























-
近年来,我国越来越重视臭氧(O3)污染,在多区域范围内O3已取代颗粒物成为首要污染物[1 − 3]. 苏皖鲁豫交界地区位于汾渭平原和长三角之间,已成为继京津冀等区域之后,新的联防联控重点控制区域[4 − 6]. 该地区位于共有22个城市,基础排放量大,空气质量改善进程总体相对滞后,臭氧浓度高且上升趋势明显,光化学污染过程呈现出与周边地区的同步性[7]. 目前针对该区域研究较少,相关监测持续时间较短,无法表征长时间的光化学污染特征变化. 亟需对苏皖鲁豫区域加以重点关注,补齐大气污染防治短板,对于该区域的O3污染治理能力提高和大气光化学污染防治策略制定具有重要的意义.
过氧乙酰硝酸酯(CH3C(O)OONO2,PAN)是光化学烟雾的关键产物[8 − 10]. PAN属于非自然过程排放的污染物,仅通过人类活动排放的前体污染物的光化学反应生成,是光化学污染的重要指示剂[11 − 12]. 大气中挥发性有机物(VOCs)在氧化过程中,可生成PA自由基(Peroxyacetyl Radical),而后PA可与NO2结合生成PAN[13 − 14]. PAN在对流层上部稳定,但在大气对流层下部具有明显的热不稳定性,与O3相比不易远距离输送,能更好地反映本地区域的光化学污染情况[15 − 17]. 在国内,包括城市、郊区、农村和偏远地区,都有PAN的时空变化及其形成机制的相关研究. Yuan等[18]研究表明,在珠三角农村地区,PAN的增加主要受气相化学控制,其次是垂直运输,而其损失主要受水平运输和干沉积的调节. Liu等[19]研究表明,高环境浓度的细颗粒物和气态亚硝酸可以显著促进PAN的形成. Xia等[20]基于稳态模型研究表明,在东南沿海地区生物质燃烧源是PA自由基的重要来源.
淮北市位于苏皖鲁豫交界地区中心位置,地处苏豫皖三省交界,经济发展迅速,近年来光化学污染频发. 本研究于2021年秋季(10月)和2022年夏季(6月)利用PAN在线监测,在淮北市开展PAN污染特征分析与来源研究,对于探究苏皖鲁豫交界地区大气光化学污染及变化规律,加强局地区域光化学污染的理解和防控,将具有积极意义.
-
本研究于2021年10月和2022年的6月,在淮北市进行PAN浓度在线监测. 用10月份的监测数据代表秋季,6月份的代表夏季,分别获得了21 d和16 d的PAN有效浓度数据. 采样地点位于淮北市(116.73°E,33.89°N)某开发区空旷地,采样高度4.5 m左右,空间位置如图1所示.
监测点周边无高大建筑遮挡,采样和数据审核符合《环境空气质量自动监测技术规范》和《国家大气光化学监测网自动监测数据审核技术指南(2021版)(试行)》中的相关要求.
本研究使用PAN大气在线监测系统(PANs-1000,聚光科技,2020.11),该分析仪包含气相色谱仪(GC-ECD)、校准仪和零气发生器. 可支持5 min分辨率的大气环境PAN浓度自动连续监测,以采样泵为动力,控制三通电磁阀选择通过空气样品或校准样品. 在采样模式下,样品通过聚四氟乙烯微孔滤膜过滤后进入系统,通过六通阀进入定量环;采样结束后,通过六通阀切换,开始进样. 在进样模式下,定量环与载气气路连接,样品随载气进入色谱柱后进行分离. 分离后的组分被带入到ECD检测器中,得到PAN检测结果. 标定时,校准仪和零气发生器配合在紫外光照下光解生成PAN标气;再与稀释气混合稀释到所需标定浓度进行分析. 为保证数据准确性,在观测期间每周绘制一条标准曲线,每条标准曲线采用6个浓度梯度(0—10)×10−9,标准曲线R2均大于0.98,重复性≤3%,最低检出限0.5×10−9.
-
本研究将利用潜在源贡献函数(potential source contribution function,PSCF)评估该监测点PAN污染最可能的来源区域. 研究首先基于美国国家环境预测中心全球资料同化系统数据GDAS(ftp://arlftp.arlhq.noaa.gov/pub/archives),使用HYSPLIT模型在监测点(500m AGL)处模拟气团的后向轨迹. 考虑到PAN热解大气寿命较短,本研究进行1 h后向轨迹计算. 结合后向轨迹与PAN的监测值,利用TrajStat软件对PSCF值进行计算. PSCF值表示特定网格中污染轨迹数(污染物浓度超过设定阈值)占通过该网格轨迹总数的概率,在0—1范围内,其数值越高,表示该网格区域对受体点污染物浓度的潜在源贡献值越大. 研究区域空间网格分辨率为1°×1°,污染标准值设置为观测期间PAN小时浓度的第50百分位数,引入了权重函数以降低不确定性[21].
-
基于国家气象科学数据中心(http://data.cma.cn),采集了58113气象站点(116.45°E,33.56°N)的温度(T)和相对湿度(RH)气象数据. 观测站点光解速率常数的监测采用大气光解速率分析仪(PFS-100,Focused Photonics Inc,2020.9),利用石英接收头收集来自各个方向的太阳辐射,可获得210—790 nm的光谱,并能计算J(O1D)的光解速率. 基于安徽省生态环境厅空气质量实时数据发布平台(https://sthjt.ah.gov.cn/site/tpl/5371),采集距PAN监测点最近(7.7 km)的烈山区政府国控站点(116.80°E,33.90°N)的地面NO2、NO、O3浓度数据. 气象数据和大气污染物浓度数据覆盖PAN的观测期间,时间分辨率为1 h.
-
2021年10月,PAN的体积分数平均值为(0.42±0.24)×10−9,远低于2022年6月夏季的浓度均值为(1.87±0.86)×10−9,主要原因是PAN作为一种典型的光化学反应二次污染物,夏季的高温强辐射都会造成PAN的形成和累积. 观测期间PAN体积分数范围在2021年10月和2022年6月分别为(0.08—1.44)×10−9和(0.61—5.72)×10−9. 淮北市PAN体积分数水平均值和最大值与国内其他重点区域城市文献观测结果对比如表1所示. 本监测点10月份的PAN均值浓度低于苏皖鲁豫交界地区的青岛市. 在其它3个重点区域中与深圳市相近,但仍高于国内外各背景站[22],说明秋季淮北市大气光化学污染问题依然存在. 夏季时段内苏皖鲁豫区域的PAN最大值浓度明显高于表中所列其它三大重点区域城市. 这表明了目前苏皖鲁豫区域内夏季的光化学污染仍处在较严重范围内,应加强对该区域内光化学污染的研究.
-
大气污染物的日变化特征可以反映源排放和大气化学反应的综合影响[14]. 观测期间PAN、O3、NO2和气象因子的日变化趋势如图2所示. 两个季节PAN日变化浓度均呈单峰型特征,在2021年秋季峰值(0.56×10−9)出现在12:00—13:00;在2022年夏季提前两小时(10:00—12:00)达到峰值,而其峰值浓度是秋季的4.88倍. PAN体积分数在上午随着太阳辐射加强而不断升高,正午达峰,夏季PAN出现较早且较高的峰值是由于较高的太阳辐射强度和温度,这促进了PAN的产生和热分解[27]. 受强烈的光化学反应条件、周边地区的区域输送和大气背景水平增加的影响,淮北市夏季的PAN浓度水平高于秋季,这和其它地区的研究一致[13,28].
淮北市PAN日变化特征与合肥市[25]和厦门市[14]相一致,而北京市[23]和青岛市[13]均为双峰型,在18时至20时出现第二个相对低的峰值. 这种情况的主要原因可能是与观测地点和时段有关,本研究监测点位于郊区,呈双峰型变化的城市监测点多数位于市区内,城市晚高峰期间前体物的排放会促使光化学反应活性升高,促进PAN浓度的二次抬升. 除此之外,相对南方城市,北方城市夏秋季夜间温度较低,不利于PAN热解,仍有较高残留.
NO2是PAN的主要前体物之一,日变化趋势呈双峰分布,秋季两个峰值出现在08:00和19:00,夏季则出现在03:00和22:00,白天的趋势符合光化学生成特征. NO两个季节最高值均出现在08:00—09:00,与早高峰汽车尾气排放有关. 氮氧化物夏季整体平均浓度低于秋季,与气象条件和输送等因素有很大关系[29]. O3日变化趋势与温度的相一致,呈单峰型,峰值出现在午后,夏季O3平均浓度高于秋季. 有研究表明,高温低湿环境条件下,更容易产生光化学污染[5 − 6].
-
PA自由基(Peroxyacetyl Radical)在光化学反应中起着非常重要的作用,目前直接测量PA自由基仍然非常困难[30 − 31]. 除化学过程外,动力和沉积过程也会影响PA的变化. 在短时间内和稳定的大气条件下,可以忽略物理过程的贡献,用d[PAN]/dt表示PAN的瞬时变化率来体现化学过程影响[32]. 假设PAN的生成过程(1)和热分解过程(2)相对平衡,PA自由基可通过与NO反应去除(3),则PA自由基浓度可使用公式(4)进行估算. PAN生成速率(RPAN)可直接反映光化学污染前体物的贡献,同时规避了PAN的热解,因此本研究拟采用Zhang等[32]的方法,使用公式(5)来进行小时分辨率的生成速率计算,用RPAN表征光化学污染生成特征,单位为10−9·h-1.
式中,k1(9.5×10-12 cm³·(molecule·s)-1、k2(2.52×1016exp-13573/T s-1)分别是反应式(1)和(2)的速率常数,T表示温度(单位:℃)[13]. 本研究中,用臭氧光解速率常数J(O1D)来近似反映近地面的光辐射强度变化趋势[26].
计算结果如图3所示,夏季CPA自由基浓度均值为(0.91±0.90)×10-12,范围为(0.02—5.02)×10-12;而秋季CPA自由基浓度均值为(0.08±0.19)×10-12,范围为(0.001—1.75)×10-12.
CPA自由基昼高夜低的日变化规律与其主要基于日间光化学反应生成相一致. RPAN变化规律与CPA自由基一致,夏季RPAN均值为(7.43±6.44)×10−9·h-1,峰值出现在13:00,与CPA自由基峰值时间一致. 秋季RPAN均值为(0.56±0.95)×10−9·h-1. 值得注意的是,秋季RPAN与夏季略有不同,除了在13:00的第一个峰值外,在20:00出现第二个较小的峰,结合图2可知与这一时段氮氧化物的排放增加有关. 夏季CPA自由基和RPAN显著高于秋季,与夏季强烈的光辐射有关. 可以看出,夏季光辐射强度达到最大值后CPA自由基和RPAN仍在升高,说明淮北市夏季光化学反应更加强烈.
-
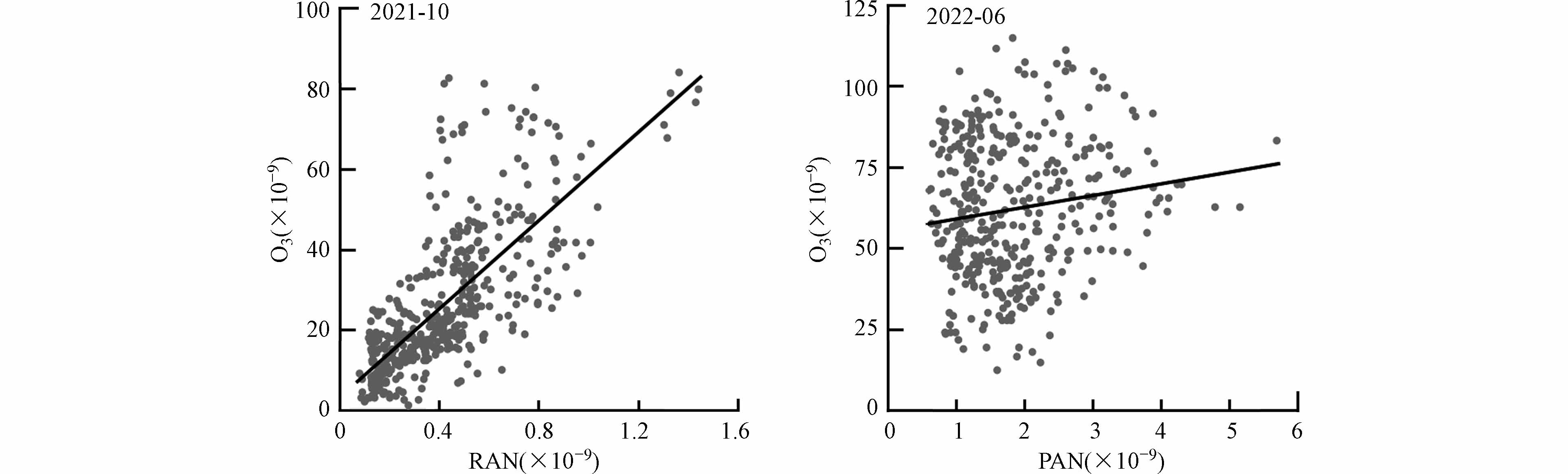
由于挥发性有机物和氮氧化物的光化学反应都产生PAN与O3,因此可通过分析PAN和O3相关性,作为判断空气质量的指标[19]. 图4给出了两个观测月份PAN与O3小时均值散点图和线性拟合. 秋季时PAN与O3呈现较好的正相关(r=0.72),表明它们很可能来自相同的光化学过程,或者来自共同的传输区域. 整个秋季观测期间PAN与O3的斜率为0.01,低于京津廊城市[22,24]和其他区域背景点[18,23],这意味着PAN相对于O3的生产效率相对较低,可能是由于挥发性有机物和氮氧化物等前体物水平下降,或者受其他地区清洁空气输送的影响. 而夏季时PAN与O3的相关性不明显,夏季温度更高,而PAN具有热不稳定性,高温时段可通过热分解反应生成PA自由基和NO2[19]. 同时这也表明夏季观测期间光化学污染物可能存在不同的来源,PAN高温条件下主要来自于本地生成,O3的来源还有区域传输,也与各自不同的去除途径有关. 这也表示秋季时苏皖鲁豫区域应控制本地光化学污染物生成,而夏季时本地生成和区域传输都应该加以重视.
-
化学反应式(1)、式(2)和式(3)表明PAN与NO2之间存在相互关系. 本研究中PAN与NO2的相关性如图5所示.
十月份二者皮尔逊积矩相关系数为0.028,六月份相关系数为0.194;两个月份的观测结果均显示相关性较差. 由反应式(1)可以看出NO2是PAN生成的重要前体物,但高浓度的NO2会和VOCs形成竞争,从而制约PA自由基的生成,这会抑制PAN的生成产生. 本研究获取的PAN与NO2的相关性与其他城市地区[23,26]的研究较为一致,但有研究[22]显示偏远地区背景站的观测中PAN与NO2的呈现很强的相关性,这可能与城市地区NO2的浓度水平较高有关.
-
众多研究[12,26]指出NO/NO2的值是影响PAN热分解速率的重要因素,从反应式(1)、式(2)和式(3)也可推导出. 本研究中PAN与NO/NO2的相关性如图6所示,两个月份80%以上的PAN散点落在NO/NO2比值0.4下方. 这和PAN的热解去除途径有关,反应式(3)中NO可通过消耗PA自由基降低PAN的生成速率,当比值较低时,更多的PA自由基参与到与NO2的反应,最终生成PAN[33]. 因此大多数高浓度的PAN通常出现在NO/NO2比值较低的时候,进一步证实了NO对PAN去除的重要作用. 本研究中两者相关性不显著,与长三角区域城市地区[26]研究结果较为一致.
-
为判断PAN的空间来源解析,研究区域输送对监测点光化学污染的影响,采用HYSPLIT模型开展监测点处500 m气团的后向轨迹聚类分析,同时利用TrajStat软件绘制PAN空间来源解析结果,结果如图7所示. 分析结果表明,2021年10月,该监测点主要受东北、北部和南部三个方向气团的影响,占比分别为34.48%、25.82%和18.06%;而2022年6月主要受东南、西南和南部气团的影响,占比分别为41.43%、17.93%和17.33%.
2021年10月PSCF高值区域主要出现在监测点南部,即所有通过该区域的气团后向轨迹的PAN浓度超过设定阈值的概率为40%—60%,证明该区域对监测点PAN浓度的潜在源贡献较高. 这可能主要源于西南方向的安庆市石化行业发达,VOCs排放量较高;淮南市的电力产业发达,氮氧化物排放量较高,都为PAN生成提供了丰富的前体物.
2022年6月PSCF高值区主要出现在北部,PSCF值达到0.7—0.9,显示了山东省西南部是PAN的潜在源贡献地区;而在17.93%的西南部气团输送情况下,该监测点西部方向亦未发现PSCF高值区,表明该气团污染较轻;41.43%东南方向的气团,表明在工业化程度较高的城市群(马鞍山、芜湖、南京)之间存在长距离运输. 有研究表明,PAN可作为NOx和自由基的临时贮存器,输送到偏远地区,重新分配NOx,并在大尺度区域范围内干预O3的生成[34 − 35].
-
(1)淮北市PAN的体积分数范围在2021年10月和2022年6月分别为(0.08—1.44)×10−9和(0.61—5.72)×10−9. 两个季节PAN日变化浓度均呈单峰型特征,东北方向的高值区显示出淮北市中心城区城市羽流的影响. 区别于O3的长距离迁移特征,秋季和夏季PAN污染主要来自当地产生和积累,但相邻城市的短程迁移可能在一定程度上导致PAN浓度较高.
(2)夏季PA自由基和PAN生成速率显著高于秋季,与夏季强烈的光辐射有关. 结合PAN和O3的观测值及其比值,发现淮北市不仅在夏季遇到光化学污染,而且秋季也存在光化学污染,体现了苏皖鲁豫区域较强的大气氧化环境.
(3)该观测处PAN浓度的潜在源,2021年10月高值区主要出现在监测点南部,可能是因为西南方向相近城市较高的前体物排放. 2022年6月高值区主要出现在北部和东南部,显示了泰安市是该监测点PAN的潜在源贡献地区;东南方向的气团,在工业化程度较高的城市群(马鞍山、芜湖、南京)之间存在长距离运输. 这证明苏皖鲁豫交界地区当前光化学污染是一个区域性问题,需要从区域尺度联防联控.
苏皖鲁豫典型城市过氧乙酰硝酸酯(PAN)污染特征分析
Analysis of photochemical pollution potential characteristics based on PAN observation in the border area of Jiangsu, Anhui, Shandong and Henan
-
摘要: 为探究苏皖鲁豫区域光化学污染特征,于2021年10月和2022年6月在淮北市开展光化学污染产物过氧乙酰硝酸酯(peroxyacetyl nitrate,PAN)的在线监测,分析了PAN浓度特征、空间来源、产生速率和变化趋势. 观测结果表明,观测处2021年10月PAN的浓度范围为(0.08—1.44)×10−9,2022年6月的浓度范围为(0.61—5.72)×10−9;PAN的峰值大部分出现在NO/NO2比值较低的时段. 结合气团后向轨迹,2021年10月观测处PAN的潜在源贡献函数(PSCF)高值区范围出现在南部方向,可能是因为西南方向相近城市较高的前体物排放;2022年6月高值区主要出现在北部和东南部,显示山东省西南部是潜在源贡献地区,东南方向的气团在工业化程度较高的城市群之间存在长距离运输.
-
关键词:
- 过氧乙酰硝酸酯(PAN) /
- 光化学污染特征 /
- 苏皖鲁豫 /
- 在线监测.
Abstract: In order to explore the potential of photochemical pollution in the regions of Jiangsu, Anhui, Shandong and Henan, the online monitoring of peroxyacetyl nitrate (PAN), a product of photochemical pollution, was carried out in Huaibei City in October 2021 and June 2022, and the concentration characteristics, spatial sources, production rate and change trend of PAN were analyzed. The observation results show that the concentration range of PAN at the observation site is (0.08—1.44)×10−9 in October 2021 and (0.61—5.72)×10−9 in June 2022. Most of the peak of PAN appeared in the period of low NO/NO2 ratio. In combination with the backward trajectory of the air mass, the high value range of the potential source contribution function (PSCF) of PAN at the observation site in October 2021 appears in the south, which may be due to the higher precursor emissions in the cities near the southwest. In June 2022, the high value areas mainly appeared in the north and southeast, indicating that the southwest of Shandong Province is a potential source contribution area, and the air mass in the southeast has long distance transportation between the urban agglomerations with a high degree of industrialization. -
反渗透(reverse osmosis, RO)作为一种膜分离技术,已经广泛应用于海水淡化、废水处理和中水回用等领域[1-3]. 在RO工艺中,为防止微生物繁殖造成的膜堵塞,杀菌灭藻是不可缺少的预处理步骤[4-5]. 游离氯(HOCl/OCl−)凭借其低廉的成本和广谱的杀菌性成为RO工艺中最常用的消毒药剂[5]. 但是,由于RO膜容易与游离氯发生反应,造成膜结构的破坏,因此,需要在RO膜前加装去除游离氯的装置[6]. 目前,常见的去除游离氯的方法包括活性炭脱氯、还原剂中和以及紫外线脱氯等[7-8]. 其中,紫外线脱氯技术不会滋生微生物,无需外加药剂,同时还能强化消毒效果[9],已经开始在市场上应用[10].
溴离子在海水中的质量浓度高达65 mg·L−1(摩尔浓度为0.81 mmol·L−1),能够与游离氯(HOCl/OCl−)反应生成游离溴(HOBr/OBr−),反应如式(1)—式(2)所示[11-12]. 因此,当RO工艺应用于海水淡化时,脱氯工艺中所需要脱除的氧化性物质为游离溴. Shemer等[12]研究表明,无论是游离氯还是游离溴都会对RO膜的结构产生破坏作用. 游离氯和游离溴在紫外照射下均可以发生分解(式(3)—式(4))[13]. 已有研究表明[11,13],游离氯和游离溴的摩尔吸光系数、量子产率以及光解的自由基反应过程均不相同. 这说明紫外照射下游离氯和游离溴的分解规律可能不同.
OCl−+Br−→OBr−+Cl−k=9.0×10−4L⋅(mol⋅s)−1 (1) HOCl+Br−→HOBr+Cl−k=6.8×103L⋅(mol⋅s)−1 (2) HOCl/OCl−+hv→HO⋅/O−⋅+Cl⋅ (3) HOBr/OBr−+hv→HO⋅/O−⋅+Br⋅ (4) 近年来,游离氯在紫外照射下的分解规律得到了较为广泛的研究. 田芳等[14]研究了中压紫外照射下游离氯的分解规律,发现游离氯分解速率受到pH、腐殖酸等水质因素的影响,但水中常见的低浓度无机阴离子对游离氯分解的影响不大. Yin等[15]利用LED光源研究了不同波长的紫外线照射对游离氯分解的影响,发现紫外波长和pH对游离氯的分解速率具有重要作用,建立了游离氯分解速率与波长和pH的三维模型. Feng等[16]发现低压紫外灯照射下,游离氯初始浓度和水中pH对游离氯的分解影响不大. 然而,游离溴在紫外照射下分解规律的研究相对较少. Guo等[11]研究发现,在低压紫外照射下,游离溴的初始浓度(20—200 μmol·L−1)对游离溴的分解影响很小. 海水基质成分复杂,无机离子和有机物的存在可能会影响游离氯和游离溴的分解速率,进而影响RO工艺中紫外线去除游离氯和游离溴的效果,但目前针对这一问题尚缺乏深入研究.
低压紫外灯是一种主要发射254 nm短波紫外线的单色光源,凭借其较高的能源转换率和强力的杀菌能力,已经在水处理领域广泛应用[17],对于游离氯和游离溴也具有分解效果[11]. Sperle等[18]研究还发现短波紫外对RO膜生物堵塞的处理也具有重要意义. 因此,本研究通过测定游离氯和游离溴在低压紫外灯照射下的浓度变化,研究了水中pH、阴离子和腐殖酸对游离氯和游离溴分解的影响,且分析了其中可能存在的原因,本研究可为紫外线去除游离氯和游离溴工艺的优化提供数据参考.
1. 实验部分(Experiment section)
1.1 主要试剂
次氯酸钠溶液、溴化钾、氯化钠、硝酸钾、硫酸钠、碳酸氢钠、氟化钠、磷酸二氢钠、磷酸氢二钠、硼酸和四硼酸钠,均为分析纯,购自国药集团化学试剂有限公司. N,N-二乙基-1,4-苯二胺硫酸盐,为分析纯,购自上海麦克林生化科技有限公司. 腐殖酸,有效含量大于82%,购自天津光复精细化工研究所. 实验中溶液均为超纯水(18.2 MΩ·cm)配置.
实际海水取自青岛近海海域,经0.45 μm滤膜过滤后使用,其基本理化指标如下:盐度为29.7‰,pH为8.02,DOC为1.92 mg·L−1. 人造海水的配置主要参考Mocledon海水配方[19],盐度为33‰,pH为8.01,内含氯离子(530 mmol·L−1)、溴离子(0.8 mmol·L−1)、硫酸根(27 mmol·L−1)、碳酸氢根(2.4 mmol·L−1)、硼酸盐(0.95 mmol·L−1)、磷酸盐(30 μmol·L−1)、硝酸根(30 μmol·L−1)和氟离子(50 μmol·L−1)等无机离子.
1.2 光照实验
游离氯和游离溴的光照实验在定制的低压紫外平行光束仪(NLC-PXGY-40W,福建新大陆环保科技有限公司)中进行,反应器为石英玻璃皿(直径d=10 cm,高度h=4 cm),放置于光束仪下方的磁力搅拌器上,实验时可以通过调节搅拌器下方的升降台高度实现反应器内光强的变化. 实验开始前对平行光束仪预热至少15 min,待光强稳定后进行实验. 由于游离氯和游离溴的初始浓度对其在低压紫外照射下的分解速率影响不大[11,16],因此将游离氯和游离溴的初始浓度固定为10 mg·L−1(约140 μmol·L−1),同时将反应器内反应液体积设定为200 mL. 进行实验时,每隔一定时间取样,测定游离氯或游离溴的浓度.
在反应器内加入190 mL的5 mmol·L−1磷酸盐缓冲液(pH=6或8)或硼酸盐缓冲液(pH=10),再加入一定体积的KBr溶液(反应液Br−终浓度为65 mg·L−1)或纯水,之后加入次氯酸钠溶液,搅拌混匀10 min,使之充分反应后进行实验,研究不同pH对游离氯和游离溴分解的影响.
在反应器内加入190 mL的5 mmol·L−1磷酸盐缓冲液(pH=8),再加入一定体积的KBr溶液(反应液Br−终浓度为65 mg·L−1)或纯水,加入次氯酸钠溶液,搅拌混匀10 min,再加入一定浓度的阴离子或腐殖酸溶液,之后进行实验,研究阴离子和腐殖酸对游离氯和游离溴分解的影响.
在反应器内加入一定体积的实际海水或人造海水,再加入次氯酸钠溶液,搅拌混匀10 min,之后进行实验,研究实际海水和人造海水对游离氯和游离溴分解的影响.
1.3 测定方法
紫外光强用LS125紫外辐照计(深圳林上科技有限公司)测定,实验中光强稳定在约0.9 mW·cm−2;pH用雷磁PHS-3C型pH计(上海仪电科学仪器股份有限公司)测定;盐度使用AZ8371型盐度计(中国台湾衡欣科技股份有限公司)测定;摩尔吸光系数、游离氯和游离溴浓度利用752N紫外可见分光光度计(上海仪电分析仪器有限公司)测定;DOC用TOC-V总有机碳分析仪(日本岛津公司)测定. 游离氯和游离溴浓度参照DPD分光光度法(HJ 586—2010)测定,以Cl2(mg·L−1)计.
1.4 计算方法
利用一级动力学方程(式(5))拟合游离氯或游离溴的分解速率.
ln(C0Ct)=KobsQ (5) 式中,C0和Ct分别为游离氯或游离溴的初始浓度和经过t时间后的质量浓度,mg·L−1;Kobs为表观一级反应速率常数,cm2·mJ−1;Q为紫外剂量,mJ·cm−2.
如果假设体系中某化合物W为唯一的吸光物质,那么其在254 nm处的光解速率可以由式(6)描述[20].
dCwdt=−ΦI0(1−10−ϵCwl)z (6) 式中,Φ为化合物W的量子产率,mol·Einstein−1;I0为光强度,mEinstein·(cm2·s)−1;ε为化合物W在254 nm处的摩尔吸光系数,L·(mol·cm)−1;l为光路长度,cm;z为溶液深度,cm;Cw为化合物W的摩尔浓度,mol·L−1. 本研究中I0为0.191×10−5 mEinstein·(cm2·s)−1,同时由于使用了平行光束仪(l=z),且实验中εCwl较小,根据泰勒一阶展开式,式(6)可近似为式(7),再经过简单变换即式(8)—式(9). 因此,游离氯或游离溴的直接光解速率常数Kact可以通过式(8)计算;表观量子产率Φobs可以通过式(9)计算.
dCwdt=−2.303ΦI0εCw=−KCw (7) Kact=2.303ΦactI0ε (8) Φobs=Kobs2.303I0ε (9) 2. 结果与讨论 (Results and discussion)
2.1 pH对游离氯和游离溴分解的影响
海水的典型pH一般在8.0左右,呈弱碱性. 在常规水处理的pH范围内,游离氯和游离溴主要是以HOX和OX−的形态存在(X=Cl, Br),其比例与pH的具体大小密切相关[16,21](图1). 在pH为6时,游离氯(pKa=7.5)中HOCl占比为96.9%,游离溴(pKa=8.8)中HOBr占比为99.8%. 在pH为8时,游离氯中OCl−占主要部分,占比76.0%;游离溴中HOBr占主要部分,占比86.3%. 在pH为10时,游离氯和游离溴中分别以OCl−和OBr−为主,占比分别为99.7%和94.1%.
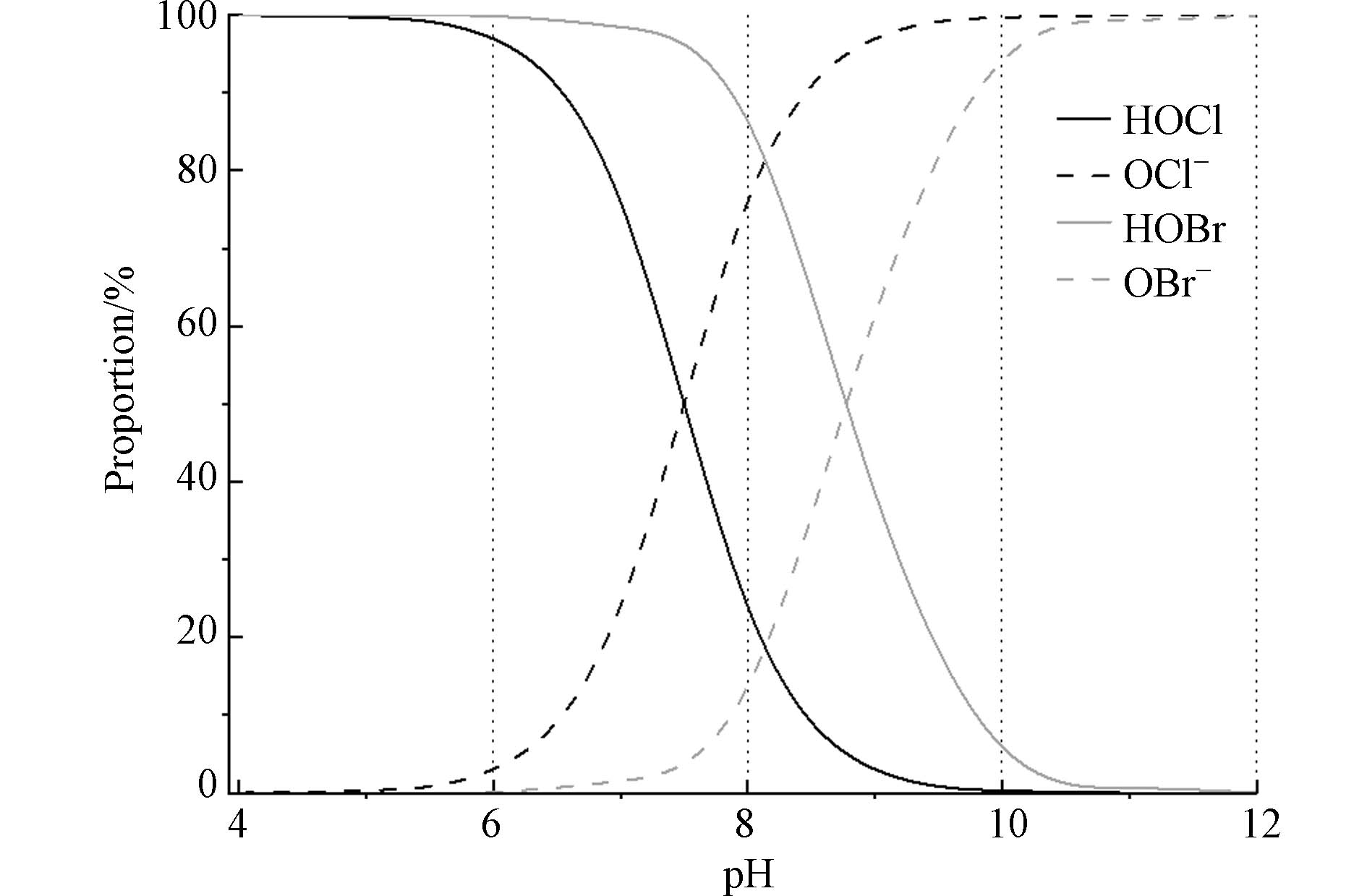 图 1 不同pH下游离氯(HOCl/OCl−)和游离溴(HOBr/OBr−)的各形态在水中所占的比例Figure 1. Proportion of various forms of free chlorine (HOCl/OCl−) and free bromine (HOBr/OBr−) in water at different pH
图 1 不同pH下游离氯(HOCl/OCl−)和游离溴(HOBr/OBr−)的各形态在水中所占的比例Figure 1. Proportion of various forms of free chlorine (HOCl/OCl−) and free bromine (HOBr/OBr−) in water at different pHpH的变化引起物质存在形态的变化,进而可能会影响物质分解速率的变化. 因此,考察了游离氯、游离溴在初始质量浓度为10 mg·L−1、不同pH条件下游离氯和游离溴的分解速率,结果如图2所示.
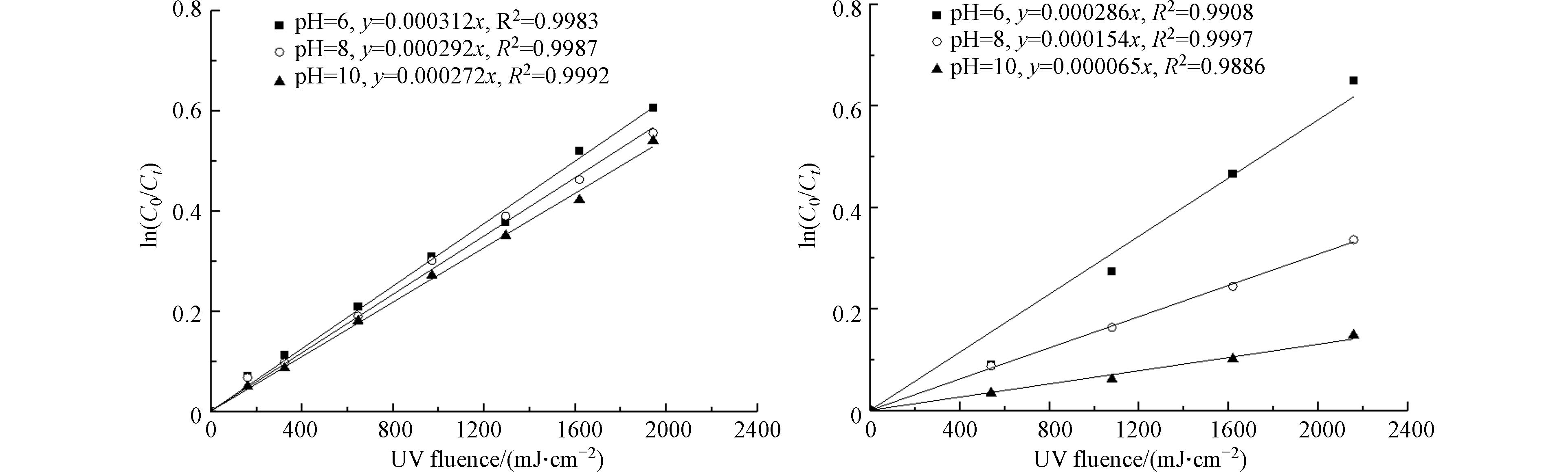 图 2 pH对游离氯(a)和游离溴(b)分解速率的影响Figure 2. Effect of pH on decomposition rates of free chlorine(a) and free bromine(b)
图 2 pH对游离氯(a)和游离溴(b)分解速率的影响Figure 2. Effect of pH on decomposition rates of free chlorine(a) and free bromine(b)由图2(a)可以看出,在低压紫外的照射下,游离氯的分解速率常数随pH下降略有上升. 当pH为10时,游离氯分解的表观一级反应速率常数Kobs为0.000272 cm2·mJ−1;当pH为6时,游离氯分解的Kobs为0.000312 cm2·mJ−1,上升了14.7%. 这表明在254 nm的紫外波长下,低pH下以HOCl为主要存在形态的游离氯相对容易分解. 由图2(b)可以看出,游离溴的分解速率随pH的下降明显上升. 当pH为10时,游离溴分解的Kobs为0.000065 cm2·mJ−1;当pH为6时,游离氯分解的Kobs为0.000286 cm2·mJ−1,上升了3.4倍. 这表明游离溴的分解速率更容易受到pH变化的影响,且低pH下以HOBr为主要存在形态的游离溴分解速率更快. 这主要是由于不同形态的游离氯和游离溴的摩尔吸光系数和量子产率以及自由基反应过程的不同[11]. 游离氯和游离溴分解的表观一级反应速率常数Kobs是由直接光解速率常数(Kact)和自由基反应速率常数(Kradical)决定的. 其中自由基反应速率常数(Kradical)是由自由基引起的间接光解速率常数(Krad-indirect)和再生成速率常数(Krad-reformation)决定的(式(10))[20].
Kobs=Kact+Kradical=Kact+Krad−indirect−Krad−reformation (10) 为了进一步验证机理,本研究测定了HOCl、OCl−、HOBr和OBr−的摩尔吸光系数(图3),并利用实验所获得的Kobs和式(9)计算相应物质的表观量子产率(Φobs),利用已有研究所报道的实际量子产率(Φact),通过计算Φact/Φobs获得物质直接光解的比例. 由表1可见,HOCl、OCl−、HOBr和OBr−直接光解分别占比54.4%、54.5%、60.6%和45.6%. 这表明无论是游离氯还是游离溴,直接光解和自由基反应对其分解的影响均不可忽视.
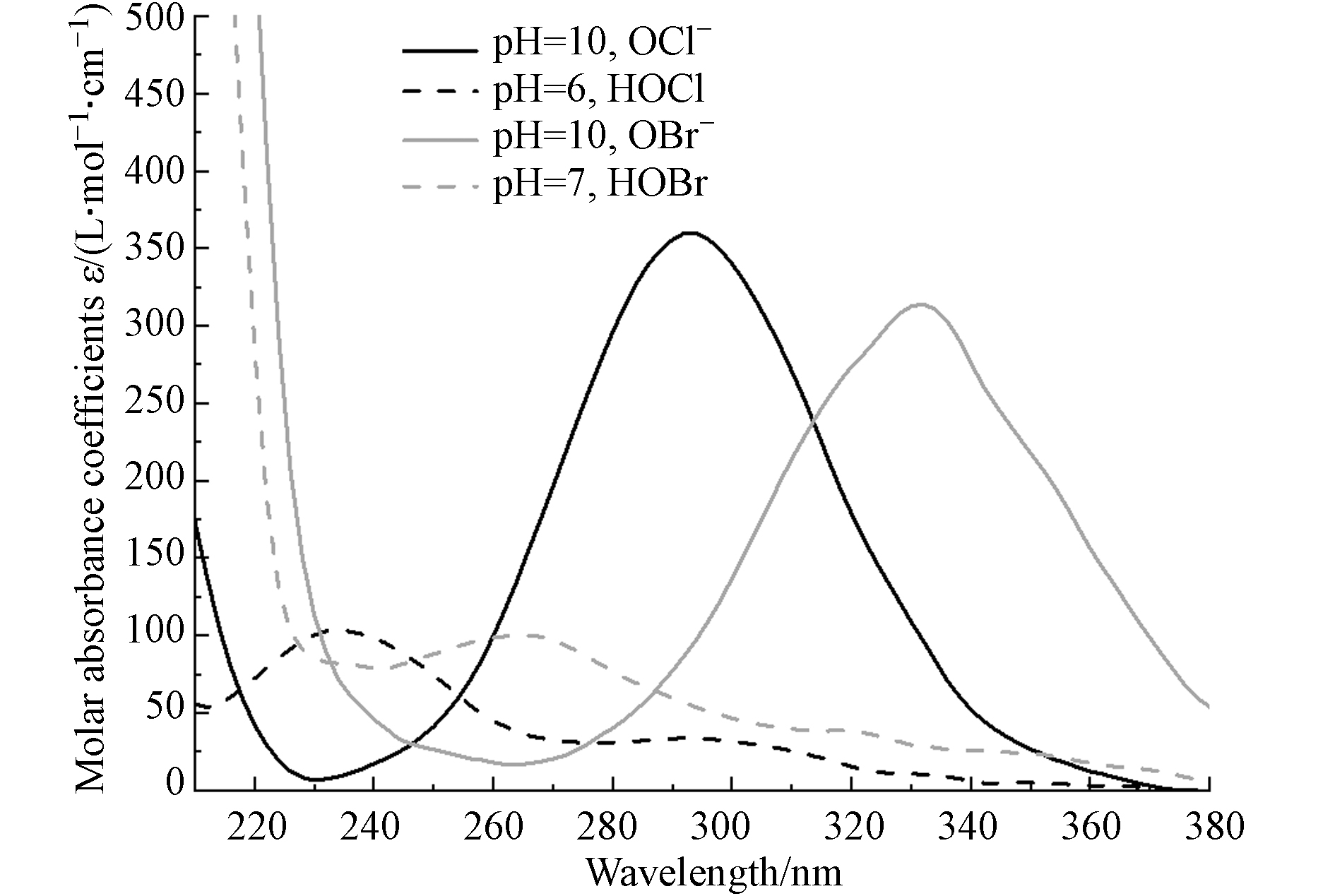 图 3 不同波长下游离氯(HOCl/OCl−)和游离溴(HOBr/OBr−)的摩尔吸光系数Figure 3. Molar absorbance coefficients of free chlorine (HOCl/OCl−) and free bromine (HOBr/OBr−) at different wavelengths表 1 游离氯和游离溴的量子产率和分解速率常数Table 1. Quantum yields and decomposition rate constants for free chlorine and free bromine
图 3 不同波长下游离氯(HOCl/OCl−)和游离溴(HOBr/OBr−)的摩尔吸光系数Figure 3. Molar absorbance coefficients of free chlorine (HOCl/OCl−) and free bromine (HOBr/OBr−) at different wavelengths表 1 游离氯和游离溴的量子产率和分解速率常数Table 1. Quantum yields and decomposition rate constants for free chlorine and free bromine氧化剂 Oxidants ε254nm /(L·(mol·cm)−1) Φobs-254nm/(mol·Es−1) Φact-254nm/(mol·Es−1) Kobs/(cm2·mJ−1) Kact/(cm2·mJ−1) Φact/Φobs HOCl 62 1.14 0.62[20] 0.000 312 0.000 169 0.544 OCl- 61 1.01 0.55[20] 0.000 272 0.000 147 0.545 HOBr 92 0.71 0.43[11] 0.000 286 0.000 174 0.606 OBr- 26 0.57 0.26[11] 0.000 065 0.000 030 0.456 | Show Table DownLoad:
CSV
DownLoad:
CSV
此外,从HOCl到OCl−转变的分解速率变化来看,Kobs下降了0.000040 cm2·mJ−1,而Kact仅下降了0.000022 cm2·mJ−1,说明pH变化不仅仅影响游离氯直接光解(55%)还影响自由基反应(45%). 类似地,从HOBr转变为OBr−,Kobs下降了0.000221 cm2·mJ−1,而Kact下降了0.000144 cm2·mJ−1,也说明了pH通过影响游离溴的直接光解(65.2%)和自由基反应(34.8%)来影响分解速率.
2.2 无机阴离子对游离氯和游离溴分解的影响
地表水,尤其是海水中,存在着各种无机阴离子. 海水中常见的无机阴离子有氯离子、硫酸根、碳酸氢根、硝酸盐、磷酸盐、硼酸盐(以H3BO3的形式存在)和氟离子等. 但这些无机阴离子在紫外照射下对游离氯,尤其是对游离溴的分解速率的影响仍不清晰. 本研究为模拟海水中无机阴离子的影响,除设置氯离子质量浓度为2 g·L−1(56 mmol·L−1)和20 g·L−1(560 mmol·L−1)外,其余离子设置为5 mmol·L−1或50 mmol·L−1进行实验. 由于海水的典型pH在8.0左右,因此实验中pH保持为8.0.
由图4(a)可见,在pH 8时,游离氯在低压紫外照射下的分解速率随着Cl−浓度的增加而降低,与空白对照相比,20 g·L−1 Cl−导致游离氯分解速率降低13.7%. 这可能是Cl−与含氯自由基(Cl·、Cl2−·、ClO·等)和羟基自由基(HO·)反应导致的[20]. 一方面,Cl−的存在能够与游离氯直接光解产生的Cl·反应生成Cl2−·(式(11)),从而减弱了Cl·与游离氯的间接光解反应(式(12)—式(13)),导致分解速率下降;另一方面,高浓度的Cl−对体系内HO·的清除反应(式(14))则抑制了HO·与游离氯的间接光解反应(式(15)—式(16)),也会导致分解速率的下降. 但是Cl−与HO·发生的主要反应(式(14)) 是可逆反应,刘宇程等[22]的理论计算研究表明,当Cl−的浓度大于50 g·L−1时,其正反应(kf)占优势而逆反应(kr)较弱. 由于海水中的Cl−浓度约为20 g·L−1,因此在该浓度条件下,Cl−不能够起到清除HO·的作用.
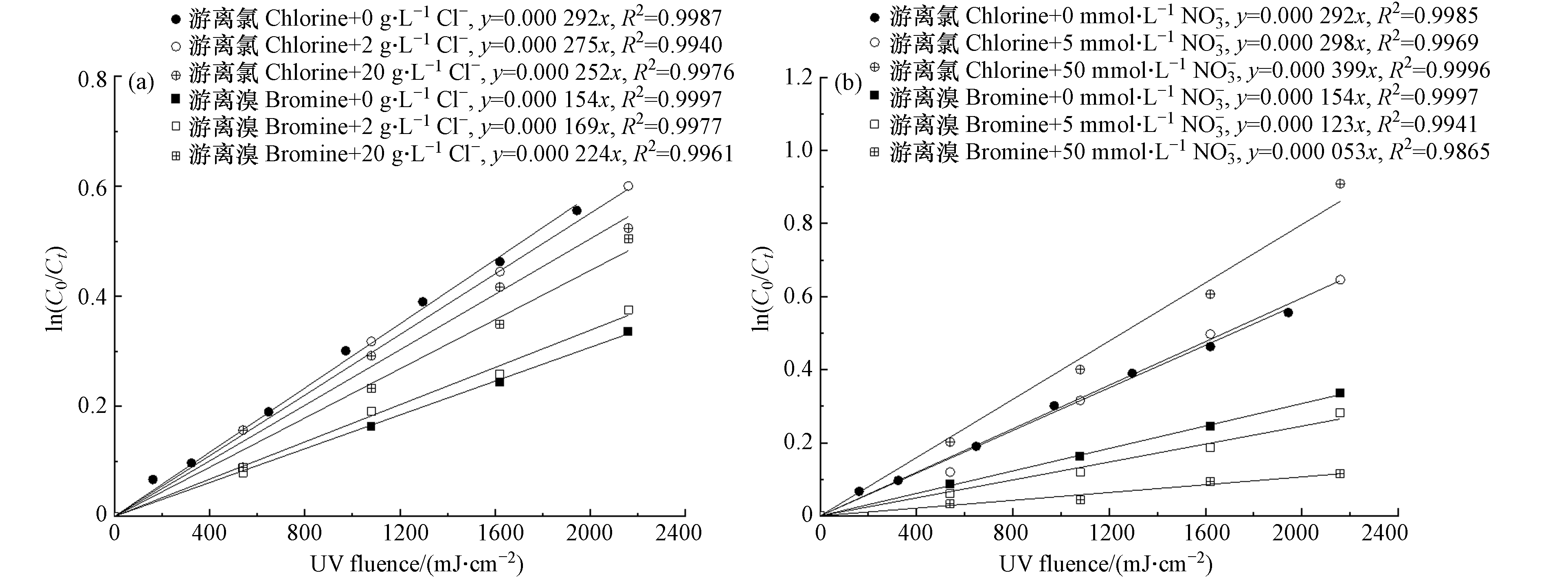 图 4 (a) 不同Cl-浓度下游离氯和游离溴的分解动力学;(b) 不同NO3-浓度下游离氯和游离溴的分解动力学Figure 4. (a) Decomposition kinetics of free chlorine and free bromine at different Cl- concentrations,(b) Decomposition kinetics of free chlorine and free bromine at different NO3- concentrations
图 4 (a) 不同Cl-浓度下游离氯和游离溴的分解动力学;(b) 不同NO3-浓度下游离氯和游离溴的分解动力学Figure 4. (a) Decomposition kinetics of free chlorine and free bromine at different Cl- concentrations,(b) Decomposition kinetics of free chlorine and free bromine at different NO3- concentrationsCl⋅+Cl−→Cl−2⋅k=8.5×109L⋅(mol⋅s)−1 (11) Cl⋅+HOCl→H++Cl−+ClO⋅k=3.0×109L⋅(mol⋅s)−1 (12) Cl⋅+OCl−→Cl−+ClO⋅k=8.2×109L⋅(mol⋅s)−1 (13) HO⋅+Cl−⇆ClOH⋅−kf=4.3×109L⋅(mol⋅s)−1,kr=6.1×109s−1 (14) HO⋅+HOCl→H2O+ClO⋅k=2.0×109L⋅(mol⋅s)−1 (15) HO⋅+OCl−→OH−+ClO⋅k=8.8×109L⋅(mol⋅s)−1 (16) 由图4(b)可见,与Cl−的影响规律相反,游离氯在紫外照射下的分解速率随着
NO−3 NO−3 NO−3 NO−3 NO−3 NO−3 NO−3+hv→NO2⋅+O−⋅ (17) O−⋅+H2O→HO⋅+OH−k=9.4×107s−1 (18) 由图4(a)可见,在pH为8时,游离溴的分解速率随着Cl−浓度的增加而上升,20 g·L−1 Cl−导致游离溴分解速率上升45.4%. 这与体系内含溴自由基(Br·、
Br⋅−2 Br⋅−2 Br⋅−2 由图4(b)可见,游离溴的分解速率随着NO3−浓度的增加而下降,50 mmol·L−1
NO−3 NO−3 NO−3 NO−3 Br⋅−2 Br⋅+Br−→Br⋅−2k=1.2×1010L⋅(mol⋅s)−1 (19) HO⋅+Br−→BrOH⋅−k=1.1×1010L⋅(mol⋅s)−1 (20) Br⋅+HOBr→BrO⋅+HBrk=1.0×108L⋅(mol⋅s)−1 (21) HO⋅+HOBr→BrO⋅+H2Ok=2.0×109L⋅(mol⋅s)−1 (22) BrOH⋅−+Br−→Br⋅−2+OH−k=1.9×108L⋅(mol⋅s)−1 (23) Br⋅−2+HO⋅→HOBr+Br−k=1.0×109L⋅(mol⋅s)−1 (24) Br⋅+Cl−→BrCl⋅−k=1.0×108L⋅(mol⋅s)−1 (25) BrCl⋅−+HO⋅→BrCl+H2Ok=1.0×109L⋅(mol⋅s)−1 (26) 由图5可见,在pH为8的条件下,水中常见的其他阴离子(
SO2−4 HCO−3 HPO2−4 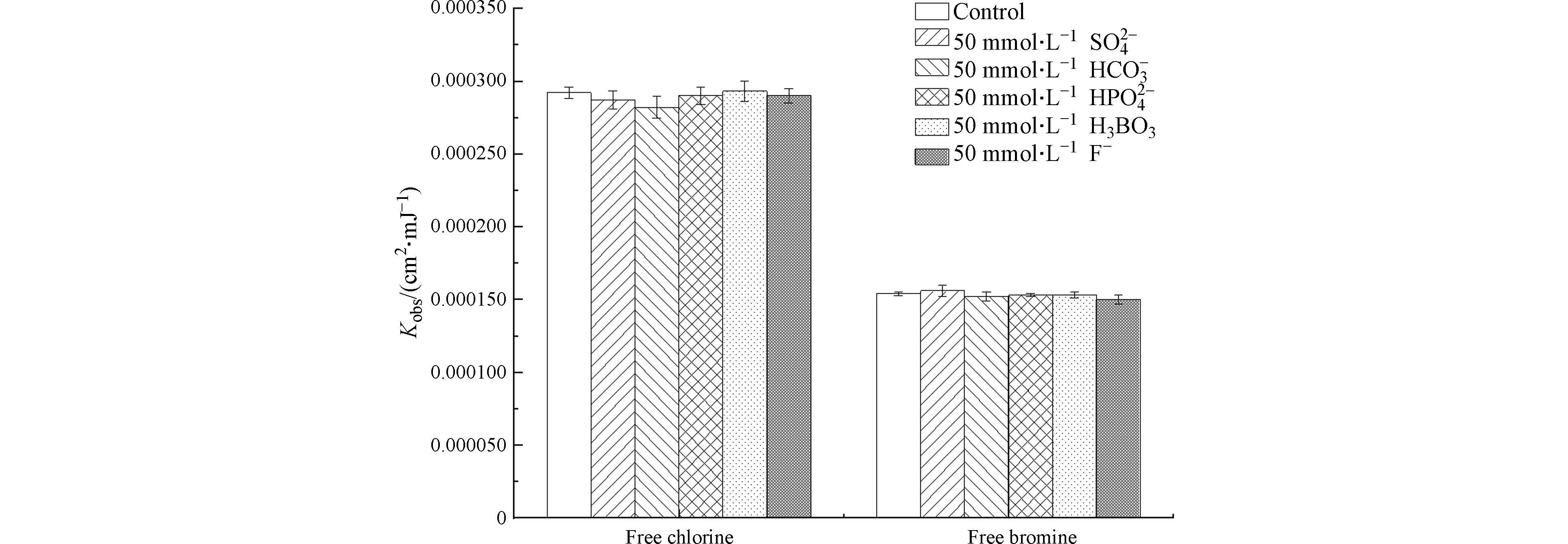 图 5 无机阴离子对游离氯和游离溴分解速率常数的影响Figure 5. Effect of inorganic anions on decomposition rate constants of free chlorine and free bromine
图 5 无机阴离子对游离氯和游离溴分解速率常数的影响Figure 5. Effect of inorganic anions on decomposition rate constants of free chlorine and free bromine2.3 腐殖酸对游离氯和游离溴分解的影响
腐殖酸是一种在天然水体中广泛存在的有机物[25],常作为水体环境中有机组分的代表物,其对紫外照射下游离氯分解的影响已经引起了诸多关注. 田芳等[14]研究了在中压紫外照射下腐殖酸对游离氯分解的影响,发现腐殖酸浓度与游离氯的分解速率常数呈线性关系,随腐殖酸浓度的升高其分解速率上升. 但是,在低压紫外照射下,腐殖酸对游离氯分解的影响尚存争议. 一方面,腐殖酸可以作为紫外滤层吸收紫外光而抑制游离氯的分解;另一方面,腐殖酸可以与游离氯反应加速其分解[26]. 同时,腐殖酸对游离溴分解的影响的研究较为缺乏. 因此,本研究以0—4.28 mg·L−1的腐殖酸(以DOC计)作为影响因素研究紫外照射下游离氯和游离溴的分解规律,以进一步明确腐殖酸对游离氯和游离溴分解的影响.
由图6(a)可见,随着腐殖酸浓度的升高,游离氯的分解速率先下降再上升,当腐殖酸浓度为0.11 mg·L−1时,游离氯的分解的Kobs为0.000250 cm2·mJ−1,与空白组相比降低了14.3%;当腐殖酸浓度为4.28 mg·L−1时,分解的Kobs达到0.000336 cm2·mJ−1,与空白组相比升高了15.1%. 由图6(b)可见,游离溴的分解速率常数随腐殖酸浓度的升高先短暂下降后迅速上升. 当腐殖酸浓度为0.11 mg·L−1时,游离溴的分解的Kobs为0.000128 cm2·mJ−1,与空白组相比降低了16.9%;当腐殖酸浓度为4.28 mg·L−1时,其分解的Kobs达到0.000525 cm2·mJ−1,是空白组的3.4倍.
 图 6 (a) 不同腐殖酸浓度下游离氯的分解动力学;(b) 不同腐殖酸浓度下游离溴的分解动力学Figure 6. (a) Decomposition kinetics of free chlorine at different humic acid concentrations, (b) Decomposition kinetics of free bromine at different humic acid concentrations
图 6 (a) 不同腐殖酸浓度下游离氯的分解动力学;(b) 不同腐殖酸浓度下游离溴的分解动力学Figure 6. (a) Decomposition kinetics of free chlorine at different humic acid concentrations, (b) Decomposition kinetics of free bromine at different humic acid concentrations由此可见,腐殖酸在低浓度时可能是以紫外滤层吸收紫外光占据主导地位,随着浓度的升高,腐殖酸作为反应物起到主要作用. 以威海乳山湾近海海域为例[27],该海域海水的DOC含量范围为0.70—3.19 mg·L−1,所以可以推测海水中的有机物主要是起到促进分解的作用.
此外,腐殖酸对游离溴分解的促进作用明显大于游离氯. 造成这种结果的可能原因是腐殖酸与游离溴更容易反应,从而促进了它的分解. 已有研究表明[28-29],相比于游离氯,某些具有特殊结构的有机物更容易被游离溴氧化,如酚类、胺类、含硫类化合物等. 其中,酚羟基是腐殖酸中普遍具有的重要官能团[30]. 为了证明这一假设,在pH为8的条件下,研究了无紫外照射下的游离氯和游离溴与腐殖酸的反应,发现游离溴与腐殖酸的反应速率明显高于游离氯与腐殖酸的反应(图7). 这说明,腐殖酸与游离溴更容易反应是腐殖酸对游离溴分解的促进作用较大的重要原因.
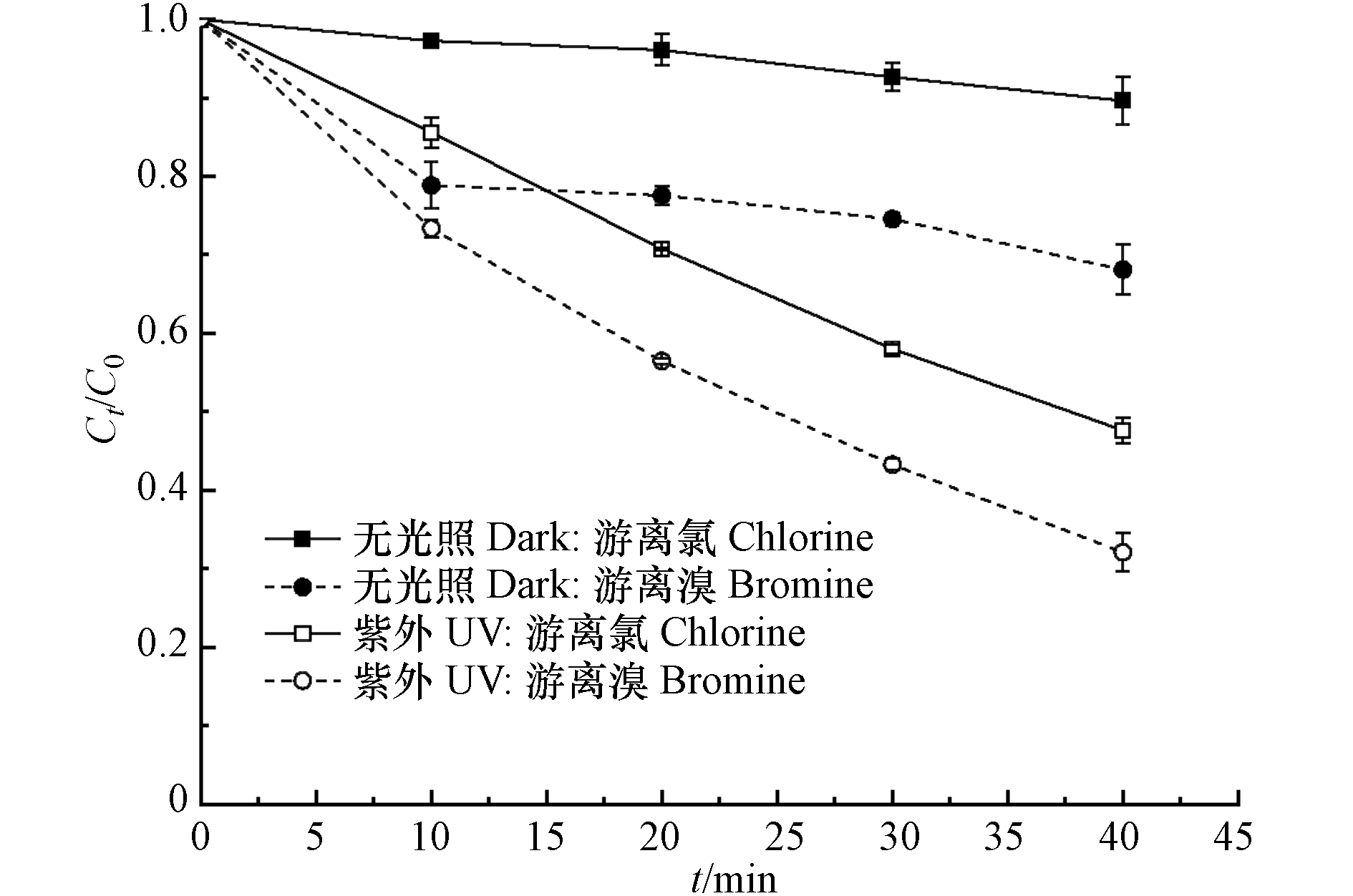 图 7 4.28 mg·L−1腐殖酸对游离氯和游离溴分解的影响Figure 7. Effect of 4.28 mg·L−1 humic acid on decomposition of free chlorine and free bromine
图 7 4.28 mg·L−1腐殖酸对游离氯和游离溴分解的影响Figure 7. Effect of 4.28 mg·L−1 humic acid on decomposition of free chlorine and free bromine2.4 实际海水对游离氯和游离溴分解的影响
实际海水中同时存在上述研究中所涉及的各类无机阴离子和有机物,能够从多个方面影响游离氯和游离溴的分解速率. 由于海水淡化的RO系统中游离氯的摩尔浓度低于海水中溴离子的摩尔浓度[31],此时系统中的游离氯能够被溴离子完全转化为游离溴. 因此,本研究设置游离溴浓度为10 mg·L−1,以实际海水和人造海水为反应液,研究低压紫外照射工艺在海水中的应用效果,为工艺的实际应用提供参考.
由图8可见,人造海水中游离溴的分解的Kobs为0.000237 cm2·mJ−1,与空白相比升高了53.9%,与添加20 g·L−1 Cl−的游离溴的分解速率差别不大(图4(a)).
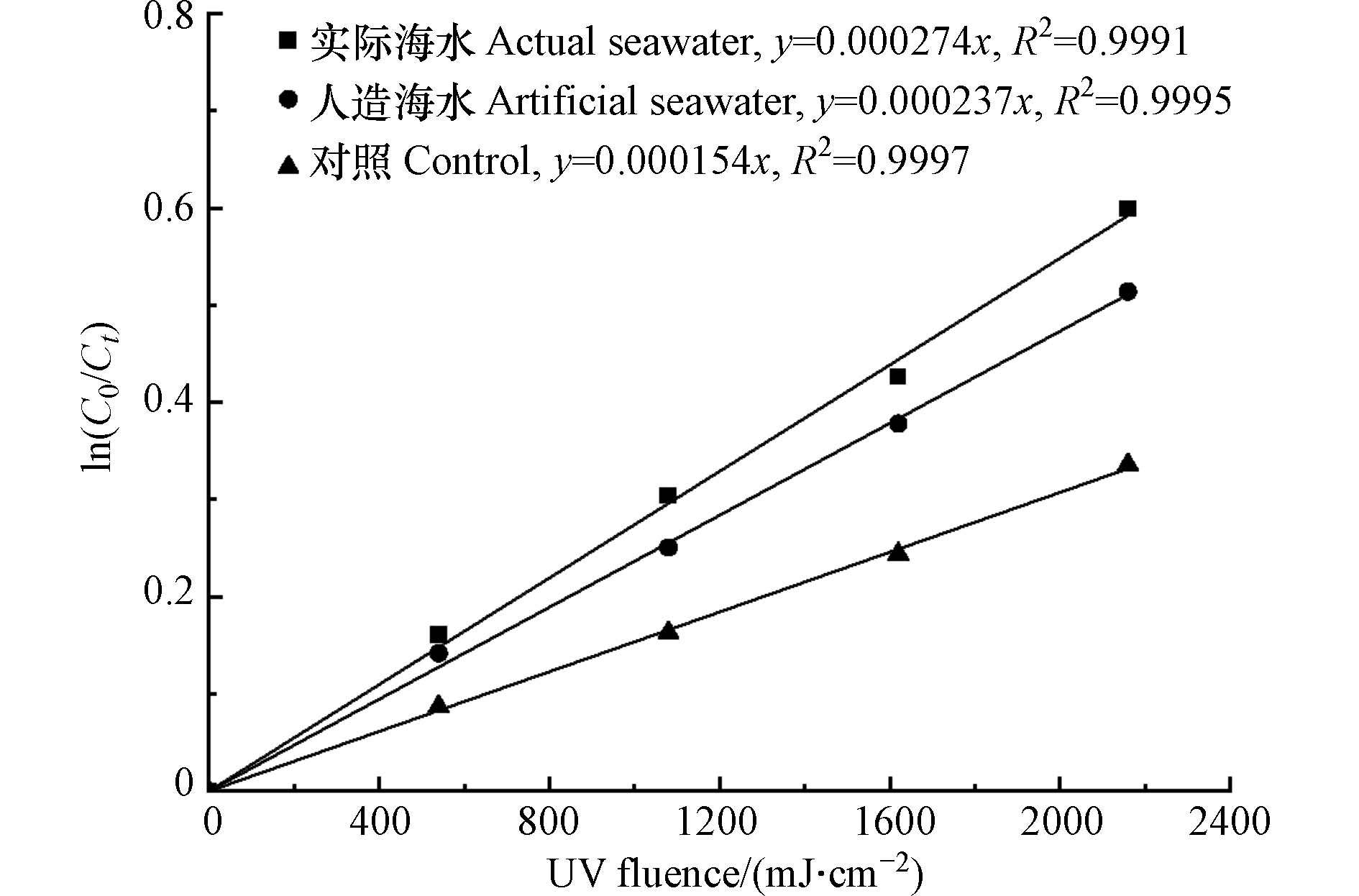 图 8 实际海水和人造海水中游离溴在紫外线照射下的分解动力学Figure 8. Decomposition kinetics of free bromine under UV irradiation in actual and artificial seawater
图 8 实际海水和人造海水中游离溴在紫外线照射下的分解动力学Figure 8. Decomposition kinetics of free bromine under UV irradiation in actual and artificial seawater这表明海水无机阴离子中Cl−对游离溴的分解起着重要的促进作用. 实际海水中游离溴的分解的Kobs为0.000274 cm2·mJ−1,与人造海水相比升高了15.6%. 这可能是由于海水中的有机物对游离溴分解起到了促进作用. Cho等[31]的研究表明,在低压紫外照射下,实际海水(DOC为2.2 mg·L−1)中游离氯(溴)的分解速率是人造海水的1.6倍. 虽然研究结果有一定的差异,但以上结果均证明在实际海水中,有机物对于游离溴在低压紫外光照射下的分解能够起到一定的促进作用. 这与腐殖酸影响实验中所得的结果基本相符.
3. 结论(Conclusion)
(1)在低压紫外照射下,随着pH降低,游离氯和游离溴的分解速率升高,其中pH降低对游离溴分解速率促进作用更大. pH通过影响直接光解和自由基反应过程影响游离氯和游离溴的分解速率. 对于海水淡化来说,通过适当降低RO工艺的进水pH,可以提高紫外线去除游离溴的效率.
(2)高浓度Cl−能够抑制游离氯的分解而促进游离溴的分解,高浓度
NO−3 SO2−4 HCO−3 HPO2−4 (3)低浓度腐殖酸可抑制游离溴和游离氯的分解,高浓度腐殖酸则会促进其分解,且对游离溴分解的促进程度强于游离氯.
(4)在实际海水中,一定浓度的Cl−和有机物的存在能够促进游离溴在紫外照射下的分解.
-

图 1 监测点(A),烈山区政府站点(B),58113气象站(C)分布图
Figure 1. The locations of observation site (A), LSQ air quality monitoring station (B), and the meteorological monitoring station numbered 58113(C)

图 2 PAN及其相关物种在秋季和夏季的日变化
Figure 2. Diurnal variations of PAN and its related species in autumn and summer
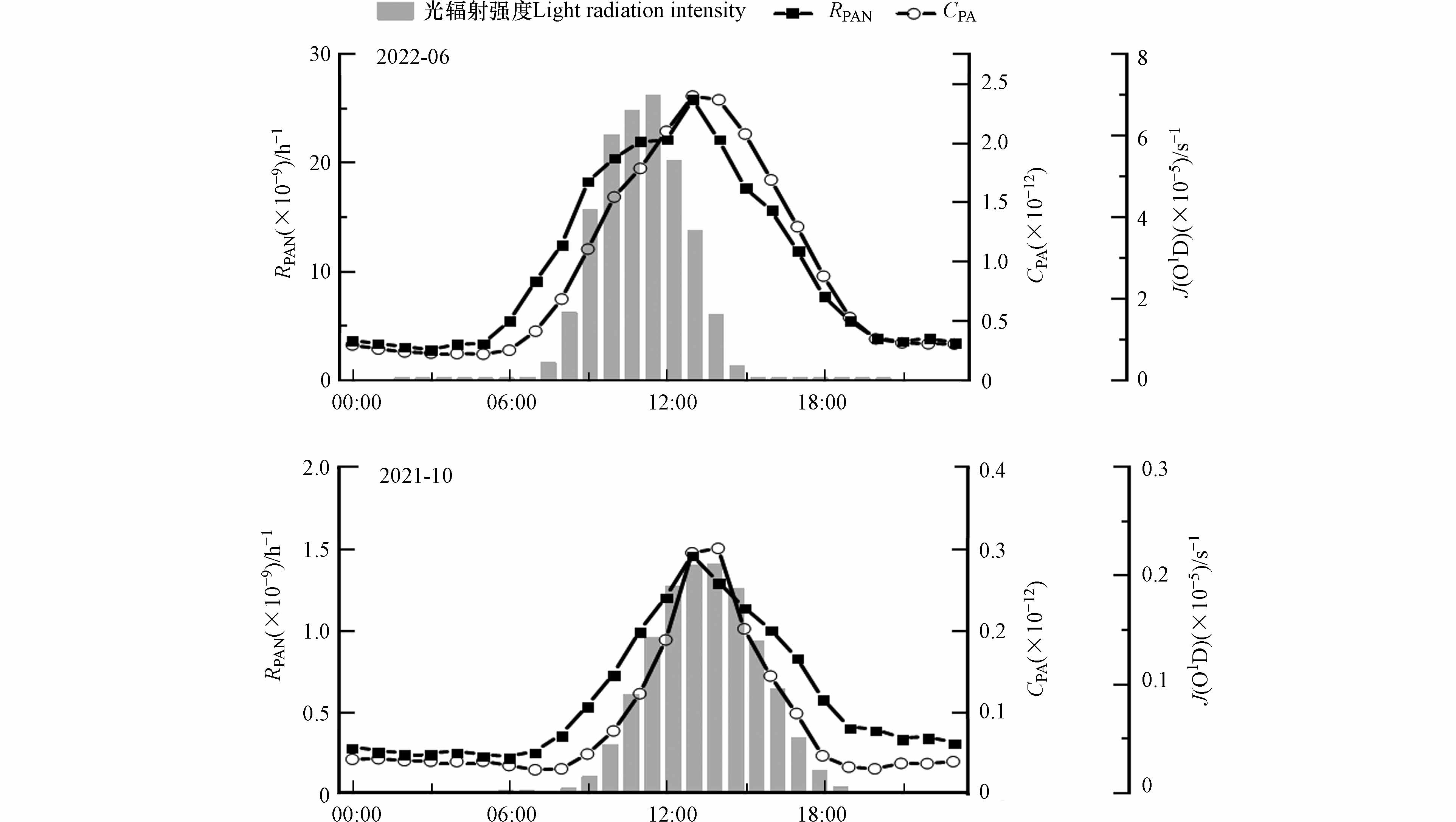
图 3 PA自由基及PAN生成速率在秋季和夏季的日变化
Figure 3. Diurnal variations of Peroxyacetyl Radical and RPAN in autumn and summer

图 5 PAN与NO2散点关系图
Figure 5. Scatter plots showing the relationships between PAN and NO2

图 6 PAN与NO/NO2的散点关系图
Figure 6. Scatterplots showing the relationships between PAN and NO/NO2

图 7 2021年10月和2022年6月淮北市气团后向轨迹聚类分析及PAN的PSCF值分布情况
Figure 7. Clustering analysis of air masses backward trajectory in Huaibei and the PSCF value distribution of PAN in October of 2021 and June of 2022
表 1 国内不同地区PAN浓度均值和最大值对比
Table 1. PAN concentrations in this study and comparison with the other sites in the urban regions
区域Region 城市City 观测时间Observation time 平均值/(×10−9)Average 最大值/(×10−9)Maximum 参考文献 京津冀 北京 2021年7月 0.89 — [23] 北京 2010年1—3月 0.70 3.51 [22] 京津冀 天津 2018年9月 0.93 — [8] 北京 2016年11月—2017年1月 1.1 7.1 [24] 苏皖鲁豫交界地区 淮北 2021年10月 0.42 1.44 本研究 淮北 2022年6月 1.87 5.72 本研究 青岛 2018年10—11月 0.75 5.83 [13] 2019年7—8月 0.81 7.82 [13] 长三角 上海 2017 0.7 7.0 [9] 合肥 2016年8月 1.10 4.65 [25] 浙江 2019年6—10月 0.66 4.35 [26] 珠三角 深圳 2018年9—10月 0.56 3.90 [20] 厦门 2020年3—11月 0.92 — [14]
下载: 导出CSV
-
[1] 柴发合. 我国大气污染治理历程回顾与展望[J]. 环境与可持续发展, 2020, 45(3): 5-15. doi: 10.19758/j.cnki.issn1673-288x.202003005 CHAI F H. Review and prospect on the atmospheric pollution control in China[J]. Environment and Sustainable Development, 2020, 45(3): 5-15 (in Chinese). doi: 10.19758/j.cnki.issn1673-288x.202003005
[2] 姜华, 高健, 李红, 等. 我国大气污染协同防控理论框架初探[J]. 环境科学研究, 2022, 35(3): 601-610. doi: 10.13198/j.issn.1001-6929.2022.01.11 JIANG H, GAO J, LI H, et al. Preliminary research on theoretical framework of cooperative control of air pollution in China[J]. Research of Environmental Sciences, 2022, 35(3): 601-610 (in Chinese). doi: 10.13198/j.issn.1001-6929.2022.01.11
[3] 张涵, 姜华, 高健, 等. PM2.5与臭氧污染形成机制及协同防控思路[J]. 环境科学研究, 2022, 35(3): 611-620. ZHANG H, JIANG H, GAO J, et al. Formation mechanism and management strategy of cooperative control of PM2.5 and O3[J]. Research of Environmental Sciences, 2022, 35(3): 611-620 (in Chinese).
[4] 花丛, 江琪, 迟茜元, 等. 我国中东部地区2015—2020年夏半年PM2.5和臭氧复合污染气象特征分析[J]. 环境科学研究, 2022, 35(3): 650-658. HUA C, JIANG Q, CHI X Y, et al. Meteorological characteristics of PM2.5-O3 air combined pollution in central and Eastern China in the summer half years of 2015-2020[J]. Research of Environmental Sciences, 2022, 35(3): 650-658 (in Chinese).
[5] 栗泽苑, 杨雷峰, 华道柱, 等. 2013—2018年中国近地面臭氧浓度空间分布特征及其与气象因子的关系[J]. 环境科学研究, 2021, 34(9): 2094-2104. doi: 10.13198/j.issn.1001-6929.2021.06.16 LI Z Y, YANG L F, HUA D Z, et al. Spatial pattern of surface ozone and its relationship with meteorological variables in China during 2013-2018[J]. Research of Environmental Sciences, 2021, 34(9): 2094-2104 (in Chinese). doi: 10.13198/j.issn.1001-6929.2021.06.16
[6] 黄小刚, 赵景波, 曹军骥, 等. 中国城市O3浓度时空变化特征及驱动因素[J]. 环境科学, 2019, 40(3): 1120-1131. HUANG X G, ZHAO J B, CAO J J, et al. Spatial-temporal variation of ozone concentration and its driving factors in China[J]. Environmental Science, 2019, 40(3): 1120-1131 (in Chinese).
[7] 汪旭颖, 严刚, 雷宇, 等. 苏皖鲁豫交界地区大气污染形势和问题分析[J]. 环境保护, 2020, 48(17): 45-48. doi: 10.14026/j.cnki.0253-9705.2020.17.008 WANG X Y, YAN G, LEI Y, et al. The status and problems of air pollution of the border area of Jiangsu, Anhui, Shandong and Henan[J]. Environmental Protection, 2020, 48(17): 45-48 (in Chinese). doi: 10.14026/j.cnki.0253-9705.2020.17.008
[8] QIU Y L, LIN W L, LI K, et al. Vertical characteristics of peroxyacetyl nitrate (PAN) from a 250-m tower in Northern China during September 2018[J]. Atmospheric Environment, 2019, 213: 55-63. doi: 10.1016/j.atmosenv.2019.05.066 [9] ZHANG G, JING S G, XU W Y, et al. Simultaneous observation of atmospheric peroxyacetyl nitrate and ozone in the megacity of Shanghai, China: Regional transport and thermal decomposition[J]. Environmental Pollution, 2021, 274: 116570. doi: 10.1016/j.envpol.2021.116570 [10] SUN M, ZHOU Y, WANG Y F, et al. Seasonal discrepancies in peroxyacetyl nitrate (PAN) and its correlation with ozone and PM2.5: Effects of regional transport from circumjacent industrial cities[J]. Science of the Total Environment, 2021, 785: 147303. doi: 10.1016/j.scitotenv.2021.147303 [11] ZHANG G, MU Y J, LIU J F, et al. Seasonal and diurnal variations of atmospheric peroxyacetyl nitrate, peroxypropionyl nitrate, and carbon tetrachloride in Beijing[J]. Journal of Environmental Sciences, 2014, 26(1): 65-74. doi: 10.1016/S1001-0742(13)60382-4 [12] ZHANG G, MU Y J, ZHOU L X, et al. Summertime distributions of peroxyacetyl nitrate (PAN) and peroxypropionyl nitrate (PPN) in Beijing: Understanding the sources and major sink of PAN[J]. Atmospheric Environment, 2015, 103: 289-296. doi: 10.1016/j.atmosenv.2014.12.035 [13] LIU Y H, SHEN H Q, MU J S, et al. Formation of peroxyacetyl nitrate (PAN) and its impact on ozone production in the coastal atmosphere of Qingdao, North China[J]. The Science of the Total Environment, 2021, 778: 146265. doi: 10.1016/j.scitotenv.2021.146265 [14] LIU T T, CHEN G J, CHEN J S, et al. Seasonal characteristics of atmospheric peroxyacetyl nitrate (PAN) in a coastal city of Southeast China: Explanatory factors and photochemical effects[J]. Atmospheric Chemistry and Physics, 2022, 22(7): 4339-4353. doi: 10.5194/acp-22-4339-2022 [15] XU X B, ZHANG H L, LIN W L, et al. First simultaneous measurements of peroxyacetyl nitrate (PAN) and ozone at Nam Co in the central Tibetan Plateau: Impacts from the PBL evolution and transport processes[J]. Atmospheric Chemistry and Physics, 2018, 18(7): 5199-5217. doi: 10.5194/acp-18-5199-2018 [16] GAO T Y, HAN L, WANG B, et al. Peroxyacetyl nitrate observed in Beijing in August from 2005 to 2009[J]. Journal of Environmental Sciences, 2014, 26(10): 2007-2017. doi: 10.1016/j.jes.2014.08.002 [17] ZHANG J, XU Z, YANG G, et al. Peroxyacetyl nitrate (PAN) and peroxypropionyl nitrate (PPN) in urban and suburban atmospheres of Beijing, China[J]. Atmospheric Chemistry and Physics, 2011, 11(11): 8173-8206. [18] YUAN J, LING Z H, WANG Z, et al. PAN–precursor relationship and process analysis of PAN variations in the Pearl River Delta region[J]. Atmosphere, 2018, 9(10): 372. doi: 10.3390/atmos9100372 [19] LIU L, WANG X F, CHEN J M, et al. Understanding unusually high levels of peroxyacetyl nitrate (PAN) in winter in Urban Jinan, China[J]. Journal of Environmental Sciences, 2018, 71: 249-260. doi: 10.1016/j.jes.2018.05.015 [20] XIA S Y, ZHU B, WANG S X, et al. Spatial distribution and source apportionment of peroxyacetyl nitrate (PAN) in a coastal region in Southern China[J]. Atmospheric Environment, 2021, 260: 118553. doi: 10.1016/j.atmosenv.2021.118553 [21] TUAZON E C, CARTER W P L, ATKINSON R. Thermal decomposition of peroxyacetyl nitrate and reactions of acetyl peroxy radicals with nitric oxide and nitrogen dioxide over the temperature range 283-313 K[J]. The Journal of Physical Chemistry, 1991, 95(6): 2434-2437. doi: 10.1021/j100159a059 [22] 廖敏萍, 龚道程, 王少霞, 等. 国庆期间南岭背景大气中PAN的浓度特征与来源[J]. 中国环境科学, 2021, 41(6): 2493-2503. doi: 10.19674/j.cnki.issn1000-6923.20210331.011 LIAO M P, GONG D C, WANG S X, et al. Concentration characteristics and sources of PAN at Nanling background station during the National Day holidays[J]. China Environmental Science, 2021, 41(6): 2493-2503 (in Chinese). doi: 10.19674/j.cnki.issn1000-6923.20210331.011
[23] 王兴锋, 魏巍, 李睿, 等. 京津廊城市气团光化学污染潜势分析[J]. 中国环境科学, 2022, 42(5): 1985-1993. doi: 10.3969/j.issn.1000-6923.2022.05.001 WANG X F, WEI W, LI R, et al. Analysis of photochemical pollution potential of air masses from various cities in Beijing-Tianjin-Langfang border[J]. China Environmental Science, 2022, 42(5): 1985-1993 (in Chinese). doi: 10.3969/j.issn.1000-6923.2022.05.001
[24] XU W Y, ZHANG G, WANG Y, et al. Aerosol promotes peroxyacetyl nitrate formation during winter in the North China plain[J]. Environmental Science & Technology, 2021, 55(6): 3568-3581. [25] 张劲松, 魏桢, 陈志强, 等. 合肥市大气过氧乙酰硝酸酯污染特征研究[J]. 环境科学与技术, 2018, 41(增刊1): 77-81. ZHANG J S, WEI Z, CHEN Z Q, et al. Research on characteristics of atmospheric peroxyacetyl nitrate pollution in Hefei[J]. Environmental Science & Technology, 2018, 41(Sup 1): 77-81 (in Chinese).
[26] 孙鑫, 唐倩, 邹巧莉, 等. 金华地区夏秋大气中过氧乙酰硝酸酯的污染特征[J]. 南京信息工程大学学报(自然科学版), 2020, 12(6): 721-728. SUN X, TANG Q, ZOU Q L, et al. Characteristics of atmospheric PAN pollution in Jinhua during summer and autumn[J]. Journal of Nanjing University of Information Science & Technology (Natural Science Edition), 2020, 12(6): 721-728 (in Chinese).
[27] XUE L K, WANG T, LOUIE P K K, et al. Increasing external effects negate local efforts to control ozone air pollution: A case study of Hong Kong and implications for other Chinese Cities[J]. Environmental Science & Technology, 2014, 48(18): 10769-10775. [28] QIU Y L, MA Z Q, LI K, et al. Markedly enhanced levels of peroxyacetyl nitrate (PAN) during COVID-19 in Beijing[J]. Geophysical Research Letters, 2020, 47(19): e2020GL089623. [29] 黄红铭, 黄增, 韦江慧, 等. 广西大气中过氧乙酰硝酸酯污染特征研究[J]. 化学工程师, 2020, 34(2): 36-39. doi: 10.16247/j.cnki.23-1171/tq.20200236 HUANG H M, HUANG Z, WEI J H, et al. Research on characteristics of atmospheric peroxyacetyl nitrate pollution in Guangxi[J]. Chemical Engineer, 2020, 34(2): 36-39 (in Chinese). doi: 10.16247/j.cnki.23-1171/tq.20200236
[30] HU Y J, FU H B, BERNSTEIN E R. Generation and detection of the peroxyacetyl radical in the pyrolysis of peroxyacetyl nitrate in a supersonic expansion[J]. The Journal of Physical Chemistry. A, 2006, 110(8): 2629-2633. doi: 10.1021/jp058196i [31] 陈瑾一, 彭月祥, 张成龙, 等. 过氧乙酰硝酸酯分析仪的光化学合成标定方法研究及应用[J]. 环境化学, 2021, 40(6): 1862-1870. doi: 10.7524/j.issn.0254-6108.2020012003 CHEN J Y, PENG Y X, ZHANG C L, et al. Study and application in calibration method of photochemical synthesis for peroxyacetyl nitrate analyzer[J]. Environmental Chemistry, 2021, 40(6): 1862-1870 (in Chinese). doi: 10.7524/j.issn.0254-6108.2020012003
[32] ZHANG H L, XU X B, LIN W L, et al. Wintertime peroxyacetyl nitrate (PAN) in the megacity Beijing: Role of photochemical and meteorological processes[J]. Journal of Environmental Sciences, 2014, 26(1): 83-96. doi: 10.1016/S1001-0742(13)60384-8 [33] GONG D C, LIAO M P, WU G C, et al. Characteristics of peroxyacetyl nitrate (PAN) in the high-elevation background atmosphere of South-Central China: Implications for regional photochemical pollution[J]. Atmospheric Environment, 2021: 118424. [34] FISCHER E V, JAFFE D A, REIDMILLER D R, et al. Meteorological controls on observed peroxyacetyl nitrate at Mount Bachelor during the spring of 2008[J]. Journal of Geophysical Research: Atmospheres, 2010, 115(D3): D03302. [35] FISCHER E V, JACOB D J, YANTOSCA R M, et al. Atmospheric peroxyacetyl nitrate (PAN): A global budget and source attribution[J]. Atmospheric Chemistry and Physics, 2014, 14(5): 2679-2698. doi: 10.5194/acp-14-2679-2014 期刊类型引用(1)
1. 吴渴,王学中,张丹丹,朱华龙,闫永馨,李凡修,毋振海,郑振威,高祺凯. 2017~2022年亳州市PM_(2.5)与O_3复合污染演变特征及典型污染过程. 环境科学. 2024(10): 5715-5728 .  百度学术
百度学术
其他类型引用(1)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 2143
- HTML全文浏览数: 2143
- PDF下载数: 55
- 施引文献: 2
