

-
由于人类活动干扰和自然过程导致大量污染物进入自然水体,所造成的环境污染已严重威胁生态安全、人类健康以及经济发展等. 而自然水体中污染物的迁移、转化以及生物可利用性强烈受到矿物水界面的地球化学过程(如吸附、沉淀、溶解以及多相催化等)影响[1-2]. 因此,通过调控矿物与环境污染物的地球化学过程以此强化矿物对环境污染物的净化能力,被认为是解决自然水体污染的有效途径之一[3-4].
铁的氢氧化物(羟基氧化铁)和氧化物(以下简称铁氧化物)在地表环境中分布广泛、资源丰富、价格低廉并具有大的比表面积、活跃氧化还原性以及电子输运能力,且与环境相容性好,是环境自净化的重要贡献者[5-7]. 在黑暗环境中,铁氧化物可通过化学吸附和静电引力吸附去除环境中的污染物,污染物的吸附量取决于铁氧化物的表面性质,如比表面积、表面羟基密度、表面电荷等,所涉及的吸附机制主要包括静电引力、配体交换、疏水作用、熵效应、氢键结合、离子架桥等[6, 8-10]. 例如,铁氧化物对砷(As)具有良好的亲和力,是自然环境中一种最重要的固定砷的吸附剂[5, 9]. Yu等的研究表明水铁矿依靠自身大的比表面积和高的活性羟基密度对As(V)有很好的吸附能力,吸附容量高达160 mg·g−1[11]. 而在光照条件下,具有半导体性质的铁氧化物(如针铁矿、赤铁矿)被光激发产生光生电子与空穴,其中光生电子转移至分子氧后可以形成O2·−,O2·−可以继续得电子和质子生成H2O2,H2O2又进一步发生反应生成·OH,所产生的活性氧物种可使有机污染物降解甚至矿化. 铁氧化物光催化降解有机污染物己被广泛研究[12-22]. 李芳柏教授团队[23]发现,土壤中的纤铁氧化物、磁赤铁矿、赤铁矿在紫外光与可见光下辐射下均可降解双酚A. 另一方面,光(<580 nm)的引入可以促使Fe(Ⅲ)光解还原成Fe(Ⅱ),而Fe(Ⅱ)在一定程度上可以通过活化分子氧产生活性氧物种[24]. 两种途径所产生的H2O2与Fe(Ⅱ)构成Fenton反应,可产生强氧化性的·OH. 虽然铁氧化物具有一定的光化学活性,但众多研究结果显示其光化学活性却很低[25- 26].
自然环境中天然有机物(如草酸、腐殖酸等)常与铁氧化物共存于自然水体和土壤,能与其结合形成可溶性络合物. 其中,草酸主要来自植物根系、根系周围微生物和真菌的分泌物,或者酚类物质的降解中间产物,自然水体中草酸浓度一般在2.5 × 10−5 mol·L−1 到4.0 × 10−3 mol·L−1之间[27]. 在暗黑环境中,草酸的存在会在一定程度上影响着铁氧化物的化学吸附和静电引力吸附能力[28]. 这是因为草酸与污染物之间存在竞争或者协同吸附,而具体的作用方式与污染物的类型以及草酸浓度有关. Lamy等[29]发现,草酸的存在能促进针铁矿对Cd的吸附,主要是由于草酸在针铁矿与Cd之间担任“架桥”角色. Flynn等[30]运用EXAFS 和ATR-FTIR分析发现草酸与Ni2+可在针铁矿和赤铁矿表面形成三元络合物,而溶解态草酸与Ni2+较容易形成二元络合物,导致针铁矿和赤铁矿吸附Ni2+的效果下降. 由此可见,草酸与铁氧化物之间的相互作用会强烈地影响着环境中的污染物迁移转化,反之亦然. 当引入光照后,光照可为草酸与铁氧化物之间的电子转移过程提供额外的光化学途径,能显著促进草酸-铁氧化物光化学体系中活性氧(O2·−、·OH、H2O2等)的生成,进而提高体系中污染物的降解效率.
目前,铁离子与草酸光化学过程研究介质涉及水体、土壤、空气等,这些研究均表明不同介质中共存的Fe(Ⅲ)与草酸均能在光源照射下活化分子氧产生活性氧物种[23, 31-32]. 铁(Ⅲ)-羧酸盐光化学体系已被证明可诱导多种持久性污染物的光降解,包括含氯除草剂、肥料、双酚、药物等[33-38]. 与此同时,自然光下草酸铁体系的有效光降解能力也已得到证明[39]. 由此,利用天然存在的草酸强化铁氧化物净化环境污染物的能力,被认为可为发展原位环境修复技术提供新契机. 近年来,针对草酸-铁氧化物光化学体系中草酸在铁氧化物表面上的吸附/溶出行为与污染物降解效率、动力学以及途径之间的相关性进行了大量报道. 已涉及的铁氧化物包括针铁矿、纤铁氧化物、赤铁矿、磁赤铁矿和磁铁矿等[26-27, 29-30],而不同结构的铁氧化物光化学活性差异巨大. Huang等[40]通过研究草酸与不同铁氧化物,如赤铁矿、针铁矿、磁铁矿以及磁赤铁矿,与草酸发生光化学反应原位产生活性氧物种降解诺氟沙星. 研究结果发现不同铁氧化物体系诺氟沙星的降解效率不同,其中针铁矿的效果最高,其次为赤铁矿,而磁铁矿与磁赤铁矿光化学效果接近.
对于草酸与铁氧化物的相互作用及光化学过程而言,主要包含以下几个重要过程:(1) 草酸在铁氧化物表面吸附,形成具有高光化学活性的Fe(Ⅲ)-草酸配合物;(2) Fe(Ⅲ)-草酸配合物在光照下,吸收光子,发生光生电荷转移及光分解;(3) 光解产物促进分子氧活化,产生活性氧物种;(4) 活性氧物种降解污染物. 因此,草酸-铁氧化物体系污染物去除效率取决于体系中活性氧的物种形式与含量,而其的产生与转化途径受控于草酸在铁氧化物表面的吸附与转化特性,即草酸与铁氧化物相互作用.
本文综合论述了草酸与铁氧化物相互作用过程含草酸在铁氧化物表面吸附过程和草酸诱导铁氧化物溶出过程、铁氧化物表面和溶于液相中的草酸铁络合物光分解过程以及活化分子氧过程以及各个过程对体系中污染物降解的影响,并对其未来研究方向和应用前景进行了总结与展望.
-
相对于草酸铁均相体系而言,草酸与铁氧化物的异相体系需先进行一个“吸附”过程,再进入光诱导反应阶段,即草酸需先吸附于铁氧化物表面,继而溶解络合形成草酸铁配体[41]. 由于草酸在铁氧化物表面的吸附配位构型制约着体系中表面光生电子的生成和传递效率,同时还影响着溶解于液相中的草酸铁配合物形式. 因此,要深入研究草酸与铁氧化物的相互作用及其环境光化学效应,首先要明确草酸在铁氧化物表面的吸附配位构型.
-
草酸根在溶液中存在电离平衡,平衡常数为 pKa1 = 1.17和 pKa2 = 4.15,分别对应有3个物种,即H2C2O4、HC2O4−和C2O42−. 草酸根物种形式对 pH 值有高度的依赖性,其关系如图1所示.
在自然水体中草酸多以阴离子形式存在,而在自然界中铁氧化物的表面大多都带正电荷,因此草酸可以很容易通过静电引力吸附到带正电的铁氧化物表面[43]. 草酸可以在铁氧化物表面上形成多种不同的络合构型 [44-45],包括氢键结合的外核络合物(草酸的H原子键合到与Fe原子相连的OH基)和几种内核络合物,例如单核双齿(草酸的2个O原子与1个Fe原子结合)、单齿单核(草酸的1个O原子与1个Fe原子结合)和双核双齿(草酸的两个O原子分别与1个Fe原子结合)[46],如图2. 利用傅立叶变换衰减全反射红外光谱法(ATR-FTIR)与DFT 计算可确定草酸在铁氧化物表面的吸附配位构型. 前人对草酸在铁氧化物表面吸附与溶出行为的研究主要聚焦于利用傅立叶变换衰减全反射红外光谱法(ATR-FTIR)分析草酸诱导铁氧化物溶解行为,并结合密度泛函理论(DFT)计算的方式探讨草酸在铁氧化物表面的吸附络合构型.
草酸在铁氧化物表面的络合构型与体系溶液的pH有关. Borowski [45]等采用ATR-FTIR结合DFT理论计算的方式研究了不同pH下草酸在纤铁氧化物表面的络合构型. 研究结果显示,草酸在纤铁氧化物表面可形成3种络合构型,分别为外核氢键构型,和单核双齿以及双核双齿的内核构型. 在pH 6时,主要形成的是外层络合物,而随着pH的下降,络合物逐渐以单核双齿的内层络合物为主. Bhandari [44]等采用ATR-FTIR/DFT手段发现草酸以单核双齿的形式吸附在水铁矿表面. Kubicki等[47]同样也发现草酸是以单核双齿的形式吸附在针铁矿表面. 目前的研究结果认为,草酸在铁氧化物如赤铁矿、针铁矿、水铁矿、纤铁氧化物表面均以单核双齿的形式存在. 与此同时,草酸在同种铁氧化物表面的吸附络合构型也受草酸与铁含量的比值影响. 在pH 4.5条件下草酸含量相对较低时(草酸/Fe < 0.1),水铁矿表面吸附态的草酸呈双核双齿构型;而当草酸含量增加时(草酸/Fe ≥ 0.1),水铁矿表面的双核双齿草酸铁络合物被单核双齿构型和氢键外层络合构型络合物替代[48].
络合构型的不同影响着草酸诱导铁氧化物溶解速率和体系中污染物的降解效率. 如纤铁氧化物的溶解速率与表面形成的单核双齿络合物含量呈正比关系,而不是草酸的总浓度[45]. 在紫外线下(365 nm),单核双齿的光分解速率高于双核双齿和外核氢键络合物,这也意味着单核双齿络合物的草酸-铁氧化物体系污染物的降解效果更好. 图3所示为草酸吸附在针铁矿、赤铁矿、磁铁矿以及磁赤铁矿表面所形成的草酸铁络合物构型与体系中诺氟沙星降解效率之间的关系[40]. 从图3可以看出,诺氟沙星的降解效率于铁氧化物表面的单核双齿构型的内核络合物的含量呈正相关,而与双核双齿构型的内核络合物以及外圈构型的络合物相关性不大. 由此可见,通过调控草酸在铁氧化物表面的吸附络合构型可以提高体系催化降解污染物的能力.
值得关注的是利用现代先进谱学技术考察不同体系中铁氧化物吸附草酸后光照下的光生电荷转移过程,分析光生电荷动力学,揭示光照下草酸在铁氧化物表面不同配位构型对吸光行为以及光生电荷转移的影响;同时结合理论计算揭示草酸修饰前后电子排布与Fe—O键变化,阐明草酸-铁氧化物光化学体系光驱动电荷转移机制. 在此基础上结合草酸在铁氧化物表面的吸附与转化特性,深入揭示草酸-铁氧化物光化学体系界面反应过程具有一定研究意义.
-
铁氧化物的光化学溶解被认为是维持人体生物铁和海洋浮游植物的生长不可替代的重要途径,铁氧化物的溶解与生物地球化学循环与环境污染修复等息息相关[42, 49]. 铁氧化物表面的Fe(Ⅲ)在光激发下,可能会发生轻微的光还原转化为Fe(Ⅱ),并参与活性氧物种的生成. 而有草酸存在时,铁氧化物的铁浸出显著提高[50-51].
对于草酸-铁氧化物体系而言,草酸的络合作用可以促使草酸铁络合物通过非还原溶解或还原溶解的形式从铁氧化物表面脱落[27, 52]. 其中,非还原溶解是指吸附在铁氧化物表面后所形成的草酸铁络合物进入到液相中,铁的价态未发生变化,过程如公式(1)所示:
这一过程具有较高的反应能,低温下进行缓慢. 在光照的条件下,铁氧化物表面的Fe(Ⅲ)-Ox络合物发生光敏反应弱化了Fe(Ⅲ)—O键,部分以Fe(Ⅲ)-Ox络合物的形式溶解进入液相.
还原溶解过程是吸附在铁氧化物表面的草酸铁络合物在光的激发下发生了配体至金属的电荷转移过程(LMCT),即草酸转移一个电子给Fe(Ⅲ)使其还原为Fe(Ⅱ),草酸则发生氧化过程分解了CO2和CO2·−. 由于Fe(Ⅱ)与O之间的作用力很弱,因此铁氧化物表面的Fe(Ⅱ)会倾向于溶解至液相中.
影响溶解机制最主要的因素有pH、温度和光照[52]. 由于草酸铁高的光化学活性,光照能为电子转移过程提供额外的光化学途径,使其可以大大促进公式(2)的反应过程,从而加快铁氧化物的光还原溶解. 在光照辐射下,草酸诱导铁氧化物溶解过程的主导反应是光化学反应而非热力学反应,光的催化作用克服了电子转移所需的活化能,且光入射波长对赤铁矿的光还原溶解影响很大[52].
草酸-铁氧化物光化学体系中有机污染物的去除效率与铁氧化物吸附草酸能力以及草酸诱导铁溶出量均呈正相关,即草酸吸附量越大、铁离子浸出量越高,则体系中污染物去除效果越好[53-54]. 液相中的铁离子主要来源于铁氧化物表面草酸铁配体的光还原溶解(Fe2+)和非还原溶解(Fe3+). 而草酸诱导铁氧化物表面铁离子溶出方式的不同造成了体系中污染物降解的决速步骤不一致. 部分研究者认为污染物的降解速率由铁氧化物光化学还原溶解至液相的Fe2+含量决定. Mazellier和Sulzberger[55]在研究针铁矿(α-FeOOH)与草酸异相光化学体系降解农药阿特拉津的时候发现,针铁矿光还原生成溶解态Fe(Ⅱ)的速率及草酸作为电子供体决定了阿特拉津在此类异相体系中的动力学. 而也有研究者认为,非还原溶解于液相中的草酸铁络合物的光还原过程才是污染物降解的关键. Huang等[56]发现,适当的延长草酸-磁铁矿体系预吸附时间,溶于液相中的草酸铁络合物含量增加,促使体系中诺氟沙星去除率提高. 当预吸附时间为120 min时,光照1 h后,诺氟沙星去除速率由0.0036 min−1提高至0.0398 min−1. 因此,适当的延长预吸附时间是提高污染物降解效率的一种有效手段.
目前判断草酸诱导铁氧化物溶出的方式主要是根据浸入到溶液中铁离子的价态. 溶液中的 Fe(Ⅱ)或表面结合的Fe(Ⅱ)可以在LMCT过程中直接产生,也可能来源于铁氧化物表面Fe(Ⅱ)的溶解释放或溶液中Fe(Ⅱ)被重新吸附回铁氧化物表面. 因此,单靠鉴别铁离子价态的方式判断草酸诱导铁氧化物溶出方式并不准确,需要结合其他的手段原位深入研究铁溶出过程. 此外,影响草酸诱导铁氧化物溶出方式如铁溶出与吸附络合构型、铁氧化物的晶体结构之间的内在联系,目前尚未解决,还需进一步研究.
-
不同形式的草酸铁配体的光化学活性存在巨大差异[57, 58],如,Fe(C2O4)+、Fe(C2O4)2−、Fe(C2O4)33−在254 nm 波长光辐射下量子产率分别为0、1.18和1.60,在436 nm下量子产率分别降低至0、1.0和0.6[59- 60]. 目前,草酸-铁氧化物光化学体系液相中草酸铁配体的形式主要根据Panias等[61]通过研究纯铁氧化物吸附草酸的热力学过程推导出的公式来确定. Lan等[53]报道磁赤铁矿(γ-Fe2O3)和草酸盐的紫外光照体系降解五氯苯酚(PCP)时根据体系中草酸浓度变化以及Fe2+和Fe3+随时间变化情况,结合Panias推算出来的公式计算出了各草酸铁络合物在反应过程中的比例变化. 结果显示不同初始草酸浓度体系,Fe(C2O4)2−和Fe(C2O4)33−均是主要的草酸铁络合物物种,而在草酸初始浓度为0.8 mmol·L−1时,Fe(C2O4)2−是主导的草酸铁络合物物种.
但是该计算方法忽略了光照对草酸诱导铁氧化物表面铁离子溶出过程的影响,难以真实地反映出草酸-铁氧化物光化学体系液相中草酸铁配体的形式. 近年来,随着谱学分析技术在金属配合物化学性质研究中的应用,可以清晰得到不同配合物结构形式的光学谱图[45],尤其是通过引入计算模拟方法,可以深层次分析液相羧酸铁配合物的结构特性[24],如表面增强拉曼光谱法(SERS)和高效液相色谱-电喷雾质谱(HPLC-ESI-MS)[62]. 研究者采用ATR-FTIR与DFT理论相结合的手段、联合SERS和HPLC-ESI-MS技术研究水铁矿-草酸体系界面反应过程,发现草酸吸附在水铁矿表面后形成了单核双齿络合物,而后该单核双齿络合物以Fe(C2O4)+的形式非还原溶解于液相中,并快速的转化为Fe(C2O4)2−. 而进入液相中Fe(C2O4)2–络合物则通过配体至金属电荷转移过程(LCMT)快速光解,其光分解速率远高于均相体系的络合物Fe(C2O4)33–[46].
草酸铁配体形式与pH、Fe(Ⅲ)与草酸盐含量比例有关[62]. 研究发现在pH小于3时,Fe(C2O4)+是唯一优势物种,对污染物的降解速度有负面影响;而在pH值为3和5时,以Fe(C2O4)2−和Fe(C2O4)33−两种主要形式并存,此时污染物的降解速率显著提升[63]. 因此,有望通过合理调控pH以及铁氧化物与草酸盐含量的比例来控制体系中草酸铁物种的形式强化草酸-铁氧化物体系的光化学性能.
-
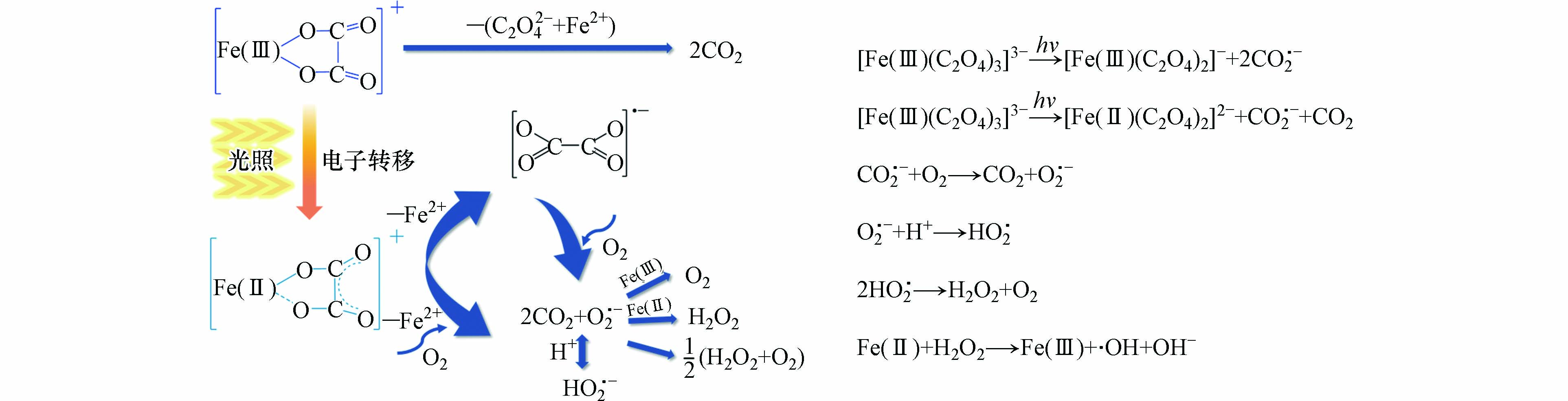
目前关于草酸铁均相体系中Fe(Ⅲ)-Ox络合物光化学分解途径有两种[57, 64-65]. 一种为分子内的配体至金属电荷转移过程(LMCT),Fe(Ⅲ)-Ox络合物在100 ps内迅速分解为Fe(Ⅱ)、CO2 和CO2·− (图4a, b, c). 另一种分解途径为光离解过程,该过程中涉及Fe—O键的断裂,但是草酸不转移电子给Fe(Ⅲ),电子全部用于生成CO2·−. 因此该过程中Fe(Ⅲ)-Ox络合物在100 ps内迅速分解的产物为Fe(Ⅲ) + 2CO2·− (图4d). 尽管该两种过程还存在争议,但是基于大部分报道的草酸铁光化学体系中产生了Fe(Ⅱ),目前普遍更接受草酸铁络合物的光分解遵循LMCT机理[40, 54, 57],具体过程如公式(2—5)所示.
这一过程为诱导阶段,溶解速率相对缓慢. 而随着溶液中Fe(Ⅱ)离子的积累,铁氧化物溶解速率加快,进入了自发溶解阶段,过程如公式(6—9)所示.
首先溶液中的Fe(Ⅱ)或Fe(Ⅱ)-Ox络合物重新被吸附回到铁氧化物表面,并在表面架桥形成Fe(Ⅲ)-C2O42−-Fe(Ⅱ)络合物;随后,Fe(Ⅲ)-C2O42−-Fe(Ⅱ)络合物中外层的Fe(Ⅱ)自发转移到电子至Fe(Ⅲ)[52, 56],产生Fe(Ⅱ)-C2O42−-Fe(Ⅲ)络合物;外层的C2O42−-Fe(Ⅲ)络合物不稳定会被重新释放溶解于水体中.
在异相铁氧化物-草酸体系中,铁氧化物表面所吸附的草酸铁配体(吸附态)与溶液中的草酸铁配体(溶解态)均参与了整个光催化过程,对目标污染物的降解均有贡献[56, 66]. 但是较少有研究很好的区分异相和均相光化学反应过程对污染物去除的贡献,最常用的手段之一是通过对比研究相同溶解态铁离子含量的均异体系中光化学去除污染物性能的差异性. Li等[66]发现当异相体系(1 g·L−1铁氧化物+1.2 mmol·L−1草酸)与均相体系(0.75 mmol·L−1 Fe3+ +1.2 mmol·L−1草酸)中的溶解态Fe(Ⅲ)浓度相近时,365 nm紫外光照射40 min后,双酚A在异相铁氧化物体系的降解率(84.0%)明显高于其在均相体系中的降解率(58.8%),这说明,固体表面所发生的草酸铁的LMCT反应显著影响整个体系的动力学. 研究者发现,通过引入预吸附的手段可以区分均相/异相反应对污染物去除的贡献,且非还原溶解液相中草酸铁络合物的LMCT光分解制约着体系中有机污染物的去除速率[56]. 预吸附时间越长,则非还原溶入液相的草酸铁络合物含量越高,体系中有机污染物的去除速率也越快.
近年来,随着原位谱学分析技术在固液界面有机配体化学性质研究中的应用,可以原位清晰观察配体含量在光反应过程中的变化. 比如,原位ATR-FTIR技术在红外光谱仪样品室加装一个含原位池的漫反射装置,能成功规避光照对红外信号的干扰,可以在光照下有效观察固体表面吸附物种变化以及光反应中间产物的生成,结合XPS等手段分析铁氧化物铁元素价态变化,可以直观草酸在铁氧化物表面光分解等界面反应过程. Xu等[62, 67]采用ATR-FTIR的手段原位观察了草酸在水铁矿、赤铁矿表面吸附后在可见光辐射下的FTIR波谱变化,并结合反应前后的Fe 2p3/2 XPS谱变化,发现草酸在水铁矿表面难以分解,而在赤铁矿表面可以通过光解离途径进行分解. 此外,通过吸附一段后时间分离固液的方法发现水铁矿体系的液相草酸铁络合物是通过典型的LMCT过程分解,而赤铁矿体系中液相草酸铁络合物同样是通过光解离途径分解. 总体而言,对于不同结构性质的铁氧化物,与草酸构成的光化学体系中的界面反应过程不同.
总体而言,铁氧化物-草酸光化学体系中的界面反应过程强烈依赖铁氧化物的结构性质,而相关的研究尚不能清晰阐明二者之间的内在联系. 这主要是由于现有的研究手段难以从原子层面深入分析,其次体系太复杂,影响因素太多,比如常规的pH、铁氧化物晶面效应、共存体系常规阴阳离子干扰、金属掺杂铁氧化物等等因素.
-
在活性氧物种(ROS)的形成过程中,分子氧在其中起着重要的作用[68]. 研究者发现,在脱氧条件下体系中无过氧化氢(H2O2)生成,难以产生有效的活性氧物种,360 min后非草隆仅为22%的轻微降解;而自由曝气条件下,非草隆在180 min内能够实现完全降解,且降解产物的毒性明显降低;强制曝气时非草隆在120 min即可完全降解[69]. 通常认为草酸-铁氧化物光化学体系中活性氧物种的生成主要有3个阶段,分别为铁氧化物表面与液相中的草酸铁配体光分解产生CO2·−、活化分子氧生成活性氧物种(O2·−、H2O2),以及光还原产生的Fe(Ⅱ)或Fe(Ⅱ)−草酸络合物与H2O2发生Fenton反应产生·OH,反应历程如图5所示.
通常而言,因·OH具有较高的氧化能力(氧化电位为2.8 eV),污染物的降解效率主要取决于整个过程中产生的·OH 的数量. 前人均认为·OH是通过两步法生成的,即为O2·−→H2O2→·OH [55, 69]. 活化分子氧产生的O2·−进一步与H+/OH−反应产生H2O2,而后H2O2与Fe(Ⅱ)进行Fenton反应产生·OH(两步法过程). 因此在已报道的铁氧化物-草酸光化学体系,研究者不仅探讨了·OH产生过程,同时也定量分析了H2O2浓度. 例如,紫外光辐射下草酸浓度为1 mmol·L−1时,磁铁矿-草酸体系中H2O2浓度最高浓度为45 μmol·L−1 [56]. 而在磁赤铁矿-草酸-UV体系[53]和赤铁矿-草酸-UV体系[69]中,H2O2浓度分别为2 mg·L−1 (58.8 μmol·L−1) 和4 mg·L−1 (117.6 μmol·L−1). 然而,水铁矿-草酸体系H2O2抑制剂CAT对·OH 的产生几乎无影响. 与此同时,在整个光化学过程中,几乎检测不到 H2O2, 表明·OH并不是来自于Fe(Ⅱ)与H2O2组成的Fenton反应过程. 然而Fe(Ⅱ)却显著影响着·OH的生成. 因此,草酸-水铁矿体系O2·−和Fe(Ⅱ)可一步产生·OH,无需通过先产生中间产物H2O2. 与两步法相比,草酸-水铁矿体系中一步法·OH的产生量远高于均相体系的两步法,因此可更快速有效地降解水体中污染物.
此外,草酸铁络合物光分解途径的不一样也会显著影响体系中活性氧物种的产生. 如上文中介绍,理论上一个Fe(Ⅲ)-Ox分子通过光解离和LMCT途径分解可分别产生一个和两个CO2·−自由基[64- 65]. 因此,光解离过程中的O2·−产生量应高于分子内LMCT过程中的产生量. Xu等[67]采用EPR技术分析了赤铁矿-草酸光化学体系和同异相体系光反应和暗反应最高溶解铁离子含量相同的均相铁离子-草酸均相光化学体系中·OH和O2·−产生情况,发现遵循光分解途径的异相体系不管是·OH还是O2·−含量均高于均相体系. 更多的活性氧物种生成量指示着污染物降解效率的提高,这在一定程度上表现出光分解途径的优越性.
草酸-铁氧化物光化学体系中ROS的产生与转化是污染物降解的关键,如何调控ROS的产生过程来提高污染物降解效率是目前的研究重点与热点. 然而,ROS的产生途径与草酸铁类型、浓度以及铁氧化物的结构性质之间的内在联系尚不明晰,还需进一步探索.
-
铁氧化物与草酸之间的协同光化学作用强烈影响着环境中污染物的迁移与转化,使其在环境领域备受关注. 而在草酸-铁氧化物体系优异的污染物降解效果得到证实之后,其光化学体系应用于环境修复的前景发现之余,深刻明晰该体系光化学反应过程及内在机制显得尤为重要. 草酸与铁氧化物表面的铁络合形成高光化学活性草酸铁配体,光辐射下可通过活化分子氧促使生成活性氧物种,进而提高体系中污染物的降解效率. 然而,分子氧活化效率除了受氧含量制约之外,还受控于草酸在铁氧化物表面上的吸附/溶出、电子转移以及氧化还原过程. 本文系统总结了光照下草酸在铁氧化物表面的吸附与转化特性、草酸铁络合物光分解以及光化学活化分子氧途径,这些成果为有机污染物的控制和原位环境修复奠定理论基础和提供技术支持. 然而由于体系的复杂性,光子被吸收后发生的光生电荷转移及铁溶出过程研究不够深入,草酸铁络合物结构性质与界面光化学分解之间的联系仍待进一步探索,有关草酸-铁氧化物体系光驱动电荷过程尚未揭示,草酸与铁氧化物界面反应机制及其环境效应仍需进一步研究讨论. 未来随着相关的环境光化学过程与机理被阐明,研发自然界中广泛存在的铁氧化物和天然小分子酸相结合的绿色、环境友好的污染控制技术,采用少量干预增强环境自净取得环境效益,将成为解决自然水体污染的有效途径之一,也是未来研发污染物控制技术的新方向.
草酸与铁氧化物相互作用及光化学活化分子氧过程的研究进展
Environmental photochemical behaviors of iron minerals and oxalate and reactive oxygen species generation: A review
-
摘要: 草酸与铁氧化物共存于自然环境中,二者之间的相互作用及光化学行为强烈影响着分子氧的活化. 而分子氧活化影响共存体系中污染物的迁移与转化,是发展绿色污染控制氧化技术的关键. 因此,探讨草酸与铁氧化物之间的相互作用与光化学活化分子氧是目前的研究热点之一. 本文系统总结了近年来围绕草酸与铁氧化物相互作用以及草酸诱导铁氧化物活化分子氧的研究成果,论述了草酸在铁氧化表面的吸附与转化特性、草酸铁络合物光化学过程以及活性氧产生与转移途径,同时探讨了上述过程对环境污染物降解的影响,借此加深理解草酸诱导铁氧化物环境光化学行为与活化分子氧原理,并对今后的研究发展方向提出了展望,以期为利用天然铁氧化物和有机质发展原位环境修复技术提供依据.Abstract: Oxalic acid or oxalate (Ox) and iron minerals coexist in the natural environment. The interaction and photochemical behavior between Ox and iron minerals strongly affect the process of molecular oxygen activation. Molecular oxygen activation affects the migration and transformation of pollutants in the coexisting system, which is the key to the development of green pollution control oxidation technology. Therefore, exploring the interaction between Ox and iron minerals and subsequent photochemical activation of molecular oxygen is one of the current research hotspots. This review systematically summarizes the recent research on the interaction between Ox and iron minerals, the adsorption behaviors of Ox on the surface of iron minerals and its conversion features, Fe(Ⅲ)-oxalate complex photolysis, and reactive oxygen species (ROS) generation. Moreover, the influence of the above process on the degradation of pollutants is also discussed. This review can deepen the understanding of the photochemical transformation of organic contaminants with the activation of molecular oxygen by iron minerals, and put forward the prospect of future research development direction. Furthermore, this review may provide a basis for the development of in situ environmental remediation technologies using natural iron minerals and organic matter.
-
Key words:
- oxalate /
- iron mineral /
- photoactivation of molecular oxygen /
- photodegradation /
- Fe-oxalate complex
-
煤矸石是煤炭开采和洗选过程中产生的一种干基灰分大于50%的岩石. 按来源可分为煤巷矸石、水洗矸石、岩巷矸石、自燃矸石、手选矸石和剥离矸石[1],故来源十分丰富,其产量约占原煤总产量的10%—25%,是煤炭工业排放量最大的固体废物,也是占地面积最大的工业固体废物之一,占全国工业固体废物的20%以上[2]. 煤矸石的排放和堆放造成了严重的资源浪费和环境污染. 露天存放的煤矸石中含有大量的有毒重金属元素,在受日晒、雨淋、风吹等自然条件的影响后,可能通过雨水渗入地表水或土壤,然后通过土壤渗入浅层地下水,这使得镉、汞等各种有毒有害元素渗入到地下,严重影响生态平衡[3 − 4]. 煤矸石相比于普通煤炭,其具有含碳量低、热值低、质地坚硬的特点,是矿山固体废弃物的一种. 其次,从化学组成来看,煤矸石主要含有无机质和有机质[5],其中无机质主要为SiO2和Al2O3,其次是Fe2O3、CaO、MgO等氧化物和单质元素. 因此,集多种有用元素于一体的特殊性质,决定了煤矸石的综合利用成为了众多学者的研究热点和重点[6].
目前,煤矸石已广泛用于有用组分回收、废水处理、建筑材料、农业生产、制备氧化铝[7 − 8]和高压电缆、发电等[9 − 12],制备煤矸石基土壤改良剂也是一种新的利用方式. 其中,煤矸石改良剂作为其资源化利用的重要方式,受到了研究者的广泛关注,然而目前煤矸石改良剂还存在一些问题,比如含硫量高、养分缺乏、重金属污染[13 − 15]等,在利用之前首先应当确定煤矸石的理化性质,通过活化改性等预处理措施,提高煤矸石与修复土壤的适配性,降低其有毒有害成分,实现煤矸石的资源化利用. 本文综述了煤矸石资源化的研究进展,对比了不同的改性方法,进一步阐述了污泥改性煤矸石在生态修复与土壤改良方面的进展,为后期煤矸石和污泥的高值化利用奠定基础.
1. 煤矸石概况及资源化利用现状(General situation and resource utilization status of coal gangue)
1.1 煤矸石组成
煤矸石由多种岩石块体组成,成分相当复杂[16]. 从物理组成来看,煤矸石所含矿物成分比较复杂,主要矿物有黏土类矿物、碳酸盐类矿物、铝土矿、黄铁矿、石英、云母、长石、煤质和植物化石等. 就化学成分而言,主要为无机物并混合有少量有机物的混合物,其中无机物为SiO2和Al2O3,还含有不同量的Fe2O3、Cao、MgO、SO3、K2O、Na2O等无机物,以及微量稀有元素(钛、钒、钴等)[17]. 表1总结了煤矸石的组成情况,从表中可以看出煤矸石样品之间成分存在一定的差异,但是绝大多数煤矸石中所含的无机质主要为SiO2和Al2O3,其次是 Fe2O3、CaO、MgO、TiO2以及其他的氧化物等.
表 1 煤矸石组成成分表(%)Table 1. Coal gangue composition table (%)来源Source SiO2 Al2O3 Fe2O3 CaO MgO TiO2 R2O 参考文献References 掘进矸石 53.10 18.40 8.10 4.50 1.50 0.85 0.70 [17] 洗选矸石 50.50 37.90 4.15 1.80 1.07 1.60 0.65 普通煤矸石 41.10 22.90 2.40 0.73 0.34 0.86 2.00 [18] 六安市煤炭开采 37.80 21.20 2.50 2.60 0.30 0.90 1.40 [19] 内蒙古准格尔 36.90 38.98 0.33 — 0.03 1.01 0.17 [20] 于贵州盘县某矿区 37.30 17.35 18.19 6.85 1.15 4.19 — [21] 普通煤矸石 38.18 18.48 12.97 2.86 2.63 4.51 0.02 [22] 高铝煤矸石 42.17 48.41 0.07 3.77 0.94 1.35 — [23] 普通煤矸石 41.47 15.95 3.53 1.23 1.79 — 1.71 [24] 注:R2O为其他氧化物,“—”未检测到相应物质. R2O is other oxide, and "—"indicates that the corresponding substance has not been detected. | Show Table DownLoad:
CSV
DownLoad:
CSV
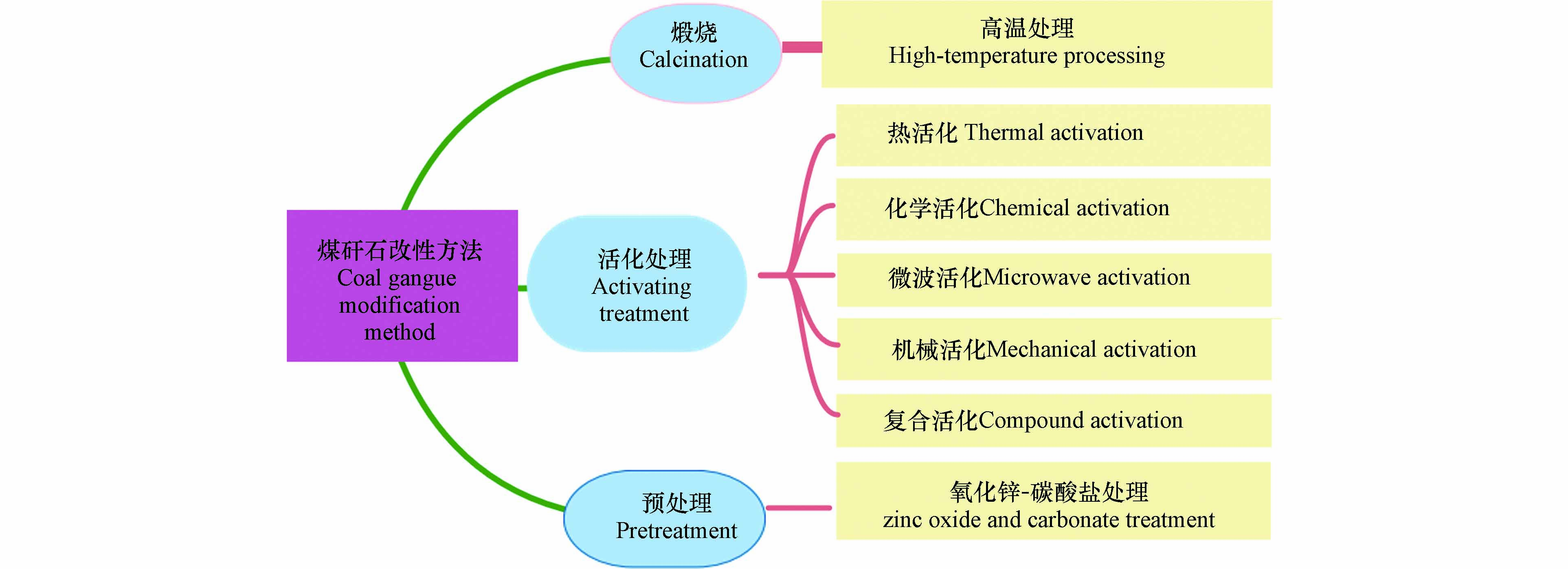
1.2 煤矸石的活化、改性及其主要用途
1.2.1 煤矸石的活化与改性
煤矸石中不具有粉煤灰和矿渣中的玻璃体等活性物质,所以一般而言未经特殊处理的煤矸石并不具有反应活性[25]. 因此,适当的活化处理措施对其反应活性的提高至关重要. 煤矸石的活化方法一般分为:热活化、化学活化、微波活化、机械活化和复合活化等[26 − 27]. 热活化是高温下煤矸石微观结构中颗粒剧烈的热运动,使煤矸石中形成大量自由端的断裂点,处于热力学不稳定状态的玻璃相结构,煤矸石经热活化后含有大量活性氧化硅以及氧化铝,从而达到活化的目的. 2012年何燕[28]研究了热活化煤矸石-水泥复合体系水化性能,结果表明煅烧温度为750 ℃,保温时间为4 h的热活化煤矸石对水泥体系的火山灰贡献率较高. 化学活化是通过引入少量的其它试剂,致使结构中共价键断裂,形成离子并进入溶液,使煤矸石中化学键不断被破坏,促使其结构解体,达到激发其活性的目的[29]. 2012年吴红等[30]利用化学方法对活化煤矸石,研究了不同激发剂对免烧砖性能的影响,实验结果表明,煤矸石基免烧砖强度在激发剂CaO和Na2SO4的激发作用下显著提高,最佳掺量为8%CaO、2%Na2SO4. 机械活化一般是指利用机械力化学原理进行活化,即通过机械能的施加使固体等物质的物理化学性质发生改变. 2011年司鹏[31]系统的研究了机械力活化过程中煤矸石的粒度、矿物结构以及反应产物活性的内在联系,发现球磨时间、方法、介质决定了煤矸石的机械力活化效果,球磨过程中煤矸石活性被充分激发. 一般情况下,上述方式单独处理时都会面临耗能大,反应不能完全进行,效率低等问题,因此出现了复合活化的方式. 有研究表明,复合活化效果通常情况下优于单一活化[32 − 33]. 2010年Li等[34]提出了一种新的复合机械-水热活化(CMHTA)技术,并以传统的机械-热激活(TMTA)技术为对比,研究结果表明,复合活化效果能显著提高粗煤矸石的活性. 2010年张晓旭等[35]采取热活化、机械活化和化学活化并用的方式将煤矸石与石灰混合活化,实验表明,当石灰和煤矸石取代水泥量的30%,其中石灰占煤矸石量的40%时,煅烧温度为675 ℃时,强度和流动性达到最佳值,与单一热活化煤矸石水泥砂浆相比强度有大幅度提高.
然而,由于成煤条件及环境不同导致煤矸石组分复杂且多样,活性成分含量低,其利用效率并不高,活化虽然能改变煤矸石的部分性能,但是对材料本身变化较小,达不到实际应用的特殊效果,为了提高煤矸石的活性,增加其特殊的性能,对煤矸石改性研究已经成为了热点. 改性方法如图1所示[36],其中酸改性可以使Al、Fe、Ca等金属更易溶解,改性后的煤矸石内部和表面产生的孔隙更多,使得吸附能力更强. 相比而言,碱处理不仅能溶解煤矸石中部分金属氧化物,而且能与煤矸石中存在的硅铝酸盐反应,从而合成沸石分子筛,其吸附能力比传统的沸石分子筛更强[37]. 煤矸石改性不仅降低化工产品的生产成本,而且能提高煤矸石的利用效率,并且产生更高的附加值,经济和环境效益显著,符合“低碳经济、绿色经济、循环经济”的国家政策.
1.2.2 煤矸石的用途
废煤矸石综合利用是坚持走资源节约型、环境友好型发展道路的必然选择,是大势所趋. 据统计[38 − 39],12%的煤矸石用于生产建材,32%用于发电,56%用于工程及其他. 随着科学技术的发展,以及对煤矸石资源化综合利用认识的不断深入,我国众多学者已经在有用组分回收、废水处理、制备建筑材料、农业生产等方面取得了显著成绩,但并未摆脱煤矸石资源化利用率低的现状[5],下面分别从几个方面对煤矸石利用情况进行综述.
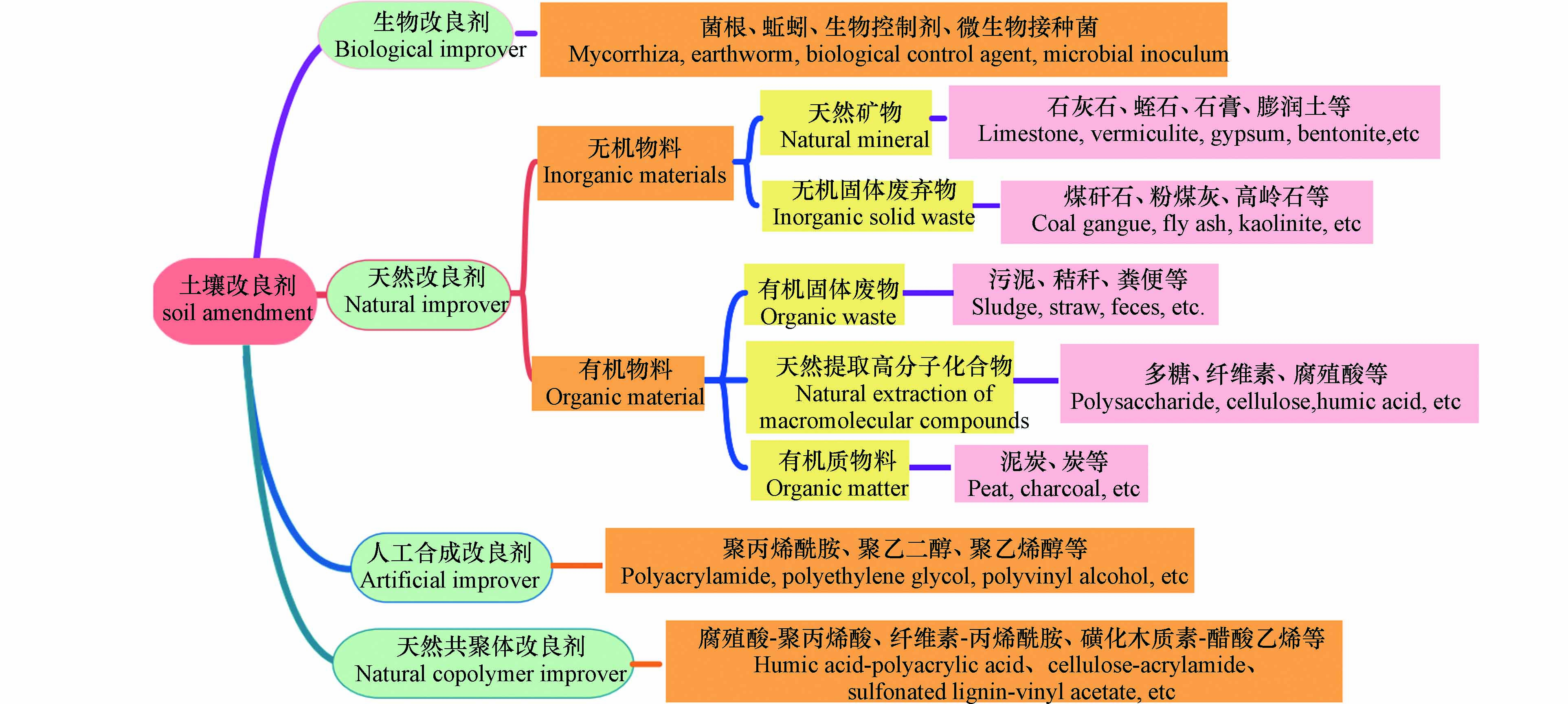
1)煤矸石作为土壤改良剂. 煤矸石中含有腐殖酸、有机质、硅、钾、铁以及多种稀有元素,能促进有益微生物的活性和植物根系的发育,因此,以煤矸石为主的土壤改良剂研究引起了学者的广泛关注. 如图2所示,目前使用的土壤改良剂,按照生产原料可以分为天然改良剂、合成改良剂、天然合成共聚物改良剂和生物改良剂[40].
煤矸石作为天然的无机固体废物,其资源十分丰富. 有研究表明,煤矸石土壤改良剂能有效治理土壤板结、沙化、盐渍化等问题,改善土壤通透性,增加土壤保水保肥能力,减少土壤水分蒸发,增加土壤耐盐碱能力,促进植物根系更好吸收微量元素[41 − 42]. 2016年王琼等[43 − 44]研究了不同的高硫煤矸石对苏打盐渍土化学性状的影响,其研究结果表明,施用高硫煤矸石对苏打盐渍土的改良有较好的效果. 2020年Li等[45]对煤矸石填筑复垦土壤重建过程进行了长达10年的研究,结果表明土壤在复垦后开始逐渐从无序状态恢复到超有序状态,为煤矸石土壤复垦技术提出了宝贵经验. 其次,煤矸石还可以作为重金属钝化剂,可以改变尾矿中锌、铅、镉、铜和铬等重金属的形态,使交换态和碳酸盐结合态转化为铁锰结合态、有机结合态和残留态,进而降低重金属的有效性[46].
2)煤矸石作为化工原料. 煤矸石中含有丰富的铝元素,是廉价易得的铝基化工原料,当煤矸石中Al2O3含量大于35%时,可利用煤矸石代替铝土矿提取和制备氧化铝、氢氧化铝和聚合氯化铝等20多种铝盐化工产品[5]. 2020年贾敏等[20]对煤矸石煅烧活化提取氧化铝技术进行研究,通过煅烧和一步酸溶工艺可以成功地生产冶金级氧化铝. 煤矸石中还含有30%—65%的氧化硅,有效回收煤矸石中的氧化硅成分可生产白炭黑、碳化硅等一系列硅系化工产品,是煤矸石高附加值利用的重要途径之一[19]. 2021年Xie等[47]研究了煤矸石中矿物的高温煅烧相变及铝硅矿物的高效分离,实验表明,煤矸石中高岭石在高温煅烧过程中发生相变,并且在相变过程中释放出活性二氧化硅矿物,可通过碱浸提将其与其他矿物分离. 煤矸石中除含有丰富的铝、硅等常见的金属外,还有少量钛、镓等元素. 通过活化煤矸石,可用来提取有价金属. 2020年辜芳等[22]通过优化pH、混合萃取剂比例、振荡时间等工艺参数,实现了混合稀土与铁、铝的有效分离. 在煤矸石的有价金属的提取过程中,还应该注重分段回收、分段磨矿,以确保有用元素的高效回收[48]. 2020年Ashfaq等[49]在低pH值下的静态和动态浸出条件下,金属离子浸出的浓度相对较高,这归因于各个金属离子配合物的溶解度积值的差异. 在目标金属离子中,金属提取率分别为30%和65%,砷和硒在静态和动态浸出测试中表现出最高的迁移率. 此外,煤矸石还可以用来制备催化剂载体[50 − 51],负载催化过硫酸盐的活性组分,制备出改性煤矸石基过硫酸盐催化剂[52 − 53],从而达到以废治废的目的.
3)煤矸石作为吸附剂. 煤矸石因其矿物成分中高岭石的存在,使得它具有一定的层间结构,同时具有优异的稳定性,通过对煤矸石进行活化和改性,或与其它吸附剂复合,可提高其比表面积和离子交换能力,制得的煤矸石基吸附材料用于去除水中氨氮、磷、有机物、重金属离子等污染物[54],也可以改善污泥中的有机物、重金属离子等具有较好的效果[5,55]. 2010年王现丽等[56]利用改性煤矸石处理味精精馏段生产废水,结果表明投加改性煤矸石对浊度、氨氮、有机污染物的去除率分别可达到70.73%,63.67%,69.81%. 2015年裴会芳等[57]以城市污泥和煤矸石为原料,进行了制备多孔陶粒的实验研究,结果表明,以煤矸石和城市污泥为原料,可以制备多孔陶粒,煤矸石和污泥中的部分重金属已溶解到硅酸盐玻璃相中,形成稳定的固溶体,不会对环境造成二次污染. 2016年Jabłońska等[58]研究了天然煤矸石和改性煤矸石的结构和表面性质,结果表明,天然煤矸石的比表面积较小,其内部主要是介孔,化学改性的煤矸石增大了总孔容和比表面积,可以用于工业废水的预处理. 2020年石凯等[59]采用两步化学活化法制得多孔煤矸石,并研究了多孔煤矸石的吸附性能,结果表明,多孔煤矸石吸附罗丹明B属于熵驱动型物理吸附. 2022年Zhang等[60]采用ZnCl2对天然煤矸石进行改性,与原煤矸石相比,改性煤矸石对含磷废水的处理能力明显提高.
4)煤矸石作为建筑材料. 煤矸石主要是碳质、泥质和砂质的混合物,在岩性上主要包含煤质、泥页岩和粉砂质泥页岩等类型[61]. 煤矸石可作为混凝土的骨料,且其矿物组成和化学成分与黏土相似,可代替黏土作为原料用于制备水泥、砖和新型墙体材料[62]. 煤矸石强度较高,经过破碎、筛分之后可以代替石子来制备混凝土. 其次,煤矸石具有良好的防腐性能,可以用作道路填料. 经检测,煤矸石的强度、抗冻性或抗裂性均满足公路技术要求,具有地基承载力高,抗滑稳定性好等优点[63]. 利用高孔隙率、强吸水性的煤矸石可生产多孔烧结砖,采用回转窑法生产煤矸石陶粒等[64]. 煤矸石是由多种矿岩组成的混合物,密实度高,荷载能力强. 同时,煤矸石具有适当的导水性、吸附特性和浸出行为,因此可将其作为充填材料用于回填复垦,不仅降低了煤矸石堆存的占地率,实现了煤矸石的就近处置,而且改善了地下采煤引起的地表沉降,具有良好的经济效益和环境效益. 2016年刘章锋等[65]通过添加煤矸石作为骨架材料,水泥作为凝胶材料,纤维作为辅助材料对污泥进行固化改性,并研究其固化机理,结果显示固化体内部黏聚力随煤矸石添加数量的增大而减小,黏聚力随煤矸石添加量的增大而增大. 2016年Hu等[66]列举了煤矸石在建筑材料、灌浆材料、空心砖等方面的应用. 砖瓦企业提供了新的发展思路和方向,是中国式的消纳污泥的重要途径,有利于行业综合利用技术水平的提升,对砖瓦行业发展低碳、环保绿色产业,加快污泥无害化、减量化、资源化综合利用有重要现实意义. 煤矸石储量大、价格低,在生产建筑材料方面有着非常好的应用前景. 但煤矸石作混凝土骨料要充分考虑其抗折强度、耐磨性、渗性和抗冻性,煤矸石制水泥要控制好煤矸石的掺入比例,煤矸石制砖要通过工或参数优化提高其可塑性,煤矸石制新型墙体材料要增加其科技投入. 同时,应当注意的是煤矸石在制备建筑材料时需要进行高温煅烧,这可能会导致煤矸石中所含有的有害微量元素以气体的形式释放到大气当中[5].
5)煤矸石作为陶瓷原料. 氧化铝和二氧化硅是陶瓷生产中常用的原料,从表1可以看出,煤矸石中的主要成分也是二氧化硅和氧化铝,而且煤矸石本身也具有大量的微孔和较高的比表面积,完全可以利用煤矸石来制备性能优异的陶瓷等材料. 然而污泥中也含有与黏土类似的硅酸盐成分,在一定条件下可以代替黏土生产陶瓷,故国内外许多学者对利用煤矸石和污泥制备多孔材料和陶粒进行了研究. 2016年支楠等[67]研究了煤矸石污泥陶粒烧结膨胀性能,结果表明,仅以污泥为原料不可能生产出合格陶粒,必须配入辅助原料,考虑到煤矸石成分等因素,故采用煤矸石为辅助原料. 2015年祁非等[68]以城市污泥和煤矸石为原料制备了陶粒,实验结果表明陶粒能有效固化城市污泥中有害重金属元素,并且不会对环境造成的二次污染. 2019年Zhou等[69]以煤矸石为原料,采用喷雾干燥烧结法制备低成本陶瓷微球吸附剂,用于水溶液中阳离子红X-5GN和阳离子蓝X-GRRL的脱除,吸附剂的吸附容量为1.044 mg·g−1和 2.170 mg·g−1,使用煤矸石陶瓷吸附剂处理有色废水可以达到以废处理废弃物的目的,煤矸石吸附剂的综合经济效益和环境效益具有广阔的应用前景. 2022年张会等[70]以煤矸石和滑石为原料,在高压使其成型,干燥后经过高温煅烧并保温,压成型法制得的试样采用XRD分析试样为堇青石,可见固体废弃物煤矸石为主要原料,可制得具有一定性能的堇青石多孔陶瓷. 同年,程冠吉等[71]利用废弃煤矸石为原料,并添加铝矾土、可溶性淀粉混合均匀成型后经烧结制成多孔莫来石陶瓷,通过测定显气孔率得到最优制作工艺.
目前,我国煤矸石综合利用方式主要有分选、采矿区充填、铺路、发电、生产空心砌块和水泥等,表2总结了煤矸石的综合利用情况. 由于废弃煤矸石中有用矿石占比较少,分选的经济成本要求较大;粒径大的煤矸石充填采矿区可能造成坍塌,粒径小的煤矸石充填又可能被雨水冲洗流失;利用煤矸石作为化工原料又会对环境造成二次污染,治理成本较高;烧制轻骨料、生产空心砌块等技术成熟,可以消纳大量煤矸石,但总体产品附加值不高. 所以急需开发一种新的方法,使得煤矸石变废为宝,提高煤矸石的可利用价值.
表 2 煤矸石的综合利用情况Table 2. Comprehensive utilization of coal gangue利用途径Utilization ways 具体方式Specific way 优点Advantage 缺点Disadvantage 分选 有用矿物 重复利用资源 有用矿石占比较少、经济成本较大 矸石 直接利用 采矿区充填 技术含量较低、操作简单、经济 充填不紧密、容易坍塌 铺路建设 耐腐蚀能力强、抗压抗剪强度大 雨水天气易打滑,存在安全隐患 生活中的应用 发电 节约能源,变废为宝 热值低,炉耗高 化工原料 硅、铝元素含量高 造成环境二次污染 建筑材料 生产水泥、砖等 节省土地和能源,变废为宝 产品受样品差异大,质量问题较多 | Show TableDownLoad:
CSV
2. 污泥、煤矸石在土壤修复改良方面的研究进展(Research progress of sludge and coal gangue in soil remediation and improvement)
煤矸石具有一定的孔隙度、透气、保水性性能,目前已被应用于制备采煤沉陷区复垦充填基质、土壤改良剂和矿物肥料等. 通过物理、化学或生物方法将煤矸石改性,或添加其他基质共同处理等方法,可以有效改良煤矸石的理化性质,解决其生物活性差的缺陷,使其形成类似土壤性质和结构的生态基质,对于改善植物生存环境,重新建立和恢复复垦区的土壤生态体系,加速煤矸石成土化具有积极作用[72]. 煤矸石与其它物质配合,克服了其持水性差、肥力不足等缺点,同时还能调节极端pH值,同时煤矸石中的营养元素匮乏限制了其作为优质土壤改良剂的能力. 城市污泥同样作为一类产量巨大的废弃物,不仅含有大量的有机质、氮及其他养分,同时含有丰富的微生物类群,能够满足植物生长的需要. 近年来,污泥已被用于土地复垦等领域[73 − 74],既能为植物提供肥料和微量元素,又可改善土壤的理化性质,促进土壤熟化,还能增加土壤微生物的活性[75],但污泥因为病原菌和重金属的潜在危险[76],其土地施用率受到严格的限制. 针对上述两种废弃物的特性,能否找到适合的方法将二者进行处理改造,使其发挥各自优势,实现优势互补,对于两种废弃物的处置和资源化利用具有重要的意义和应用前景. 表3总结了近年来污泥和煤矸石在土壤修复利用的情况,证明了污泥耦合煤矸石治理污染土壤的可行性. 2018年包红旭等[77]公开了一种适用于无土草坪的煤矸石混合培养基质及应用,该发明是将采煤过程中产生的煤矸石、生活污水处理厂中的剩余活性污泥以及农作物秸秆作为材料配制而成. 但是,由于煤矸石和污泥中均存在一定量的重金属,作为基质应用时还是存在重金属环境风险,后续进行环境风险评估也是煤矸石和污泥作为土壤基质使用的必要条件.
表 3 污泥、煤矸石修复土壤利用情况Table 3. Utilization of soil remediation by sludge and coal gangue研究对象Research objects 研究方法Research method 结论Conclusion 参考文献References 污泥、煤矸石 混合复配,采用高羊茅盆栽试验进行验证 煤矸石粒径越小,基质黏粒含量越高;污泥堆肥能显著提高基质黏粒百分比,有利于保水保肥;植物堆肥则能提高砂粒占比,有利于透水透气 [81] 污泥、煤矸石 利用ZnCl2、盐酸对污泥、煤矸石复合基改性并对废水厌氧消化 污泥和煤矸石制备的复合基活性炭表面孔状结构发达,官能团种类增加,可改变厌氧微生物群落结构,优势菌种得到富集 [82] 优良城市污泥、煤矸石 混合复配,淋滤盆栽实验 可以有效钝化煤矸石中重金属元素,淋溶出煤矸石中的重金属含量低,且能有效提升渗滤液的pH,可以作为优良基质 [83] 城市污泥、煤矸石以及土壤 城市污泥、煤矸石土壤混合基质盆栽试验 植物-土壤系统可以逐渐降低生长介质中有害物质的浓度 [84] 污泥、煤矸石、粉煤灰以及土壤 污泥、煤矸石、粉煤灰混合后加入到土壤中进行盆栽试验 有利于植物的生长,而且复合基质中重金属污染水平处于清洁状态 [85] 污泥、煤矸石以及土壤 不同处理的污泥和煤矸石混合后加入到土壤中进行盆栽试验 能促进部分植物地下部分的生长 [86] 污泥、煤矸石、粉煤灰以及土壤 污泥、煤矸石和粉煤灰混合后加入到土壤中进行盆栽试验 土壤的有机质、全氮、有效磷及速效钾含量均达到了土壤等级的一级标准,土壤的营养成分均得到了改善 [87] 污泥、煤矸石、粉煤灰以及土壤 污泥、煤矸石和粉煤灰混合后加入到土壤中进行梯田试验 可以实现固体废弃物的资源化利用,变废为宝,同时又增加煤矸石山复垦中土壤的肥力 [88] | Show TableDownLoad:
CSV
煤矸石容重高于一般土壤且毛管孔隙少,将其掺入土壤可以增强土壤透气性和疏松度,具有一定的营养成分,但是难以形成土壤团聚体且缺少活性微生物;污泥农用的透气性不佳但是可提高土壤中氮磷元素、有机质的含量以及微生物,调节土壤孔隙,改善土壤的团粒结构[13]. 城市污泥直接应用于煤矸石边坡生态修复,有利于植物生长[78]. 土壤和植物的混合体系使生长基质中的有害物质逐渐减少. 对地表水环境的负影响主要是氮、磷的富营养化,但环境安全总体可控[79]. 所以将用污泥来改性煤矸石可以改善煤矸石营养元素匮乏的缺点[80],使基质具有良好的营养水平,获得理想的植物营养效果,并且在保有其良好透气性的基础上,增加保水保肥性.
3. 污泥煤矸石基质改良土壤的因素及其机制(Factors and mechanism of soil improvement by sludge coal gangue substrate)
3.1 pH对污泥煤矸石基质改良土壤的影响及机制
pH是影响污泥改性煤矸石的关键影响因素之一. 在不同的pH下,污泥和煤矸石所发生的反应也存在差异. 在常温的情况下,酸性煤矸石中普遍含有较高的硫化物及其他有害金属元素,黄铁矿(FeS2)等硫化物遇降水和氧气就会氧化产生酸矿水[89](公式1、2). 煤矸石山中的有毒有害金属元素就会随酸矿水溶解排出,导致矿区周围土壤和水体的污染,并且酸矿水的排出,会影响土壤的pH,严重时会对土壤微生物以及植物造成抑制作用. 2023年卢欢等[90]研究了pH对煤矸石中重金属和SO42−释放行为的影响,初始淋溶液pH越低越有利于SO42−的释放,同时产生大量H+,体系pH降低至1.31—1.52,这是酸性矿山废水生成的根源所在. 在pH值较低的酸性环境中,溶液中有大量氢离子,吸附剂也吸附较多氢离子,造成吸附剂表面负电荷的减少,所以污泥煤矸石复合基质的材料对土壤改良的效果较好.
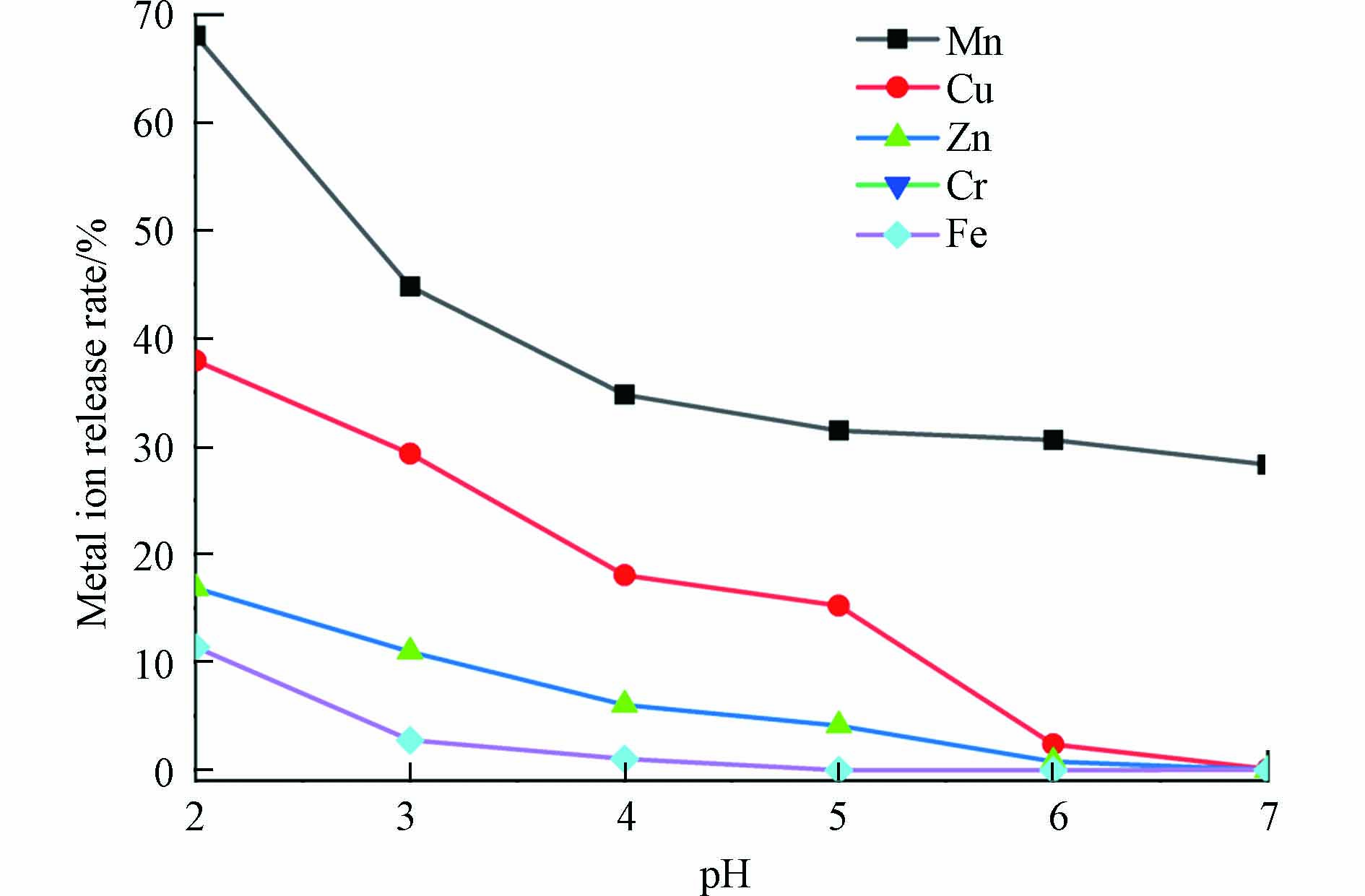
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) 不同的污泥成分不同,pH也存在一定的差异. 2015年田卉宇等[86]研究了不同的污泥和煤矸石理化性质,结果显示pH由小到大分别为冻融污泥(pH7.08)、腐熟污泥(pH7.12)、脱水污泥(pH7.65)、煤矸石(pH8.73),并且提出污泥的添加可以有效降低煤矸石基质的pH值,有利于植物的生长. 2014年马保国等[91]利用污泥和酸性煤矸石做成淋溶柱开展柱淋溶模拟试验,从淋溶第1天至第41天,pH从2.0升至7.5并保持平衡,而单独的酸性煤矸石在淋溶模拟试验中pH迅速从2.5下降至1.5左右并保持平衡,对淋溶液进行分析后发现污泥覆盖煤矸石不仅能降低淋滤液酸性,而且能控制重金属淋溶迁移. 2022年周新华等[92]研究了pH值对碱性煤矸石碱度和重金属释放规律影响研究,如图3所示,pH值对碱性煤矸石中碱度释放具有较大影响,当pH值逐渐增大时,碱性煤矸石向水体释放的碱度随pH变大而降低,从环境污染防控角度考虑,应尽量控制碱性煤矸石堆体环境体系pH值大于6.
3.2 配比对污泥煤矸石基质改良土壤的影响及机制
煤矸石、污泥以及其他废弃物按照不同的比例混合后,对土壤的修复效果也有所不同. 2011年Qian等[93]道报了煤矸石-粉煤灰-污泥混合物用于矿山废弃地复垦的盆栽试验研究,结果表明,煤矸石-粉煤灰-污泥比例为2:6:2是最佳的混合比例,能最大限度地减少有毒元素,提供足够的养分,3种基质的适当配比可以有效地促进植物的再生利用,增加植物对养分的吸收,为煤矸石、粉煤灰和污泥的生态利用提供参考. 2015年刘荷芳等[94]研究了生污泥、腐熟污泥和冻融污泥3种污泥在煤矸石山复垦中的应用效果,综合考虑污泥腐熟和运输的成本,最终确定煤矸石、污泥和粉煤灰的质量配比为60:30:10的混合基质可作为煤矸石山复垦的最佳选择. 2017年王迁等[87]将矿区脱水煤矸石与污泥、粉煤灰和土壤按不同质量混合,结果表明土壤中添加不同基质的混合物均不同程度地改善了土壤养分含量,满足植物生长需要,最终确定土壤、污泥、煤矸石和粉煤灰配比为60%:5%:15%:20%效果最佳. 2018年周昊等[95]以煤矿区粉煤灰、生活污泥以及煤矸石为原料,按照不同的配比添加至土壤中,最终选择污泥、煤矸石、粉煤灰和土壤质量配比为5%:15%:20%:60%为种植白三叶的最佳方案,并指出在土壤改良和植物修复过程中应着重控制其相应掺杂比,以提高改良效果及修复效率,其研究结果和王迁一致. 2018年段超等[96]发明了一种改良盐碱地的生物质废弃物土壤调理剂,其中污泥和煤矸石在调理剂中占比为75%左右,该方法成本低,能加快盐碱地改良的进展,提高改良效果. 2020年方娜[88]将污泥、煤矸石和粉煤灰按一定比例混合,用于煤矸石山复垦,结果表明,在一定程度的调整生污泥的复混比例,可以达到良好的复垦效果. 加入不同比例的煤矸石和污泥可以改善混合基质的部分性能. 煤矸石中有机质含量较污泥低,混合基质中加入煤矸石比例越高,有机质含量相对降低,速效氮、磷含量会随之降低,反之,混合基质中加入污泥比例越高,有机质含量升高,速效氮、磷含量随之升高,混合基质的性能得到改善. 污泥、煤矸石耦合过程中,配比直接影响到反应物的比例,极有可能影响整个反应.
3.3 粒径对污泥煤矸石基质改良土壤的影响及机制
煤矸石的表面存在孔隙、附着小颗粒,且包含的赤铁矿、沸石、黄铁矿、绿泥石、高岭石和磁铁矿等矿物容易溶解. 煤矸石粒径较大,养分不足且保水性差,污泥的加入起到了填充保持作用. 煤矸石粒径越小,固液接触界面的面积越大,导致污染物溶解释放总量呈增加的趋势. 污泥粒径越小其吸附效果越好,初始离子浓度对不同重金属元素吸附效果影响有差异,吸附效果升高或降低,主要取决于活性污泥中的微生物对不同重金属元素的生物敏感性[97]. 2018年孔涛等[98]探讨了煤矸石对盐碱地绿化的改良效果及其对土壤微生物的影响,结果表明,煤矸石可以改善盐碱地的生态质量,不同粒径的煤矸石混合基质可以作为盐碱地绿化的改良剂. 2020年狄军贞等[99]研究了不同地区煤矸石污染组分溶解释放规律与粒径的关系,结果表明煤矸石粒径越小,污染物越容易溶解释放,并且建议煤矸石的粒径大于0.18—0.25 mm,如图4所示,在煤矸石粒径为0.25—6.70 mm时,SO42−、Fe2+、Mn2+的质量浓度均相对较低,可以减少煤矸石在复垦过程中离子的释放. 2021年易名儒等[100]研究了不同粒径污泥的结构稳定性,结果显示不同粒径的污泥具有不同的结构稳定性,其对污染物降解能力差异较大. 煤矸石粒径大小≤8目并与城市污泥以体积比1:1混合制成植生基质改良后的沙土理化性质有了明显改善且可以保证植物的健康生长[101]. 粒径小的颗粒基质表面能较高,同时颗粒间接触面积小,导致总表面积很大,处于较高能量状态. 2021年Han等[102]利用煤矸石覆盖矿区土壤,探究了粒径对土壤修复性能的影响,作者认为粒径小的煤矸石与空气和水接触更多,并且可以形成更窄的毛细孔,增强持水能力,最终确定覆盖用煤矸石粒径范围为0.5—2 cm效果最佳.
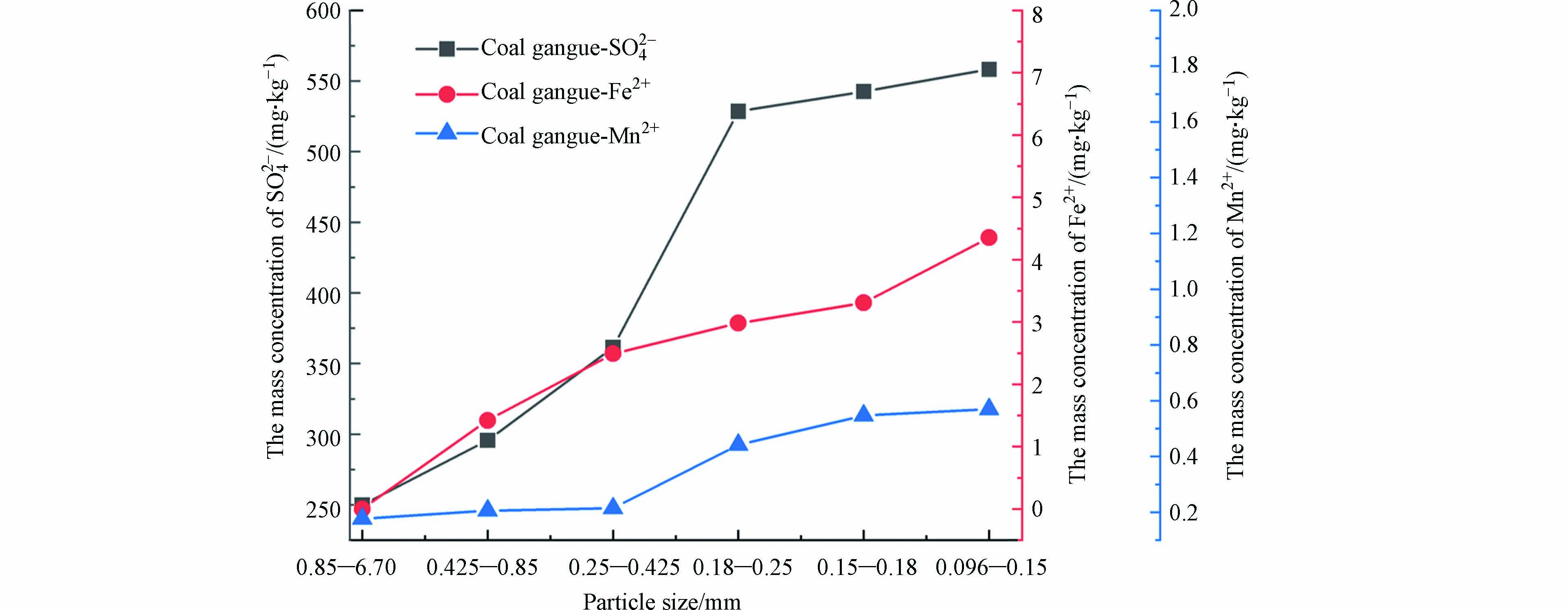
3.4 其他因素对污泥煤矸石基质改良土壤的影响
在污泥、煤矸石耦合的过程中,除了上述的温度、配比、粒径外,还有其他的因素会影响污泥改性煤矸石的性能,比如污泥和煤矸石本身的组成、水分、灰分、压力等,其中水分、灰分会随着掺杂量的改变而改变. 含水率也会影响到反应,表面含水率小于内部的含水率,产生了湿度差,由于内层相对于表层湿度较大,于是水分就从内层向表层移动. 由于污泥和煤矸石本身组成成分差异,当植物基质中的煤矸石比例逐步提高后,土壤中的磷元素会面临短缺问题,当煤矸石比例偏低时,可能出现富营养化现象,因为煤矸石与污泥的磷元素差异比有机质含量差异和氮元素含量差异要显著得多,如果煤矸石和污泥成分所造成过高的碳磷比会影响植物体内的RNA转录[103],影响植物体内的蛋白质合成,过高的氮磷比会降低植物的固氮量.
4. 展望(Prospects)
煤矸石的资源化利用一直是国内外学者们研究的重点,也是国家可持续发展的重大需求. 由于污泥和煤矸石的产量巨大,国家对煤矸石、污泥资源化利用给予了较大的政策支持.
(1)煤矸石、污泥资源化处理技术虽取得了快速发展,但其配套设备、技术的成熟度仍需进一步完善. 污泥和煤矸石联合后相比单类物质具有更大的优势. 尽管很多学者在煤矸石和污泥修复改良土壤方面做了大量的研究,包括煤矸石基质、土壤微生态的长期稳定性以及对植被的影响方面,但是由于煤矸石和污泥均属于固体废弃物,在资源化利用的同时,还需要考虑其对对环境的安全性.
(2)煤矸石、污泥资源化还应当关注其用量大、成本低、效益高、具有较强的针对性等特点,加强煤矸石和污泥作为土壤改良剂的产品标准和改良工程规范标准,使煤矸石、污泥土壤改良剂生产工艺、产品标准和改良过程规范化,使得煤矸石、污泥土壤改良剂能够大规模应用和推广. 因此,未来几年应加强煤矸石、污泥土壤改良剂基础理论研究及配套设备开发,进一步拓展利用途径.
(3)我国煤矸石、污泥资源化利用率虽然在逐年增加,但由于煤矸石、污泥产量巨大,资源化利用途径依然有待提高. 交叉领域的兴起为煤矸石、污泥资源化利用带来了曙光,可以加大煤矸石、污泥在其他领域的资源化应用,提倡“以废治废”的观点,解决煤矸石、污泥以及土壤带来的环境污染,实现社会经济和环境效益的协调发展.
-
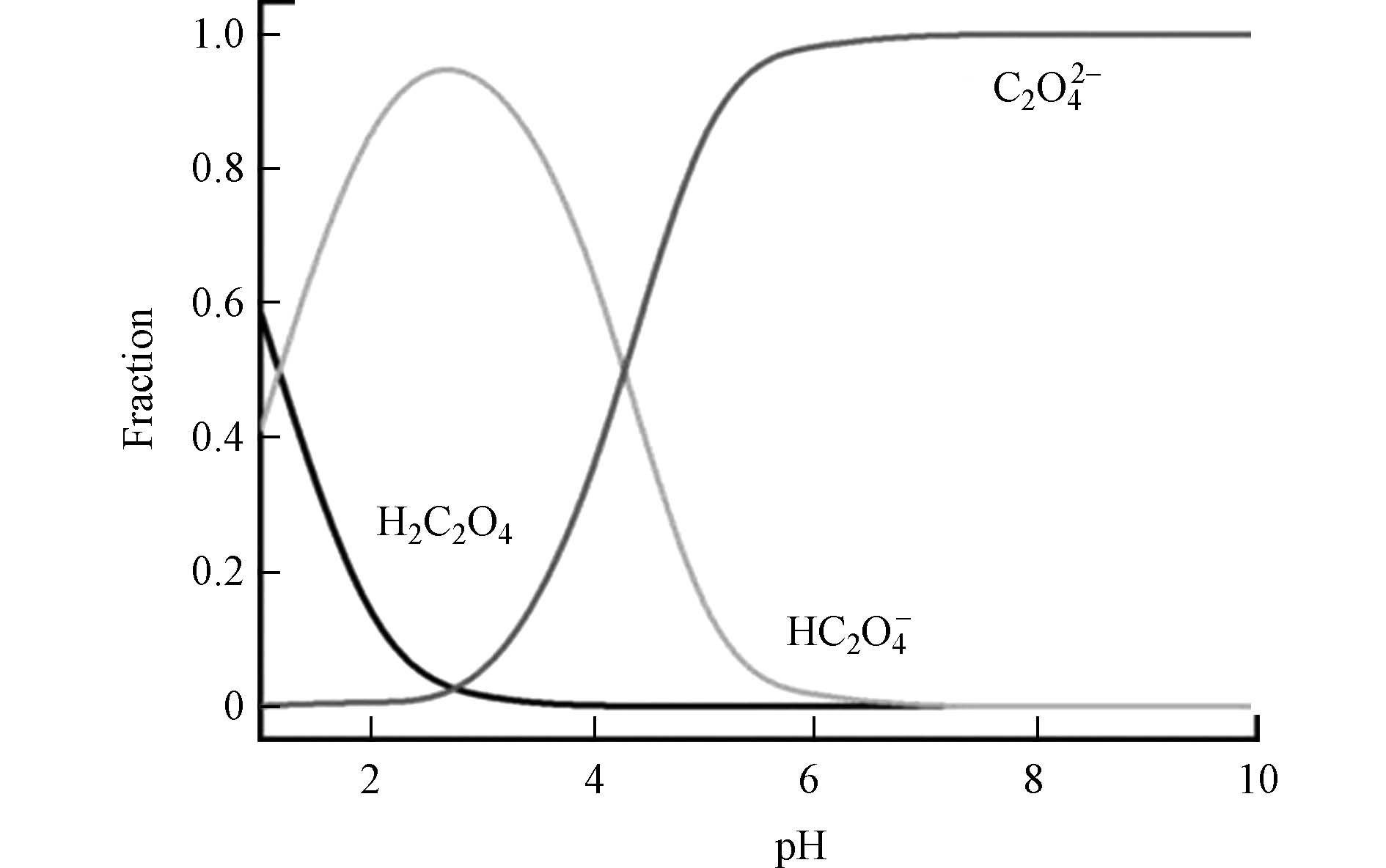
图 1 草酸在不同pH溶液中的物种形式[42]
Figure 1. Species forms of oxalic acid in solutions of different pH
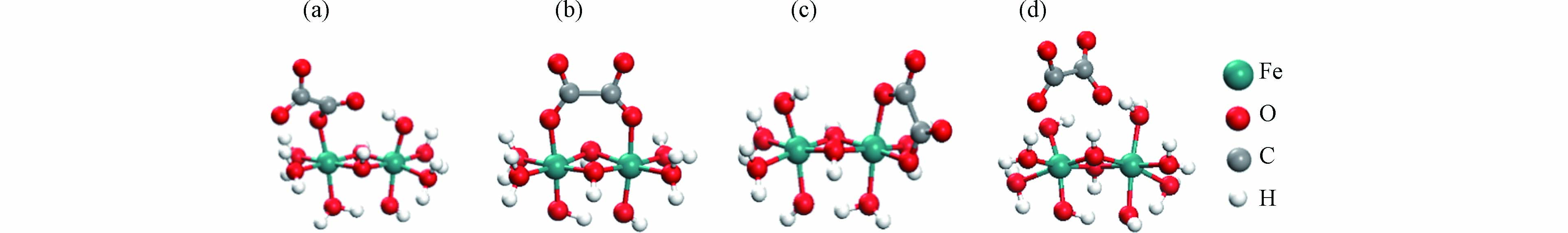
图 2 草酸根在铁簇上的理论络合构型[46]
Figure 2. Optimized Ox surface complex geometries on iron clusters
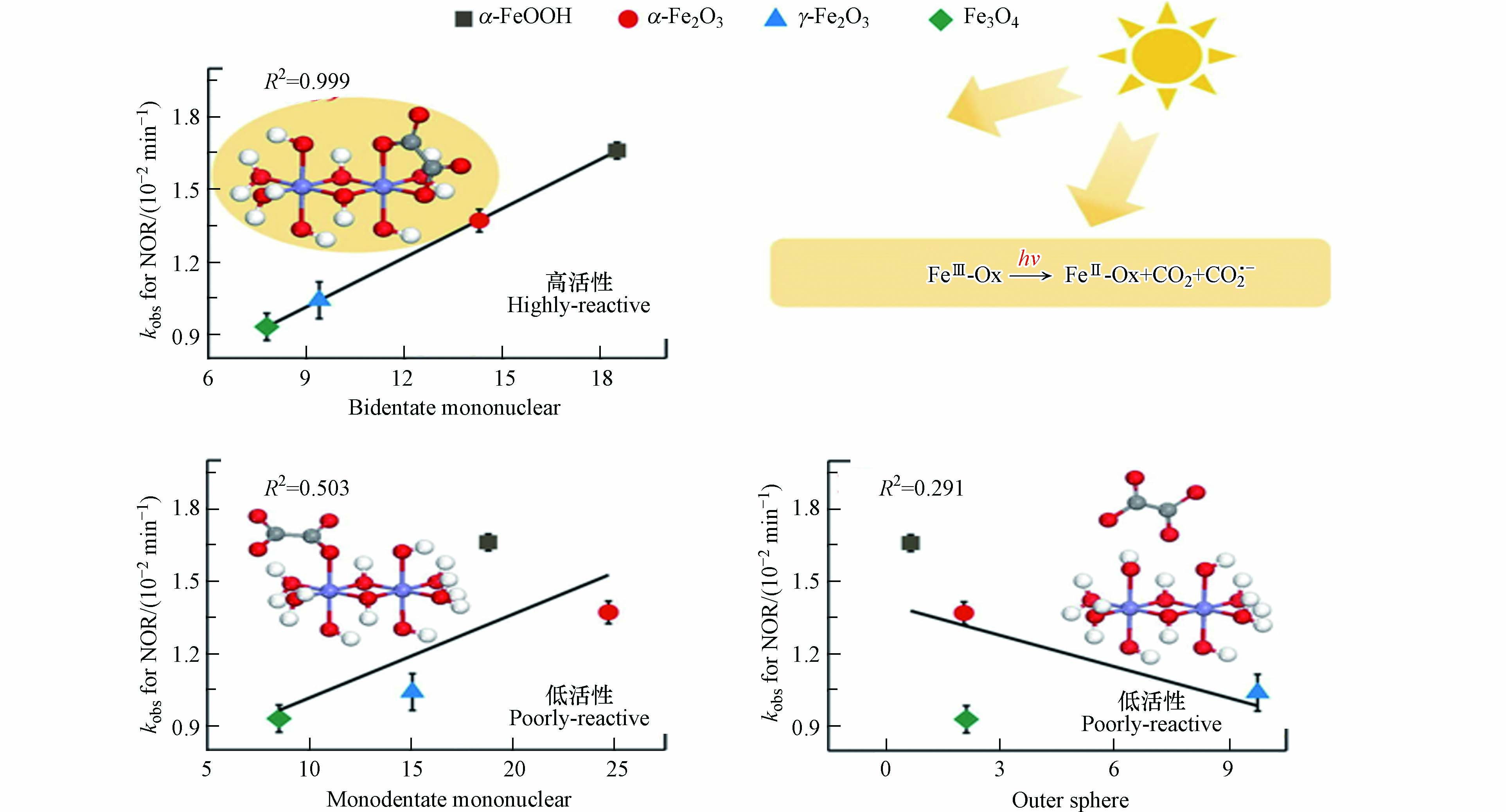
图 3 草酸在铁氧化物表面形成的络合物构型与诺氟沙星降解速率的相关性[40]
Figure 3. Correlation between Ox surface complex geometries on iron oxides and the degradation rate of norfloxacin
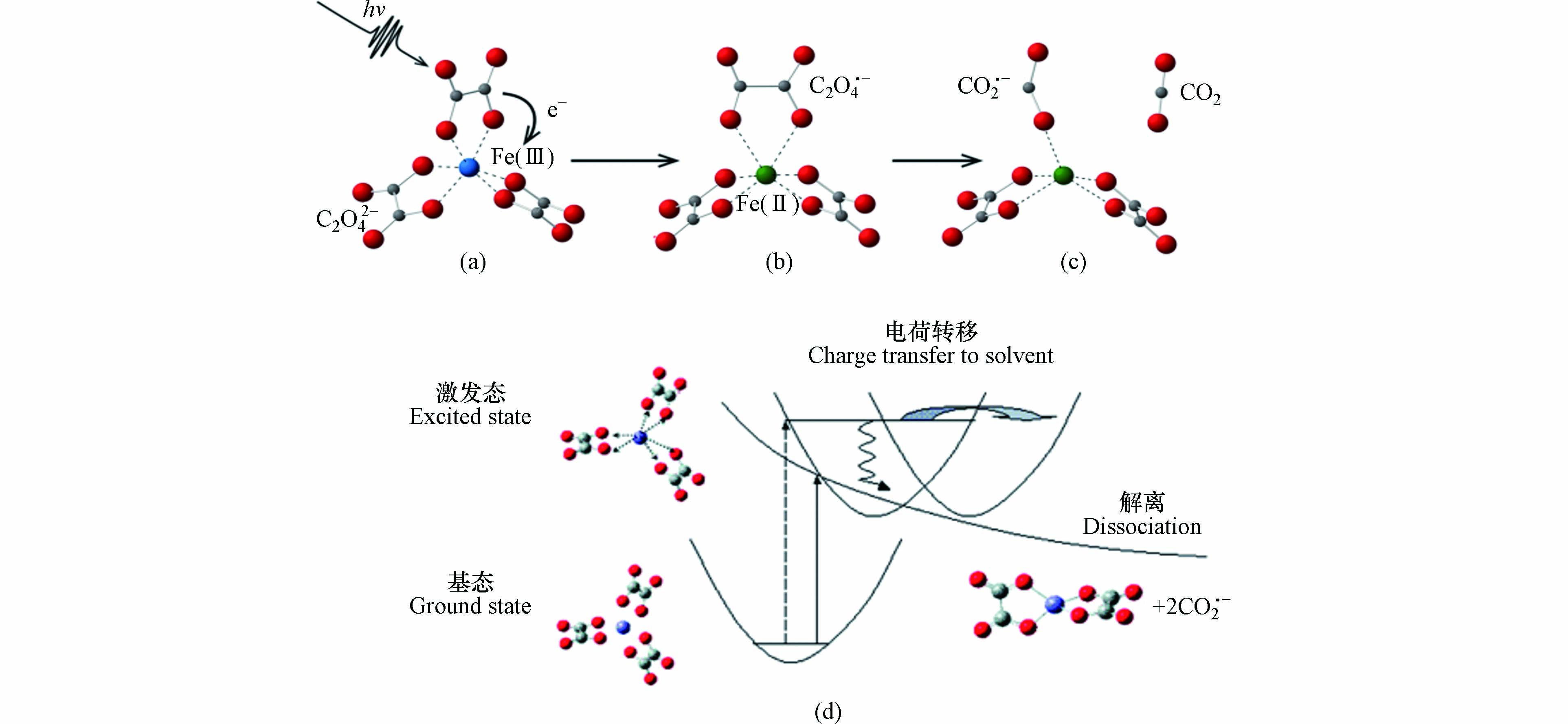
图 4 草酸铁络合物分子内电荷转移过程
Figure 4. Intramolecular charge transfer processes of Fe-oxalate complex
-
[1] ZHAO Z J, YAO L W, LI J, et al. Determination of interactions of ferrihydrite-humic acid-Pb (Ⅱ) system [J]. Environmental Science and Pollution Research, 2022, 29(15): 21561-21575. doi: 10.1007/s11356-021-17258-z [2] ZHAO X P, LI Z P, TANG W J, et al. Competitive kinetics of Ni(Ⅱ)/Co(Ⅱ) and Cr(VI)/P(Ⅴ) adsorption and desorption on goethite: A unified thermodynamically based model [J]. Journal of Hazardous Materials, 2022, 423: 127028. doi: 10.1016/j.jhazmat.2021.127028 [3] 卜庆伟, 曹红梅, 贺小凡, 等. 交互作用对有机-矿质复合体吸附四环素的影响 [J]. 环境化学, 2020, 39(12): 3552-3561. doi: 10.7524/j.issn.0254-6108.2020042904 BU Q W, CAO H M, HE X F, et al. The impact of interaction on organic-mineral complexes adsorb tetracycline [J]. Environmental Chemistry, 2020, 39(12): 3552-3561(in Chinese). doi: 10.7524/j.issn.0254-6108.2020042904
[4] QIU X R, DING L, ZHANG C, et al. Exposed facets mediated interaction of polystyrene nanoplastics (PSNPs) with iron oxides nanocrystal [J]. Journal of Hazardous Materials, 2022, 435: 128994. doi: 10.1016/j.jhazmat.2022.128994 [5] GAO M S, SU Y, GAO J B, et al. Arsenic speciation transformation in soils with high geological background: New insights from the governing role of Fe [J]. Chemosphere, 2022, 302: 134860. doi: 10.1016/j.chemosphere.2022.134860 [6] SHI M Q, MIN X B, KE Y, et al. Recent progress in understanding the mechanism of heavy metals retention by iron (oxyhydr)oxides [J]. Science of the Total Environment, 2021, 752: 141930. doi: 10.1016/j.scitotenv.2020.141930 [7] 朱剑锋, 王艳琼, 王红武. 铁氧化物促进微生物直接种间电子传递的机理及其研究现状 [J]. 环境化学, 2022, 41(6): 1856-1868. doi: 10.7524/j.issn.0254-6108.2021112501 ZHU J F, WANG Y Q, WANG H W. A review on enhancement of direct interspecies electron transfer induced by iron oxides and its mechanism [J]. Environmental Chemistry, 2022, 41(6): 1856-1868(in Chinese). doi: 10.7524/j.issn.0254-6108.2021112501
[8] WANG Y Q, LIU W, WANG T, et al. Arsenate adsorption onto Fe-TNTs prepared by a novel water-ethanol hydrothermal method: Mechanism and synergistic effect [J]. Journal of Colloid and Interface Science, 2015, 440: 253-262. doi: 10.1016/j.jcis.2014.10.036 [9] REN H T, JI Z Y, WU S H, et al. Photoreductive dissolution of schwertmannite induced by oxalate and the mobilization of adsorbed As(V) [J]. Chemosphere, 2018, 208: 294-302. doi: 10.1016/j.chemosphere.2018.05.187 [10] QIN X P, LIU F, ZHAO L, et al. Adsorption of levofloxacin to goethite: Batch and column studies [J]. Environmental Engineering Science, 2016, 33(4): 235-241. doi: 10.1089/ees.2015.0379 [11] YU B, JIA S Y, LIU Y, et al. Mobilization and re-adsorption of arsenate on ferrihydrite and hematite in the presence of oxalate [J]. Journal of Hazardous Materials, 2013, 262: 701-708. doi: 10.1016/j.jhazmat.2013.09.010 [12] ZHANG P, YANG X Y, ZHAO Z B, et al. One-step synthesis of flowerlike C/Fe2O3 nanosheet assembly with superior adsorption capacity and visible light photocatalytic performance for dye removal [J]. Carbon, 2017, 116: 59-67. doi: 10.1016/j.carbon.2017.01.087 [13] WANG Y N, WANG J M, DENG R P, et al. Preparation and photocatalytic property of porous α-Fe2O3 nanoflowers [J]. Materials Research Bulletin, 2018, 107: 94-99. doi: 10.1016/j.materresbull.2018.07.013 [14] WANG Y H, SHI H H, CUI K, et al. Hierarchical hematite/TiO2 nanorod arrays coupled with responsive mesoporous silica nanomaterial for highly sensitive photoelectrochemical sensing [J]. Biosensors and Bioelectronics, 2018, 117: 515-521. doi: 10.1016/j.bios.2018.06.030 [15] SEO J H, CHOI K, NAM J, et al. Synergetic donor-donor codoping strategy for enhanced photoelectrochemical activity of hematite [J]. Applied Catalysis B:Environmental, 2020, 260: 118186. doi: 10.1016/j.apcatb.2019.118186 [16] LI G, WANG C, YAN Y P, et al. Highly enhanced degradation of organic pollutants in hematite/sulfite/photo system [J]. Chemical Engineering Journal, 2020, 386: 124007. doi: 10.1016/j.cej.2019.124007 [17] KEERTHANA S, YUVAKKUMAR R, RAVI G, et al. A strategy to enhance the photocatalytic efficiency of α-Fe2O3 [J]. Chemosphere, 2021, 270: 129498. doi: 10.1016/j.chemosphere.2020.129498 [18] NGUYEN N T T, NGUYEN A Q K, KIM M S, et al. Effect of Fe3+ as an electron-transfer mediator on WO3-induced activation of peroxymonosulfate under visible light [J]. Chemical Engineering Journal, 2021, 411: 128529. doi: 10.1016/j.cej.2021.128529 [19] THARANI K, JEGATHA CHRISTY A, SAGADEVAN S, et al. Photocatalytic and antibacterial performance of iron oxide nanoparticles formed by the combustion method [J]. Chemical Physics Letters, 2021, 771: 138524. doi: 10.1016/j.cplett.2021.138524 [20] LAI C, SHI X X, LI L, et al. Enhancing iron redox cycling for promoting heterogeneous Fenton performance: A review [J]. Science of the Total Environment, 2021, 775: 145850. doi: 10.1016/j.scitotenv.2021.145850 [21] KIFLE G A, HUANG Y, XIANG M H, et al. Heterogeneous activation of peroxygens by iron-based bimetallic nanostructures for the efficient remediation of contaminated water. A review [J]. Chemical Engineering Journal, 2022, 442: 136187. doi: 10.1016/j.cej.2022.136187 [22] YOU Y Y, HUANG S B, CHEN M S, et al. Hematite/selenium disulfide hybrid catalyst for enhanced Fe(III)/Fe(II) redox cycling in advanced oxidation processes [J]. Journal of Hazardous Materials, 2022, 424: 127376. doi: 10.1016/j.jhazmat.2021.127376 [23] LI F B, CHEN J J, LIU C S, et al. Effect of iron oxides and carboxylic acids on photochemical degradation of bisphenol A [J]. Biology and Fertility of Soils, 2006, 42(5): 409-417. doi: 10.1007/s00374-006-0084-7 [24] LUO H W, ZENG Y F, CHENG Y, et al. Activation of peroxymonosulfate by iron oxychloride with hydroxylamine for ciprofloxacin degradation and bacterial disinfection [J]. Science of the Total Environment, 2021, 799: 149506. doi: 10.1016/j.scitotenv.2021.149506 [25] GAJOVIĆ A, SILVA A M T, SEGUNDO R A, et al. Tailoring the phase composition and morphology of Bi-doped goethite-hematite nanostructures and their catalytic activity in the degradation of an actual pesticide using a photo-Fenton-like process [J]. Applied Catalysis B:Environmental, 2011, 103(3/4): 351-361. [26] KHAGHANI S, GHANBARI D. Magnetic and photo-catalyst Fe3O4-Ag nanocomposite: Green preparation of silver and magnetite nanoparticles by garlic extract [J]. Journal of Materials Science:Materials in Electronics, 2017, 28(3): 2877-2886. doi: 10.1007/s10854-016-5872-8 [27] REICHARD P U, KRETZSCHMAR R, KRAEMER S M. Dissolution mechanisms of goethite in the presence of siderophores and organic acids [J]. Geochimica et Cosmochimica Acta, 2007, 71(23): 5635-5650. doi: 10.1016/j.gca.2006.12.022 [28] JIN X H, LI X F, GUO C L, et al. Fate of oxalic-acid-intervened arsenic during Fe(Ⅱ)-induced transformation of As(Ⅴ)-bearing jarosite [J]. Science of the Total Environment, 2020, 719: 137311. doi: 10.1016/j.scitotenv.2020.137311 [29] LAMY I, DJAFER M, TERCE M. Influence of oxalic acid on the adsorption of cadmium at the goethite surface [J]. Water, Air, and Soil Pollution, 1991, 57(1): 457-465. [30] FLYNN E D, CATALANO J G. Competitive and cooperative effects during nickel adsorption to iron oxides in the presence of oxalate [J]. Environmental Science & Technology, 2017, 51(17): 9792-9799. [31] LEE C H, KEENAN C R, SEDLAK D L. Polyoxometalate-enhanced oxidation of organic compounds by nanoparticulate zero-valent iron and ferrous ion in the presence of oxygen [J]. Environmental Science & Technology, 2008, 42(13): 4921-4926. [32] GONZÁLEZ A G, BIANCO A, BOUTORH J, et al. Influence of strong iron-binding ligands on cloud water oxidant capacity [J]. Science of the Total Environment, 2022, 829: 154642. doi: 10.1016/j.scitotenv.2022.154642 [33] BATISTA A P S, COTTRELL B A, NOGUEIRA R F P. Photochemical transformation of antibiotics by excitation of Fe(Ⅲ)-complexes in aqueous medium [J]. Journal of Photochemistry and Photobiology A:Chemistry, 2014, 274: 50-56. doi: 10.1016/j.jphotochem.2013.09.017 [34] POZDNYAKOV I, SHERIN P, BAZHIN N, et al. Fe(Ox)3]3- complex as a photodegradation agent at neutral pH: Advances and limitations [J]. Chemosphere, 2018, 195: 839-846. doi: 10.1016/j.chemosphere.2017.12.096 [35] WAN D, ZUO J L, CHEN Y, et al. Photodegradation of amitriptyline in Fe(Ⅲ)-citrate-oxalate binary system: Synergistic effect and mechanism [J]. Chemosphere, 2018, 210: 224-231. doi: 10.1016/j.chemosphere.2018.07.006 [36] WAN D, ZHANG G F, CHEN Y, et al. Photogeneration of hydroxyl radical in Fe(Ⅲ)-citrate-oxalate system for the degradation of fluconazole: Mechanism and products [J]. Environmental Science and Pollution Research, 2019, 26(9): 8640-8649. doi: 10.1007/s11356-019-04348-2 [37] YAN R, YANG W J, YOU D, et al. Photoinduced evolution of optical properties and compositions of methoxyphenols by Fe(III)-carboxylates complexes in atmospheric aqueous phase [J]. Chemosphere, 2022, 295: 133860. doi: 10.1016/j.chemosphere.2022.133860 [38] EROKHIN S E, SNYTNIKOVA O A, NOVIKOV M V, et al. Probing reactions between imipramine and hydroxyl radical with the photolysis of iron(III) oxalate: Implications for the indirect photooxidation of tricyclic antidepressants in waters [J]. Journal of Photochemistry and Photobiology A:Chemistry, 2022, 422: 113559. doi: 10.1016/j.jphotochem.2021.113559 [39] BI W L, DONG W B. The degradation of oxytetracycline with ferrous oxalate under different light irradiation [J]. Environmental Technology, 2021, 42(7): 1084-1091. doi: 10.1080/09593330.2019.1652698 [40] HUANG M J, XIANG W, ZHOU T, et al. The critical role of the surface iron-oxalate complexing species in determining photochemical degradation of norfloxacin using different iron oxides [J]. Science of the Total Environment, 2019, 697: 134220. doi: 10.1016/j.scitotenv.2019.134220 [41] 兰青, 莫家乐, 曹美苑. 铁-多羧基有机酸光化学体系研究进展 [J]. 生态环境学报, 2018, 27(10): 1972-1980. LAN Q, MO J L, CAO M Y. Research progress on the photochemistry system of the Fe-organic acids with multi-carboxyls: A review [J]. Ecology and Environmental Sciences, 2018, 27(10): 1972-1980(in Chinese).
[42] ZHAN G M, FANG Y M, ZHANG M, et al. Oxalate promoted iron dissolution of hematite via proton coupled electron transfer [J]. Environmental Science:Nano, 2022, 9(5): 1770-1779. doi: 10.1039/D1EN01190A [43] YAO Q, GUO C L, LI X F, et al. Synergy of oxalic acid and sunlight triggered Cr(III)-bearing Schwertmannite transformation: Reaction mechanism, Cr and C spatial distribution and speciation on the nano scale [J]. Geochimica et Cosmochimica Acta, 2022, 329: 70-86. doi: 10.1016/j.gca.2022.05.018 [44] BHANDARI N, HAUSNER D B, KUBICKI J D, et al. Photodissolution of ferrihydrite in the presence of oxalic acid: An In situ ATR-FTIR/DFT study [J]. Langmuir, 2010, 26(21): 16246-16253. doi: 10.1021/la101357y [45] BOROWSKI S C, BISWAKARMA J, KANG K, et al. Structure and reactivity of oxalate surface complexes on lepidocrocite derived from infrared spectroscopy, DFT-calculations, adsorption, dissolution and photochemical experiments [J]. Geochimica et Cosmochimica Acta, 2018, 226: 244-262. doi: 10.1016/j.gca.2018.01.024 [46] XU T Y, ZHU R L, SHANG H, et al. Photochemical behavior of ferrihydrite-oxalate system: Interfacial reaction mechanism and charge transfer process [J]. Water Research, 2019, 159: 10-19. doi: 10.1016/j.watres.2019.04.055 [47] KUBICKI J D, TUNEGA D, KRAEMER S. A density functional theory investigation of oxalate and Fe(II) adsorption onto the (010) goethite surface with implications for ligand- and reduction-promoted dissolution [J]. Chemical Geology, 2017, 464: 14-22. doi: 10.1016/j.chemgeo.2016.08.010 [48] LI F Y, KOOPAL L, TAN W F. Roles of different types of oxalate surface complexes in dissolution process of ferrihydrite aggregates [J]. Scientific Reports, 2018, 8: 2060. doi: 10.1038/s41598-018-20401-5 [49] VOELZ J L, JOHNSON N W, CHUN C L, et al. Quantitative dissolution of environmentally accessible iron residing in iron-rich minerals: A review [J]. ACS Earth and Space Chemistry, 2019, 3(8): 1371-1392. doi: 10.1021/acsearthspacechem.9b00012 [50] WANG Z Z, FU H B, ZHANG L W, et al. Ligand-promoted photoreductive dissolution of goethite by atmospheric low-molecular dicarboxylates [J]. The Journal of Physical Chemistry. A, 2017, 121(8): 1647-1656. doi: 10.1021/acs.jpca.6b09160 [51] 黄荃莅, 黄魁, 卢远桓, 等. 草酸浸出和太阳光催化回收赤泥中的铁和铝 [J]. 环境工程, 2021, 39(12): 199-205. HUANG Q L, HUANG K, LU Y H, et al. Recovery of iron and aluminum from red mud by oxalic acid leaching and solar photocatalysis [J]. Environmental Engineering, 2021, 39(12): 199-205(in Chinese).
[52] SIFFERT C, SULZBERGER B. Light-induced dissolution of hematite in the presence of oxalate. A case study [J]. Langmuir, 1991, 7(8): 1627-1634. doi: 10.1021/la00056a014 [53] LAN Q, LI F B, LIU C S, et al. Heterogeneous photodegradation of pentachlorophenol with maghemite and oxalate under UV illumination [J]. Environmental Science & Technology, 2008, 42(21): 7918-7923. [54] LAN Q, LI F B, SUN C X, et al. Heterogeneous photodegradation of pentachlorophenol and iron cycling with goethite, hematite and oxalate under UVA illumination [J]. Journal of Hazardous Materials, 2010, 174(1/2/3): 64-70. [55] MAZELLIER P, SULZBERGER B. Diuron degradation in irradiated, heterogeneous iron/oxalate systems: The rate-determining step [J]. Environmental Science & Technology, 2001, 35(16): 3314-3320. [56] HUANG M J, ZHOU T, WU X H, et al. Distinguishing homogeneous-heterogeneous degradation of norfloxacin in a photochemical Fenton-like system (Fe3O4/UV/oxalate) and the interfacial reaction mechanism [J]. Water Research, 2017, 119: 47-56. doi: 10.1016/j.watres.2017.03.008 [57] WANG Z H, XIAO D X, LIU J S. Diverse redox chemistry of photo/ferrioxalate system [J]. RSC Advances, 2014, 4(84): 44654-44658. doi: 10.1039/C4RA07153K [58] 兰青, 叶志钧, 陈熠熠, 等. 异相草酸铁光降解五氯酚过程中的铁物种分配 [J]. 环境化学, 2017, 36(2): 336-344. doi: 10.7524/j.issn.0254-6108.2017.02.2016040506 LAN Q, YE Z J, CHEN Y Y, et al. Distribution of Fe species during the photodegradation of pentachlorophenol in heterogeneous Fe-oxalate system [J]. Environmental Chemistry, 2017, 36(2): 336-344(in Chinese). doi: 10.7524/j.issn.0254-6108.2017.02.2016040506
[59] CHEN Y, WU F, LIN Y X, et al. Photodegradation of glyphosate in the ferrioxalate system [J]. Journal of Hazardous Materials, 2007, 148(1/2): 360-365. [60] FAUST B C, ZEPP R G. Photochemistry of aqueous iron(III)-polycarboxylate complexes: Roles in the chemistry of atmospheric and surface waters [J]. Environmental Science & Technology, 1993, 27(12): 2517-2522. [61] PANIAS D, TAXIARCHOU M, DOUNI I, et al. Thermodynamic analysis of the reactions of iron oxides: Dissolution in oxalic acid [J]. Canadian Metallurgical Quarterly, 1996, 35(4): 363-373. doi: 10.1179/cmq.1996.35.4.363 [62] XIAO D X, GUO Y G, LOU X Y, et al. Distinct effects of oxalate versus malonate on the iron redox chemistry: Implications for the photo-Fenton reaction [J]. Chemosphere, 2014, 103: 354-358. doi: 10.1016/j.chemosphere.2013.11.069 [63] SERAGHNI N, DEKKICHE B A, BELATTAR S, et al. Role of Fe(III) and oxalic acid in the photo-Fenton system for 3-methylphenol degradation in aqueous solution under natural and artificial light [J]. Int J Chem React Eng, 2018, 16(9): 20170211. doi: 10.1515/ijcre-2017-0211 [64] CHEN J, ZHANG H, TOMOV I V, et al. Electron transfer mechanism and photochemistry of ferrioxalate induced by excitation in the charge transfer band [J]. Inorganic Chemistry, 2008, 47(6): 2024-2032. doi: 10.1021/ic7016566 [65] MANGIANTE D M, SCHALLER R D, ZARZYCKI P, et al. Mechanism of ferric oxalate photolysis [J]. ACS Earth and Space Chemistry, 2017, 1(5): 270-276. doi: 10.1021/acsearthspacechem.7b00026 [66] LI F B, LI X Z, LIU C S, et al. Effect of oxalate on photodegradation of bisphenol A at the interface of different iron oxides [J]. Industrial & Engineering Chemistry Research, 2007, 46(3): 781-787. [67] XU T Y, FANG Y M, TONG T Y, et al. Environmental photochemistry in hematite-oxalate system: Fe(III)-Oxalate complex photolysis and ROS generation [J]. Applied Catalysis B:Environmental, 2021, 283: 119645. doi: 10.1016/j.apcatb.2020.119645 [68] WU B D, ZHANG G Y, ZHANG L, et al. Key factors in the ligand effects on the photo redox cycling of aqueous iron species [J]. Geochimica et Cosmochimica Acta, 2020, 281: 1-11. doi: 10.1016/j.gca.2020.05.004 [69] KRIBÉCHE M E A, SEHILI T, LESAGE G, et al. Insight into photochemical oxidation of Fenuron in water using iron oxide and oxalate: The roles of the dissolved oxygen [J]. Journal of Photochemistry and Photobiology A:Chemistry, 2016, 329: 120-129. doi: 10.1016/j.jphotochem.2016.06.021 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5054
- HTML全文浏览数: 5054
- PDF下载数: 154
- 施引文献: 0
