

-
挥发性有机化合物(volatile organic compounds,VOCs)是一种广泛存在于环境中的有毒物质[1],根据世界卫生组织(WHO)的定义常温下沸点为50—260 ℃的各种有机物,主要来自于石油、表面涂层和电子制造等工业生产[2],长时间处在含有VOCs的环境中会对人体存在极大的危害. 目前VOCs的处理技术主要有吸附回收技术、生物法、等离子体技术、蓄热燃烧和紫外光催化氧化降解技术[3-9]. 其中紫外光催化氧化技术因其技术成本低、绿色环保等优点吸引着大量学者的关注[10-13]. 紫外光催化氧化技术的原理是在单独紫外光照射或紫外光协同催化剂的条件下,利用紫外光波段的光子轰击空气中的H2O和O2,使其解离成羟基自由基和游离的氧原子等活性氧基团,与有机物反应并将有机物降解为无毒无害的小分子[14]. 研究表明,相对湿度是紫外光催化氧化降解有机物的重要因素之一[12,15-17],存在较佳的反应相对湿度,在此相对湿度下有机物的直接转化率最高,能达到90%以上[18-19]. 这些研究中大多只使用有机物的直接转化率作为有机物降解程度的判断标准,但在最佳直接转化率的工况下有机物的矿化率并不一定最大,且其数值往往低于20%[11,15],这说明有机物经紫外光催化氧化降解后并没有完成转化为CO2和H2O,所以仅使用直接转化率作为有机物降解效果的判断依据是不全面的.
不同相对湿度条件下紫外光催化氧化降解有机物分子的程度不一,导致降解后的尾气中还存在着相当复杂的中间有机物. 而且从有机物降解产物的成分分析来看,在最佳转化率工况下有机物的降解方向也并不完全朝着生成小分子方向进行,这也是近年来紫外光催化氧化技术发展的瓶颈问题[11, 15]. 因此,确定紫外光催化氧化技术最佳的工艺条件,需要结合直接转化率、矿化率以及尾气成分来综合分析. 然而有机物种类众多,产物成分与降解路径复杂,需要进行大量的实验研究. 量子化学密度泛函理论(density functional theory,DFT)是一种主要以电子密度分布函数为基础的电子基态的结构理论,可以从分子水平上研究通过实验方法难以阐释的反应机制. 通过量子化学计算获得不同相对湿度条件下的有机物的降解路径,对于理解光催化氧化降解有机物、确定较佳的操作工况具有积极的指导意义.
目前理论计算研究还主要集中在对有机物氧化降解机理的猜测上[12, 17],针对不同相对湿度条件下的反应路径的研究报道较少. 本文在紫外光氧化反应实验系统中开展了有机物苯的降解实验,研究了相对湿度对苯的直接转化效率、矿化率以及对应的尾气成分的影响. 并基于量子化学密度泛函理论对不同相对湿度条件下中间有机物的生成机理进行理论计算,提出较为合理的降解路径.
-
紫外光氧化降解实验系统示意图如图1所示,主要包括(a)配气系统、(b)紫外光反应系统(c)检测系统和(d)尾气处理系统四大部分组成. 紫外光反应器为自制不锈钢折流板式,其目的是为了增大有机分子与壁面的接触面积,且反应器的内壁敷设有活性炭纤维布用来增加对有机物苯分子的吸附能力,通过与其他没有在反应器内部敷设活性炭纤维吸附材料的学者研究对比发现,添加活性炭纤维布对后续反应路径的量子化学研究不存在影响. 反应器的高度为250 mm,截面宽度为68 mm,有效体积为3.27 L,4只紫外灯管(雪莱特)均匀置于反应器中,紫外灯管发出波长为(185+254)nm的紫外光,功率均为8 W,发光强度约为28 μW·cm−2.
芳香烃作为VOCs中的一类有机物,苯可以作为其代表物质,同时大多芳香烃在完全氧化降解的过程中都会经历到苯环的开环氧化过程. 本实验以苯作为目标污染物,采用苯的标气(氮气填充)、N2和O2,通过气体质量流量计控制柜1调节3种气体的比例配制苯初始浓度为100 mg·m−3,氧气含量为21%的混合气体. 混合后的气体分为两路,一路经过玻璃转子流量计2和恒温水浴箱3并与另一路气体混合,通过调节两路气流的比例以及恒温水浴箱的温度来调节实验气体的温湿度从而控制混合气体的相对湿度,进入反应器的气体温湿度由温湿度仪4实时检测. 气相色谱仪10可以对反应器进出口气体中的有机物连续检测,有机溶剂8和活性炭颗粒9是为了对反应后尾气中残留的有机物和臭氧等二次污染物进行处理.
-
所采集的气体样品采用GC-MS联用仪(2010 PLUS,日本岛津)测定,采用的色谱柱为DB-WAX色谱柱. 柱温首先在50 ℃下保持1 min,然后以5 ℃·min−1的升温速率加热到200 ℃,再以10 ℃·min−1的升温速率加热到280 ℃并维持10 min. 质谱离子源温度为200 ℃,扫描方式为全扫描,质量数范围为35—450. 分析结果通过VOCs图谱与NIST05数据库进行对比确定最终产物.
式中,ηD 为苯的直接转化效率,C0为苯进气口浓度(mg.m−3),Ct为苯经过紫外光反应器处理一定时间后的浓度(mg·m−3).
式中,ηM 为苯的矿化效率,C1理论上苯转化后全部矿化出口CO2浓度(mg·m−3),C2实际测得的出口CO2浓度(mg·m−3).
为了从理论上研究二者的竞争氧化过程,本研究采用密度泛函(DFT)的方法进行理论计算,所有的量子化学计算均使用Gaussian16[20]程序进行. 对于几何构型优化及过渡态搜索,均采用M062X/6-311+G(d,p)[18, 21],并且确认所有过渡态只有一个虚频,在此虚频的振动方向连接着反应物与产物,其他分子均没有虚频. 得到过渡态构型后,继续进行内禀反应坐标(IRC)计算,沿能量降低方向找寻过渡态所对应的反应势能面的能量最低点,确认两端对应着反应物与产物,同时对每一步反应的反应物、过渡态和产物进行频率计算得到各自的热力学能,再对其作差得到每一步反应的能垒.
-
图2为相对湿度对有机物苯的直接转化效率和矿化率的影响. 由图2可见,随着相对湿度从40%增加到50%,苯的直接转化效率由60%增加到82%,这主要是因为随着相对湿度的增加,体系的H2O分子逐渐增加,产生的羟基自由基也不断增加,因此氧化效率增强;当相对湿度由50%增加到80%,苯的直接转化效率由82%显著下降到48%,这主要是因为体系的光子量恒定,随着相对湿度的继续增加,过多的水分子会和苯分子一起竞争吸收光子,从而导致苯的直接转化率开始降低. 然而,当体系的相对湿度从40%增加到80%,苯的矿化率由15%急剧降低至2%,较低的矿化率表明苯并没有完全转化为CO2和H2O,这与Kang等[11]和陈越平[15]的研究结果相同. 尤其特别的是,当相对湿度为50%时的矿化率并不是最大值,且相比相对湿度为40%,矿化率显著下降了40%. 因此,对相对湿度的选择需要继续结合尾气成分进行分析.
图3为3个相对湿度(40%、50%和80%)条件下的尾气经GC-MS检测后的谱图,每条谱线上的信号峰与标准数据库对比分析后所得到的有机物如表1所示.
本文对GC-MS谱图进行分析遵循两个原则:一是对峰的筛选以峰的强度为标准,因为强度较大的峰可以说明该有机物的含量较多,而含量多的有机物更能说明是苯分子氧化降解的主要产物;二是根据每一个峰与标准库对照后的可信度,本文只对可信度SI在90—95及以上的峰进行指认. 由图3可见,3种不同相对湿度条件下尾气的成分中都含有P1(二氧化碳),说明在本次紫外光氧化降解的实验中有一部分的苯分子可以被完全降解;尾气中都生成了P2(丙酮),丙酮的峰面积相较于其它几种有机物更大,说明丙酮是本研究中主要的降解产物,这与王子东[22]的研究结果是一样的. 除此之外,不同相对湿度条件下尾气中所含有机物的种类差异较大,其中低相对湿度条件下有机物的种类和数量最为复杂,包括降解生成的P3(丙酮醇)、芳香类有机物P11(苯甲醛)和P10(苯乙酮),碳原子的个数分别为六、八、九的P7(己醛)、P8(辛醛)和P9(壬醛),这与张宇飞[12]的研究结果相似. 高相对湿度条件下尾气中有机物成分还包括P11(苯甲醛)以及非苯环类的环状有机物P4(2,2,3-三甲基-3-氧杂环丁醇).
当体系中的相对湿度达到50%时,苯的直接转化效率最大,经反应后尾气中有机物的种类最少,但是尾气中却存在更大相对分子质量且毒性更大的有机物P5(1-甲基萘)和P6(2-甲基萘). 这表明较高的苯直接转化率所对应的工况并不一定是最好的工艺条件.
-
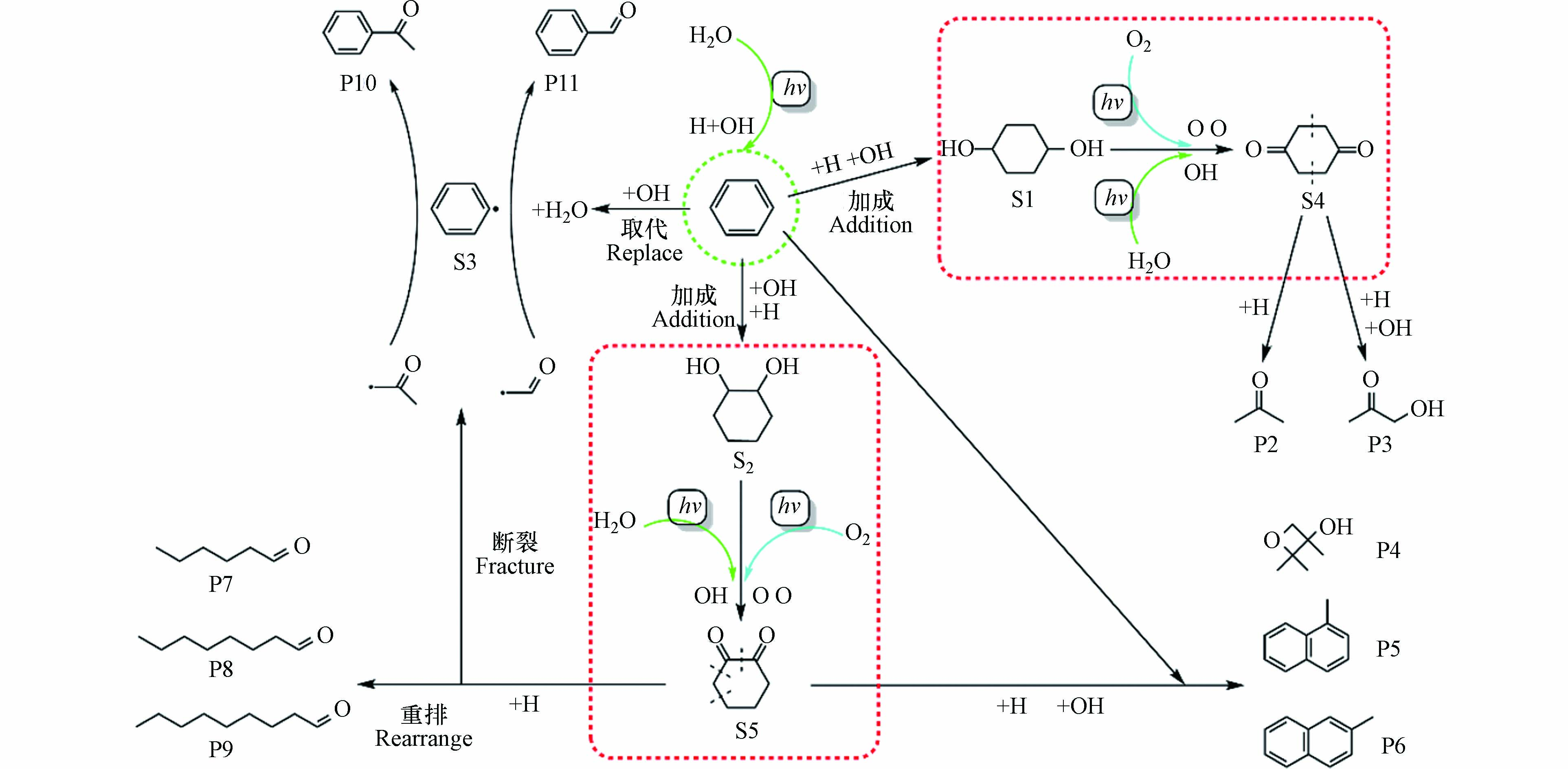
苯环平面上的π电子云是富电子基团,类似烯烃,可与缺电子的基团(亲电试剂)发生亲电取代反应. 在本研究中,亲电试剂是来自于水分子和氧气分子经紫外光照射后分解产生的游离H、OH和O自由基[10]. 苯分子的降解可能路径如图4所示,一部分苯与体系中游离的H和OH自由基发生完全亲电加成反应,这是因为在紫外光的照射条件下,苯的加成反应是在不饱和键上同时进行的[23],本文选择加成产物S1(1,4-环己二醇)和S2(1,2-环己二醇)作为中间物的代表[24-25]. 另一部分苯分子会与游离的OH自由基发生亲电取代反应,OH自由基与苯分子上的一个H原子反应后生成游离的中间物苯自由基S3和一个水分子.
中间物S1两端的羟基会被体系中的活性氧自由基O和OH继续氧化为羰基生成S4(1,4-环己二酮),S4的结构不稳定,会从对称轴处断裂形成两分子相同的基团—CH2COCH2—,该基团会与体系中的H以及OH自由基结合生成尾气中的产物P2(丙酮)或 P3(丙酮醇). 同理,中间物S2上的两个羟基也会被体系中的活性氧自由基O和OH继续氧化为羰基生成S5(1,2-环己二酮). 由于两个邻位羰基的作用使得S5很不稳定,所以会在体系其它原子的作用下四分五裂为小片段的游离基,其中一部分会与未参加反应的苯分子以及游离H、O和OH自由基作用生成P4(2,2,3-三甲基-3-氧杂环丁醇)、P5(1-甲基萘)和P6(2-甲基萘),另一部分会加氢后重排形成长链醛类有机物P7(己醛)、P8(辛醛)和P9(壬醛). 同时生成的游离乙酮基(—COCH3)和甲醛基(—COH)会与上述产生的中间物苯自由基S3结合形成产物P10(苯乙酮)和P11(苯甲醛). 由于在整个苯分子降解的过程中,中间物S1继续氧化为S4以及S2继续氧化为S5是最关键同时也是最为复杂的中间过程,在这一过程中,起到氧化作用的自由基主要来自空气中的氧气分子裂解而成的游离氧原子与不同湿度条件下产生的羟基自由基.
-
游离氧原子与羟基自由基引发的S1氧化为S4反应能量变化如图5所示,S1氧化反应主要分为四步,分别是羟基上的氢原子以及与羟基相连碳上的氢原子依次被氧化脱除,相对应的过渡态结构以及虚频值如图6所示.
实验中混合气体的氧气含量为21%,氧气分子含量多于由相对湿度带来的水分子含量,所以在波长为(185、254)nm的紫外光照射下,S1发生氧化反应过程中起主导作用的活性氧基团为游离的氧原子. 如图5,由氧原子引发的S1氧化为S4的四步反应能垒值依次为10.8、48.1、10.5、0.1 kcal·mol−1, 其中能垒最大的为第二步反应,说明该步反应对整个氧化过程起着关键作用. 由羟基自由基引发的S1氧化为S4四步反应的能垒值依次为9.4、22.9、9.3、44.1 kcal·mol−1,可以看到羟基自由基的引入能明显的降低S1氧化反应中的反应能垒,其中第一步到第三步反应的能垒分别降低了13.0%、11.4%和52.4%,这说明随着体系相对湿度的增加,有利于S1的氧化进行. 图3中也可以看到,相对湿度为80%的条件下出现了相对湿度为40%和50%没有的中间产物P3(丙酮醇),结合图4可以知道P3(丙酮醇)是中间物S4的后续反应形成的,同时P3(丙酮醇)的形成相较于P2(丙酮)需要更多的羟基自由基,这些都说明了,随着相对湿度的增加,羟基自由基的增加促进了S1的氧化.
-
与S1到S4氧化过程类似,氧原子与羟基自由基竞争作用下的S2到S5氧化过程中起主导作用的活性氧基团为游离的氧原子. 游离氧原子与羟基自由基引发的S2氧化为S5反应能量变化如图7所示,S2氧化反应主要分为四步,分别是羟基上的氢原子以及与羟基相连的碳上面的氢原子依次被氧化,相对应的过渡态结构以及虚频值如图8所示.
由氧原子引发的S2氧化为S5四步反应的能垒值依次为15.3、1.2、11.6、4.0 kcal·mol−1,其中能垒最大的为第三步反应,说明该步反应对整个氧化过程起着关键作用. 由羟基自由基引发的S2氧化为S5四步反应的能垒值依次为10.1、4.6、16.0、6.8 kcal·mol−1. 可以看到,羟基自由基的引入仅在第一步反应中降低了34.0%的能垒值,而对于后三步的反应均有不同程度的增加,这表明随着体系相对湿度的增加,羟基自由基的引入也是有利于S2的氧化.
仅由氧原子引发的S1氧化为S4以及S2氧化为S5的四步反应中能垒最大值分别为48.1 kcal·mol−1和15.3 kcal·mol−1,所以在相对湿度较低的条件下,苯的主要氧化路径是S2氧化为S5. 结合图3和图4也可以看到,当体系的相对湿度为40%时,尾气中出现产物P7(己醛)、P8(辛醛)和P9(壬醛)、P10(苯乙酮)和P11(苯甲醛),这与所猜测的苯分子降解路径完全一致. 此时苯的直接转化率与矿化率分别为60%和15%,虽然此时苯降解的矿化效果最好,但是较低的直接转化率以及尾气中生成比苯分子质量更大的有机物.
羟基自由基的引入使得S1与S2氧化的四步反应中能垒最大值从之前的48.1 、15.3 kcal·mol−1分别下降到22.9 、11.6 kcal·mol−1,大大缩减了S1氧化为S4以及S2氧化为S5的难易程度差距,使得在较高相对湿度的条件下,苯的氧化路径是由S1氧化为S4以及S2氧化为S5共同决定的. 随着相对湿度的增加,尾气中出现产物P3(丙酮醇)、P4 (2,2,3-三甲基-3-氧杂环丁醇)、P5 (1-甲基萘)和P6 (2-甲基萘). 当体系的相对湿度为80%时,苯的直接转化率48%和矿化率2%均为最低,但此时尾气中的产物成分较少.
根据以上的分析表明,羟基自由基的引入对S1氧化为S4以及S2氧化为S5的过程都有积极的作用,同时改变了苯分子的降解路径,从而促进苯分子的氧化降解. 苯的氧化降解应结合直接转化率、矿化率以及尾气成分来综合分析,在相对湿度较高与较低的条件下,均出现了各自的利弊,所以对于特定的工艺氧化降解路径还需要谨慎的考虑相对湿度的调节.
-
(1)苯的直接转化率随着相对湿度的增加出现先升高后降低的趋势,在相对湿度为50%时,直接转化率达到最大值82%,但矿化率随相对湿度增加由15%急剧降低至2%.
(2)在相对湿度为40%的条件下,苯分子的氧化降解路径主要是1,2-环己二醇氧化为1,2-环己二酮,路径的四步反应中最大的能垒值为15.3 kcal·mol−1,苯降解的矿化效果最好,直接转化效率最低,尾气中出现分子质量更大的有机物. 随着相对湿度的增加,苯的氧化路径是由1,4-环己二醇氧化为1,4-环己二酮与1,2-环己二醇氧化为1,2-环己二酮共同决定,羟基自由基的引入使得1,4-环己二醇与1,2-环己二醇氧化的四步反应中能垒最大值从48.1 kcal·mol−1和15.3 kcal·mol−1分别下降到22.9 kcal·mol−1和11.6 kcal·mol−1,苯的矿化率和直接转化率急剧降低,尾气中的产物成分减少.
羟基自由基与游离氧原子竞争作用下的光氧化降解苯反应路径
Study on the reaction path of photo-oxidative degradation of benzene under the competition of hydroxyl radicals and free oxygen atoms
-
摘要: 本文实验研究了相对湿度对光氧化降解苯的直接转化效率、矿化率以及对应的尾气成分的影响,并利用量子化学DFT(Density functional theory)的方法讨论了不同相对湿度条件下羟基自由基与游离氧原子竞争作用下的光氧化降解苯反应路径. 结果表明,苯的直接转化效率随着体系相对湿度的增加出现先升高后降低的趋势,相对湿度为50%时直接转化效率最高,为82%,矿化率随相对湿度的增加显著降低,最低可达到2%,且不同相对湿度条件下的尾气成分差异较大. 在相对湿度为40%的条件下,苯分子的氧化降解路径主要是1, 2-环己二醇氧化为1,2-环己二酮,此时最大的反应能垒值为15.3 kcal·mol−1,有利于提高苯降解的矿化效果,生成了P10苯乙酮和P11苯甲醛等分子质量更大的有机物;随着相对湿度由50%提高到80%,最大的反应能垒值由48.1 kcal·mol−1降低到22.9 kcal·mol−1,此时苯的氧化路径由1,4-环己二醇氧化为1,4-环己二酮与1,2-环己二醇氧化为1,2-环己二酮共同决定,羟基自由基的引入大大降低了1,4-环己二醇与1,2-环己二醇氧化的难度,苯的矿化率和直接转化率急剧降低. 确定最佳的相对湿度需要综合分析直接转化效率、矿化率以及尾气成分.Abstract: The effects of relative humidity on the direct conversion efficiency, mineralization rate and components of waste gas from benzene photooxidation degradation were studied. Quantum chemical DFT method was used to analyze the reaction path of benzene photooxidation degradation under the competition of hydroxyl radical and oxygen atoms at different relative humidity. The results showed that the direct conversion efficiency of benzene increased first and then decreased with increasing relative humidity. The maximum direct conversion efficiency was 82% appearing at relative humidity of 50%. The mineralization rate decreased significantly at higher relative humidity, and the smallest value is 2%. The components of waste gas varied much at different relative humidity. At relative humidity of 40%, the oxidative degradation path of benzene was determined by oxidation of 1,2-cyclohexanediol to 1,2-cyclohexanedione, and the maximum reaction energy barrier is 15.3 kcal·mol−1, which is beneficial to improving the mineralization effect and generating some organic compounds with higher molecular weight such as P10 acetophenone and P11 benzaldehyde; As the relative humidity increased from 50% to 80%, the maximum reaction energy barrier decreased from 48.1 kcal·mol−1 to 22.9 kcal·mol−1. At this time, oxidative degradation path of benzene was determined by oxidation of 1, 4-cyclohexanediol to 1,4-cyclohexanedione and oxidation of 1,2-cyclohexanediol to 1,2-cyclohexanedione. The introduction of hydroxyl radicals greatly decreased the oxidation difficulty of 1,4-cyclohexanediol and 1,2-cyclohexanediol, resulting in sharply decreasing mineralization rate and direct conversion efficiency. To determine the optimal relative humidity, it is necessary to comprehensively analyze the direct conversion efficiency, mineralization rate, and components of waste gas.
-
Key words:
- photooxidation /
- benzene /
- relative humidity /
- quantum chemistry /
- DFT
-
截至2019年,我国已建成5 000多座城镇污水处理厂[1],其中有数百座污水处理厂被城市发展区域包围成为“城中厂”[2]。污水处理厂在运行中会产生大量恶臭物质,由此带来的恶臭污染问题日益严重[3]。《恶臭污染物排放标准》(GB14554-1993)中规定了恶臭污染物的排放标准,要求污水处理企业应严格限制相关污染物的排放。与其他恶臭气体去除方法相比,生物除臭法具有投资少、操作简便、运行成本低、二次污染小等优点[4],因而近年来获得迅速发展。据统计,生物除臭专利近5 a的数量已占历年除臭专利总量的75%以上[5],故有必要对污水处理厂产生恶臭物质及生物除臭技术应用中的关键影响因素进行梳理。本文系统总结了污水处理厂恶臭污染的特点,并分析生物技术应用中的关键影响因素及工程应用案例,以期为污水处理厂兴建或改造现有除臭设备、解决恶臭污染问题提供参考。
1. 污水处理厂恶臭污染及生物除臭方法
1.1 恶臭物质产生的处理单元及其特点
污水处理厂每个处理单元的工艺运行条件不同,使得各处理单元的污水性质存在差异,因此,各处理单元产生的恶臭物质在总量、组分和排放强度上亦不同。
恶臭浓度是基于嗅觉器官实验法对恶臭强度进行数量化表征的指标[6]。一般将无臭清洁空气对单位体积恶臭物质进行连续稀释,直至嗅辨员阈值时的稀释倍数定义为恶臭浓度,单位为OU·m−3。为评价各处理单元的恶臭排放强度,将各单元恶臭浓度乘以恶臭气体的排放量定义为恶臭散发率(图1(a))[6]。其中,排放量用气泵抽取某一密闭空间内恶臭气体的流量(m3·h−1)来表示。为评估各处理单元对污水处理厂恶臭污染的贡献,将各单元恶臭散发率乘以恶臭散发表面积(即暴露在外的水面面积,单位为m2)得到的结果定义为各处理单元产生的恶臭比例[6](图1(b))。
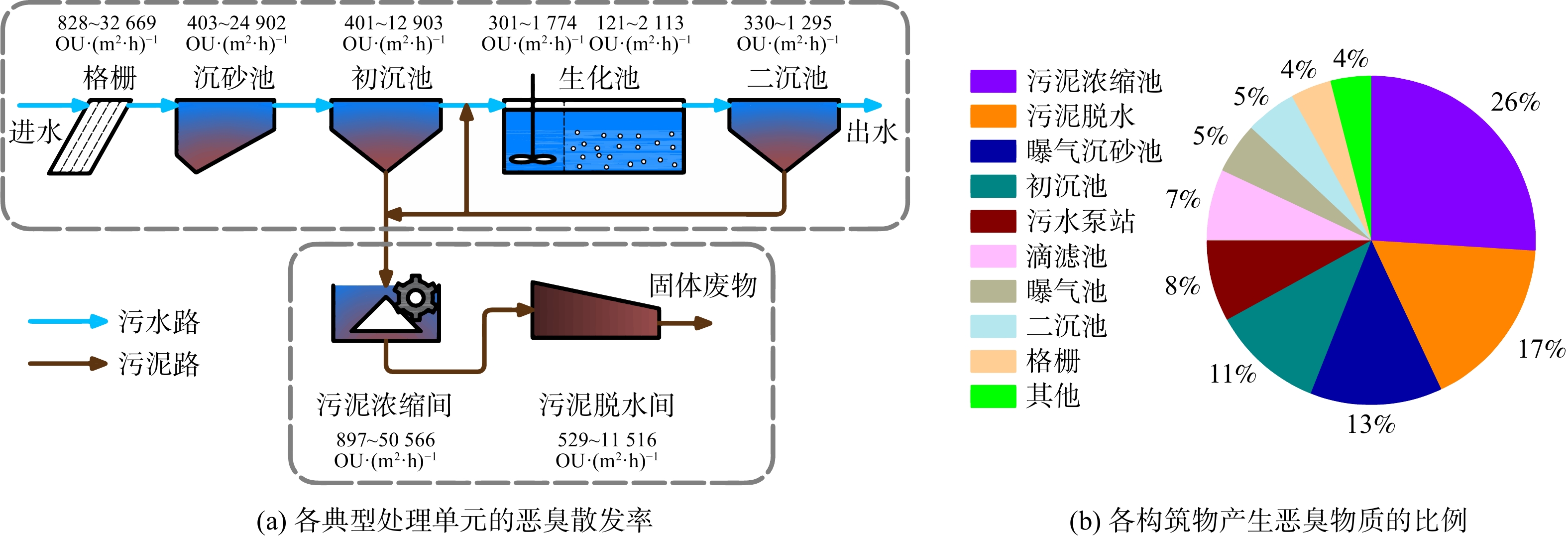 图 1 城镇污水处理厂各处理单元恶臭物质分布Figure 1. Distribution of malodorous substances in each treatment unit of urban sewage treatment plant注:(b)中恶臭物质比例为恶臭散发率等于恶臭浓度乘以其流量。
图 1 城镇污水处理厂各处理单元恶臭物质分布Figure 1. Distribution of malodorous substances in each treatment unit of urban sewage treatment plant注:(b)中恶臭物质比例为恶臭散发率等于恶臭浓度乘以其流量。污水处理厂中产生恶臭物质的处理单元包括2部分:污水处理的进水区(进水泵站、格栅、沉砂池等)与污泥处理区(污泥浓缩间、污泥脱水间等)[7]。大部分恶臭物质的产生是由于污水和污泥中氧含量较低所致,通常厌氧处理工艺产生的恶臭强度比好氧处理工艺的更大[8]。长污泥龄污水处理工艺,如氧化沟,所产生的恶臭浓度高于短泥龄处理工艺,如曝气池。恶臭物质的排放受温度影响亦较为明显。如曝气沉砂池和粗格栅产生的恶臭存在明显的季节差异:夏、秋季节较高,冬、春季节较低。这是由于在不同季节污水温度差别很大,温度会影响恶臭物质生成反应的进行程度和反应速率[7]。另外,污水水质对恶臭产生的影响亦较为明显。当污水化学需氧量和硫酸盐质量浓度更高时,处理单元产生的恶臭浓度也会更高[9]。因此,对治理污水处理厂恶臭污染之前,应首先考虑污水处理工艺及各处理单元的恶臭物质排放特征,结合具体环境因素(水质、温度、氧气条件及污泥龄等),重点控制污泥处理区和污水进水区产生的恶臭物质。
1.2 恶臭物质的组成
污水处理厂各处理单元所产生的恶臭物质组成及浓度不尽相同。还原硫化合物(reduced sulfur compound, RSC)是污水、污泥处理过程中产生最重要的一类恶臭物质[10],包括H2S、甲硫醇、甲硫醚、二硫化碳、二甲基二硫醚等。RSC主要来源于厌氧条件下污水中的硫酸盐还原及含硫有机物的脱硫作用。AOKI等[11]测定了污水处理厂9个不同位置的21种恶臭物质质量浓度(表1),发现RSC是造成污水处理厂上游构筑物(进水井、格栅沉砂池、初沉池等)恶臭污染的主要污染物。JIANG等[12]基于24项不同研究的结果,总结了污水处理厂沉砂池、格栅、初沉池和生化池的环境空气中RSC特征。其中,H2S质量浓度为0.1~20 480 μg·m−3,甲硫醇质量浓度为0.4~2.4 μg·m−3,甲硫醚质量浓度为0.4~5 450 μg·m−3,二硫化碳质量浓度为3.06~9.82 μg·m−3,二甲基二硫醚质量浓度为0.62~1 600 μg·m−3。污泥处理单元的构筑物,如储泥池和污泥脱水间,所释放的恶臭物质主要是RSC及含氮化合物,其中含氮化合物主要是氨、胺、吲哚和甲基吲哚等[13]。综上所述,污水处理厂排放的恶臭物质中,约90%以上是含硫化合物,其中H2S是最主要的污染物[9-10,14-15]。
表 1 污水处理厂各处理单元环境空气中恶臭物质的质量浓度[16]Table 1. The mass concentration of malodorous substances in the air of each sewage treatment unit10−2 mg·L−1 处理单元 硫化氢 甲硫醇 甲硫醚 二甲基二硫醚 氨 乙醛 丙酸 正丁酸 正戊酸 异戊酸 莰烯 提升泵房 1 244.6 85.71 16.33 31.96 0.76 10.61 0.2 0.28 0.0027 0.09 1.4 配水井 3 187.5 200 55.36 13.88 1.59 17.29 0.1 0.04 — — 3.83 浓缩池上清液回流井 212.5 65.71 110.71 28.18 3.72 29.46 0.69 0.28 0.0014 0.05 1.46 初沉池进水口 607.14 9.86 0.83 0.17 — 60.89 0.03 — — — — 生化池进水口 22.77 0.71 0.55 0.13 0.01 18.07 0.03 — — — — 缺氧段末端 3.04 1.27 3.88 0.42 — 23.57 — — — — 0.55 好氧段末端 4.71 0.2 2.77 0.34 — 29.46 — — — — 0.36 最终沉淀池出水 0.35 0.31 0.83 — — 17.68 — — — — — 出水口 0.05 — — — — 19.45 — — — — — 注:—代表结果低于检测限。 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.3 生物除臭技术的关键影响因素
约81%~91%的城镇污水处理厂安装了恶臭物质处理系统,其中生物法占78%,化学洗涤法占11%,活性炭吸附占2%,另有9%的污水处理厂将恶臭物质引入曝气池进行处理[17]。生物处理是最常用的恶臭处理技术。该技术的本质是以恶臭物质为底物驯化适宜的微生物菌群,使恶臭污染物在微生物同化、异化的过程中被去除掉(图2)。生物除臭技术的关键影响因素如下。
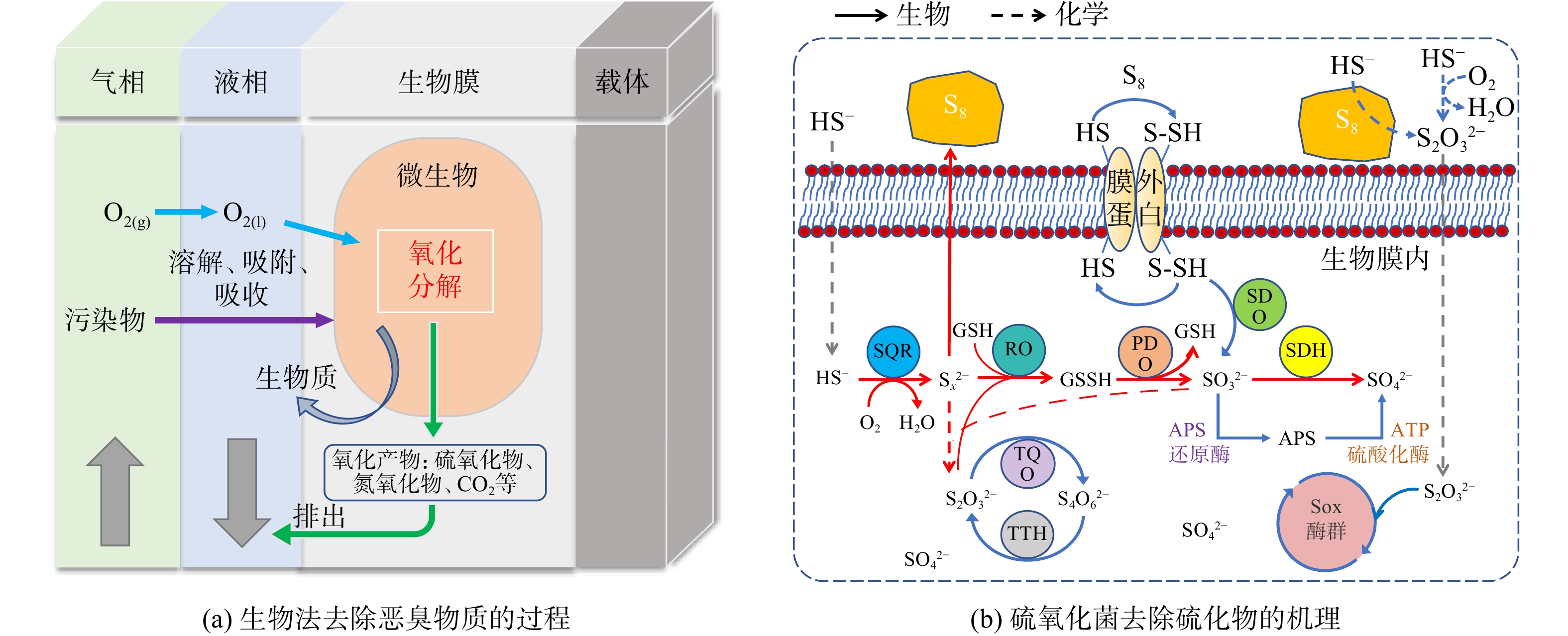 图 2 生物法去除恶臭污染物的过程示意图Figure 2. Schematic diagram of biological removal of odorous pollutants注:SQR为硫化物-醌氧化还原酶(sulfide- quinone oxidoreductase);RO为硫氰酸酶(rhodanese);PDO为过硫化物加双氧酶(persulfide dioxygenase);SDH为亚硫酸盐脱氢酶(sulfite dehydrogenase);GSH为谷胱甘肽(glutathione);GSSH为谷胱甘肽过硫化物(glutathione persulfide);TQO为硫代硫酸盐-醌氧化还原酶(thiosulfate- quinone oxidoreductase);TTH为四硫化物水解酶(tetrathionate hydrolase);APS为腺苷酰硫酸(adenosine-5’-phosphosulfate);ATP为三磷酸腺苷(adenosine triphosphate);Sox为硫氧化(sulfur oxidation)。
图 2 生物法去除恶臭污染物的过程示意图Figure 2. Schematic diagram of biological removal of odorous pollutants注:SQR为硫化物-醌氧化还原酶(sulfide- quinone oxidoreductase);RO为硫氰酸酶(rhodanese);PDO为过硫化物加双氧酶(persulfide dioxygenase);SDH为亚硫酸盐脱氢酶(sulfite dehydrogenase);GSH为谷胱甘肽(glutathione);GSSH为谷胱甘肽过硫化物(glutathione persulfide);TQO为硫代硫酸盐-醌氧化还原酶(thiosulfate- quinone oxidoreductase);TTH为四硫化物水解酶(tetrathionate hydrolase);APS为腺苷酰硫酸(adenosine-5’-phosphosulfate);ATP为三磷酸腺苷(adenosine triphosphate);Sox为硫氧化(sulfur oxidation)。1) 微生物。微生物是生物除臭过程的引擎,亦是去除污染物的执行者[18]。城镇污水处理厂的含硫恶臭物质主要靠硫氧化微生物去除(表2)。硫氧化微生物的营养类型可分为4种:专性无机营养、专性光合营养、兼性无机营养和专性有机营养[19]。专性无机营养的菌种有Thiobacillus denitrificans、Acidithiobacillus thiooxidans和 Chlorobium limicola,可在中性和酸性条件下高效去除含H2S的恶臭物质[20-21]。专性有机营养的菌种包括:Xanthomonas、Pseudomonas和Hyphomicrobium,这些菌株亦可高效去除H2S及含硫有机恶臭物质[22-23]。此类微生物通常适宜在中性条件下生存。硫氧化微生物的接种来源非常广泛,包括污水处理厂的活性污泥、被相关污染物污染过的土壤或水体、实验室中驯化可降解相关污染物的菌群以及菌群中分离获得的纯种菌[24-25]等。
表 2 脱硫微生物的特征Table 2. The characteristics of desulphurization microorganisms微生物名称 营养类型 底物 最佳pH 最佳温度/℃ 硫氧化酶 Acidianus ambivalens 专性无机营养 S2-, S0 2.5 94 硫氧合酶/还原酶 Acidianus brierleyi 专性无机营养 S2-, S0, S2O32-, S4O62- 1.8 70 硫氧合酶 Oscillatoria limnetica 专性光合营养 S2- 7.5 35 硫化氢:醌还原酶 Chlorobaculum thiosulfatophilum 专性光合营养 S2-, S0, S4O62- 6.8 25~35 硫化细胞色素c;硫代硫酸盐:细胞色素c还原酶 Thiocapsa roseopersicina 兼性光合营养 S2-, S0, S2O32- 7.3 20~35 亚硫酸盐:受体氧化还原酶 Paracoccus denitrificans 兼性无机营养 S2-, S0, S2O32- 8.0 30 亚硫酸盐脱氢酶;硫代硫酸盐氧化酶系统 Paracoccus versutus 兼性无机营养 S2-, S0, S2O32- 8.0 30 亚硫酸盐脱氢酶;硫代硫酸盐氧化酶系统 Thiobacillus denitrificans 专性无机营养 S2-, S0, S2O32-, S4O62-, SO32- 7.0 30 腺苷酰硫酸还原酶;亚硫酸盐氧化酶 Acidithiobacillus ferrooxidans 专性无机营养 S2-, S0, S2O32-, S4O62-, SO32- 2.5 30 硫:铁(III)氧化还原酶;硫化物:铁(III)氧化还原酶 Acidithiobacillus thiooxidans 专性无机营养 S2-, S0, S2O32-, S4O62-, SO32- 2.5 30 亚硫酸盐:细胞色素c氧化还原酶 | Show TableDownLoad:
CSV
2)营养液。为使微生物处于最佳代谢状态,通常需要满足的条件有:适当的水分、营养、温度、pH等。流动液相可为微生物生长提供水分和矿物质营养,并可提供控制运行条件的手段。液相通常富含基本矿物营养物质,包括氮、磷、钾和微量元素等组成的盐类,例如:KH2PO4、Na2HPO4、KNO3、(NH4)2SO4、NH4Cl、NH4HCO3、CaCl2、MgSO4、MnSO4、FeSO4、Na2MoO4和维生素[26-27]。在研究实践中,污水处理厂处理的生活污水即可作为微生物的营养液[28]。此外,营养液还可发挥冲洗微生物代谢产物并调节pH的作用。
3) pH。稳定的pH是实现高效去除恶臭污染物、避免发生抑制作用的前提条件。大部分硫氧化微生物只在特定的pH范围内具有活性,其适宜的pH一般为2.0~10.0。异养硫氧化微生物的最适宜pH接近中性,高于或低于该范围均会使其活性受到抑制。在不同pH条件下还可能演化出不同微生物群体,亦会影响微生物系统的去除性能[29]。另外,pH在减少酸性气体气液传质阻力方面具有显著作用,较高的pH有利于酸性气体溶解并提高酸性气体的去除率。在处理含硫恶臭气体时,通常会使填料床pH下降。为增强系统的缓冲能力,可通过添加缓冲材料,如碳酸钙、白云石或粉碎的贝壳来稳定系统pH,也可通过喷洒具有缓冲作用的营养液来控制pH,如Ca(OH)2、K2HPO4、NH4Cl和尿素等。
4)填料。填料床是生物反应器的核心组件,可为微生物的附着和气液接触提供场所[30],甚至提供营养及pH缓冲能力[31]。理想的填料应满足如下特点:低成本、大比表面积、高孔隙率、高化学稳定性和高结构强度、低密度,并易于微生物附着。沸石、陶粒、珍珠岩、熔岩是常用填料[32-33],具有比表面积大与孔隙率高,且利于生物膜的固定等优势。泰勒花环、聚丙烯鲍尔环、聚氨酯泡沫等塑料材料的化学性能稳定、成本低、孔隙率高,因此,这些材料在许多研究和大规模应用中被成功应用[34-36]。当污染物浓度较低时,采用大比表面积的填料可有利于克服传质阻力[37-38]。堆肥、泥炭、土壤等天然填料本身携带着丰富的微生物群体,可为微生物提供营养,然而天然填料的使用寿命有限,通常使用2~3 a后就需要更换[39]。
2. 恶臭生物处理工艺典型案例
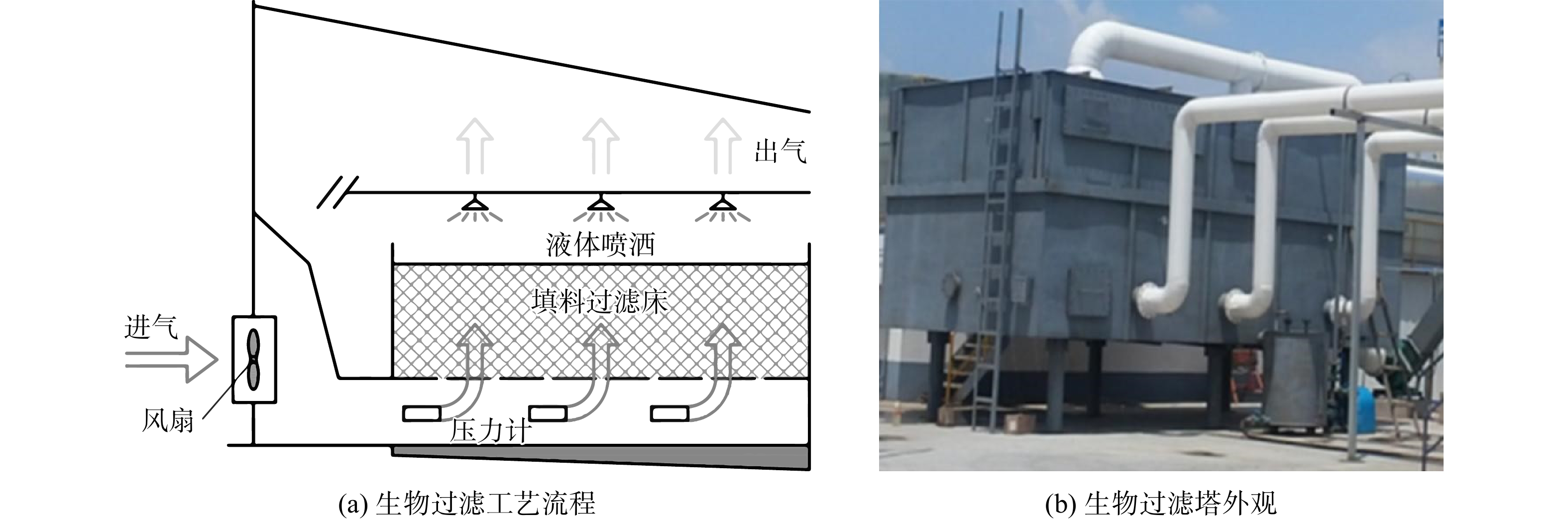
2.1 生物过滤法案例
生物过滤法宜用于去除流量大但浓度低的废气,对水溶性差的污染物去除效果较好,可用于去除无量纲亨利系数等于或低于10的气态污染物[40]。付妍等的研究[41]即为利用生物过滤法处理天津某污水处理厂的恶臭气体的典型案例(图3)。该生物过滤系统包括1个生物过滤塔、1个空气增压泵和1个水泵。待处理污染物为复合型恶臭气体,来自于该厂的混合池、曝气池和污泥区,废气总量11 000 m3·h−1,平均空床停留时间(empty bed residence time, EBRT)为15 s。该案例中的恶臭物质主要有H2S及致臭挥发性有机物(volatile organic compounds, VOCs)2类,其平均进气质量浓度分别为2.5~3.5 mg·m−3和200~300 mg·m−3,平均去除率分别为100%和>90%。处理过程中反应器的平均压降为0.45 kPa·m−1。生物过滤塔采用的填料为TJ-003填料,具有大比表面积、结构稳定、耐腐蚀及易于微生物附着等特点,其填充体积和高度分别为45 m3和1.5 m。研究者根据恶臭物质类型筛选得到复合优势菌群,并将其接种于填料上。该菌群的单位填料接种生物量为4.44 kg·m−3,对此污水处理厂恶臭物质的去除效果良好,可有效降低微生物系统的启动时间。生物过滤塔所需补充水为污水处理厂进水。通过位于填料床上方的喷头对填料床进行间歇洒水以实现补水,并维持反应器内湿度;同时,污水处理厂进水含有的丰富营养元素(碳氮磷等)可为微生物提供充分的营养以维持其活性,系统的最小补水量为159 L·(m3·d)−1。然而,污水处理厂进水中丰富的营养易导致填料中微生物过度增殖,使得反应器内部堵塞频率增多。生物过滤塔的反应器结构简单、除臭效率高、建设及运行成本低;然而该反应器无连续流动液相,仅靠定期补水来维持滤床内的湿度和pH,因此,在处理高浓度含硫气体时,滤床填料易酸化腐败导致滤床堵塞,需要定期更换填料。综上所述,在生物过滤工艺运行中,应避免系统内生物过度增殖,并确保实现pH的灵活调节,以保证系统的长期稳定运行。
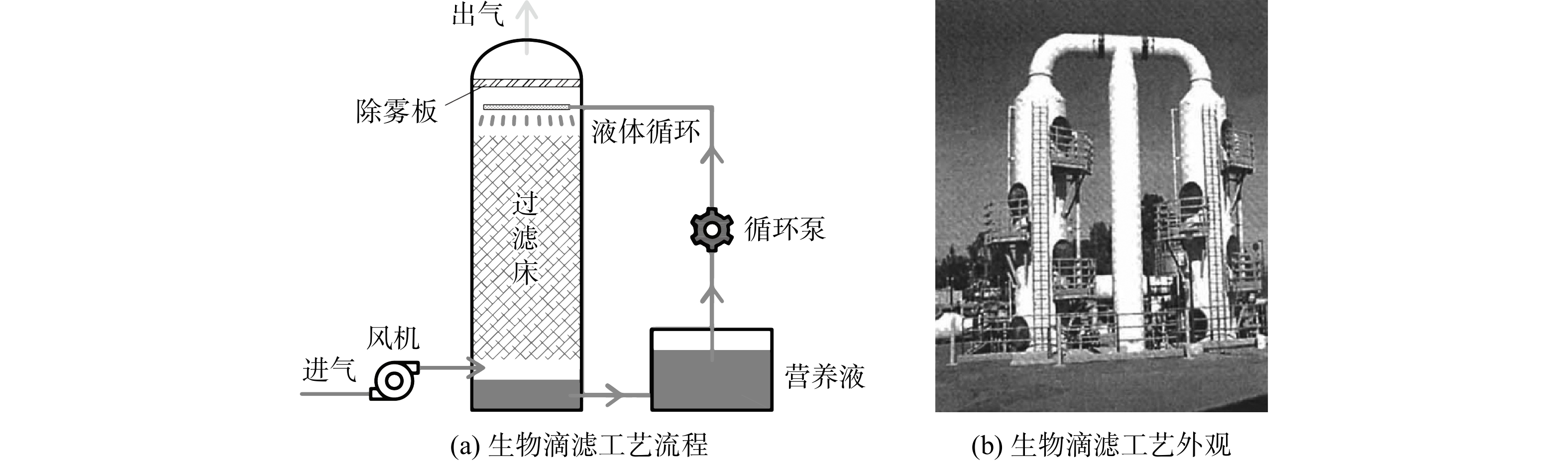
2.2 生物滴滤法案例
生物滴滤法适用于无量纲亨利系数等于或低于1的废气处理[40]。生物滴滤塔内保持着不断循环的液相,其工艺参数,如pH、营养物、盐含量和有毒降解产物等,易于被连续监测和控制。因此,对于易导致生物系统酸化的含硫气体,生物滴滤池的处理效果比生物过滤池更优。GABRIIL等[42]将美国加利福尼亚州某污水处理厂内处理废气的化学洗涤塔改造为生物滴滤塔(图4),用于处理含有H2S和致臭VOCs的复合恶臭气体。该污水处理厂恶臭污染物的恶臭浓度约为(1 980 ± 480)OU·m−3。改造后的生物滴滤塔内径为1.8 m、高9.7 m;塔内填料为聚氨酯泡沫(比表面积 600 m2·m−3,孔隙率97%),堆积高度为3.7 m。聚氨酯海绵拥有较大的比表面积,更利于微生物附着及气液接触。填料上的微生物来自该污水处理厂的活性污泥,降低了基础建设成本,但需要较长的启动时间来驯化除臭微生物。利用该污水处理厂的二沉池出水为生物滴滤塔的循环液,可为微生物提供水分和所需营养,且不易引起微生物过度增殖。循环液的流量为1.8 m3·(m2·h)−1。定期更新循环液将其pH维持在1.5~2.3,以避免发生微生物活性抑制的现象。恶臭气体为收集自进水下水道干管和螺杆泵外壳的臭味空气。气体流量为16 300 m3·h−1,塔内平均EBRT为2 s。实验结果表明:当H2S质量浓度为45.5 mg·m−3时,滴滤塔对H2S的去除率高达95%,去除负荷(以H2S质量计)为105 g·(m3·h)−1;当H2S的质量浓度为91.1 mg·m−3时,去除率可达90%。RSC的平均去除率为15%~40%、VOCs的去除率约为30%,恶臭浓度去除率约为65%。相比于生物过滤法,生物滴滤系统的结构更简单,建设和运行成本更低,但生物滴滤法对水溶性较差的废气去除率稍差。因此,在处理VOCs时,其质量浓度不宜高于500 mg·m−3,同时还需增加EBRT,以延长气液接触时间,从而保证系统的去除效果。
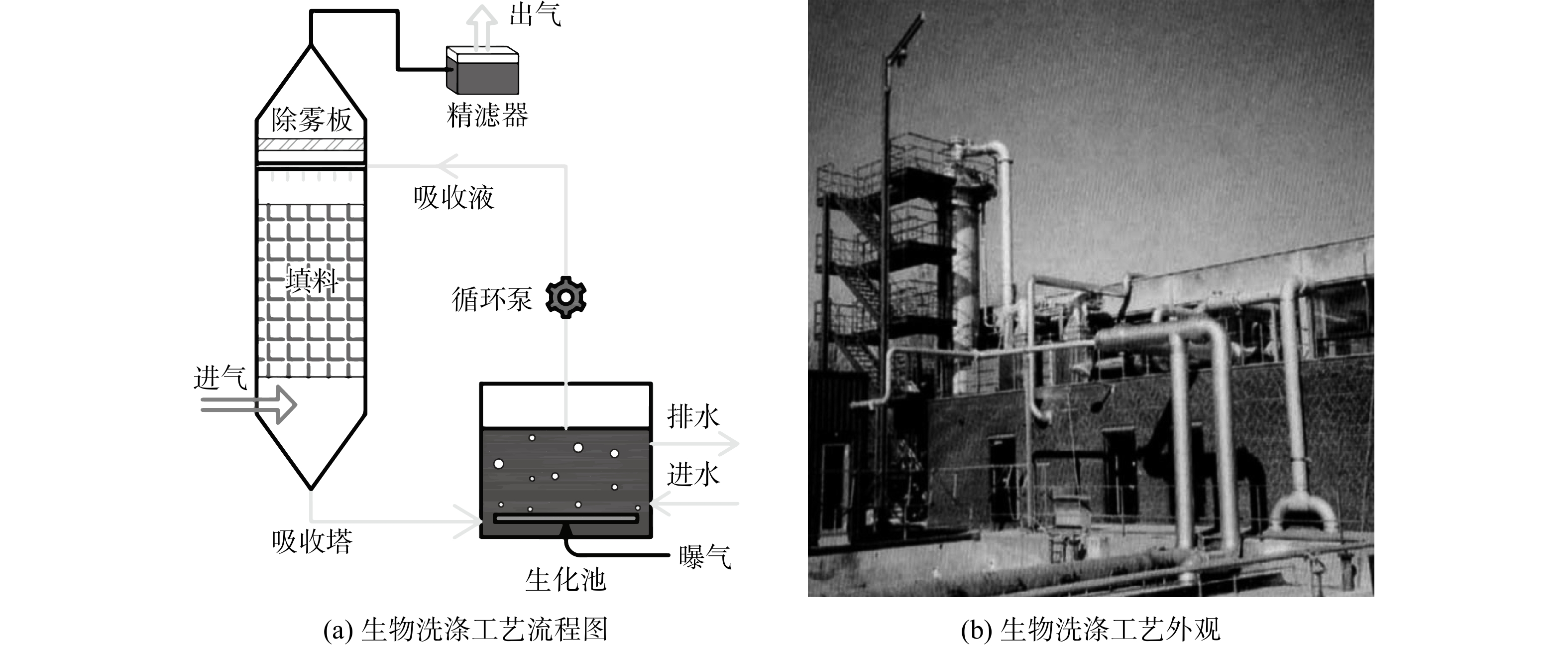
2.3 生物洗涤法案例
在处理污染物质量浓度高(>500 mg·m−3),且无量纲亨利系数为0.01及以下的亲水性污染物(如醇、醛、脂肪酸和二醇)时,生物洗涤法的性价比更佳[40]。HANSEN等[43]采用生物洗涤器处理丹麦哥本哈根某污水处理厂的恶臭气体,就是其中的一个典型案例。恶臭气体收集自该厂的沉砂池、油水分离池和格栅等处理单元,气体流量为6 000 m3·h−1。其中,H2S的年平均质量浓度约为10 mg·m−3,夏季和秋季平均质量浓度为25~35 mg·m−3,有机硫化合物浓度较小(<0.1 mg·m−3)。该生物洗涤器由1座填充塑料填料的吸收塔和1座基于活性污泥工艺的生物氧化池构成(图5)。吸收塔的直径为1.6 m、高12 m。采用比表面积较大且结构强度高的塑料空心环和金属混合物作为吸收塔填料,在增大气液接触面积的同时可避免因填料压缩造成的堵塞。吸收塔填料填充体积为12 m3,生物氧化池体积为11 m3。生物反应器以污水处理厂的曝气池活性污泥为菌源;同时,生物反应器中的活性污泥又被用作吸收塔的吸收液,液体流量为70 m3·h−1。生物反应器的pH通过添加化学试剂的方式(以每千克H2S气体添加的NaOH质量计,约2.4 kg·kg−1)调节,维持在8.5~9.0,以确保微生物具备良好的活性并减少酸性气体的传质阻力。研究结果表明,生物洗涤器对H2S及有机硫化合物的去除率大于99%[44]。运行过程中洗涤段的空气压降稳定,塑料填料上也未出现微生物增殖不受控制的情况。与生物过滤法和生物滴滤法相比,生物洗涤法可避免生物质生长堵塞滤床的风险。生物洗涤设备同样具备结构简单、体积小、建设成本低,运行稳定性高且运行参数的易于控制等优点。然而,为了维持系统pH的稳定,需要频繁投加碱性试剂,这使得系统运行更复杂,运行成本更高。另外,生物洗涤法很难去除水溶性较差的污染物,可考虑在液相中加入有机吸收剂、固体吸收剂、吸附剂和生物表面活性剂等,以促进对生物洗涤法对疏水化合物的去除。
3. 问题与展望
目前,生物除臭技术应用中存在如下问题。1)生物反应器内载体表面生物膜的生长会降低滤床的孔隙率,导致压降过高,甚至滤床堵塞[45]。化学清洗,定期切换流动方向以及高速水流冲洗[37]等方法已被尝试用于防止生物质的积累,但这些方法大都停留在实验室规模的装置中,尚未有效应用于实际工程中的生物反应器。2)生物过滤法处理含高浓度H2S时,如污泥发酵产生的H2S质量浓度可达6 071 mg·m−3[46],产生的硫酸会导致反应器内环境被迅速酸化。如何避免微生物活性因pH降低而受到抑制,同时保证对H2S的高效去除是生物过滤法应用中的难点。3)生物洗涤法处理恶臭气体时需克服气液传质阻力将污染物从气相转移至液相中,这使其对水溶性较差的污染物处理效果欠佳。因此,实现对疏水性污染物的有效去除是生物洗涤法进一步优化的方向。
生物除臭技术未来发展可集中解决如下问题。1)探究功能微生物所需的最佳营养液成分,以富集高丰度的功能微生物,从而以提高生物系统的处理性能和应对冲击负荷的能力。2)研发结构强度大、比表面积高且易于微生物附着的新型填料,以改善生物反应器内的传质效果,从而提高生物系统对恶臭污染物的去除性能。3)探究维持生物反应器内pH长期稳定且更加经济可行的策略,以维持功能微生物的活性。4)探究高效去除VOCs且同时维持生物量长期平衡的策略,以避免生物反应器内部发生堵塞。5)富集耐极酸性的除臭微生物,以提高生物系统应对系统酸化的能力。6)开发有机吸收剂、固体吸收剂、吸附剂和生物表面活性剂,促进对烷烃等疏水气态化合物的吸收。7)利用基因工程手段培育特种微生物,从而提高对难降解有机物的去除效率。
-
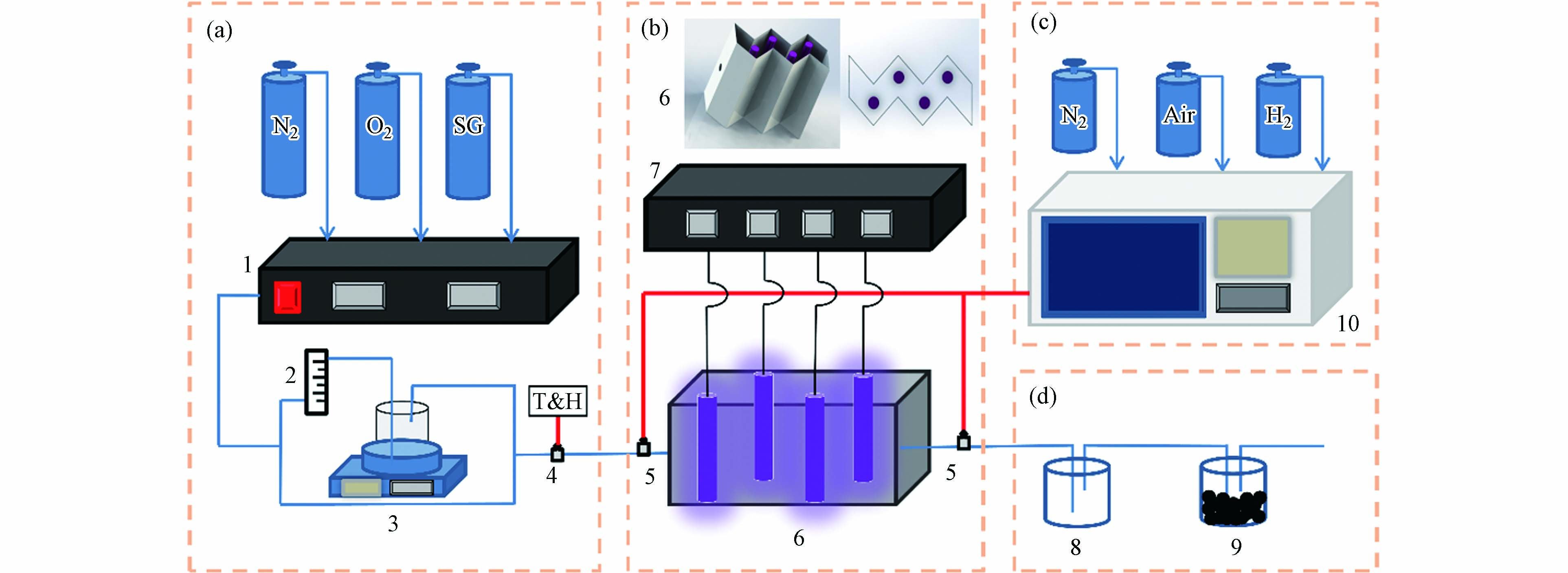
图 1 紫外光氧化降解实验系统示意图
Figure 1. Schematic diagram of UV photooxidative degradation experimental system
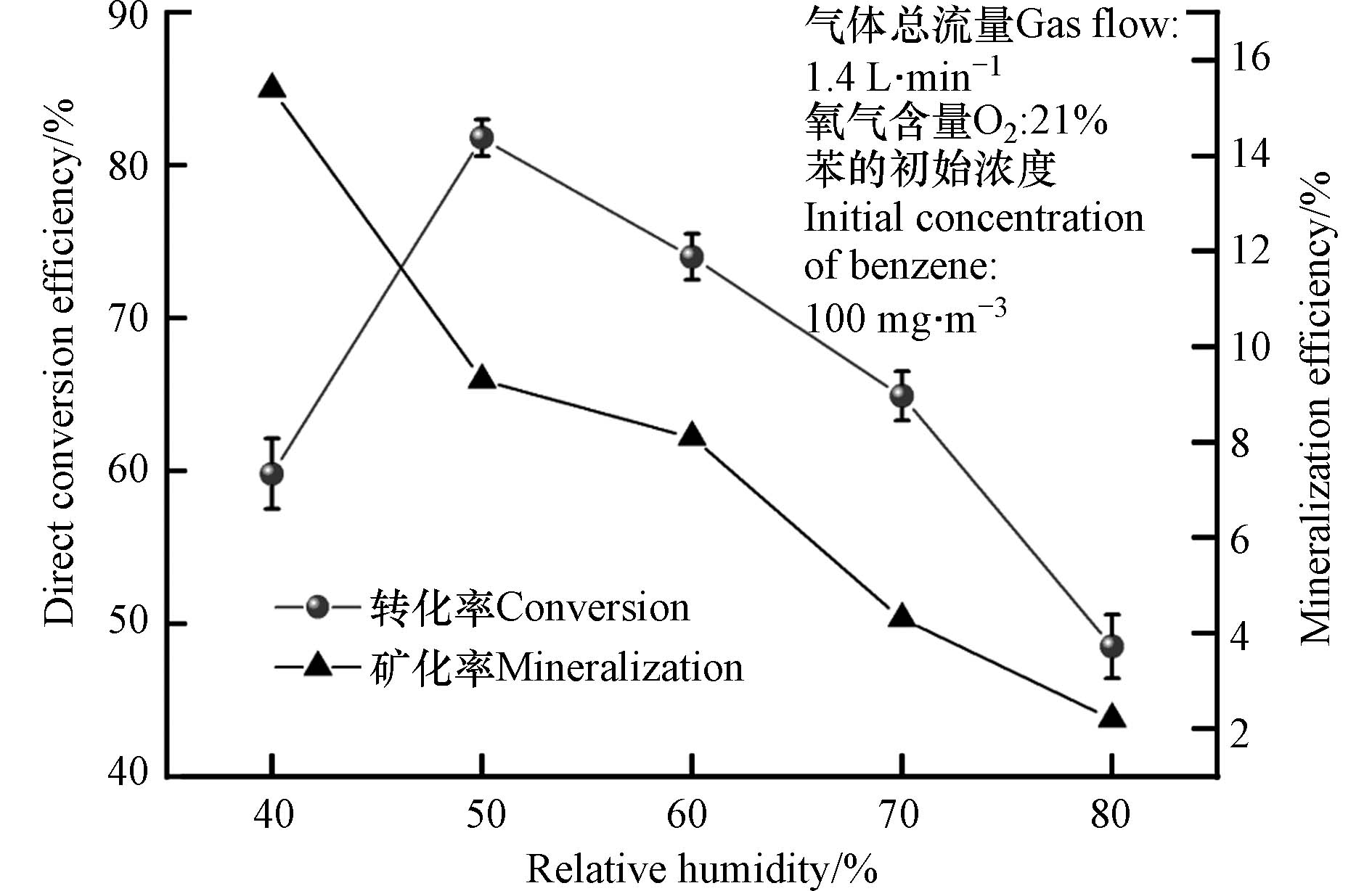
图 2 不同相对湿度下苯的直接转化率与矿化率
Figure 2. Direct conversion efficiency and mineralization rate of benzene at different relative humidity
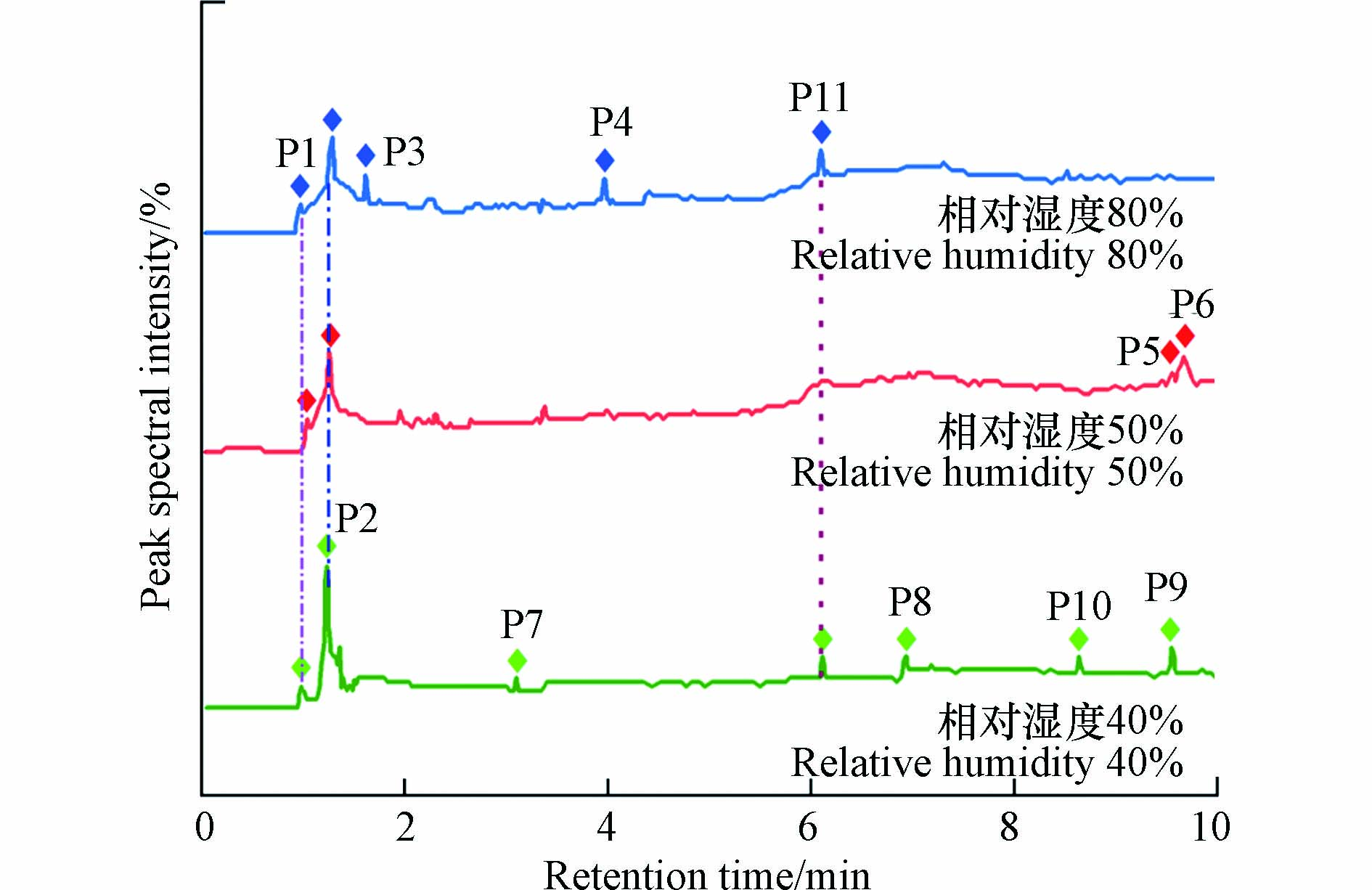
图 3 不同相对湿度下尾气成分GC-MS谱图
Figure 3. GC-MS spectra of exhaust gas components under different relative humidity
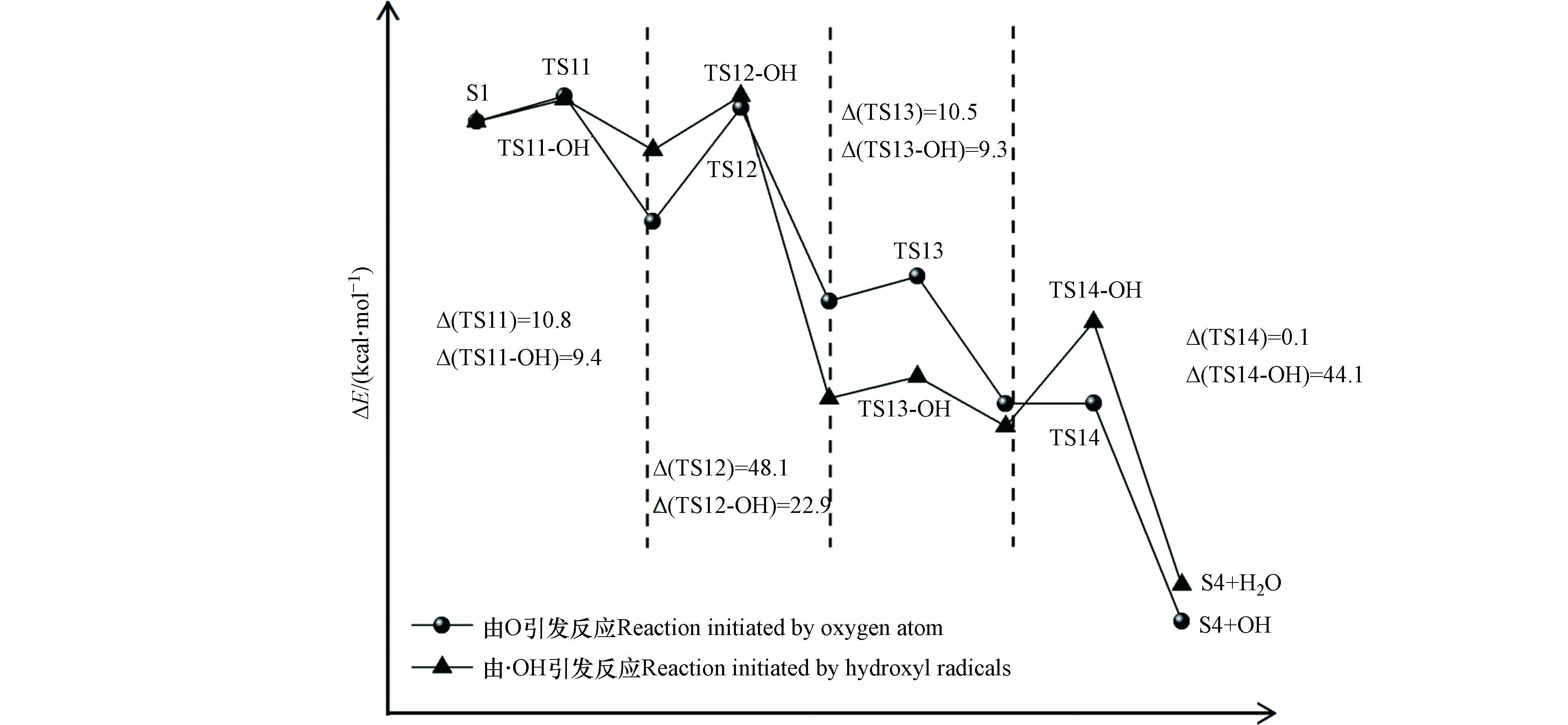
图 5 氧原子与羟基自由基引发的S1氧化为S4的反应
Figure 5. Oxidation of S1 to S4 Initiated by Oxygen Atoms and Hydroxyl Radicals
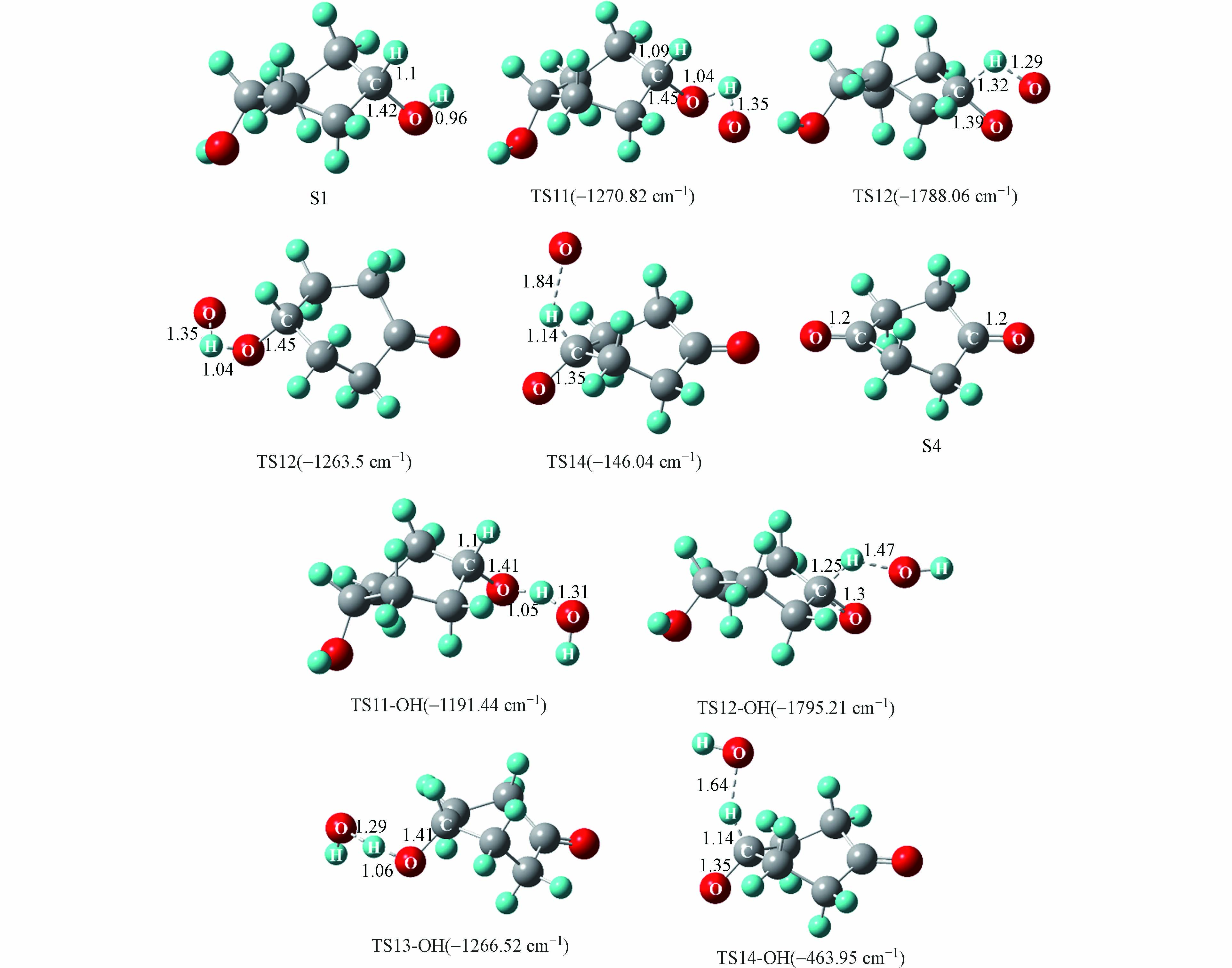
图 6 氧原子与羟基自由基引发的S1氧化为S4的反应过渡态结构
Figure 6. Transition state structure of the oxidation of S1 to S4 induced by oxygen atoms and hydroxyl radicals
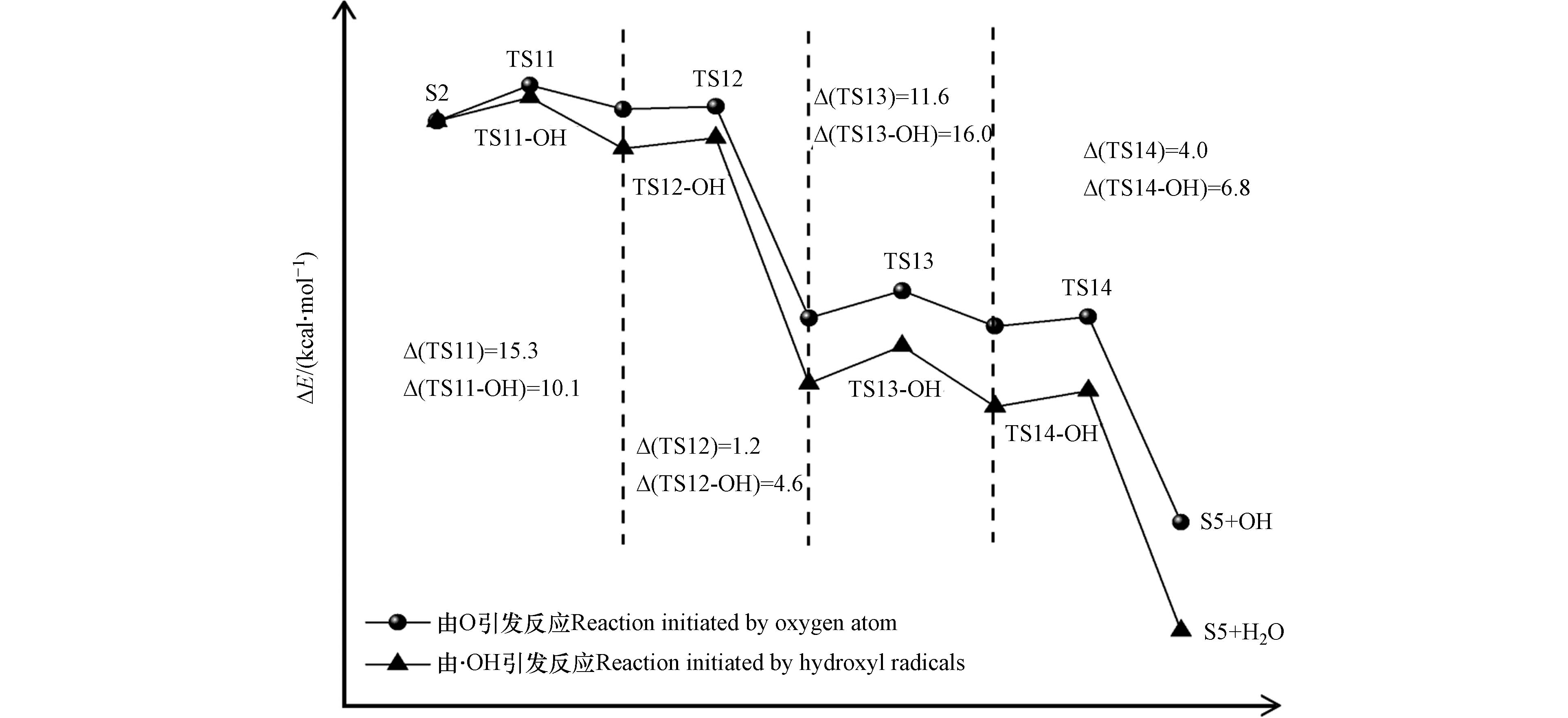
图 7 氧原子与羟基自由基引发的S2氧化为S5的反应
Figure 7. Oxidation of S2 to S5Initiated by Oxygen Atoms and Hydroxyl Radicals
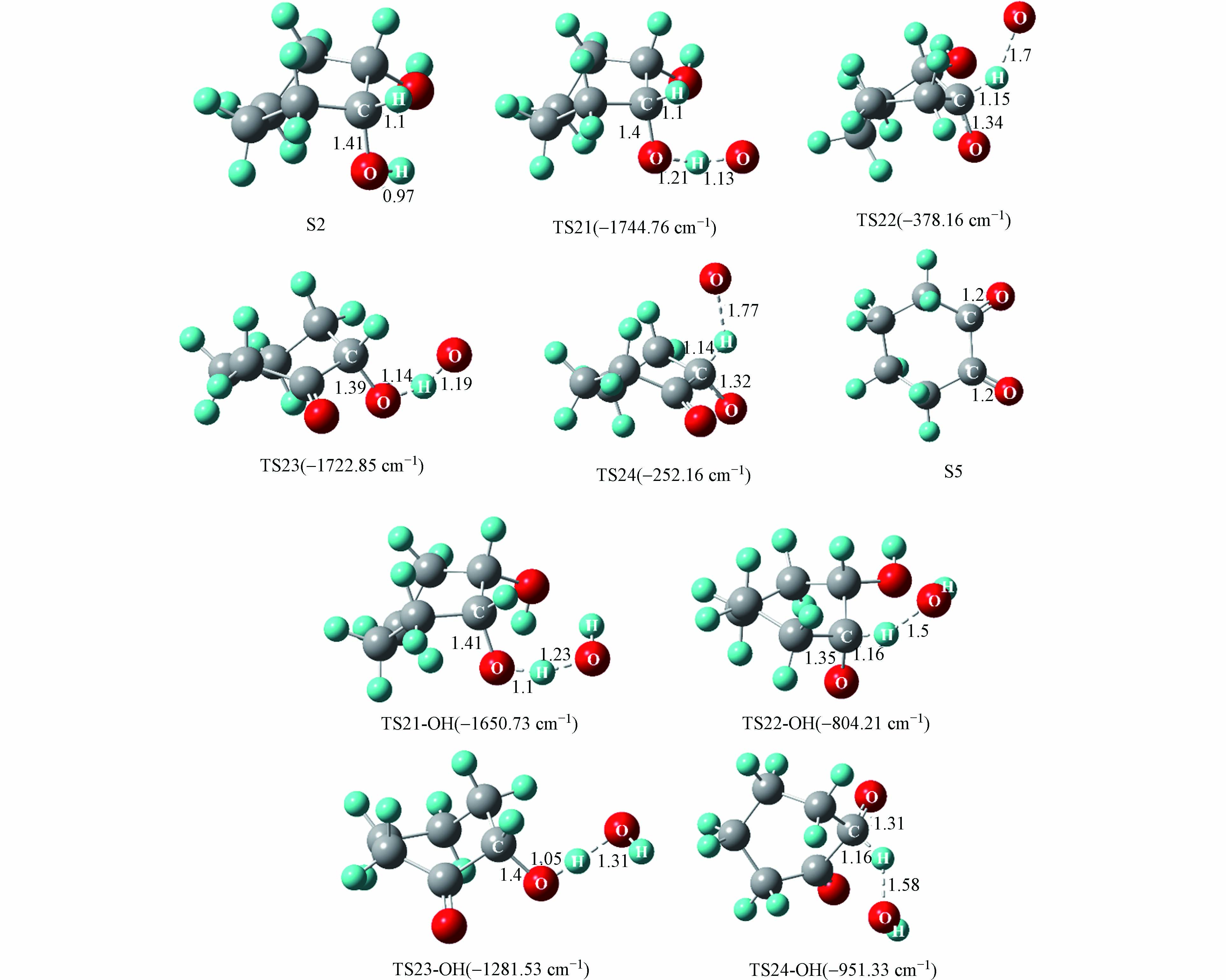
图 8 氧原子与羟基自由基引发的S2氧化为S5的反应过渡态结构
Figure 8. Transition state structure of the oxidation of S2 to S5 induced by oxygen atoms and hydroxyl radicals
表 1 不同相对湿度下尾气GC-MS检测主要成分
Table 1. Detection of main components in exhaust gas by GC-MS under different relative humidity
编号 Serial number 名称 Name CAS 分子式 Molecular formula 保留时间/min Retention time P1 二氧化碳 124-38-9 CO2 0.975 P2 丙酮 67-64-1 
1.233 P3 丙酮醇 116-06-6 1.325 P4 2,2,3-三甲基-3-氧杂环丁醇 25910-96-7 1.617 P5 1-甲基萘 90-12-0 9.675 P6 2-甲基萘 91-12-0 9.675 P7 己醛 66-25-1 3.100 P8 辛醛 124-13-0 7.075 P9 壬醛 124-19-6 9.558 P10 苯乙酮 98-86-2 8.642 P11 苯甲醛 100-52-7 6.117
下载: 导出CSV
-
[1] 吴健, 高松, 陈曦, 等. 涂料制造行业挥发性有机物排放成分谱及影响 [J]. 环境科学, 2020, 41(4): 1582-1588. WU J, GAO S, CHEN X, et al. Source profiles and impact of volatile organic compounds in the coating manufacturing industry [J]. Environmental Science, 2020, 41(4): 1582-1588(in Chinese).
[2] PATERSON C A, SHARPE R A, TAYLOR T, et al. Indoor PM2.5, VOCs and asthma outcomes: A systematic review in adults and their home environments [J]. Environmental Research, 2021, 202: 111631. doi: 10.1016/j.envres.2021.111631 [3] 廖正祝, 田红. 煤化工VOCs吸附处理技术研究进展及展望 [J]. 洁净煤技术, 2021, 27(1): 155-168. LIAO Z Z, TIAN H. Research progress and prospect of coal chemical VOCs adsorption treatment technology [J]. Clean Coal Technology, 2021, 27(1): 155-168(in Chinese).
[4] DU Z H, LIN X. Research progress in treatment of VOCs by dielectric barrier plasma cooperating catalyst [J]. IOP Conference Series:Earth and Environmental Science, 2020, 508(1): 012132. doi: 10.1088/1755-1315/508/1/012132 [5] 郭海倩, 缪晶晶, 姜理英, 等. 低温等离子体-生物耦合系统对复合CVOCs的降解 [J]. 环境科学, 2018, 39(2): 640-647. GUO H Q, MIAO J J, JIANG L Y, et al. Composite CVOCs removal in a combined system of nonthermal plasma and a biotrickling filter [J]. Environmental Science, 2018, 39(2): 640-647(in Chinese).
[6] 孙昕, 史路肖, 张燚, 等. 真空紫外/过二硫酸盐去除饮用水中嗅味物质 [J]. 环境科学, 2018, 39(5): 2195-2201. SUN X, SHI L X, ZHANG Y, et al. Removal of odorants in drinking water using VUV/persulfate [J]. Environmental Science, 2018, 39(5): 2195-2201(in Chinese).
[7] 庄媛, 刘杰民, 曲琛, 等. 芬顿催化氧化VOCs过程中的传质增强及协同作用研究进展 [J]. 环境化学, 2021, 40(11): 3307-3315. doi: 10.7524/j.issn.0254-6108.2021033108 ZHUANG Y, LIU J M, QU C, et al. Mass transfer enhancement and synergistic effect during VOCs removal by Fenton oxidation [J]. Environmental Chemistry, 2021, 40(11): 3307-3315(in Chinese). doi: 10.7524/j.issn.0254-6108.2021033108
[8] 党小庆, 王琪, 曹利, 等. 吸附法净化工业VOCs的研究进展 [J]. 环境工程学报, 2021, 15(11): 3479-3492. doi: 10.12030/j.cjee.202011052 DANG X Q, WANG Q, CAO L, et al. Research progress on purification of VOCs in industrial gas by adsorption [J]. Chinese Journal of Environmental Engineering, 2021, 15(11): 3479-3492(in Chinese). doi: 10.12030/j.cjee.202011052
[9] 梁文俊, 李坚, 李依丽, 等. 低温等离子体法去除苯和甲苯废气性能研究 [J]. 环境污染治理技术与设备, 2005(5): 51-55. LIANG W J, LI J, LI Y L, et al. Degradation of benzene and toluene with cold plasma [J]. Techniques and Equipment for Environmental Pollution Control, 2005(5): 51-55(in Chinese).
[10] 黄雪燕, 庄晶晶, 胡学靖, 等. 室内VOCs光催化法处理研究进展 [J]. 环境保护与循环经济, 2021, 41(3): 29-32. HUANG X Y, ZHUANG J J, HU X J, et al. Research progress in photocatalytic treatment of indoor VOCs [J]. Environmental Protection and Circular Economy, 2021, 41(3): 29-32(in Chinese).
[11] KANG I S, XI J Y, HU H Y. Photolysis and photooxidation of typical gaseous VOCs by UV Irradiation: Removal performance and mechanisms[J]. 中国环境科学与工程前沿: 英文版, 2018(3): 107-120. KANG I S, XI J Y, HU H Y. Photolysis and photooxidation of typical gaseous VOCs by UV Irradiation: Removal performance and mechanisms [J]. Frontiers of Environmental Science & Engineering, 2018(3): 107-120.
[12] 张宇飞, 朱燕群, 王树荣, 等. 甲苯的光氧化降解试验研究 [J]. 环境科学学报, 2015, 35(9): 2759-2765. ZHANG Y F, ZHU Y Q, WANG S R, et al. Experimental study on the degradation of toluene by photo-oxidation [J]. Acta Scientiae Circumstantiae, 2015, 35(9): 2759-2765(in Chinese).
[13] de LUIS A M, LOMBRAÑA J I, MENÉNDEZ A, et al. Analysis of the toxicity of phenol solutions treated with H2O2/UV and H2O2/Fe oxidative systems [J]. Industrial & Engineering Chemistry Research, 2011, 50(4): 1928-1937. [14] BRASLAVSKY S, ACUNA A U, ADAM W, et al. Glossary of terms used in photochemistry, 3rd edition (IUPAC Recommendations 2006) [J]. Pure & Applied Chemistry, 2007, 79(3): 293-465. [15] 陈越平. H2O2强化紫外光催化降解低浓度二甲苯废气的研究[D]. 杭州: 浙江工业大学, 2016. CHEN Y P. Research on low concentration xylene degradation by H2O2 enhanced UV photocatalysis[D]. Hangzhou: Zhejiang University of Technology, 2016(in Chinese).
[16] 周灵浚, 卜岩枫, 成卓韦, 等. 真空紫外光解乙苯废气的工艺特性及转化机制研究 [J]. 环境污染与防治, 2014, 36(6): 13-19. ZHOU L J, BU Y F, CHENG Z W, et al. Conversion characteristics and mechanism analysis of gaseous ethylbenzene degraded by vacuum ultraviolet photodecomposition [J]. Environmental Pollution & Control, 2014, 36(6): 13-19(in Chinese).
[17] 张春洋, 马永亮. UV254nm+185nm光照降解气态甲苯的实验研究 [J]. 中国环境科学, 2011, 31(6): 898-903. ZHANG C Y, MA Y L. Experimental study on UV254nm+185nm photodegradation of gaseous toluene [J]. China Environmental Science, 2011, 31(6): 898-903(in Chinese).
[18] CHEN J Y, HE Z G, JI Y M, et al. OH radicals determined photocatalytic degradation mechanisms of gaseous styrene in TiO2 system under 254 nm versus 185 nm irradiation: Combined experimental and theoretical studies [J]. Applied Catalysis B:Environmental, 2019, 257: 117912. doi: 10.1016/j.apcatb.2019.117912 [19] DANESHVAR N, BEHNAJADY M A, MOHAMMADI M K A, et al. UV/H2O2 treatment of Rhodamine B in aqueous solution: Influence of operational parameters and kinetic modeling [J]. Desalination, 2008, 230(1/2/3): 16-26. [20] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 16 Rev. B. 01 [M]. Wallingford, CT. 2016. [21] BICZYSKO M, PANEK P, SCALMANI G, et al. Harmonic and anharmonic vibrational frequency calculations with the double-hybrid B2PLYP method: Analytic second derivatives and benchmark studies [J]. Journal of Chemical Theory and Computation, 2010, 6(7): 2115-2125. doi: 10.1021/ct100212p [22] 王子东, 马永亮. 利用UVC去除低浓度苯的实验研究 [J]. 环境工程学报, 2009, 3(7): 1284-1288. WANG Z D, MA Y L. Experimental study on removal of low concentration benzene with UVC [J]. Chinese Journal of Environmental Engineering, 2009, 3(7): 1284-1288(in Chinese).
[23] 周政. 基础有机化学[M]. 北京: 高等教育出版社, 1990.263-264. ZHOU Z. Basic organic chemistry [M]. Beijing: Higher Education Press, 1990.263-264(in Chinese).
[24] ZHANG W P, LI G Y, LIU H L, et al. Photocatalytic degradation mechanism of gaseous styrene over Au/TiO2@CNTs: Relevance of superficial state with deactivation mechanism [J]. Applied Catalysis B:Environmental, 2020, 272: 118969. doi: 10.1016/j.apcatb.2020.118969 [25] ZHANG W P, LI G Y, WANG W J, et al. Enhanced photocatalytic mechanism of Ag3PO4 nano-sheets using MS2 (M = Mo, W)/rGO hybrids as co-catalysts for 4-nitrophenol degradation in water [J]. Applied Catalysis B:Environmental, 2018, 232: 11-18. doi: 10.1016/j.apcatb.2018.03.006 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2526
- HTML全文浏览数: 2526
- PDF下载数: 101
- 施引文献: 0
