

 下载:
下载:
-
自20世纪以来,抗生素对医药的发展具有重要的贡献,极大地保护了人类和动植物免受细菌性疾病的危害[1]. 由于抗生素在临床和养殖业上的滥用,加剧了细菌抗生素抗性(耐药性)的进化和传播[2]. 抗生素抗性基因(antibiotic resistance genes, ARGs)作为编码细菌抗生素耐药性的功能基因,于2006年被定义为一种新型“污染物”[3]. 致病菌可以通过水平基因转移(horizontal gene transfer, HGT)获得外源性抗生素抗性基因,并通过食物链或环境接触等途径进入人体,使抗生素药效明显降低,对人类健康造成极大威胁[4].
自然环境中的一些微生物可以生物合成抗生素,抑制其它微生物生长并获得更多的营养物质,这些微生物必然携带抗生素抗性基因以免受抗生素的影响[5]. 此外,与外排泵耐药机理相关的蛋白也参与信号分子的运输或中间代谢产物的排放等过程[6]. 在深海、高原等几乎没有人类活动干扰的地区能检测到抗生素抗性基因[7-9]. 然而,在人类活动影响严重的水、土壤、大气等环境介质中均检测到多样性和丰度更高的抗生素抗性基因[10-13]. 上述研究表明,抗生素耐药性是一种自然现象,但人类活动,特别是抗生素的生产和使用,加速了抗生素抗性基因在环境中的传播和富集.
大陆边缘海是陆源污染物向远洋传输的重要过渡区域. 渤海是处于半封闭状态的内海,水体更新周期较长,易于富集陆源污染物,是中国污染最严重的海域之一[14- 15]. 黄海和南海海域也受到不同程度的人类活动影响[16-19]. 目前,对中国边缘海中抗生素抗性基因和致病菌的研究较少,且缺乏边缘海沉积物中抗生素抗性基因和致病菌的完整图谱信息. 因此,研究我国边缘海中抗生素抗性基因和致病菌的多样性和丰度,不仅有助于认识海洋环境中的抗生素抗性基因的特征,而且对进一步评价陆源污染对边缘海的影响有着重要的意义. 本研究旨在使用宏基因组学方法分析我国边缘海沉积物中抗生素抗性基因的组成、丰度及其潜在致病菌群落,探讨抗生素抗性基因和致病菌的组成特征和污染现状.
-
本研究的两个珠江口沉积物样品是于2011年6月在狮子洋通道水域(PRE1)与珠江口中部(PRE2)采集,代表人类活动干扰严重的河口区域. 4个南海沉积物样品(SCS1至SCS4)来源于2011年8月的南海北部航次. 2016年7月采集了3个黄海沉积物样品(YS1至YS3)以及靠近渤海海峡处的1个渤海沉积物样品(BS1). 所有沉积物样品均使用表层沉积物采样器采集,在采集后都储存在无菌的密封聚乙烯塑料袋中并在4 ℃冰箱保存,运送至实验室后立即进行DNA的提取.
-
使用FastDNA Spin Kit for Soil(MP Biomedical,Santa Ana,CA)试剂盒提取沉积物中的DNA,提取方法见试剂盒使用手册[20]. 由于海洋沉积物中的DNA丰度低,为减少批次效应或样品异质性产生的误差,对每个沉积物样品进行多次DNA提取后合并. 使用Thermo Scientific NanoDrop 2000分光光度计测定DNA的纯度和浓度. 每个样品约5 μg的DNA被超声剪切后使用T4 DNA聚合酶与Klenow酶进行末端修复,随后使用T4多核苷酸激酶进行修复末端的磷酸化. 使用T4 DNA连接酶将接头与DNA片段连接,并通过凝胶电泳得到长度合适的DNA目标片段,随后使用PCR扩增目标片段,以此构建样品的DNA文库. DNA在安诺优达基因科技有限公司(浙江,中国)使用Illumina HiSeq 2500平台完成测序. 所有样品总宏基因组数据约60 Gb,每个样本的测序数据约为2—6 Gb不等. 对所有原始测序数据进行数据过滤,去除低质量碱基序列(含有测序质量值低于20的碱基或者包含不明确碱基的序列).
-
使用BLAST(Linux版本2.2.29)程序将测序数据与相关的数据库进行数据比对[21]. 使用BLASTN将宏基因组测序数据与SILVA数据库(SILVA_132_SSURef_Nr99)进行比对,识别样品中的16S rRNA基因,E-value阈值为10-20[22]. 随后,使用MEGAN(MEGAN community 6.21.16)最低共同祖先(Lowest Common Ancestor)算法来注释沉积物中的细菌群落,其中BLAST的绝对截止值为50[23].
从NCBI RefSeq数据库(NCBI Reference Sequence Database)下载质粒序列并建立质粒数据库,从INTEGRALL数据库中挑选整合酶及基因盒序列建立整合子数据库. 将样品DNA序列与质粒和整合子数据库进行了比对,识别这两种可移动遗传元件(Mobile genetic elements, MGEs)相关的基因序列. 识别质粒序列的阈值是比对长度大于90%且相似度大于95%[24];识别整合子序列的阈值是比对长度大于70%且相似度大于90%[25].
DeepARG数据库(deepARG-DB,v1.0.2)是将CARD(The Comprehensive Antibiotic Resistance Database)、ARDB(Antibiotic Resistance Genes Database)以及UNIPROT(Universal Protein)合并后去除重复序列得到的抗生素抗性基因数据库[26]. 使用BLASTX将样品测序数据与deepARG数据库进行比对,抗生素抗性基因序列的识别阈值是序列相似度大于90%且比对长度大于75%的测序读长. 此外,从测序宏基因组数据中提取抗生素抗性基因序列,再与质粒数据库比对,识别质粒携带的抗生素抗性基因[27]. 通过PHI-base和VFDB提供的致病菌分类和NCBI参考序列,从SILVA数据库中提取致病菌16S rRNA基因序列,建立非冗余的致病菌16S rRNA基因数据库. 使用BLASTN算法将样品测序数据与致病菌16S rRNA基因数据库进行比对,识别致病菌16S rRNA序列的E-value阈值为10-20,比对结果也使用MEGAN进行物种注释.
-
使用RStudio(版本4.1.3)中的vegan软件包(版本2.6-2)进行Pearson相关性分析与ANOSIM检验,置换检验次数为999次,P < 0.05被认为具有统计学意义. 使用OriginPro 2021(版本9.8.0.200)绘制直方图、韦恩图与主成分分析图.
-
通过数据比对和MEGAN注释中国边缘海沉积物样品中的16S rRNA基因,并输出在门、种水平上的细菌分类结果,如图1所示. 图1A是各采样点种水平上的细菌多样性,所有中国边缘海沉积物中共发现542种细菌,各样品的检出率从12.7%(SCS2和SCS4,69种)到28.4%(YS2,154种)不等. 南海的细菌物种多样性显著低于其他边缘海,可能由于南海沉积物的采样深度大,深海极端环境中细菌多样性较低. 在我国边缘海沉积物中占比最高的是变形菌门(Proteobacteria, 42.5%),随后是浮霉菌门(Planctomycetes, 8.0%),拟杆菌门(Bacteroidetes,7.8%),厚壁菌门(Firmicutes,5.8%),绿弯菌门(Chloroflexi,5.2%),如图1B所示.
有研究报道,渤海沉积物中变形菌门、拟杆菌门、放线菌门(Actinobacteria)和浮霉菌门是渤海海域的优势菌门[28]. 也有研究发现,变形菌门与浮霉菌门在渤海、黄海和南海的沉积物中相对丰度都是最高的[29]. 这些研究与本研究的分析结果一致,说明变形菌门和浮霉菌门是中国边缘海沉积物中的优势菌门. 在沉积物细菌群落的相对组成上,黄渤海的拟杆菌门占比显著高于其他海域,南海的浮霉菌门占比显著高于其他海域. 绿弯菌门是一类通过光合作用产能的细菌,该菌门在珠江口沉积物的占比明显高于边缘海沉积物.
-
各海域沉积物中的抗生素抗性基因总丰度以及比对上的抗性基因参考序列数和亚型数如图2A所示. 南海沉积物中抗生素抗性基因总丰度约是黄渤海的2倍. 与珠江口相比,边缘海沉积物中的抗生素抗性基因总丰度较低. 沉积物中抗生素抗性基因亚型与序列多样性在各区域的分布趋势与其总丰度相似. 其中,5个抗生素抗性基因亚型在渤海、黄海和南海沉积物中均被检出,包括多黏菌素(arnA)、多重耐药(mexF, rpoB2, smeR)、硝基咪唑(msbA)抗性基因(图2B). 同时,上述5个抗生素抗性基因亚型在珠江口沉积物中也均有检出.
各个区域沉积物中的抗生素抗性基因按照抗性类型分类,相对百分比如图2C所示. 在我国边缘海沉积物中多重耐药基因的丰度最高,其平均相对占比为89.1%(南海:85.9%;黄海:93.6%;渤海:88.0%). 在人类活动干扰严重的珠江口水域,沉积物中多重耐药基因的相对丰度仅为64.4%,但对人类常用抗生素(如磺胺类、氨基糖苷类、β-内酰胺类、四环素类等)相关的抗性基因相对占比显著提高,与人类常用抗生素相关的抗性基因普遍具有单一耐药特性. 比较我国边缘海和珠江口,发现珠江口沉积物中表达单一抗性的抗性基因平均相对占比是边缘海沉积物的约3倍,平均总丰度比边缘海高约13倍.
在几乎不受到人类活动影响的海洋环境中,微生物抗生素抗性基因组有其自身特征,这些抗性基因除抗生素耐药性之外,还具有其他生物学功能. 例如,在我国边缘海沉积物中都检出的rpoB2基因所编码的蛋白能参与碳和氮循环,mexF和mexW两种多重耐药基因编码的蛋白可参与磷循环[30]. 相比自然环境,受到人类活动影响大的区域中对人类常用抗生素具有单一抗性的抗性基因丰度大,占比高,且出现了许多抗生素的新型抗性基因[31-32]. 这一差异可能是由于抗生素的使用污染周边环境,诱导产生新型抗生素抗性基因并加速抗性基因在微生物之间传播[33]. 区域性抗生素抗性基因的组成特征说明了人类活动和环境抗性基因污染之间的关系,其中一些抗生素抗性基因亚型可以作为基因标志物指示环境受人类影响的程度[34].
-
在中国边缘海沉积物中可广泛检测到可移动遗传元件相关的基因,包括质粒和整合子基因. 如图3A所示,中国边缘海沉积物中整合子基因丰度在1.44 × 10−6—5.31 × 10−6之间,显著低于珠江口沉积物中整合子基因的平均丰度(1.75 × 10−5);中国边缘海沉积物中质粒基因丰度远高于整合子基因,其丰度范围是2.29 × 10−4—8.68 × 10−4,质粒基因丰度在中国边缘海和珠江口之间差异并不显著. 此外,中国边缘海沉积物中可能由质粒携带的抗生素抗性基因如图3B所示. 结果表明,各边缘海沉积物中检出的质粒携带的抗性基因极少,并且主要是多重耐药基因;珠江口沉积物中可能由质粒携带的抗生素抗性基因在所有抗性基因中的占比约是20%,主要与磺胺类、氨基糖苷类、大环内酯-林可酰胺-链阳菌素类(MLS)和四环素类等常用抗生素相关,其比例和丰度均显著高于边缘海(P < 0.01).
质粒、整合子等可移动遗传元件是功能基因在微生物之间水平转移的重要载体[35]. 在自然环境中(如西藏高原和南极)均可检测到与质粒、整合子相关的基因片段,结合中国边缘海沉积物的分析结果,说明可移动遗传元件在环境中是广泛存在的[9,36]. 但是,自然环境中的可移动遗传元件与抗生素抗性基因传播的关系不大,在中国边缘海沉积物中极少检出可能由质粒携带的抗生素抗性基因. 而珠江口沉积物中可能由质粒携带的抗生素抗性基因的多样性和丰度的显著提高,表明在污染环境中可移动遗传元件有助于抗生素抗性基因的传播,即抗生素抗性基因的高传播性是其在污染环境中的一个重要特征.
-
中国边缘海沉积物中人类致病菌的群落结构如图4所示. 图4A是各采样点沉积物中门水平上致病菌的相对组成,属于变形菌门的致病菌在中国边缘海沉积物中平均相对占比最高(60.5%),其次是厚壁菌门(27.5%)、放线菌门(7.6%)等. 图4B是各采样点沉积物种水平上致病菌的相对百分比. 在种水平上,肺炎链球菌(Streptococcus pneumoniae)是中国边缘海沉积物中相对丰度最高的致病菌种(24.4%),随后是肺炎克雷伯菌(Klebsiella pneumoniae, 19.9%)、无乳链球菌(Streptococcus agalactiae, 9.2%)、铜绿假单胞菌(Pseudomonas aeruginosa, 6.9%)和牙龈卟啉单胞菌(Porphyromonas gingivalis, 3.4%). 渤海的肺炎克雷伯菌、无乳链球菌、铜绿假单胞菌和牙龈卟啉单胞菌占比均高于其他区域. 中国边缘海沉积物中检出的相对占比超过1%的致病菌共有16种,其中有3种是生物安全等级为Ⅲ级的高风险致病菌,分别是贝氏柯克斯体(Coxiella burnetii)、土拉弗朗西斯菌(Francisella tularensis)以及结核分枝杆菌(Mycobacterium tuberculosis). 研究报道,海洋沉积物中有高丰度的肺炎链球菌、肺炎克雷伯菌和铜绿假单胞菌,亚穆纳河中也发现了大量肺炎链球菌和铜绿假单胞菌,这些研究与本文的分析结果一致[37-38].
使用16S rRNA基因总丰度对人类致病菌进行归一化处理,并对中国边缘海沉积物样品进行主成分分析,如图4C所示. 前两个主成分可以解释不同区域间人类致病菌总方差的65.7%. 根据人类致病菌的组成特征,沉积物样品可以聚为3类,即黄渤海、南海和珠江口. 与其它区域相比,4种致病菌(包括肺炎链球菌、肺炎克雷伯菌、无乳链球菌和铜绿假单胞菌)在黄渤海沉积物中的占比相对较高. 肺炎链球菌是引起败血症、脑膜炎、肺炎等侵袭性疾病的重要病原体,无论是在发达国家还是欠发达国家,其给幼儿以及老年人带来的疾病负担都是最高的[39]. 南海沉积物中的特征人类致病菌是沙眼衣原体(Chlamydia trachomatis)和土拉弗朗西斯菌. 土拉弗朗西斯菌作为生物安全等级为Ⅲ级的高风险人类致病菌,可以通过空气传播,具有高度传染性[40]. 珠江口沉积物中的特征人类致病菌则为副溶血性弧菌(Vibrio parahaemolyticus)和空肠弯曲杆菌(Campylobacter jejuni). 研究表明河口副溶血性弧菌丰度显著高于近海[41]. 空肠弯曲杆菌能引起食源性细菌感染,可以由水生动物通过食物链感染人类[42]. 空肠弯曲杆菌具有微需氧、适宜中等盐度、低温下难以存活的特性,这可能导致其在珠江口沉积物中含量比边缘海高[43]. 总的来说,各区域人类致病菌群落结构的差异可归因于人类活动、环境因子等多方面因素共同作用[44].
-
中国边缘海沉积物中广泛检测到抗生素抗性基因,其中南海沉积物中的抗性基因丰度和多样性比黄渤海高,这些边缘海环境中抗生素抗性基因水平传播能力低. 与边缘海沉积物相比,珠江口沉积物中的抗生素抗性基因具有丰度、多样性以及传播能力高的特征,说明抗生素的使用能引起抗生素抗性基因的进化和传播. 不同区域沉积物中人类致病菌群落结构显著不同,边缘海沉积物中发现3种高风险致病菌,包括贝氏柯克斯体、土拉弗朗西斯菌等,将产生较高的潜在环境健康风险.
中国边缘海沉积物中抗生素抗性基因和致病菌的组成特征
Compositional characteristics of antibiotic resistance genes and putative pathogenic bacteria in sediments of Chinese marginal seas
-
摘要: 抗生素抗性基因作为一种新型“污染物”已经受到广泛关注. 研究边缘海中抗生素抗性基因和人类致病菌的组成特征,有助于深入认识海洋环境中抗生素抗性基因的起源和海洋致病菌的潜在健康风险. 使用基于高通量测序的宏基因组学分析方法在中国边缘海沉积物中发现了多种抗生素抗性基因,主要以多重耐药基因为主,其中南海沉积物中抗生素抗性基因的总丰度约是黄渤海的2倍. 与中国边缘海相比,珠江口沉积物中抗生素抗性基因丰度更高,与人类常用抗生素(如磺胺类、氨基糖苷类、β-内酰胺类、四环素类等)的耐药性更为相关. 此外,中国边缘海沉积物中几乎没有发现质粒携带的抗性基因,而珠江口有约20%的抗性基因由质粒携带. 中国边缘海沉积物中主要人类致病菌种包括肺炎链球菌(24.4%)、肺炎克雷伯菌(19.9%)、无乳链球菌(9.2%)、铜绿假单胞菌(6.9%)等. 珠江口沉积物中致病菌群落组成与中国边缘海显著不同,副溶血性弧菌和空肠弯曲杆菌的丰度相对更高. 上述研究表明,中国边缘海沉积物中存在高多样性的抗生素抗性基因和人类致病菌,人类活动干扰可导致其污染水平提高.Abstract: Antibiotic resistance genes (ARGs) are highly concerned as a group of emerging “contaminants”. Characterization of ARGs and putative human pathogenic bacteria (HPB) in the marginal sea is essential for understanding the origin of ARGs and the potential health risks posed by oceanic pathogenic bacteria. Multiple ARGs were found in the sediments of Chinese marginal seas using high-throughput sequencing-based metagenomic approaches, with multidrug being the predominant resistance type. Herein, total ARG abundance in the sediments of South China Sea were approximately 2 times higher than those in the sediments collected from the Bohai Sea and Yellow Sea. Compared to Chinese marginal seas, ARGs in the sediments of the Pearl River Estuary (PRE) are more abundant and closely associated with anthropogenic antibiotics such as sulfonamides, aminoglycosides, β-lactams, tetracyclines, etc. ARGs possibly carried by plasmids were hardly detected in the sediments of Chinese marginal seas, but could account for approximately 20% of the total ARGs in the PRE sediments. The prevalent putative HPB in the sediments of Chinese marginal seas were Streptococcus pneumoniae (24.4%), Klebsiella pneumoniae (19.9%), Streptococcus agalactiae (9.2%), Pseudomonas aeruginosa (6.9%), etc. The community composition of putative HPB in the PRE sediments was substantially different from those in the Chinese marginal seas, which were featured with the relatively higher ratios of Vibrio parahaemolyticus and Campylobacter jejuni. Our results suggest that diverse ARGs and putative HPB occur in the sediments of Chinese marginal seas, and anthropogenic impacts may increase their pollution levels.
-
Key words:
- Chinese marginal seas /
- antibiotic resistance genes /
- pathogenic bacteria /
- metagenomics /
- sediments.
-
自20世纪以来,抗生素对医药的发展具有重要的贡献,极大地保护了人类和动植物免受细菌性疾病的危害[1]. 由于抗生素在临床和养殖业上的滥用,加剧了细菌抗生素抗性(耐药性)的进化和传播[2]. 抗生素抗性基因(antibiotic resistance genes, ARGs)作为编码细菌抗生素耐药性的功能基因,于2006年被定义为一种新型“污染物”[3]. 致病菌可以通过水平基因转移(horizontal gene transfer, HGT)获得外源性抗生素抗性基因,并通过食物链或环境接触等途径进入人体,使抗生素药效明显降低,对人类健康造成极大威胁[4].
自然环境中的一些微生物可以生物合成抗生素,抑制其它微生物生长并获得更多的营养物质,这些微生物必然携带抗生素抗性基因以免受抗生素的影响[5]. 此外,与外排泵耐药机理相关的蛋白也参与信号分子的运输或中间代谢产物的排放等过程[6]. 在深海、高原等几乎没有人类活动干扰的地区能检测到抗生素抗性基因[7-9]. 然而,在人类活动影响严重的水、土壤、大气等环境介质中均检测到多样性和丰度更高的抗生素抗性基因[10-13]. 上述研究表明,抗生素耐药性是一种自然现象,但人类活动,特别是抗生素的生产和使用,加速了抗生素抗性基因在环境中的传播和富集.
大陆边缘海是陆源污染物向远洋传输的重要过渡区域. 渤海是处于半封闭状态的内海,水体更新周期较长,易于富集陆源污染物,是中国污染最严重的海域之一[14- 15]. 黄海和南海海域也受到不同程度的人类活动影响[16-19]. 目前,对中国边缘海中抗生素抗性基因和致病菌的研究较少,且缺乏边缘海沉积物中抗生素抗性基因和致病菌的完整图谱信息. 因此,研究我国边缘海中抗生素抗性基因和致病菌的多样性和丰度,不仅有助于认识海洋环境中的抗生素抗性基因的特征,而且对进一步评价陆源污染对边缘海的影响有着重要的意义. 本研究旨在使用宏基因组学方法分析我国边缘海沉积物中抗生素抗性基因的组成、丰度及其潜在致病菌群落,探讨抗生素抗性基因和致病菌的组成特征和污染现状.
1. 材料与方法(Materials and methods)
1.1 样品采集
本研究的两个珠江口沉积物样品是于2011年6月在狮子洋通道水域(PRE1)与珠江口中部(PRE2)采集,代表人类活动干扰严重的河口区域. 4个南海沉积物样品(SCS1至SCS4)来源于2011年8月的南海北部航次. 2016年7月采集了3个黄海沉积物样品(YS1至YS3)以及靠近渤海海峡处的1个渤海沉积物样品(BS1). 所有沉积物样品均使用表层沉积物采样器采集,在采集后都储存在无菌的密封聚乙烯塑料袋中并在4 ℃冰箱保存,运送至实验室后立即进行DNA的提取.
1.2 沉积物DNA提取和高通量测序
使用FastDNA Spin Kit for Soil(MP Biomedical,Santa Ana,CA)试剂盒提取沉积物中的DNA,提取方法见试剂盒使用手册[20]. 由于海洋沉积物中的DNA丰度低,为减少批次效应或样品异质性产生的误差,对每个沉积物样品进行多次DNA提取后合并. 使用Thermo Scientific NanoDrop 2000分光光度计测定DNA的纯度和浓度. 每个样品约5 μg的DNA被超声剪切后使用T4 DNA聚合酶与Klenow酶进行末端修复,随后使用T4多核苷酸激酶进行修复末端的磷酸化. 使用T4 DNA连接酶将接头与DNA片段连接,并通过凝胶电泳得到长度合适的DNA目标片段,随后使用PCR扩增目标片段,以此构建样品的DNA文库. DNA在安诺优达基因科技有限公司(浙江,中国)使用Illumina HiSeq 2500平台完成测序. 所有样品总宏基因组数据约60 Gb,每个样本的测序数据约为2—6 Gb不等. 对所有原始测序数据进行数据过滤,去除低质量碱基序列(含有测序质量值低于20的碱基或者包含不明确碱基的序列).
1.3 生物信息学分析
使用BLAST(Linux版本2.2.29)程序将测序数据与相关的数据库进行数据比对[21]. 使用BLASTN将宏基因组测序数据与SILVA数据库(SILVA_132_SSURef_Nr99)进行比对,识别样品中的16S rRNA基因,E-value阈值为10-20[22]. 随后,使用MEGAN(MEGAN community 6.21.16)最低共同祖先(Lowest Common Ancestor)算法来注释沉积物中的细菌群落,其中BLAST的绝对截止值为50[23].
从NCBI RefSeq数据库(NCBI Reference Sequence Database)下载质粒序列并建立质粒数据库,从INTEGRALL数据库中挑选整合酶及基因盒序列建立整合子数据库. 将样品DNA序列与质粒和整合子数据库进行了比对,识别这两种可移动遗传元件(Mobile genetic elements, MGEs)相关的基因序列. 识别质粒序列的阈值是比对长度大于90%且相似度大于95%[24];识别整合子序列的阈值是比对长度大于70%且相似度大于90%[25].
DeepARG数据库(deepARG-DB,v1.0.2)是将CARD(The Comprehensive Antibiotic Resistance Database)、ARDB(Antibiotic Resistance Genes Database)以及UNIPROT(Universal Protein)合并后去除重复序列得到的抗生素抗性基因数据库[26]. 使用BLASTX将样品测序数据与deepARG数据库进行比对,抗生素抗性基因序列的识别阈值是序列相似度大于90%且比对长度大于75%的测序读长. 此外,从测序宏基因组数据中提取抗生素抗性基因序列,再与质粒数据库比对,识别质粒携带的抗生素抗性基因[27]. 通过PHI-base和VFDB提供的致病菌分类和NCBI参考序列,从SILVA数据库中提取致病菌16S rRNA基因序列,建立非冗余的致病菌16S rRNA基因数据库. 使用BLASTN算法将样品测序数据与致病菌16S rRNA基因数据库进行比对,识别致病菌16S rRNA序列的E-value阈值为10-20,比对结果也使用MEGAN进行物种注释.
1.4 统计分析
使用RStudio(版本4.1.3)中的vegan软件包(版本2.6-2)进行Pearson相关性分析与ANOSIM检验,置换检验次数为999次,P < 0.05被认为具有统计学意义. 使用OriginPro 2021(版本9.8.0.200)绘制直方图、韦恩图与主成分分析图.
2. 结果与讨论(Results and discussion)
2.1 细菌群落组成和结构
通过数据比对和MEGAN注释中国边缘海沉积物样品中的16S rRNA基因,并输出在门、种水平上的细菌分类结果,如图1所示. 图1A是各采样点种水平上的细菌多样性,所有中国边缘海沉积物中共发现542种细菌,各样品的检出率从12.7%(SCS2和SCS4,69种)到28.4%(YS2,154种)不等. 南海的细菌物种多样性显著低于其他边缘海,可能由于南海沉积物的采样深度大,深海极端环境中细菌多样性较低. 在我国边缘海沉积物中占比最高的是变形菌门(Proteobacteria, 42.5%),随后是浮霉菌门(Planctomycetes, 8.0%),拟杆菌门(Bacteroidetes,7.8%),厚壁菌门(Firmicutes,5.8%),绿弯菌门(Chloroflexi,5.2%),如图1B所示.
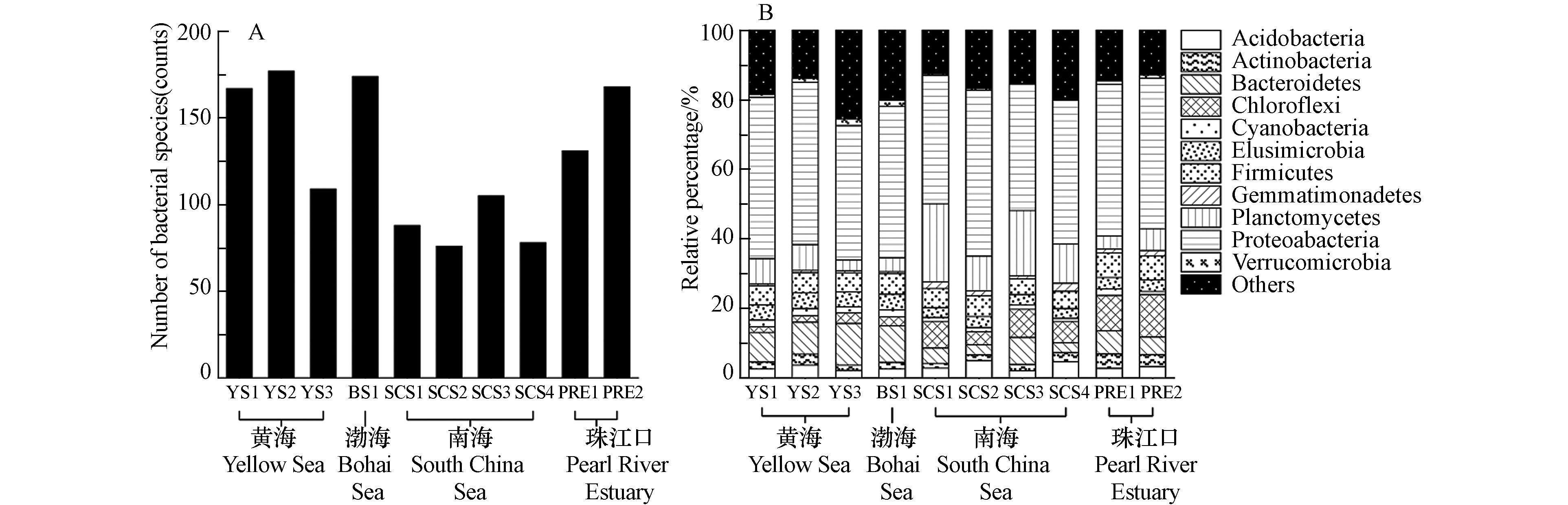 图 1 中国边缘海沉积物中细菌的多样性(A)和群落结构(B)Figure 1. The diversity (A) and structure (B) of bacterial communities in the sediments of Chinese marginal seas
图 1 中国边缘海沉积物中细菌的多样性(A)和群落结构(B)Figure 1. The diversity (A) and structure (B) of bacterial communities in the sediments of Chinese marginal seas有研究报道,渤海沉积物中变形菌门、拟杆菌门、放线菌门(Actinobacteria)和浮霉菌门是渤海海域的优势菌门[28]. 也有研究发现,变形菌门与浮霉菌门在渤海、黄海和南海的沉积物中相对丰度都是最高的[29]. 这些研究与本研究的分析结果一致,说明变形菌门和浮霉菌门是中国边缘海沉积物中的优势菌门. 在沉积物细菌群落的相对组成上,黄渤海的拟杆菌门占比显著高于其他海域,南海的浮霉菌门占比显著高于其他海域. 绿弯菌门是一类通过光合作用产能的细菌,该菌门在珠江口沉积物的占比明显高于边缘海沉积物.
2.2 抗生素抗性基因的丰度和多样性
各海域沉积物中的抗生素抗性基因总丰度以及比对上的抗性基因参考序列数和亚型数如图2A所示. 南海沉积物中抗生素抗性基因总丰度约是黄渤海的2倍. 与珠江口相比,边缘海沉积物中的抗生素抗性基因总丰度较低. 沉积物中抗生素抗性基因亚型与序列多样性在各区域的分布趋势与其总丰度相似. 其中,5个抗生素抗性基因亚型在渤海、黄海和南海沉积物中均被检出,包括多黏菌素(arnA)、多重耐药(mexF, rpoB2, smeR)、硝基咪唑(msbA)抗性基因(图2B). 同时,上述5个抗生素抗性基因亚型在珠江口沉积物中也均有检出.
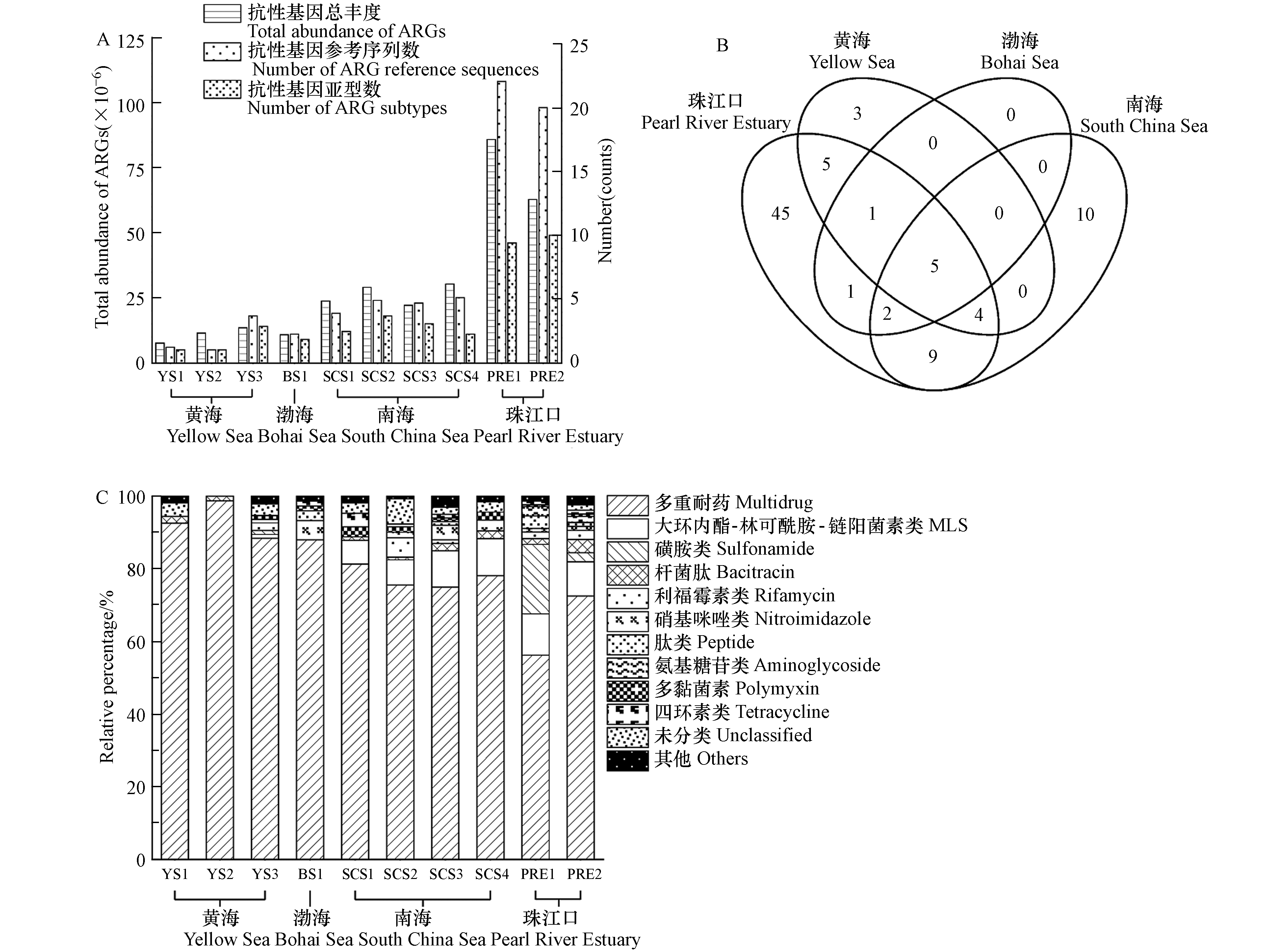 图 2 中国边缘海沉积物中抗生素抗性基因的丰度和多样性(A)、抗生素抗性基因亚型数韦恩图(B)以及抗生素抗性基因的相对组成(C)Figure 2. Total abundance and diversity of ARGs (A), Venn diagram of ARG subtypes (B) and relative composition of ARGs (C) in the sediments of Chinese marginal seas
图 2 中国边缘海沉积物中抗生素抗性基因的丰度和多样性(A)、抗生素抗性基因亚型数韦恩图(B)以及抗生素抗性基因的相对组成(C)Figure 2. Total abundance and diversity of ARGs (A), Venn diagram of ARG subtypes (B) and relative composition of ARGs (C) in the sediments of Chinese marginal seas各个区域沉积物中的抗生素抗性基因按照抗性类型分类,相对百分比如图2C所示. 在我国边缘海沉积物中多重耐药基因的丰度最高,其平均相对占比为89.1%(南海:85.9%;黄海:93.6%;渤海:88.0%). 在人类活动干扰严重的珠江口水域,沉积物中多重耐药基因的相对丰度仅为64.4%,但对人类常用抗生素(如磺胺类、氨基糖苷类、β-内酰胺类、四环素类等)相关的抗性基因相对占比显著提高,与人类常用抗生素相关的抗性基因普遍具有单一耐药特性. 比较我国边缘海和珠江口,发现珠江口沉积物中表达单一抗性的抗性基因平均相对占比是边缘海沉积物的约3倍,平均总丰度比边缘海高约13倍.
在几乎不受到人类活动影响的海洋环境中,微生物抗生素抗性基因组有其自身特征,这些抗性基因除抗生素耐药性之外,还具有其他生物学功能. 例如,在我国边缘海沉积物中都检出的rpoB2基因所编码的蛋白能参与碳和氮循环,mexF和mexW两种多重耐药基因编码的蛋白可参与磷循环[30]. 相比自然环境,受到人类活动影响大的区域中对人类常用抗生素具有单一抗性的抗性基因丰度大,占比高,且出现了许多抗生素的新型抗性基因[31-32]. 这一差异可能是由于抗生素的使用污染周边环境,诱导产生新型抗生素抗性基因并加速抗性基因在微生物之间传播[33]. 区域性抗生素抗性基因的组成特征说明了人类活动和环境抗性基因污染之间的关系,其中一些抗生素抗性基因亚型可以作为基因标志物指示环境受人类影响的程度[34].
2.3 可移动基因元件丰度和质粒携带的抗生素抗性基因
在中国边缘海沉积物中可广泛检测到可移动遗传元件相关的基因,包括质粒和整合子基因. 如图3A所示,中国边缘海沉积物中整合子基因丰度在1.44 × 10−6—5.31 × 10−6之间,显著低于珠江口沉积物中整合子基因的平均丰度(1.75 × 10−5);中国边缘海沉积物中质粒基因丰度远高于整合子基因,其丰度范围是2.29 × 10−4—8.68 × 10−4,质粒基因丰度在中国边缘海和珠江口之间差异并不显著. 此外,中国边缘海沉积物中可能由质粒携带的抗生素抗性基因如图3B所示. 结果表明,各边缘海沉积物中检出的质粒携带的抗性基因极少,并且主要是多重耐药基因;珠江口沉积物中可能由质粒携带的抗生素抗性基因在所有抗性基因中的占比约是20%,主要与磺胺类、氨基糖苷类、大环内酯-林可酰胺-链阳菌素类(MLS)和四环素类等常用抗生素相关,其比例和丰度均显著高于边缘海(P < 0.01).
 图 3 中国边缘海沉积物中质粒基因和整合子基因的丰度(A)和质粒携带的抗生素抗性基因丰度(B)Figure 3. Total abundance of plasmid and integron genes (A) and ARGs possibly carried by plasmids (B) in the sediments of Chinese marginal seas
图 3 中国边缘海沉积物中质粒基因和整合子基因的丰度(A)和质粒携带的抗生素抗性基因丰度(B)Figure 3. Total abundance of plasmid and integron genes (A) and ARGs possibly carried by plasmids (B) in the sediments of Chinese marginal seas质粒、整合子等可移动遗传元件是功能基因在微生物之间水平转移的重要载体[35]. 在自然环境中(如西藏高原和南极)均可检测到与质粒、整合子相关的基因片段,结合中国边缘海沉积物的分析结果,说明可移动遗传元件在环境中是广泛存在的[9,36]. 但是,自然环境中的可移动遗传元件与抗生素抗性基因传播的关系不大,在中国边缘海沉积物中极少检出可能由质粒携带的抗生素抗性基因. 而珠江口沉积物中可能由质粒携带的抗生素抗性基因的多样性和丰度的显著提高,表明在污染环境中可移动遗传元件有助于抗生素抗性基因的传播,即抗生素抗性基因的高传播性是其在污染环境中的一个重要特征.
2.4 致病菌的多样性和相对组成
中国边缘海沉积物中人类致病菌的群落结构如图4所示. 图4A是各采样点沉积物中门水平上致病菌的相对组成,属于变形菌门的致病菌在中国边缘海沉积物中平均相对占比最高(60.5%),其次是厚壁菌门(27.5%)、放线菌门(7.6%)等. 图4B是各采样点沉积物种水平上致病菌的相对百分比. 在种水平上,肺炎链球菌(Streptococcus pneumoniae)是中国边缘海沉积物中相对丰度最高的致病菌种(24.4%),随后是肺炎克雷伯菌(Klebsiella pneumoniae, 19.9%)、无乳链球菌(Streptococcus agalactiae, 9.2%)、铜绿假单胞菌(Pseudomonas aeruginosa, 6.9%)和牙龈卟啉单胞菌(Porphyromonas gingivalis, 3.4%). 渤海的肺炎克雷伯菌、无乳链球菌、铜绿假单胞菌和牙龈卟啉单胞菌占比均高于其他区域. 中国边缘海沉积物中检出的相对占比超过1%的致病菌共有16种,其中有3种是生物安全等级为Ⅲ级的高风险致病菌,分别是贝氏柯克斯体(Coxiella burnetii)、土拉弗朗西斯菌(Francisella tularensis)以及结核分枝杆菌(Mycobacterium tuberculosis). 研究报道,海洋沉积物中有高丰度的肺炎链球菌、肺炎克雷伯菌和铜绿假单胞菌,亚穆纳河中也发现了大量肺炎链球菌和铜绿假单胞菌,这些研究与本文的分析结果一致[37-38].
 图 4 中国边缘海沉积物中人类致病菌在门水平(A)和种水平(B)的群落结构以及主成分分析(C)Figure 4. Community structure of putative human pathogenic bacteria (HPB) in the sediments of Chinese marginal seas at the levels of phylum (A) and species (B), and principal component analysis (C)
图 4 中国边缘海沉积物中人类致病菌在门水平(A)和种水平(B)的群落结构以及主成分分析(C)Figure 4. Community structure of putative human pathogenic bacteria (HPB) in the sediments of Chinese marginal seas at the levels of phylum (A) and species (B), and principal component analysis (C)使用16S rRNA基因总丰度对人类致病菌进行归一化处理,并对中国边缘海沉积物样品进行主成分分析,如图4C所示. 前两个主成分可以解释不同区域间人类致病菌总方差的65.7%. 根据人类致病菌的组成特征,沉积物样品可以聚为3类,即黄渤海、南海和珠江口. 与其它区域相比,4种致病菌(包括肺炎链球菌、肺炎克雷伯菌、无乳链球菌和铜绿假单胞菌)在黄渤海沉积物中的占比相对较高. 肺炎链球菌是引起败血症、脑膜炎、肺炎等侵袭性疾病的重要病原体,无论是在发达国家还是欠发达国家,其给幼儿以及老年人带来的疾病负担都是最高的[39]. 南海沉积物中的特征人类致病菌是沙眼衣原体(Chlamydia trachomatis)和土拉弗朗西斯菌. 土拉弗朗西斯菌作为生物安全等级为Ⅲ级的高风险人类致病菌,可以通过空气传播,具有高度传染性[40]. 珠江口沉积物中的特征人类致病菌则为副溶血性弧菌(Vibrio parahaemolyticus)和空肠弯曲杆菌(Campylobacter jejuni). 研究表明河口副溶血性弧菌丰度显著高于近海[41]. 空肠弯曲杆菌能引起食源性细菌感染,可以由水生动物通过食物链感染人类[42]. 空肠弯曲杆菌具有微需氧、适宜中等盐度、低温下难以存活的特性,这可能导致其在珠江口沉积物中含量比边缘海高[43]. 总的来说,各区域人类致病菌群落结构的差异可归因于人类活动、环境因子等多方面因素共同作用[44].
3. 结论(Conclusion)
中国边缘海沉积物中广泛检测到抗生素抗性基因,其中南海沉积物中的抗性基因丰度和多样性比黄渤海高,这些边缘海环境中抗生素抗性基因水平传播能力低. 与边缘海沉积物相比,珠江口沉积物中的抗生素抗性基因具有丰度、多样性以及传播能力高的特征,说明抗生素的使用能引起抗生素抗性基因的进化和传播. 不同区域沉积物中人类致病菌群落结构显著不同,边缘海沉积物中发现3种高风险致病菌,包括贝氏柯克斯体、土拉弗朗西斯菌等,将产生较高的潜在环境健康风险.
-
图 1 中国边缘海沉积物中细菌的多样性(A)和群落结构(B)
Figure 1. The diversity (A) and structure (B) of bacterial communities in the sediments of Chinese marginal seas
图 2 中国边缘海沉积物中抗生素抗性基因的丰度和多样性(A)、抗生素抗性基因亚型数韦恩图(B)以及抗生素抗性基因的相对组成(C)
Figure 2. Total abundance and diversity of ARGs (A), Venn diagram of ARG subtypes (B) and relative composition of ARGs (C) in the sediments of Chinese marginal seas
图 3 中国边缘海沉积物中质粒基因和整合子基因的丰度(A)和质粒携带的抗生素抗性基因丰度(B)
Figure 3. Total abundance of plasmid and integron genes (A) and ARGs possibly carried by plasmids (B) in the sediments of Chinese marginal seas
-
[1] NEU H C. The crisis in antibiotic resistance [J]. Science, 1992, 257(5073): 1064-1073. doi: 10.1126/science.257.5073.1064 [2] LIVERMORE D M. Minimising antibiotic resistance [J]. The Lancet Infectious Diseases, 2005, 5(7): 450-459. doi: 10.1016/S1473-3099(05)70166-3 [3] PRUDEN A, PEI R T, STORTEBOOM H, et al. Antibiotic resistance genes as emerging contaminants: Studies in northern Colorado [J]. Environmental Science & Technology, 2006, 40(23): 7445-7450. [4] WEI Z Y, FENG K, WANG Z J, et al. High-throughput single-cell technology reveals the contribution of horizontal gene transfer to typical antibiotic resistance gene dissemination in wastewater treatment plants [J]. Environmental Science & Technology, 2021, 55(17): 11824-11834. [5] NEWMAN D J, CRAGG G M, SNADER K M. Natural products as sources of new drugs over the period 1981-2002 [J]. Journal of Natural Products, 2003, 66(7): 1022-1037. doi: 10.1021/np030096l [6] MARTÍNEZ J L. Natural antibiotic resistance and contamination by antibiotic resistance determinants: The two ages in the evolution of resistance to antimicrobials [J]. Frontiers in Microbiology, 2012, 3: 1. [7] CHEN B W, YANG Y, LIANG X M, et al. Metagenomic profiles of antibiotic resistance genes (ARGs) between human impacted estuary and deep ocean sediments [J]. Environmental Science & Technology, 2013, 47(22): 12753-12760. [8] D'COSTA V M, KING C E, KALAN L, et al. Antibiotic resistance is ancient [J]. Nature, 2011, 477(7365): 457-461. doi: 10.1038/nature10388 [9] CHEN B W, YUAN K, CHEN X, et al. Metagenomic analysis revealing antibiotic resistance genes (ARGs) and their genetic compartments in the Tibetan environment [J]. Environmental Science & Technology, 2016, 50(13): 6670-6679. [10] KARKMAN A, DO T T, WALSH F, et al. Antibiotic-resistance genes in waste water [J]. Trends in Microbiology, 2018, 26(3): 220-228. doi: 10.1016/j.tim.2017.09.005 [11] GHOSH S, LAPARA T M. The effects of subtherapeutic antibiotic use in farm animals on the proliferation and persistence of antibiotic resistance among soil bacteria [J]. The ISME Journal, 2007, 1(3): 191-203. doi: 10.1038/ismej.2007.31 [12] XIE J W, JIN L, LUO X S, et al. Seasonal disparities in airborne bacteria and associated antibiotic resistance genes in PM2.5 between urban and rural sites [J]. Environmental Science & Technology Letters, 2018, 5(2): 74-79. [13] PEI R T, KIM S C, CARLSON K H, et al. Effect of river landscape on the sediment concentrations of antibiotics and corresponding antibiotic resistance genes (ARG) [J]. Water Research, 2006, 40(12): 2427-2435. doi: 10.1016/j.watres.2006.04.017 [14] GAO X L, CHEN C T A. Heavy metal pollution status in surface sediments of the coastal Bohai Bay [J]. Water Research, 2012, 46(6): 1901-1911. doi: 10.1016/j.watres.2012.01.007 [15] 李淑文. 环渤海污染问题的原因和对策 [J]. 经济研究导刊, 2007(3): 159-161. doi: 10.3969/j.issn.1673-291X.2007.03.063 LI S W. The reason and counterplan of the pollution around Bohai [J]. Economic Research Guide, 2007(3): 159-161(in Chinese). doi: 10.3969/j.issn.1673-291X.2007.03.063
[16] 潘家华, 庄贵阳. 中国黄海海域污染的态势与控制方略浅析 [J]. 太平洋学报, 1998, 6(1): 48-54. PAN J H, ZHUANG G Y. Analysis on Pollution Situation and Control Strategy of Yellow Sea Area [J]. Pacific Journal, 1998, 6(1): 48-54(in Chinese).
[17] 梁亚荣, 吴鹏. 论南海海洋环境保护公众参与制度的完善 [J]. 法学杂志, 2010, 31(1): 22-24,28. doi: 10.16092/j.cnki.1001-618x.2010.01.009 LIANG Y R, WU P. On the systematic improvement of public participation in oceanic environmental protection of Southern China Sea [J]. Law Science Magazine, 2010, 31(1): 22-24,28(in Chinese). doi: 10.16092/j.cnki.1001-618x.2010.01.009
[18] SU H C, HU X J, XU W J, et al. Diversity, abundances and distribution of antibiotic resistance genes and virulence factors in the South China Sea revealed by metagenomic sequencing [J]. Science of the Total Environment, 2022, 814: 152803. doi: 10.1016/j.scitotenv.2021.152803 [19] ZHANG Y P, NIU Z G, ZHANG Y, et al. Occurrence of intracellular and extracellular antibiotic resistance genes in coastal areas of Bohai Bay (China) and the factors affecting them [J]. Environmental Pollution, 2018, 236: 126-136. doi: 10.1016/j.envpol.2018.01.033 [20] GUO F, ZHANG T. Biases during DNA extraction of activated sludge samples revealed by high throughput sequencing [J]. Applied Microbiology and Biotechnology, 2013, 97(10): 4607-4616. doi: 10.1007/s00253-012-4244-4 [21] ALTSCHUL S F, MADDEN T L, SCHÄFFER A A, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs [J]. Nucleic Acids Research, 1997, 25(17): 3389-3402. doi: 10.1093/nar/25.17.3389 [22] QUAST C, PRUESSE E, YILMAZ P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools [J]. Nucleic Acids Research, 2012, 41(D1): 590-596. doi: 10.1093/nar/gks1219 [23] HUSON D H, AUCH A F, QI J, et al. MEGAN analysis of metagenomic data [J]. Genome Research, 2007, 17(3): 377-386. doi: 10.1101/gr.5969107 [24] YANG Y, LI B, JU F, et al. Exploring variation of antibiotic resistance genes in activated sludge over a four-year period through a metagenomic approach [J]. Environmental Science & Technology, 2013, 47(18): 10197-10205. [25] MOURA A, SOARES M, PEREIRA C, et al. INTEGRALL: a database and search engine for integrons, integrases and gene cassettes [J]. Bioinformatics, 2009, 25(8): 1096-1098. doi: 10.1093/bioinformatics/btp105 [26] ARANGO-ARGOTY G, GARNER E, PRUDEN A, et al. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data [J]. Microbiome, 2018, 6(1): 23. doi: 10.1186/s40168-018-0401-z [27] ZHANG T, ZHANG X X, YE L. Plasmid metagenome reveals high levels of antibiotic resistance genes and mobile genetic elements in activated sludge [J]. PLoS One, 2011, 6(10): e26041. doi: 10.1371/journal.pone.0026041 [28] WANG S P, YAN Z G, WANG P Y, et al. Comparative metagenomics reveals the microbial diversity and metabolic potentials in the sediments and surrounding seawaters of Qinhuangdao mariculture area [J]. PLoS One, 2020, 15(6): e0234128. doi: 10.1371/journal.pone.0234128 [29] ZHANG J, CHEN M, HUANG J F, et al. Diversity of the microbial community and cultivable protease-producing bacteria in the sediments of the Bohai Sea, Yellow Sea and South China Sea [J]. PLoS One, 2019, 14(4): e0215328. doi: 10.1371/journal.pone.0215328 [30] ZHANG Z Y, ZHANG Q, WANG T Z, et al. Assessment of global health risk of antibiotic resistance genes [J]. Nature Communications, 2022, 13: 1553. doi: 10.1038/s41467-022-29283-8 [31] ZHANG Y J, HU H W, YAN H, et al. Salinity as a predominant factor modulating the distribution patterns of antibiotic resistance genes in ocean and river beach soils [J]. The Science of the Total Environment, 2019, 668: 193-203. doi: 10.1016/j.scitotenv.2019.02.454 [32] ZHANG H K, WANG Y B, LIU P Y, et al. Unveiling the occurrence, hosts and mobility potential of antibiotic resistance genes in the deep ocean [J]. Science of the Total Environment, 2022, 816: 151539. doi: 10.1016/j.scitotenv.2021.151539 [33] BAQUERO F, NEGRI M C, MOROSINI M I, et al. Antibiotic-selective environments [J]. Clinical infectious diseases, 1998, 27(Suppl 1): S5-11. [34] CHEN B W, LIANG X M, HUANG X P, et al. Differentiating anthropogenic impacts on ARGs in the Pearl River Estuary by using suitable gene indicators [J]. Water Research, 2013, 47(8): 2811-2820. doi: 10.1016/j.watres.2013.02.042 [35] FROST L S, LEPLAE R, SUMMERS A O, et al. Mobile genetic elements: The agents of open source evolution [J]. Nature Reviews Microbiology, 2005, 3(9): 722-732. doi: 10.1038/nrmicro1235 [36] YUAN K, YU K, YANG R Q, et al. Metagenomic characterization of antibiotic resistance genes in Antarctic soils [J]. Ecotoxicology and Environmental Safety, 2019, 176: 300-308. doi: 10.1016/j.ecoenv.2019.03.099 [37] YANG J, WANG C, SHU C, et al. Marine sediment bacteria harbor antibiotic resistance genes highly similar to those found in human pathogens [J]. Microbial Ecology, 2013, 65(4): 975-981. doi: 10.1007/s00248-013-0187-2 [38] DAS B K, BEHERA B K, CHAKRABORTY H J, et al. Metagenomic study focusing on antibiotic resistance genes from the sediments of River Yamuna [J]. Gene, 2020, 758: 144951. doi: 10.1016/j.gene.2020.144951 [39] BOGAERT D, de GROOT R, HERMANS P. Streptococcus pneumoniae colonisation: The key to pneumococcal disease [J]. The Lancet Infectious Diseases, 2004, 4(3): 144-154. doi: 10.1016/S1473-3099(04)00938-7 [40] OYSTON P C F, SJÖSTEDT A, TITBALL R W. Tularaemia: bioterrorism defence renews interest in Francisella tularensis [J]. Nature Reviews Microbiology, 2004, 2(12): 967-978. doi: 10.1038/nrmicro1045 [41] PENG Z, ZHUANG Z, HUANG R, et al. Distribution of pathogen in the Bohai sea in spring and summer [J]. African Journal of Microbiology Research, 2010, 4(13): 1383-1390. [42] YOUNG K T, DAVIS L M, DIRITA V J. Campylobacter jejuni: Molecular biology and pathogenesis [J]. Nature Reviews Microbiology, 2007, 5(9): 665-679. doi: 10.1038/nrmicro1718 [43] SNELLING W J, MATSUDA M, MOORE J E, et al. Campylobacter jejuni [J]. Letters in Applied Microbiology, 2005, 41(4): 297-302. doi: 10.1111/j.1472-765X.2005.01788.x [44] LEVIN L A, ETTER R J, REX M A, et al. Environmental influences on regional deep-sea species diversity [J]. Annual Review of Ecology and Systematics, 2001, 32: 51-93. doi: 10.1146/annurev.ecolsys.32.081501.114002 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3641
- HTML全文浏览数: 3641
- PDF下载数: 89
- 施引文献: 0
