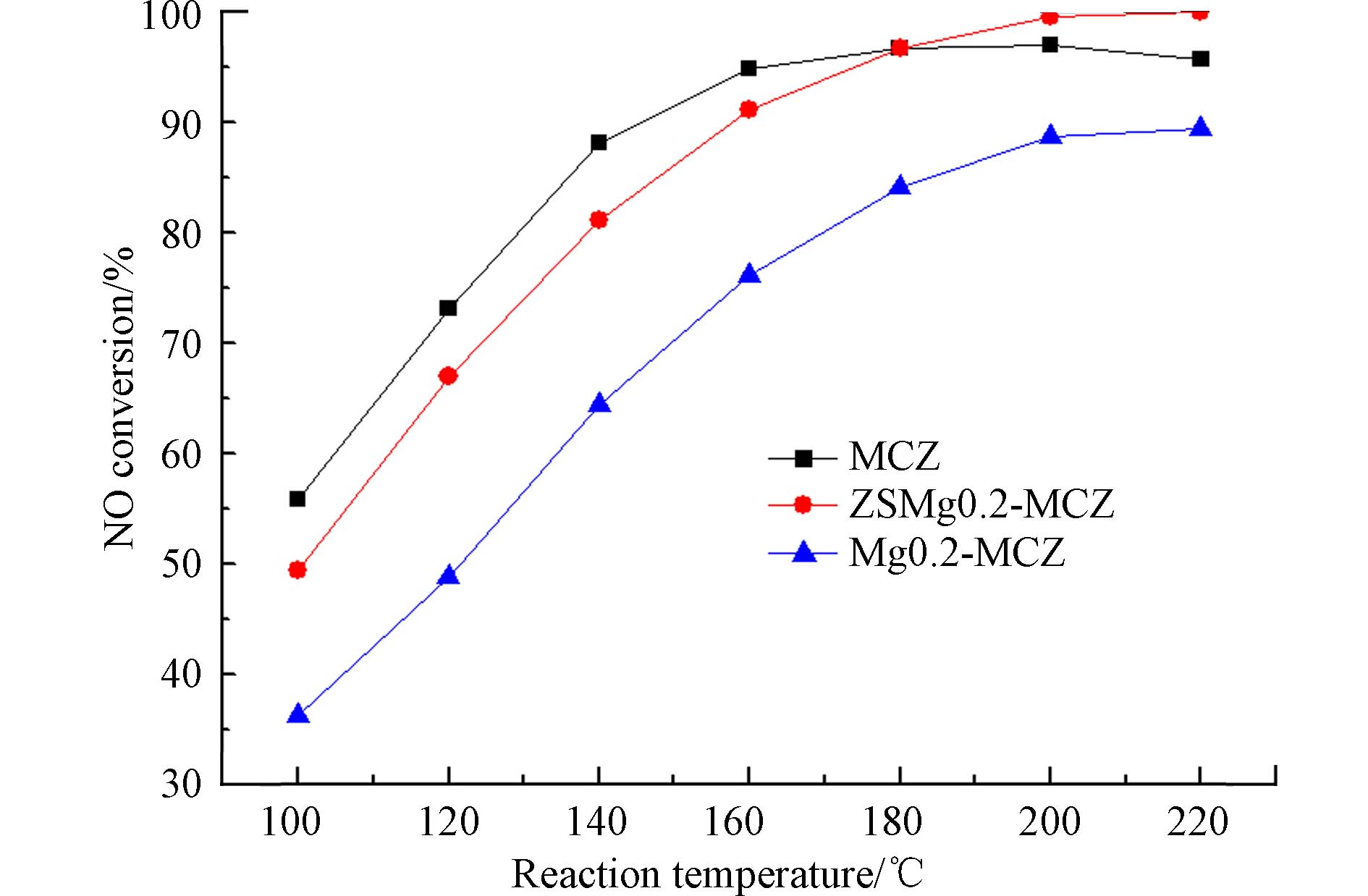
中毒催化剂酸洗再生后的脱硝活性对比曲线如图8所示. 可以看出,中毒催化剂经0.5 mol·L−1的H2SO4溶液酸洗后,极大程度的恢复了脱硝活性,在100—140 °C范围内,脱硝效率比新鲜催化剂低10%左右,160 °C之后与新鲜催化剂的脱硝效率差异逐渐减小,在180 °C时脱硝活性完全恢复,随着温度的继续升高,催化剂的脱硝效率甚至超过了新鲜催化剂,在220 °C时脱硝效率达到了100%. 结合表征分析可得出,酸洗可以去除中毒催化剂表面及孔洞内的碱土金属,增加催化剂的酸性,而且能够在催化剂表面形成新的硫酸盐组分,与吸附的水相互作用生成Lewis酸性位点,增加NH3的吸附量,从而提高脱硝效率.
-
(1)MnOx/Ce-ZrO2低温SCR催化剂具有较宽的活性窗口,在160—220 °C之间能保持90%以上的脱硝效率,在200 °C时,效率最高达到了97%.
(2)碱土金属Mg的添加会导致MnOx/Ce-ZrO2催化剂中毒,且负载量越大,催化活性越低,中毒程度越深. BET、XRD、H2-TPR、NH3-TPD、XPS等表征结果表明,虽然Mg中毒后的催化剂晶型未发生变化,但其比表面积和Lewis酸位酸量降低、氧化还原能力减弱、表面吸附氧浓度减少,这些都导致了催化剂的失活.
(3)对Mg负载量为0.2的催化剂的再生实验表明,酸洗后极大程度地恢复了脱硝活性,在180 ℃之后催化剂的脱硝效率甚至超过了新鲜催化剂,在220 °C时脱硝效率达到了100%. BET、NH3-TPD表征结果显示酸洗之后中毒催化剂的比表面积、表面酸度和化学吸附氧浓度基本恢复到新鲜催化剂的水平,说明酸洗可以去除中毒催化剂表面及孔洞内的碱土金属,增加催化剂的酸性,而且能够在催化剂表面形成新的硫酸盐组分,与吸附的水相互作用生成Lewis酸性位点,增加NH3的吸附量,从而提高脱硝效率. 这是催化剂活性恢复的主要原因.
(4)本文通过对中毒催化剂的酸洗再生进行初步的研究和表征机理分析,为催化剂中毒和再生处理提供了一定的理论基础,对延长催化剂的使用寿命,降低生产成本,避免环境污染等具有重要的现实意义.
MnOx/Ce-ZrO2低温SCR催化剂的镁中毒及再生实验
Experimental study on magnesium poisoning and regeneration of MnOx / Ce-ZrO2 low-temperature SCR catalyst
-
摘要: 锰基氧化物在低温下去除NO的高活性和高选择性得到了较多的研究,MnOx/Ce-ZrO2低温催化剂具有很好的抗H2O和SO2性能,但烟气中存在的碱土金属(Mg、Ca)会造成催化剂的中毒. 为了深入探究碱土金属(Mg)对催化剂性能的影响和中毒催化剂的再生机理,并进一步对中毒催化剂进行再生实验,探究其再生机理,采用共沉淀法制备了MnOx/Ce-ZrO2催化剂,并用浸渍法模拟不同负载量的碱土金属Mg对催化剂中毒程度的影响. 结果表明,Mg的添加会导致催化剂中毒,且负载量越大,中毒程度越深. 运用BET、XRD、H2-TPR、NH3-TPD、XPS等手段对新鲜催化剂和中毒催化剂进行了表征. 结果表明,虽然Mg中毒后的催化剂晶型未发生变化,但其比表面积和Lewis酸位酸量降低、氧化还原能力减弱、表面吸附氧浓度减少,导致催化剂的失活. 对Mg负载量为0.2的催化剂的再生实验表明,酸洗后脱硝活性有了很大程度的恢复,在180 ℃之后甚至超过了新鲜催化剂的效率. BET和NH3-TPD结果表明,酸洗之后的中毒催化剂的比表面积和表面酸度基本恢复到新鲜催化剂的水平,这是催化剂活性恢复的主要原因.Abstract: The high activity and high selectivity of manganese-based oxides in removing NO at low temperature have been studied. MnOx/Ce-ZrO2 low temperature catalyst has good anti-H2O and SO2 performance, but the alkali soil metals (Mg, Ca) existing in flue gas will cause the poisoning of catalyst.In order to further explore the influence of alkali soil metal (Mg) on the performance of catalyst, and the regeneration mechanism of poisoning catalyst, and further explore the regeneration mechanism, MnOx/Ce-ZrO2 catalyst was prepared by precipitation method, and the influence of alkali soil metal Mg on the degree of poisoning.The results showed that the addition of Mg can cause catalyst poisoning, and the deeper the load is. BET, XRD, by other means, H2-TPR, NH3-TPD, XPS and so on. The results showed that although the crystal form of the catalyst did not change after Mg poisoning, its specific surface area and Lewis acid amount, reduced redox capacity, and reduced surface adsorbed oxygen concentration led to the inactivation of the catalyst. Regeneration experiments with the catalyst with a Mg load of 0.2 showed a substantial recovery of denitrification activity after acid wash, even exceeding the efficiency of the fresh catalyst after 180℃. BET and NH3-TPD results show that the specific surface area and surface acidity of the poisoned catalyst are basically restored to the level of fresh catalyst, which is the main reason for the recovery of catalyst activity.
-
Key words:
- SCR catalyst /
- deactivation /
- poisoning /
- regeneration /
- alkaline-earth metals
-
氨氮一般指水中以游离氨(
NH3 NH+4 废水脱氨主要方法有生物法、化学沉淀法、吹脱法、吸附法[5]。但是生物法流程长,反应器大,占地多,常需要额外投加碳源,能耗大,成本高;化学沉淀法成本高,再生难,有二次污染;吹脱法能耗大,有二次污染,出水氨氮浓度仍然偏高。吸附法具有除污效率高、操作便捷、适用范围广、吸附剂可重复使用等优点,在氨氮废水处理中逐渐得到了广泛关注。ALSHAMERI等[6]对比了高岭石、埃洛石、蒙脱石、海泡石等六种天然粘土矿物对
NH+4 NH+4 目前,大部分吸附材料多以微纳米颗粒的形式存在,直接与废水混合能够充分利用其吸附性能,但也存在微纳米材料使用中流失和回收困难等问题。通过微滤或超滤膜辅助回收,将会增加额外能耗和成本,影响其进一步的推广应用。也有研究将吸附材料制备成毫米级微球,通过柱吸附实现污染物去除,但大颗粒吸附材料吸附容量低于微纳米颗粒[7]。因此,如何既能保证吸附容量,又能实现微尺度吸附材料的低碳回收,已经成为吸附法应用中的新困难。动态膜技术具有成本低、操作压力小、清洗简单、微颗粒分离效果好等优点,为微尺度颗粒材料回收提供了绿色低碳新方法[8]。但目前多数研究围绕在动态膜系统过滤特性和应用效能,沸石吸附脱氨效能和优化改性,鲜有对动态膜成膜材料吸附性能的拓展和深度耦合探索,及对动态膜系统和成膜材料作用的独立解析,考虑到沸石吸附脱氨和动态膜分离的优点和不足,本研究尝试构建吸附与动态膜耦合系统,同步实现废水中氨氮去除和颗粒吸附材料回收,从而为吸附法进一步走向工程应用提供新方案。
本研究围绕吸附脱氨和动态膜回收2个方面,通过静态吸附实验,系统探究沸石吸附脱氨效能和投加量、初始氨氮浓度对脱氨效能的影响,揭示沸石吸附氨氮的动力学特性,阐明吸附-动态膜系统的脱氨效果和成膜特征,为沸石吸附脱氨和沸石颗粒回收的进一步应用提供参考。
1. 材料与方法
1.1 材料和实验用水
主要使用的实验材料包括:74~150 μm沸石,硅藻土,0.45 μm微孔滤膜,38 μm尼龙网,压力计等。实验所需试剂主要有氯化铵、酒石酸钾钠、纳氏试剂,均为分析纯。
在容量瓶中按照标准配置1 000 mg·L−1的氨氮储备溶液,配置完成后将溶液移入细口试剂瓶待用;使用时,按需求量取一定体积的氨氮储备溶液,按比例稀释为10 mg·L−1氨氮模拟废水。氨氮测定方法参考《纳氏试剂分光光度法》HJ 535-2009。
1.2 实验装置
为了保证同步进行氨氮去除和吸附材料的回收,使用自制循环吸附-动态膜系统进行成膜研究。循环吸附-动态膜系统和实验装置见图1,该装置由反应池、蠕动泵、自制膜组件、压力计、电动搅拌器组成。实验过程中,反应池中加入2 L的模拟废水及吸附剂,搅拌加速混合,通过蠕动泵将混合液送入自制膜组件,在膜组件中截留吸附剂并形成动态膜,膜分离后的混合液回流至反应池中。
1.3 动力学实验
称取0.4 g沸石移至150 mL锥形瓶中,取1 mL氨储备溶液于100 mL容量瓶中定容,配制模拟氨氮污水,初始质量浓度为10 mg·L−1,再分别转入锥形瓶;将锥形瓶同时放入恒温摇床中振荡(25 ℃,180 r min−1),反应进行至5、10、30 min和1、3、6、12、24 h时取出并取样7 mL左右于10 mL离心管中待滤;经0.45 μm滤膜过滤后测量滤液中氨氮浓度,进行准一级与准二级动力学拟合。
1.4 吸附等温实验
分别称取0.4 g沸石移至锥形瓶中,分别取1、2、4、6、8、10 mL氨储备溶液于100 mL容量瓶中定容,配制10、20、40、60、80、100 mg·L−1的模拟氨氮废水,按顺序移入锥形瓶中;将锥形瓶同时放入恒温摇床中振荡(25 ℃,180 r min−1),反应进行至5、10、30 min和1、3、6、12、24 h时取出并取样7 mL;经0.45 μm滤膜过滤后测氨氮浓度,计算去除率与吸附量,进行Langmuir与Freundlich拟合。
2. 结果与讨论
2.1 沸石吸附脱氨性能及吸附特性
1)沸石投加量对氨氮去除的影响。沸石投加量对氨氮的去除率及吸附量的影响结果如图2所示。可见,在100 mL体系中、25 ℃、180 r min−1恒温振荡箱和初始氨氮质量浓度10 mg·L−1反应条件下,沸石对模拟废水中氨氮的去除率随其投加量的增加逐渐增加,在投加量为10 g·L−1时氨氮去除率可到达67%左右,而当沸石投加量从1 g·L−1增加到10 g·L−1时,吸附量由1.48 mg·g−1降至0.64 mg·g−1。这是因为随着沸石投加量的增加,沸石可以提供更多的吸附位点和更大的比表面积,因此,氨氮的去除率随之增加,但吸附剂投加量的增加也会造成部分吸附位点被覆盖,吸附位点之间产生竞争,故单位质量的沸石的吸附效率随着吸附剂投加量的增大而逐渐降低。在低氨氮质量浓度为10 mg·L−1下开展的投加量优化实验中,沸石对氨氮去除效果较好。这是因为沸石是一种骨架状铝硅酸盐,具有离子交换的特性,对于
NH+4 NH+4 NH+4 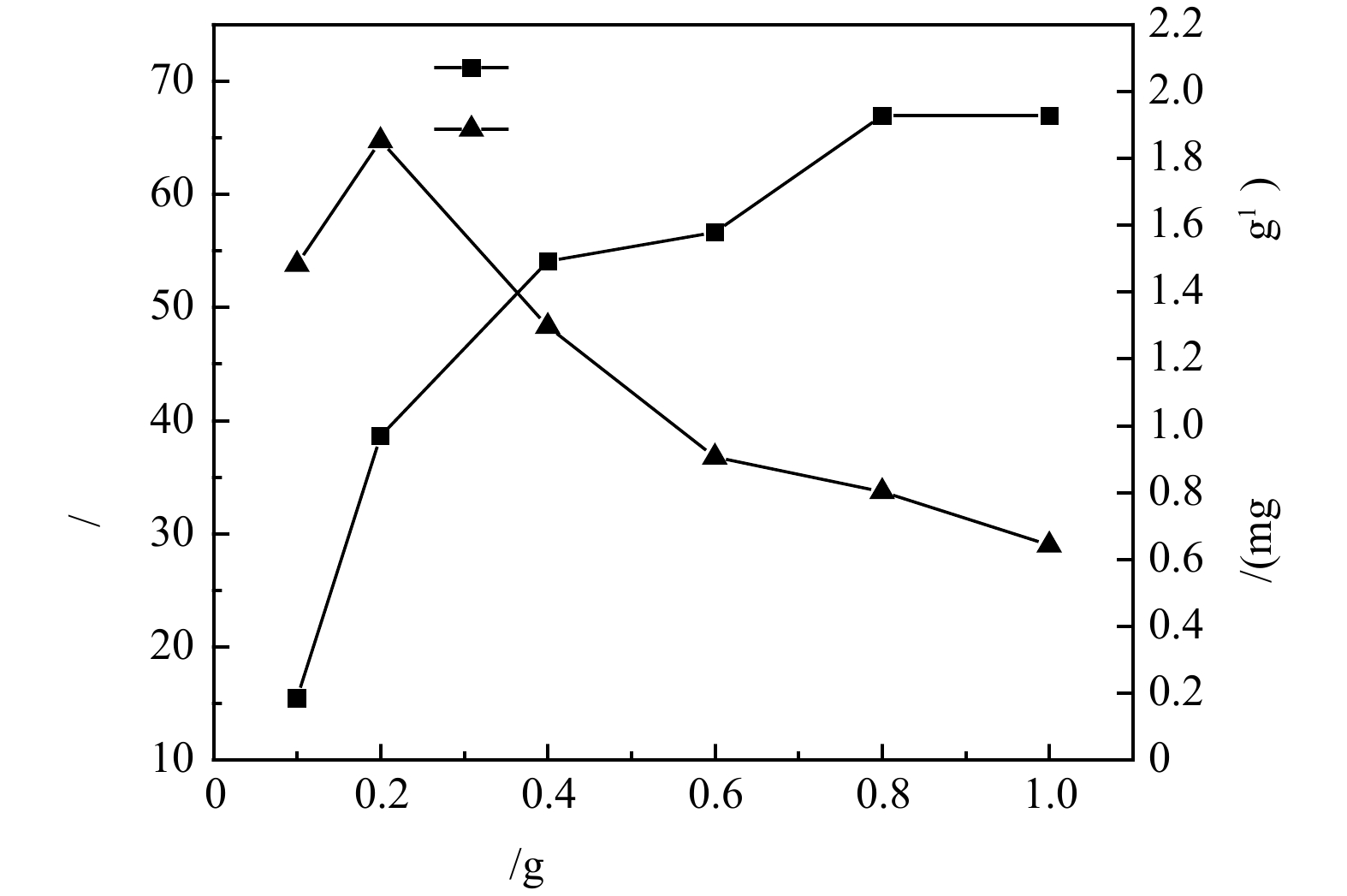 图 2 氨氮去除率和吸附量随沸石投加量的变化规律Figure 2. Variations of the removal rate and adsorption capacity of ammonia with zeolite dosage
图 2 氨氮去除率和吸附量随沸石投加量的变化规律Figure 2. Variations of the removal rate and adsorption capacity of ammonia with zeolite dosage2)初始氨氮浓度对沸石脱氨效果的影响。沸石对氨氮的去除率及吸附量随初始氨氮浓度变化规律如图3 所示。可见,沸石对氨氮的去除率随氨氮初始浓度的增加而下降,而吸附量却随之增加,当沸石投加量为4 g·L−1,初始氨氮质量浓度为100 mg·L−1时,沸石对氨氮吸附量可提升到3.89 mg·g−1,但氨氮去除率也降至17%。随着氨氮浓度的增加,体系中的
NH+4 NH+4  图 3 氨氮去除率和吸附量随氨氮初始浓度的变化规律Figure 3. Variations of the removal rate and adsorption capacity of ammonia with initial ammonia concentration.
图 3 氨氮去除率和吸附量随氨氮初始浓度的变化规律Figure 3. Variations of the removal rate and adsorption capacity of ammonia with initial ammonia concentration.3)沸石对氨氮的吸附动力学研究。采用准一级和准二级动力学模型,对沸石投加量为4 g·L−1,初始氨氮质量浓度为10 mg·L−1的24 h吸附动力学实验数据进行拟合分析。由图4可以看出,在吸附开始的1 h内,沸石对氨氮吸附速率较快,1 h后吸附量缓慢上升,12 h吸附趋于平衡,平衡吸附量在1.3 mg·g−1左右。在吸附初期,沸石颗粒上吸附位点较多,氨氮浓度高,吸附较快,而随着吸附进行,氨氮浓度降低,吸附位点被占用,吸附速率下降。此外,沸石的表面物理吸附及
NH+4 NH+4 qt t 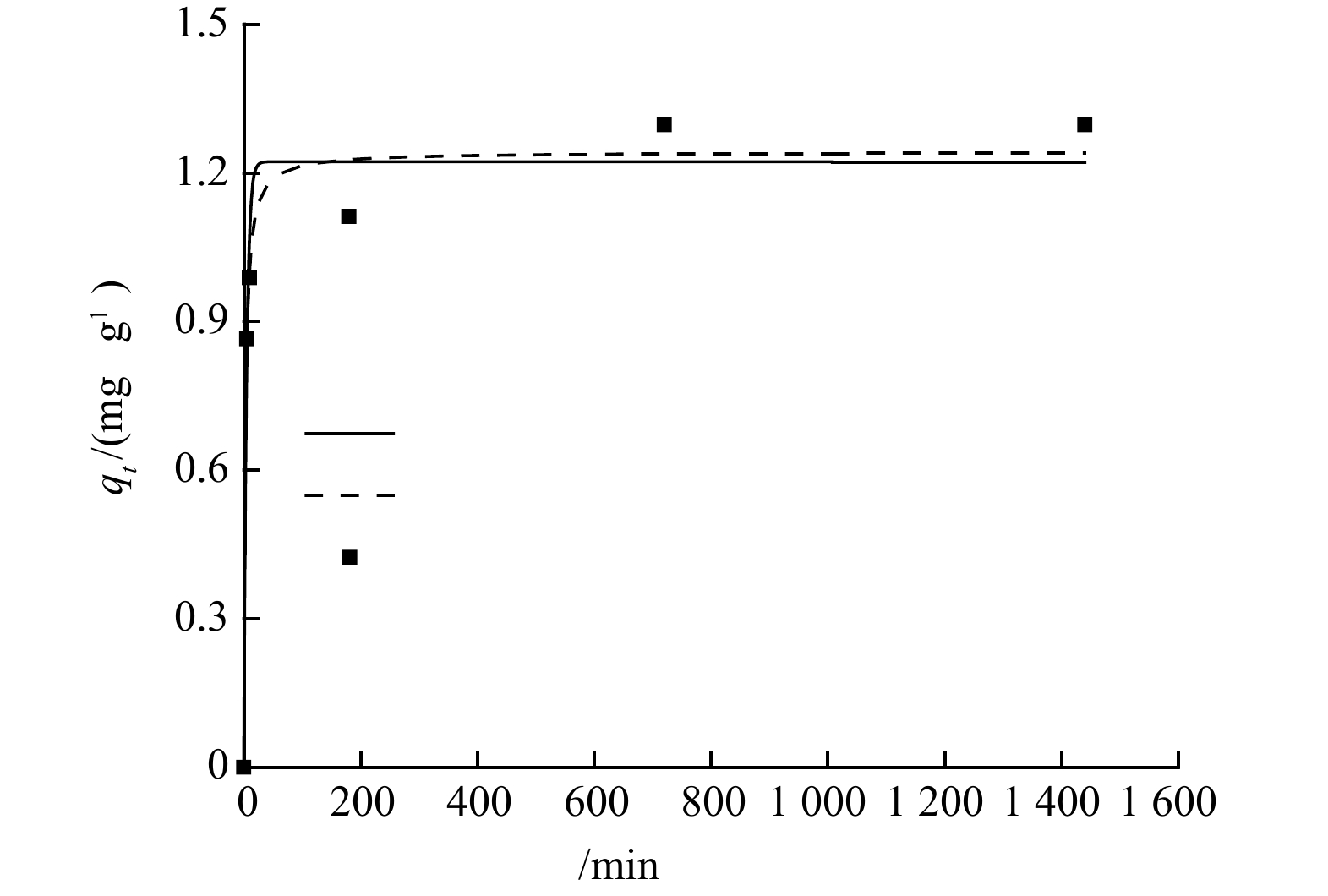 图 4 沸石吸附氨氮的准一级与准二级动力学模型拟合曲线Figure 4. Pseudo-first-order model and pseudo-second-order model curves for ammonia adsorption onto zeolite表 1 准一级与准二级动力学方程拟合参数及相关系数Table 1. Fitting parameters and correlation coefficients of pseudo-first order kinetics and pseudo second order kinetics
图 4 沸石吸附氨氮的准一级与准二级动力学模型拟合曲线Figure 4. Pseudo-first-order model and pseudo-second-order model curves for ammonia adsorption onto zeolite表 1 准一级与准二级动力学方程拟合参数及相关系数Table 1. Fitting parameters and correlation coefficients of pseudo-first order kinetics and pseudo second order kineticsqe,exp /(mg·g−1) 准一级动力学 准二级动力学 qe,celc/(mg·g−1) k1/min−1 R2 qe,celc/(mg·g−1) k2/(g·(mg·min)−1) R2 1.30 1.22 0.21 0.969 1.24 0.34 0.982 | Show Table DownLoad:
CSV
DownLoad:
CSV
4)沸石对氨氮的吸附等温线。在25 ℃的条件下,测定4 g·L−1沸石在不同初始氨氮浓度下的平衡吸附量,进行吸附等温研究,结果见图5。吸附等温模型是用于描述在特定实验温度下,吸附剂与吸附质之间的相互作用[13]。本研究使用Langmuir和Freundlich模型(表2) 对数据进行分析,以准确地研究氨氮在沸石上的吸附机理。首先分别以
1/qe 1/Ce lgqe lgCe n > 1 0.1 < 1/n < 0.5 1/n 1/n > 2 1/n 表 2 Langmuir与Freundlich吸附等温模型拟合参数及相关系数Table 2. Fitting parameters and correlation coefficients of adsorption isotherm models of Langmuir and Freundlich温度/ ℃ Langmuir Freundlich qm/(mg·g−1) b/(L·mg−1) R2 Kf/(mg(1−1/n) g−1 L1/n) 1/n R2 25 4.12 0.33 0.995 0.69 0.40 0.960 | Show TableDownLoad:
CSV
2.2 沸石-动态膜系统脱氨效能和成膜特征研究
1) 沸石动态膜系统脱氨效果。在初始氨氮质量浓度10 mg·L−1,2 L体系,电动搅拌器室温搅拌1 h条件下,吸附动态膜系统中沸石对氨氮的去除率及吸附量随沸石投加量的变化规律见图6。由图6可以看出,与在100 mL体系中、25 ℃、180 r min−1恒温振荡箱和初始氨氮质量浓度10 mg·L−1反应条件(图2)相比,该条件下沸石对氨氮的去除率与吸附量稍有降低,基本能够充分发挥沸石的吸附脱氨效能,不会造成沸石动态膜系统内沸石吸附位点的浪费。当沸石投加量达到10 g·L−1以后,对氨氮去除率的提升非常有限,基本在55%~60%,吸附量随沸石投加量的增加逐渐下降。为了保证吸附效果,循环吸附-动态膜系统中沸石的投加量以10 g·L−1为宜,此时的去除率与吸附量分别为56%和0.59 mg·g−1。本系统中,沸石吸附容量与NaCl改性沸石[15]吸附容量(0.56 mg·g−1,初始氨氮质量浓度20 mg·L−1,投加量30 g·L−1,室温30 ℃)较为接近,但氨氮去除率明显低于改性4A沸石分子筛(91%)[14] 和NaCl改性沸石(83%)[15],氨氮去除率的差距可能是由于沸石投加量增大和初始浓度差异引起的,后续沸石动态膜系统可根据实际脱氨需要进行沸石改性优化。
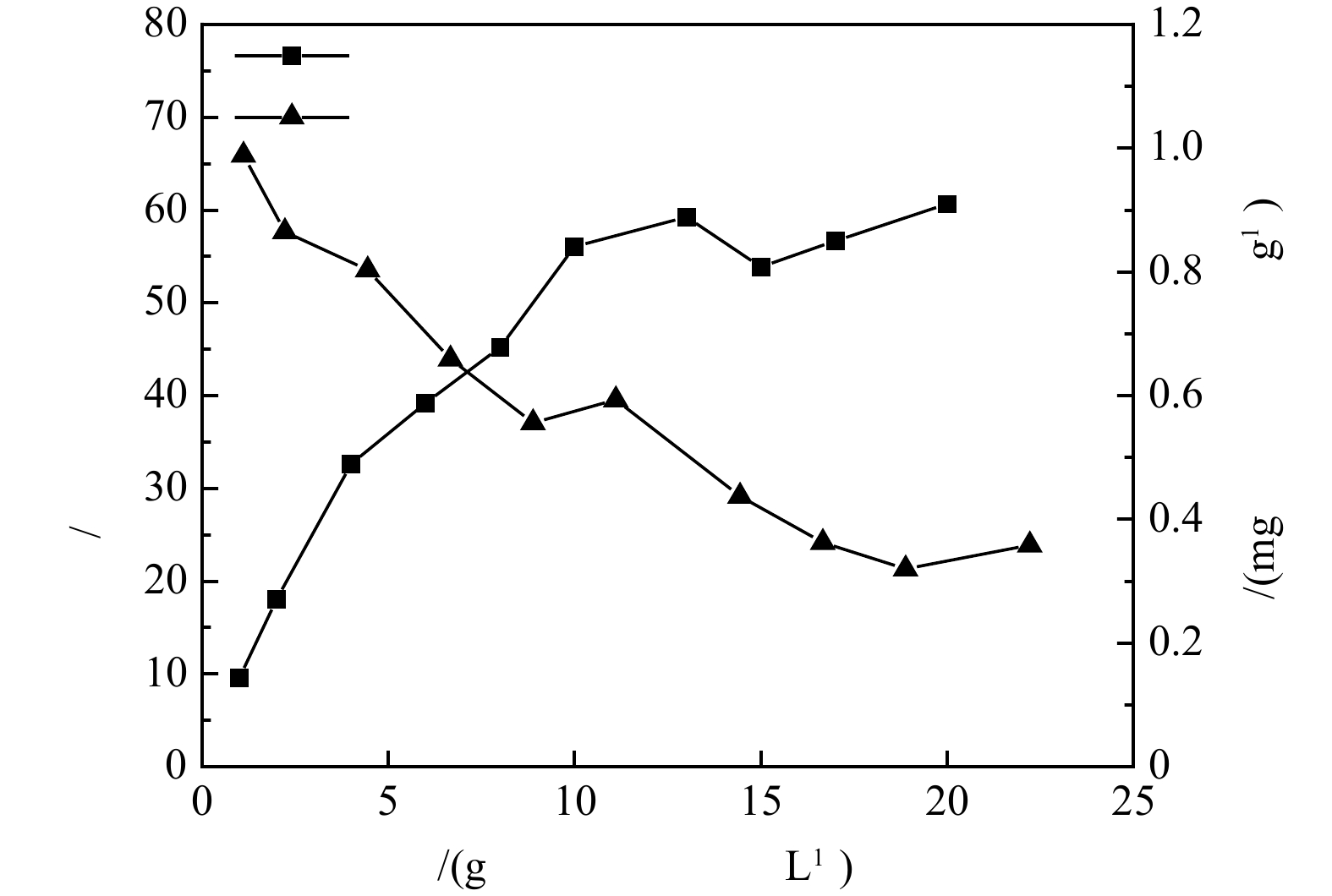 图 6 沸石-动态膜系统中氨氮去除率及吸附量随沸石投加量的变化规律Figure 6. Variations of ammonia nitrogen removal rate and adsorption amount with zeolite dosage in the zeolite-dynamic membrane system
图 6 沸石-动态膜系统中氨氮去除率及吸附量随沸石投加量的变化规律Figure 6. Variations of ammonia nitrogen removal rate and adsorption amount with zeolite dosage in the zeolite-dynamic membrane system2)沸石动态膜系统的成膜特征。结果表明,在沸石粉投加量为1 g·L−1,蠕动泵流量为40 mL·min−1和38 μm支撑膜的条件下,沸石动态膜的形成效果最佳,其浊度与跨膜压的变化规律如图7(a)所示。从图7(a)中可知,动态膜出水浊度在 5 min即从最初的145.5 NTU降低到25.3 NTU,10 min时降至7.52 NTU,此时跨膜压开始上升,出水浊度也开始缓慢波动上升。动态膜整体形成过程维持了约45 min,而跨膜压在40 min内上升至约95 kPa。在38 μm支撑膜截留作用下,沸石动态膜系统出水浊度逐渐下降,随着动态膜层不断密实,沸石动态膜对小粒径沸石截留效果也在逐渐提升,但实验观察到出水浊度经过下降阶段后,随压力的上升呈现出不断缓慢上升趋势。推测可能是在动态膜形成过程中,进水的冲击及沸石颗粒性质造成动态膜滤饼层并不稳定,在跨膜压作用下小颗粒沸石穿透膜层导致浊度升高。实验中发现在此条件下沸石形成的动态膜层较薄,不太均匀(图7(b)),说明沸石动态膜的稳定性有待进一步提升。若以浊度值去除率近似估算沸石回收率,沸石动态膜系统对沸石颗粒的回收率最高可达到94.8%。
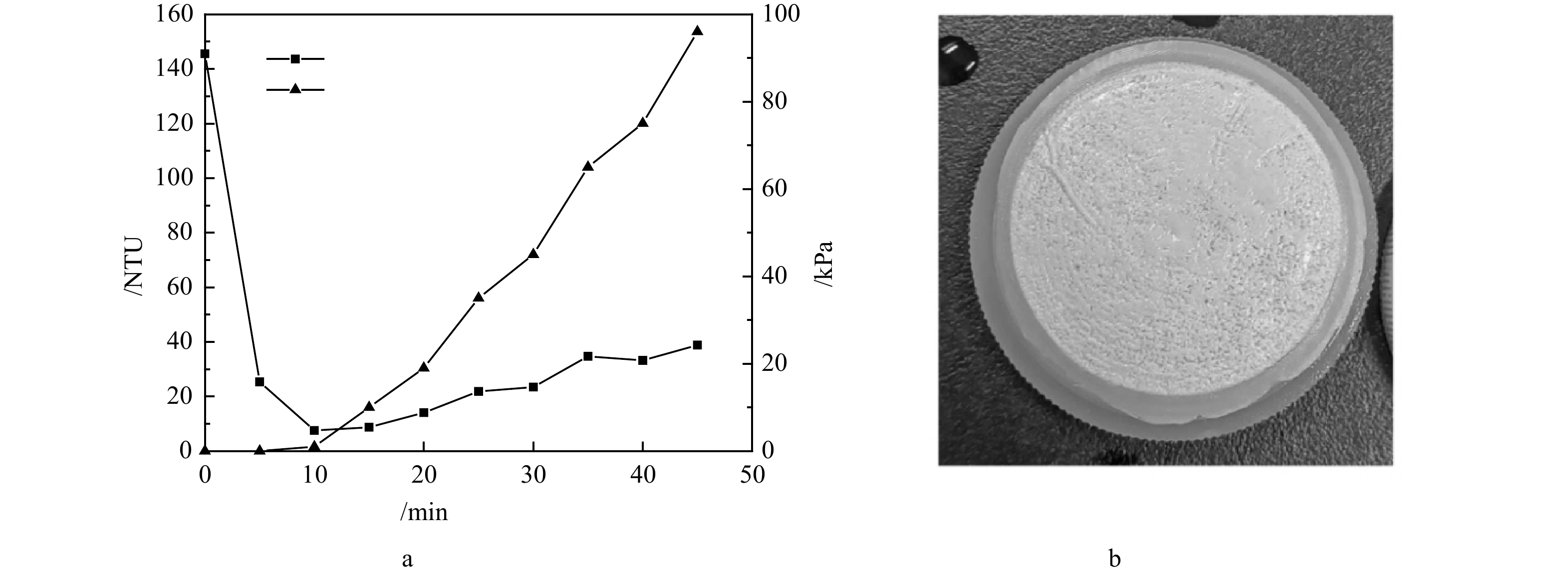 图 7 沸石动态膜成膜过程中出水浊度与跨膜压的变化规律和成膜图像Figure 7. Changes in effluent turbidity and transmembrane pressure during the zeolite DM formation and DM picture
图 7 沸石动态膜成膜过程中出水浊度与跨膜压的变化规律和成膜图像Figure 7. Changes in effluent turbidity and transmembrane pressure during the zeolite DM formation and DM picture3)复合硅藻土沸石动态膜系统成膜特征和脱氨效果。为了有效提升沸石动态膜稳定性,同时保证动态膜对氨氮的去除效果,后续实验选用硅藻土作为辅助成膜材料,以1:1质量比与沸石进行复配。为了避免动态膜形成过快增加动态膜形成过程的监测困难,先投加10 g·L−1沸石与10 g·L−1硅藻土于2 L 10 mg·L−1氨氮废水中,搅拌1 h以充分混合,然后取200 mL混合液稀释10倍后开始构建复合硅藻土沸石动态膜,在动态膜形成过程中出水浊度与跨膜压随时间的变化情况如图8(a)所示。沸石与硅藻土按质量比1:1复配形成动态膜时,出水浊度下降很快,5 min即由81.6 NTU下降至1.23 NTU,10 min后一直稳定在1 NTU以下,表明动态膜已稳定形成,且连续运行55 min直至跨膜压上升至82 kPa。硅藻土的加入使出水浊度的稳定性得到明显改善,相比于沸石动态膜,沸石与硅藻土混合复配吸附动态膜形成快速且稳定,实验结束后发现复合硅藻土沸石动态膜更加致密而均匀(图8(b)),可以有效截留微尺度沸石。从跨膜压的变化规律来看,沸石与硅藻土复配也一定程度上延长了装置的运行时间。因此,沸石与硅藻土复配形成的动态膜可以有效回收微尺度沸石。若以浊度值去除率近似估算硅藻土和沸石的回收率,复合硅藻土沸石动态膜系统回收率可达到98.77%,高于动态膜对微尺度改性活性炭的回收率(63%~87%)[16]。
 图 8 复合硅藻土沸石成膜过程中出水浊度与跨膜压的变化规律及成膜图像Figure 8. Variations of effluent turbidity and TMP during the DM formation with zeolite and diatomite and DM picture
图 8 复合硅藻土沸石成膜过程中出水浊度与跨膜压的变化规律及成膜图像Figure 8. Variations of effluent turbidity and TMP during the DM formation with zeolite and diatomite and DM picture沸石与硅藻土混合复配成膜过程中氨氮浓度的变化规律如图9所示。由图9可知,在反应开始时,在初始氨氮质量浓度为10 mg·L−1时,经过60 min后剩余氨氮质量浓度为4.4 mg·L−1,去除率可达56%,与单独投加沸石时对氨氮的去除效果相近。硅藻土本身对氨氮的吸附效果不佳,该吸附过程主要还是以沸石吸附为主。60 min稀释后开始形成动态膜时,系统内与出水的氨氮质量浓度(折算为稀释前)在6.5 mg·L−1左右,比稀释前略有提升,可能是因为稀释过程导致了氨氮吸附平衡的移动,沸石吸附的氨氮部分转移到了稀释后溶液中。由复合硅藻土沸石动态膜出水浊度的变化规律可知,系统运行5 min即可形成稳定的动态膜,随着动态膜的持续形成和过滤,动态膜出水氨氮浓度略低于反应系统内氨氮浓度。这可能是因为在动态膜成膜前系统混合搅拌过程中,沸石对氨氮的吸附已基本趋于平衡,而氨氮废水经过动态膜时与沸石接触时间较短,无法进行充分接触,因此,复合硅藻土沸石动态膜层对氨氮的去除效果有限。复合硅藻土-沸石-动态膜系统实现了废水中氨氮的吸附去除,同时回收了微颗粒沸石,可为吸附材料的再生和回收利用提供参考。
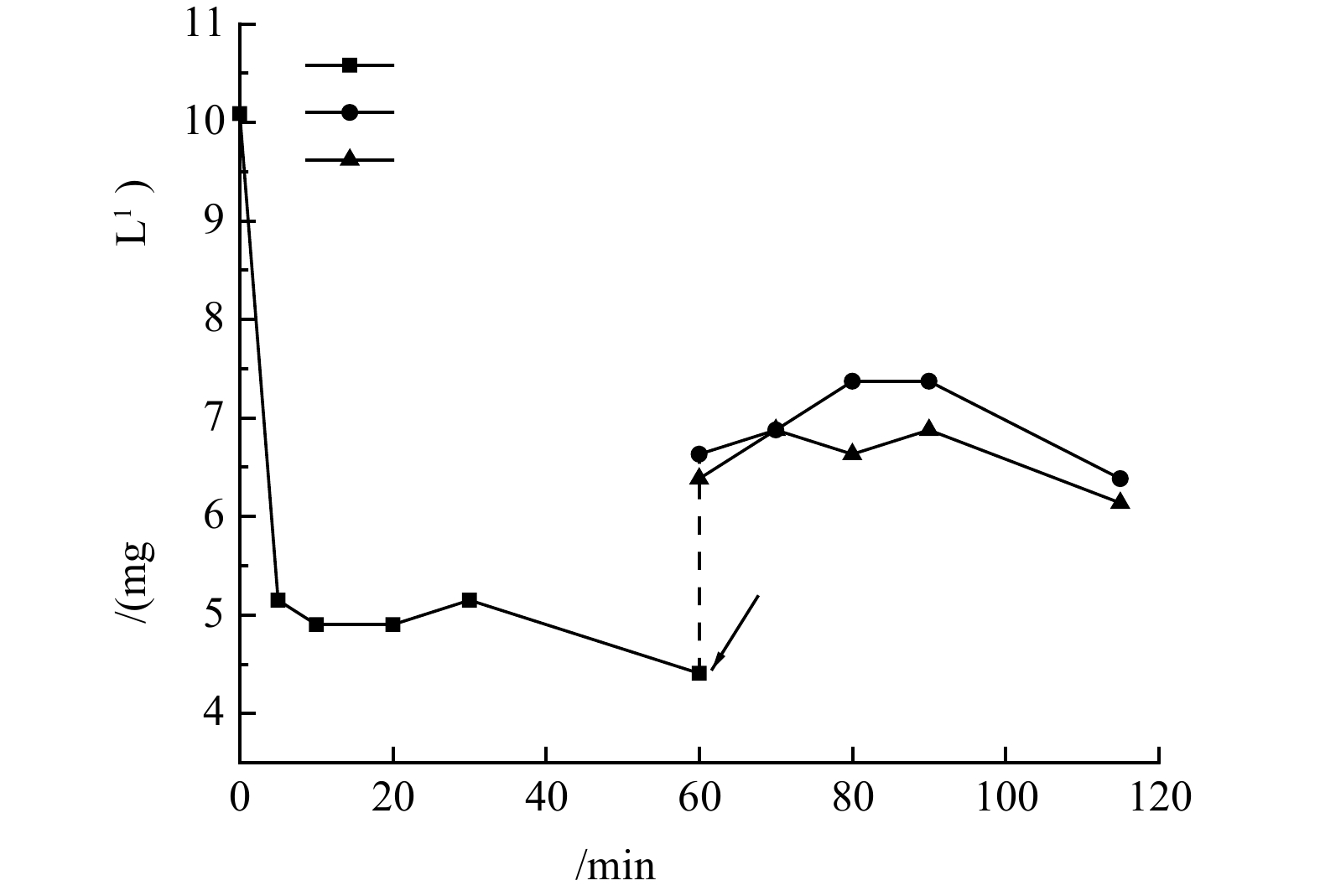 图 9 复合硅藻土沸石动态膜形成过程中氨氮浓度的变化规律Figure 9. Variation of ammonia nitrogen concentration during the DM formation with zeolite and diatomite
图 9 复合硅藻土沸石动态膜形成过程中氨氮浓度的变化规律Figure 9. Variation of ammonia nitrogen concentration during the DM formation with zeolite and diatomite整体来看,沸石静态吸附与沸石动态膜系统在氨氮去除性能上的表现相差不大,但沸石动态膜系统的优势在于可以对沸石吸附颗粒进行有效回收。尽管单纯沸石构建动态膜系统出现了出水浊度波动上升和沸石颗粒穿透,但通过复合硅藻土沸石动态膜系统能够保证了沸石颗粒的有效回收,从而提升了动态膜系统稳定性,也同步实现了废水中氨氮的去除。
3. 结论
1)在沸石静态吸附研究中,在初始氨氮质量浓度为10 mg·L−1,沸石投加量为4 g·L−1时,沸石对氨氮去除率和吸附量分别可达54%和1.3 mg·g−1;当初始氨氮质量浓度增至100 mg·L−1时,沸石对氨氮吸附量可提升至3.89 mg·g−1,但氨氮去除率也降至17%。
2)沸石脱氨过程更符合准二级动力学过程;Langmuir吸附等温模型对沸石吸附脱氨拟合效果更优,理论最大平衡吸附量为4.12 mg·g−1。
3)在10 g·L−1沸石投加量下,沸石动态膜系统对氨氮的去除率为56%,出水浊度可降至7.52 NTU,沸石动态膜的稳定性有待进一步提升。
4)沸石与硅藻土按照1:1的质量比复配,10 min即可快速构建复合硅藻土沸石动态膜,出水浊度稳定在1 NTU以下,氨氮去除率可达到56%,在脱氨的同时能够实现沸石的有效回收,并显著提升了复合硅藻土沸石动态膜系统的稳定性。沸石吸附是复合沸石动态膜系统脱氨的主要作用机制。
-
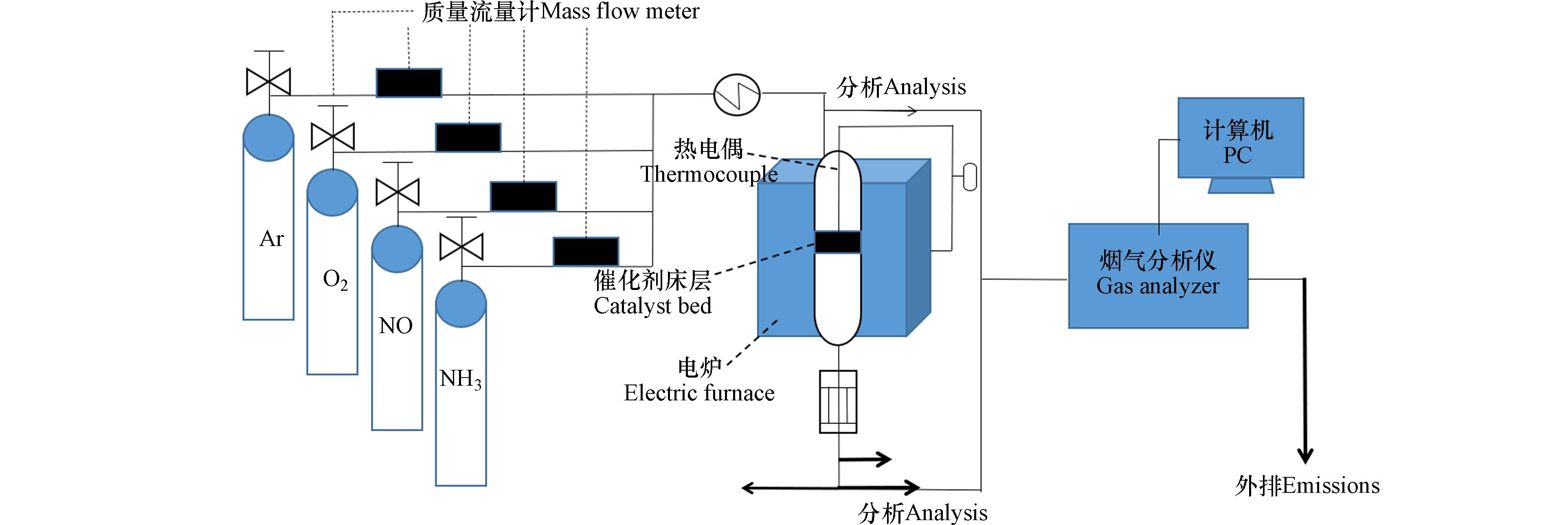
图 1 选择性催化还原实验装置示意图
Figure 1. Schematic representation of the experimental setup for selective catalytic reduction
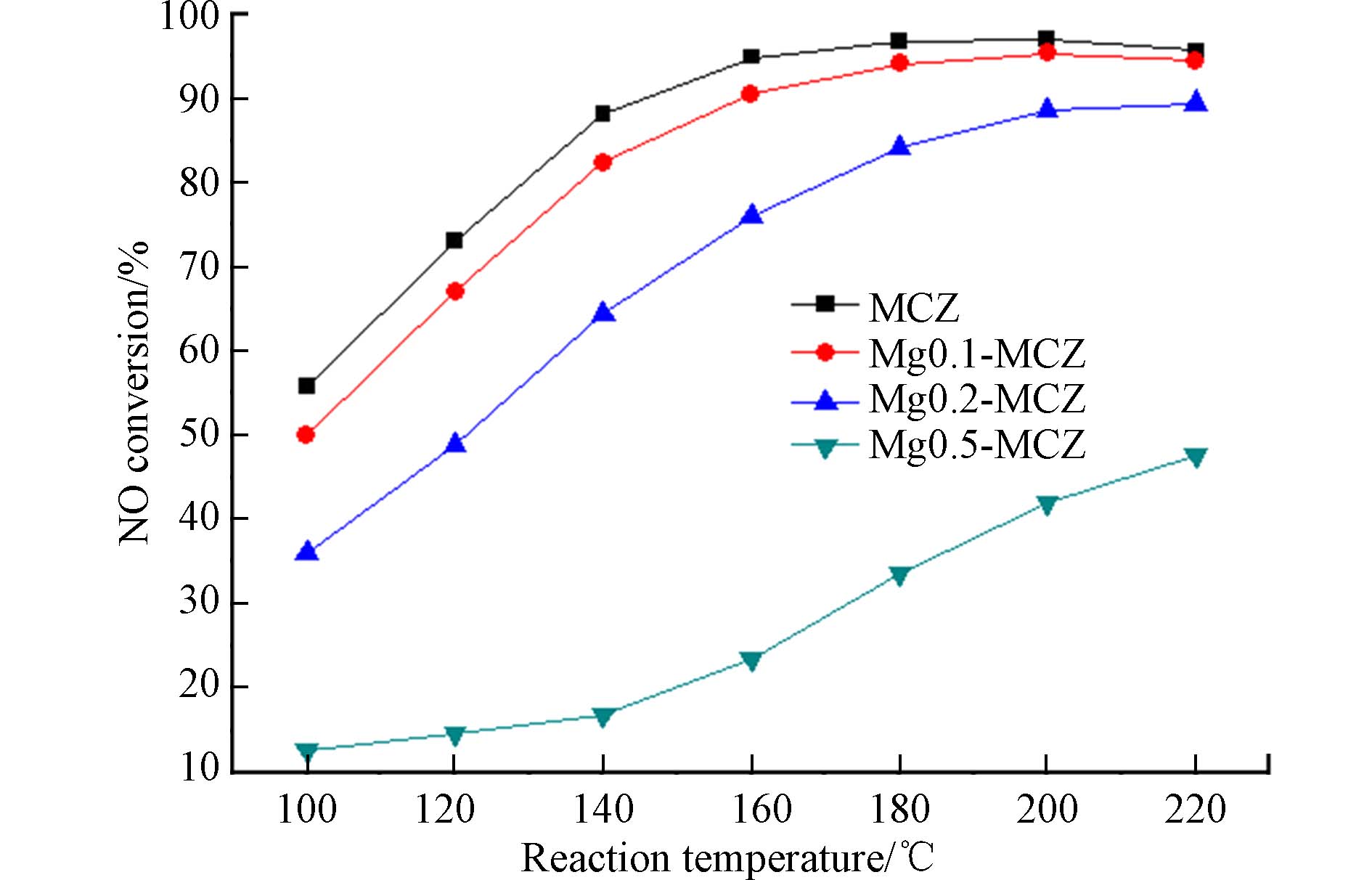
图 6 不同温度下Mg对催化剂脱硝效率的影响
Figure 6. Effect of Mg on catalyst denitration efficiency at different temperatures
表 1 四种催化剂的质构特性
Table 1. Structural properties of the four catalysts
样品Samples BET比表面积/ (m2·g−1)BET surface area 总孔隙体积/ (cm3·g−1)Total pore volume 平均孔径/ nmAverage pore diameter MCZ 51.572 0.082 3.817 Mg0.1-MCZ 46.092 0.081 3.814 Mg0.2-MCZ 44.093 0.079 3.816 Mg0.5-MCZ 38.564 0.072 3.814
下载: 导出CSV
表 2 两种不同氧原子的比值
Table 2. Ratios of the two different oxygen atoms
样品Samples 晶格氧Oα 化学吸附氧Oβ 羟基氧Oγ MCZ 0.48 0.47 0.05 Mg0.1-MCZ 0.61 0.32 0.07 Mg 0.2-MCZ 0.62 0.28 0.10 Mg 0.5-MCZ 0.71 0.23 0.07
下载: 导出CSV
表 3 三种催化剂的质构特性
Table 3. The textural properties of three catalysts
样品Samples BET比表面积/ (m2·g-1)BET surface area 总孔隙体积 / (cm3·g−1)Total pore volume 平均孔径/ nmAverage pore diameter MCZ 51.572 0.082 3.817 ZSMg0.2-MCZ 50.835 0.083 3.825 Mg0.2-MCZ 44.093 0.079 3.816
下载: 导出CSV
表 4 三种催化剂的表面酸度
Table 4. Surface acidity of the three catalysts
样品Samples 表面酸度/(mmol·g−1)Surface acidity MCZ 3.79 ZSMg0.2-MCZ 3.71 Mg0.2-MCZ 3.40
下载: 导出CSV
-
[1] 郑长乐. 脱硝催化剂再生技术研究进展[J]. 电力科技与环保, 2019, 35(1): 10-12. ZHENG C L. Research progress on the regeneration of de NOx catalyst[J]. Electric Power Technology and Environmental Protection, 2019, 35(1): 10-12 (in Chinese).
[2] 陆超, 朱文韬, 郭博闻, 等. 火电厂SCR脱硝催化剂工艺特性评价试验[J]. 电力科技与环保, 2019, 35(1): 7-9. LU C, ZHU W T, GUO B W, et al. Evaluation test of technology characteristic of SCR denitration catalyst of thermal power plant[J]. Electric Power Technology and Environmental Protection, 2019, 35(1): 7-9 (in Chinese).
[3] 郭丽颖, 朱林, 侯健, 等. 燃气轮机NOx控制技术及催化剂应用的现状与展望[J]. 电力科技与环保, 2019, 35(1): 13-15. GUO L Y, ZHU L, HOU J, et al. Present situation and expectation of gas turbine nitrogen oxide control technology and catalyst applications[J]. Electric Power Technology and Environmental Protection, 2019, 35(1): 13-15 (in Chinese).
[4] LIU Z M, IHL WOO S. Recent advances in catalytic DeNOXScience and technology[J]. Catalysis Reviews, 2006, 48(1): 43-89. doi: 10.1080/01614940500439891 [5] CHEN L, LI J H, GE M F. The poisoning effect of alkali metals doping over nano V2O5–WO3/TiO2 catalysts on selective catalytic reduction of NO x by NH3[J]. Chemical Engineering Journal, 2011, 170(2/3): 531-537. [6] LONG R Q, YANG R T, CHANG R. Low temperature selective catalytic reduction (SCR) of NO with NH3 over Fe–Mn based catalysts[J]. Chemical Communications, 2002(5): 452-453. doi: 10.1039/b111382h [7] GUO R T, ZHOU Y, PAN W G, et al. Effect of preparation methods on the performance of CeO2/Al2O3 catalysts for selective catalytic reduction of NO with NH3[J]. Journal of Industrial and Engineering Chemistry, 2013, 19(6): 2022-2025. doi: 10.1016/j.jiec.2013.03.010 [8] SULTANA A, SASAKI M, HAMADA H. Influence of support on the activity of Mn supported catalysts for SCR of NO with ammonia[J]. Catalysis Today, 2012, 185(1): 284-289. doi: 10.1016/j.cattod.2011.09.018 [9] BAIDYA T, GUPTA A, DESHPANDEY P A, et al. High oxygen storage capacity and high rates of CO oxidation and NO reduction catalytic properties of Ce1– xSn xO2 and Ce0.78Sn0.2Pd0.02O2- δ[J]. The Journal of Physical Chemistry C, 2009, 113(10): 4059-4068. doi: 10.1021/jp8060569 [10] WU Z B, JIN R B, WANG H Q, et al. Effect of ceria doping on SO2 resistance of Mn/TiO2 for selective catalytic reduction of NO with NH3 at low temperature[J]. Catalysis Communications, 2009, 10(6): 935-939. doi: 10.1016/j.catcom.2008.12.032 [11] SHEN B X, YAO Y, MA H Q, et al. Ceria modified MnOx/TiO2-pillared clays catalysts for the selective catalytic reduction of NO with NH3 at low temperature[J]. Chinese Journal of Catalysis, 2011, 32(11/12): 1803-1811. [12] KAMBUR A, POZAN G S, BOZ I. Preparation, characterization and photocatalytic activity of TiO2–ZrO2 binary oxide nanoparticles[J]. Applied Catalysis B:Environmental, 2012, 115/116: 149-158. doi: 10.1016/j.apcatb.2011.12.012 [13] LIU Z M, WANG K C, ZHANG X Y, et al. Study on methane selective catalytic reduction of NO on Pt/Ce0.67Zr0.33O2 and its application[J]. Journal of Natural Gas Chemistry, 2009, 18(1): 66-70. doi: 10.1016/S1003-9953(08)60076-6 [14] SI Z C, WENG D, WU X D, et al. Lattice oxygen mobility and acidity improvements of NiO–CeO2–ZrO2 catalyst by sulfation for NO x reduction by ammonia[J]. Catalysis Today, 2013, 201: 122-130. doi: 10.1016/j.cattod.2012.05.001 [15] CAO F, XIANG J, SU S, et al. The activity and characterization of MnOx–CeO2–ZrO2/γ-Al2O3 catalysts for low temperature selective catalytic reduction of NO with NH3[J]. Chemical Engineering Journal, 2014, 243: 347-354. doi: 10.1016/j.cej.2014.01.034 [16] YANG D, LI J H, WEN M F, et al. Enhanced activity of Ca-doped Cu/ZrO2 for nitrogen oxides reduction with propylene in the presence of excess oxygen[J]. Catalysis Today, 2008, 139(1/2): 2-7. [17] 张晓鹏. 基于Mn/Ce-ZrO2催化剂的低温NH3-SCR脱硝性能研究[D]. 天津: 南开大学, 2013. ZHANG X P. Research of Mn/Ce-ZrO2 for selective catalytic reduction of NOx with NH3 at low temperature[D]. Tianjin: Nankai University, 2013 (in Chinese).
[18] 杨志琴. 以纳米ZrO2为载体的低温SCR催化剂的制备及性能研究[D]. 南京: 南京师范大学, 2011. YANG Z Q. Preparation and properties of low-temperature SCR catalyst supported on nano-ZrO2[D]. Nanjing: Nanjing Normal University, 2011 (in Chinese).
[19] 周凯, 庄柯, 姚杰, 等. 燃气电厂蜂窝式SCR脱硝催化剂性能检测与评估[J]. 电力科技与环保, 2021, 37(1): 18-23. ZHOU K, ZHUANG K, YAO J, et al. Performance assessment and analysis of honeycomb SCR denitration catalyst in gas power plant[J]. Electric Power Technology and Environmental Protection, 2021, 37(1): 18-23 (in Chinese).
[20] SONG H, OZKAN U S. Changing the oxygen mobility in Co/ceria catalysts by Ca incorporation: Implications for ethanol steam reforming[J]. The Journal of Physical Chemistry. A, 2010, 114(11): 3796-3801. doi: 10.1021/jp905608e [21] TANG X L, HAO J M, XU W G, et al. Low temperature selective catalytic reduction of NO x with NH3 over amorphous MnO x catalysts prepared by three methods[J]. Catalysis Communications, 2007, 8(3): 329-334. doi: 10.1016/j.catcom.2006.06.025 [22] 沈伯雄, 陈建宏, 姚燕, 等. 碱土金属对MnOx-CeO2/ZrO2-PILC催化剂SCR活性影响研究[J]. 燃料化学学报, 2012, 40(12): 1487-1491. SHEN B X, CHEN J H, YAO Y, et al. Effects of alkali metals on catalyst of MnO x-CeO2/ZrO2-PILC in the low-temperature selective catalytic reduction[J]. Journal of Fuel Chemistry and Technology, 2012, 40(12): 1487-1491 (in Chinese).
[23] ETTIREDDY P R, ETTIREDDY N, MAMEDOV S, et al. Surface characterization studies of TiO2 supported manganese oxide catalysts for low temperature SCR of NO with NH3[J]. Applied Catalysis B:Environmental, 2007, 76(1/2): 123-134. [24] JIN R B, LIU Y, WU Z B, et al. Low-temperature selective catalytic reduction of NO with NH3 over MnCe oxides supported on TiO2 and Al2O3: A comparative study[J]. Chemosphere, 2010, 78(9): 1160-1166. doi: 10.1016/j.chemosphere.2009.11.049 [25] WANG H Q, CHEN X B, GAO S, et al. Deactivation mechanism of Ce/TiO2 selective catalytic reduction catalysts by the loading of sodium and calcium salts[J]. Catalysis Science & Technology, 2013, 3(3): 715-722. [26] JING L Q, XU Z L, SUN X J, et al. The surface properties and photocatalytic activities of ZnO ultrafine particles[J]. Applied Surface Science, 2001, 180(3/4): 308-314. 期刊类型引用(1)
1. 何应钦. 基于活性炭吸附的自来水中THMs去除技术研究. 山西化工. 2024(10): 264-266 .  百度学术
百度学术
其他类型引用(0)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 1045
- HTML全文浏览数: 1045
- PDF下载数: 8
- 施引文献: 1



