






-
芬顿技术(Fenton)是一种高级氧化技术,产生的羟基自由基(·OH)氧化能力极强(E=2.8 V),能同时降解有机和无机污染物,在环境领域具有良好的应用前景[1-3]. 但由于使用的试剂过氧化氢(H2O2)在水体环境中不能稳定存在,易分解成H2O和O2,利用率较低[4-5]. 为克服传统Fenton技术的不足,学者们提出利用过氧化钙(CaO2)代替H2O2参与反应的类Fenton技术. CaO2可以在环境水体中缓慢释放H2O2,具有试剂有效期长、H2O2利用率高、pH适用范围广等传统Fenton技术没有的优点,目前已被学者和工程人员广泛研究和应用(如反应式1—4所示)[6]. 然而,现有研究大多是针对Fe2+和CaO2的反应体系,关于Fe3+和CaO2反应的研究相对较少[7-8]. 而且CaO2缓释H2O2的过程中会产生Ca(OH)2,提高溶液的pH,使铁离子沉淀析出影响催化效果,因此铁离子利用率较低. 如何促进铁离子溶解,强化Fe3+/Fe2+循环,对实现铁离子高效利用、增强体系降解性能具有重要的实际意义. 一些学者发现,Fe3+与乙二胺四乙酸盐(EDTA)、三聚磷酸盐(STPP)、草酸盐(OA)等配体络合可以抑制铁沉淀的生成[9-11]. 此外,引入配体也可以有效降低Fe3+/Fe2+的氧化还原电位,增强电子传递速率,促进Fe2+催化CaO2产生更多的·OH,提高反应效果[12].
EDTA是一种十分常用的代表性螯合剂,能和碱金属、稀土元素和过渡金属等形成稳定的水溶性络合物,其与Fe3+形成络合物后可以抑制铁离子沉淀,提高铁离子的利用率. 与其他配体相比,EDTA具有更强的螯合能力,其与Fe2+和Fe3+络合的稳定常数可以达到14.27和24[13]. 本文选用EDTA作为CaO2类Fenton体系的配体,以苯酚为目标污染物,研究了不同因素(EDTA浓度、Fe3+浓度、CaO2投加量、初始pH值)对Fe3+/EDTA/CaO2体系降解苯酚的影响及作用机理,揭示了EDTA增强铁离子溶解及循环、CaO2加快反应体系启动的重要作用,并确定了体系中活性自由基的产生及作用机制,为CaO2类Fenton体系在水环境修复中的应用提供一定的理论依据.
-
六水合三氯化铁、乙二胺四乙酸二钠、过氧化氢、钼酸铵、二水合磷酸二氢钠、十二水合磷酸氢二钠、邻菲啰啉、乙酸、乙酸铵(分析纯,天津光复精细化工研究所);过氧化钙、抗坏血酸(分析纯,国药集团化学试剂有限公司);苯酚(分析纯,北京化工).
-
分析天平(PL203,梅特勒-托利多仪器公司);pH计(PHB-4,上海雷磁);磁力搅拌器(HJ-6A,金坛市医疗仪器厂);紫外-可见分光光度仪(EVOLUTION 201,赛默飞世尔科技公司);高效液相色谱仪(Agilent1100,美国Agilent);气相色谱-质谱联用仪(Agilent 6890/5973,美国Agilent).
-
实验在250 mL血清瓶中进行,向200 mL 50 mmol·L−1的磷酸缓冲溶液(pH=6.5)中加入一定量的苯酚,使溶液中的苯酚浓度为50 mg·L−1. 投加不同体积100 mmol·L−1 Fe3+-EDTA(物质的量比为1:0.5)混合溶液(反应溶液的体积变化可以忽略不计),混合均匀后,投加一定量的CaO2,实验计时开始. 反应过程中,在既定时间取出0.5 mL样品,然后迅速加入0.5 mL无水乙醇终止反应,用0.22 µm滤头过滤到安捷伦样品瓶中. 如无特殊说明,体系中Fe3+浓度为1 mmol·L−1,EDTA浓度为0.5 mmol·L−1,CaO2浓度为0.75 g·L−1,反应过程中体系的pH保持在6.5. 每组设3个平行样取平均值,实验数据的标准误差不超过5%.
-
运用高效液相色谱法测定苯酚的含量[14]. 色谱柱型号为SB-C18(ϕ4.6 mm×250 mm,5 µm),采用等度洗脱,流动相体积比为乙腈:水=40:60. 进样量为10 µL,流速为1 mL·min−1,柱温为30 ℃,VWD波长为270 nm,出峰时间为3.1 min左右.
-
采用气相色谱-质谱联用法(GC-MS)以及液相色谱-质谱联用法(LC-MS) 测定中间产物,样品前处理过程如下[15]:在不同的反应时间(0、10、30、60、120 min)收集100 mL样品,加入乙酸乙酯进行萃取,将上层清液取出加入适量无水硫酸钠干燥脱水,将脱水后的溶液迅速倒入65 ℃的旋转蒸发仪中,蒸发浓缩后用0.22 µm滤膜过滤得到GC-MS所需的样品. 测试条件如下: 以不分流模式注入1 μL样品,色谱柱温度以20 ℃·min−1的速度从40 ℃升到200 ℃保持4 min,以10 ℃·min−1的速度从200 ℃升到250 ℃保持5 min. 在EI模式下获得质谱,电子能量为70 eV; 将待测样品以 20 μL 的进样量注入LC-MS系统,洗脱液为乙腈,流速为0.2 mL·min−1,采用负离子扫描模式在m/z范围30—150获得质谱.
-
选用钼酸铵法测定溶液中的H2O2浓度[16]. 取出适量样品后用0.22 µm滤头过滤,将1 mL过滤后的样品和1 mL 2.4 mmol·L−1的钼酸铵溶液加入到10 mL离心管,用去离子水稀释至10 mL. 显色15 min后用紫外-可见分光光度计在450 nm处检测样品的吸光度. 根据标准曲线可以得到H2O2的浓度.
-
Fe2+浓度采用1,10-菲咯啉显色法测定[17]. Fe2+在pH=3—9时与1,10-菲咯啉反应生成稳定的橙红色络合物,橙红色络合物的吸光度与浓度的关系符合朗伯-比尔定律,可通过测定橙红色络合物在510 nm处的吸光度换算出溶液中Fe2+的浓度. 加入抗坏血酸将Fe3+还原为Fe2+,由此可得到总铁离子的浓度.
-
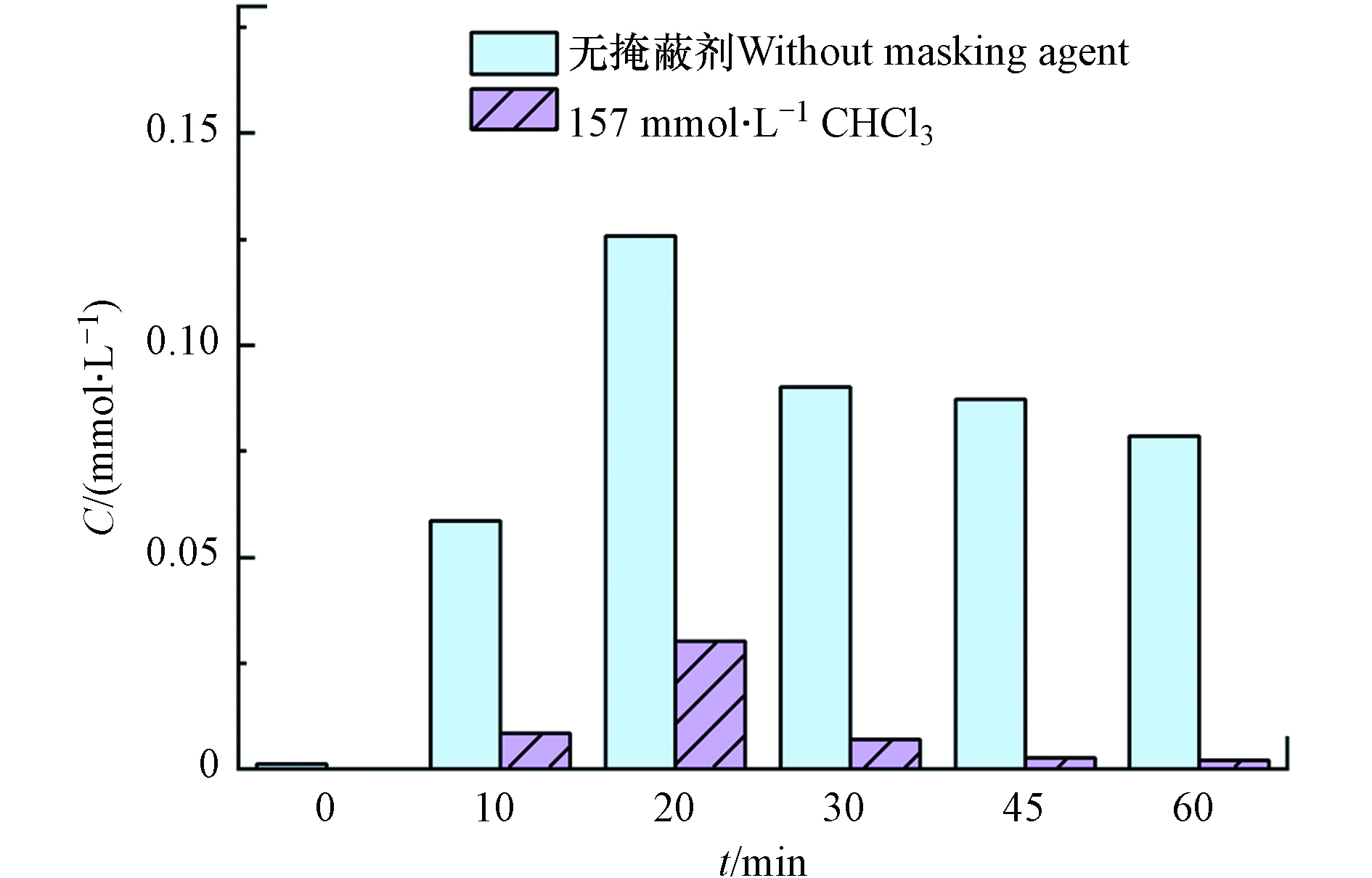
·OH探针实验[18]:测定反应过程中·OH的累积浓度. 苯甲酸与·OH反应生成邻-羟基苯甲酸、间-羟基苯甲酸、对-羟基苯甲酸的3种异构体,且它们的产量比为邻-羟基苯甲酸∶间-羟基苯甲酸∶对-羟基苯甲酸=1.7∶2.3∶1.2,利用高效液相色谱仪检测对-羟基苯甲酸的产量,进而可知体系生成·OH的产量. 掩蔽实验:分别利用氯仿(CHCl3,157 mmol·L−1)、叔丁醇(TBA,63 mmol·L−1)分别作·O2−、·OH的掩蔽剂. 后续通过高效液相色谱仪检测反应过程中苯酚浓度的变化情况.
-
为了探究体系中各组分对苯酚降解的作用,考察了不同组分体系中苯酚的降解效果,结果如图1(a)所示. 在单独CaO2、EDTA/CaO2和Fe3+/CaO2的条件下,苯酚浓度在120 min内均没有明显变化,说明单独CaO2的氧化能力不足以氧化苯酚,添加EDTA不能直接对CaO2起到作用,而缺少EDTA的Fe3+/CaO2也因为铁离子易沉淀、电子传递速率慢的原因,而无法有效降解苯酚;在Fe3+/EDTA/CaO2中,苯酚的降解分为初始缓慢降解阶段和随后的快速降解阶段,120 min时苯酚的降解率在95%以上. 结合不同组分条件下H2O2浓度的变化(图1(b))可知,在单独CaO2、EDTA/CaO2和Fe3+/CaO2的条件下H2O2浓度在前20 min内迅速升高,之后几乎没有变化,而在Fe3+/EDTA/CaO2中前20 min内H2O2浓度的上升速度明显减缓,这说明有一部分H2O2是被缓慢消耗的,20 min后H2O2消耗速率加快,苯酚降解速率也随之加快,直到120 min时苯酚完全降解,说明苯酚降解与H2O2有关,H2O2消耗速率越快苯酚降解效果越好. 综合以上结果可以分析出:只有在Fe3+、EDTA、CaO2 等3种组分均在时,CaO2水解释放的H2O2才能与Fe3+快速地发生类Fenton反应氧化降解污染物.
-
图2(a)显示了EDTA浓度对Fe3+/CaO2降解苯酚的影响:没有投加EDTA时,Fe3+/CaO2几乎不降解苯酚,而分别投加了0.125、0.25、0.5、1、2 mmol·L−1的EDTA后,120 min时苯酚去除率分别提高至55.7%、72.2%、100%、100%和87.2%. 由此可见,投加EDTA可以有效地促进Fe3+/CaO2氧化降解苯酚,当EDTA浓度小于0.5 mmol·L−1时EDTA浓度越高苯酚的降解效果越好,但随着EDTA浓度从0.5 mmol·L−1增加至2 mmol·L−1,苯酚的降解速率并没有进一步加快甚至反而受到抑制,这可能是因为过量的EDTA会与苯酚竞争·OH,导致苯酚的降解效果变差,因此在本研究中选取0.5 mmol·L−1为EDTA的最佳浓度. 对体系中EDTA浓度为0.125、0.25、0.5 mmol·L−1时的苯酚降解曲线进行动力学拟合(图2(b)),拟合结果表明,3种EDTA浓度条件下的苯酚降解曲线均符合拟一级动力学,且随着EDTA浓度增大,拟合曲线斜率(kobs)也显著增大,说明体系内EDTA浓度是反应的限制性因素.
图3显示了Fe3+/CaO2和Fe3+/EDTA/CaO2不同组分条件下,降解苯酚的过程中溶解态Fe2+和总溶解态铁离子(TFe)的浓度变化. 在Fe3+/CaO2中,10 min内TFe浓度迅速降低至0.34 mmol·L−1,30 min 时TFe仅剩0.02 mmol·L−1,几乎检测不到溶解态Fe2+,这是由于初始条件处于中性环境,且CaO2水解会产生大量Ca(OH)2,游离态铁离子会迅速转化为沉淀,因此该组分条件下不易发生类Fenton反应,无法有效降解苯酚.
而在Fe3+/EDTA/CaO2中,整个反应过程中Fe3+浓度始终维持在0.85 mmol·L−1以上,同时也能测到少量的溶解态Fe2+,这是因为EDTA与Fe3+形成稳定的络合物会抑制Fe3+沉淀,促进类Fenton反应顺利进行.
因此,EDTA的加入可以明显增加铁离子在中性pH环境中的溶解度,促使铁离子高效催化CaO2降解污染物.
-
为了进一步探究CaO2促进苯酚降解的原因,对比了Fe3+/EDTA/H2O2和Fe3+/EDTA/CaO2两种体系对苯酚的降解效果,结果如图4(a)所示. 在Fe3+/EDTA/H2O2体系中,前30 min苯酚几乎没有降解,30 min后苯酚开始被缓慢降解,120 min时苯酚降解率达到48%;在Fe3+/EDTA/CaO2体系中,120 min时苯酚的降解率在95%以上,与Fe3+/EDTA/H2O2体系相比,Fe3+/EDTA/CaO2体系在反应前期能更快地启动反应,随后对体系中苯酚的降解速率也明显更高. 结合不同组分体系中H2O2的浓度变化(图4(b))可以发现,Fe3+/EDTA/CaO2体系在前20 min缓慢消耗H2O2,20 min后H2O2消耗速率加快,苯酚降解速率也随之加快,而在Fe3+/EDTA/H2O2体系中前30 min几乎没有消耗H2O2,30 min后H2O2才被缓慢消耗. 对比两个体系在反应120 min时的H2O2消耗量以及苯酚降解率可以发现,Fe3+/EDTA/CaO2体系的消耗量更多,苯酚降解率更高. 在Fe3+/EDTA/CaO2体系中用H2O2酶掩蔽H2O2后还可以测到少量的Fe2+产生(图5),而掩蔽了Fe3+/EDTA/H2O2体系中的H2O2后却未检测到Fe2+产生,说明在Fe3+/EDTA/CaO2体系中更易产生Fe2+,这可能是因为Fe3+和EDTA形成的络合物Fe3+-EDTA易于吸附到CaO2的固体表面,EDTA起到桥接Fe3+和CaO2的作用,促进电子从CaO2转移到Fe3+,将Fe3+还原为Fe2+. 因此,引入EDTA的Fe3+/EDTA/CaO2体系可以加快Fe3+/Fe2+循环,促进类Fenton反应的进行和苯酚的降解. Pan等[19]在Fe3+-EDTA促进CaO2类Fenton反应的研究中也发现,Fe3+-EDTA倾向于吸附在CaO2的固体表面,形成[Fe3+-EDTA···CaO2]−的过渡态,电子从CaO2转移到Fe3+-EDTA,促进Fe3+-EDTA还原,与本文结论一致.
-
为确定反应过程中活性自由基的种类,在0 min和30 min时利用ERP光谱和DMPO自旋捕获加合物进行分析,结果如图6所示. 0 min时未出现特征峰,而在30 min时检测到·OH 的1:2:2:1四重态的特征峰(图6(a)),表明体系中有·OH产生;30 min时也观察到1:1:1:1的四重态特征峰(图6(b)),证明体系中存在
⋅O−2 . 分别利用叔丁醇(TBA)和氯仿(CHCl3)掩蔽·OH和⋅O−2 后,苯酚的降解效果如图7(a)所示,掩蔽·OH后体系几乎没有降解效果;掩蔽⋅O−2 后,反应过程中苯酚的降解速率比无掩蔽体系的降解速率慢,但120 min时苯酚降解率也能达到82.2%. 由此可知,·OH对体系中苯酚的降解起主要作用,⋅O−2 在降解过程中起到一定的促进作用. 反应过程中·OH的累积浓度如图7(b)所示,·OH的产生速率与苯酚的降解速率变化一致,证明是由·OH直接氧化降解苯酚.图8为无掩蔽体系和掩蔽
⋅O−2 体系中Fe2+浓度的变化,掩蔽了⋅O−2 后产生的Fe2+浓度低于无掩蔽体系中的浓度,说明⋅O−2 可以将体系中Fe3+还原为Fe2+. 本实验在Pan等[19]的研究结果基础上又进一步发现在体系中⋅O−2 的作用: Fe3+/EDTA/CaO2体系中除CaO2之外,⋅O−2 也起到了还原Fe3+的作用,二者共同加速Fe3+向Fe2+的转化,促进苯酚降解. -
固定EDTA∶Fe3+物质的量比为0.5∶1,不同Fe3+浓度条件下苯酚的降解效果如图9(a)所示. 随着Fe3+浓度由0.25 mmol·L−1提高到2 mmol·L−1,在120 min的反应时间内苯酚的降解率从20.1%上升至100%. 可见Fe3+浓度对苯酚降解效果至关重要.
-
图9(b)为CaO2投加量对苯酚降解的影响. 由图9可知,随着CaO2投加量不断增加,苯酚的降解率也随之增加,当CaO2投加量增加至0.75 g·L−1时,反应120 min后苯酚的降解率增加至100%. 这主要是由于提高CaO2投加量会释放更多的H2O2,但CaO2投加量进一步増加至1.5 g·L−1时,苯酚的降解会受到抑制,反应120 min时降解率从100%下降至43.4%. 这是因为过量的CaO2与水反应后生成大量的Ca(OH)2,体系中的pH值会逐渐上升超过11,超过磷酸盐的缓冲能力,铁离子沉淀,类Fenton反应受到抑制,影响苯酚的降解效果.
-
图9(c)所示为不同初始pH条件下Fe3+/EDTA/CaO2体系对苯酚的降解效果,结果表明,体系在初始pH=3—7内均有很好的降解效果,在pH=8时苯酚降解效果较差,这可能是因为CaO2在碱性环境中易分解成O2而不是H2O2,且·OH在碱性环境中的活性较低,因此体系在酸性和中性环境下的降解效果更好.
-
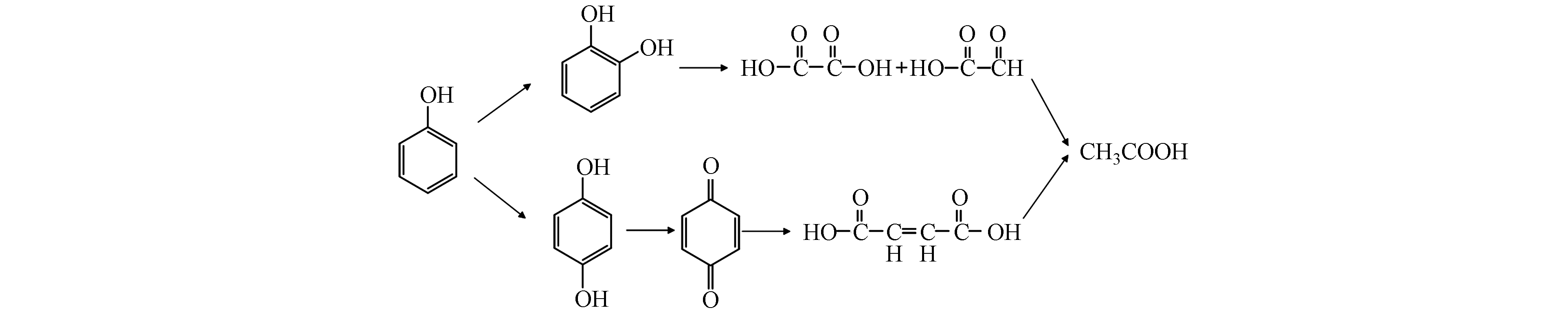
通过GC-MS对Fe3+/EDTA/CaO2体系降解苯酚的氧化产物进行测定,结果如表1所示,测得的苯酚降解中间产物主要包括对苯二酚、邻苯二酚、对苯醌、乙醛酸. 通过LC-MS对氧化产物进行进一步测定,检测到草酸(乙二酸)、马来酸(顺丁烯二酸)、乙酸,通过以上产物推测体系可能发生的降解途径如图10所示,苯酚被氧化成邻苯二酚、对苯二酚和对苯醌,中间产物进一步开环形成小分子酸.
-
(1)Fe3+/EDTA/CaO2体系可以高效降解苯酚,EDTA的加入可以明显增加铁离子在中性环境中的溶解度,而且EDTA可以桥接CaO2和Fe3+,加快二者之间的电子转移速率,促进Fe3+/Fe2+循环,改善苯酚的降解效果. 与Fe3+/EDTA/CaO2体系相比,Fe3+/EDTA/CaO2体系能更快地启动反应,提高反应速率.
(2)Fe3+/EDTA/CaO2体系产生活性自由基降解苯酚,起主要作用的是·OH,
⋅O−2 起到还原Fe3+的作用,加快体系中苯酚的降解速率.(3)Fe3+/EDTA/CaO2体系中EDTA: Fe3+的最佳比例为0.5:1;Fe3+浓度越高,苯酚降解速率越快;随着CaO2投加量的增大,苯酚的降解效果越好,但过量反而会抑制苯酚降解.
Fe3+-EDTA催化CaO2类Fenton体系降解苯酚的机制及效果
Mechanism and effect of phenol degradation by Fe3+-EDTA catalyzing CaO2 Fenton-like system
-
摘要: 基于CaO2的类Fenton体系具有缓释H2O2、高效降解污染物、有效作用周期长等优点,在环境修复领域得到广泛应用. 但目前研究主要针对Fe2+/CaO2体系,关于Fe3+/CaO2的研究较少,而如何提高Fe3+活化CaO2的能力,是铁离子高效循环利用、污染物持续降解的关键. 本研究有针对性地建立了Fe3+/EDTA/CaO2体系,对其降解能力、作用机制、关键因素进行了深入分析,结果表明,Fe3+/EDTA/CaO2体系在中性条件下对苯酚的降解率在95%以上,EDTA的加入可以明显增加铁离子在中性环境中的溶解度,而且EDTA可以桥接CaO2和Fe3+,加快二者之间的电子转移速率,促进Fe3+/Fe2+循环,改善苯酚的降解效果;活性自由基测定的实验表明,降解苯酚起主要作用的活性自由基是羟基自由基(·OH),
⋅O−2 起到还原Fe3+的作用,促进Fe3+/Fe2+循环. 本研究对CaO2类Fenton体系的应用具有一定的理论意义.-
关键词:
- 过氧化钙(CaO2) /
- 三价铁(Fe3+) /
- 乙二胺四乙酸二钠(EDTA) /
- 类芬顿 /
- 苯酚
Abstract: CaO2-based Fenton-like systems are widely applied in environmental remediation with the advantages of slow release of H2O2, high efficiency for pollutant degradation, and long term of effectiveness. However, the current studies focus on Fe2+/CaO2 system, and limited studies were reported regarding to Fe3+/CaO2 system. Whereas the enhancement of Fe3+ on the activation of CaO2 is critical in Fe cyclic utilization and continuous degradation of pollutants. This study established Fe3+/EDTA/CaO2 system, and evaluated the degradation ability, interaction mechanisms, and key factors. The results indicate the degradation efficiency of phenol was greater than 95% by Fe3+/EDTA/CaO2 system under neutral pH conditions. The addition of EDTA enhanced the solubility of Fe3+ under neutral pH conditions; EDTA also bridged CaO2 and Fe3+ and enhanced the circulation of Fe2+/Fe3+ to improve the degradation of phenol. The analysis of the active free radicals indicate hydroxyl radical (·OH) played a key role in phenol degradation, and⋅O−2 enhanced Fe2+/Fe3+ circulation through the reduction of Fe3+. The study provides theoretical foundation in the application of Fenton-like systems.-
Key words:
- calcium peroxide /
- ferric ion /
- ethylenediaminetetraacetic acid disodium salt /
- fenton-like /
- phenol.
-
羰基化合物, 即醛类和酮类化合物, 广泛存在于日常生活环境中, 对人体健康有重要影响. 研究发现, 空气中的羰基化合物不仅对人体的嗅觉产生影响, 还会刺激人体免疫系统, 长时间接触羰基化合物可能会出现不良健康影响. 另外羰基化合物还会引起染色体分裂而造成遗传物质损害或断裂, 如姐妹染色体交换及染色体变异等, 使羰基化合物具有致癌性和致突变性[1-4]. 人们在室内生活工作的时间超过80%, 室内成为越来越多的人最直接和最经常接触的环境[5]. 室内空气中大多数装饰和装修材料包括建筑材料[6]、新家具(尤其是由胶合板制成的家具[7]、木地板[8])、家居用品(如胶水、油漆和涂料[9])、香烟烟雾[10]、室内烹饪[3]以及一些生活用品(如消毒剂、化妆品和空气清新剂)等都会向室内空气环境直接释放羰基化合物[11-13]. 越来越密闭的建筑结构往往又会使室内产生的化学污染物不断积累[14]. 还有些羰基化合物作为一些污染物与臭氧反应的次级产物出现在室内空气环境中[15]. 比如, 臭氧与头发和衣服上残留的人体皮肤油反应也可作为室内空气中羰基化合物的来源[16-17]. 此外, 城区的车辆尾气和工业厂房的燃料燃烧也会产生羰基化合物, 然后通过空气渗透和自然通风进入室内环境中[18-19].
许多国内学者对室内羰基化合物的污染特征、来源情况以及健康影响开展相关研究工作. 黄晓影[20]研究发现, 包头市城区住宅羰基化合物采暖季污染更严重, 其中甲醛和乙醛浓度均与换气次数呈负相关, 较高的相对湿度是甲醛、乙醛和丙醛暴露的危险因素. Pu 等[21]研究发现, 北京城区住宅12种羰基化合物中甲醛污染最严重, 乙醛、丙酮和己醛次之, 其中甲醛受沙发材料、吸烟情况、家庭位置影响, 乙醛受吸烟情况和厨房结构影响, 丙酮受厨房结构和盆栽植物影响, 己醛受盆栽植物影响. Huang 等[22]研究发现, 室内甲醛暴露可能会增加儿童患普通感冒的风险. 由于环境中气态羰基化合物的分析方法比较繁琐以及相对较高的分析费用, 多数文献针对少数几种羰基化合物展开研究, 很少关注气味污染严重的高分子量醛(己醛、庚醛、辛醛、壬醛和癸醛)以及能够诱导细胞损伤和产生晚期糖基化终产物的乙二醛和甲基乙二醛等其它羰基化合物的污染情况及其对人体健康的影响, 这不足以充分了解我国城市住宅室内外空气中羰基化合物的污染特征及健康效应. 本研究旨在通过供暖季与非供暖季在城市居民住宅开展室内外同步观测, 分析季节变化对不同羰基污染物的影响,并评估室内羰基污染物对人群的健康风险及其对嗅觉的污染情况, 以期为进一步研究住宅室内痕量羰基污染物特征及其影响提供一定的科学依据.
1. 材料与方法(Materials and methods)
1.1 样品采集
本研究于2016 年供暖季(1月28日—30日)和非供暖季(9月23日—26日)在西安市雁塔区某住宅区5户住宅开展室内羰基化合物的同步观测, 为便于比较, 另选一住宅的露天阳台同步采集了室外样品. 采样点周围无明显工业污染源, 属于城市住宅区, 各住户家庭信息如表1所示. 采样期间, 所有仪器均放置在客厅中心位置(离地面1.0—1.5 m), 避开通风口. 采样头选用Sep-Pak DNPH(2,4-二硝基苯肼)-silica Gel cartridge + Ozone scrubber (Waters Corporation, USA), 流量为0.6—0.8 L·min−1的真空泵(Thomas)进行主动采样, 采集时间为 8 h(9:30—17:30), 采集后的DNPH样品用铝箔包裹后, 置于4 ℃以下冰箱保存, 防止样品被污染. 供暖季共采集DNPH样品18个(室内每户3个+室外3个), 均为工作日2个+周末1个; 非供暖季共采集24个(室内每户4个+室外4个), 均为工作日2个+周末2个. 此外, 采用 CO2分析仪(LI-820, LI-COR, USA)监测室内通风换气次数.
表 1 各采样住户家庭信息Table 1. Household information of each sampled household采样点Site 房屋年龄/aAge of house 面积/m2Area 装修后放置时间/monthStorage time after decoration 地板材质Floor material 壁纸Wall-paper 取暖燃料Fuel for heating 烹饪燃料Fuel for cooking 每天烹饪次数Cooking frequency 通风方式Ventilation 吸烟Smoking QJ-1 5 100 6—12 客厅+卧室复合木地板 是 天然气 天然气 > 3 半开 否 > 1 h QJ-2 5 100 6—12 客厅+卧室瓷砖 否 天然气 液化石油气 > 3 半开 否 > 1 h QJ-3 5 102 6—12 客厅+卧室复合木地板 否 天然气 天然气 1 半开 否 电 < 1 h QJ-4 5 100 3—6 客厅+卧室复合竹地板 是 天然气 天然气 3 半开 否 < 1 h QJ-5 5 148 3—6 客厅瓷砖卧室复合木地板 否 天然气 天然气 3 全开 否 < 1 h | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 样品分析及质控
DNPH样品经乙腈洗提, 利用高效液相色谱仪(HPLC, Aglient 1200 LC)分析. 在25 ℃柱温、流速2.0 mL·min−1、检测波长为360 nm、390 nm和420 nm、进样量为20 μL的条件下, 利用梯度洗脱程序进行分析, 可定量检测20种羰基化合物, 分别为甲醛(C1)、乙醛(C2)、丙酮(A3K)、丙醛(n-C3)、2-丁酮(MEK)、丁醛(i,n-C4)、苯甲醛(Benz)、异戊醛(i-C5)、正戊醛(n-C5)、邻甲苯甲醛(o-tol)、间甲苯甲醛(m-tol)、对甲苯甲醛(p-tol)、2,5-二甲基苯甲醛(2,5-DB)、己醛(C6)、庚醛(C7)、辛醛(C8)、壬醛(C9)、癸醛(C10)、乙二醛(Gly)和甲基乙二醛(mGly), 其中己醛、庚醛、辛醛、壬醛和癸醛, 统称为高分子量羰基化合物(C6—C10), 各物质的方法检出限如表2所示. 本方法其他的详细介绍及质量控制参见文献[23].
表 2 供暖季室内外环境中羰基化合物浓度Table 2. Concentrations of carbonyl compounds in indoor and outdoor environments in heating season羰基化合物Carbonyl compounds 方法检测限/(µg·mL−1)Method detection limit 平均值±标准偏差 (AVG±SD)/( μg·m−3) QJ-1 QJ-2 QJ-3 QJ-4 QJ-5 QJ-O 甲醛(C1) 0.002 86.7 ± 10.8 23.2 ± 5.1 40.7 ± 2.8 30.9 ± 6.4 46.3 ± 6.9 9.7 ± 1.3 乙醛(C2) 0.004 38.4 ± 6.1 24.1 ± 5.7 48.5 ± 8.9 25.1 ± 5.0 31.9 ± 7.9 12.2 ± 2.6 丙酮(A3K) 0.006 46.5 ± 11.9 35.5 ± 13.9 47.8 ± 5.9 32.2 ± 10.3 31.2 ± 4.5 14.9 ± 4.4 丙醛(C3) 0.004 4.9 ± 0.7 3.5 ± 1.0 4.6 ± 0.5 3.1 ± 0.8 4.3 ± 0.2 2.3 ± 0.4 2-丁酮(MEK) 0.005 3.5 ± 0.6 3.2 ± 0.9 5.3 ± 1.0 3.3 ± 1.1 4.5 ± 0.3 3.1 ± 0.6 丁醛(i,n-C4) 0.003 2.9 ± 0.4 2.4 ± 0.7 3.0 ± 0.3 2.4 ± 0.6 2.5 ± 0.1 1.4 ± 0.3 苯甲醛(Benz) 0.007 1.9 ± 0.2 1.8 ± 0.4 2.9 ± 0.3 2.2 ± 0.7 1.9 ± 0.2 1.0 ± 0.2 异戊醛(i-C5) 0.004 3.4 ± 0.6 2.7 ± 0.7 3.0 ± 0.3 2.6 ± 0.6 3.6 ± 0.3 1.7 ± 0.4 正戊醛(n-C5) 0.005 2.4 ± 0.4 4.5 ± 1.4 3.0 ± 0.3 2.2 ± 0.7 2.1 ± 0.3 0.6 ± 0.2 邻甲苯甲醛(o-tol) 0.006 0.5 ± 0.1 0.6 ± 0.3 0.4 ± 0.1 0.5 ± 0.4 0.7 ± 0.2 0.1 ± 0.1 间甲苯甲醛(m-tol) 0.004 0.5 ± 0.1 0.5 ± 0.1 0.5 ± 0.2 0.5 ± 0.2 0.5 ± 0.2 0.5 ± 0.1 对甲苯甲醛(p-tol) 0.003 0.3 ± 0.1 0.6 ± 0.2 0.5 ± 0.2 0.3 ± 0.2 0.4 ± 0.2 0.2 ± 0.0 2,5-二甲基苯甲醛(2,5-DB) 0.003 0.6 ± 0.1 0.7 ± 0.2 0.6 ± 0.2 0.6 ± 0.1 0.5 ± 0.2 0.5 ± 0.2 己醛(C6) 0.007 11.8 ± 2.5 30.0 ± 7.7 14.9 ± 1.6 10.8 ± 4.6 12.6 ± 2.5 0.9 ± 0.2 庚醛(C7) 0.011 4.7 ± 1.0 4.5 ± 1.2 4.6 ± 0.1 3.6 ± 0.7 3.9 ± 0.8 0.8 ± 0.3 辛醛(C8) 0.016 5.6 ± 1.0 5.9 ± 1.5 5.5 ± 0.5 5.8 ± 1.7 5.8 ± 1.5 0.9 ± 0.3 壬醛(C9) 0.017 16.4 ± 2.8 18.6 ± 5.1 15.0 ± 1.1 14.7 ± 3.2 18.9 ± 5.9 2.1 ± 0.5 癸醛(C10) 0.001 2.9 ± 0.5 3.5 ± 2.2 2.8 ± 0.8 5.4 ± 0.8 3.3 ± 0.9 0.6 ± 0.2 乙二醛(Gly) 0.001 0.4 ± 0.1 0.7 ± 0.1 0.4 ± 0.1 0.4 ± 0.1 0.4 ± 0.1 0.8 ± 0.4 甲基乙二醛(mGly) 0.001 0.6 ± 0.2 0.9 ± 0.2 0.7 ± 0.2 0.8 ± 0.2 1.1 ± 0.2 1.0 ± 0.2 总和(SUM) 235.1±33.9 167.3 ± 38.7 204.7 ± 11.6 147.2 ± 33.1 176.3 ± 25.7 55.3 ± 9.4 | Show TableDownLoad:
CSV
1.3 数据分析
绘图使用Origin 9.0软件, 统计分析用SPSS 26和 Excel 2021 软件. 样本正态性检验采用 Kolmogorov-Smirnov 检验. 若研究变量为非正态分布, 则进行非参数双样本Mann-Whitney U检验和多样本Kruskal-Wallis H检验, 比较不同住户的羰基化合物浓度差异. 所有统计分析结果在 P <0.05 时,被认为具有统计学意义.
1.4 健康风险评价方法
与成人相比, 儿童因更高的呼吸频率、更多的脆弱性和更长的暴露时间[24-25], 更易受到环境污染物的影响, 因此有必要了解室内空气中羰基化合物污染对儿童健康的影响. 本研究采用美国环境保护署(U.S. Environmental Protection Agency, EPA)提出的个人吸入暴露的计算方法评估0—6岁儿童健康风险[26], 计算公式如下:
Eij=Cij×IRj×tj (1) 式中, Eij 为微环境 (j) 中污染物(i) 的日吸入剂量, μg·d−1; Cij为微环境 (j) 中污染物 (i) 的浓度, 即室内羰基化合物的实测浓度, μg·m−3; IRj 是微环境中的吸入率 (j) , m3·h−1, 2岁以下儿童IRj为0.22 m3·h−1, 2—6岁儿童为0.33 m3·h−1; tj 是在微环境 (j) 中的暴露时间, 即儿童每天在室内度过的时间, h·d−1, 2岁以下儿童tj为21.37 h·d−1 , 2—6岁儿童为20.15 h·d−1. IRj和tj 数据均来源于中国人群暴露参数手册(儿童卷)概要[27].
根据最近的研究中采用的方法[26, 28], 对儿童而言, 需要调整无显著风险水平(no significant risk levels, NSRLs) [26] , 调整后的儿童特定NSRLs: 2 岁以下儿童, 甲醛、乙醛分别为 0.52 μg·d−1、1.16 μg·d−1; 2—6岁儿童, 甲醛、乙醛分别为 3.15 μg·d−1、7.08 μg·d−1. 此外, 用风险商(risk quotients, RQ)来表示健康风险, RQ 的计算方法是将儿童每日吸入剂量除以儿童特定的 NSRLs. RQ >1 表明儿童吸入剂量超过了 10−5 终生致癌风险阈值, 即儿童面临潜在的癌症风险.
1.5 气味污染评价方法
除了对人体健康造成影响外, 羰基化合物还会产生气味污染. 羰基化合物的质量浓度高并不意味着其气味强度也大, 而是与气味阈值共同呈比例地影响人体对气味的感知. 为了将污染物的浓度和气味强度联系起来, 需要对其气味进行污染评价. 气味污染评价中羰基化合物的气味强度用气味活性值(odor activity value, OAV)表示[13, 29]:
OAVi=CmiCti (2) 式中, OAVi 为污染物 i 的气味活性值, 无量纲; Cmi为污染物 i 的质量浓度, μg·m−3; Cti为污染物 i 的气味阈值, μg·m−3, 其取自日本环境卫生中心对有害气体污染物气味阈值的研究结果[30].
污染物的气味活性值以大多数正常人的感官为准, OAVi ≤1 表示一般人无法感知到污染物 i 的气味; OAVi >1可感知到污染物 i 所散发的气味, 且 OAVi 越大, 能感知到的气味越大, 即其气味强度越大.
2. 结果与讨论 (Results and discussion)
2.1 羰基化合物的浓度水平
室内环境中羰基化合物的质量浓度为(144.3±56.2) μg·m−3, 其中甲醛(25.5%)、丙酮(19.1%)和乙醛(15.0%)是含量最高的3种物质, 其平均浓度分别为(36.8±21.0) μg·m−3、(27.5±14.3) μg·m−3、(21.7±14.4) μg·m−3, 均明显高于室外浓度(图1). 这3类羰基化合物的来源与居住者的生活息息相关, 可从建筑材料、环境烟草烟雾、烹饪油烟、香味消费品、个人护理产品、清洁等人为活动或通过室内VOCs(尤其是萜烯)的氧化排放[31-33]. 室内高分子量羰基化合物(C6—C10)占比也比较大, 约27.7%(15.9%—45.9%), C6—C10平均浓度(39.9 μg·m−3)是室外(9.0 μg·m−3)的4.4倍. 研究表明, 己醛不仅包含在香料和香精中, 还存在于含有植物化学物质的食物和办公家具[34]. 辛醛有很强的水果香味, 可用作食用香料, 也可作肥皂洗涤剂的香料使用. 壬醛具有玫瑰、柑橘等香气, 存在于红茶、绿茶中. 室外大气环境中羰基化合物的质量浓度为(63.0±21.8) μg·m−3, 其中丙酮浓度最高((14.4±4.8) μg·m−3), 其次是甲醛((13.5±6.8) μg·m−3)和乙醛((11.0±4.3) μg·m−3), 这3类化合物约占室外总羰基化合物浓度的61.8%. I/O 比值(室内/室外)能更好地推断室内羰基化合物的可能影响因素(室内源或室外渗透). 由图1可知, 乙二醛(Gly)和甲基乙二醛(mGly)的I/O 比值均低于1.0, 表明这两类典型的α-二羰基物质可能更多的来自室外环境的渗透. 研究发现, 室外大气环境中乙二醛和甲基乙二醛主要来源于生物质燃烧和挥发性有机化合物(如异戊二烯、芳烃和烯烃)的氧化[35-36]. 值得注意的是,低分子量的羰基化合物甲醛、乙醛、丙酮和高分子量羰基化合物(C6—C10)的 I/O 比值明显较高(1.9—11.8),说明室内排放源对这些羰基化合物有显著贡献.
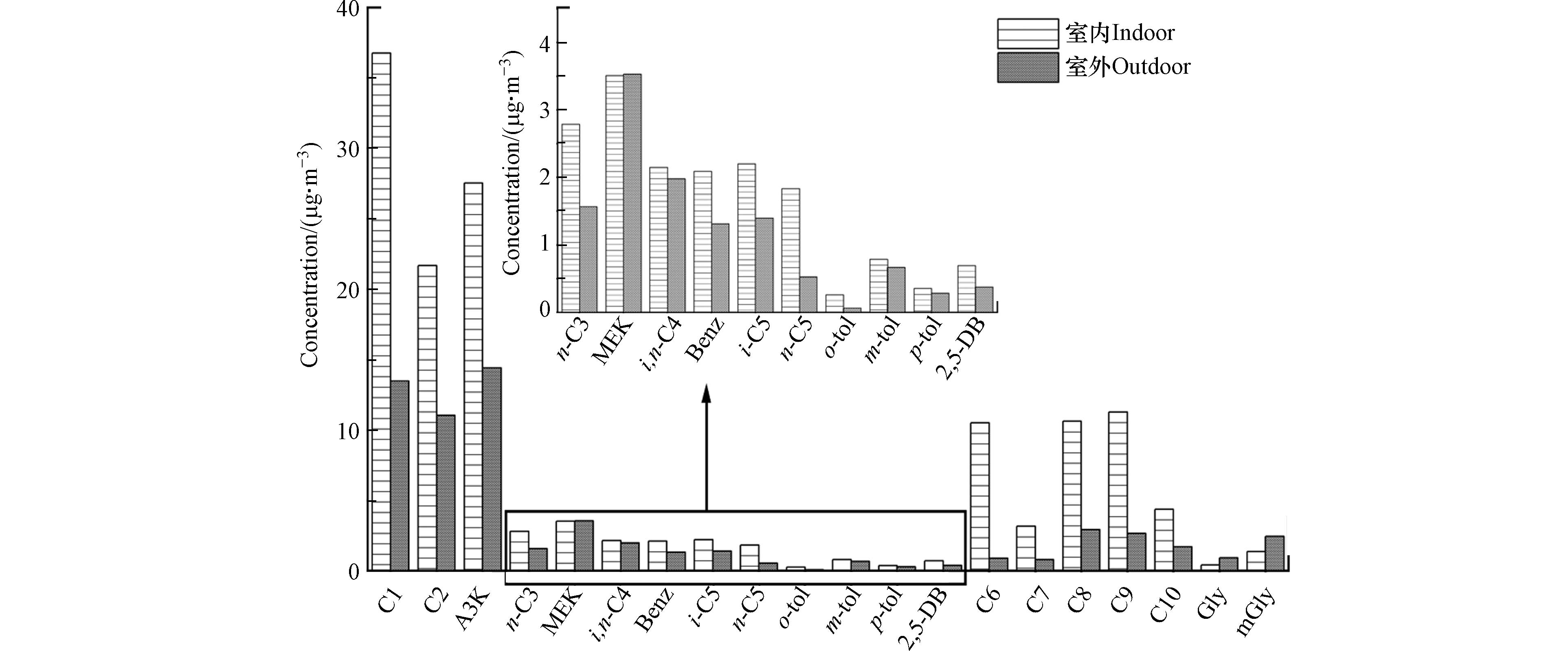 图 1 住宅室内外环境中羰基化合物浓度Figure 1. Concentrations of carbonyl compounds in indoor and outdoor environment of residential buildings
图 1 住宅室内外环境中羰基化合物浓度Figure 1. Concentrations of carbonyl compounds in indoor and outdoor environment of residential buildings从表2和表3中可以看出, 室内环境中非供暖季浓度最高的5种羰基化合物为甲醛、丙酮、辛醛、乙醛和壬醛, 其浓度范围分别为(17.6—49.9) μg·m−3、(13.1—18.3) μg·m−3、(8.3—19.3) μg·m−3、(7.6—12.9) μg·m−3和(4.0—8.8) μg·m−3. 而供暖季则为甲醛、丙酮、乙醛、壬醛和己醛, 其浓度范围分别为(23.2—86.7) μg·m−3、(31.2—47.8) μg·m−3、(24.1—48.5) μg·m−3、(14.9—18.9) μg·m−3和(10.8—30.0) μg·m−3, 均明显高于非供暖季. 该结果与有关包头市城区住宅羰基化合物污染水平的研究结果一致, 可能与供暖季住宅内温湿度、通风换气次数以及室外环境状况等因素有关[20]. 整体来看,无论供暖季还是非供暖季, 住宅室内环境中最丰富的3类羰基化合物均为甲醛、乙醛和丙酮, 这与国内其它城市(北京、大连、上海、武汉和长沙等)的研究结果一致[26]. 另外, 本文所关注的3类高分子量羰基化合物(己醛、辛醛和壬醛)占比也较高(14.4%—33.0%)(图2). Feng 等[37]研究发现,餐饮油烟可能是造成空气中高分子量羰基化合物浓度偏高的原因之一. 由此可见, 室内高分子量的羰基化合物可能来源于住户的烹饪活动. 室外大气环境中羰基化合物浓度表现为非供暖季(68.9±28.2) μg·m−3明显高于供暖季(55.3±9.4) μg·m−3, 其中甲醛浓度增量最大(6.6 μg·m−3), 其次是辛醛(3.5 μg·m−3)和甲基乙二醛(2.5 μg·m−3), 这归因于非供暖季温度偏高, 太阳辐射较强, 有利于大气有机物发生光化学反应, 同时非供暖季植物排放对羰基化合物有重要的贡献.
表 3 非供暖季室内外环境中羰基化合物浓度Table 3. Concentrations of carbonyl compounds in indoor and outdoor environments in non-heating season羰基化合物Carbonyl compounds 平均值±标准偏差 (AVG±SD)/(μg·m−3) QJ-1 QJ-2 QJ-3 QJ-4 QJ-5 QJ-O 甲醛(C1) 49.9 ± 11.1 19.9 ± 2.3 22.4 ± 3.1 32.2 ± 7.9 17.6 ± 2.1 16.3 ± 8.2 乙醛(C2) 12.9 ± 2.8 8.7 ± 1.5 9.7 ± 2.9 12.1 ± 4.1 7.6 ± 1.9 10.1 ± 5.4 丙酮(A3K) 17.8 ± 4.1 18.3 ± 3.9 17.0 ± 6.7 16.8 ± 6.2 13.1 ± 3.6 14.1 ± 5.7 丙醛(C3) 2.1 ± 0.7 1.3 ± 0.0 1.4 ± 0.4 2.1 ± 0.6 0.9 ± 0.2 1.0 ± 0.2 2-丁酮(MEK) 2.8 ± 0.7 2.4 ± 0.4 2.5 ± 0.5 3.7 ± 2.1 4.0 ± 2.7 3.9 ± 1.4 丁醛(i,n-C4) 1.3 ± 0.2 1.1 ± 0.1 1.4 ± 0.1 1.5 ± 0.5 3.1 ± 4.1 2.5 ± 2.3 苯甲醛(Benz) 2.3 ± 0.6 2.1 ± 0.8 1.9 ± 0.8 2.7 ± 1.1 1.2 ± 0.2 1.5 ± 1.0 异戊醛(i-C5) 1.7 ± 0.4 1.2 ± 0.3 0.9 ± 0.1 2.3 ± 1.8 1.0 ± 0.2 1.2 ± 0.3 正戊醛(n-C5) 0.9 ± 0.3 1.1 ± 0.3 0.8 ± 0.3 1.0 ± 0.5 0.4 ± 0.2 0.5 ± 0.1 邻甲苯甲醛(o-tol) bda bd bd bd bd bd 间甲苯甲醛(m-tol) 1.1 ± 0.4 1.1 ± 0.5 1.1 ± 0.6 0.9 ± 0.2 1.0 ± 0.5 0.8 ± 0.5 对甲苯甲醛(p-tol) 0.4 ± 0.0 0.2 ± 0.0 0.3 ± 0.1 0.2 ± 0.0 0.3 ± 0.2 0.4 ± 0.2 2,5-二甲基苯甲醛(2,5-DB) 1.2 ± 0.7 0.7 ± 0.2 0.8 ± 0.4 0.9 ± 0.2 0.4 ± 0.1 0.3 ± 0.1 己醛(C6) 6.1 ± 1.9 8.1 ± 2.4 5.1 ± 2.4 5.0 ± 1.7 1.4 ± 0.2 0.9 ± 0.2 庚醛(C7) 3.5 ± 1.6 1.9 ± 0.5 1.6 ± 0.7 2.7 ± 1.1 0.9 ± 0.2 0.8 ± 0.3 辛醛(C8) 15.8 ± 5.0 19.3 ± 5.6 19.2 ± 10.4 13.6 ± 4.5 8.3 ± 2.1 4.4 ± 1.2 壬醛(C9) 8.8 ± 3.4 5.6 ± 1.1 4.2 ± 1.9 8.4 ± 3.0 4.0 ± 0.9 3.1 ± 1.5 癸醛(C10) 7.2 ± 2.8 4.6 ± 0.9 3.4 ± 1.6 7.0 ± 2.5 3.3 ± 0.7 2.5 ± 1.3 乙二醛(Gly) 0.3 ± 0.1 0.3 ± 0.2 0.4 ± 0.4 0.3 ± 0.2 0.5 ± 0.5 1.0 ± 0.9 甲基乙二醛(mGly) 1.7 ± 0.5 1.9 ± 0.7 1.9 ± 1.0 2.0 ± 0.6 2.0 ± 0.9 3.5 ± 2.2 总和 137.7 ± 35.0 100.0 ± 17.2 95.8 ± 29.5 115.4 ± 37.6 70.9 ± 10.8 68.9 ± 28.2 注: a bd 低于检测下限。represents below limit of detection. | Show TableDownLoad:
CSV
室内环境中甲醛、乙醛、丙酮的浓度范围分别为(17.6—86.7) μg·m−3、(7.6—48.5) μg·m−3、(13.1—47.8) μg·m−3, 不同住户室内羰基化合物浓度差异明显, 这可能与不同的生活习惯以及装修样式等因素有关[20-21]. 具体来看, 供暖季QJ-1室内甲醛平均浓度((86.7±10.8) μg·m−3)明显超过2022年7月11日国家市场监督管理总局(国家标准化管理委员会)批准发布的新版标准(GB/T 18883—2022) [38]中提到的室内甲醛1小时平均浓度(80 μg·m−3), 可能与该住宅在采样前期新增家具(床和衣柜)有关. QJ-3供暖季室内乙醛浓度((48.5±8.9) μg·m−3)比日本厚生劳动省出台的室内乙醛浓度限值(48 μg·m−3)略高[39]; 整体来看, QJ-1—QJ-5室内丙醛浓度均未超过加拿大卫生部报道的暴露限值(8 μg·m−3), 丙醛达到该限值的临界效应表现为嗅觉上皮萎缩[40].
图2为供暖季与非供暖季住宅室内外空气中羰基化合物的浓度占比. 从图2可以看出, 低分子量的甲醛、乙醛和丙酮对室内外空气中羰基化合物的贡献较大, 占总羰基化合物浓度的47.0%—73.0%, 且供暖季的占比显著高于非供暖季, 其中乙醛占比全部表现为供暖季 > 非供暖季; 结合表2和表3浓度数据发现, 室内外环境中的乙醛浓度也均表现为供暖季 > 非供暖季. 值得注意的是, QJ-2无论是供暖季还是非供暖季, 己醛、辛醛和壬醛这3类羰基化合物的占比之和都超过 30%, 而甲醛、乙醛和丙酮占比之和较其它住宅偏低,推测是因为该住宅内选择瓷砖铺设且家具量较少. 此外, 与其它住户相比, QJ-2选用液化石油气作为烹饪燃料可能对室内羰基化合物的污染情况也有影响,需进一步研究.
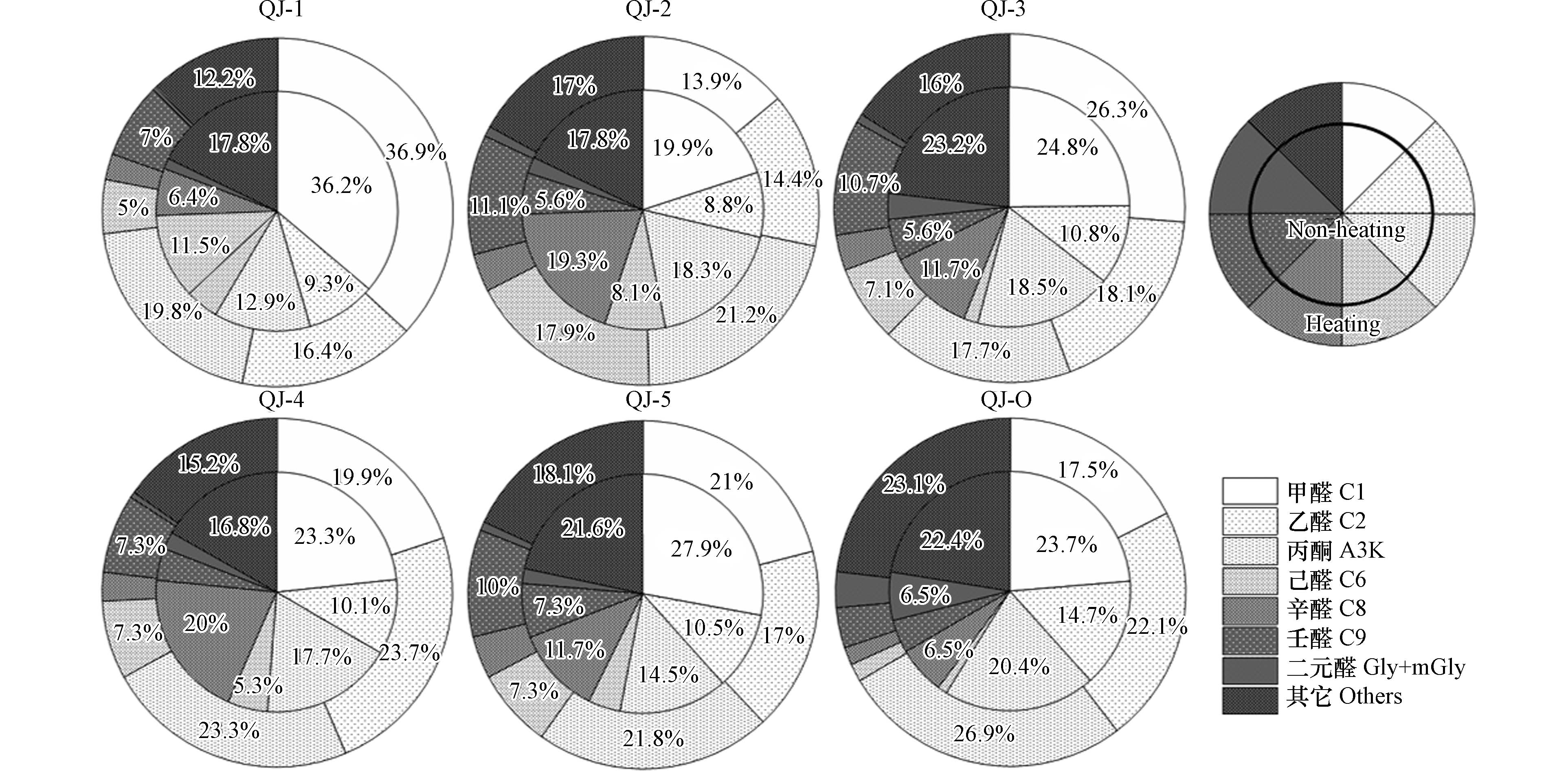 图 2 住宅室内外环境中羰基化合物占比Figure 2. The proportion of carbonyl compounds in the indoor and outdoor environment of residential buildings
图 2 住宅室内外环境中羰基化合物占比Figure 2. The proportion of carbonyl compounds in the indoor and outdoor environment of residential buildings2.2 羰基化合物的影响因素
2.2.1 通风换气次数
诸多研究表明, 通风换气次数越大, 室内污染物浓度越小[20,29]. 为了解室内的通风状况, 本研究采用CO2示踪气体衰减法[29,41]计算得到各住宅供暖季和非供暖季的通风换气次数(表4). 该方法以CO2为示踪气体, 当室内CO2浓度比室外高时, 由于室内外的空气交换,室内CO2浓度会呈指数衰减, 式(3)为该过程中的质量守恒方程, 经推导可将CO2浓度与换气次数的关系写成式(4), 再通过线性回归计算, 即可求得室内的换气次数[29].
表 4 供暖季与非供暖季住户通风换气次数Table 4. The air exchange rate of households in heating season and non-heating season采样点Site 供暖季Heating season 非供暖季Non-Heating season 范围/h−1Range 平均值±标准偏差/h−1Mean±SD 范围/h−1Range 平均值±标准偏差/h−1Mean±SD QJ-1 0.1—0.7 0.3 ± 0.2 2.6—23.5 8.5 ± 8.6 QJ-2 0.4—0.8 0.6 ± 0.2 4.2—13.4 7.0 ± 3.8 QJ-3 0.2—0.6 0.4 ± 0.2 3.2—34.5 12.1 ± 15.0 QJ-4 0.2—0.5 0.4 ± 0.1 3.0—21.6 10.3 ± 8.8 QJ-5 0.1—0.6 0.3 ± 0.2 5.8—48.6 16.7 ± 18.0 | Show TableDownLoad:
CSV
VdCindτ=G(Cout−Cin) (3) ln(Cin−Cout)=ln(Cin,0−Cout)−ACHτ (4) 式中, Cin为室内CO2实时浓度, mg·m−3; Cout为室外CO2浓度, mg·m−3; G为通风量, m3·h−1; ACH为每小时换气次数, h−1; τ为时间, h; Cin,0表示室内CO2初始浓度, mg·m−3.
如表4所示, 供暖季室内通风换气次数为(0.1—0.8) h−1, 非供暖季为(2.6—48.6) h−1, 非供暖季换气次数比供暖季高1—2个数量级. 显然, 高频率的通风换气次数是非供暖季室内环境中羰基化合物浓度显著低于供暖季的原因. 但是, 室内羰基化合物的浓度还受室内温湿度、烹饪活动、人员情况(人员数量、人员类型等)、生活习性(个人护理产品、卫浴产品使用等)以及室外污染状况等其它诸多因素影响[31-33,37]. 此外, 室内外环境中二元醛(Gly+mGly)浓度则表现为非供暖季高于供暖季, 而且室内二元醛浓度随着室外浓度的增加而增加, 进一步表明室外源对室内贡献明显.
2.2.2 周末效应
“周末效应”的概念在 1974 年被Cleveland 等[42]首次提出. 研究表明, 大气污染物质量浓度变化在工作日和周末存在明显差异, 这种变化被称为“周末效应”[43-44]. 周末效应源于工作和休息模式的节律性, 与人类活动密切相关. 本文采用如下方式量化周末效应(ΔWE)[44-46]:
ΔWE=[(C周末−C工作日)/C工作日] (5) 式中, C工作日 表示工作日的浓度均值, C周末 表示周末的浓度均值. 为保持数据一致性, 本研究中C工作日 采用周五的污染物浓度, C周末 采用周六的污染物浓度.
图3绘制了供暖季(a)和非供暖季(b)住宅室内外空气中羰基化合物浓度的相对偏差. 从图3可以看出, 供暖季, 不同住宅室内环境中羰基化合物的周末效应并不一致, 除QJ-5外, 其余4户住宅室内甲醛和丙酮浓度周末相比工作日增加, 分别增加了12.3%—32.3%、15.9%—69.4%; 高分子量羰基化合物(C6—C10)也表现出周末浓度高于工作日的“周末效应”. 室外大气环境中甲醛和乙醛浓度周末相比工作日减少, 分别减少了15.3% 和13.7%, 而丙酮则增加了26.7%; 高分子量的羰基化合物(C6—C10)浓度降幅超过30%. 非供暖季, “周末效应”更明显, 无论室内环境还是室外大气, 甲醛、乙醛、丙酮、壬醛、癸醛、乙二醛和甲基乙二醛均表现出工作日浓度高于周末的“周末效应”. 室内环境中甲醛、乙醛和丙酮周末浓度相较于工作日分别减少了15.9%—34.6%、30.7%—45.6%、30.6%—55.1%; 室外大气中这三类污染物浓度同样是周末低于工作日, 分别减少了51.0%、53.1%和53.6%.
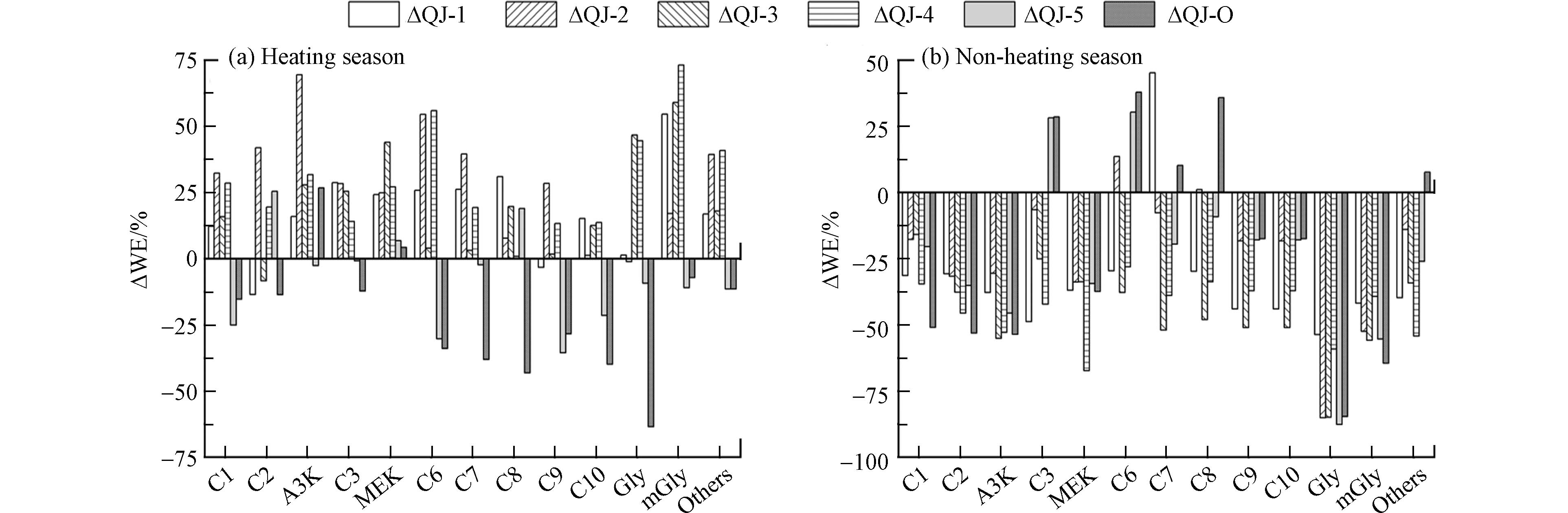 图 3 室内外空气中周末与周中羰基化合物浓度的相对偏差Figure 3. Deviations of concentration of carbonyl compounds during weekend and week in indoor and outdoor environments
图 3 室内外空气中周末与周中羰基化合物浓度的相对偏差Figure 3. Deviations of concentration of carbonyl compounds during weekend and week in indoor and outdoor environments综上所述, 住宅室内环境中, 供暖季多数羰基化合物呈周末高于工作日的“周末效应”, 非供暖季则呈工作日高于周末的“周末效应”; 这归因于供暖季室外较低的温度, 居民周末更多选择室内活动以及低频的通风换气次数导致室内环境中多数羰基化合浓度周末高于工作日; 而非供暖季相对较好的通风条件使室内外羰基化合物浓度的“周末效应”变化一致, 均表现为周末低于工作日. 室外环境中羰基化合物浓度不受季节变化影响, 均呈现工作日高于周末的“周末效应”. 研究发现, 在典型的城市地区PM2.5具有明显的工作日偏高, 周末偏低的“周末效应”[46]. 由此可见, 室外羰基化合物受到人类活动、污染排放和气象等各种因素的综合影响.
2.3 健康风险评估
表5总结了各住户室内儿童每日吸入甲醛和乙醛的剂量. 2岁以下儿童的甲醛和乙醛日吸入量最高值分别为313.9 μg·d−1和121.1 μg·d−1, 2—6岁的儿童甲醛和乙醛日最高吸入量分别为457.5 μg·d−1和176.5 μg·d−1. 可发现随着年龄的增长, 身体不断发育, 儿童羰基污染物吸入剂量增加. 供暖季室内甲醛、乙醛浓度高于非供暖季, 因此供暖季节室内羰基化合物污染情况更值得关注.
表 5 各住户室内儿童每日吸入的甲醛和乙醛剂量及风险商Table 5. Daily inhalation doses and risk quotients of formaldehyde and acetaldehyde by children in each household甲醛Formaldehyde 乙醛Acetaldehyde 采样点Site 2岁以下Birth to < 2 years 2—6 岁2 years to < 6 years 2岁以下Birth to < 2 years 2—6 岁2 years to < 6 years 吸入剂量/(μg·d−1) Daily inhalation dose 风险商Risk Quotients 吸入剂量/(μg·d−1) Daily inhalation dose 风险商Risk Quotients 吸入剂量/(μg·d−1) Daily inhalation dose 风险商Risk Quotients 吸入剂量/(μg·d−1) Daily inhalation dose 风险商Risk Quotients QJ-1 313.9 609.0 457.5 145.4 117.9 101.6 171.9 24.3 QJ-2 99.2 192.4 144.5 45.9 75.4 65.1 109.9 15.5 QJ-3 140.2 272.1 204.4 64.9 121.1 104.4 176.5 24.9 QJ-4 145.0 281.3 211.3 67.1 85.5 73.7 124.6 17.6 QJ-5 137.4 266.6 200.3 63.6 82.9 71.5 120.9 17.1 | Show TableDownLoad:
CSV
儿童特定的无显著风险水平用于表征儿童在家中接触甲醛和乙醛的健康风险[26]. 如表5所示,每户儿童接触甲醛和乙醛的风险商均远大于1, 表明各住户儿童皆面临潜在的癌症风险. 2岁以下儿童甲醛暴露RQ值为192.4—609.0, 2—6岁儿童甲醛暴露RQ值为45.9—145.4, 即儿童的甲醛暴露量是“安全限”水平的数十至数百倍. 2岁以下儿童乙醛暴露RQ值为65.1—104.4, 2—6岁儿童乙醛暴露RQ值为15.5—24.9. 与乙醛相比, 儿童在家接触甲醛对其健康的影响相对更大. 这与Bradman 等[28]研究中甲醛暴露的 RQ 值(12.0—51.7)高于乙醛暴露的 RQ 值(2.3—9.8)的结果一致.
需指出, 在计算儿童吸入甲醛和乙醛的剂量时, 只考虑了儿童在住宅中接触羰基化合物的情况, 如果还考虑到儿童在学校等其它室内环境接触羰基化合物, 患癌风险会更大. 因此, 室内羰基化合物污染对儿童健康的影响, 需要居民和标准制定者高度重视. 此外, 由于缺乏指南和风险指数, 其它含量较高的羰基化合物(例如己醛和壬醛)的化学危害潜力或癌症风险无法系统地评估.
2.4 气味污染评价
各住户室内环境中不同污染物对应的气味活性值如表6所示. 在分析各住宅所散发的羰基化合物成分时, 检测到较高浓度的甲醛、乙醛和丙酮, 但由于甲醛和丙酮的气味阈值相对较高, 因此这两种高浓度污染物的气味活性要低得多, 只有乙醛的气味活性更明显, 其气味活性值均大于5. 各住宅室内环境中高分子量的己醛、庚醛、辛醛、壬醛和癸醛的气味活性值均大于1, 尤其是辛醛最大, 均大于100, 其次是己醛. 对于广泛用作装修和家居的木质材料释放的气味物质——辛醛具有肥皂和柑橘味, 己醛具有香草味. 吴可[29]研究发现, 装修完工后室内环境中的气味污染主要来自于庚醛、辛醛、壬醛和己醛等醛类污染物. 而且这些高分子量的醛类污染物释放量在臭氧的影响下还会随时间而增加[47]. 因此, 室内环境中羰基化合物的气味污染更多应关注高分子量羰基化合物. 已有研究表明, 室内不良的气味感知是病态建筑综合征(sick building syndrome, SBS)的危险因素, 高分子量的羰基化合物对SBS的影响相当明显, 己醛对SBS的一般症状的危险性达到了显著性水平, 癸醛和辛醛对SBS的皮肤症状的危险性也达到了显著性水平[48]. 所以, 考虑针对室内环境中高分子量羰基化合物规定限值亦十分必要.
表 6 各住宅室内污染物的气味活性值Table 6. Odor activity values of indoor pollutants in various residences羰基化合物Carbonyl compounds 气味阈值/(μg·m−3)Odor threshold 气味活性值Odor activity value QJ-1 QJ-2 QJ-3 QJ-4 QJ-5 甲醛 670.3 0.10 0.03 0.05 0.05 0.04 乙醛 3.0 8.7 5.6 8.9 6.3 6.1 丙酮 108900.0 <0.01 <0.01 <0.01 <0.01 <0.01 丙醛 2.6 1.4 0.9 1.1 1.0 0.9 2-丁酮 1416.4 <0.01 <0.01 <0.01 <0.01 <0.01 丁醛 1.6 1.3 1.1 1.3 1.2 1.7 异戊醛 0.4 6.7 5.1 4.7 6.3 5.4 正戊醛 1.6 1.1 1.8 1.1 1.0 0.7 己醛 1.3 7.1 15.2 7.4 6.3 4.9 庚醛 0.8 5.1 4.0 3.5 3.9 2.7 辛醛 0.06 187.8 220.4 232.1 169.1 126.3 壬醛 2.2 5.8 5.6 4.0 5.4 4.8 癸醛 2.8 1.8 1.5 1.1 2.2 1.2 | Show TableDownLoad:
CSV
3. 结论(Conclusion)
本研究选择5户城市住宅进行同步观测, 对室内外空气环境中痕量羰基化合物的浓度水平、影响因素、健康风险及气味污染展开研究. 通过分析, 得出以下结论:
(1)居民住宅室内外环境中总羰基化合物浓度变化表现为: 室内 > 室外; 室内供暖季 > 室内非供暖季; 室外供暖季 < 室外非供暖季; 室内外空气中甲醛、乙醛和丙酮3类羰基化合物比重高(47.0%—73.0%), 室内高分子量的羰基化合物(C6—C10)总浓度(39.9 μg·m−3)是室外(9.0 μg·m−3)的4倍多. 季节变化表明, 通风换气次数对室内环境中羰基化合物污染影响显著.
(2)供暖季室内多数羰基化合物呈周末高于工作日的“周末效应”, 非供暖季则呈工作日高于周末的“周末效应”. 室外环境中羰基化合物浓度不受季节影响均呈现工作日高于周末的“周末效应”.
(3)健康风险评估显示各住户儿童甲醛和乙醛的吸入剂量均远超过OEHHA 给出的暴露剂量安全限值, 表明各家庭的儿童都面临潜在的癌症风险. 住宅室内环境中主要气味污染来自于气味阈值较低的高分子量羰基化合物, 包括己醛、庚醛、辛醛、壬醛和癸醛.
需指出, 本研究中样本量并不是很充足, 但室内外同步观测结果也能反映城市住宅室内外空气中痕量羰基化合物不同季节的污染情况; 此外, 不同住户家庭成员的数量以及不同的生活习惯等因素对室内羰基污染物的影响各不相同, 关于这部分讨论还有待于进一步的研究. 尽管如此, 这些痕量羰基化合物的测定对我国室内空气质量监管的来源分配和数据保存仍具有价值.
-
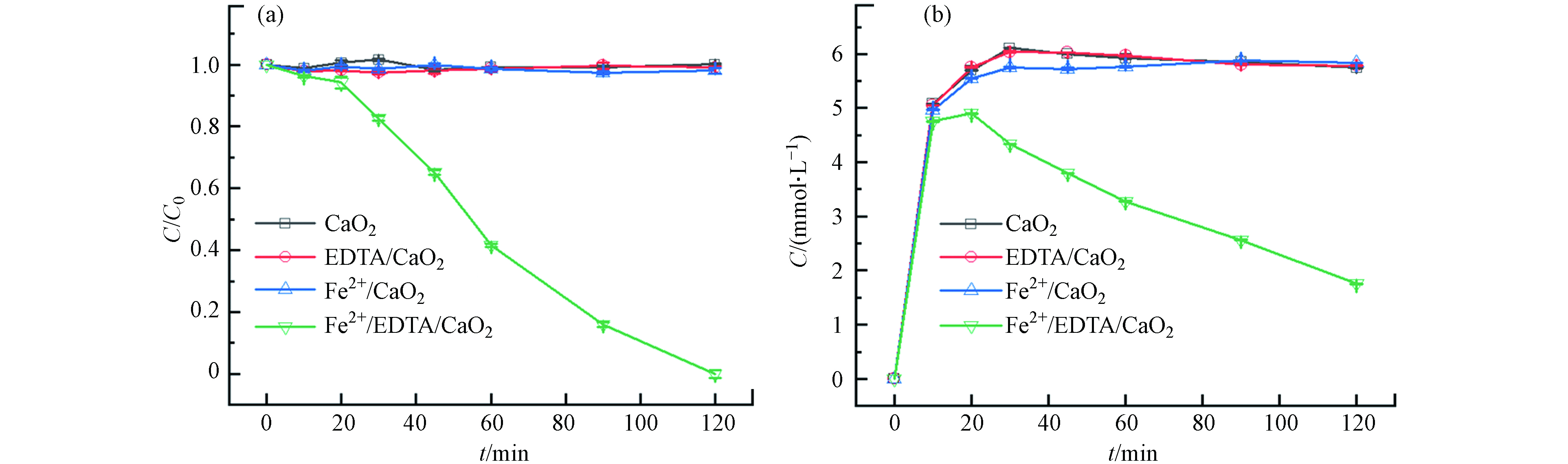
图 1 不同组分体系中苯酚的降解效果(a)及H2O2的浓度变化(b)
Figure 1. The degradation of phenol(a)and the change of H2O2 concentration(b)in different component systems
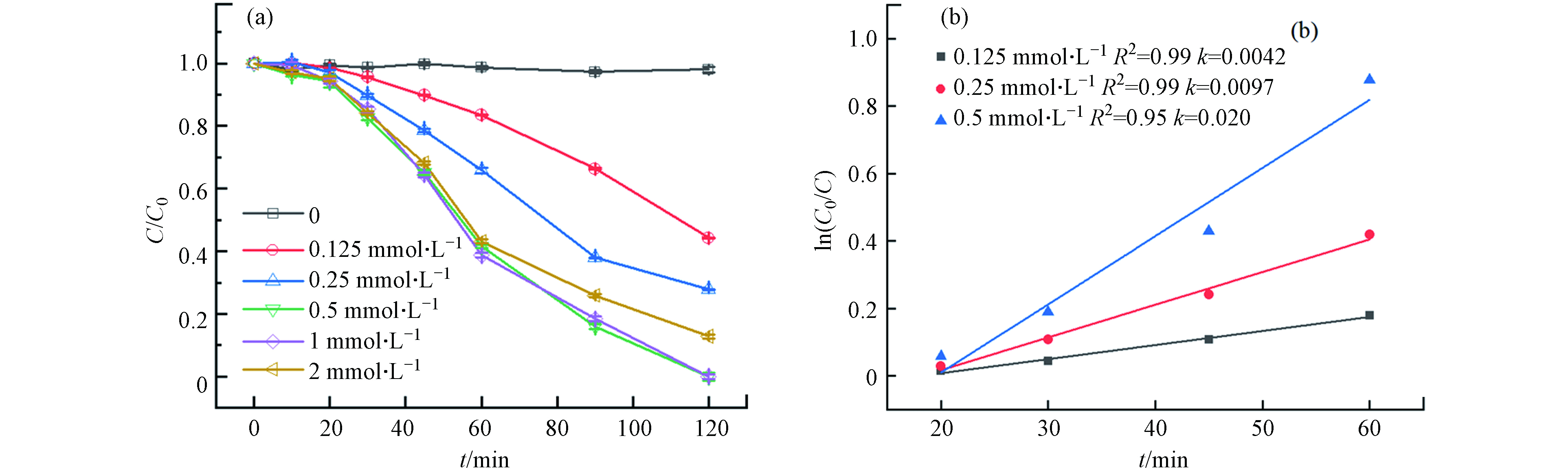
图 2 EDTA浓度对Fe3+/EDTA/CaO2体系降解苯酚的影响(a)及动力学拟合(b)
Figure 2. Effect of EDTA concentration on the degradation of phenol(a)by Fe3+/EDTA/CaO2 system and kinetic fitting(b)
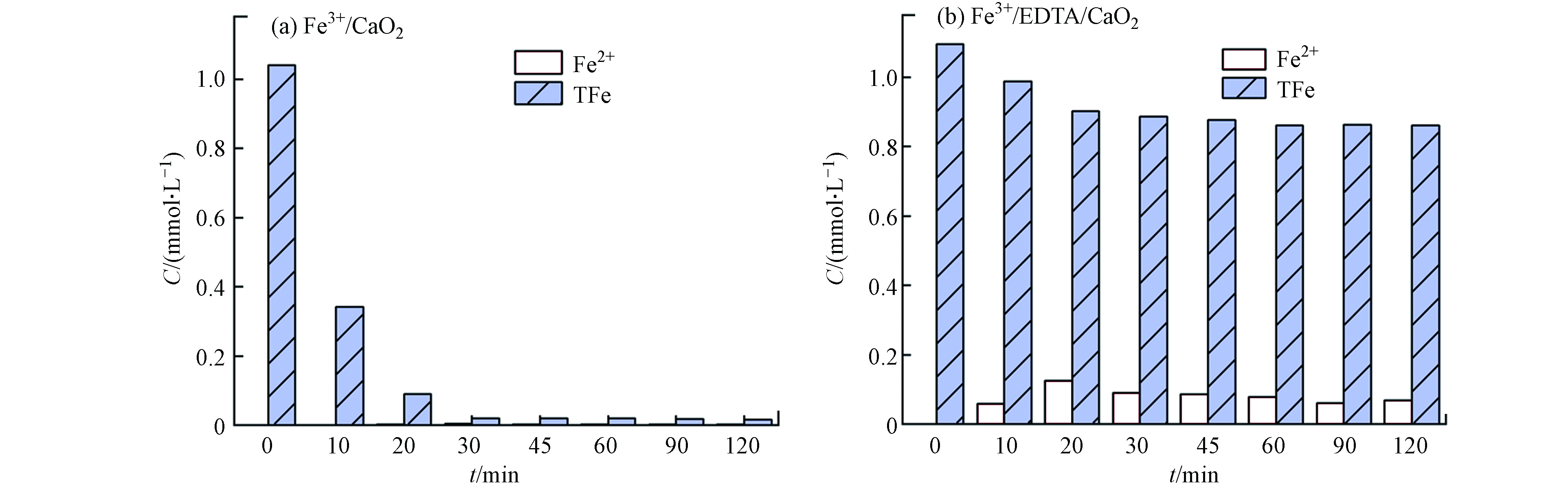
图 3 Fe3+/CaO2和Fe3+/EDTA/CaO2体系中游离态铁离子的浓度变化
Figure 3. Changes in the concentration of free state iron ions in the Fe3+/CaO2 and Fe3+/EDTA/CaO2 systems
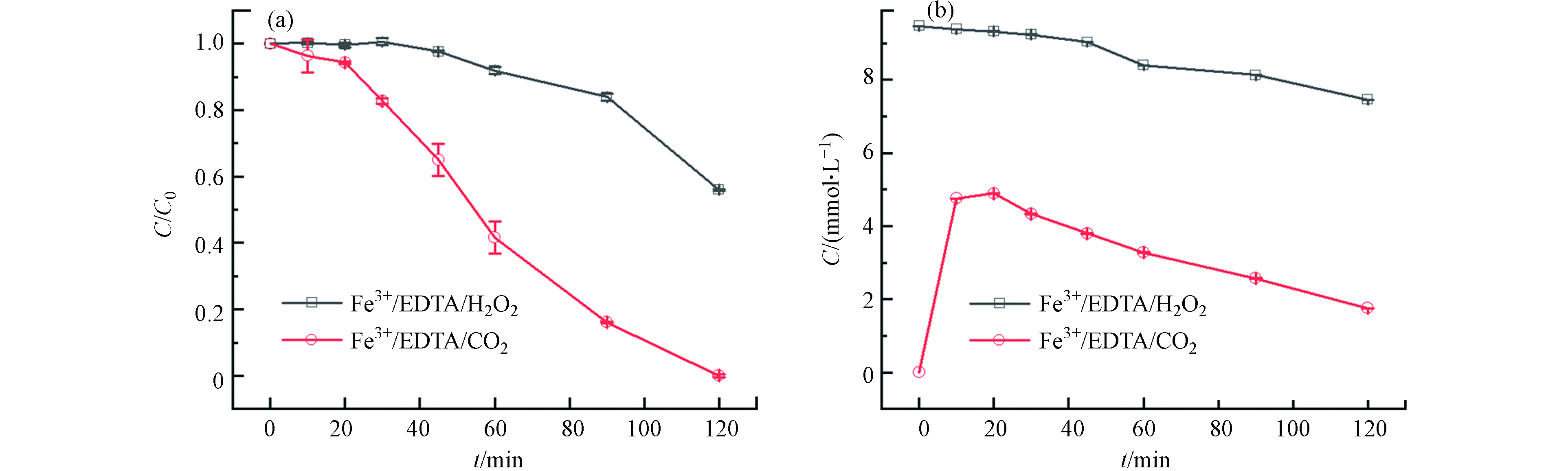
图 4 不同组分体系中的苯酚降解效果(a)及H2O2浓度变化(b)
Figure 4. The degradation of phenol(a)in different component systems(b)
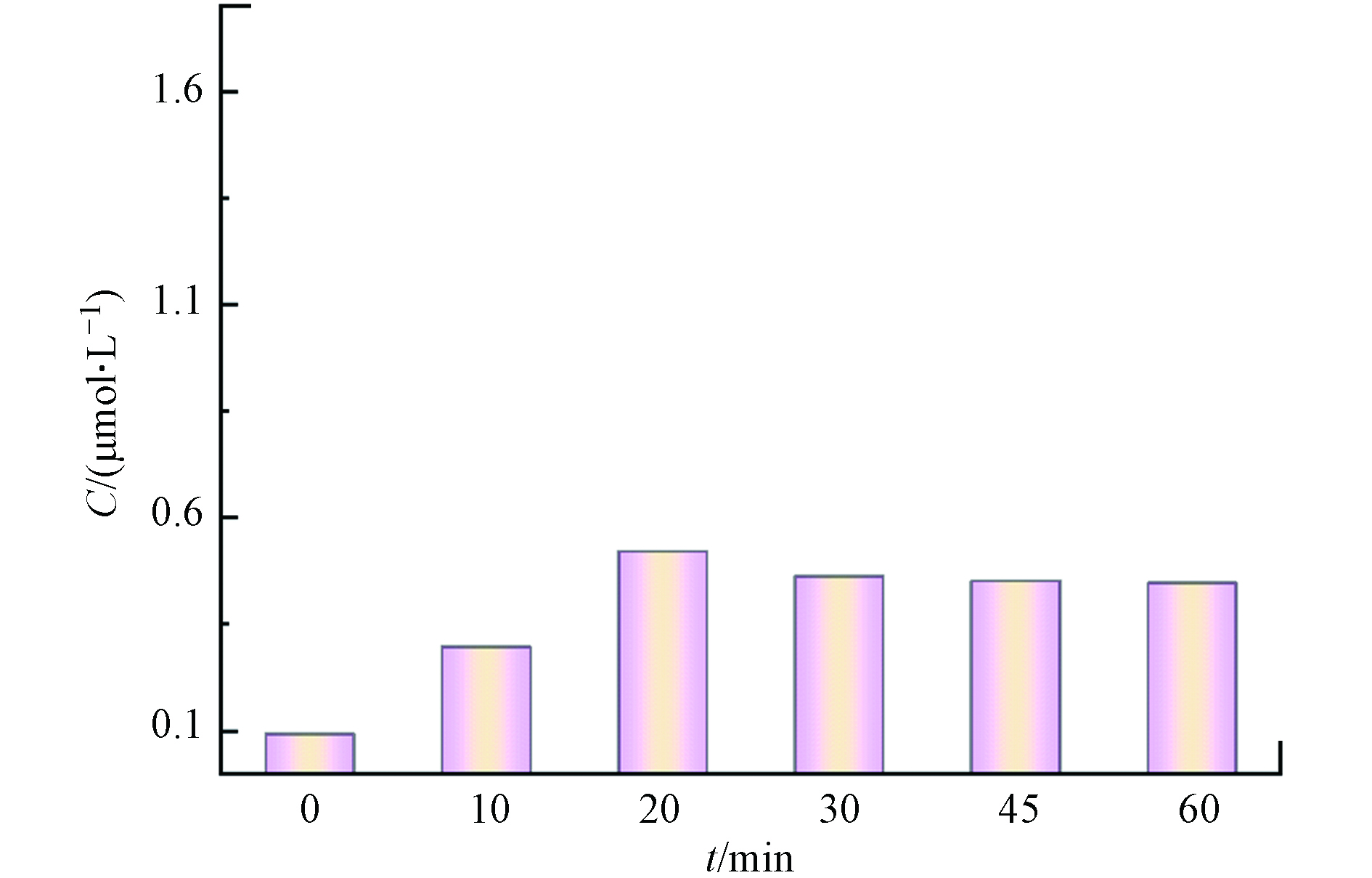
图 5 掩蔽H2O2后Fe3+/EDTA/CaO2体系中Fe2+的浓度变化
Figure 5. The change of Fe2+ concentration after clearing H2O2 in the Fe3+/EDTA/CaO2 system
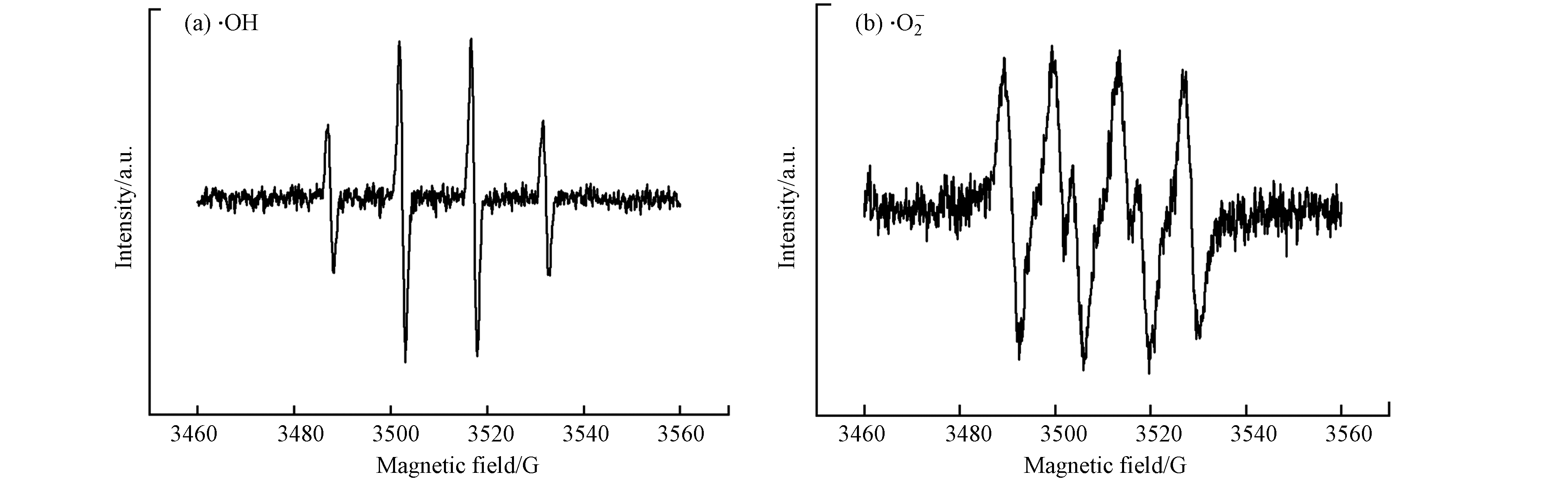
图 6 30 min时体系中活性自由基的EPR图
Figure 6. EPR of reactive free radicals in the system at 30 min
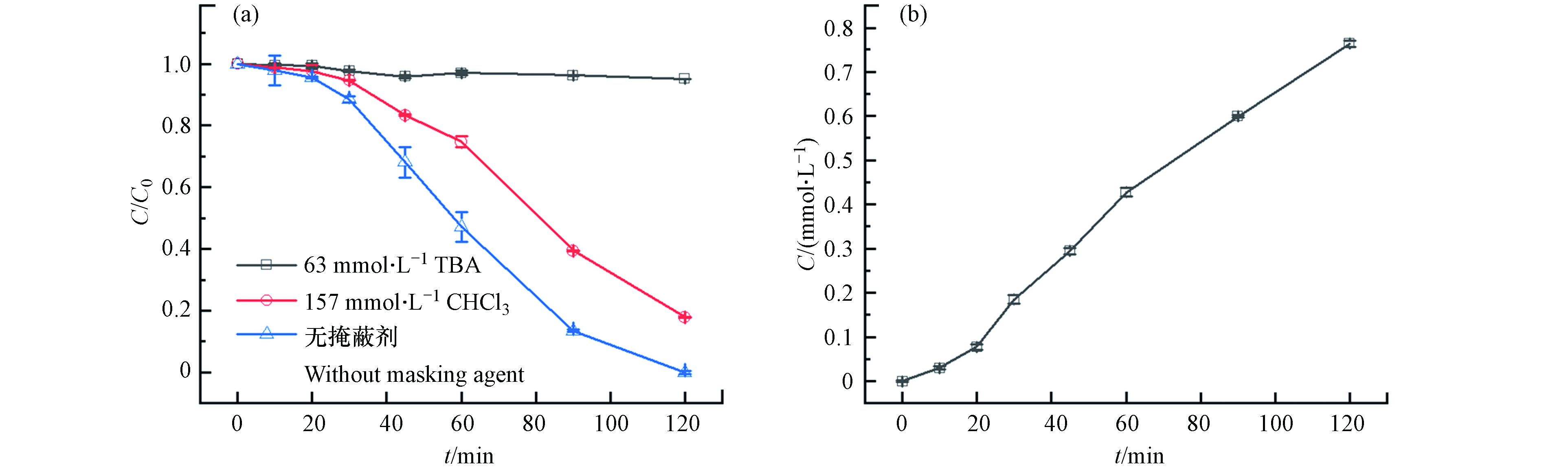
图 7 活性自由基的掩蔽(a)与探针实验(b)
Figure 7. Clearing (a) and probe experiments (b) of reactive free radicals
表 1 GC-MS测得苯酚降解的中间产物
Table 1. Intermediate products of phenol degradation measured by GC-MS
仪器出峰时间/min Peak time 化合物 Compound 分子结构 Molecular Structure 取样时间/min Sampling time 0 10 30 60 120 6.73 苯酚 
√ √ √ √ 12.5 对苯二酚 √ √ √ √ 12.1 邻苯二酚 √ √ √ √ 6.7 对苯醌 √ √ √ 3.5 乙醛酸 √ √
下载: 导出CSV
-
[1] FENTON H J H. Oxidation of tartaric acid in presence of iron [J]. Journal of the Chemical Society, Transactions, 1894, 65: 899-910. doi: 10.1039/CT8946500899 [2] NEYENS E, BAEYENS J. A review of classic Fenton's peroxidation as an advanced oxidation technique [J]. Journal of Hazardous Materials, 2003, 98(1/2/3): 33-50. [3] ALEKSIĆ M, KUŠIĆ H, KOPRIVANAC N, et al. Heterogeneous Fenton type processes for the degradation of organic dye pollutant in water—The application of zeolite assisted AOPs [J]. Desalination, 2010, 257(1/2/3): 22-29. [4] SAFARZADEH-AMIRI A, BOLTON J R, CATER S R. The use of iron in advanced oxidation processes [J]. Journal of Advanced Oxidation Technologies, 1996, 1(1): 18-26. [5] KOCHANY J, LIPCZYNSKA-KOCHANY E. Application of the EPR spin-trapping technique for the investigation of the reactions of carbonate, bicarbonate, and phosphate anions with hydroxyl radicals generated by the photolysis of H2O2 [J]. Chemosphere, 1992, 25(12): 1769-1782. doi: 10.1016/0045-6535(92)90018-M [6] 方兴斌. CaO2催化氧化水中双酚A及化学缓释供氧的研究[D]. 上海: 华东理工大学, 2017. FANG X B. Bisphenol A degradation using Fe3+-activated CaO2 and O2 supply through CaO2 slow relasing in microbial systems[D]. Shanghai: East China University of Science and Technology, 2017(in Chinese).
[7] 章琴琴, 丁世敏, 封享华, 等. Fenton法降解邻苯二甲酸二乙酯的动力学特征及其影响因素研究 [J]. 环境化学, 2020, 39(11): 3009-3016. doi: 10.7524/j.issn.0254-6108.2019082201 ZHANG Q Q, DING S M, FENG X H, et al. Study on the degradation kinetic characteristics and influencing factors of diethyl phthalate by Fenton treatment [J]. Environmental Chemistry, 2020, 39(11): 3009-3016(in Chinese). doi: 10.7524/j.issn.0254-6108.2019082201
[8] 钱婧, 李威, 张银龙, 等. Fenton法/草酸钠-Fenton法降解典型抗癌药5-氟尿嘧啶 [J]. 环境化学, 2014, 33(7): 1229-1234. doi: 10.7524/j.issn.0254-6108.2014.07.016 QIAN J, LI W, ZHANG Y L, et al. Degradation of anticancer drug 5-fluorouracil by Fenton and oxalic-Fenton process [J]. Environmental Chemistry, 2014, 33(7): 1229-1234(in Chinese). doi: 10.7524/j.issn.0254-6108.2014.07.016
[9] LI T Y, ZHANG C W, ZHANG J Y, et al. Remediation of 2, 4-dichlorophenol-contaminated groundwater using nano-sized CaO2 in a two-dimensional scale tank [J]. Frontiers of Environmental Science & Engineering, 2021, 15(5): 87. [10] ZHANG X, GU X G, LU S G, et al. Degradation of trichloroethylene in aqueous solution by calcium peroxide activated with ferrous ion [J]. Journal of Hazardous Materials, 2015, 284: 253-260. doi: 10.1016/j.jhazmat.2014.11.030 [11] WANG H F, ZHAO Y S, SU Y, et al. Fenton-like degradation of 2, 4-dichlorophenol using calcium peroxide particles: Performance and mechanisms [J]. RSC Advances, 2017, 7(8): 4563-4571. doi: 10.1039/C6RA26754H [12] LI Y M, WANG J, ZHANG A, et al. Enhancing the quantity and quality of short-chain fatty acids production from waste activated sludge using CaO2 as an additive [J]. Water Research, 2015, 83: 84-93. doi: 10.1016/j.watres.2015.06.021 [13] ENGELMANN M D, BOBIER R T, HIATT T, et al. Variability of the Fenton reaction characteristics of the EDTA, DTPA, and citrate complexes of iron [J]. Biometals, 2003, 16(4): 519-527. doi: 10.1023/A:1023480617038 [14] 陈东洋, 冯家力, 张昊, 等. 固相萃取/高效液相色谱法测定饮用水中苯并(a)芘及双酚A [J]. 分析测试学报, 2015, 34(7): 848-851. doi: 10.3969/j.issn.1004-4957.2015.07.017 CHEN D Y, FENG J L, ZHANG H, et al. Determination of benzo(a) pyrene and bisphenol A in drinking water by solid phase extraction/high performance liquid chromatography [J]. Journal of Instrumental Analysis, 2015, 34(7): 848-851(in Chinese). doi: 10.3969/j.issn.1004-4957.2015.07.017
[15] 张成武. 基于Fe(Ⅱ)-STPP配合物活化分子氧的高级氧化体系降解对硝基酚的效果及机理[D]. 长春: 吉林大学, 2019. ZHANG C W. Reserch on the effect and mechanism of P-nitrophenol degradation by advanced oxidation technology based on activation of molecular oxygen by Fe(Ⅱ)-STPP complex[D]. Changchun: Jilin University, 2019(in Chinese).
[16] LING Y H, LONG M C, HU P D, et al. Magnetically separable core-shell structural γ-Fe2O3@Cu/Al-MCM-41 nanocomposite and its performance in heterogeneous Fenton catalysis [J]. Journal of Hazardous Materials, 2014, 264: 195-202. doi: 10.1016/j.jhazmat.2013.11.008 [17] BIAGLOW J E, KACHUR A V. The generation of hydroxyl radicals in the reaction of molecular oxygen with polyphosphate complexes of ferrous ion [J]. Radiation Research, 1997, 148(2): 181-187. doi: 10.2307/3579576 [18] JOO S H, FEITZ A J, SEDLAK D L, et al. Quantification of the oxidizing capacity of nanoparticulate zero-valent iron [J]. Environmental Science & Technology, 2005, 39(5): 1263-1268. [19] PAN Y, SU H R, ZHU Y T, et al. CaO2 based Fenton-like reaction at neutral pH: Accelerated reduction of ferric species and production of superoxide radicals [J]. Water Research, 2018, 145: 731-740. doi: 10.1016/j.watres.2018.09.020 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3923
- HTML全文浏览数: 3923
- PDF下载数: 88
- 施引文献: 0










