


-
染料污染是纺织、橡胶、造纸等工业废水中常见的一种污染物. 由具有较高的毒性和色度,会对水生态系统造成危害和潜在的致癌效应[1]. 过去的研究报道了多种去除染料的方法,如生物处理、膜过滤、吸附等[2]. 其中,基于硫酸根自由基(SO4−·)的高级氧化技术(AOPs)具有氧化性强,操作简单等优点被研究人员广泛关注[3]. 此外,太阳光作为一种可以长期使用的清洁能源,半导体光催化在去除有机污染物方面也显示出了极大的优势和广阔的应用背景[4].
近年来,铋基金属化合物由于其良好的稳定性、无毒性和可见光活性,被广泛应用于可持续光催化中[5]. 钼酸铋(Bi2MoO6)由于具有较窄的带隙(2.6—2.9 eV)、较高的热稳定性和化学稳定性以及容易产生丰富的自由基物种被广泛研究,然而由于其窄带隙和光生电子-空穴对易复合,限制了其光催化活性[6]. 氧化铜(CuO)作为一种金属氧化物和半导体材料具有合理带隙(1.2—1.8 eV)和独特的物理化学性质,能够展示出优良稳定性和的可见光捕获能力,并且被证明能够有效地激活过一硫酸盐(PMS)[7]. 然而CuO的光催化活性同样易收到光生电子-空穴对复合的影响. 因此将Bi2MoO6−与CuO构成二元异质结可以有效抑制光生电子-空穴对的复合,提高可见光下对污染物的光催化降解性能.
本研究采用水热法和浸渍煅烧法成功制备出在可见光照射下具有良好催化活性的CuO/Bi2MoO6复合材料. 对其形貌、结构等进行分析,研究了CuO/Bi2MoO6复合材料作为PMS的活化剂在可见光照射下对AO7的降解效果,并探究了体系中产生的活性物质和降解机理.
-
二水合钼酸钠(Na2MoO4·2H2O)购于天津恒兴化学试剂有限公司;十六烷基三甲基溴化铵(CTAB,C19H42BrN)购于上海润捷化学试剂有限公司;五水合硝酸铋(Bi(NO3)3·5H2O)、金橙Ⅱ(AO7)(C16H11N2NaO4S)、过氧单磺酸钾(PMS)(KH3S4O18)、乙醇(C2H5OH)、乙二醇(CH2OH)2、亚硝酸钠(NaNO2)、糠醇(C5H6O2)、对苯醌(C6H4O2)、异丙醇(C3H8O)、叔丁醇(C4H10O)、氯化钠(NaCl)、腐殖酸均购于国药集团化学试剂有限公司,以上药品均为分析纯. 实验用水为去离子水.
-
Bi2MoO6的制备:将4 mmol Bi(NO3)3·5H2O添加到30 mL乙二醇溶液,超声30 min使其分散,将2 mmol Na2MoO4·2H2O添加到30 mL乙二醇溶液,置于磁力搅拌器搅拌30 min,然后将硝酸铋溶液缓慢加入钼酸钠溶液中. 将0.3 g CTAB加入上述溶液,继续40℃恒温搅拌1 h. 随后将混合溶液移至100 mL聚四氟乙烯高压反应釜中,放入烘箱160℃水热反应24 h. 自然条件下冷却并过滤,用无水乙醇和去离子水洗涤数次,放入烘箱在60 ℃干燥10 h,得到BMO.
CuO/Bi2MoO6复合材料的制备:将上述制备的0.5 g Bi2MoO6添加到150 mL去离子水中与酒精的混合溶液中(去离子水:酒精=2:1)超声30 min使其分散,并加入一定量的Cu(NO3)3·3H2O. 将悬浊液放入80 ℃水浴锅中,边搅拌边蒸发至完全蒸干(约4 h),收集粉体并且300 ℃煅烧2 h. 然后用去离子水和无水乙醇洗涤数次,最后在60℃烘箱中干燥10 h,得到复合材料CuO/Bi2MoO6. 根据Cu(NO3)3·3H2O的添加量,制备出1%wt CuO/Bi2MoO6、3%wt CuO/Bi2MoO6、5%wt CuO/Bi2MoO6,记1-CBM、3-CBM、5-CBM.
-
通过对AO7的可见光降解,考察了催化剂的光催化活性. 选用14 W的日光灯模拟可见光. 反应在烧杯中进行,搅拌速度恒定,水浴加热,反应温度保持在(25±2)℃. 日光灯置于烧杯上方,将一定量的催化剂和1 mmol·L−1的PMS同时加入100 mL 0.1 mmol·L−1 AO7溶液中开始反应. 反应过程中,样品在预定时间用移液管采集,通过0.45 μL滤头过滤,并加入1 mL 0.2 mol·L−1的亚硝酸钠猝灭剂. 过滤液用紫外可见分光光度计在波长为484 nm测试AO7浓度变化.
-
利用X射线衍射仪(德国Bruker D8 Advance)来分析材料的晶相结构;利用X射线光电子能谱仪(Thermo Scientific ESCALAB Xi+)来分析材料的元素组成;利用扫描电子显微镜(Hitachi Regulus8100)来观察材料的形貌结构;利用紫外/可见/红外漫反射仪(日本Shimadzu UV-3600i Plus)来测试材料的吸收波长;利用ICP OES(PerkinElmer ICP 2100)来测试溶液中浸出Cu2+的浓度.
-
通过XRD对Bi2MoO6和CuO/Bi2MoO6复合材料进行表征,如图1(a)所示. BMO的2θ的衍射峰约为10.9°、23.5°、28.3°、32.5°、32.6°、33.1°、47.1°、55.6°、58.4°. 所有的衍射峰均与正交相的Bi2MoO6标准卡片(JCPDS#21-0102)一致,对应的(020)、(111)、(131)、(200)、(002)、(060)、(062)、(133)和(262)晶格面. CuO/Bi2MoO6复合材料的XRD结果与BMO的衍射峰基本相同,为观察到明显的CuO衍射特征峰,这主要是由于复合材料中CuO的含量过低所致. 但可以发现CuO负载后的复合材料,这些峰的强度增强,半宽度变窄,峰尖变尖,这表明CuO/Bi2MoO6复合材料的表面结构和结晶度增加.
-
采用XPS分析方法测定5-CB复合材料的化学状态和元素组成. 图1(b)为5-CB的XPS全谱图,以284.5 eV处C 1s峰的结合能作为标定的参考,5-CB中同时检测到Cu 2p、Bi 4f和Mo 3d,这表明Bi2MoO6与CuO共存. 由图2可以看出,Bi 4f7/2、Bi 4f5/2、Mo 3d3/2、Mo 3d5/2和O 1s的结合能分别为158.8、164.1、232.1、235.3、529.7 eV. Cu 2p3/2和Cu 2p1/2的峰均为单峰,分别对应933.0 eV和953.6 eV的结合能. 在941.2 eV和964.1 eV处观察到的电子结合能峰对应与Cu2+的卫星峰,这证明了Cu是以+2氧化态存在[8]. 与BMO相比,5-CB复合材料XPS峰的向更高的结合能方向移动,这主要是由于CuO/Bi2MoO6异质结的形成引起的能量的变化[9]. XPS的结果揭示了Bi2MoO6与CuO建立了较强的界面效应和结合,促进了载体的转移.
-
通过SEM可以看出CuO/Bi2MoO6复合材料的形貌特征. 如图3所示,可以看出BMO是由大量纳米片组成直径约为1—2 μm的3D微球. CuO纳米颗粒很好的生长在BMO微球上,与XPS中表明的CuO与BMO所共存结果一致,这说明成功合成了CuO/Bi2MoO6异质结. 图3(b)可以看出,由于1-CBM的中CuO的质量占比很少,纳米颗粒均匀的分布在微球表面. 随着CuO的负载逐渐增多,Bi2MoO6的片状结构表面被致密的CuO纳米颗粒所覆盖,如图3(c, d)所示,两者紧密接触,有利于光生电荷载流子在之中迁移[10].
-
所制备的BMO和5-CBM复合材料的紫外可见漫反射光谱如图4所示. 由图4(a)可以看出,BMO的可见光响应有限,只能吸收小于470 nm的光. 与BMO相比,5-CBM表现出较强的可见光吸收边. 表明异质结形成后,复合材料对于可见光的吸收能力增强. 利用Kubelka-Munk方程可以计算半导体的禁带宽度(Eg):
其中α为吸收系数,h为普朗克常数,ν为光频率,A为比例常数. 图4(b)是转换DRS后的BMO和5-CBM的图,可以确定其Eg分别为2.751 eV和2.508 eV. 结果表明,电子跃迁所需的能量降低,表明复合材料在可见光区具有较高的活性,提高了光催化性能[11].
-
为了探究复合材料、PMS、可见光在降解AO7的降解效率. 由图5(a)可以看出,在可见光与PMS体系中,AO7的降解活性可以忽略不计,仅去除了4%的AO7. 单独投加5-CBM可以看出,复合材料具有良好的吸附性能,主要是由于钼酸铋微球具有合适的比表面积和的氧空位浓度. 在可见光下,5-CBM对AO7的降解效率相对于BMO有所提高,表明复合材料异质结的形成可以提高光催化活性. 在黑暗条件下,PMS的引入使AO7的降解效率有明显的提高,可以说明5-CBM除了作为光催化剂外,同样可以很好地活化PMS. 在上述情况下,引入可见光可以进一步提高降解活性,30 min内去除率达到99%. 由上述分析可知,5-CBM复合材料形成的异质结、PMS的引入以及可见光的照射都能提高降解活性. 将本文制备的CuO/Bi2MoO6复合材料的催化活性与其它文献中报道的进行了比较,如表1所示. 对比结果表明,CuO/Bi2MoO6复合材料可以在较短时间内比表1中的其他材料具有更好的降解效率,这说明本文合成的催化剂是一种高效的催化材料.
在可见光照射下,通过PMS降解AO7,考察了不同负载CuO催化剂的催化性能. 如图5(b)所示,30 min辐照时间内BMO对AO7的降解率为90.6%. CuO的加入显著的提高了AO7的降解效率. 随着CuO负载量的增加,AO7的降解速率逐渐增加,主要是因为CuO的增加,协同体系降解过程中产生的含氧活性物种增多. 本文选用5-CBM作为典型的催化剂,在接下来的实验中使用.
-
在不改变其他实验参数的前提下,考察了催化剂用量对AO7降解的影响. 图6(a)显示了当5-CBM用量分别为0.1、0.25、0.4 g·L−1时协同体系对AO7的降解率. 从图6可以看出,随着催化剂投加量的增加,体系对AO7的降解能力逐渐提高. 但投加量从0.25 g·L−1增加至0.4 g·L−1时,降解速率减慢,说明催化剂的投加量不是显著影响协同体系催化性能的关键因素.
溶液的pH会影响体系中自由基的种类,从而影响AO7的降解效率. 通过缓慢滴加H2SO4和NaOH溶液分别将pH调制3、5、7、9和11. 图6(b)可以看出,pH=7、9条件下有利于降解. 通常催化剂表面的电负性随pH的降低而降低,电负性较低的HSO5−更容易吸附在催化剂表面,促进自由基的产生,但高浓度的H+对SO4·−和·OH有明显的清除作用(式2—3)[17]. 此外,pH值对溶液中催化剂的粒径也有显著影响,粒径越小,暴露的比表面积越大[18].
在降解过程中发现,反应开始后pH值会有所下降,是因为PMS本身具有酸性. 因此,碱性初始条件有利于降解过程中的pH在适当范围之内. 初始pH=9体系,在加入PMS后,整个降解过程中pH保持在5.5左右,此时催化剂的电负性较低,颗粒半径较小. 当溶液的酸性过高时,协同体系中PMS主要以H2SO5的形式存在(式4),不利于催化剂与其反应生成SO4·−,并且从Cu离子在不同pH降解条件下的浸出情况看出(表2),较酸的条件容易导致Cu2+的浸出,降低了催化活性[19]. 当pH>11时,会导致催化剂的电负性较高,粒径大,此时催化剂表面的负电荷会阻碍PMS的吸附,并且溶液中的PMS主要以SO52−存在,但由于SO52−的活化效果比HSO5−要弱,导致催化活性较低[12].
-
印染过程中为了使染料更好地附着在纤维上,通常需要需要高浓度的NaCl来屏蔽纤维表面的电荷,实际印染废水中往往存在大量Cl−,因此研究Cl−浓度的影响非常重要[20]. 如图7(a)所示,当协同体系内Cl−的浓度逐渐增加时,AO7的降解效率也逐渐增加. Cl−对AO7的降解有积极的作用主要是因为:①Cl−具有空穴清除能力,可以有效地阻止光生电子-空穴对的复合(式5)[21]. ②Cl−可以与·OH和SO4·−发生一系列反应生成Cl·、Cl2·−和HClO共同降解AO7,加快了反应速率(式6—10)[22].
腐殖酸(HA)作为自然有机质(NOM)的模型化合物,在反应体系中加入腐殖酸来模拟实际水体中AO7降解效率的影响. 图7(b)可以看出,在协同体系中加入5 mg·L−1 HA时,AO7的降解速率没有受到多大影响. 随着HA的浓度增加到25 mg·L−1时,降解效率有了明显的下降. 根据前人的研究,低浓度的HA可以刺激PMS产生·OH和SO4·−,这可能是HA中醌基团引起的. 在较高的HA浓度下,由于过量HA大分子和丰富的电子起到清除剂的作用,从而会抑制污染物的降解[23].
-
为了进一步了解CuO/Bi2MoO6复合材料协同降解体系中AO7的变化过程,进行了紫外-可见光谱扫描测试. 图8显示了AO7的光谱变化情况,AO7的主要结构特征峰有229、310、484 nm,分别对应苯环、萘环和偶氮键发色基团. 图8可以看出,随着反应的进行484 nm处可见区的吸收逐渐减少,最终消失,这表明了偶氮键发色基团被氧化而断裂. 310 nm处特征峰的下降意味着萘环的破裂. 由于测试仪器吸收波的上限为3.0,因此没法明显看出229 nm处特征峰的下降,但是在30 min的特征峰低于3.0,这说明同样具有氧化苯环的能力. 最终AO7和中间产物的芳香官能团被降解,并具有将AO7矿化为CO2和H2O的能力.
-
为研究AO7可能存在的活性物种,在体系中加入足量的异丙醇(IPA)、叔丁醇(TBA)、糠醇(FFA)、对苯醌(p-BQ)、乙二胺四乙酸二钠(EDTA-2Na)分别作为·OH/SO4·−、·OH、1O2、·O2−和h+的猝灭剂来进行自由基的捕获. 如图9所示,当加入IPA、TBA后,30 min内AO7的降解效率有所降低. 当加入FFA、p-BQ和EDTA-2Na后发现对AO7的降解起到明显的抑制作用. 结果表明,协同体系在降解AO7的过程中产生的活性物质有·OH、SO4·−、h+、·O2−和1O2,其中,·OH和SO4·−在降解AO7过程中的贡献率远低于h+、·O2−和1O2.
根据复合材料的结构特性和降解性能,可以得出协同体系可能存在的降解途径,如图10所示. 在可见光照射下,Bi2MoO6在负载CuO后,Bi2MoO6导带上的电子可以自发移动到CuO的导带上,被激发的电子通过Cu2+/Cu+的转换,使得电子空穴对有效分离,避免复合(式11)[24]. 随后,光生电子容易捕捉水中的氧,产生超氧自由基,PMS的引入也可以与光生电子反应产生SO4·−,有效地降解污染物(式12—13)[25]. 另外,价带空穴也会与H2O/OH−生成·OH(式14)[26]. 1O2在AO7的降解过程中也起到了非常重要的作用. 已有研究报道,1O2是PMS活化过程中的一种选择性活性氧,1O2可以通过PMS自身分解产生,也可以通过与超氧自由基重组形成(式15—17)[27]. 此外,CuO在活化PMS的过程中,可以实现Cu2+/Cu+的相互转化生成SO4·−和SO5·−(式18—19). 最终,AO7在多种活性物种的作用下,被分解为一些小分子物质甚至矿化为CO2和H2O(式20).
-
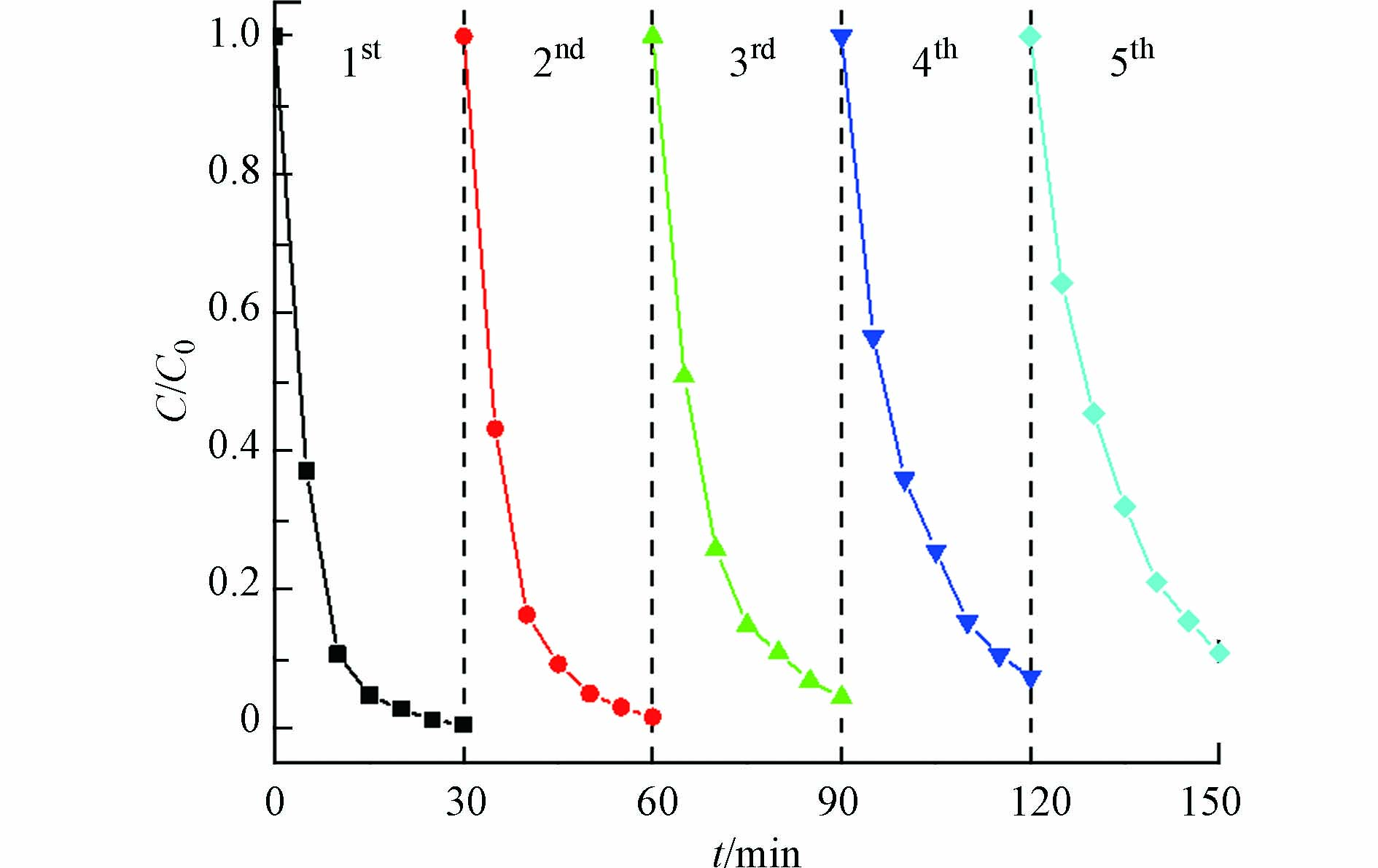
研究催化剂的可重复性和稳定性,具有长远的实际应用研究价值. 在相同的实验条件下,对5-CB复合材料的稳定性进行了5次重复性实验. 如图11所示,30 min内AO7的降解率分别为99.5%、98.4%、95.6%、92.6%和89.2%. 实验结果表明,5-CB复合材料具有较高的稳定性.
-
(1)本研究采用水热法与浸渍煅烧法成功制备CuO/Bi2MoO6异质结复合材料. 在可见光下,催化剂用量为0.25 g·L−1,PMS为1 mmol·L−1,AO7可以在30 min内达到99%以上的降解率. 由于异质结的构建和PMS的激活,可以有效抑制光生电子-空穴对的复合,产生具有较高氧化性能的活性物质.
(2)CuO/Bi2MoO6复合材料协同降解体系可以在更为宽泛的pH内高效的降解污染物. Cl−的引入可以提高AO7的降解效率,而较高浓度的HA会对AO7的降解产生的一定的抑制作用. 5-CBM在被重复利用5次以后,30 min仍保持这89.2%的降解率. 说明CuO/Bi2MoO6复合材料协同降解体系具有更高的适用范围以及稳定性.
(3)CuO/Bi2MoO6复合材料协同降解体系可以产生多个活性物种(h+、·O2−、1O2、·OH和SO4·−)共同降解AO7,其中·O2−、1O2和h+对降解AO7起主要作用.
CuO/Bi2MoO6复合材料在可见光下协同过一硫酸盐降解AO7
Efficient degradation of AO7 by CuO/Bi2MoO6 composites synergistically with peroxymonosulfate under visible light
-
摘要: 本文采用水热法和浸渍煅烧法成功合成CuO/Bi2MoO6异质结,并建立了CuO/Bi2MoO6复合材料在可见光下活化过一硫酸盐降解AO7的协同催化体系. 采用X射线衍射(XRD)、X射线光电子能谱(XPS)、扫描电子显微镜(SEM)和紫外可见漫反射光谱(UV-vis)对复合材料的晶相结构、元素组成、形貌结构和吸收波长进行了表征,并探讨了在不同条件下的对AO7的去除性能. 结果表明,CuO的引入增强了Bi2MoO6的光催化性能,同时增强了对PMS的活化. 当AO7的浓度为0.1 mmol·L−1,催化剂为0.25 g·L−1,PMS为1 mmol·L−1条件下,AO7的降解率可以在30 min内达到99%以上. 提出了AO7一种可能存在的降解机理,并通过自由基消除实验进行了验证. 复合材料在5次降解循环后仍能保持活化性能,具有较高的稳定性.Abstract: In this paper, CuO/Bi2MoO6 heterojunctions were successfully synthesized by hydrothermal method and impregnation calcination method, and a synergistic catalytic system of CuO/Bi2MoO6 composites activated by peroxymonosulfate to degrade AO7 under visible light was established. X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM) and ultraviolet-visible diffuse reflectance spectroscopy (UV-vis) were used to investigate the crystal phase structure, elemental composition, morphological structure and absorption of the composites The wavelengths were characterized and the removal performance of AO7 under different conditions was discussed. The results showed that the introduction of CuO enhanced the photocatalytic performance of Bi2MoO6 and enhanced the activation of PMS at the same time. When the concentration of AO7 is 0.1 mmol·L−1, the catalyst is 0.25 g·L−1, and the PMS is 1 mmol·L−1, the degradation rate of AO7 can reach more than 99% within 30 min. A possible degradation mechanism of AO7 was proposed and verified by free radical elimination experiments. The composites can still maintain the activation performance after 5 degradation cycles and have high stability.
-
Key words:
- bismuth molybdate /
- copper oxide /
- composite material /
- photocatalytic /
- peroxymonosulfate.
-
染料污染是纺织、橡胶、造纸等工业废水中常见的一种污染物. 由具有较高的毒性和色度,会对水生态系统造成危害和潜在的致癌效应[1]. 过去的研究报道了多种去除染料的方法,如生物处理、膜过滤、吸附等[2]. 其中,基于硫酸根自由基(SO4−·)的高级氧化技术(AOPs)具有氧化性强,操作简单等优点被研究人员广泛关注[3]. 此外,太阳光作为一种可以长期使用的清洁能源,半导体光催化在去除有机污染物方面也显示出了极大的优势和广阔的应用背景[4].
近年来,铋基金属化合物由于其良好的稳定性、无毒性和可见光活性,被广泛应用于可持续光催化中[5]. 钼酸铋(Bi2MoO6)由于具有较窄的带隙(2.6—2.9 eV)、较高的热稳定性和化学稳定性以及容易产生丰富的自由基物种被广泛研究,然而由于其窄带隙和光生电子-空穴对易复合,限制了其光催化活性[6]. 氧化铜(CuO)作为一种金属氧化物和半导体材料具有合理带隙(1.2—1.8 eV)和独特的物理化学性质,能够展示出优良稳定性和的可见光捕获能力,并且被证明能够有效地激活过一硫酸盐(PMS)[7]. 然而CuO的光催化活性同样易收到光生电子-空穴对复合的影响. 因此将Bi2MoO6−与CuO构成二元异质结可以有效抑制光生电子-空穴对的复合,提高可见光下对污染物的光催化降解性能.
本研究采用水热法和浸渍煅烧法成功制备出在可见光照射下具有良好催化活性的CuO/Bi2MoO6复合材料. 对其形貌、结构等进行分析,研究了CuO/Bi2MoO6复合材料作为PMS的活化剂在可见光照射下对AO7的降解效果,并探究了体系中产生的活性物质和降解机理.
1. 实验部分 (Experimental section)
1.1 实验药品
二水合钼酸钠(Na2MoO4·2H2O)购于天津恒兴化学试剂有限公司;十六烷基三甲基溴化铵(CTAB,C19H42BrN)购于上海润捷化学试剂有限公司;五水合硝酸铋(Bi(NO3)3·5H2O)、金橙Ⅱ(AO7)(C16H11N2NaO4S)、过氧单磺酸钾(PMS)(KH3S4O18)、乙醇(C2H5OH)、乙二醇(CH2OH)2、亚硝酸钠(NaNO2)、糠醇(C5H6O2)、对苯醌(C6H4O2)、异丙醇(C3H8O)、叔丁醇(C4H10O)、氯化钠(NaCl)、腐殖酸均购于国药集团化学试剂有限公司,以上药品均为分析纯. 实验用水为去离子水.
1.2 催化剂的制备
Bi2MoO6的制备:将4 mmol Bi(NO3)3·5H2O添加到30 mL乙二醇溶液,超声30 min使其分散,将2 mmol Na2MoO4·2H2O添加到30 mL乙二醇溶液,置于磁力搅拌器搅拌30 min,然后将硝酸铋溶液缓慢加入钼酸钠溶液中. 将0.3 g CTAB加入上述溶液,继续40℃恒温搅拌1 h. 随后将混合溶液移至100 mL聚四氟乙烯高压反应釜中,放入烘箱160℃水热反应24 h. 自然条件下冷却并过滤,用无水乙醇和去离子水洗涤数次,放入烘箱在60 ℃干燥10 h,得到BMO.
CuO/Bi2MoO6复合材料的制备:将上述制备的0.5 g Bi2MoO6添加到150 mL去离子水中与酒精的混合溶液中(去离子水:酒精=2:1)超声30 min使其分散,并加入一定量的Cu(NO3)3·3H2O. 将悬浊液放入80 ℃水浴锅中,边搅拌边蒸发至完全蒸干(约4 h),收集粉体并且300 ℃煅烧2 h. 然后用去离子水和无水乙醇洗涤数次,最后在60℃烘箱中干燥10 h,得到复合材料CuO/Bi2MoO6. 根据Cu(NO3)3·3H2O的添加量,制备出1%wt CuO/Bi2MoO6、3%wt CuO/Bi2MoO6、5%wt CuO/Bi2MoO6,记1-CBM、3-CBM、5-CBM.
1.3 降解实验
通过对AO7的可见光降解,考察了催化剂的光催化活性. 选用14 W的日光灯模拟可见光. 反应在烧杯中进行,搅拌速度恒定,水浴加热,反应温度保持在(25±2)℃. 日光灯置于烧杯上方,将一定量的催化剂和1 mmol·L−1的PMS同时加入100 mL 0.1 mmol·L−1 AO7溶液中开始反应. 反应过程中,样品在预定时间用移液管采集,通过0.45 μL滤头过滤,并加入1 mL 0.2 mol·L−1的亚硝酸钠猝灭剂. 过滤液用紫外可见分光光度计在波长为484 nm测试AO7浓度变化.
1.4 催化剂的表征
利用X射线衍射仪(德国Bruker D8 Advance)来分析材料的晶相结构;利用X射线光电子能谱仪(Thermo Scientific ESCALAB Xi+)来分析材料的元素组成;利用扫描电子显微镜(Hitachi Regulus8100)来观察材料的形貌结构;利用紫外/可见/红外漫反射仪(日本Shimadzu UV-3600i Plus)来测试材料的吸收波长;利用ICP OES(PerkinElmer ICP 2100)来测试溶液中浸出Cu2+的浓度.
2. 结果与讨论 (Results and discussion)
2.1 催化剂的表征分析
2.1.1 XRD分析
通过XRD对Bi2MoO6和CuO/Bi2MoO6复合材料进行表征,如图1(a)所示. BMO的2θ的衍射峰约为10.9°、23.5°、28.3°、32.5°、32.6°、33.1°、47.1°、55.6°、58.4°. 所有的衍射峰均与正交相的Bi2MoO6标准卡片(JCPDS#21-0102)一致,对应的(020)、(111)、(131)、(200)、(002)、(060)、(062)、(133)和(262)晶格面. CuO/Bi2MoO6复合材料的XRD结果与BMO的衍射峰基本相同,为观察到明显的CuO衍射特征峰,这主要是由于复合材料中CuO的含量过低所致. 但可以发现CuO负载后的复合材料,这些峰的强度增强,半宽度变窄,峰尖变尖,这表明CuO/Bi2MoO6复合材料的表面结构和结晶度增加.
 图 1 (a)Bi2MoO6与CuO/Bi2MoO6的XRD图谱; (b) Bi2MoO6与CuO/Bi2MoO6的XPS全谱Figure 1. (a)XRD patterns of Bi2MoO6 and CuO/Bi2MoO6; (b) XPS full spectrum of Bi2MoO6 and CuO/Bi2MoO6
图 1 (a)Bi2MoO6与CuO/Bi2MoO6的XRD图谱; (b) Bi2MoO6与CuO/Bi2MoO6的XPS全谱Figure 1. (a)XRD patterns of Bi2MoO6 and CuO/Bi2MoO6; (b) XPS full spectrum of Bi2MoO6 and CuO/Bi2MoO62.1.2 XPS能谱分析
采用XPS分析方法测定5-CB复合材料的化学状态和元素组成. 图1(b)为5-CB的XPS全谱图,以284.5 eV处C 1s峰的结合能作为标定的参考,5-CB中同时检测到Cu 2p、Bi 4f和Mo 3d,这表明Bi2MoO6与CuO共存. 由图2可以看出,Bi 4f7/2、Bi 4f5/2、Mo 3d3/2、Mo 3d5/2和O 1s的结合能分别为158.8、164.1、232.1、235.3、529.7 eV. Cu 2p3/2和Cu 2p1/2的峰均为单峰,分别对应933.0 eV和953.6 eV的结合能. 在941.2 eV和964.1 eV处观察到的电子结合能峰对应与Cu2+的卫星峰,这证明了Cu是以+2氧化态存在[8]. 与BMO相比,5-CB复合材料XPS峰的向更高的结合能方向移动,这主要是由于CuO/Bi2MoO6异质结的形成引起的能量的变化[9]. XPS的结果揭示了Bi2MoO6与CuO建立了较强的界面效应和结合,促进了载体的转移.
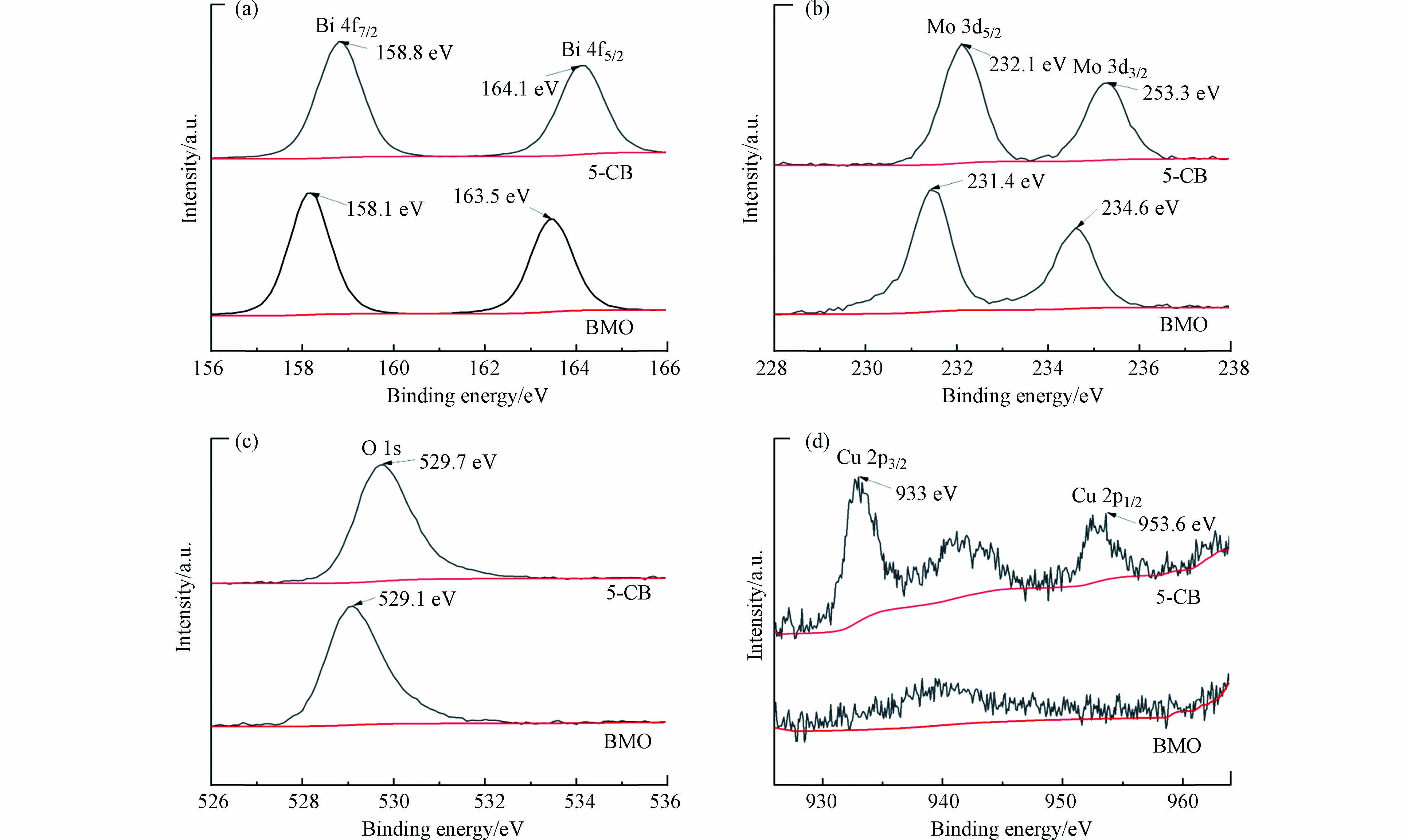 图 2 (a) Bi2MoO6与CuO/Bi2MoO6的XPS图谱Figure 2. XPS spectra of Bi2MoO6 and CuO/Bi2MoO6(a)Bi 4f; (b)Mo 3d; (c)O 1s; (d)Cu 2p
图 2 (a) Bi2MoO6与CuO/Bi2MoO6的XPS图谱Figure 2. XPS spectra of Bi2MoO6 and CuO/Bi2MoO6(a)Bi 4f; (b)Mo 3d; (c)O 1s; (d)Cu 2p2.1.3 SEM分析
通过SEM可以看出CuO/Bi2MoO6复合材料的形貌特征. 如图3所示,可以看出BMO是由大量纳米片组成直径约为1—2 μm的3D微球. CuO纳米颗粒很好的生长在BMO微球上,与XPS中表明的CuO与BMO所共存结果一致,这说明成功合成了CuO/Bi2MoO6异质结. 图3(b)可以看出,由于1-CBM的中CuO的质量占比很少,纳米颗粒均匀的分布在微球表面. 随着CuO的负载逐渐增多,Bi2MoO6的片状结构表面被致密的CuO纳米颗粒所覆盖,如图3(c, d)所示,两者紧密接触,有利于光生电荷载流子在之中迁移[10].
 图 3 Bi2MoO6与CuO/Bi2MoO6的SEM图Figure 3. SEM images of Bi2MoO6 and CuO/Bi2MoO6(a)BMO; (b)1-CBM; (c)3-CBM; (d)5-CBM
图 3 Bi2MoO6与CuO/Bi2MoO6的SEM图Figure 3. SEM images of Bi2MoO6 and CuO/Bi2MoO6(a)BMO; (b)1-CBM; (c)3-CBM; (d)5-CBM2.1.4 UV-vis DRS分析
所制备的BMO和5-CBM复合材料的紫外可见漫反射光谱如图4所示. 由图4(a)可以看出,BMO的可见光响应有限,只能吸收小于470 nm的光. 与BMO相比,5-CBM表现出较强的可见光吸收边. 表明异质结形成后,复合材料对于可见光的吸收能力增强. 利用Kubelka-Munk方程可以计算半导体的禁带宽度(Eg):
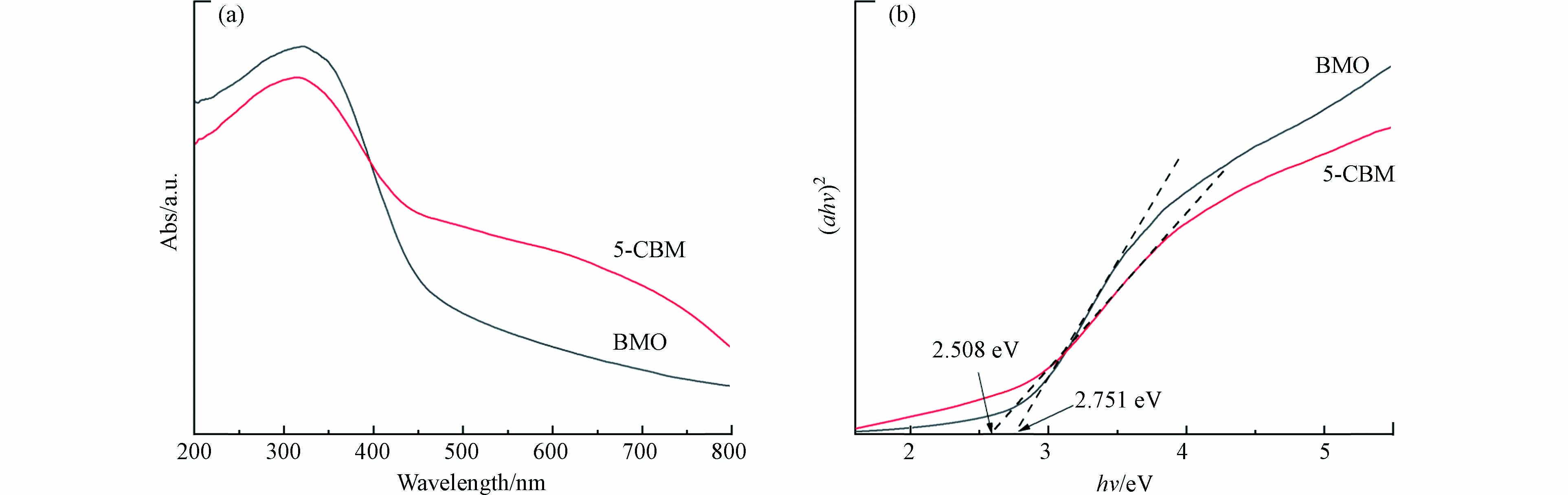 图 4 (a)Bi2MoO6与CuO/Bi2MoO6的UV-vis图谱; (b)能带谱Figure 4. (a) UV-vis spectra of Bi2MoO6 and CuO/Bi2MoO6; (b) Band spectrum
图 4 (a)Bi2MoO6与CuO/Bi2MoO6的UV-vis图谱; (b)能带谱Figure 4. (a) UV-vis spectra of Bi2MoO6 and CuO/Bi2MoO6; (b) Band spectrumαhv=A(hv−Eg)1/2 (1) 其中α为吸收系数,h为普朗克常数,ν为光频率,A为比例常数. 图4(b)是转换DRS后的BMO和5-CBM的图,可以确定其Eg分别为2.751 eV和2.508 eV. 结果表明,电子跃迁所需的能量降低,表明复合材料在可见光区具有较高的活性,提高了光催化性能[11].
2.2 催化剂的降解性能分析
2.2.1 不同体系和不同样品对AO7的降解效率
为了探究复合材料、PMS、可见光在降解AO7的降解效率. 由图5(a)可以看出,在可见光与PMS体系中,AO7的降解活性可以忽略不计,仅去除了4%的AO7. 单独投加5-CBM可以看出,复合材料具有良好的吸附性能,主要是由于钼酸铋微球具有合适的比表面积和的氧空位浓度. 在可见光下,5-CBM对AO7的降解效率相对于BMO有所提高,表明复合材料异质结的形成可以提高光催化活性. 在黑暗条件下,PMS的引入使AO7的降解效率有明显的提高,可以说明5-CBM除了作为光催化剂外,同样可以很好地活化PMS. 在上述情况下,引入可见光可以进一步提高降解活性,30 min内去除率达到99%. 由上述分析可知,5-CBM复合材料形成的异质结、PMS的引入以及可见光的照射都能提高降解活性. 将本文制备的CuO/Bi2MoO6复合材料的催化活性与其它文献中报道的进行了比较,如表1所示. 对比结果表明,CuO/Bi2MoO6复合材料可以在较短时间内比表1中的其他材料具有更好的降解效率,这说明本文合成的催化剂是一种高效的催化材料.
 图 5 (a)不同体系中AO7的降解效果; (b)不同质量比对AO7的降解效果的影响Figure 5. (a) Degradation effect of AO7 in different systems; (b) Effects of different mass ratios on the degradation of AO7表 1 本文催化体系与其他相关研究的有效性对比Table 1. Comparison of the effectiveness of the catalytic system in this paper with other related studies
图 5 (a)不同体系中AO7的降解效果; (b)不同质量比对AO7的降解效果的影响Figure 5. (a) Degradation effect of AO7 in different systems; (b) Effects of different mass ratios on the degradation of AO7表 1 本文催化体系与其他相关研究的有效性对比Table 1. Comparison of the effectiveness of the catalytic system in this paper with other related studies相似催化体系Similar catalytic system 污染物Pollutant 降解率/%Removal 时间/ minTime 文献Literature CuO/Bi2MoO6+PMS+vis AO7 99 30 本文 Bi2MoO6+PMS+vis ATZ 99 60 [12] Bi2MoO6+PS+vis AO7 99.4 120 [13] Co-BiVO4+PMS+vis AO7 98 80 [14] Bi2MoO6/CuBi2O4+vis CIP 90.2 180 [15] Bi2MoO6/Bi2WO6/AgI/Ag+vis MG 90.5 180 [16] | Show Table DownLoad:
CSV
DownLoad:
CSV
在可见光照射下,通过PMS降解AO7,考察了不同负载CuO催化剂的催化性能. 如图5(b)所示,30 min辐照时间内BMO对AO7的降解率为90.6%. CuO的加入显著的提高了AO7的降解效率. 随着CuO负载量的增加,AO7的降解速率逐渐增加,主要是因为CuO的增加,协同体系降解过程中产生的含氧活性物种增多. 本文选用5-CBM作为典型的催化剂,在接下来的实验中使用.
2.2.2 催化剂用量和初始pH的影响
在不改变其他实验参数的前提下,考察了催化剂用量对AO7降解的影响. 图6(a)显示了当5-CBM用量分别为0.1、0.25、0.4 g·L−1时协同体系对AO7的降解率. 从图6可以看出,随着催化剂投加量的增加,体系对AO7的降解能力逐渐提高. 但投加量从0.25 g·L−1增加至0.4 g·L−1时,降解速率减慢,说明催化剂的投加量不是显著影响协同体系催化性能的关键因素.
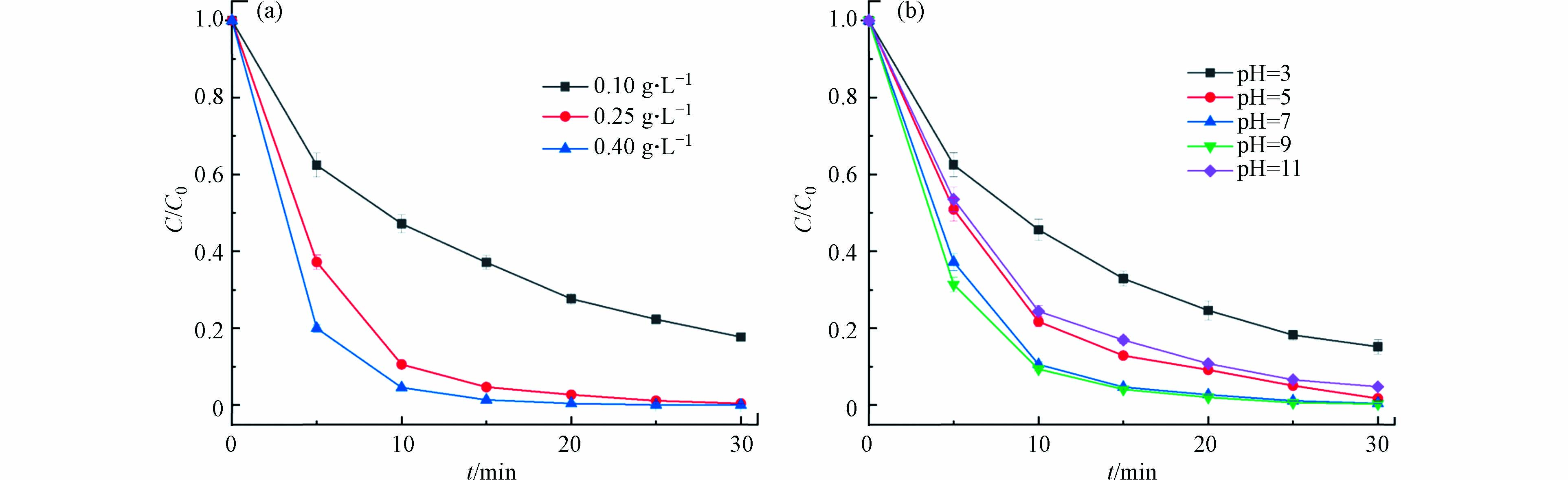 图 6 (a)催化剂投加量对AO7降解效果的影响; (b) pH对AO7降解效果的影响Figure 6. (a) Effects of catalyst dosage on the degradation of AO7; (b)Effects of pH on the degradation of AO7
图 6 (a)催化剂投加量对AO7降解效果的影响; (b) pH对AO7降解效果的影响Figure 6. (a) Effects of catalyst dosage on the degradation of AO7; (b)Effects of pH on the degradation of AO7溶液的pH会影响体系中自由基的种类,从而影响AO7的降解效率. 通过缓慢滴加H2SO4和NaOH溶液分别将pH调制3、5、7、9和11. 图6(b)可以看出,pH=7、9条件下有利于降解. 通常催化剂表面的电负性随pH的降低而降低,电负性较低的HSO5−更容易吸附在催化剂表面,促进自由基的产生,但高浓度的H+对SO4·−和·OH有明显的清除作用(式2—3)[17]. 此外,pH值对溶液中催化剂的粒径也有显著影响,粒径越小,暴露的比表面积越大[18].
⋅OH+H++e−→H2O (2) SO⋅−4+H++e−→HSO−4 (3) 在降解过程中发现,反应开始后pH值会有所下降,是因为PMS本身具有酸性. 因此,碱性初始条件有利于降解过程中的pH在适当范围之内. 初始pH=9体系,在加入PMS后,整个降解过程中pH保持在5.5左右,此时催化剂的电负性较低,颗粒半径较小. 当溶液的酸性过高时,协同体系中PMS主要以H2SO5的形式存在(式4),不利于催化剂与其反应生成SO4·−,并且从Cu离子在不同pH降解条件下的浸出情况看出(表2),较酸的条件容易导致Cu2+的浸出,降低了催化活性[19]. 当pH>11时,会导致催化剂的电负性较高,粒径大,此时催化剂表面的负电荷会阻碍PMS的吸附,并且溶液中的PMS主要以SO52−存在,但由于SO52−的活化效果比HSO5−要弱,导致催化活性较低[12].
表 2 不同pH条件下Cu2+的浸出Table 2. Leaching of Cu2+ at different pHpH 3 5 7 9 11 Cu2+/(mg·L−1) 2.54 0.36 0.04 0.05 0.41 | Show TableDownLoad:
CSV
HSO−5+H+→H2SO5 (4) 2.2.3 Cl−和腐殖酸(HA)的影响
印染过程中为了使染料更好地附着在纤维上,通常需要需要高浓度的NaCl来屏蔽纤维表面的电荷,实际印染废水中往往存在大量Cl−,因此研究Cl−浓度的影响非常重要[20]. 如图7(a)所示,当协同体系内Cl−的浓度逐渐增加时,AO7的降解效率也逐渐增加. Cl−对AO7的降解有积极的作用主要是因为:①Cl−具有空穴清除能力,可以有效地阻止光生电子-空穴对的复合(式5)[21]. ②Cl−可以与·OH和SO4·−发生一系列反应生成Cl·、Cl2·−和HClO共同降解AO7,加快了反应速率(式6—10)[22].
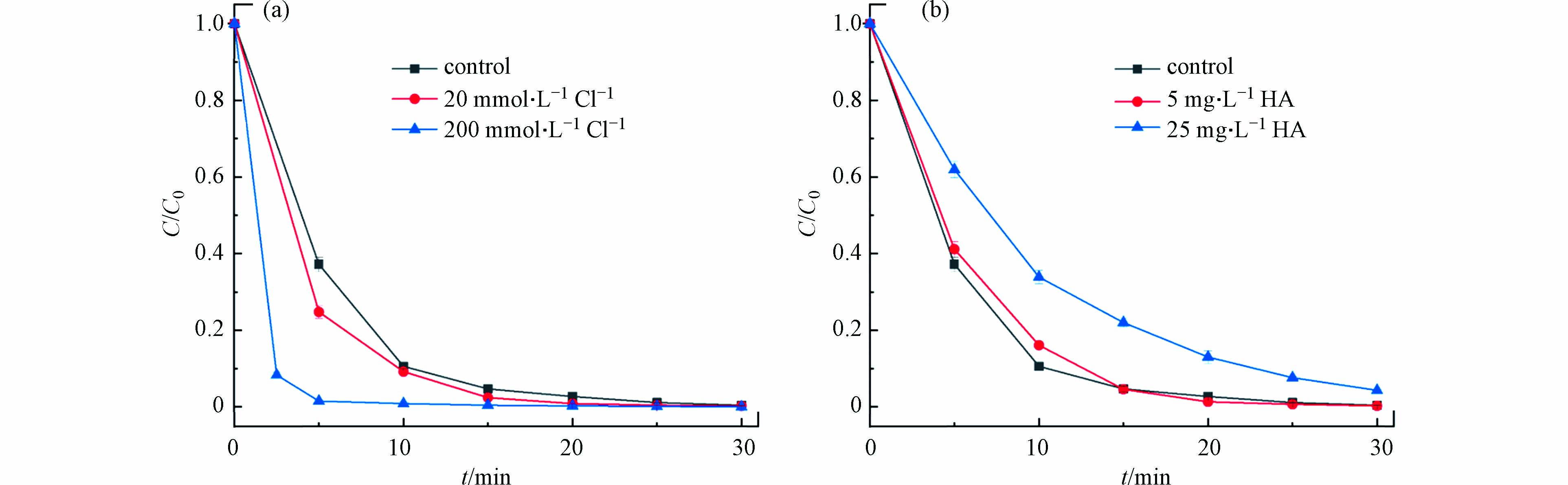 图 7 (a)Cl-的对AO7降解效果影响; (b)腐殖酸对AO7降解效果的影响Figure 7. (a) Effects of Cl- on the degradation of AO7; (b) Effects of HA on the degradation of AO7
图 7 (a)Cl-的对AO7降解效果影响; (b)腐殖酸对AO7降解效果的影响Figure 7. (a) Effects of Cl- on the degradation of AO7; (b) Effects of HA on the degradation of AO72Cl−+2h+VB→Cl2 (5) Cl−+SO⋅−4→SO2−4+Cl⋅ (6) Cl⋅+Cl−→Cl⋅−2 (7) Cl−+⋅OH→OH−+Cl⋅ (8) Cl⋅+Cl⋅−2→Cl2+Cl− (9) Cl2+H2O→HClO+HCl (10) 腐殖酸(HA)作为自然有机质(NOM)的模型化合物,在反应体系中加入腐殖酸来模拟实际水体中AO7降解效率的影响. 图7(b)可以看出,在协同体系中加入5 mg·L−1 HA时,AO7的降解速率没有受到多大影响. 随着HA的浓度增加到25 mg·L−1时,降解效率有了明显的下降. 根据前人的研究,低浓度的HA可以刺激PMS产生·OH和SO4·−,这可能是HA中醌基团引起的. 在较高的HA浓度下,由于过量HA大分子和丰富的电子起到清除剂的作用,从而会抑制污染物的降解[23].
2.2.4 AO7的紫外-可见光谱变化
为了进一步了解CuO/Bi2MoO6复合材料协同降解体系中AO7的变化过程,进行了紫外-可见光谱扫描测试. 图8显示了AO7的光谱变化情况,AO7的主要结构特征峰有229、310、484 nm,分别对应苯环、萘环和偶氮键发色基团. 图8可以看出,随着反应的进行484 nm处可见区的吸收逐渐减少,最终消失,这表明了偶氮键发色基团被氧化而断裂. 310 nm处特征峰的下降意味着萘环的破裂. 由于测试仪器吸收波的上限为3.0,因此没法明显看出229 nm处特征峰的下降,但是在30 min的特征峰低于3.0,这说明同样具有氧化苯环的能力. 最终AO7和中间产物的芳香官能团被降解,并具有将AO7矿化为CO2和H2O的能力.
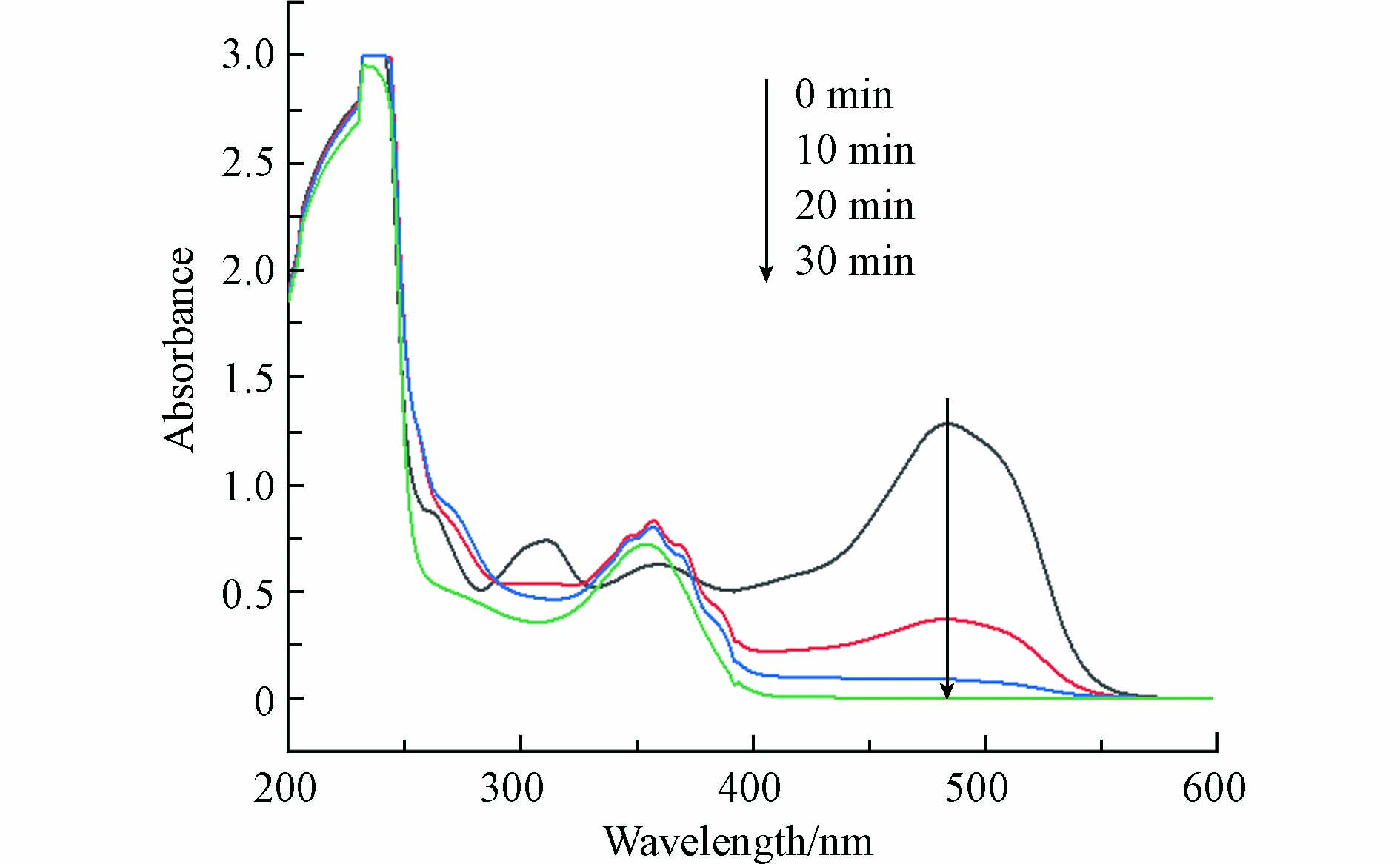 图 8 协同体系降解过程中AO7的紫外-可见光谱变化Figure 8. UV-Vis spectral changes of AO7 during synergistic system degradation
图 8 协同体系降解过程中AO7的紫外-可见光谱变化Figure 8. UV-Vis spectral changes of AO7 during synergistic system degradation2.2.5 AO7可能的降解机理
为研究AO7可能存在的活性物种,在体系中加入足量的异丙醇(IPA)、叔丁醇(TBA)、糠醇(FFA)、对苯醌(p-BQ)、乙二胺四乙酸二钠(EDTA-2Na)分别作为·OH/SO4·−、·OH、1O2、·O2−和h+的猝灭剂来进行自由基的捕获. 如图9所示,当加入IPA、TBA后,30 min内AO7的降解效率有所降低. 当加入FFA、p-BQ和EDTA-2Na后发现对AO7的降解起到明显的抑制作用. 结果表明,协同体系在降解AO7的过程中产生的活性物质有·OH、SO4·−、h+、·O2−和1O2,其中,·OH和SO4·−在降解AO7过程中的贡献率远低于h+、·O2−和1O2.
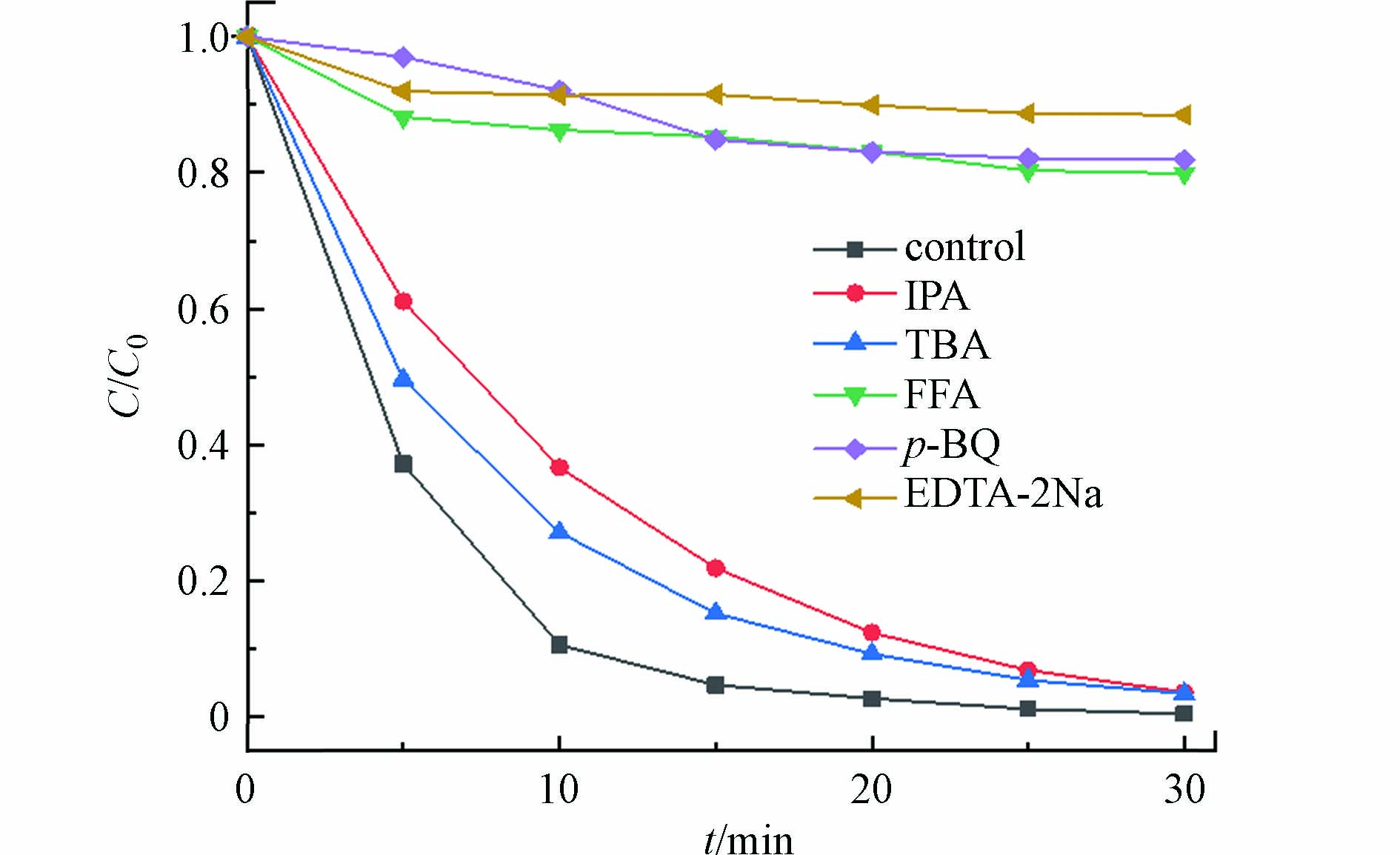 图 9 不同猝灭剂对AO7降解效果的影响Figure 9. (a) Effects of different quenchers on the degradation of AO7
图 9 不同猝灭剂对AO7降解效果的影响Figure 9. (a) Effects of different quenchers on the degradation of AO7根据复合材料的结构特性和降解性能,可以得出协同体系可能存在的降解途径,如图10所示. 在可见光照射下,Bi2MoO6在负载CuO后,Bi2MoO6导带上的电子可以自发移动到CuO的导带上,被激发的电子通过Cu2+/Cu+的转换,使得电子空穴对有效分离,避免复合(式11)[24]. 随后,光生电子容易捕捉水中的氧,产生超氧自由基,PMS的引入也可以与光生电子反应产生SO4·−,有效地降解污染物(式12—13)[25]. 另外,价带空穴也会与H2O/OH−生成·OH(式14)[26]. 1O2在AO7的降解过程中也起到了非常重要的作用. 已有研究报道,1O2是PMS活化过程中的一种选择性活性氧,1O2可以通过PMS自身分解产生,也可以通过与超氧自由基重组形成(式15—17)[27]. 此外,CuO在活化PMS的过程中,可以实现Cu2+/Cu+的相互转化生成SO4·−和SO5·−(式18—19). 最终,AO7在多种活性物种的作用下,被分解为一些小分子物质甚至矿化为CO2和H2O(式20).
 图 10 CuO/Bi2MoO6复合材料可见光协同体系降解AO7的机理示意图Figure 10. Schematic diagram of the mechanism of CuO/Bi2MoO6 composites degrading AO7 by visible light synergistic system
图 10 CuO/Bi2MoO6复合材料可见光协同体系降解AO7的机理示意图Figure 10. Schematic diagram of the mechanism of CuO/Bi2MoO6 composites degrading AO7 by visible light synergistic systemCatalyst+hv→e−+h+ (11) O2+e−→⋅O−2 (12) e−+HSO−5→SO⋅−4+OH− (13) h++H2O/OH−→⋅OH (14) ⋅O−2+⋅O−2+2H+→1O2+H2O2 (15) ⋅O−2+H2O2→1O2+OH−+⋅OH (16) ⋅O−2+⋅OH→1O2+OH− (17) Cu2++HSO−5→Cu++SO⋅−5+H+ (18) Cu++HSO−5→Cu2++SO⋅−4+OH− (19) (⋅OH,SO⋅−4,h+,⋅O−2,1O2)+AO7→Pruducts (20) 2.2.6 催化剂的重复利用性及稳定性
研究催化剂的可重复性和稳定性,具有长远的实际应用研究价值. 在相同的实验条件下,对5-CB复合材料的稳定性进行了5次重复性实验. 如图11所示,30 min内AO7的降解率分别为99.5%、98.4%、95.6%、92.6%和89.2%. 实验结果表明,5-CB复合材料具有较高的稳定性.
3. 结论 (Conclusion)
(1)本研究采用水热法与浸渍煅烧法成功制备CuO/Bi2MoO6异质结复合材料. 在可见光下,催化剂用量为0.25 g·L−1,PMS为1 mmol·L−1,AO7可以在30 min内达到99%以上的降解率. 由于异质结的构建和PMS的激活,可以有效抑制光生电子-空穴对的复合,产生具有较高氧化性能的活性物质.
(2)CuO/Bi2MoO6复合材料协同降解体系可以在更为宽泛的pH内高效的降解污染物. Cl−的引入可以提高AO7的降解效率,而较高浓度的HA会对AO7的降解产生的一定的抑制作用. 5-CBM在被重复利用5次以后,30 min仍保持这89.2%的降解率. 说明CuO/Bi2MoO6复合材料协同降解体系具有更高的适用范围以及稳定性.
(3)CuO/Bi2MoO6复合材料协同降解体系可以产生多个活性物种(h+、·O2−、1O2、·OH和SO4·−)共同降解AO7,其中·O2−、1O2和h+对降解AO7起主要作用.
-
图 1 (a)Bi2MoO6与CuO/Bi2MoO6的XRD图谱; (b) Bi2MoO6与CuO/Bi2MoO6的XPS全谱
Figure 1. (a)XRD patterns of Bi2MoO6 and CuO/Bi2MoO6; (b) XPS full spectrum of Bi2MoO6 and CuO/Bi2MoO6
图 4 (a)Bi2MoO6与CuO/Bi2MoO6的UV-vis图谱; (b)能带谱
Figure 4. (a) UV-vis spectra of Bi2MoO6 and CuO/Bi2MoO6; (b) Band spectrum
图 5 (a)不同体系中AO7的降解效果; (b)不同质量比对AO7的降解效果的影响
Figure 5. (a) Degradation effect of AO7 in different systems; (b) Effects of different mass ratios on the degradation of AO7
图 6 (a)催化剂投加量对AO7降解效果的影响; (b) pH对AO7降解效果的影响
Figure 6. (a) Effects of catalyst dosage on the degradation of AO7; (b)Effects of pH on the degradation of AO7
图 7 (a)Cl-的对AO7降解效果影响; (b)腐殖酸对AO7降解效果的影响
Figure 7. (a) Effects of Cl- on the degradation of AO7; (b) Effects of HA on the degradation of AO7
图 8 协同体系降解过程中AO7的紫外-可见光谱变化
Figure 8. UV-Vis spectral changes of AO7 during synergistic system degradation
图 9 不同猝灭剂对AO7降解效果的影响
Figure 9. (a) Effects of different quenchers on the degradation of AO7
图 10 CuO/Bi2MoO6复合材料可见光协同体系降解AO7的机理示意图
Figure 10. Schematic diagram of the mechanism of CuO/Bi2MoO6 composites degrading AO7 by visible light synergistic system
表 1 本文催化体系与其他相关研究的有效性对比
Table 1. Comparison of the effectiveness of the catalytic system in this paper with other related studies
下载: 导出CSV
表 2 不同pH条件下Cu2+的浸出
Table 2. Leaching of Cu2+ at different pH
pH 3 5 7 9 11 Cu2+/(mg·L−1) 2.54 0.36 0.04 0.05 0.41
下载: 导出CSV
-
[1] TU Y M, SHAO G Y, ZHANG W J, et al. The degradation of printing and dyeing wastewater by manganese-based catalysts [J]. Science of the Total Environment, 2022, 828: 154390. doi: 10.1016/j.scitotenv.2022.154390 [2] GHANBARI F, MORADI M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: Review [J]. Chemical Engineering Journal, 2017, 310: 41-62. doi: 10.1016/j.cej.2016.10.064 [3] 李春琴, 邹亚辰, 贾小宁. 过硫酸盐高级氧化技术活化方法及降解机理的研究进展 [J]. 化学与生物工程, 2022, 39(6): 1-6,27. doi: 10.3969/j.issn.1672-5425.2022.06.001 LI C Q, ZOU Y C, JIA X N. Research progress in activation methods of persulfate and degradation mechanism of organic pollutants by persulfate advanced oxidation process [J]. Chemistry & Bioengineering, 2022, 39(6): 1-6,27(in Chinese). doi: 10.3969/j.issn.1672-5425.2022.06.001
[4] FANG B, XING Z P, SUN D D, et al. Hollow semiconductor photocatalysts for solar energy conversion [J]. Advanced Powder Materials, 2022, 1(2): 100021. doi: 10.1016/j.apmate.2021.11.008 [5] 县涛, 高宇姝, 孙小锋, 等. 修饰AuAg合金纳米颗粒对BiOBr纳米片光催化降解和还原性能的提高 [J]. 材料导报, 2022, 36(13): 54-61. XIAN T, GAO Y S, SUN X F, et al. Enhanced photocatalytic degradation and reduction activity of BiOBr nanoplates by the decoration of AuAg alloy nanoparticles [J]. Materials Reports, 2022, 36(13): 54-61(in Chinese).
[6] ZHENG Y, ZHOU T F, ZHAO X D, et al. Atomic interface engineering and electric-field effect in ultrathin Bi2MoO6 nanosheets for superior lithium ion storage [J]. Advanced Materials, 2017, 29(26): 1700396. doi: 10.1002/adma.201700396 [7] ZHANG R H, CHEN M L, XIONG Z K, et al. Highly efficient degradation of emerging contaminants by magnetic CuO@FexOy derived from natural mackinawite (FeS) in the presence of peroxymonosulfate [J]. Chinese Chemical Letters, 2022, 33(2): 948-952. doi: 10.1016/j.cclet.2021.07.029 [8] GAO X M, FEI J, DAI Y, et al. Hydrothermal synthesis of series Cu-doped Bi2WO6 and its application in photo-degradative removal of phenol in wastewater with enhanced efficiency [J]. Journal of Molecular Liquids, 2018, 256: 267-276. doi: 10.1016/j.molliq.2018.02.044 [9] MENG X Y, HAO M J, SHI J Z, et al. Novel CuO/Bi2WO6 heterojunction with enhanced visible light photoactivity [J]. Advanced Powder Technology, 2017, 28(12): 3247-3256. doi: 10.1016/j.apt.2017.09.036 [10] SHONEYE A, TANG J W. Highly dispersed FeOOH to enhance photocatalytic activity of TiO2 for complete mineralisation of herbicides [J]. Applied Surface Science, 2020, 511: 145479. doi: 10.1016/j.apsusc.2020.145479 [11] PELAEZ M, NOLAN N T, PILLAI S C, et al. A review on the visible light active titanium dioxide photocatalysts for environmental applications [J]. Applied Catalysis B:Environmental, 2012, 125: 331-349. doi: 10.1016/j.apcatb.2012.05.036 [12] SHEN Z Y, ZHOU H Y, PAN Z C, et al. Degradation of atrazine by Bi2MoO6 activated peroxymonosulfate under visible light irradiation [J]. Journal of Hazardous Materials, 2020, 400: 123187. doi: 10.1016/j.jhazmat.2020.123187 [13] ZHU B Y, CHENG H, MA J F, et al. Bi2MoO6 microspheres for the degradation of orange II by heterogeneous activation of persulfate under visible light [J]. Materials Letters, 2020, 261: 127099. doi: 10.1016/j.matlet.2019.127099 [14] CHEN X, ZHOU J B, ZHANG T L, et al. Enhanced degradation of tetracycline hydrochloride using photocatalysis and sulfate radical-based oxidation processes by Co/BiVO4 composites [J]. Journal of Water Process Engineering, 2019, 32: 100918. doi: 10.1016/j.jwpe.2019.100918 [15] LI Z, ZHENG R J, DAI S J, et al. In-situ mechanochemical fabrication of p-n Bi2MoO6/CuBi2O4 heterojunctions with efficient visible light photocatalytic performance [J]. Journal of Alloys and Compounds, 2021, 882: 160681. doi: 10.1016/j.jallcom.2021.160681 [16] PIAO C C, CHEN L, LIU Z Y, et al. Construction of solar light-driven dual Z-scheme Bi2MoO6/Bi2WO6\AgI\Ag photocatalyst for enhanced simultaneous degradation and conversion of nitrogenous organic pollutants [J]. Separation and Purification Technology, 2021, 274: 119140. doi: 10.1016/j.seppur.2021.119140 [17] YANG S J, QIU X J, JIN P K, et al. MOF-templated synthesis of CoFe2O4 nanocrystals and its coupling with peroxymonosulfate for degradation of bisphenol A [J]. Chemical Engineering Journal, 2018, 353: 329-339. doi: 10.1016/j.cej.2018.07.105 [18] GUO D, WANG Y Q, CHEN C, et al. A multi-structural carbon nitride co-modified by Co, S to dramatically enhance mineralization of Bisphenol f in the photocatalysis-PMS oxidation coupling system [J]. Chemical Engineering Journal, 2021, 422: 130035. doi: 10.1016/j.cej.2021.130035 [19] 丁丽丹, 周家斌, 刘文博, 等. CuO/Bi2O3光催化耦合过一硫酸盐氧化降解盐酸四环素 [J]. 环境工程学报, 2021, 15(3): 898-910. DING L D, ZHOU J B, LIU W B, et al. Oxidative degradation of tetracycline hydrochloride by CuO/Bi2O3 photocatalysis coupling with peroxymonosulfate [J]. Chinese Journal of Environmental Engineering, 2021, 15(3): 898-910(in Chinese).
[20] 王柯晴, 徐劼, 陈家斌, 等. 氧基氯化铁非均相活化过一硫酸盐降解金橙Ⅱ [J]. 中国环境科学, 2020, 40(8): 3385-3393. doi: 10.19674/j.cnki.issn1000-6923.2020.0378 WANG K Q, XU J, CHEN J B, et al. Degradation of AO7 by PMS activated by FeOCl [J]. China Environmental Science, 2020, 40(8): 3385-3393(in Chinese). doi: 10.19674/j.cnki.issn1000-6923.2020.0378
[21] MAHMOODI N M, ARAMI M, LIMAEE N Y, et al. Photocatalytic degradation of agricultural N-heterocyclic organic pollutants using immobilized nanoparticles of titania [J]. Journal of Hazardous Materials, 2007, 145(1/2): 65-71. [22] 竺哲欣, 马晓吉, 尚志国, 等. 氯离子对模拟太阳光活化过一硫酸氢钾氧化卡马西平的影响规律 [J]. 环境科学学报, 2021, 41(6): 2131-2137. doi: 10.13671/j.hjkxxb.2021.0099 ZHU Z X, MA X J, SHANG Z G, et al. Effect of chloride ion on oxidation of carbamazepine by simulated sunlight activation of peroxymonosulfate [J]. Acta Scientiae Circumstantiae, 2021, 41(6): 2131-2137(in Chinese). doi: 10.13671/j.hjkxxb.2021.0099
[23] FANG G D, GAO J, DIONYSIOU D D, et al. Activation of persulfate by quinones: Free radical reactions and implication for the degradation of PCBs [J]. Environmental Science & Technology, 2013, 47(9): 4605-4611. [24] CHEN L Q, LEI J S, TIAN L J, et al. One-pot in situ fabrication of Cu-coupled rugby-shaped BiVO4 sosoloid for enhancing photocatalytic activity with SPR effect via ultrasonic hydrothermal strategy [J]. Ceramics International, 2021, 47(16): 23001-23013. doi: 10.1016/j.ceramint.2021.05.014 [25] SHEIKHMOHAMMADI A, ASGARI E, NOURMORADI H, et al. Ultrasound-assisted decomposition of metronidazole by synthesized TiO2/Fe3O4 nanocatalyst: Influencing factors and mechanisms [J]. Journal of Environmental Chemical Engineering, 2021, 9(5): 105844. doi: 10.1016/j.jece.2021.105844 [26] JI Y F, KONG D Y, LU J H, et al. Cobalt catalyzed peroxymonosulfate oxidation of tetrabromobisphenol A: Kinetics, reaction pathways, and formation of brominated by-products [J]. Journal of Hazardous Materials, 2016, 313: 229-237. doi: 10.1016/j.jhazmat.2016.04.033 [27] ZHANG X, CHEN S H, LIAN X Y, et al. Efficient activation of peroxydisulfate by g-C3N4/Bi2MoO6 nanocomposite for enhanced organic pollutants degradation through non-radical dominated oxidation processes [J]. Journal of Colloid and Interface Science, 2022, 607: 684-697. doi: 10.1016/j.jcis.2021.08.198 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2322
- HTML全文浏览数: 2322
- PDF下载数: 95
- 施引文献: 0
