











































-
铅是有毒的环境污染物,被世界卫生组织列为十大危害公共卫生安全的化学物质之一. 环境铅污染主要来源于工业生产和应用. 尽管从20世纪末开始,全世界各个国家先后禁止使用含铅汽油以及含铅钢管和硬聚氯乙烯(PCV)塑料管材,但是由于社会工业的快速发展,铅的使用量仍然逐年增加. 根据2021年3月Statista研究部门的数据统计,全球铅消费量从2004年至2020年由7297万吨增加至11545万吨[1],增长了58.2%,其主要应用领域是铅蓄电池生产,是铅总消费量的80%左右[2]. 铅蓄电池的生产及回收过程使得铅以蒸气和烟尘形式逸散在空气中,并通过呼吸摄入方式进入人体肺部,进一步进入血液循环系统. 当血液中铅含量超过正常范围(400 mg·L−1)[3],可引起铅中毒.
铅中毒已被认为是重大的公共健康问题,尤其在发展中国家. 铅可以随着血液循环转移至身体全身的各个组织器官[4-5],引起神经系统、造血系统、肾脏系统、心血管系统、生殖系统以及骨骼系统等病变[6]. 大脑的中枢神经系统是铅毒性最敏感的器官,与神经退行性疾病(比如阿尔兹海默症)的发生密切相关[7]. 铅暴露对儿童的大脑发育系统具有更显著的毒性效应. 大量研究表明,儿童铅中毒可引起大脑发育障碍、认知能力和智商降低等症状,且这种毒性效应具有持久性甚至永久性[8-9]. 铅对生殖系统具有毒性损伤作用,对男性常见的危害包括降低精子运动性能、减少精子数量、损伤染色体,导致不育、前列腺功能异常和血清睾酮变化等,对女性的危害包括患不孕症、流产、胎膜早破、妊娠高血压和早产等[10]. 此外,孕期铅暴露可直接影响胎儿的正常发育,可能会改变大脑中髓鞘形成的时间,对儿童后期的学习或其他认知功能产生长期的有害影响[11-12]. 综上,铅中毒可严重危害人体健康.
驱铅治疗可以缓解铅中毒的损伤效应. 依地酸二钠钙(CaNa2EDTA)是目前常用的驱铅治疗药物,其原理是利用EDTA与铅离子发生络合反应,形成稳定的水溶性依地酸铅,从而通过肾脏系统随尿液排出[13]. 但是目前临床上铅中毒患者在治疗过程中铅的脱除效率较低,血液中仅部分铅通过尿液排出[14]. 此外,该方法存在血液中铅(血铅)浓度回升的现象,即在治疗初期血铅水平降低,但是一段时间后血铅浓度回升,导致治疗效果较差. 影响铅中毒患者治疗效果的因素来自多方面,可能包括用药因素和个体的生理状态等,具体的影响因素目前还有待研究.
本文以使用CaNa2EDTA进行驱铅治疗的铅中毒患者作为研究对象,分别在用药前、用药后24 h、用药后72 h对全血、血浆以及尿液中的铅含量进行检测,同时检测尿液的pH值,分析铅中毒患者在治疗过程中,血铅、血浆铅和尿铅的变化趋势以及他们的铅脱除率,进一步分析不同体系中铅脱除率与尿液pH值的相关性,进而探究驱铅治疗效果的影响因素.
-
本试验共收集24名来中国广东惠州市的铅中毒患者。患者在惠州市职业病防治院接受驱铅治疗,治疗方法为静脉注射CaNa2EDTA,同时口服补充维生素C,其中有3名患者接受了两次驱铅治疗. 分别在患者用药前24 h、用药后24 h、用药后72 h收集血液和尿液样品,共收集了81份. 以上实验由惠州市职业病防治院的伦理委员会批准,并且征得所有患者的同意. 铅中毒患者的信息见表1.
-
实验仪器:离心机(安徽中科中佳科学仪器有限公司);电感耦合等离子体质谱(ICP-MS 8800;安捷伦科技有限公司);石墨烯微波消解仪(JRY-X350;湖南金蓉园仪器设备有限公司);pH测量仪(SevenCompact;梅特勒托利多);超低温冰箱(赛默飞世尔科技公司).
试剂:65%浓硝酸(EMPARTA;德国Merck);30%过氧化氢(优级纯;国药集团化学试剂有限公司);铅标准溶液(Multi-element calibration standard 2A;美国安捷伦科技有限公司);内标溶液(ICP-MS Internal Std Mix;安捷伦科技有限公司).
-
血液样品:使用肝素钠抗凝管采集铅中毒患者的血液样品(2 mL),常温放置(25 ℃左右),6 h内进行以下前处理:取0.5 mL全血至1.5 mL离心管,用于全血中总铅含量检测,保存于-80 ℃冰箱;对剩余全血样品(1.5 mL)进行离心处理(500 r·min−1,10 min),转移上层血浆至新的1.5 mL离心管中,再次离心(3000 r·min−1,10 min),将上层血浆转移至新的1.5 mL离心管,保存于-80 ℃冰箱,用于血浆中铅含量的检测.
尿液样品:将收集的尿液样品转移至15 mL离心管中,直接冻存于-80 ℃冰箱,用于尿液中铅含量的检测和尿液pH值检测.
-
全血和血浆样本:取200 μL样品,加200 μL浓硝酸消解和60 μL 30%的过氧化氢,95 ℃加热2 h,冷却后使用去离子水定容至10 mL,使用滤网(0.22 μm)进行过滤处理,最后使用ICP-MS测定铅浓度.
尿液样本:使用含2%硝酸的水溶液直接将尿液样品稀释10倍,再使用滤网(0.22 μm)进行过滤处理,最后使用ICP-MS测定铅浓度.
样品检测过程中采用方法空白、空白加标、平行测试来控制测试质量,该方法测定铅浓度的回收率为111.5% ± 1.4%.
-
pH校准:使用3种pH标准缓冲溶液(pH4.01、pH7.00、pH9.21)依次进行pH校准,并保存校准结果. 校正不同标准缓冲溶液之间,使用去离子水冲洗电极,并用无尘纸吸取电极表面残留的水分.
尿液样品的pH测定:使用去离子水冲洗电极,使用无尘纸吸取电极表面残留的水分,将电极放入样品中,按读数键开始测量,在读数稳定后,记录数据;随后进行下一个样品的测定. 按照此方法测定所有尿液样品的pH值.
-
使用Excel(Microsoft Office 2016)进行数据处理和分析,使用Graphpad软件(GraphPad Prism 8)进行图表绘制,具有显著性差异结果的标准是P < 0.05.
-
铅中毒患者血铅含量较高,通过使用驱铅药物,可以促进血铅通过肾脏系统随尿液排出体外,从而降低人体血液系统中的铅含量. 本次铅中毒患者在治疗过程中,血铅浓度呈现明显的下降趋势(图1A). 铅中毒患者的血铅含量分布范围广,在用药前其浓度范围为276.00—727.29 μg·L−1;用药后24 h和72 h,血铅浓度范围分别为227.20—615.20 μg·L−1和188.00—510.40 μg·L−1. 从用药前至用药后24 h,血铅浓度显著降低(P < 0.01);从用药后24 h至用药后72 h,血铅浓度也显著下降(P < 0.001). 大部分病人的血浆铅浓度也具有明显的连续降低趋势,而少部分病人的血浆铅浓度先降低后升高(图1B). 在用药前、用药后24 h和用药后72 h,血浆铅的浓度范围分别为1.76—17.18 μg·L−1、1.10—9.84 μg·L−1和0.67—16.13 μg·L−1. 在驱铅治疗的不同阶段血浆铅的变化趋势不具备统一性,这可能与不同病人具有个体差异有关. 此外,血浆铅的浓度是血铅的1.2%左右,说明在血液系统中仅有少部分铅存在于血浆中,而大部分铅赋存于血细胞中,这与之前相关的研究结果是一致的[15]. 几乎所有铅中毒患者的尿铅浓度,在治疗过程中均呈现先升高后降低的趋势(图1C). 在用药前、用药后24 h和用药后72 h,尿铅的浓度范围分别为2.92—230.04 μg·L−1、17.68—1294.26 μg·L−1和10.13—454.91 μg·L−1. 从用药前至用药后24 h,尿铅浓度显著升高(P < 0.001),从用药后24 h至用药后72 h,尿铅浓度显著下降(P < 0.01),说明在用药后24 h,通过尿液排出体外的铅含量比在用药后72 h高. 这可能是因为在用药前期,CaNa2EDTA的浓度较高,其通过络合反应结合的铅的含量较高. CaNa2EDTA是乙二胺四酸(EDTA)的二钠钙盐,可以与多种金属结合生成稳定的可溶络合物. 上述结果显示使用CaNa2EDTA治疗铅中毒患者,血铅和血浆铅的浓度均逐渐降低,同时尿铅含量增加.
-
尽管用药具有一定的驱铅效果,但是不同病人的铅脱除率参差不齐,且在不同治疗阶段的铅脱除率不同. 本文定义铅脱除率为相对于用药前的铅浓度降低的百分比,定义铅排出比例为用药后尿铅浓度与用药前尿铅浓度的比值. 在用药后24 h和72 h,血铅中的脱除率分别为(15.92 ± 8.30)%和(30.02 ± 10.32)%,且具有显著差异(图2A). 在用药后24 h和72 h,血浆铅中的脱除率分别为(37.51 ± 39.27)%和(50.62 ± 37.36)%(图2B). 在用药后24 h和72 h,铅的排出比例分别为5.52 ± 2.82和2.01 ± 1.34,且具有显著差异(图2C). 上述结果显示,铅中毒患者治疗过程中,血液系统中铅的整体脱除效果较差,最高的平均脱除率只有50.62%. 铅脱除率可以直接反应铅中毒患者的治疗效果. 上述结果说明,铅中毒患者的驱铅治疗效果较差. 这可能是因为大部分铅中毒患者属于慢性铅中毒,超过90%铅赋存于骨骼系统或其他软组织中[16]. 慢性铅中毒患者在使用驱铅药物后,尽管血铅可以随尿液排出体外,但体内其他部位(骨骼、软组织)的铅可能又转移至血液系统中,从而导致血铅的脱除率较低. 因此,慢性铅中毒患者的驱铅治疗更具挑战性.
进一步研究发现,血铅和血浆铅的脱除率具有显著差异. 在用药后24 h,血浆铅的脱除率显著高于血铅,同样在用药后72 h,也存在类似的差别(图3). 说明血浆铅与EDTA的络合效率高于血细胞中铅. 血细胞中的铅主要与红细胞中的血红蛋白结合. 尽管EDTA与铅的络合物(结合常数为18.0)比血红蛋白与铅的络合物(结合常数为4.08)稳定[17],但是由于红细胞膜的空间隔离作用,EDTA较难与红细胞中的铅接触发生反应,而更易与血浆铅发生络合,因此,使得血浆铅的脱除率较高.
-
进一步研究显示,尿铅与血浆铅的相关性较强,而与血铅的相关性较弱(图4). 在用药后72 h,血浆铅和尿铅的皮尔逊相关系数高达0.88(P < 0.0001). 对比不同治疗阶段,用药后24 h和72 h的血浆铅与尿铅的相关性比用药前的强,这说明用药治疗促使血浆铅与尿铅的关联性增强. 用药后,EDTA主要与血浆铅发生络合反应,致使血浆铅优先通过尿液排出,因此血浆铅与尿铅的相关性增强. 已有研究显示,血铅和血浆铅之间以及血铅和尿铅之间均呈对数关系[18],这也说明血浆铅与尿铅具有线性相关性. 尽管血浆铅含量较低,但是其流动性和健康危害更大,可以穿过血脑屏障进入大脑组织,也可能穿过胎盘屏障进入胎儿体内. 研究表明,在母体血铅水平升高的情况下,发育中的胎儿可能因母体血浆铅升高而暴露于铅的风险高于全血铅水平所预测的风险[19]. 上述结果说明血浆铅可能是尿铅的直接来源,血浆铅的含量及变化是影响铅中毒患者治疗效果的重要因素.
血浆铅可以与其他组织器官直接进行物质交换,通过环境暴露从肺部或肠道吸收的铅,首先被吸收进入血浆,进一步可能被血细胞吸收,可能直接从血浆转移至骨骼、肝脏、脾脏或大脑,也可能进入肾脏,通过尿液排出体外. 同时,对于长期铅暴露人群,铅可能从骨骼或其他组织转移至血浆,成为血浆铅[20]. 血浆铅就如一个交换池,血浆铅的易变性也直接影响了铅中毒患者的治疗效果,比如急性铅暴露人群的血浆铅在较短时间内迅速升高,而后逐渐转移至骨骼或其他组织器官,因此对于急性暴露人群的治疗,越快接受治疗,其铅脱除效率可能越好. 铅在血浆和血细胞中的分配平衡可以显著影响血浆铅的含量及其变化. 由于血铅和血浆铅呈现对数关系,因此,当血铅含量较低时,血浆铅随血铅的升高而增加;当血铅含量较高时,血浆铅随着血铅的升高而迅速增加[18],这是因为血细胞中铅的结合接近或处于饱和状态,增加的血铅几乎全部分配于血浆中. 由此,可以推断当血铅含量超过血细胞的铅饱和状态时,铅中毒患者的治疗效果较好.
-
溶液pH是影响EDTA与Pb络合的重要参数. 尿液pH值也是重要的生物化学指标,反映肾脏调节体液酸碱平衡的能力,其变化与多种因素有关,包括每日摄入的食物酸碱性、糖尿病、肾炎等. 实验进一步测定了尿液的pH值,探究了尿液pH与铅中毒患者治疗效果的关系. 研究结果显示,尿液pH值与血铅的脱除率具有关联性,用药后24 h,血铅脱除率与pH值具有显著正相关(r = 0.53,P < 0.05)(图5),说明尿液pH增加可以促进血液中的铅排出体外. 已有较多研究工作探究了土壤pH对重金属的吸附以及生物利用性的影响,一般来说,随着pH值的增加,铅在土壤中的吸附量均呈现明显的上升趋势[21]. 在溶液的酸碱度呈碱性时,铅离子形成氢氧化铅沉淀[22]. 但目前关于人体体液pH值与铅含量变化的研究较少,pH值是影响人体生物化学反应的重要参数,也是评估人体健康的重要指标,因此pH值对铅脱除的影响值得更多的关注和研究.
驱铅治疗的效果与多种因素有关[23-24]. 血铅可以与其他组织器官发生铅交换. 当体内生理状态发生改变时,比如怀孕、骨骼中酸碱环境变化等,铅可能从骨骼中释放,转移至血液系统或其他软组织器官中[25]. 此外,CaNa2EDTA与铅的络合反应与多种生理因素有关,比如血液的酸碱度、血脂含量、其他二价金属含量等. 当血液中酸碱度发生变化,EDTA与铅的络合反应效率会发生变化;当其他二价金属含量较高时,也会与EDTA发生络合反应,从而降低了铅脱除率.
-
铅是广泛分布于环境中的有毒重金属,长期铅暴露可导致铅中毒. 本文对铅中毒患者治疗过程中的血铅、血浆铅、尿铅以及尿液pH值进行测定,分析铅浓度的变化趋势以及铅的脱除效率,进一步探究了铅中毒患者治疗效果的影响因素. 本次铅中毒患者在治疗过程中(用药前、用药后24 h、用药后72 h),血铅浓度呈现明显的下降趋势;大部分病人的血浆铅浓度也具有明显的连续降低趋势,而少部分血浆铅浓度先降低后升高;尿铅浓度呈现先升高后降低的趋势. 血液中铅的脱除率整体较低,不超过50%,血浆铅的脱除率显著高于血铅的脱除率. 血浆铅和尿液酸碱度是是影响铅中毒病人驱铅治疗效果的重要因素. 研究发现,尿铅与血浆铅的相关性较强,而与血铅的相关性较弱. 在用药后72 h,血浆铅和尿铅的皮尔逊相关系数高达0.88(P < 0.0001). 尿液pH值与血铅的脱除率具有关联性. 在用药后24 h,血铅脱除率与pH值具有显著正相关(r = 0.53,P < 0.05). 本研究可为提高铅中毒病人的驱铅治疗效果提供科学依据.
铅中毒患者驱铅治疗效果的影响因素
Factors on the effect of lead-elimination treatment in lead-poisoned patients
-
摘要: 铅是有毒的环境污染物,铅中毒严重危害人体健康. 静脉注射依地酸二钠钙(CaNa2EDTA)是目前临床上最常使用的驱铅治疗方法,但其治疗效果不令人满意,治疗效果的影响因素有待进一步探究. 本文以铅中毒患者作为研究对象,使用CaNa2EDTA进行驱铅治疗,分别在用药前、用药后24 h以及用药后72 h测定全血、血浆和尿液中的铅含量以及尿液的酸碱度(pH值),分析了铅中毒患者在驱铅治疗过程中,血铅、血浆铅和尿铅的变化趋势和铅脱除率,并进一步探究了影响铅脱除率的因素. 实验结果显示,随着用药治疗,血铅和血浆铅逐渐降低,同时尿铅升高;血浆铅的脱除率显著高于血铅,说明血浆铅与EDTA的络合效率高于血细胞中铅;尿铅与血浆铅呈正相关关系,而与血铅的相关性较弱,说明血浆铅是影响驱铅治疗效果的重要因素;用药后24 h,尿液pH值与血铅脱除率呈显著正相关关系,说明尿液酸碱度可能是影响驱铅治疗效果的重要参数. 本研究为提高铅中毒患者驱铅治疗效果提供了重要依据.Abstract: Lead (Pb) is a toxic environmental pollutant, and Pb poisoning seriously threatens human health. The most commonly used Pb-eliminating treatment in clinical practice is intravenous injection of calcium disodium edetate (CaNa2EDTA). However, the therapeutic effect is unsatisfactory, and the factors that influence it need to be investigated further. Pb-poisoned patients were used as research subjects in this paper and CaNa2EDTA was used in the Pb-eliminating treatment. The Pb concentration in whole blood, plasma, urine, and urine pH were all measured before medication, as well as 24 and 72 hours later. The changing trend of blood Pb, plasma Pb, urine Pb, and their Pb elimination rate during Pb-eliminating treatment and the factors influencing Pb elimination rate were investigated. The results showed that the elimination rate of plasma Pb was significantly higher than that of blood Pb, indicating that the complexation efficiency of plasma Pb and EDTA was higher than that of Pb in blood cells. Urine Pb was found to be positively related to plasma Pb, whereas the correlation between urine Pb and blood Pb was weak, indicating that plasma Pb is an important factor influencing the treatment effect of Pb elimination. The urine pH was found to be positively correlated with the blood Pb elimination rate 24 hours after medication, indicating that urine pH could be an important factor influencing the therapeutic effect of Pb elimination. This study provides an important basis for improving the therapeutic effect of Pb poisoning.
-
Key words:
- Pb poisoning /
- treatment effect /
- factors /
- plasma Pb /
- blood Pb /
- urine Pb /
- pH.
-
海洋中的溶解氧对于维持海洋生物的生命活动及海洋生态系统的稳定具有重要作用[1-4]. 海洋溶解氧的收支受物理、生物和化学等过程的共同影响,且这些过程所造成的氧同位素分馏效应是不同的(图1)[5-8]. 海洋溶解氧的主要来源为海洋表层的大气溶解氧和浮游植物光合作用产氧. 当混合层中的氧气达到溶解平衡后,其氧同位素组成(δ18O)大约为+0.7‰,而光合作用产生的氧气其δ18O与源海水相同,即δ18O约为-23.5‰[7]. 大洋混合层中溶解氧的δ18O为二者的混合值,δ18O越低指征该海域生产力越高[9]. 河口或受冰川融水影响的海域海水的δ18O值偏低,如受长江冲淡水影响的长江口及其毗邻海域[10]. 当夏季海洋表层的生产力较高时,长江口海洋溶解氧的δ18O值可能会低至-5‰[11]. 氧气的消耗过程主要为生物呼吸作用耗氧. 由于生物在呼吸过程中倾向于消耗较轻的氧同位素(16O),使得水体中剩余的溶解氧更富集较重的氧同位素(18O). 因此,随着呼吸作用的进行,海洋溶解氧浓度降低,且剩余溶解氧的δ18O升高. 海洋中的呼吸耗氧遵循封闭系统瑞利分馏的原则,与氧浓度呈现非线性的变化关系(图1)[12]. 不管是在开阔大洋还是在河口海岸的陆架区域,生物呼吸作用不仅能发生在水柱中(water column respiration, WCR),也发生在底层沉积物中(sediment oxygen respiration, SOR)[13],且这两种呼吸作用显示出不同的氧同位素分馏效应(图1). 观测到的海洋浮游生物WCR氧同位素分馏系数范围为-26‰至-14‰[14],而培养实验得到的SOR氧同位素分馏系数为-5‰至-1‰[15]. 因此,两种呼吸过程会造成迥异的溶解氧δ18O演化趋势,且普遍认为SOR对于水体中溶解氧的氧同位素分馏效应较小. 利用这个特征,前人区分及量化分析了海洋耗氧过程中水柱呼吸与沉积物呼吸的占比[11,16]. 另外,洋流导致的物理输运,如平流、扩散等,也会影响海洋溶解氧浓度和同位素组成. 但物理作用引起的水团混合对δ18O和氧浓度的影响是线性的,与呼吸作用不同(图1). 基于溶解氧同位素在氧循环各过程中的特性,尤其是在不同耗氧过程中分馏效应的差异,使得其成为研究海洋溶解氧收支平衡的重要指标之一[6].
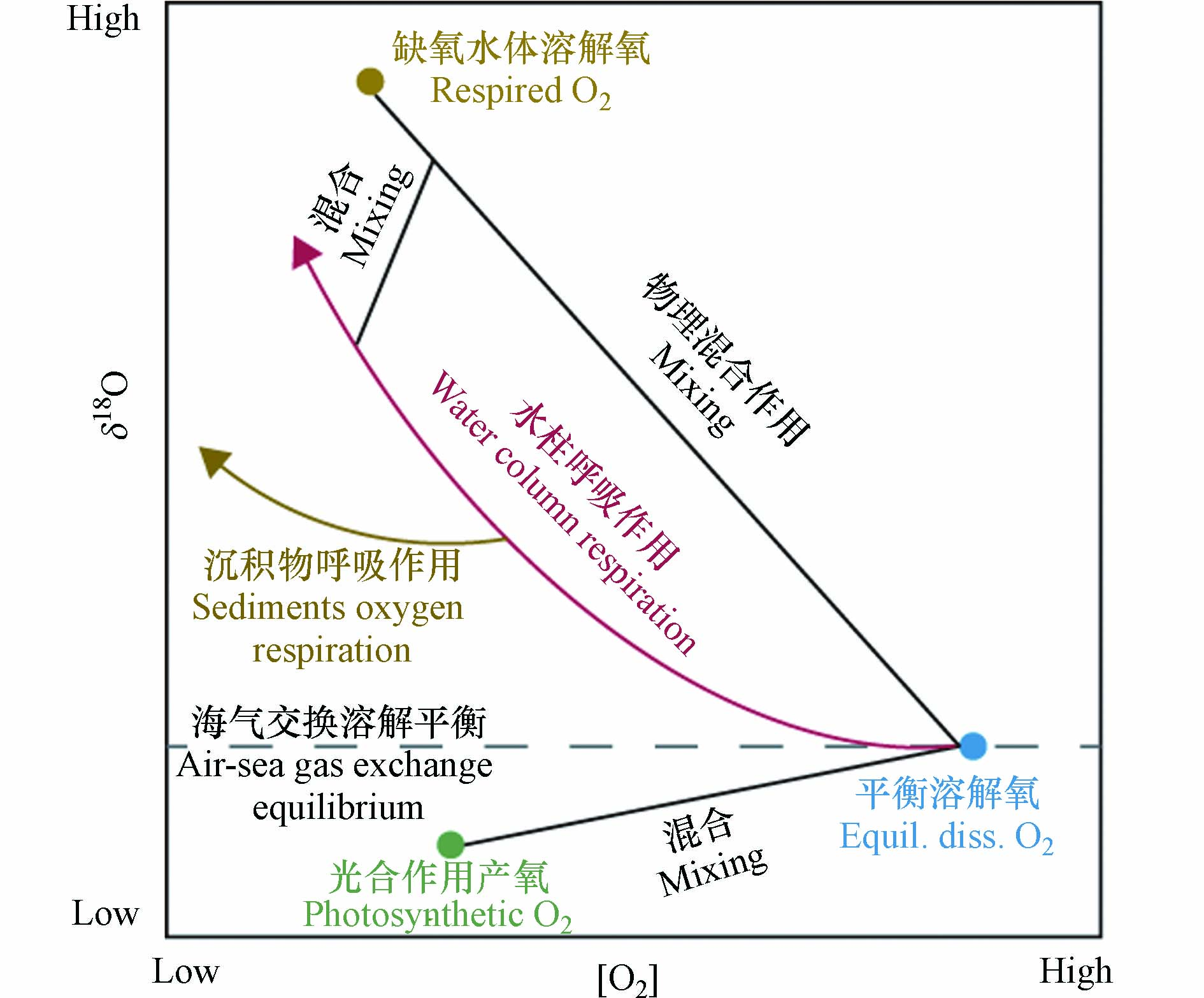 图 1 海洋氧循环过程中的同位素效应示意图Figure 1. Schematic diagram of isotopic fractionations during marine oxygen cycling
图 1 海洋氧循环过程中的同位素效应示意图Figure 1. Schematic diagram of isotopic fractionations during marine oxygen cycling由于人类活动的影响,河口海岸区域的营养盐输入激增,导致长江口及其周边海域富营养化,引发海洋缺氧事件,且近年来观测到的缺氧区面积也在逐渐扩大[17-20]. 长江口缺氧区的形成机制复杂,海水的层化、海底地形及富营养化引起的耗氧过程等都会加剧底层海水的缺氧程度[21-24]. 由于长江口海域的整体水深较浅,底层沉积物中富含海洋自生及陆源输入的有机质[25],底层水受沉积物再悬浮的影响也会造成额外的有机质分解耗氧[26]. 因此,生物通过呼吸作用降解有机物消耗氧气的过程不仅发生在水柱中,沉积物中的生物呼吸也是不容忽视的过程[27]. 探究WCR和SOR对长江口缺氧的贡献,对于探讨该海域缺氧区的形成机制具有重要意义.
测量海洋溶解氧的同位素组成需要较为严格的采样、提取和纯化条件,具有一定的难度. 利用溶解氧同位素探究海洋氧循环的研究多集中于美国东西岸及墨西哥湾等海域[28-30],并没有在各大海域得到广泛的应用.
本文详细描述了海洋溶解氧的采集、实验室提取和纯化及呼吸作用暗培养的方法,并阐述了氧同位素的质谱仪测量和数据校正的具体过程. 同时,利用2020年于长江口采集的混合层海水和底层沉积物,分别进行了暗培养实验,确定了长江口特定的WCR和SOR的氧同位素分馏系数,并估算了水柱呼吸和沉积物呼吸在总耗氧量中的占比,分析了长江口缺氧区占主导的耗氧机制.
1. 实验部分(Experimental section)
1.1 氧同位素的定义及公式
自然界中氧的稳定同位素有16O、17O、18O,丰度分别为99.76%、0.04%和0.20%[31]. 氧同位素组成及氧气氩气之间的气体比值通常用相对丰度δ值(‰)来表示.
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) 其中,18R = 18O/16O,即18O的丰度与16O丰度的比值,O2/Ar指的是气体中氧气与氩气的含量比,下标sam和std分别代表样品和标样,δ值的单位为千分比(‰). 本研究以空气为标样,即所有样品的δ18O和δO2/Ar值都是相对于空气相应值的偏差. 因此根据定义空气本身的δ18O和δO2/Ar值为0‰.
微生物在呼吸作用中倾向于先消耗轻的氧同位素(16O),使剩余的溶解氧中富集较重的氧同位素(18O). 呼吸作用过程中造成的氧同位素分馏程度用分馏系数α或ε来表示 [12,32],定义如下:
stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) 其中,respired代表生物呼吸作用消耗的氧气,residual代表剩余的氧气. 呼吸作用的α值通常为略小于1的数值. 对于海洋生物来说,其观测的范围为0.974–0.986,对应的ε值为-26‰至-14‰[12,14],ε是α的千分比形式(‰). 海洋生物的呼吸作用遵循封闭系统瑞利分馏的原则,即[33]:
stringUtils.convertMath(!{formula.content}) (5) 其中,residual代表呼吸作用之后剩余的溶解氧,initial代表初始的溶解氧,f是呼吸作用后剩余溶解氧占初始溶解氧的比例,即
[O2]/[O2]0 stringUtils.convertMath(!{formula.content}) (6) 结合公式(1)和(4)及
103ln(1+X)≈X stringUtils.convertMath(!{formula.content}) (7) 通过观测呼吸作用过程中不同时间节点的氧浓度和δ18O值,利用δ18O和
ln([O2]/[O2]0) 1.2 溶解氧的采集方法
大气中氧气的δ18O(0‰)与溶解氧的δ18O截然不同,因此溶解氧采集过程中需要严格防止大气污染. 为保证采集过程中的真空条件,制作了带有真空阀门(LouwersHanique®)的容积为500 mL的样品瓶(图2a). 采样前,清洗过后的样品瓶需根据采集海水的量加入氯化汞(HgCl2)饱和溶液. 最终采集的海水样品中氯化汞浓度需大于2×10−5 kg∙L−1,以确保氯化汞阻断所有生命活动,达到固定海水溶解氧的目的. 加完氯化汞的样品瓶需在低温(< 40°C)、通风的条件下缓慢烘干,待饱和氯化汞溶液凝结为晶体后,再将样品瓶移至实验室真空管线上抽真空(图3). 当样品瓶中的压力小于10−3 Pa后,关闭瓶上方的真空阀门,并在阀门支管中注入蒸馏水、盖上橡胶帽. 阀门支管内的水可辅助真空阀门的O型圈保持样品瓶的真空度. 样品瓶在取样前后都应将阀门支管内注满水. 如此,样品瓶的真空度最长可保持6个月左右.
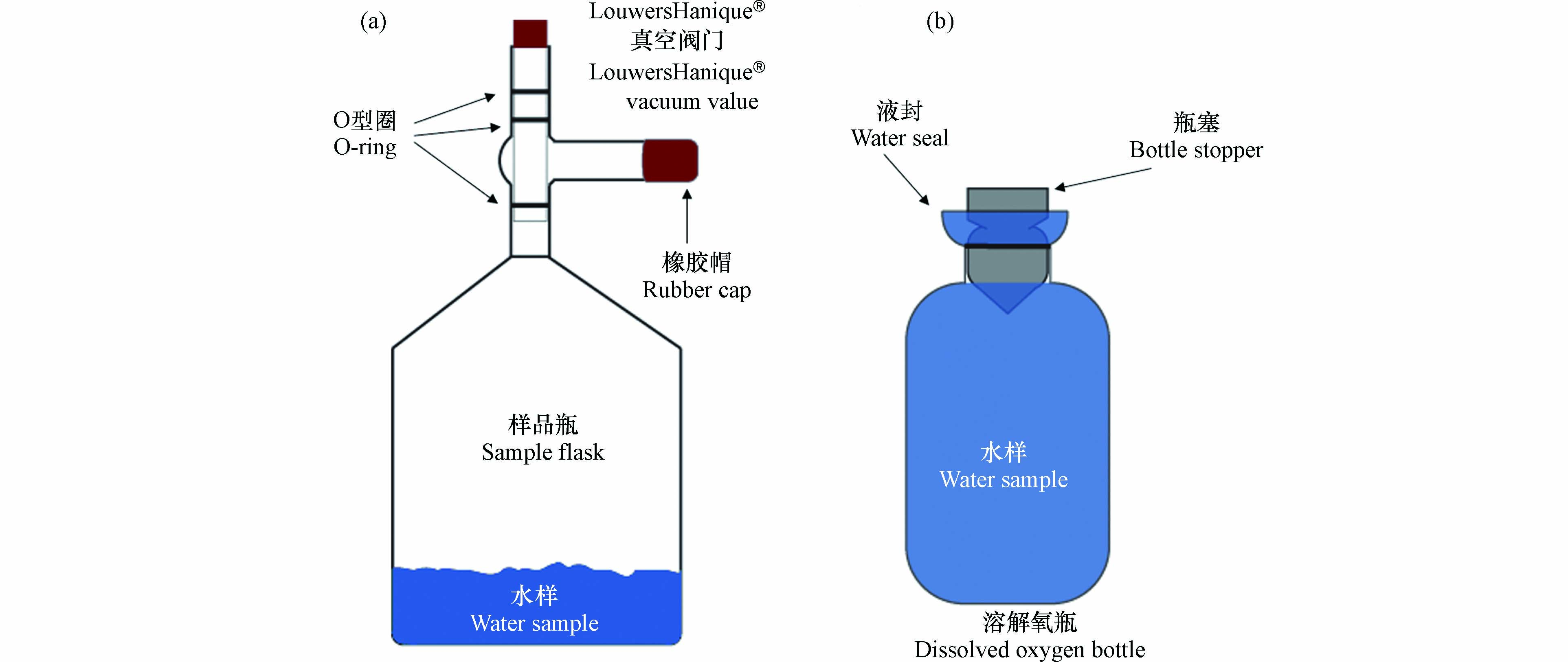 图 2 采集海洋溶解氧(a)的真空样品瓶和实验室呼吸作用暗培养所需的溶氧瓶(b).Figure 2. Vacuum sample flask (a) for dissolved O2 collection, and dissolved oxygen bottle for dark incubation experiment (b).
图 2 采集海洋溶解氧(a)的真空样品瓶和实验室呼吸作用暗培养所需的溶氧瓶(b).Figure 2. Vacuum sample flask (a) for dissolved O2 collection, and dissolved oxygen bottle for dark incubation experiment (b).采集不同深度的原位海水需借助于船载的温度盐度探测仪及海水收集装置(Sea-bird Costal Hydrocat, CTD). 为防止大气污染,海水溶解氧样品需第一个于CTD上采样. 利用橡胶软管将CTD中的水引入样品瓶的阀门支管内,冲洗5–10 s,直至支管内原先注满的蒸馏水被替换为海水. 之后,在保证支管内有水溢出的状态下缓慢打开真空阀门至最下方的O型圈解除密封,海水样品会沿阀门下方的管壁进入真空瓶内. 此时可以观察到大量的气泡从海水中溢出,进入到样品瓶的顶空中. 在质谱仪上测量溶解氧δ18O需约2×10−5 mol O2,因此一般需采集约100 mL的海水. 但对于缺氧区等溶解氧含量极低的海水,可根据其溶解氧浓度适当增加采集的海水量,确保溶解气体中的O2达到质谱仪的检测限. 但总的来说,采集的海水体积最好不超过样品瓶体积的50%,此时残留在溶解态中的气体占比小于2%,对顶空中气态氧气δ18O值的影响可以忽略. 如果样品瓶顶空体积过小,导致其气压过高,将会阻碍溶解气体从海水中的释放. 此时,需要根据N2、O2、Ar的溶解度和分压对气态和溶解态的气体同位素组成及气体比值进行校正.
1.3 呼吸作用暗培养实验
为确定WCR和SOR的氧同位素分馏系数,本研究在实验室中利用海水和海洋沉积物进行了生物呼吸作用暗培养实验. 依托2020年夏季长江口共享航次(NORC2020-03),本研究于“浙渔科2”号科考船上通过CTD和箱式采泥器分别采集了原位的混合层海水和底层沉积物(122.56°E,29.4472°N)用于实验室暗培养.
1.3.1 海水呼吸作用暗培养
根据公式(7)可知,计算呼吸作用分馏系数ε需要初始、及不同时间节点的氧浓度和δ18O值,以得到δ18O–
ln([O2]/[O2]0) 为获得准确度较高的δ18O–
ln([O2]/[O2]0) 1.3.2 沉积物呼吸作用培养实验
为得到纯粹的沉积物呼吸作用分馏系数,排除海水中微生物的影响,沉积物培养的溶液为实验室配制的人工海水. 人工海水的盐度是35.00‰,其组成为每升Milli-Q水中加入35g NaCl和0.5 g NaHCO3. 配制后的人工海水需加入磁力转子,在磁力搅拌器的作用下与大气平衡约24 h,使人工海水中的溶解气体与大气达到溶解平衡.
沉积物培养实验的流程与海水培养实验大致相同,具体操作为:于溶解氧瓶中加入5 g沉积物,再加入溶解平衡后的人工海水并注满,塞上磨口玻璃塞并液封保存. 当培养达到目标时间后将水样取至真空瓶中保存,供后续同位素测量. 需特别说明的是,沉积物培养实验的t = 0样品为达到溶解平衡的人工海水,此时的溶解气体未受到任何沉积物呼吸的影响,可视作溶解氧的初始状态. 由于沉积物中的生物含量及有机质含量较高,因此沉积物培养实验的呼吸速率比海水培养的呼吸速率快. 沉积物呼吸暗培养的水样分别在培养开始后的第1天、第2天、第3天取样完成.
1.4 溶解氧的提取和纯化
溶解氧的提取与纯化方法参照了气体溶解平衡取样法[34]. 首先,取样完成后的真空样品瓶需在摇床上(2 r·s−1)摇晃至少48 h,使顶空中的气体与残留在液态中的溶解气体达到同位素平衡. 之后,将样品瓶连接到图3a所示的溶解氧提取线路上. 打开真空泵(Pfeiffer Vacuum Duo 3),抽真空5 min左右. 然后缓慢打开样品瓶上的阀门,样品瓶中的水会在内外压力差的作用下被排出到融水收集瓶内. 注意,排水至最终阶段时需留存约1 mL的液体没过阀门最下端的O型圈,此时快速关闭阀门,以防止瓶内的气体被真空泵抽走.
排水后的样品瓶内即为需要纯化和收集的溶解气体. 将样品瓶连接到溶解氧纯化线路上(图3b),瓶身底部没入-30°C的乙醇浴中,将剩余在样品瓶中的水全部冻结. 在真空管线的另一端接入样品收集阀,收集阀中填充有干燥并去气的硅胶颗粒(45–60 mesh, Sigma). 打开真空管线上所有的阀门使样品瓶与真空泵之间形成通路并抽真空,最终管线的真空度需达到10−3 Pa. 之后,关闭图3b中的V1, V2阀门,使管线与真空泵隔绝,并将两个冷肼及样品收集阀都没入到液氮(-196°C)中. 此时打开样品瓶上方的真空阀门,使溶解气体扩散至真空管线内. 溶解气体中的水蒸气、二氧化碳会经由两个液氮冷肼去除,纯化后的气体为氧气、氮气和氩气,最终被收集于样品阀中的硅胶颗粒上,整个过程持续约45 min.
1.5 溶解氧的同位素测量和校正
溶解气体的同位素及气体比值是在双路进样的气体同位素质谱仪(Thermo Fisher Delta V Plus)上进行的. 将样品收集阀连接到质谱仪双路进样系统的样品气缸侧,加热硅胶10min使气体充分混合. 之后打开样品收集阀,纯化后的氧气、氮气、氩气即扩散进入质谱仪的样品气缸. 参考气侧为氮气、氧气、氩气占比(N2:O2:Ar)分别为63.93%、34.35%、1.72%的混合气体(纯度均为99.999%). 该气体比值与平衡溶解气体的N2:O2:Ar比值接近,会减小因参考气与样品之间气体比值不同而对氧同位素值造成的质量干扰. 开始同位素测量后,设置质谱仪的测定时间为26 s, 循环15次为一次测量,共测定3次,总耗时约45 min. 质谱仪配备有质量数为28、29、 32、 34、40的法拉第杯,因此可同时测定溶解气体的δ15N、δ18O、δO2/Ar的组成. 由同位素质谱仪得到的初始数据为相对于参考气的同位素及气体比值的数据(
δvs.ref 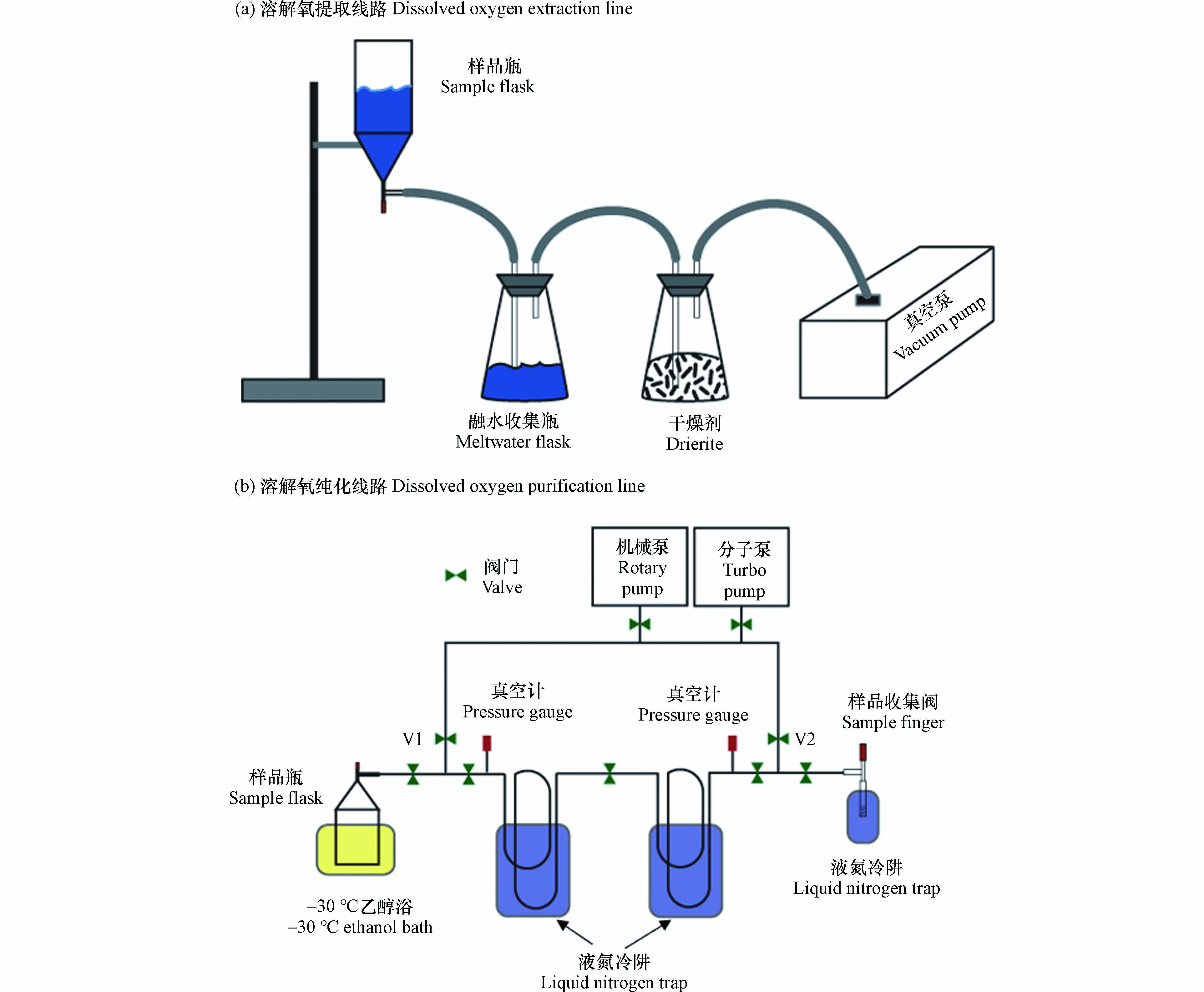 图 3 溶解氧的提取(a)和纯化(b)线路.Figure 3. Dissolved oxygen extraction line (a) and purification line (b).
图 3 溶解氧的提取(a)和纯化(b)线路.Figure 3. Dissolved oxygen extraction line (a) and purification line (b).高精度高准确度的同位素及气体比值数据需遵循严格的同位素校正流程. 具体需要四个步骤:零点校正、质量干扰校正、空气标样校正和第二标样对比验证[35].
(1)零点校正(zero enrichment):在样品气缸和参考气气缸中都引入参考气,对比测量. 若两侧的气缸及毛细管流速都能够在长时间测量中保持平衡,且没有明显的偏移,则最终零点校正的δ18O, δO2/Ar平均值都应接近于0. 在2020.06 —2020.10和2020.10—2021.03两个测量周期(每根离子源灯丝的寿命期为一个测量周期)共做了79次零点校正测量,观测到的δ18O, δO2/Ar均接近于0(表1). 之后,将所有样品的δ18O和δO2/Ar值都减掉零点的δ18O和δO2/Ar平均值,即完成零点校正. 如果长时间、多次观测到偏离于零点较大的δ18O, δO2/Ar值,应及时检查仪器状态是否正常.
表 1 氧同位素测量校正参数及标样精度Table 1. Isotopic correction parameters and precisions2020.06 — 2020.10 2020.10—2021.03 零点校正aZero enrichment Nd 66 13 δ18O/‰ -0.005 -0.005 δO2/Ar/‰ +0.018 -0.011 质量干扰斜率Mass interference slope δ18O – δΝ2/O2 −5.514×10−5 −2.112×10−3 δO2/Ar – δΝ2/O2 −7.958×10−3 −1.341×10−2 空气标样精度bPrecision of air standards Nd 60 54 δ18O /‰ ±0.035 ±0.035 δO2/Ar /‰ ±0.4 ±0.5 平衡溶解气标样cDissolved gases Nd 7 5 δ18O /‰ 0.753 0.846 δO2/Ar /‰ -92.7 -90.8 a测量周期中所有零点校正的平均值;b空气标样精度为实验室测量的外部精度(1σ);c 实验室所制的平衡溶解气的氧同位素及气体比值平均值;d N代表标样数量 a Mean values of zero enrichments during the same analytical session; b External precisions (1σ) are established on air standards; c Mean values of lab equilibrated dissolved gases; d N represents numbers of analyses | Show Table DownLoad:
CSV
DownLoad:
CSV
(2)质量干扰校正(mass interference correction or “chemical slope” correction):由于样品的N2:O2:Ar比会随着氧气组分的不同而改变,因此会与参考气的N2:O2:Ar产生差异. 样品与参考气气体组分的差异会引起质量干扰(mass interference),观测的结果表明氮气越多,实测的δ18O值越低(表1). 因此,在每个测量周期中,都应该首先确定质量干扰的斜率. 具体操作方式为,两侧都加入参考气,并在样品气缸中少量多次加入高纯氮气,改变样品侧的N2:O2:Ar比值,并测定每个节点δΝ2/O2值对应的δ18O和δO2/Ar值,即可分别得到δ18O – δΝ2/O2和δO2/Ar – δΝ2/O2的质量干扰斜率(m). 校正公式为:
stringUtils.convertMath(!{formula.content}) (8) 其中
δMI−corr δZE−corr (3)空气标样校正(correction versus air standards):由于各实验室所用的参考气组成各不相同,因此同位素及气体比值数据需相对于同期测定的国际标样空气的数据进行标准化. 即,将公式(1), (2)中的分母由参考气的18R值替换为空气的18R值. 由于空气的存留时间远大于其大气混合时间,因此全球大气是混合均匀的,其氧同位素及气体比值组成也具有全球均一性. 在本研究测样过程中,每天都会于实验室外采集空气,在真空管线上干燥、纯化,并在质谱仪上测量其δ18O和δO2/Ar值. 在2020.06 —020.10和2020.10—2021.03两个测量周期共分别测量了60次和54次空气标样,δ18O和δO2/Ar的测量精度如表1所示,均与国际上同等的气体氧同位素实验室的精度达到一致[36-38]. 观测到的空气标样精度即为本实验室同位素测量的外部精度(external precision). 之后,需要将质谱仪上得到的相对于参考气的样品数据转换为相对于空气标样的数据,公式为:
stringUtils.convertMath(!{formula.content}) (9) 其中
δMI-corr δair δvs.air (4)第二标样对比验证:本研究以实验室制作的平衡溶解气体为第二标样,验证了溶解氧提取、纯化方法及同位素矫正流程的准确性. 取1 L 去离子水,加入400 μL饱和氯化汞溶液阻断生命活动. 在磁力搅拌器的作用下使溶解气与大气在室温(22°C)平衡至少24 h,注意磁力搅拌器转速不能过快,以防造成溶液的过饱和状态. 后续取样和提取纯化过程与“1.3”和“1.4”中描述的类似,即利用软管和虹吸作用将水取至真空样品瓶中,并于真空管线上完成提取和纯化,最终于质谱仪上测量. 基于本方法测得的平衡溶解气的δ18O和δO2/Ar值如表1所示,与22 °C下的理论值0.73‰和-93‰接近[39-41],证明本研究所述的溶解氧同位素及气体比值的测定方法是准确可靠的.
2. 结果与讨论 (Results and discussion)
2.1 呼吸作用氧同位素分馏系数
通过对长江口采集的混合层海水和底层沉积物进行呼吸作用暗培养,分别测定了长江口水柱呼吸作用(WCR)和沉积物呼吸作用(SOR)的氧同位素分馏系数
ϵWCR ϵSOR ln([O2]/[O2]0) 表 2 培养实验取样时间及同位素数据Table 2. Sampling information and isotopic data of dark incubation experiments采样时间Sampling time δ18O / ‰ δO2/Ar / ‰ 溶解氧浓度/ (μmol∙kg−1)Dissolved oxygen concentration 混合层海水呼吸作用暗培养 2020.08.06a −3.858 29.9 259.4 2020.08.18 7.474 −401.4 150.8 2020.08.24 10.638 −484.7 129.8 2020.09.07 11.768 −514.0 122.4 沉积物呼吸作用暗培养 2020.09.15a 0.835 −125.9 210.1 2020.09.16 2.848 −293.0 169.9 2020.09.17 4.547 −406.3 142.7 2020.09.17 4.810 −427.3 137.7 2020.09.18 5.529 −490.9 122.4 2020.09.18 6.488 −530.8 112.8 a 该组培养实验的t0样品. a Initial incubation samples (t0) | Show TableDownLoad:
CSV
各时间节点的溶解氧浓度可通过对应的δO2/Ar值计算得到. 氩气与氧气的物理性质较为相似,比如溶解度、扩散速率、对温度的敏感度等[42]. 但是Ar是惰性气体,海洋中Ar的含量主要受物理因素如压力变化、海水温度变化和气泡注入等影响,而不受生物过程的影响[43]. 在整个培养实验过程中,温盐条件已知,Ar浓度保持不变,即为其饱和溶解度. 因此,可以通过海水的温度和盐度,精确计算出Ar的浓度,再根据O2/Ar比值,计算得到海水中氧气的浓度. Ar浓度(μmol∙kg−1)计算公式为[39]:
stringUtils.convertMath(!{formula.content}) (10) 其中,
TS=ln(298.15−t273.15+t) A0 A1 A2 A3 B0 B1 B2 stringUtils.convertMath(!{formula.content}) (11) 最后,通过对δ18O–
ln([O2]/[O2]0) ϵWCR ϵSOR αWCR αSOR αWCR αSOR=√αWCR=0.9894 αSOR= 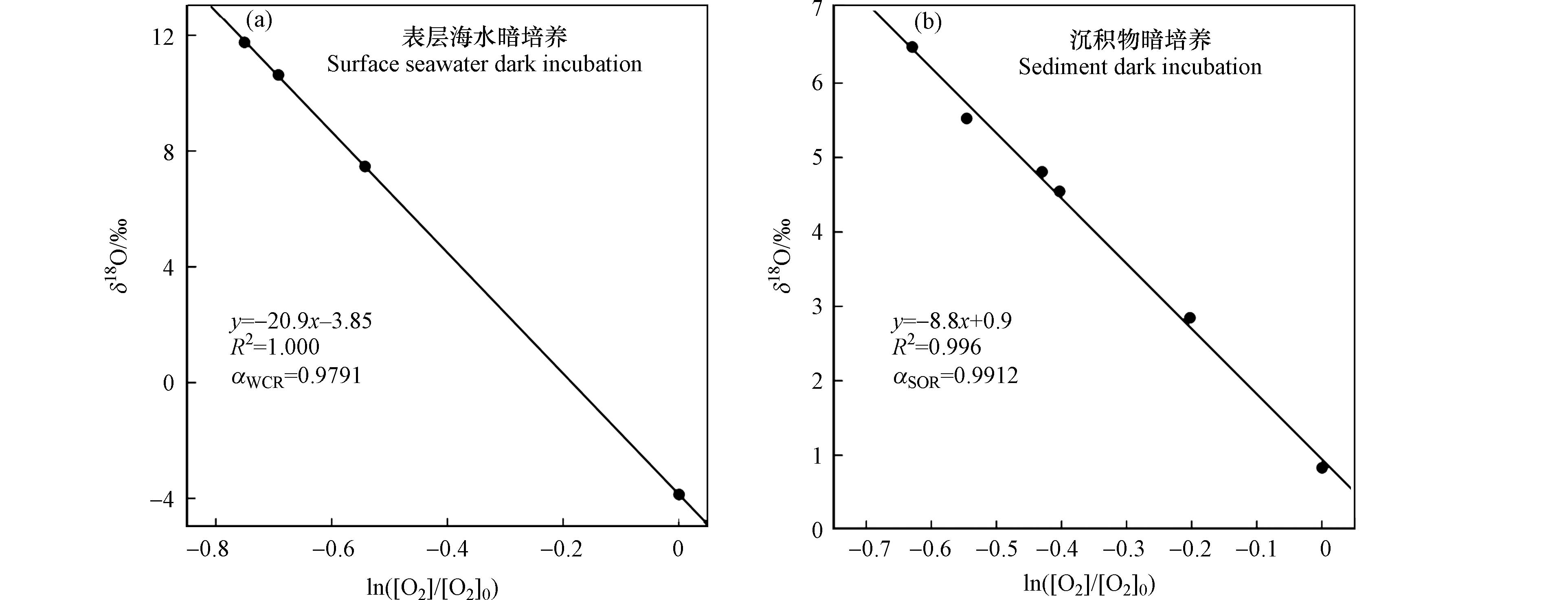 图 4 水柱呼吸和沉积物呼吸的氧同位素分馏系数Figure 4. Oxygen isotope fractionation factor of water column respiration and sediment oxygen respiration
图 4 水柱呼吸和沉积物呼吸的氧同位素分馏系数Figure 4. Oxygen isotope fractionation factor of water column respiration and sediment oxygen respiration综上,本实验观测到的
αWCR αSOR 2.2 利用氧同位素分馏系数量化分析耗氧过程
过去对大洋溶解氧同位素的研究表明,大洋当中的氧同位素与氧浓度之间的表观分馏系数往往大于0.980,有时甚至接近或高于纯水柱呼吸作用氧同位素分馏系数的最高值. 这是由于大洋中的溶解氧同时还受到物理混合及沉积物呼吸作用的影响. 对于长江口沿岸,水体的缺氧往往伴随着表层的水华作用. 大量的有机质由海洋表层沉降至底层,水柱和沉积物中的海洋生物通过呼吸作用快速地分解有机物消耗氧气,造成水体缺氧. 若以混合层溶解氧的δ18O或δO2/Ar为初始值,以底层溶解氧的δ18O或δO2/Ar为呼吸消耗后的值,即可计算出特定站位由表层至底层溶解氧同位素变化的表观分馏系数
αAPP stringUtils.convertMath(!{formula.content}) (12) fWCR fSOR fWCR+fSOR=1 stringUtils.convertMath(!{formula.content}) (13) 以Zhou 等[22]汇报的F6站位(126.00°E,30.60°N)为例,其底层海水的δ18O值分别为6.834‰,由氧氩比计算的氧浓度为160 μmol∙L−1. 假设该底层海水的初始溶解氧来源为等密度层海水在海洋混合层与大气达到平衡时的氧气,其δ18O值和氧浓度分别为0.7‰和264 μmol∙L−1. 根据混合层和底层的δ18O与相应的溶解氧饱和度的对数[
ln([O2]/[O2]0) ϵAPP= αAPP= αWCR αSOR 3. 结论(Conclusion)
本研究详细描述了海洋溶解氧的提取、纯化和同位素测量的流程. 介绍了氧同位素及气体比值的具体校正方式,并用实验室制备的第二标样验证了方法的准确性和可靠性. 通过对海水和沉积物进行暗培养,本研究测量出长江口特定的水柱呼吸和沉积物呼吸的分馏系数别为-20.9‰和-8.8‰. 二者之间的差异可被用作衡量这两种耗氧过程的端元值,结合同位素质量守恒的原则,即可实现对长江口缺氧区耗氧过程的量化分析. 基于已发表的数据和氧同位素质量守恒,推算长江口缺氧区的耗氧过程为水柱呼吸主导. 溶解氧同位素在各耗氧过程中的独特性质,使其成为示踪海洋氧循环复杂过程的重要指标之一,具有较大的应用前景.
-
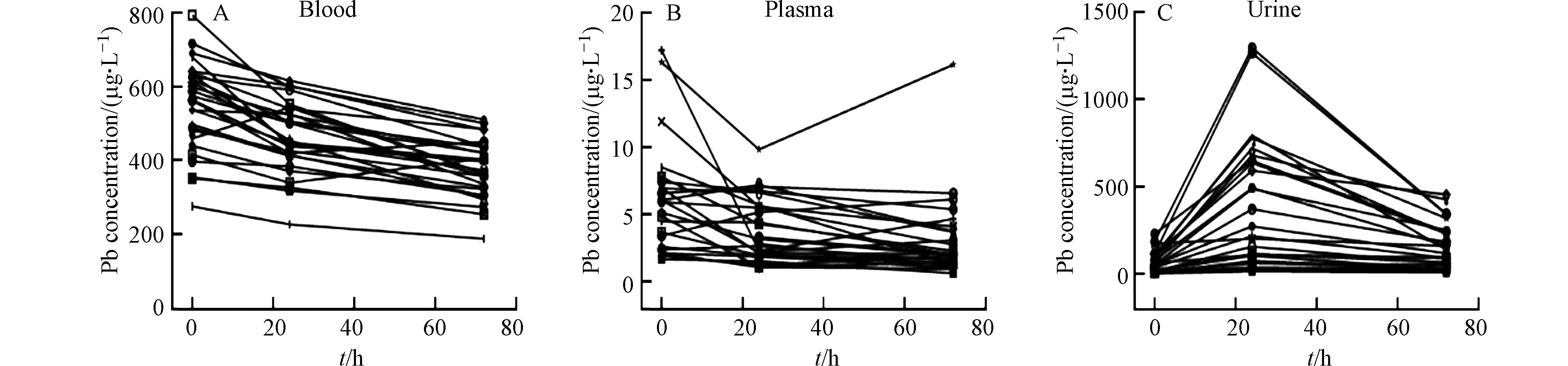
图 1 铅中毒患者在驱铅治疗过程中不同样品中铅浓度的变化趋势
Figure 1. The trend of Pb concentration in different samples during medical treatment.
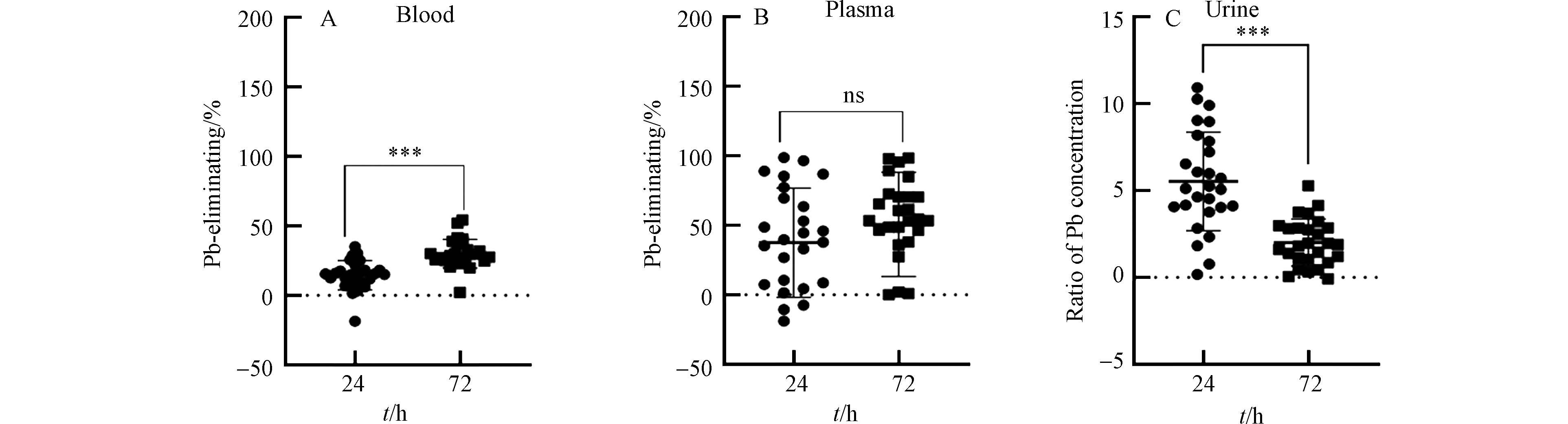
图 2 铅中毒患者在驱铅治疗过程中不同样品中的铅脱除效率
Figure 2. The removal efficiency of Pb in different samples during medical treatment
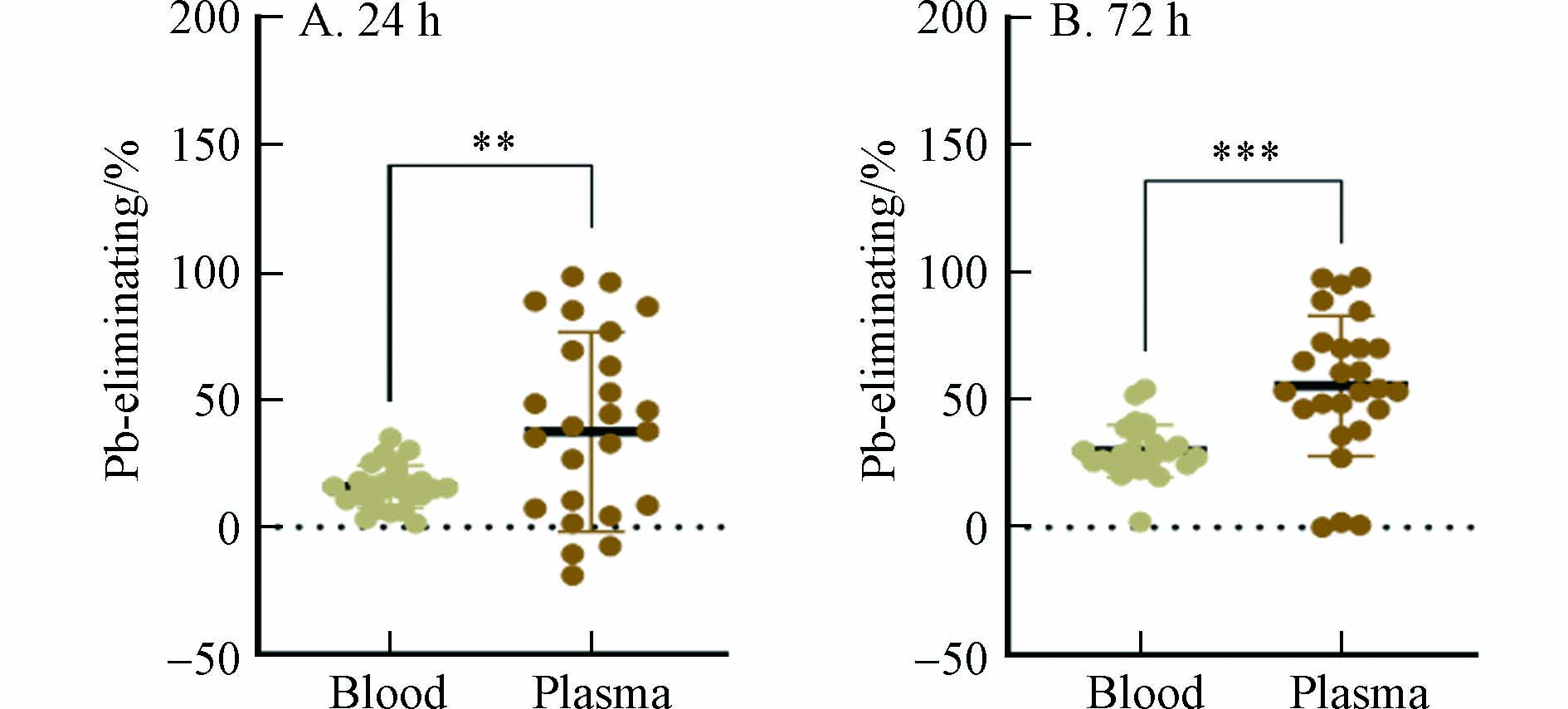
图 3 铅中毒患者在驱铅治疗过程中,不同阶段全血和血浆中的铅脱除率对比
Figure 3. Comparison of Pb removal rates in whole blood and plasma at different stages during medical treatment.
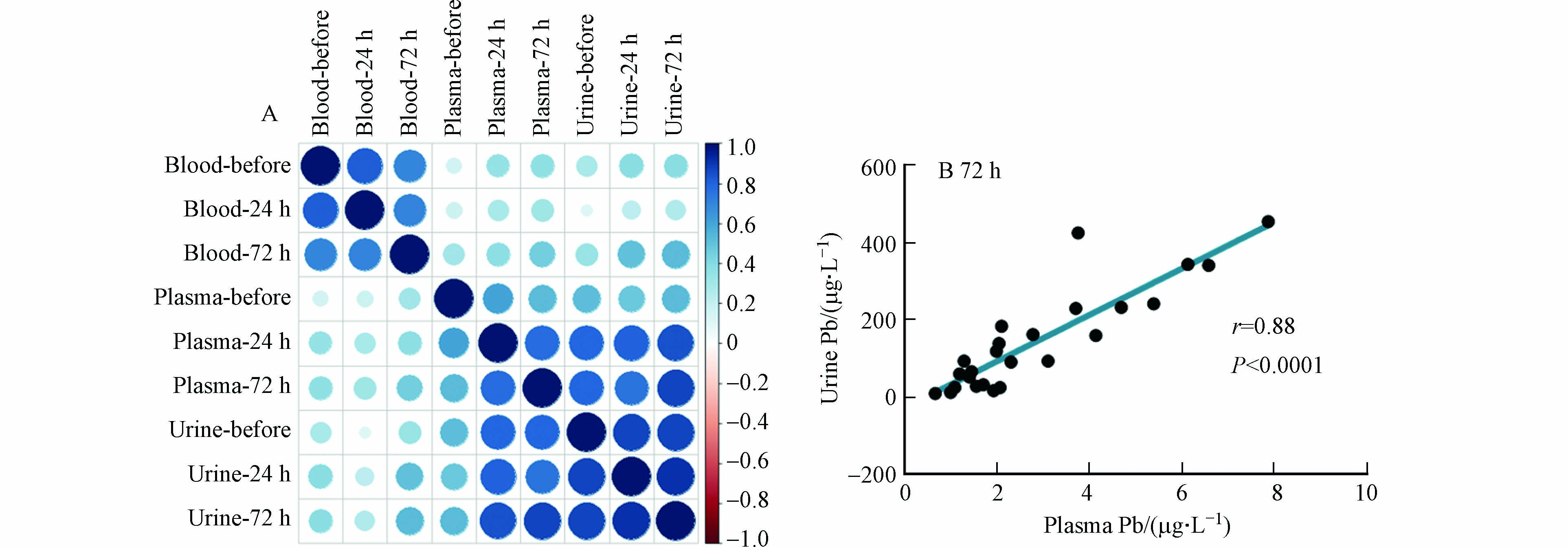
图 4 铅中毒患者在驱铅治疗过程不同样品中铅浓度的相关性分析
Figure 4. The correlation of Pb concentration in different samples during medical treatment
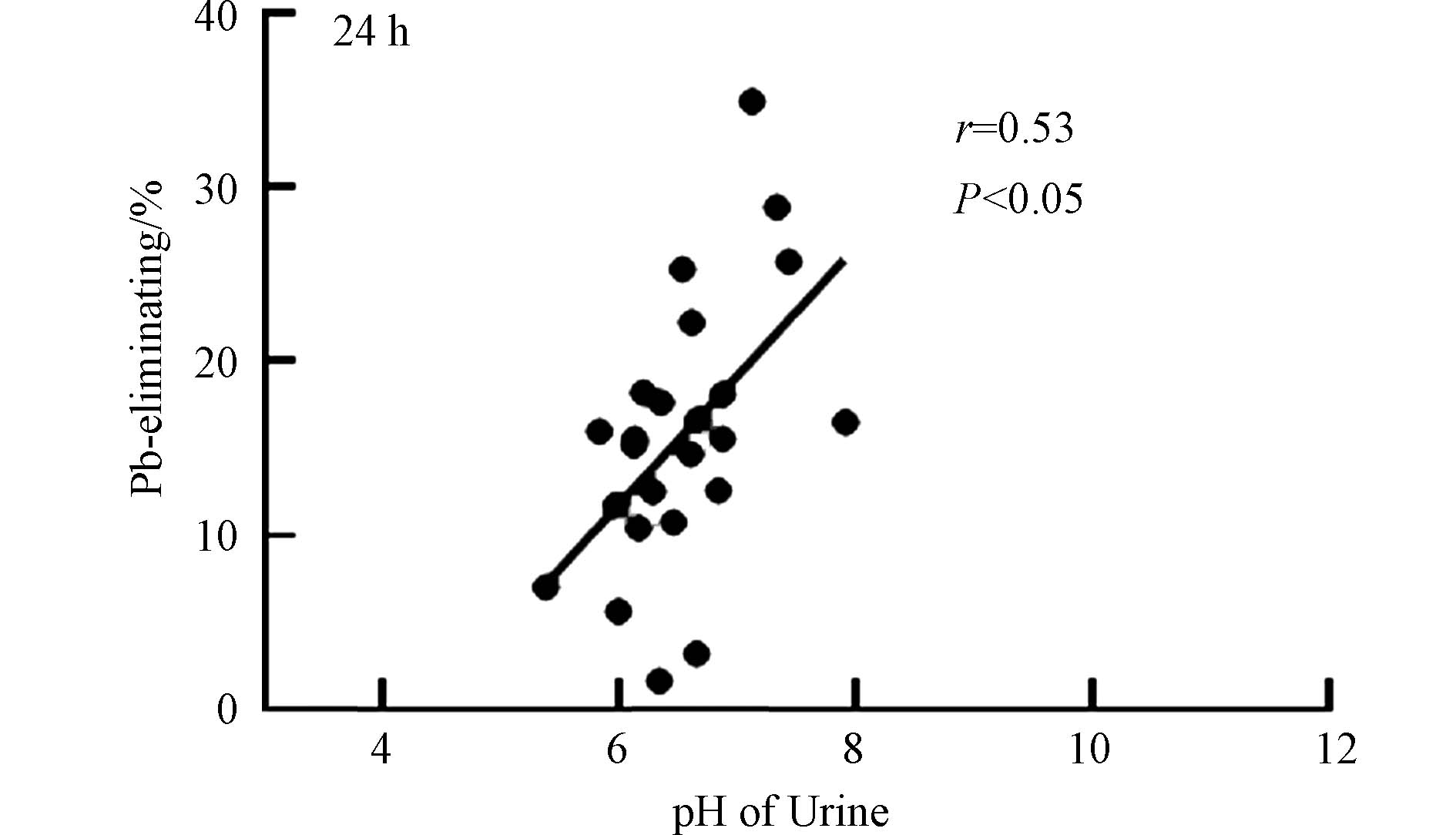
图 5 用药后24 h全血中的铅脱除率与尿液的酸碱度的相关性分析
Figure 5. The Pearson correlation of Pb removal rates in whole blood and Ph in urine at 24 h after medication
表 1 铅中毒患者的统计数据
Table 1. Demographic data of the participants
年龄Age 身体质量指数/(kg·m−2)Body mass index 男Male 女Female 40.2 ± 7.2 22.4 ± 2.3 19 (79.2%) 5 (20.8%)
下载: 导出CSV
-
[1] World lead consumption 2004-2020[EB/OL]. [2022-3-4] Statista Research Department, 2021. [2] World lead consumption 2004-2020[EB/OL]. [2022-3-4]. Statista Research Department, 2012. [3] 王玺凯, 刘媛, 沈敏, 等. 铅接触工人血铅水平对职业健康检查指标的影响 [J]. 临床医学研究与实践, 2019, 4(11): 78-79. WANG X K, LIU Y, SHEN M, et al. Effect of blood lead level on occupational health examination indicators in lead exposed workers [J]. Clinical Research and Practice, 2019, 4(11): 78-79(in Chinese).
[4] 窦建瑞. 职业性铅中毒的预防 [J]. 劳动保护, 2020(8): 74-76. doi: 10.3969/j.issn.1000-4335.2020.08.033 DOU J R. Prevention of occupational lead poisoning [J]. Labour Protection, 2020(8): 74-76(in Chinese). doi: 10.3969/j.issn.1000-4335.2020.08.033
[5] KASTURY F, SMITH E, LOMBI E, et al. Dynamics of lead bioavailability and speciation in indoor dust and X-ray spectroscopic investigation of the link between ingestion and inhalation pathways [J]. Environmental Science & Technology, 2019, 53(19): 11486-11495. [6] FLORA G, GUPTA D, TIWARI A. Toxicity of lead: A review with recent updates [J]. Interdisciplinary Toxicology, 2012, 5(2): 47-58. doi: 10.2478/v10102-012-0009-2 [7] ZHOU C C, GAO Z Y, WANG J, et al. Lead exposure induces Alzheimers's disease (AD)-like pathology and disturbes cholesterol metabolism in the young rat brain [J]. Toxicology Letters, 2018, 296: 173-183. doi: 10.1016/j.toxlet.2018.06.1065 [8] WAN C, PAN S X, LIN L F, et al. DNA methylation biomarkers of IQ reduction are associated with long-term lead exposure in school aged children in Southern China [J]. Environmental Science & Technology, 2021, 55(1): 412-422. [9] 张晴, 张斌, 赵静, 等. 环境相关浓度铅暴露诱导斑马鱼仔鱼神经行为毒性 [J]. 环境化学, 2018, 37(3): 445-452. doi: 10.7524/j.issn.0254-6108.2017080905 ZHANG Q, ZHANG B, ZHAO J, et al. Neurobehavioral toxicity of zebrafish larvae caused by lead exposure at environmentally relevant concentrations [J]. Environmental Chemistry, 2018, 37(3): 445-452(in Chinese). doi: 10.7524/j.issn.0254-6108.2017080905
[10] FLORA S J S, AGRAWAL S. Chapter 31-Arsenic, Cadmium, and Lead//GUPTA R C, ed. Reproductive and Developmental Toxicology (Second Edition) [M]. Academic Press, 2017: 537-566. [11] SILVER M K, LI X Q, LIU Y H, et al. Low-level prenatal lead exposure and infant sensory function [J]. Environmental Health:a Global Access Science Source, 2016, 15(1): 65. [12] LANPHEAR B P, RAUCH S, AUINGER P, et al. Low-level lead exposure and mortality in US adults: A population-based cohort study [J]. The Lancet Public Health, 2018, 3(4): e177-e184. doi: 10.1016/S2468-2667(18)30025-2 [13] 乔增运, 李昌泽, 周正, 等. 铅毒性危害及其治疗药物应用的研究进展 [J]. 毒理学杂志, 2020, 34(5): 416-420. doi: 10.16421/j.cnki.1002-3127.2020.05.016 QIAO Z Y, LI C Z, ZHOU Z, et al. Progress in lead toxicity research and therapeutic drug development [J]. Journal of Toxicology, 2020, 34(5): 416-420(in Chinese). doi: 10.16421/j.cnki.1002-3127.2020.05.016
[14] WAN M M, XU T T, CHI B, et al. A safe and efficient strategy for the rapid elimination of blood lead In Vivo based on a capture-fix-separate mechanism [J]. Angewandte Chemie International Edition, 2019, 58(31): 10582-10586. doi: 10.1002/anie.201904044 [15] KWON S Y, BAE O N, NOH J Y, et al. Erythrophagocytosis of lead-exposed erythrocytes by renal tubular cells: Possible role in lead-induced nephrotoxicity [J]. Environmental Health Perspectives, 2015, 123(2): 120-127. doi: 10.1289/ehp.1408094 [16] HU H, RABINOWITZ M, SMITH D. Bone lead as a biological marker in epidemiologic studies of chronic toxicity: Conceptual paradigms [J]. Environmental Health Perspectives, 1998, 106(1): 1-8. doi: 10.1289/ehp.981061 [17] BARLTROP D, SMITH A. Lead binding to human haemoglobin [J]. Experientia, 1972, 28(1): 76-77. doi: 10.1007/BF01928273 [18] SOMMAR J N, HEDMER M, LUNDH T, et al. Investigation of lead concentrations in whole blood, plasma and urine as biomarkers for biological monitoring of lead exposure [J]. Journal of Exposure Science & Environmental Epidemiology, 2014, 24(1): 51-57. [19] LAMADRID-FIGUEROA H, MARÍA TÉLLEZ-ROJO M, HERNÁNDEZ-CADENA L, et al. Biological markers of fetal lead exposure at each stage of pregnancy [J]. Journal of Toxicology and Environmental Health, Part A, 2006, 69(19): 1781-1796. doi: 10.1080/15287390600630195 [20] SILBERGELD E K, SAUK J, SOMERMAN M, et al. Lead in bone: Storage site, exposure source, and target organ [J]. Neurotoxicology, 1993, 14(2/3): 225-236. [21] YOU X J, LIU S G, DAI C M, et al. Effects of EDTA on adsorption of Cd(II) and Pb(II) by soil minerals in low-permeability layers: Batch experiments and microscopic characterization [J]. Environmental Science and Pollution Research International, 2020, 27(33): 41623-41638. doi: 10.1007/s11356-020-10149-9 [22] KIM S H, CHUNG H, JEONG S, et al. Identification of pH-dependent removal mechanisms of lead and arsenic by basic oxygen furnace slag: Relative contribution of precipitation and adsorption [J]. Journal of Cleaner Production, 2021, 279: 123451. doi: 10.1016/j.jclepro.2020.123451 [23] POTULA V, KAYE W. The impact of menopause and lifestyle factors on blood and bone lead levels among female former smelter workers: The Bunker Hill Study [J]. American Journal of Industrial Medicine, 2006, 49(3): 143-152. doi: 10.1002/ajim.20262 [24] WEYERMANN M, BRENNER H. Factors affecting bone demineralization and blood lead levels of postmenopausal women—A population-based study from Germany [J]. Environmental Research, 1998, 76(1): 19-25. doi: 10.1006/enrs.1997.3780 [25] LIU N, HUANG Y S, ZHANG H Z, et al. Unified probability distribution and dynamics of lead contents in human erythrocytes revealed by single-cell analysis [J]. Environmental Science & Technology, 2021, 55(6): 3819-3826. 期刊类型引用(0)
其他类型引用(2)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 3278
- HTML全文浏览数: 3278
- PDF下载数: 52
- 施引文献: 2
