


 下载:
下载:
-
羟基化多溴联苯醚(OH-PBDEs)是一类与母体多溴联苯醚(PBDEs)结构类似的化合物,由于其具有较强的内分泌干扰效应,因而受到了广泛的关注[1-3]. 研究表明,OH-PBDEs能够通过酶或者金属氧化物介导的氧化耦合反应发生转化[4-7]. 其中,由于酶介导氧化耦合反应能够生成土壤有机质等,因此在自然腐殖化过程中扮演者重要的角色[6, 8]. 在众多氧化酶中,漆酶是一组结合了多个铜原子的内源性多酚氧化酶,由多种真菌以及高等植物以胞外分泌物的形式产出,广泛存在于自然界中,因此漆酶介导的氧化耦合反应过程对有机污染物在环境中的迁移、转化过程起着重要的作用[6, 9].
漆酶作为氧化还原酶,可以直接氧化酚类污染物. 但是由于自身氧化还原电位较低(通常在0.5—0.8 V),导致其对于氧化还原电位高的物质氧化效果不够理想[10]. 例如,仅仅依靠反应体系中单一的漆酶无法完成木质素、多环芳烃等大分子的转化过程[11]. 研究发现一些小分子化合物可以充当介体,使得漆酶在介体的辅助作用下,通过介导电子的转移过程,生成具有较高氧化还原电位的活性物种,进而实现对其他非酚类物质的高效催化氧化[11-12]. 目前,常用的介体主要是人工合成介体,如2, 2-联氮-二(3-乙基-苯并噻唑-6-磺酸)二铵盐(ABTS)、1-羟基苯并三唑(HBT)、紫尿酸(VIO)、N-羟基-N-乙酰基-苯胺(NHA)等,其中ABTS是目前应用最广泛的介体[13-14]. 而漆酶与这些介体构成的系统成为漆酶-介体系统(laccase-mediator-system, LMS ),能够实现高效、快速的降解高氧化还原电位或不能进入漆酶活性中心的底物分子[11].
尽管对漆酶催化转化酚类化合物的动力学过程已经有了较好认识,但是这些认识主要集中在单一酶催化体系[9, 15]. 例如,已有研究报道了四溴双酚A、三氯生、对氨基苯酚、雌激素等微污染物在单一漆酶作用体系下的转化动力学过程[9, 15-17]. 然而,在实际环境中,无论是人工合成的介体还是天然的介体都将与漆酶形成LMS[11]. 此外,其他共存物质,如天然有机质,也将不可避免参与这些污染物的转化过程[18-19]. 因此,探究污染物在LMS中的动力学过程对认识其在真实自然环境下的转化过程十分重要. 然而,目前关于这一方面的仍然较为缺乏.
本研究选取3-OH-BDE-7为典型的OH-PBDEs,系统探究了3-OH-BDE-7在基于ABTS的LMS中的转化动力学过程,考察了ABTS投加量、漆酶投加量、pH值和温度对LMS介导3-OH-BDE-7转化过程的影响. 此外,还探究了天然有机质(natural organic matter, NOM)对LMS介导3-OH-BDE-7转化动力学过程的影响,并揭示了NOM抑制LMS转化3-OH-BDE-7的内在机制. 研究结果不仅对认识羟基化多溴联苯醚在自然环境中的转化过程具有重要意义,而且为合理设计基于LMS的废水处理工艺具有重要参考价值.
-
漆酶(Laccase,分离自Trametes versicolor)、2, 2-联氮-二(3-乙基-苯并噻唑-6-磺酸)二铵盐(ABTS,纯度>98%)、3-OH-BDE-7(纯度>97%)购自美国Sigma-Aldrich公司. Suwannee天然有机质(natural organic matter,NOM购自国际腐殖质协会(IHSS). 磷酸氢二钠、磷酸二氢钠购自南京化学试剂有限公司. 有机试剂(甲醇、乙酸)均为色谱纯,购自Tedia公司. 其他实验室常用试剂为分析纯. 实验所用水均来自Milli-Q纯化系统的去离子水(18.25 MΩ·cm).
-
漆酶的酶活性通过ABTS法测定[9]. 其中1个漆酶酶活性单位(U)定义为每分钟催化氧化1 μmol ABTS所需要的漆酶酶量. 具体测定反应体系如下:2.8 mL醋酸钠缓冲溶液(10 mmol·L−1,pH = 5.0). 1 mL ABTS (20 mmol·L−1)溶液,以及0.1 mL稀释的漆酶储备溶液,反应启动后,通过紫外分光光度计(Cary 100,Varian,USA)监测溶液在420 nm的吸光度在3 min内随时间的变化,重复3次,取其斜率k的平均值。酶活性通过公式(1)计算可得[20].
-
LMS系统催化氧化3-OH-BDE-7的反应在8 mL玻璃反应管中进行. 反应体系设置如下:反应体系总体积为2.0 mL,其中3-OH-BDE-7的浓度为2.0 μmol·L−1,初始漆酶活性设置为0.02、0.04、0.08、0.1、0.2、0.3、0.4、0.5 U·mL−1,ABTS的浓度分别设定为0、1.0、1.5、2.0、4.0 μmol·L−1. 反应溶液为10 mmol·L−1的磷酸钠缓冲溶液(pH=6.0). 反应管置于恒温振荡培养箱中(转速为150 r·min−1),在不同的反应时间点(0、1、5、10、20、30、60 min)下将反应管从培养箱中取出,并向其中加入2 mL的甲醇终止反应. 取1 mL的反应液置于离心机进行离心,离心条件设置为:转速20000 r·min−1,离心时间10 min. 离心后,取上清液置于棕色液相小瓶中,用高效液相色谱分析测定上清液中剩余的3-OH-BDE-7浓度. 每个实验样品至少设置3个重复.
为探究溶液pH值和温度对LMS系统催化氧化3-OH-BDE-7的影响,使用同样的反应装置进行实验. 实验条件设置为:反应体系总体积为2 mL,其中3-OH-BDE-7的浓度为2.0 μmol·L−1,初始漆酶活性为0.1 U·mL−1,ABTS的浓度为2.0 μmol·L−1. 溶液pH设置为3.0、4.0、5.0、6.0、7.0、8.0、9.0和10.0. 反应温度控制在25 ℃,反应时间为60 min. 对于温度的影响,实验条件设置为:反应体系总体积为2 mL,其中3-OH-BDE-7的浓度为2 μmol·L−1,初始漆酶活性为0.1 U·mL−1,ABTS的浓度为2.0 μmol·L−1,溶液pH控制在6.0,反应温度控制在4、10、25、35、45、55、65 ℃,反应时间为60 min. 反应结束后,按照以上步骤取样,并用高效液相色谱分析测定测定溶液中剩余的3-OH-BDE-7浓度. 每个实验样品至少设置3个重复.
为探究NOM对LMS系统催化氧化3-OH-BDE-7的影响,使用同样的反应装置进行实验. 实验条件设置为:反应体系总体积为2 mL,其中3-OH-BDE-7的浓度为2.0 μmol·L−1,初始漆酶活性为0.1 U·mL−1,ABTS的浓度为2.0 μmol·L−1,NOM的浓度为2.0、4.0、6.0、8.0、10.0 mg·L−1,溶液pH设置为6.0,温度控制在25 ℃. 在不同的反应时间点(0、1、5、10、20、30、60 min)下将反应管从培养箱中取出,按照以上步骤取样,并用高效液相色谱分析测定溶液中剩余3-OH-BDE-7的浓度. 每个实验样品至少设置3个重复.
-
安捷伦高效液相色谱仪(HPLC,Agilent 1200)用以分离及定量检测溶液中3-OH-BDE-7的浓度. 具体分析测定条件如下:流动相为甲醇-水(85∶15,V∶V),流速设定为1 mL·min−1;Eclipse XDB-C18 柱(250 mm×4 mm, 5 μm) (安捷伦,美国);检测器为可变波长扫描紫外检测器(VWD),检测波长为230 nm,进样量设置为20 μL,柱温为25 ℃. 降解产物的液相色谱-质谱联用(liquid chromatograph mass spectrometer,LC/MS)分析条件:采用 Thermo LCQ 质谱检测器(Quest LCQ Duo, USA),通过XDB C18色谱柱(150 mm×4.6 mm, 5 µm)进行分离. 流动相组成为甲醇和水(85∶15,V∶V),流速设置为0.2 mL·min−1,进样量为10 µL. ESI源参数设置为:毛细管和锥管电压分别设置为4.5 kV和25 V,去溶剂和离子源温度设置为300 ℃ 和 120 ℃. N2作为雾化气和辅助气,流速分别为350 L·h−1和50 L·h−1.
-
首先探究3-OH-BDE-7在只有漆酶存在下的降解过程,结果如图1A所示. 从图1可以看出,漆酶能够转化3-OH-BDE-7,且随着漆酶投加量的增加,3-OH-BDE-7的去除率也不断增加. 但是,即使当漆酶浓度增加到0.5 U·mL−1时,去除率依然只有~40%。此外,当漆酶含量从0.4 U·mL−1增加到0.5 U·mL−1时,反应60 min后,3-OH-BDE-7的去除率只增加了2%左右. 这可能是由于漆酶自身氧化还原电位较低(通常在0.5—0.8V),导致其对于3-OH-BDE-7氧化效果较差[10]. 3-OH-BDE-7在介体ABTS存在下的降解动力学如图1A所示,在只有漆酶(0.1 U·mL−1)的反应体系中,反应60 min后,只有8.5%的 3-OH-BDE-7被去除. 而加入介体ABTS (2 μmol·L−1)后,3-OH-BDE-7的去除速率显著提高. 例如,反应30 min后,62.5%的3-OH-BDE-7被去除,而反应60 min后,3-OH-BDE-7去除率高达84.7%. 值得注意的是,在只含有介体ABTS的条件下,3-OH-BDE-7几乎不会被去除. 以上研究结果表明介体ABTS能够显著促进漆酶催化氧化3-OH-BDE-7.
此外,从图1A发现,随着反应的进行,溶液中的3-OH-BDE-7的浓度持续降低. 根据公式(2),以ln(C0/Ct)对反应时间作图,如图1B所示. 结果发现,无论是否存在ABTS,都呈线性关系,这说明3-OH-BDE-7的去除过程符合假一级反应动力学规律,之前的研究也发现了类似的酶催化反应的特征[9].
式中,kobs为表观一级反应动力学常数,C0为底物起始浓度,Ct为反应t时刻后溶液中剩余的底物浓度,t为反应时间. 通过计算可知,只有漆酶存在的条件下,假一级动力学常数为0.0016 min−1;加入ABTS后,假一级动力学常数为0.0318 min−1. 因此,加入ABTS的漆酶体系催化氧化3-OH-BDE-7的速率约是单纯漆酶体系下的20倍.
ABTS促进漆酶催化氧化3-OH-BDE-7的作用机理如图2所示. 在只有漆酶的反应体系下,漆酶与氧气反应后形成氧化态漆酶,然后氧化态漆酶再对底物进行氧化. 而在介体存在情况下,介体首先被漆酶氧化形成活性高且具有一定稳定性的氧化态介体,氧化态介体再作为氧化剂去氧化底物. 因此,在本研究中,漆酶-ABTS介体系统催化氧化3-OH-BDE-7的机制为:ABTS优先于3-OH-BDE-7迅速与漆酶发生反应,产生ABTS+和ABTS++,然后ABTS+和ABTS++再高效催化氧化3-OH-BDE-7,与此同时自身被还原为ABTS[14].
-
为进一步明确ABTS在反应体系中的作用,本文探究了不同浓度ABTS对漆酶催化氧化3OH-BDE-7的影响. 如图3A所示,随着ABTS投加量的增加,3-OH-BDE-7的去除率不断增加. 例如,当ABTS投加量为1 μmol·L−1时,反应60 min后,3-OH-BDE-7的去除率为46%;而当ABTS投加量增加到4 μmol·L−1时,反应60 min后,3-OH-BDE-7的去除率为高达97%. 尽管研究结果已经证明了ABTS能够显著促进漆酶催化氧化3-OH-BDE-7,但是仍然有必要定量评估3-OH-BDE-7在漆酶-ATBS介体系统下的去除动力学过程. 从图3B可以看出,3-OH-BDE-7的去除过程在不同ABTS投加量的条件下依然符合假一级动力学规律. 通常情况下,根据假一级动力学规律,在酶浓度一定的情况下,3-OH-BDE-7的反应速率仅和其自身浓度相关,而与溶液中的酶浓度无关,这符合酶作为催化剂的规律,同时也证明了酶活性在反应过程中保持稳定[15]. 但是在漆酶-介体系统下,3-OH-BDE-7的降解过程依然符合这一规律,这说明ABTS的加入并不改变酶催化氧化3-OH-BDE-7的特征规律. 此外,本文进一步分析了kobs与ABTS浓度之间的相关关系,结果如图2C所示. 在漆酶浓度固定的条件下,kobs与ABTS浓度之间具有较好的正相关关系. 考虑到漆酶-ABTS介体系统介导3-OH-BDE-7氧化的速率远远高于漆酶单独氧化3-OH-BDE-7的速率(图1), 因此推测在ABTS存在下,漆酶-ABTS组成了新的催化活性中心,即ABTS将优先与漆酶发生反应产生ABTS+和ABTS++,然后ABTS+和ABTS++再介导3-OH-BDE-7的转化过程.
-
为了验证以上推测,本文进一步探究了在ABTS投加量固定的情况下,漆酶投加量对LMS系统催化氧化3-OH-BDE-7的影响. 从图4A可以看出,在ABTS固定的情况下,随着漆酶投加量的增加,3-OH-BDE-7的去除率逐渐增加. 例如,当漆酶浓度为0.02 U·mL−1时,kobs为0.006 min−1;而当漆酶浓度增加到0.5 U·mL−1时,kobs增加到0.040 min−1. 此外,3-OH-BDE-7的去除过程仍然符合假一级动力学规律,这也验证了之前的推测.
本文进一步分析了kobs与酶活性之间的相关关系. 如图4B所示,在整个漆酶投加量范围内(0.02—0.5 U·mL−1),kobs与酶活性之间并不存在正相关关系,这明显不符合酶催化反应的特征. 基于对LMS系统的作用机理的分析,本文推测漆酶与介体ABTS之间存在一个最佳配比. 由于ABTS也是漆酶的一个潜在反应底物,因此ABTS的投加量并不是越多越好,这是因为过多的ABTS会跟底物形成竞争关系,从而导致底物的去除率降低. 本文进一步对实验数据进行了分段拟合,结果如图4B所示. 漆酶投加量在0—0.1 U·mL−1和0.1—0.5 U·mL−1范围内时,kobs分别与漆酶的活性均呈现出了较好的线性关系. 在第一阶段,即漆酶投加量在0—0.1 U·mL−1范围内时,漆酶含量是3-OH-BDE-7降解的主要限速因素,由于ABTS本身不与底物发生反应,因此随着酶浓度的增加3-OH-BDE-7的去除率快速增加. 而当漆酶投加量在0.1—0.5 U·mL−1范围内时,ABTS的含量是3-OH-BDE-7降解的主要限速因素,因为此时漆酶含量足够多,但是由于漆酶自身的氧化还原电位较低,导致其直接与3-OH-BDE-7反应的速率低,所以即使再增大漆酶投加量,3-OH-BDE-7的降解速率也不会增加太多. 综上所述,这些结果进一步证明了本文前面的假设,即ABTS优先于3-OH-BDE-7迅速与漆酶发生反应,产生ABTS+和ABTS++,形成新的催化活性物种去介导底物的去除过程.
-
由图5A可以看出,在pH为4—7范围内,LMS体系对3-OH-BDE-7的去除率均可达80%以上,保持了较高水平. 当pH值增大或者减小时,3-OH-BDE-7的去除率都有所降低. 例如在pH为3和10的条件下,反应60 min后,3-OH-BDE-7的去除率分别只有63%和35%. pH会影响漆酶的活性是因为反应介质的pH值影响漆酶表面的电化学特性或反应中心,不同的pH值能够影响漆酶的稳定性,从而进一步影响漆酶的活性. 通常情况下,每种漆酶都有自身适宜的pH值范围. 从图5B可以看出,本实验所用的漆酶活性的最佳pH值是5.0. 当溶液pH值高于或低于5.0时,漆酶的活性都显著降低. 例如当溶液pH值为3时,漆酶的活性相比于pH等于5.0时降低了36%. 然而从图5A的去除率上来看,3-OH-BDE-7的去除率在pH为3时只降低了约20%, 类似的现象在之前的研究中也有被发现[21]. 有研究表明在漆酶-ABTS介体系统下,起主要作用是ABTS++[14],而ABTS++在酸性环境下更容易保持其稳定性. 这也就解释了为什么在pH为3时,酶活性降低了36%,而3-OH-BDE-7的去除率只降低了约20%. 此外,对比图5A和5B,可以发现ABTS介体的加入,可以缓解pH改变致酶失活而对降低对底物去除率的趋势. 因此,ABTS的加入扩大了漆酶催化氧化底物的pH适用范围.
温度也是影响酶活性的关键因素之一. 从图5C可以看出,本研究中LMS的反应最佳温度范围在25—35 ℃. 而当温度升高或者降低时,LMS对3-OH-BDE-7的去除率都显著降低. 例如,在温度为4 ℃时,3-OH-BDE-7去除率只有约20%. 通过对不同温度下酶活性的测定,可以发现本实验中所用的漆酶的最佳温度范围也在25—35 ℃. 此外,通过对比图5C和5D,可以发现不同温度下LMS对3-OH-BDE-7的去除率变化趋势与温度对酶活性的影响趋势基本一致,这说明ABTS的加入并不会改变漆酶催化氧化底物的温度适用范围.
-
NOM的主要成分是溶解性的腐殖质,广泛存在于多个环境介质中,例如自然水体和土壤中. 因此,酶催化氧化污染的动力学过程不可避免的受到NOM的影响[9, 18]. 本文进一步探究了不同浓度的NOM对漆酶-ABTS介体系统催化氧化3-OH-BDE-7的影响. 从图6A可以看出,NOM的存在对LMS催化氧化3-OH-BDE-7具有一定的抑制作用,并且随着NOM浓度的增加,抑制程度越来越大. 木质素过氧化物酶(Lip)以及漆酶在催化去除雌激素和四溴双酚A的反应过程中也发现了类似的现象[9, 22]. 此外,3-OH-BDE-7的降解过程在NOM存在的情况下,同样呈现出假一级动力学规律,结果如图6B所示. 通过公式(2)计算可知,当NOM浓度为2.0、4.0、6.0、8.0、10.0 mg·L−1时,其kobs分别为0.0122、0.0082、0.0058、0.0047、0.0038 min−1.
有研究发现NOM中的羟基和羧基能和漆酶活性中心的铜离子发生配位反应,从而使得漆酶的活性降低[23]. 但是,本研究中NOM的存在对漆酶的活性并没有影响,如图6C所示,这与Xia等的研究结果一致,他们也发现腐殖酸并不会影响漆酶的活性[24]. 因此本实验中NOM对酶活性的影响不是其抑制作用的主要原因.
其次,NOM也可以作为底物,与酶发生反应,这主要是因为NOM分子结构中有许多酚类官能团[18]. 由于NOM对漆酶催化反应的影响主要取决于酚类官能团的含量及其周围的电子或空间微环境[24]. 因此,不同来源的NOM可能对酶催化反应表现出不同的影响. 例如,从泥炭中提取的NOM组分对漆酶反应具有一定的抑制作用,而来源于IHSS的NOM则对漆酶反应具有一定的促进作用[18]. 为探究本实验中NOM的LMS的影响,本文进一步利用紫外分光光度计表征了NOM在LMS下的吸光度变化. 如图6D所示,NOM的吸光度值并没有发生显著的变化,这表明NOM与LMS之间不发生反应,类似的现象在Lu等[25]的研究中也观测到. 此外,NOM也可能吸附3-OH-BDE-7,进而抑制其在LMS下的去除. 为探究NOM对3-OH-BDE-7的吸附作用对其去除率的影响,本文将NOM与3-OH-BDE-7先共同孵育24 h后,再进行降解实验,结果如图6E所示. 从图6E可以看出,共同孵育条件下3-OH-BDE-7的去除率与同时投加条件下的去除率并没有显著性差异,这说明NOM对3-OH-BDE-7的吸附作用并不是NOM抑制作用的主要原因.
有研究报道,腐殖质能够逆转漆酶介导的底物氧化过程[25]. 在漆酶催化氧化底物的过程中,分为两个阶段. 第一个阶段是底物(S)首先被酶氧化成活性自由基中间体(S*),第二个阶段是活性自由基中间体(S*)发生耦合进而生成耦合产物(P). 这两个阶段很大程度上是不可逆的,然而在NOM存在的情况,生成的活性自由基中间体(S*)可能与NOM发生反应重新转化成底物(S).
假设S*达到相对稳态,即ds/dt≈0,则根据公式,可知:
代入公式(8),可知
假设S*的浓度非常小,并且变化慢,则产物的生成速率与反应物的消失速率近似相等. 则
因此,
公式变换后,
因此,1/kobs应该与NOM的浓度呈正相关,结果如图6E所示. 1/kobs与NOM的浓度之间具有较好的线性关系(R2=0.9990),这说明NOM可以与LMS中形成的底物有机自由基发生反应,使其返回到初始状态,所以NOM才表现出对3-OH-BDE-7转化的抑制作用.
-
最后,利用LC-MS对3-OH-BDE-7在LMS下的中间产物进行了鉴定。通过分析LC-MS信息,并结合溴代待物的特征质谱图,识别出了3种降解产物,结果如图7A所示。根据鉴定到的中间产物,对3-OH-BDE-7的降解路径进行了推测,结果如图7B所示。在LMS作用下,3-OH-BDE-7失去一个电子生成3-OH-BDE-7自由基(R1)。3-OH-BDE-7自由基经过电荷重排后依次形成自由基R2和R3,R3中C—O断裂后生成自由基R4。路径1是自由基R4与水反应生成2,4-二溴酚(产物1);路径2是自由基R1与自由基R2发生耦合反应1-(2,4-二苯氧基)-2,4-二溴苯甲醚(产物2);路径3是两个自由基R4发生耦合反应生成1,3-二(2,4-二苯氧基)苯醚(产物3)。这与本课题组之前的研究结果类似,即漆酶催化反应过程中,生成的有机自由基容易发生耦合反应生成二聚物或者三聚物[9]。
-
(1) ABTS能够显著促进漆酶催化氧化去除3-OH-BDE-7的速率. 加入ABTS (2 μmol·L−1)的漆酶体系催化氧化3-OH-BDE-7的速率约是单纯漆酶体系下的20倍.
(2) 当漆酶投加量固定时,随着ABTS浓度的增加,3-OH-BDE-7的去除率也不断提高,并且假一级动力学常数与ABTS的浓度呈正相关.
(3) 当ABTS投加量固定时,随着漆酶浓度的增加,尽管3-OH-BDE-7的去除率也不断提高,但是增加的程度在不断降低.
(4) ABTS的加入扩大了漆酶催化氧化3-OH-BDE-7的pH适用范围, 但是并不会改变漆酶催化氧化3-OH-BDE-7的温度适用范围.
(5) NOM能够抑制3-OH-BDE-7在LMS系统的降解,并且随着NOM浓度的增加,抑制作用逐渐增强. NOM抑制3-OH-BDE-7降解的主要原因是NOM能够淬灭催化反应过程中的底物有机自由基.
漆酶/ABTS介体系统催化氧化羟基化多溴联苯醚
Transformation of hydroxylation polybrominateddiphenyl ethers in laccase-ABTS system
-
摘要: 本研究选取3-OH-BDE-7为典型的羟基化多溴联苯醚(OH-PBDEs),系统探究了3-OH-BDE-7在基于二(3-乙基-苯并噻唑-6-磺酸)二铵盐(ABTS)的漆酶介体系统中的转化动力学过程,考察了ABTS投加量、漆酶投加量、pH值和温度以及天然有机质对酶介体系统介导3-OH-BDE-7转化过程的影响。结果表明,3-OH-BDE-7在单一漆酶体系下去除率较低,加入2.0 μmol·L−1ABTS后,3-OH-BDE-7去除率提高了约20倍。3-OH-BDE-7在漆酶介体系统的转化过程符合假一级动力学过程,其假一级动力学常数分别与ABTS投加量和漆酶投加量呈正相关。漆酶介体系统催化氧化3-OH-BDE-7的适宜pH值范围为3—8,适宜的温度范围为25—35 ℃。此外,天然有机质能够抑制3-OH-BDE-7在漆酶介体系统中的转化过程,主要原因归结于天然有机质能够与底物有机自由基发生反应,使得底物有机自由基重新回到初始状态。本研究不仅对认识羟基化多溴联苯醚在自然环境中的转化过程具有重要意义,而且为合理设计基于漆酶介体系统的废水处理工艺具有重要参考价值。Abstract: In this study, the transformation kinetics of 3-OH-BDE-7, a typical kind of hydroxylation polybrominateddiphenyl ethers (OH-PBDEs), in laccase-mediator-system (LMS) based on 2,2'-azino-bis [3-ethyl-benzothiazoline-6-sulfonic acid] diammonium salt (ABTS) was systematically investigated. The influence of ABTS dosage, laccase dosage, solution pH, temperature and natural organism matter (NOM) on the transformation of 3-OH-BDE-7 was studied. The results showed the removal of 3-OH-BDE-7 in the system containing only laccase was very low, while with the presence of ABTS, the removal of 3-OH-BDE-7 increased about 20 times. Moreover, the removal of 3-OH-BDE-7 followed pseudo-first-order kinetics, and the first-order rate constant was proportional to the dosage of ABTS and laccase, respectively. The optimal pH was between 3 and 8, and the optimal temperature was between 25 ℃ and 35 ℃.In addition, the presence of natural organic matter significantly inhibited the removal of 3-OH-BDE-7. This was mainly because NOM acted as the quenching agent of the radical intermediates and converted the intermediates back to their original forms. Taken together, this study is not only of key importance in understanding the significance of LMS catalyzed oxidation in the conversion of phenolic compounds in natural environment, but also of great value in the design of treatment processes based on LMS.
-
Key words:
- OH-PBDEs /
- laccase /
- mediator /
- kinetics /
- environmental factors
-
羟基化多溴联苯醚(OH-PBDEs)是一类与母体多溴联苯醚(PBDEs)结构类似的化合物,由于其具有较强的内分泌干扰效应,因而受到了广泛的关注[1-3]. 研究表明,OH-PBDEs能够通过酶或者金属氧化物介导的氧化耦合反应发生转化[4-7]. 其中,由于酶介导氧化耦合反应能够生成土壤有机质等,因此在自然腐殖化过程中扮演者重要的角色[6, 8]. 在众多氧化酶中,漆酶是一组结合了多个铜原子的内源性多酚氧化酶,由多种真菌以及高等植物以胞外分泌物的形式产出,广泛存在于自然界中,因此漆酶介导的氧化耦合反应过程对有机污染物在环境中的迁移、转化过程起着重要的作用[6, 9].
漆酶作为氧化还原酶,可以直接氧化酚类污染物. 但是由于自身氧化还原电位较低(通常在0.5—0.8 V),导致其对于氧化还原电位高的物质氧化效果不够理想[10]. 例如,仅仅依靠反应体系中单一的漆酶无法完成木质素、多环芳烃等大分子的转化过程[11]. 研究发现一些小分子化合物可以充当介体,使得漆酶在介体的辅助作用下,通过介导电子的转移过程,生成具有较高氧化还原电位的活性物种,进而实现对其他非酚类物质的高效催化氧化[11-12]. 目前,常用的介体主要是人工合成介体,如2, 2-联氮-二(3-乙基-苯并噻唑-6-磺酸)二铵盐(ABTS)、1-羟基苯并三唑(HBT)、紫尿酸(VIO)、N-羟基-N-乙酰基-苯胺(NHA)等,其中ABTS是目前应用最广泛的介体[13-14]. 而漆酶与这些介体构成的系统成为漆酶-介体系统(laccase-mediator-system, LMS ),能够实现高效、快速的降解高氧化还原电位或不能进入漆酶活性中心的底物分子[11].
尽管对漆酶催化转化酚类化合物的动力学过程已经有了较好认识,但是这些认识主要集中在单一酶催化体系[9, 15]. 例如,已有研究报道了四溴双酚A、三氯生、对氨基苯酚、雌激素等微污染物在单一漆酶作用体系下的转化动力学过程[9, 15-17]. 然而,在实际环境中,无论是人工合成的介体还是天然的介体都将与漆酶形成LMS[11]. 此外,其他共存物质,如天然有机质,也将不可避免参与这些污染物的转化过程[18-19]. 因此,探究污染物在LMS中的动力学过程对认识其在真实自然环境下的转化过程十分重要. 然而,目前关于这一方面的仍然较为缺乏.
本研究选取3-OH-BDE-7为典型的OH-PBDEs,系统探究了3-OH-BDE-7在基于ABTS的LMS中的转化动力学过程,考察了ABTS投加量、漆酶投加量、pH值和温度对LMS介导3-OH-BDE-7转化过程的影响. 此外,还探究了天然有机质(natural organic matter, NOM)对LMS介导3-OH-BDE-7转化动力学过程的影响,并揭示了NOM抑制LMS转化3-OH-BDE-7的内在机制. 研究结果不仅对认识羟基化多溴联苯醚在自然环境中的转化过程具有重要意义,而且为合理设计基于LMS的废水处理工艺具有重要参考价值.
1. 实验部分(Experimental section)
1.1 材料与试剂
漆酶(Laccase,分离自Trametes versicolor)、2, 2-联氮-二(3-乙基-苯并噻唑-6-磺酸)二铵盐(ABTS,纯度>98%)、3-OH-BDE-7(纯度>97%)购自美国Sigma-Aldrich公司. Suwannee天然有机质(natural organic matter,NOM购自国际腐殖质协会(IHSS). 磷酸氢二钠、磷酸二氢钠购自南京化学试剂有限公司. 有机试剂(甲醇、乙酸)均为色谱纯,购自Tedia公司. 其他实验室常用试剂为分析纯. 实验所用水均来自Milli-Q纯化系统的去离子水(18.25 MΩ·cm).
1.2 漆酶活性的测定
漆酶的酶活性通过ABTS法测定[9]. 其中1个漆酶酶活性单位(U)定义为每分钟催化氧化1 μmol ABTS所需要的漆酶酶量. 具体测定反应体系如下:2.8 mL醋酸钠缓冲溶液(10 mmol·L−1,pH = 5.0). 1 mL ABTS (20 mmol·L−1)溶液,以及0.1 mL稀释的漆酶储备溶液,反应启动后,通过紫外分光光度计(Cary 100,Varian,USA)监测溶液在420 nm的吸光度在3 min内随时间的变化,重复3次,取其斜率k的平均值。酶活性通过公式(1)计算可得[20].
漆酶酶活性(U⋅mL−1)=k(cm−1⋅min−1)×30/36((mmol⋅L−1)−1⋅min−1) (1) 1.3 LMS系统对3-OH-BDE-7的降解实验
LMS系统催化氧化3-OH-BDE-7的反应在8 mL玻璃反应管中进行. 反应体系设置如下:反应体系总体积为2.0 mL,其中3-OH-BDE-7的浓度为2.0 μmol·L−1,初始漆酶活性设置为0.02、0.04、0.08、0.1、0.2、0.3、0.4、0.5 U·mL−1,ABTS的浓度分别设定为0、1.0、1.5、2.0、4.0 μmol·L−1. 反应溶液为10 mmol·L−1的磷酸钠缓冲溶液(pH=6.0). 反应管置于恒温振荡培养箱中(转速为150 r·min−1),在不同的反应时间点(0、1、5、10、20、30、60 min)下将反应管从培养箱中取出,并向其中加入2 mL的甲醇终止反应. 取1 mL的反应液置于离心机进行离心,离心条件设置为:转速20000 r·min−1,离心时间10 min. 离心后,取上清液置于棕色液相小瓶中,用高效液相色谱分析测定上清液中剩余的3-OH-BDE-7浓度. 每个实验样品至少设置3个重复.
为探究溶液pH值和温度对LMS系统催化氧化3-OH-BDE-7的影响,使用同样的反应装置进行实验. 实验条件设置为:反应体系总体积为2 mL,其中3-OH-BDE-7的浓度为2.0 μmol·L−1,初始漆酶活性为0.1 U·mL−1,ABTS的浓度为2.0 μmol·L−1. 溶液pH设置为3.0、4.0、5.0、6.0、7.0、8.0、9.0和10.0. 反应温度控制在25 ℃,反应时间为60 min. 对于温度的影响,实验条件设置为:反应体系总体积为2 mL,其中3-OH-BDE-7的浓度为2 μmol·L−1,初始漆酶活性为0.1 U·mL−1,ABTS的浓度为2.0 μmol·L−1,溶液pH控制在6.0,反应温度控制在4、10、25、35、45、55、65 ℃,反应时间为60 min. 反应结束后,按照以上步骤取样,并用高效液相色谱分析测定测定溶液中剩余的3-OH-BDE-7浓度. 每个实验样品至少设置3个重复.
为探究NOM对LMS系统催化氧化3-OH-BDE-7的影响,使用同样的反应装置进行实验. 实验条件设置为:反应体系总体积为2 mL,其中3-OH-BDE-7的浓度为2.0 μmol·L−1,初始漆酶活性为0.1 U·mL−1,ABTS的浓度为2.0 μmol·L−1,NOM的浓度为2.0、4.0、6.0、8.0、10.0 mg·L−1,溶液pH设置为6.0,温度控制在25 ℃. 在不同的反应时间点(0、1、5、10、20、30、60 min)下将反应管从培养箱中取出,按照以上步骤取样,并用高效液相色谱分析测定溶液中剩余3-OH-BDE-7的浓度. 每个实验样品至少设置3个重复.
1.4 分析方法
安捷伦高效液相色谱仪(HPLC,Agilent 1200)用以分离及定量检测溶液中3-OH-BDE-7的浓度. 具体分析测定条件如下:流动相为甲醇-水(85∶15,V∶V),流速设定为1 mL·min−1;Eclipse XDB-C18 柱(250 mm×4 mm, 5 μm) (安捷伦,美国);检测器为可变波长扫描紫外检测器(VWD),检测波长为230 nm,进样量设置为20 μL,柱温为25 ℃. 降解产物的液相色谱-质谱联用(liquid chromatograph mass spectrometer,LC/MS)分析条件:采用 Thermo LCQ 质谱检测器(Quest LCQ Duo, USA),通过XDB C18色谱柱(150 mm×4.6 mm, 5 µm)进行分离. 流动相组成为甲醇和水(85∶15,V∶V),流速设置为0.2 mL·min−1,进样量为10 µL. ESI源参数设置为:毛细管和锥管电压分别设置为4.5 kV和25 V,去溶剂和离子源温度设置为300 ℃ 和 120 ℃. N2作为雾化气和辅助气,流速分别为350 L·h−1和50 L·h−1.
2. 结果与讨论 (Results and discussion)
2.1 ABTS促进漆酶催化氧化3-OH-BDE-7
首先探究3-OH-BDE-7在只有漆酶存在下的降解过程,结果如图1A所示. 从图1可以看出,漆酶能够转化3-OH-BDE-7,且随着漆酶投加量的增加,3-OH-BDE-7的去除率也不断增加. 但是,即使当漆酶浓度增加到0.5 U·mL−1时,去除率依然只有~40%。此外,当漆酶含量从0.4 U·mL−1增加到0.5 U·mL−1时,反应60 min后,3-OH-BDE-7的去除率只增加了2%左右. 这可能是由于漆酶自身氧化还原电位较低(通常在0.5—0.8V),导致其对于3-OH-BDE-7氧化效果较差[10]. 3-OH-BDE-7在介体ABTS存在下的降解动力学如图1A所示,在只有漆酶(0.1 U·mL−1)的反应体系中,反应60 min后,只有8.5%的 3-OH-BDE-7被去除. 而加入介体ABTS (2 μmol·L−1)后,3-OH-BDE-7的去除速率显著提高. 例如,反应30 min后,62.5%的3-OH-BDE-7被去除,而反应60 min后,3-OH-BDE-7去除率高达84.7%. 值得注意的是,在只含有介体ABTS的条件下,3-OH-BDE-7几乎不会被去除. 以上研究结果表明介体ABTS能够显著促进漆酶催化氧化3-OH-BDE-7.
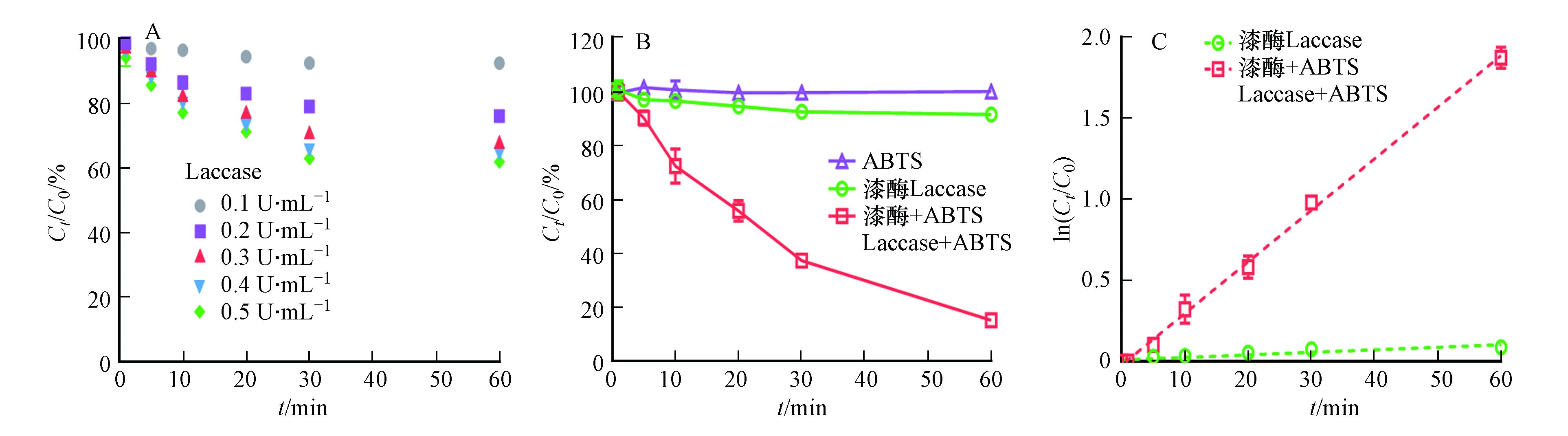 图 1 (A)3-OH-BDE-7在不同浓度漆酶(无ABTS)下的去除率;(B)3-OH-BDE-7在不同反应条件下(只有ABTS、只有漆酶和漆酶+ABTS)的去除率;(C)漆酶在有无ABTS介体条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线实验条件:[Laccase]0=0.1 U·mL−1、[ABTS]=2.0 μmol·L−1Figure 1. (A)The removal of 3-OH-BDE-7 mediated by Laccase with different dosage; (B)3-OH-BDE-7 removal efficiencies at various reaction conditions (only ABTS, only Laccase and Laccase-ABTS); (C)Pseudo first-order rate plots for 3-OH-BDE-7 removal mediated by Laccase with and without the presence of ABTS Experimental condition: [Laccase]0=0.1 U·mL−1, [ABTS]=2.0 μmol·L−1
图 1 (A)3-OH-BDE-7在不同浓度漆酶(无ABTS)下的去除率;(B)3-OH-BDE-7在不同反应条件下(只有ABTS、只有漆酶和漆酶+ABTS)的去除率;(C)漆酶在有无ABTS介体条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线实验条件:[Laccase]0=0.1 U·mL−1、[ABTS]=2.0 μmol·L−1Figure 1. (A)The removal of 3-OH-BDE-7 mediated by Laccase with different dosage; (B)3-OH-BDE-7 removal efficiencies at various reaction conditions (only ABTS, only Laccase and Laccase-ABTS); (C)Pseudo first-order rate plots for 3-OH-BDE-7 removal mediated by Laccase with and without the presence of ABTS Experimental condition: [Laccase]0=0.1 U·mL−1, [ABTS]=2.0 μmol·L−1此外,从图1A发现,随着反应的进行,溶液中的3-OH-BDE-7的浓度持续降低. 根据公式(2),以ln(C0/Ct)对反应时间作图,如图1B所示. 结果发现,无论是否存在ABTS,都呈线性关系,这说明3-OH-BDE-7的去除过程符合假一级反应动力学规律,之前的研究也发现了类似的酶催化反应的特征[9].
lnC0Ct=kobst (2) 式中,kobs为表观一级反应动力学常数,C0为底物起始浓度,Ct为反应t时刻后溶液中剩余的底物浓度,t为反应时间. 通过计算可知,只有漆酶存在的条件下,假一级动力学常数为0.0016 min−1;加入ABTS后,假一级动力学常数为0.0318 min−1. 因此,加入ABTS的漆酶体系催化氧化3-OH-BDE-7的速率约是单纯漆酶体系下的20倍.
ABTS促进漆酶催化氧化3-OH-BDE-7的作用机理如图2所示. 在只有漆酶的反应体系下,漆酶与氧气反应后形成氧化态漆酶,然后氧化态漆酶再对底物进行氧化. 而在介体存在情况下,介体首先被漆酶氧化形成活性高且具有一定稳定性的氧化态介体,氧化态介体再作为氧化剂去氧化底物. 因此,在本研究中,漆酶-ABTS介体系统催化氧化3-OH-BDE-7的机制为:ABTS优先于3-OH-BDE-7迅速与漆酶发生反应,产生ABTS+和ABTS++,然后ABTS+和ABTS++再高效催化氧化3-OH-BDE-7,与此同时自身被还原为ABTS[14].
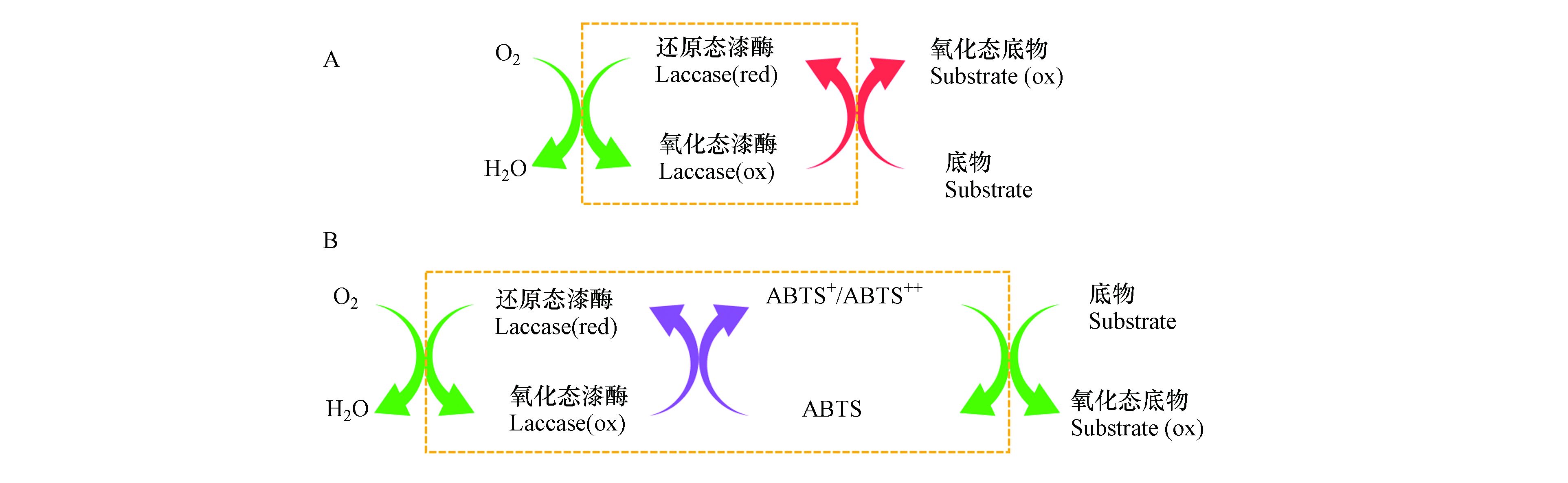 图 2 漆酶直接(A)和间接(B)催化氧化反应示意图Figure 2. Schematic diagram of direct and indirect catalytic oxidation reaction by laccase
图 2 漆酶直接(A)和间接(B)催化氧化反应示意图Figure 2. Schematic diagram of direct and indirect catalytic oxidation reaction by laccase2.2 ABTS浓度对LMS系统催化氧化3-OH-BDE-7的影响
为进一步明确ABTS在反应体系中的作用,本文探究了不同浓度ABTS对漆酶催化氧化3OH-BDE-7的影响. 如图3A所示,随着ABTS投加量的增加,3-OH-BDE-7的去除率不断增加. 例如,当ABTS投加量为1 μmol·L−1时,反应60 min后,3-OH-BDE-7的去除率为46%;而当ABTS投加量增加到4 μmol·L−1时,反应60 min后,3-OH-BDE-7的去除率为高达97%. 尽管研究结果已经证明了ABTS能够显著促进漆酶催化氧化3-OH-BDE-7,但是仍然有必要定量评估3-OH-BDE-7在漆酶-ATBS介体系统下的去除动力学过程. 从图3B可以看出,3-OH-BDE-7的去除过程在不同ABTS投加量的条件下依然符合假一级动力学规律. 通常情况下,根据假一级动力学规律,在酶浓度一定的情况下,3-OH-BDE-7的反应速率仅和其自身浓度相关,而与溶液中的酶浓度无关,这符合酶作为催化剂的规律,同时也证明了酶活性在反应过程中保持稳定[15]. 但是在漆酶-介体系统下,3-OH-BDE-7的降解过程依然符合这一规律,这说明ABTS的加入并不改变酶催化氧化3-OH-BDE-7的特征规律. 此外,本文进一步分析了kobs与ABTS浓度之间的相关关系,结果如图2C所示. 在漆酶浓度固定的条件下,kobs与ABTS浓度之间具有较好的正相关关系. 考虑到漆酶-ABTS介体系统介导3-OH-BDE-7氧化的速率远远高于漆酶单独氧化3-OH-BDE-7的速率(图1), 因此推测在ABTS存在下,漆酶-ABTS组成了新的催化活性中心,即ABTS将优先与漆酶发生反应产生ABTS+和ABTS++,然后ABTS+和ABTS++再介导3-OH-BDE-7的转化过程.
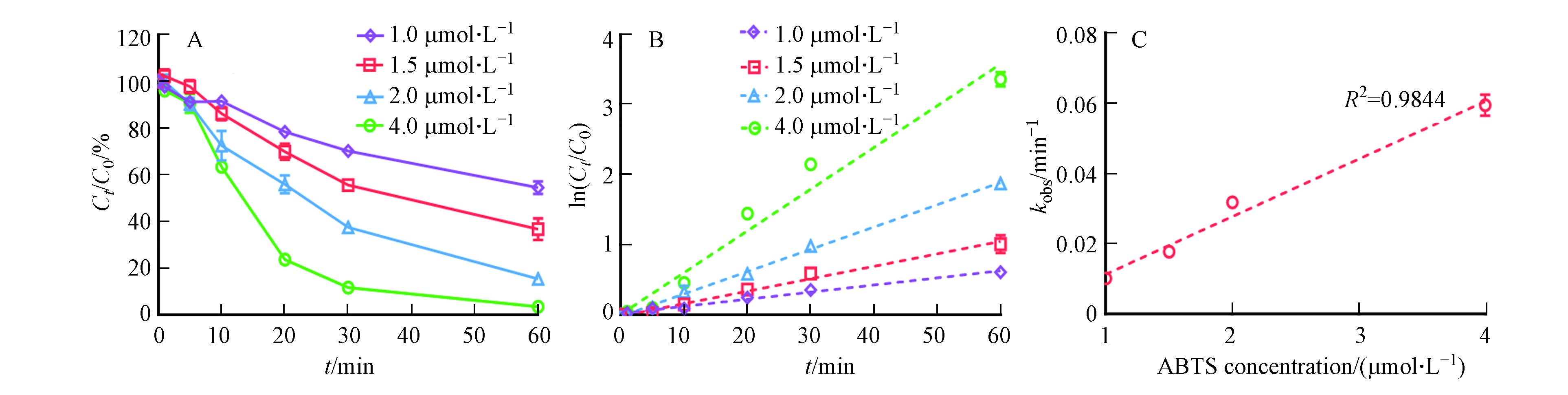 图 3 (A)ABTS投加浓度对LMS系统催化氧化3-OH-BDE-7的影响。(B)LMS在不同ABTS投加量条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线。(C)假一级动力学常数与ABTS投加量之间的关系Figure 3. (A)Influence of ABTS dosage on the removal of 3-OH-BDE-7. (B)Pseudo first-order rate plots for 3-OH-BDE-7 removal at different ABTS dosages. (C)Relationship between pseudo first-order rate constants and ABTS dosage
图 3 (A)ABTS投加浓度对LMS系统催化氧化3-OH-BDE-7的影响。(B)LMS在不同ABTS投加量条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线。(C)假一级动力学常数与ABTS投加量之间的关系Figure 3. (A)Influence of ABTS dosage on the removal of 3-OH-BDE-7. (B)Pseudo first-order rate plots for 3-OH-BDE-7 removal at different ABTS dosages. (C)Relationship between pseudo first-order rate constants and ABTS dosage2.3 漆酶投加量对LMS系统催化氧化3-OH-BDE-7的影响
为了验证以上推测,本文进一步探究了在ABTS投加量固定的情况下,漆酶投加量对LMS系统催化氧化3-OH-BDE-7的影响. 从图4A可以看出,在ABTS固定的情况下,随着漆酶投加量的增加,3-OH-BDE-7的去除率逐渐增加. 例如,当漆酶浓度为0.02 U·mL−1时,kobs为0.006 min−1;而当漆酶浓度增加到0.5 U·mL−1时,kobs增加到0.040 min−1. 此外,3-OH-BDE-7的去除过程仍然符合假一级动力学规律,这也验证了之前的推测.
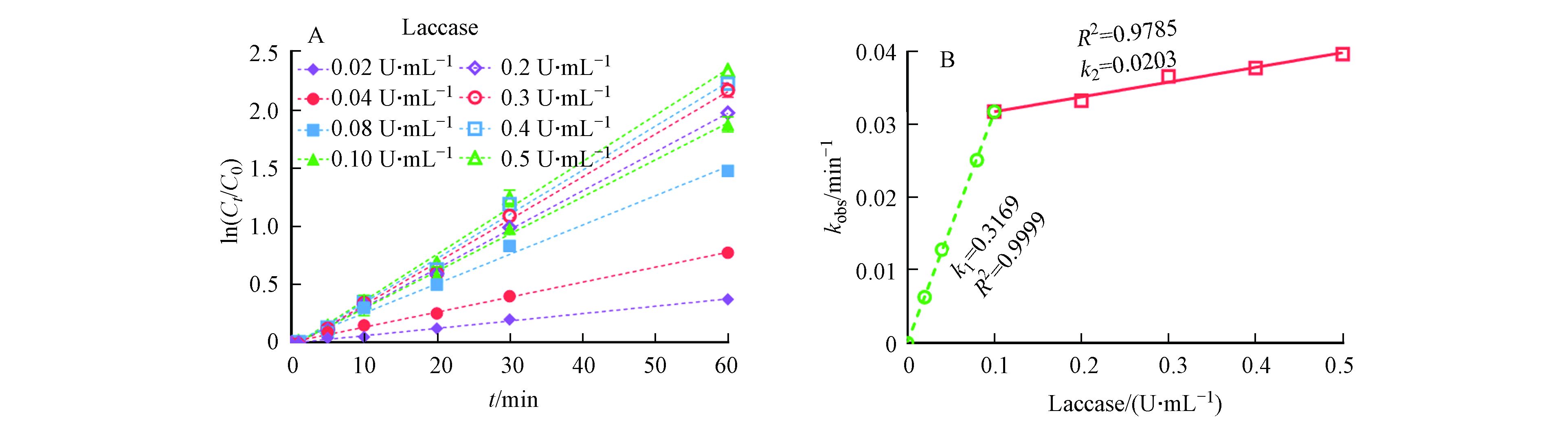 图 4 (A)LMS在不同漆酶投加量条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线;(B)假一级动力学常数与漆酶投加量之间的关系Figure 4. (A)Pseudo first-order rate plots for 3-OH-BDE-7 removal at different Laccase dosages; (B)Relationship between pseudo first-order rate constants and ABTS dosage
图 4 (A)LMS在不同漆酶投加量条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线;(B)假一级动力学常数与漆酶投加量之间的关系Figure 4. (A)Pseudo first-order rate plots for 3-OH-BDE-7 removal at different Laccase dosages; (B)Relationship between pseudo first-order rate constants and ABTS dosage本文进一步分析了kobs与酶活性之间的相关关系. 如图4B所示,在整个漆酶投加量范围内(0.02—0.5 U·mL−1),kobs与酶活性之间并不存在正相关关系,这明显不符合酶催化反应的特征. 基于对LMS系统的作用机理的分析,本文推测漆酶与介体ABTS之间存在一个最佳配比. 由于ABTS也是漆酶的一个潜在反应底物,因此ABTS的投加量并不是越多越好,这是因为过多的ABTS会跟底物形成竞争关系,从而导致底物的去除率降低. 本文进一步对实验数据进行了分段拟合,结果如图4B所示. 漆酶投加量在0—0.1 U·mL−1和0.1—0.5 U·mL−1范围内时,kobs分别与漆酶的活性均呈现出了较好的线性关系. 在第一阶段,即漆酶投加量在0—0.1 U·mL−1范围内时,漆酶含量是3-OH-BDE-7降解的主要限速因素,由于ABTS本身不与底物发生反应,因此随着酶浓度的增加3-OH-BDE-7的去除率快速增加. 而当漆酶投加量在0.1—0.5 U·mL−1范围内时,ABTS的含量是3-OH-BDE-7降解的主要限速因素,因为此时漆酶含量足够多,但是由于漆酶自身的氧化还原电位较低,导致其直接与3-OH-BDE-7反应的速率低,所以即使再增大漆酶投加量,3-OH-BDE-7的降解速率也不会增加太多. 综上所述,这些结果进一步证明了本文前面的假设,即ABTS优先于3-OH-BDE-7迅速与漆酶发生反应,产生ABTS+和ABTS++,形成新的催化活性物种去介导底物的去除过程.
2.4 pH和温度对LMS系统催化氧化3-OH-BDE-7的影响
由图5A可以看出,在pH为4—7范围内,LMS体系对3-OH-BDE-7的去除率均可达80%以上,保持了较高水平. 当pH值增大或者减小时,3-OH-BDE-7的去除率都有所降低. 例如在pH为3和10的条件下,反应60 min后,3-OH-BDE-7的去除率分别只有63%和35%. pH会影响漆酶的活性是因为反应介质的pH值影响漆酶表面的电化学特性或反应中心,不同的pH值能够影响漆酶的稳定性,从而进一步影响漆酶的活性. 通常情况下,每种漆酶都有自身适宜的pH值范围. 从图5B可以看出,本实验所用的漆酶活性的最佳pH值是5.0. 当溶液pH值高于或低于5.0时,漆酶的活性都显著降低. 例如当溶液pH值为3时,漆酶的活性相比于pH等于5.0时降低了36%. 然而从图5A的去除率上来看,3-OH-BDE-7的去除率在pH为3时只降低了约20%, 类似的现象在之前的研究中也有被发现[21]. 有研究表明在漆酶-ABTS介体系统下,起主要作用是ABTS++[14],而ABTS++在酸性环境下更容易保持其稳定性. 这也就解释了为什么在pH为3时,酶活性降低了36%,而3-OH-BDE-7的去除率只降低了约20%. 此外,对比图5A和5B,可以发现ABTS介体的加入,可以缓解pH改变致酶失活而对降低对底物去除率的趋势. 因此,ABTS的加入扩大了漆酶催化氧化底物的pH适用范围.
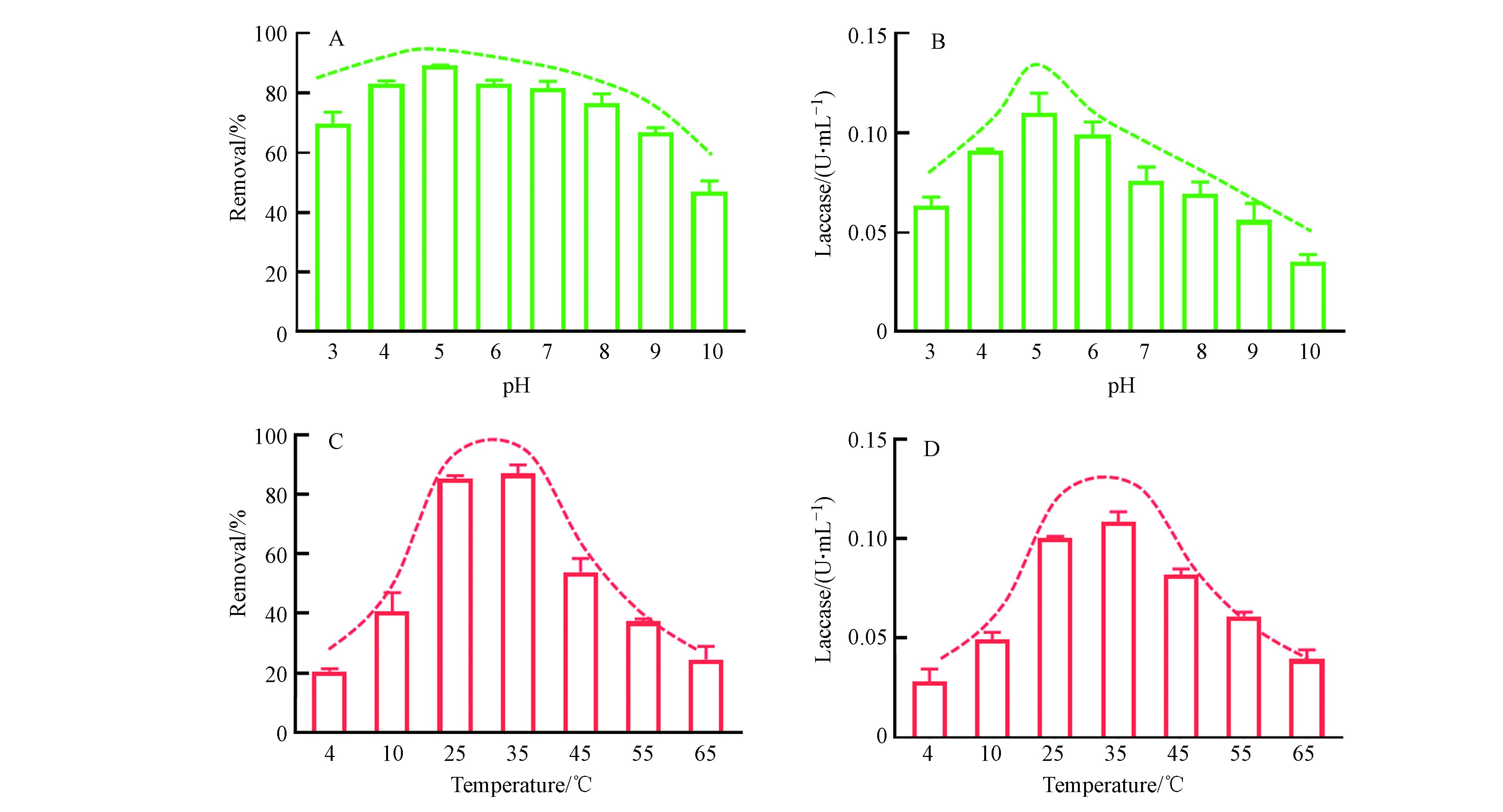 图 5 (A)溶液pH对LMS系统催化氧化3-OH-BDE-7的影响, (B)溶液pH对漆酶活性的影响,(C)温度对LMS系统催化氧化3-OH-BDE-7的影响, (D)温度对漆酶活性的影响Figure 5. (A)Influence of solution pH on the removal of 3-OH-BDE-7, (B)Influence of solution pH on the activity of Laccase ,(C)Influence of temperature on the removal of 3-OH-BDE-7, (D)Influence of temperature on the activity of Laccase
图 5 (A)溶液pH对LMS系统催化氧化3-OH-BDE-7的影响, (B)溶液pH对漆酶活性的影响,(C)温度对LMS系统催化氧化3-OH-BDE-7的影响, (D)温度对漆酶活性的影响Figure 5. (A)Influence of solution pH on the removal of 3-OH-BDE-7, (B)Influence of solution pH on the activity of Laccase ,(C)Influence of temperature on the removal of 3-OH-BDE-7, (D)Influence of temperature on the activity of Laccase温度也是影响酶活性的关键因素之一. 从图5C可以看出,本研究中LMS的反应最佳温度范围在25—35 ℃. 而当温度升高或者降低时,LMS对3-OH-BDE-7的去除率都显著降低. 例如,在温度为4 ℃时,3-OH-BDE-7去除率只有约20%. 通过对不同温度下酶活性的测定,可以发现本实验中所用的漆酶的最佳温度范围也在25—35 ℃. 此外,通过对比图5C和5D,可以发现不同温度下LMS对3-OH-BDE-7的去除率变化趋势与温度对酶活性的影响趋势基本一致,这说明ABTS的加入并不会改变漆酶催化氧化底物的温度适用范围.
2.5 NOM对LMS系统催化氧化3-OH-BDE-7的影响
NOM的主要成分是溶解性的腐殖质,广泛存在于多个环境介质中,例如自然水体和土壤中. 因此,酶催化氧化污染的动力学过程不可避免的受到NOM的影响[9, 18]. 本文进一步探究了不同浓度的NOM对漆酶-ABTS介体系统催化氧化3-OH-BDE-7的影响. 从图6A可以看出,NOM的存在对LMS催化氧化3-OH-BDE-7具有一定的抑制作用,并且随着NOM浓度的增加,抑制程度越来越大. 木质素过氧化物酶(Lip)以及漆酶在催化去除雌激素和四溴双酚A的反应过程中也发现了类似的现象[9, 22]. 此外,3-OH-BDE-7的降解过程在NOM存在的情况下,同样呈现出假一级动力学规律,结果如图6B所示. 通过公式(2)计算可知,当NOM浓度为2.0、4.0、6.0、8.0、10.0 mg·L−1时,其kobs分别为0.0122、0.0082、0.0058、0.0047、0.0038 min−1.
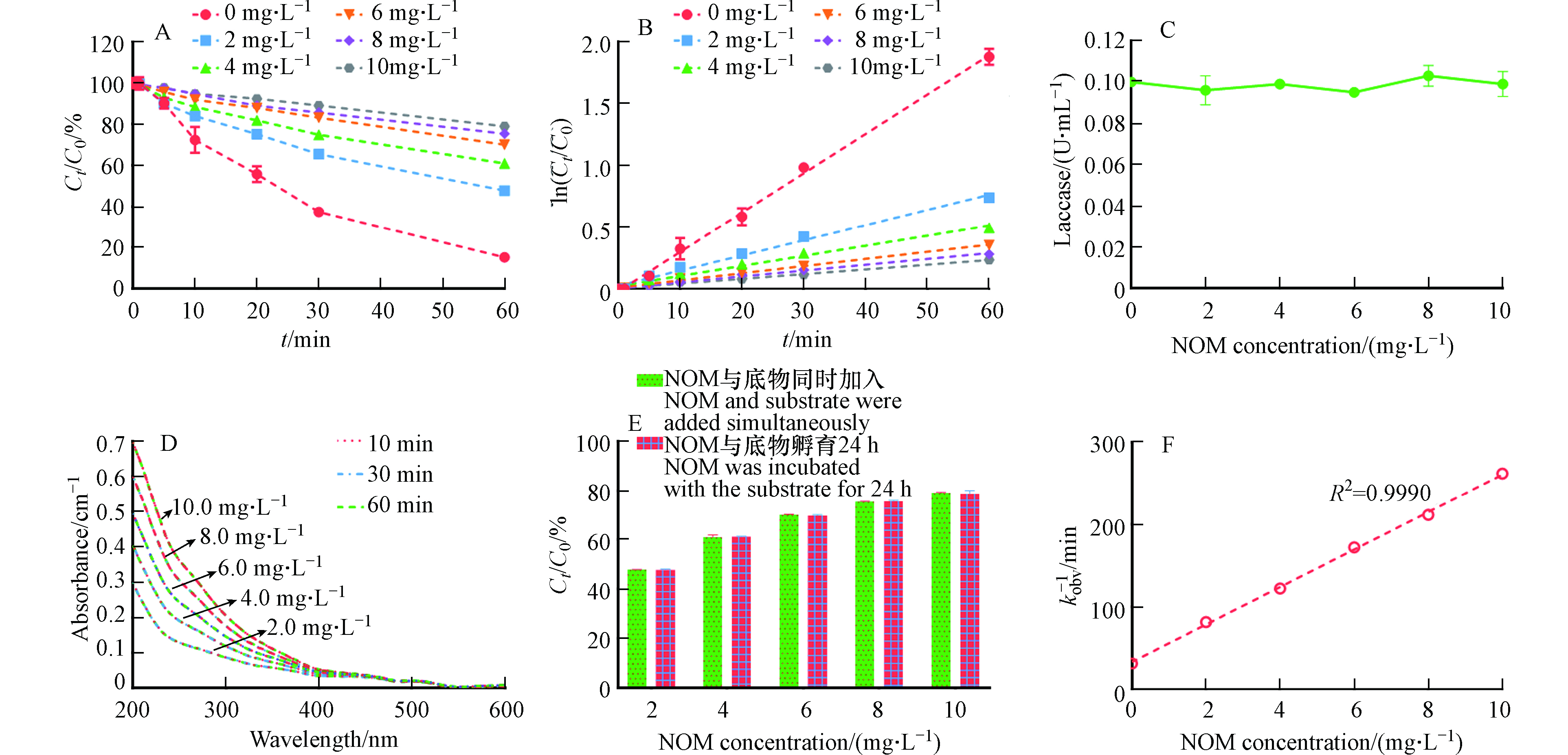 图 6 (A)不同浓度NOM对LMS催化氧化3-OH-BDE-7的影响; (B)LMS在不同NOM投加量条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线; (C)NOM对漆酶活性的影响; (D)不同浓度NOM分别LMS下随时间的吸光度变化; (E)NOM的吸附作用对3-OH-BDE-7去除率的影响; (F)假一级动力学常数与NOM投加量之间的关系Figure 6. (A)Influence of NOM dosage on the removal of 3-OH-BDE-7;(B)Pseudo first-order rate plots for 3-OH-BDE-7 removal at different NOM dosages; (C)Influence of NOM on the activity of laccase;(D)Time based UV absorbance of NOM treated by LMS;(E)The effect of NOM adsorption on the removal of 3-OH-BDE-7 mediated by LMS;(F)Relationship between pseudo first-order rate constants and NOM dosage
图 6 (A)不同浓度NOM对LMS催化氧化3-OH-BDE-7的影响; (B)LMS在不同NOM投加量条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线; (C)NOM对漆酶活性的影响; (D)不同浓度NOM分别LMS下随时间的吸光度变化; (E)NOM的吸附作用对3-OH-BDE-7去除率的影响; (F)假一级动力学常数与NOM投加量之间的关系Figure 6. (A)Influence of NOM dosage on the removal of 3-OH-BDE-7;(B)Pseudo first-order rate plots for 3-OH-BDE-7 removal at different NOM dosages; (C)Influence of NOM on the activity of laccase;(D)Time based UV absorbance of NOM treated by LMS;(E)The effect of NOM adsorption on the removal of 3-OH-BDE-7 mediated by LMS;(F)Relationship between pseudo first-order rate constants and NOM dosage有研究发现NOM中的羟基和羧基能和漆酶活性中心的铜离子发生配位反应,从而使得漆酶的活性降低[23]. 但是,本研究中NOM的存在对漆酶的活性并没有影响,如图6C所示,这与Xia等的研究结果一致,他们也发现腐殖酸并不会影响漆酶的活性[24]. 因此本实验中NOM对酶活性的影响不是其抑制作用的主要原因.
其次,NOM也可以作为底物,与酶发生反应,这主要是因为NOM分子结构中有许多酚类官能团[18]. 由于NOM对漆酶催化反应的影响主要取决于酚类官能团的含量及其周围的电子或空间微环境[24]. 因此,不同来源的NOM可能对酶催化反应表现出不同的影响. 例如,从泥炭中提取的NOM组分对漆酶反应具有一定的抑制作用,而来源于IHSS的NOM则对漆酶反应具有一定的促进作用[18]. 为探究本实验中NOM的LMS的影响,本文进一步利用紫外分光光度计表征了NOM在LMS下的吸光度变化. 如图6D所示,NOM的吸光度值并没有发生显著的变化,这表明NOM与LMS之间不发生反应,类似的现象在Lu等[25]的研究中也观测到. 此外,NOM也可能吸附3-OH-BDE-7,进而抑制其在LMS下的去除. 为探究NOM对3-OH-BDE-7的吸附作用对其去除率的影响,本文将NOM与3-OH-BDE-7先共同孵育24 h后,再进行降解实验,结果如图6E所示. 从图6E可以看出,共同孵育条件下3-OH-BDE-7的去除率与同时投加条件下的去除率并没有显著性差异,这说明NOM对3-OH-BDE-7的吸附作用并不是NOM抑制作用的主要原因.
有研究报道,腐殖质能够逆转漆酶介导的底物氧化过程[25]. 在漆酶催化氧化底物的过程中,分为两个阶段. 第一个阶段是底物(S)首先被酶氧化成活性自由基中间体(S*),第二个阶段是活性自由基中间体(S*)发生耦合进而生成耦合产物(P). 这两个阶段很大程度上是不可逆的,然而在NOM存在的情况,生成的活性自由基中间体(S*)可能与NOM发生反应重新转化成底物(S).
S+LMSk1→S∗ (3) S∗k2→P (4) S∗+NOMk3→S (5) d[S]dt=−k1[S]+k−1[S∗]CNOM (6) d[S∗]dt=k1[S]−k−1[S∗]CNOM−k2[S∗] (7) d[P]dt=k2[S∗] (8) 假设S*达到相对稳态,即ds/dt≈0,则根据公式,可知:
[S∗]=k1[S]k−1CNOM+k2 (9) 代入公式(8),可知
d[P]dt=k2k1[S]k−1CNOM+k2 (10) 假设S*的浓度非常小,并且变化慢,则产物的生成速率与反应物的消失速率近似相等. 则
d[S]dt=−d[P]dt=−k1k2[S]k−1CNOM+k2 (11) 因此,
kobs=k1k2k−1CNOM+k2 (12) 公式变换后,
1kobs=k−1k1k2CNOM+1k1 (13) 因此,1/kobs应该与NOM的浓度呈正相关,结果如图6E所示. 1/kobs与NOM的浓度之间具有较好的线性关系(R2=0.9990),这说明NOM可以与LMS中形成的底物有机自由基发生反应,使其返回到初始状态,所以NOM才表现出对3-OH-BDE-7转化的抑制作用.
2.6 3-OH-BDE-7的降解产物及降解路径
最后,利用LC-MS对3-OH-BDE-7在LMS下的中间产物进行了鉴定。通过分析LC-MS信息,并结合溴代待物的特征质谱图,识别出了3种降解产物,结果如图7A所示。根据鉴定到的中间产物,对3-OH-BDE-7的降解路径进行了推测,结果如图7B所示。在LMS作用下,3-OH-BDE-7失去一个电子生成3-OH-BDE-7自由基(R1)。3-OH-BDE-7自由基经过电荷重排后依次形成自由基R2和R3,R3中C—O断裂后生成自由基R4。路径1是自由基R4与水反应生成2,4-二溴酚(产物1);路径2是自由基R1与自由基R2发生耦合反应1-(2,4-二苯氧基)-2,4-二溴苯甲醚(产物2);路径3是两个自由基R4发生耦合反应生成1,3-二(2,4-二苯氧基)苯醚(产物3)。这与本课题组之前的研究结果类似,即漆酶催化反应过程中,生成的有机自由基容易发生耦合反应生成二聚物或者三聚物[9]。
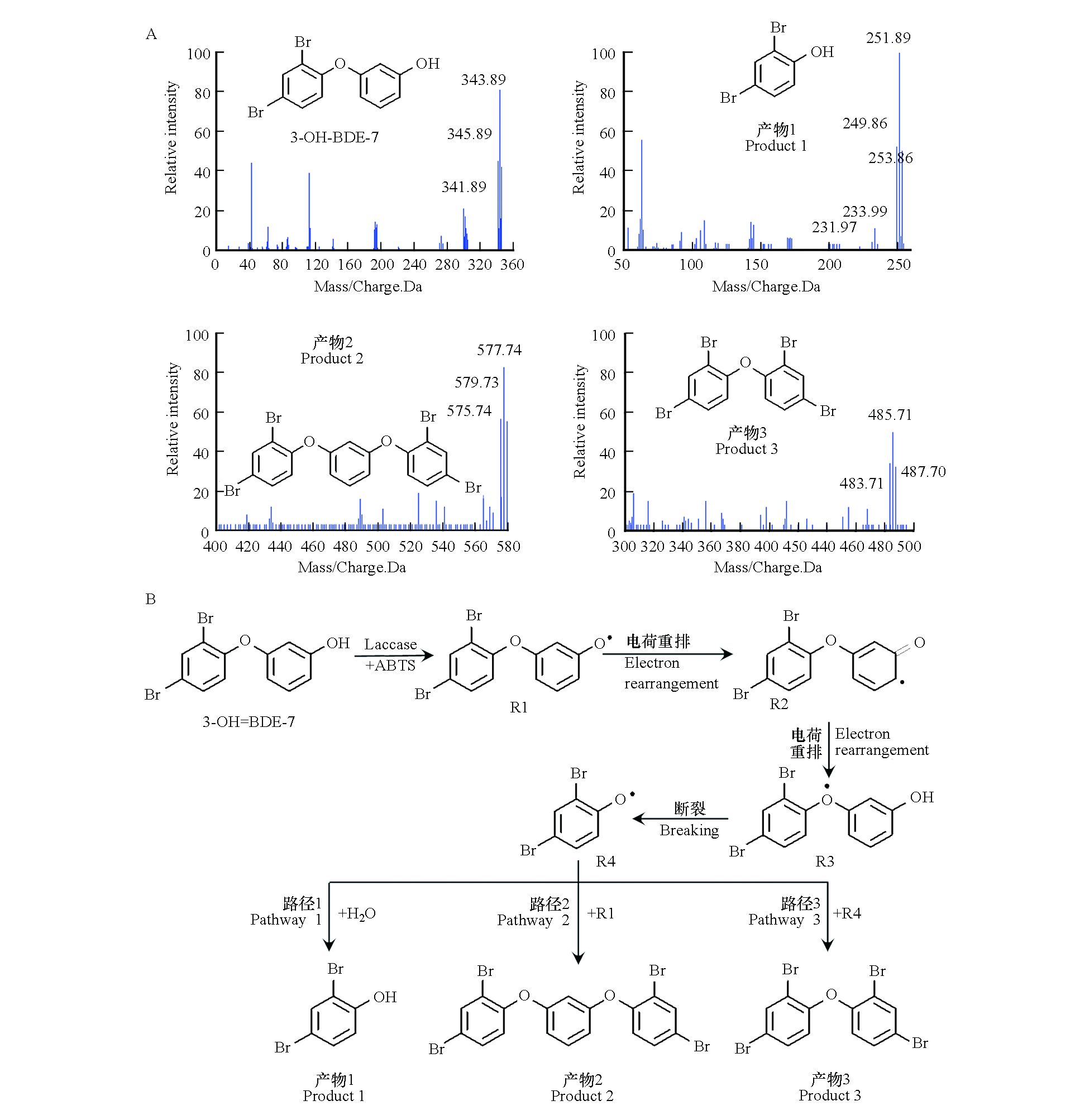 图 7 (A)3-OH-BDE-7及其转化产物的LC-MS分析结果. (B)3-OH-BDE-7在漆酶/ABTS反应体系下可能的转化路径Figure 7. (A)Mass spectra of 3-OH-BDE-7 and its products resulting from LC-MS analysis. (B)Possible reaction pathways of 3-OH-BDE-7 in laccase/ABTS mediated reaction system
图 7 (A)3-OH-BDE-7及其转化产物的LC-MS分析结果. (B)3-OH-BDE-7在漆酶/ABTS反应体系下可能的转化路径Figure 7. (A)Mass spectra of 3-OH-BDE-7 and its products resulting from LC-MS analysis. (B)Possible reaction pathways of 3-OH-BDE-7 in laccase/ABTS mediated reaction system3. 结论(Conclusion)
(1) ABTS能够显著促进漆酶催化氧化去除3-OH-BDE-7的速率. 加入ABTS (2 μmol·L−1)的漆酶体系催化氧化3-OH-BDE-7的速率约是单纯漆酶体系下的20倍.
(2) 当漆酶投加量固定时,随着ABTS浓度的增加,3-OH-BDE-7的去除率也不断提高,并且假一级动力学常数与ABTS的浓度呈正相关.
(3) 当ABTS投加量固定时,随着漆酶浓度的增加,尽管3-OH-BDE-7的去除率也不断提高,但是增加的程度在不断降低.
(4) ABTS的加入扩大了漆酶催化氧化3-OH-BDE-7的pH适用范围, 但是并不会改变漆酶催化氧化3-OH-BDE-7的温度适用范围.
(5) NOM能够抑制3-OH-BDE-7在LMS系统的降解,并且随着NOM浓度的增加,抑制作用逐渐增强. NOM抑制3-OH-BDE-7降解的主要原因是NOM能够淬灭催化反应过程中的底物有机自由基.
-
图 1 (A)3-OH-BDE-7在不同浓度漆酶(无ABTS)下的去除率;(B)3-OH-BDE-7在不同反应条件下(只有ABTS、只有漆酶和漆酶+ABTS)的去除率;(C)漆酶在有无ABTS介体条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线实验条件:[Laccase]0=0.1 U·mL−1、[ABTS]=2.0 μmol·L−1
Figure 1. (A)The removal of 3-OH-BDE-7 mediated by Laccase with different dosage; (B)3-OH-BDE-7 removal efficiencies at various reaction conditions (only ABTS, only Laccase and Laccase-ABTS); (C)Pseudo first-order rate plots for 3-OH-BDE-7 removal mediated by Laccase with and without the presence of ABTS Experimental condition: [Laccase]0=0.1 U·mL−1, [ABTS]=2.0 μmol·L−1
图 2 漆酶直接(A)和间接(B)催化氧化反应示意图
Figure 2. Schematic diagram of direct and indirect catalytic oxidation reaction by laccase
图 3 (A)ABTS投加浓度对LMS系统催化氧化3-OH-BDE-7的影响。(B)LMS在不同ABTS投加量条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线。(C)假一级动力学常数与ABTS投加量之间的关系
Figure 3. (A)Influence of ABTS dosage on the removal of 3-OH-BDE-7. (B)Pseudo first-order rate plots for 3-OH-BDE-7 removal at different ABTS dosages. (C)Relationship between pseudo first-order rate constants and ABTS dosage
图 4 (A)LMS在不同漆酶投加量条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线;(B)假一级动力学常数与漆酶投加量之间的关系
Figure 4. (A)Pseudo first-order rate plots for 3-OH-BDE-7 removal at different Laccase dosages; (B)Relationship between pseudo first-order rate constants and ABTS dosage
图 5 (A)溶液pH对LMS系统催化氧化3-OH-BDE-7的影响, (B)溶液pH对漆酶活性的影响,(C)温度对LMS系统催化氧化3-OH-BDE-7的影响, (D)温度对漆酶活性的影响
Figure 5. (A)Influence of solution pH on the removal of 3-OH-BDE-7, (B)Influence of solution pH on the activity of Laccase ,(C)Influence of temperature on the removal of 3-OH-BDE-7, (D)Influence of temperature on the activity of Laccase
图 6 (A)不同浓度NOM对LMS催化氧化3-OH-BDE-7的影响; (B)LMS在不同NOM投加量条件下催化氧化3-OH-BDE-7的假一级动力学拟合曲线; (C)NOM对漆酶活性的影响; (D)不同浓度NOM分别LMS下随时间的吸光度变化; (E)NOM的吸附作用对3-OH-BDE-7去除率的影响; (F)假一级动力学常数与NOM投加量之间的关系
Figure 6. (A)Influence of NOM dosage on the removal of 3-OH-BDE-7;(B)Pseudo first-order rate plots for 3-OH-BDE-7 removal at different NOM dosages; (C)Influence of NOM on the activity of laccase;(D)Time based UV absorbance of NOM treated by LMS;(E)The effect of NOM adsorption on the removal of 3-OH-BDE-7 mediated by LMS;(F)Relationship between pseudo first-order rate constants and NOM dosage
-
[1] ZHAO Q, ZHAO H M, QUAN X, et al. Photochemical formation of hydroxylated polybrominated diphenyl ethers (OH-PBDEs) from polybrominated diphenyl ethers (PBDEs) in aqueous solution under simulated solar light irradiation [J]. Environmental Science & Technology, 2015, 49(15): 9092-9099. [2] SUN H Z, LI Y M, HAO Y F, et al. Bioaccumulation and trophic transfer of polybrominated diphenyl ethers and their hydroxylated and methoxylated analogues in polar marine food webs [J]. Environmental Science & Technology, 2020, 54(23): 15086-15096. [3] PENG Y, XIA P, ZHANG J J, et al. Toxicogenomic assessment of 6-OH-BDE47-induced developmental toxicity in chicken embryos [J]. Environmental Science & Technology, 2016, 50(22): 12493-12503. [4] LIN K D, YAN C, GAN J. Production of hydroxylated polybrominated diphenyl ethers (OH-PBDEs) from bromophenols by manganese dioxide [J]. Environmental Science & Technology, 2014, 48(1): 263-271. [5] LI J H, ZHANG Y, DU Z K, et al. Biotransformation of OH-PBDEs by pig liver microsomes: Investigating kinetics, identifying metabolites, and examining the role of different CYP isoforms [J]. Chemosphere, 2016, 148: 354-360. doi: 10.1016/j.chemosphere.2016.01.056 [6] LIN K D, ZHOU S Y, CHEN X, et al. Formation of hydroxylated polybrominated diphenyl ethers from laccase-catalyzed oxidation of bromophenols [J]. Chemosphere, 2015, 138: 806-813. doi: 10.1016/j.chemosphere.2015.08.014 [7] 冯义平, 毛亮, 董仕鹏, 等. 过氧化物酶催化去除水体中酚类内分泌干扰物的研究进展 [J]. 环境化学, 2013, 32(7): 1218-1225. doi: 10.7524/j.issn.0254-6108.2013.07.014 FENG Y P, MAO L, DONG S P, et al. Peroxidase-catalyzed removal of phenolic endocrine disrupting chemicals from water [J]. Environmental Chemistry, 2013, 32(7): 1218-1225(in Chinese). doi: 10.7524/j.issn.0254-6108.2013.07.014
[8] LU K, HUANG Q G, WANG P, et al. Physicochemical changes of few-layer graphene in peroxidase-catalyzed reactions: Characterization and potential ecological effects [J]. Environmental Science & Technology, 2015, 49(14): 8558-8565. [9] FENG Y P, COLOSI L M, GAO S X, et al. Transformation and removal of tetrabromobisphenol A from water in the presence of natural organic matter via laccase-catalyzed reactions: Reaction rates, products, and pathways [J]. Environmental Science & Technology, 2013, 47(2): 1001-1008. [10] LUO Q, YAN X F, LU J H, et al. Perfluorooctanesulfonate degrades in a laccase-mediator system [J]. Environmental Science & Technology, 2018, 52(18): 10617-10626. [11] HILGERS R, VINCKEN J P, GRUPPEN H, et al. Laccase/mediator systems: Their reactivity toward phenolic lignin structures [J]. ACS Sustainable Chemistry & Engineering, 2018, 6(2): 2037-2046. [12] LOU Q, WU Y X, DING H J, et al. Degradation of sulfonamides in aquaculture wastewater by laccase-syringaldehyde mediator system: Response surface optimization, degradation kinetics, and degradation pathway [J]. Journal of Hazardous Materials, 2022, 432: 128647. doi: 10.1016/j.jhazmat.2022.128647 [13] LI D E, XU C, YEAGER C M, et al. Molecular interaction of aqueous iodine species with humic acid studied by I and C K-edge X-ray absorption spectroscopy [J]. Environmental Science & Technology, 2019, 53(21): 12416-12424. [14] DONG S P, XIAO H F, HUANG Q G, et al. Graphene facilitated removal of labetalol in laccase-ABTS system: Reaction efficiency, pathways and mechanism [J]. Scientific Reports, 2016, 6: 21396. doi: 10.1038/srep21396 [15] LU J H, SHI Y Y, JI Y F, et al. Transformation of triclosan by laccase catalyzed oxidation: The influence of humic acid-metal binding process [J]. Environmental Pollution, 2017, 220: 1418-1423. doi: 10.1016/j.envpol.2016.10.092 [16] SHI H H, PENG J B, LI J H, et al. Laccase-catalyzed removal of the antimicrobials chlorophene and dichlorophen from water: Reaction kinetics, pathway and toxicity evaluation [J]. Journal of Hazardous Materials, 2016, 317: 81-89. doi: 10.1016/j.jhazmat.2016.05.064 [17] LU J H, HUANG Q G, MAO L. Removal of acetaminophen using enzyme-mediated oxidative coupling processes: I. Reaction rates and pathways [J]. Environmental Science & Technology, 2009, 43(18): 7062-7067. [18] LU J H, HUANG Q G. Removal of acetaminophen using enzyme-mediated oxidative coupling processes: Ⅱ. cross-coupling with natural organic matter [J]. Environmental Science & Technology, 2009, 43(18): 7068-7073. [19] HU J Y, LU K, DONG S P, et al. Inactivation of laccase by the attack of As (Ⅲ) reaction in water [J]. Environmental Science & Technology, 2018, 52(5): 2945-2952. [20] 龚志敏, 王佳豪, 刘鹏, 等. 在不同土壤体系下漆酶催化降解对乙酰氨基酚的差异研究[J]. 环境化学, 2022, 41(7): 2256-2263. GONG Z M, WANG J H, LIU P, et al. Effects of laccase degradation on acetaminophen in different soil systems[J]. Environmental Chemistry, 2022, 41(7): 2256-2263(in Chinese).
[21] MARGOT J, COPIN P J, VON GUNTEN U, et al. Sulfamethoxazole and isoproturon degradation and detoxification by a laccase-mediator system: Influence of treatment conditions and mechanistic aspects[J]. Biochemical Engineering Journal, 2015, 103: 47-59. [22] MAO L, HUANG Q G, LUO Q, et al. Ligninase-mediated removal of 17β-estradiol from water in the presence of natural organic matter: Efficiency and pathways [J]. Chemosphere, 2010, 80(4): 469-473. doi: 10.1016/j.chemosphere.2010.03.054 [23] LORENZO M, MOLDES D, COUTO S R, et al. Inhibition of laccase activity from Trametes versicolor by heavy metals and organic compounds [J]. Chemosphere, 2005, 60(8): 1124-1128. doi: 10.1016/j.chemosphere.2004.12.051 [24] XIA Q, KONG D Y, LIU G Q, et al. Removal of 17 β -estradiol in laccase catalyzed treatment processes [J]. Frontiers of Environmental Science & Engineering, 2014, 8(3): 372-378. [25] LU J H, SHAO J, LIU H, et al. Formation of halogenated polyaromatic compounds by laccase catalyzed transformation of halophenols [J]. Environmental Science & Technology, 2015, 49(14): 8550-8557. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2988
- HTML全文浏览数: 2988
- PDF下载数: 115
- 施引文献: 0
