















-
纳米金属氧化物(MeONPs)因具有良好的光学、磁性和电子性能,广泛应用于催化剂、传感器、光学材料、电气材料和磁性存储器等行业[1-4]. MeONPs可以长期漂浮在空气中,容易通过肺部吸入进入人体,并沉积在肺部深处,即肺泡区域[5-8]. 在肺泡区域,MeONPs可以直接与肺部表面活性物质(PS)接触[8-9]. PS由脂质(90%)和蛋白质(10%)组成,在哺乳动物中含量最多的是二棕榈酰磷脂酰胆碱(DPPC),约占PS总质量的40%[10-11]. PS在与MeONPs接触后,会吸附在MeONPs表面形成脂质冠. 已有研究表明,对于聚苯乙烯纳米粒子,随着其表面吸附PS的增加,扁平型肺泡Ⅱ型上皮细胞对纳米粒子的细胞摄取量也呈现增加趋势[12]. 此外也有研究表明,由于金红石型 TiO2( TiO2-R) 比锐钛矿型 TiO2 (TiO2-A)对脂质的吸附能力更强,使TiO2-R更易破坏小鼠巨噬细胞的溶酶体膜进而导致细胞调亡[13]. 因此,MeONPs对脂质的吸附量将会影响MeONPs的细胞摄取和毒性[5,12].
近年来,有关纳米粒子对脂质吸附能力影响因素的研究备受关注. Konduru等[14]的研究表明,由于CeO2纳米粒子的疏水性高于Si-CeO2、ZnO和BaSO4纳米粒子,使得其对小鼠PS吸附量高于其他3种纳米粒子. Luo等[15]通过分子动力学研究发现,随着石墨烯纳米片长度的增加,其对PS的吸附量呈线性增加. 此外,Luo等[16]还发现与立方体和球形碳纳米粒子相比,长方体和四面体碳纳米粒子吸附PS量显著增加,并导致PS层破裂. 因此,MeONPs的疏水性、尺寸、形状等理化性质都是影响脂质吸附能力的重要因素.
目前关于MeONPs对脂质吸附的定量研究十分匮乏,前人研究涉及的MeONPs种类非常有限,仅包括:TiO2-R 、TiO2-A 、CeO2、Si-CeO2、ZnO、Fe2O3[13-14,17-18]. 但由于合成MeONPs种类(化学组成、晶体构型和尺寸上具有差异)不断增多,逐一进行吸附实验测定成本高且耗时. 因此,需要建立一种定量预测MeONPs对脂质吸附量的模型,阐明MeONPs与脂质吸附的相互作用机制.
本研究将利用超声分散法制备的DPPC囊泡与25种不同晶型和粒径的MeONPs孵育,达到吸附平衡后,通过高效液相色谱串联质谱联用仪(LC-MS/MS)定量测定了DPPC的吸附量,分析了影响DPPC吸附的关键因素. 引入MeONPs对磷酸盐的吸附量作为描述符,并结合MeONPs实验测定理化性质及元素周期表描述符,构建了MeONPs对脂质吸附量的定量预测模型,揭示了影响MeONPs对脂质吸附量的关键因素.
-
25种不同晶型粒径的MeONPs包括:氧化钇(Y2O3,50 nm)、氧化钕(Nd2O3,40 nm)、氧化铁(Fe2O3,30 nm)、氧化镨(Pr6O11,50 nm)、氧化钐(Sm2O3,40 nm)、氧化镝(Dy2O3,40 nm)、氧化钆(Gd2O3,100 nm)、氧化钬(Ho2O3,100 nm)、氧化镍(NiO,30 nm)、氧化镍(NiO,50 nm)、氧化钴(Co3O4,50 nm)、氧化钴(Co3O4,100 nm)、氧化铟(In2O3,50 nm)、氧化铟(In2O3,100 nm)、氧化铝(Al2O3,10 nm)、氧化铝(Al2O3,20 nm)、氧化铝(Al2O3,30 nm)、氧化锌(ZnO,30 nm)、氧化锌(ZnO,50 nm)、氧化锌(ZnO,90 nm)、二氧化锡(SnO2,60 nm)、三氧化二铬(Cr2O3,100 nm)、二氧化钛(金红,TiO2-R,100 nm)、二氧化钛(锐钛,TiO2-A,10 nm)、二氧化钛(锐钛,TiO2-A,40 nm). 上述材料均纯度 > 99%,购买自上海阿拉丁生化科技股份有限公司. DPPC(纯度 ≥ 99%)购买自美国Avanti Polar Lipids公司. Hepes缓冲溶液购买自美国西格玛奥德里奇公司.
-
本研究采用超声分散法制备脂质囊泡,具体制备过程如下:取25 mg的DPPC溶于10 mL甲醇,充分涡旋后,制得2.5 mg·mL−1的DPPC储备液,于 −20 ℃下冷冻保存. 取400 µL DPPC储备液于玻璃容器中,加入适量甲醇并涡旋后,将溶液氮吹至干. 向玻璃容器中加入10 mL Hepes缓冲溶液(1 mmol·L−1 NaCl,pH 7.4)水化脂质膜,并加盖在室温下静置1 h后,涡旋形成乳白色的溶液. 将该溶液转移至50 mL圆底塑料管中,在冰浴条件下使用超声波细胞破碎仪(JY92-IIDN,中国)分散囊泡. 超声波细胞破碎仪的条件设置为温度20 ℃,有效超声时间为50%(超声5 s,间隙5 s),超声至溶液澄清,得到的溶液即为10 µg·mL−1的DPPC溶液,于4 ℃的冰箱保存[19].
-
取300 µL的DPPC溶液(10 µg·mL−1)和150 µL的MeONPs溶液(10 mg·L−1)于Hepes缓冲溶液中孵育,每种MeONPs设置3组平行实验. 同时设置空白组实验,孵育条件同上. 将所有孵育溶液置于25 ℃恒温振荡箱中振荡4 h.
-
4 h后将孵育溶液在14500 g的条件下离心50 min后,取100 µL上清液加入装有900 µL甲醇的液相小瓶中,加入20 µL 2.5 mg·L−1的油酰磷脂酰胆碱(POPC,母离子:760.8 m/z;子离子:598.7 m/z)作为内标,利用高效液相色谱串联质谱联用仪(LC-MS/MS)(UPLC I-Class-Waters Xevo TQS,美国)定量测定DPPC(母离子:734.7 m/z;子离子:184.1 m/z). LC-MS/MS的条件如下:流动相A:水,流动相B:甲醇;流动相A和B各包含0.01%甲酸和5 mmol·L−1乙酸铵;流动相A : B = 2 : 98;流速:0.3 mL·min−1;进样体积:5 µL;离子源:电喷雾电离(正离子模式);驻留时间:0.003 s;锥孔电压:50 V;碰撞能量:30 V. MeONPs对DPPC吸附量的计算公式为:
式中,Qm为MeONPs对DPPC的吸附量,Q0为空白组上清液中DPPC含量,Q为MeONPs与DPPC孵育溶液上清液中DPPC含量.
-
MeONPs(80 mg·L−1)与磷酸二氢钾(85 mg·L−1),在4 mL水溶液中孵育24 h,将孵育后的溶液经0.45 µm的尼龙膜(聚醚砜,津腾)过滤后,取1 mL溶液于具塞比色管中,加入去离子水定容至50 mL. 每种MeONPs设置3组平行实验,并设置3组空白实验.
向所有具塞比色管中加入1 mL 10%抗坏血酸溶液并混匀,30 s后加入2 mL钼酸盐溶液充分混匀,静置15 min. 使用紫外/可见光谱仪(Agilent, Cary 100, Malaysia),在700 nm的波长下测定溶液吸光度. MeONPs对磷酸盐吸附量的计算公式为:
式中,
Q′m 为MeONPs对磷酸盐的吸附量,Q′0为空白组中磷酸盐含量,Q′ 为MeONPs与磷酸盐反应溶液中磷酸盐的含量. -
利用透射电子显微镜(TEM)(FEI, Tecnai G2F30 STWIN, 美国)测定MeONPs的初始粒径. 结合TEM图像,对每个MeONPs随机计数300个粒子计算平均粒径. 以Hepes缓冲溶液为溶剂配制50 µg·mL−1 MeONPs溶液,使用马尔文纳米粒度仪(Malvern,Nano-ZS90,英国)测定MeONPs的水动力学直径和Zeta电势,每种材料测量3次取平均值. 使用接触角测量仪(KINO,SL 200KB,美国)测量MeONPs接触角,每种材料测量3次计平均值.
-
本文定量测定了25种不同晶型和粒径的MeONPs对DPPC的吸附量,以每克MeONPs吸附的DPPC量作为评价MeONPs对DPPC吸附能力的指标.
-
本研究选取每克MeONPs对磷酸盐的吸附量、MeONPs理化性质和元素周期表描述符,构建预测模型. 其中4个理化性质描述符分别为:MeONPs的初始粒径、水动力学直径、Zeta电势和接触角. 4个元素周期表描述符分别为:MeONPs的分子量、阳离子电荷、金属元素电负性以及金属元素质量百分比.
-
将数据集按4∶1随机分为训练集和验证集,训练集包含20个数据点,验证集包含5个数据点. 使用SIMCA 13.0软件中的偏最小二乘(PLS)回归建立预测MeONPs对脂质吸附量的模型[20].
模型拟合能力和稳健性通过调整后的决定系数(
R2adj )、均方根误差(RMSE)以及去一法计算得到的交叉验证系数(Q2cv )进行评价[21-23]. 外部预测能力采用验证集的均方根误差(RMSEext)和外部验证系数(Q2ext )评价[21-23].采用基于标准残差(δ)和杠杆值(hi)绘制的Williams图表征模型的应用域,δ和hi的计算公式如下所示:
式中,yi 和 y分别为第i个化合物活性数据的实测值和预测值,n为训练集化合物个数,m为模型中描述符的个数.
式中,xi 和 xT i分别为描述符向量及其转置,X为描述符矩阵,XT是X的转置,h*为杠杆警戒值,m为模型中描述符的个数,n为训练集中化合物的个数. 当化合物|δ|>3.0 时被视为模型离群点. 当化合物 |δ|<3.0 且 hi<h* 时,表明模型预测效果较好. 当化合物 |δ|<3.0 且 hi>h*时,若出现在训练集,表明在模型数据集中该类化合物较少,但该化合物提高了模型的准确性和稳定性;若该结果出现在验证集中,说明模型具有一定的外推能力[24-25].
-
本研究以Gd2O3例,测定纳米颗粒在3种孵育浓度下(0.5、1、2 mg·L−1)对DPPC(2 mg·L−1)的吸附量,发现3种孵育浓度下Gd2O3对DPPC的吸附量均为1.17 g·g−1(图1a),表明这3种体系中DPPC分子的数量相较于Gd2O3的吸附位点来说是足够的.
因此,本研究后续实验中DPPC的孵育浓度设为:2 mg·L−1;同时为确保吸附能力强于Gd2O3的纳米颗粒能够达到饱和吸附且区分不同MeONPs对DPPC的吸附能力,将MeONPs的孵育浓度设为:1 mg·L−1. 此外,基于预实验结果,本研究以吸附量相对较多的Gd2O3和吸附量相对较少的Co3O4为例,进行吸附动力学实验(图1b). 通过测定Gd2O3和Co3O4在不同孵育时间对DPPC的吸附量,发现孵育4 h后,两种MeONPs对DPPC的吸附量趋于平衡,因此本研究设置孵育时间为4 h.
-
本研究测定的10 nm和40 nm TiO2-A对DPPC 的吸附量分别为0.24 g·g−1和0.28 g·g−1(图2 ). Yu等[13]测定了平均粒径为27—28 nm的TiO2-A对磷脂的吸附量(0.4 g·g−1),该研究的磷脂中DPPC占比50%,表明TiO2-A对DPPC的吸附量约为0.2 g·g−1,与本研究测定的TiO2-A的吸附结果相似.
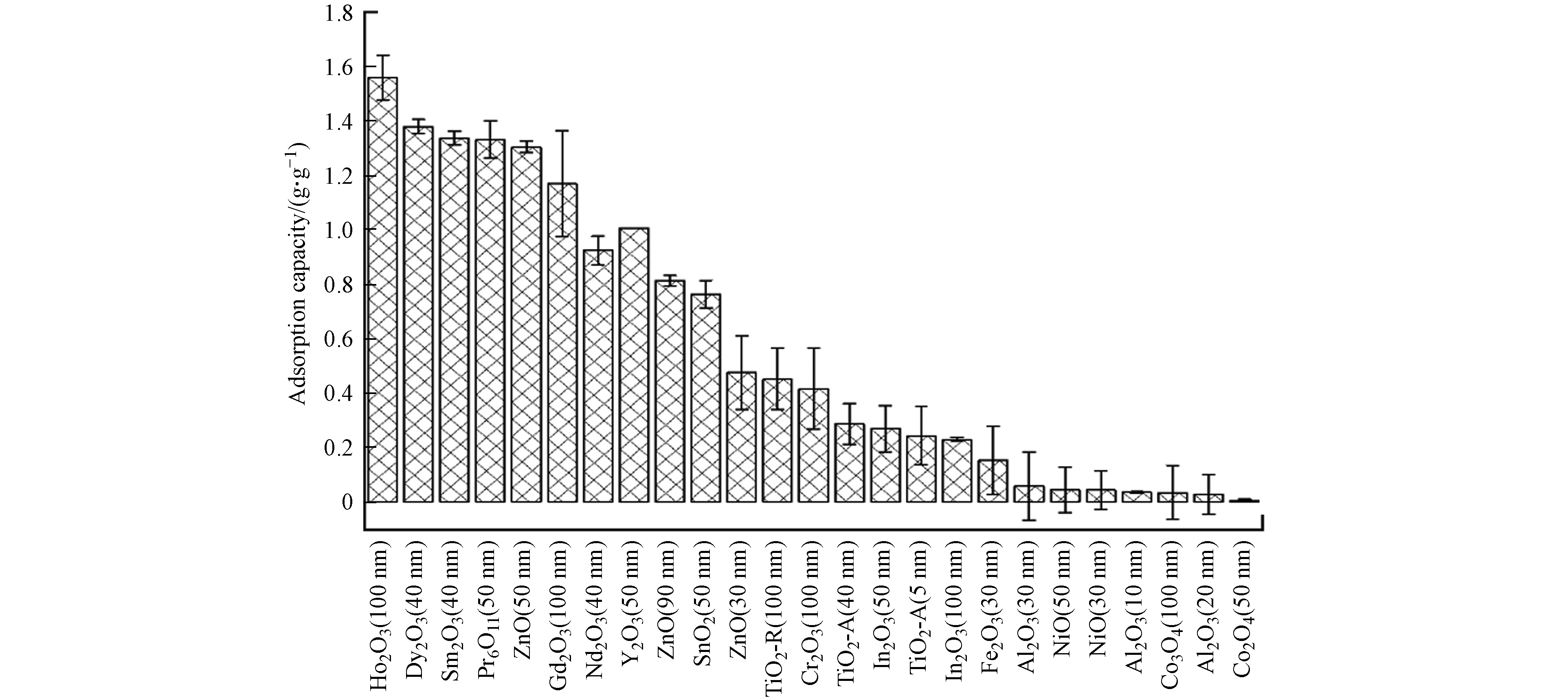
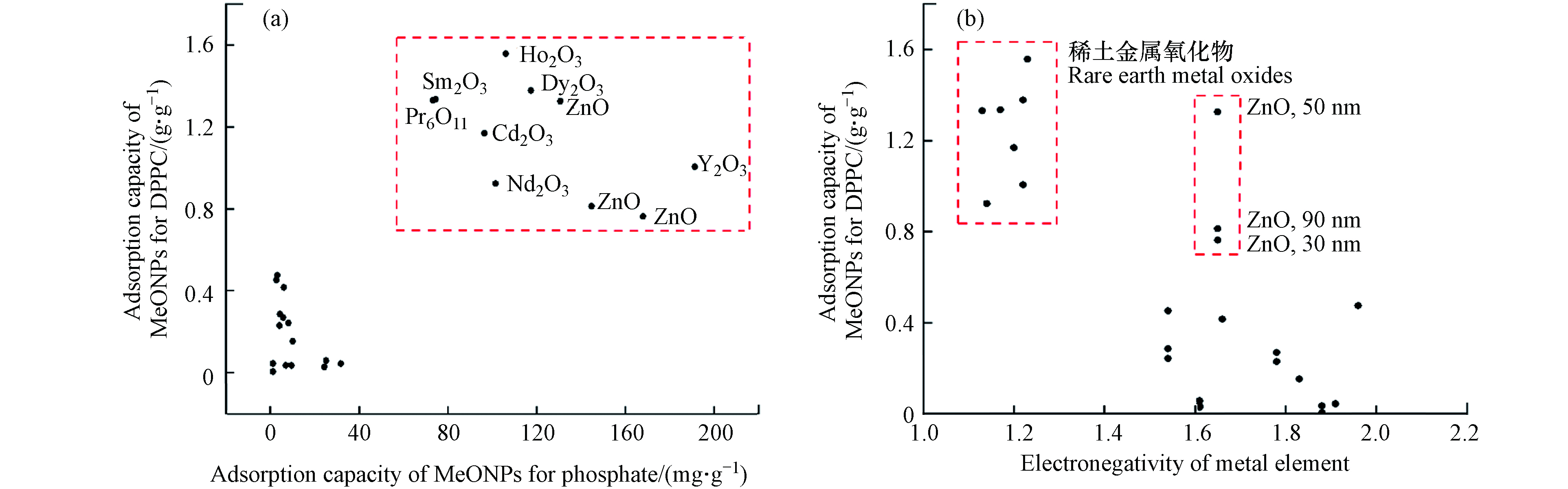
25种不同晶型和粒径的MeONPs对DPPC吸附量的定量结果如图2所示. 与本研究其他MeONPs相比,稀土金属氧化物(Ho2O3、Dy2O3、Sm2O3、Pr6O11、Gd2O3、Nd2O3、Y2O3)对DPPC有相对较高的吸附量(>1 g·g−1),说明稀土金属元素有利于MeONPs对DPPC的吸附.
本研究中涉及的TiO2-A、In2O3、Al2O3、NiO和Co3O4的粒径在10—100 nm范围内变化. 这些纳米材料对DPPC的吸附量不随粒径的变化而改变(P>0.05),表明粒径不是影响DPPC吸附的关键因素. Thorley等[12]的研究也表明对于50 nm和100 nm的聚苯乙烯纳米粒子,其对人支气管肺泡灌洗液的吸附量无显著性差异. 由此可知,粒径在10—100 nm范围内的纳米颗粒,无论对于吸附PS还是DPPC,粒径并不是影响吸附的关键因素.
本研究中50 nm ZnO对DPPC的吸附量显著高于 90 nm和30 nm ZnO(P<0.05). 这可能是由于本研究中50 nm ZnO的疏水性(接触角17°)强于90 nm (接触角7.62°)和30 nm ZnO(接触角6.27°),使其更易与DPPC疏水的尾部发生吸附. Raesch等[8]的研究也表明,与聚乳酸-羟基乙酸共聚物磁性纳米粒子和脂质包裹的磁铁矿纳米粒子相比,较为亲水的聚乙二醇磁性纳米粒子吸附较少的猪PS. 上述结果表明,接触角大的强疏水性纳米颗粒更易与磷脂的尾部发生吸附.
-
将数据集按4:1随机分为训练集和验证集,其中训练集包含20个数据点,验证集包含5个数据点. 基于PLS回归建立了预测MeONPs对脂质吸附量的模型,表达式如下:
式中, Q为每克MeONPs对DPPC的吸附量,
Q′ 为每克MeONPs对磷酸盐的吸附量,χme 为金属元素电负性,M(%)为金属元素质量百分比.模型性能评价结果表明,该模型具有良好的拟合度 (
R2adj = 0.79,RMSEtra = 0.23)、稳健性 (Q2cv = 0.74) 和预测能力 (Q2ext = 0.86). MeONPs对DPPC吸附能力的实测值和模型预测值的拟合结果见图3a. 采用 Williams 图对模型的应用域进行表征(图3b). 从图中可以看出,所有MeONPs的 |δ| < 3.0,表明模型没有离群点. 验证集中有两种MeONPs的杠杆值大于杠杆警戒值(hi > h*),说明这两种MeONPs是对模型有显著影响的化合物. 且这两种MeONPs的|δ| < 3.0,说明模型对此类远离描述符空间中心的化合物具有较好的泛化能力. 综上,本研究构建的模型可以用于预测MeONPs对DPPC的吸附量. -
模型结果表明,MeONPs吸附磷酸盐的能力是影响MeONPs对DPPC吸附的关键因素. 如图4a所示,对于DPPC吸附量相对较高的MeONPs(> 0.7 g·g−1),其对磷酸盐也有较高的吸附量(> 73 mg·g−1). 在这些吸附量较高的MeONPs中,除3种不同粒径的ZnO外,均为稀土金属氧化物. 该结果表明,DPPC头部的磷酸基团是稀土金属氧化物对DPPC吸附的重要位点.
MeONPs金属元素电负性也是DPPC的吸附重要影响因素. 如图4b所示,当MeONPs金属元素电负性较低(< 1.22)时,更易与DPPC发生吸附. 这可能是由于电负性较低的金属元素均为稀土金属,与稀土金属氧化物发生吸附的DPPC头部磷酸基团的电负性为2.87,更易与电负性较小的金属原子发生作用. 因此,当MeONPs元素电负性较高时(> 1.22),相对不利于其对磷酸基团的吸附.
此外,模型中金属元素质量百分比的回归系数为正,这反映MeONPs的金属元素质量百分比越大,其对DPPC的吸附量越大. 本研究中3种不同粒径的ZnO,其Zn元素的电负性(1.65)较大,但它们对DPPC的吸附量却相对较高(图4b),这可能与ZnO的金属元素质量百分比较大有关. ZnO的金属元素质量百分比为80.34,其值大于其他非稀土金属氧化物的金属元素质量百分比(52.92—78.77). 这进一步说明了金属元素质量百分比较大的纳米颗粒更有利于DPPC的吸附.
-
本研究以25种不同晶型和粒径的MeONPs为研究对象,将DPPC囊泡与其孵育4 h后,利用LC-MS/MS定量测定了其对DPPC的吸附量,并建立了可以快速、低成本地预测MeONPs对DPPC吸附量的模型. 吸附实验结果显示,本研究中的稀土金属氧化物(Ho2O3、Dy2O3、Sm2O3、Pr6O11、Gd2O3、Nd2O3、Y2O3)对DPPC有相对较高的吸附量(> 1 g·g−1),说明稀土金属元素促进了MeONPs对DPPC的吸附. 对于10—100 nm范围内具有不同粒径的MeONPs(包括TiO2-A、In2O3、Al2O3、NiO和Co3O4),粒径不是影响这些纳米颗粒对DPPC吸附的关键因素. 针对3种不同粒径ZnO的吸附量与其接触角的分析发现,接触角大的强疏水性纳米颗粒更易与磷脂的尾部发生吸附. 模型结果显示,对DPPC吸附量相对较高的MeONPs,其对磷酸盐的吸附量也较高,且这些纳米颗粒大部分为稀土金属氧化物,表明DPPC头部的磷酸基团是稀土金属氧化物对DPPC的重要吸附位点. 金属元素电负性及金属元素质量百分比共同影响MeONPs对DPPC的吸附,当MeONPs金属元素电负性较低(< 1.22)时,更易与DPPC发生吸附. 本研究为纳米颗粒对脂质吸附量的定量评价提供了关键数据,并有助于拓展对纳米颗粒与脂质发生吸附作用机制的理解.
纳米金属氧化物对脂质吸附能力的定量预测模型
Prediction model of the adsorption capacity of metal oxide nanoparticles for lipids
-
摘要: 纳米金属氧化物(MeONPs)对肺部表面活性物质(PS)的主要成分脂质的吸附量,是影响呼吸暴露生物效应的重要因素. 目前关于MeONPs对脂质吸附的定量研究十分有限. MeONPs种类繁多,有必要构建MeONPs对脂质吸附量的预测模型. 本研究利用二棕榈酰磷脂酰胆碱(DPPC)囊泡模拟PS,测定25种MeONPs对DPPC的吸附量,建立了可预测MeONPs对DPPC吸附量的模型. 结果显示,预测模型具有良好的拟合度、稳健性和预测能力(
R2adj = 0.79,Q2cv = 0.74,Q2ext = 0.86). 机理解释表明,DPPC头部的磷酸基团是稀土金属氧化物吸附DPPC的重要位点,且MeONPs的金属元素电负性和金属元素质量百分比共同影响DPPC的吸附. 本研究建立的预测模型不仅为MeONPs对脂质吸附能力评价提供了基础数据,还拓展了对脂质吸附机制的理解.Abstract: Adsorption capacity of metal oxide nanoparticles (MeONPs) for lipids that are the main components of pulmonary surfactant (PS), is an important factor affecting the biological effect of nanoparticles entering the human body through the respiratory tract. At present, studies on the adsorption capacity of MeONPs for lipids are very limited. Due to the increasing variety of synthetic MeONPs, it is necessary to establish a model to predict the adsorption capacity of MeONPs for lipids. In this study, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) vesicles were used to simulate PS, and 25 kinds of MeONPs were incubated with DPPC to determine the adsorption capacity of MeONPs for DPPC. The model for predicting the adsorption capacity of MeONPs for DPPC was established based on the experimental data. The results show that the prediction model exhibits satisfactory goodness of fit, robustness and predictive ability (R2adj = 0.79,Q2cv = 0.74,Q2ext = 0.86). The explanation of the mechanism suggests that the phosphoric groups in the head of DPPC are important adsorption sites of rare earth metal oxides for DPPC, and the electronegativity of metal elements and the mass percentage of metal elements jointly affect the adsorption capacity of MeONPs for DPPC. The prediction model established in this study not only provides basic data for the evaluation of the adsorption capacity of MeONPs for lipids, but also expands the understanding of the adsorption mechanism of MeONPs for lipids.-
Key words:
- metal oxide nanoparticles /
- lipids /
- pulmonary surfactant /
- adsorption /
- prediction model.
-
低温等离子体(non-thermal plasma, NTP)具有反应条件温和(常温常压)、适应性广、反应快速等优点,通过产生大量活性物种(O、·OH、O3等)将VOCs降解,受到了广泛关注,然而高的能耗及大量副产物生成限制了该技术工业化应用. 为了克服活性物种寿命短而导致的NTP降解VOCs效率不高、副产物生成的缺点,近年来,研究者开发了多种具有多孔结构的催化剂与NTP协同降解VOCs,通过延长污染物在放电区的停留时间,有效利用副产物O3产生原子氧、过氧自由基等,达到减少副产物的产生、提高能量密度、降低能耗、提高碳平衡等效果[1-7]. 已经研究的与NTP联合的催化剂包括铁电体材料、半导体催化剂、贵金属催化剂以及分子筛[8-11].
金属有机骨架(metal organic frameworks, MOFs)是一种由有机配体和金属离子或金属簇组合成的多孔催化吸附材料,由于其具有可调规整的孔道、大的比表面积和高的孔隙率,应用前景广泛. MOFs材料在获得一定能量时,由于轨道离域而呈现出半导体的特性[12],介质阻挡放电(dielectric barrier discharge, DBD)等离子体中高能电子的能量可达1—20 eV,可以提供足够的能量活化MOFs产生新的活性自由基(电子-空穴对),具有吸“拟光催化过程”和催化效果,从而促进污染物的降解[13];另外,MOFs材料高的比表面积使其具有优异的吸附性能,可有效延长VOCs在反应区的停留时间,提高处理效率,逐渐应用在等离子体和气体吸附中. 例如,Wang等[14]通过水热法制备了MIL-101(Cr)并用于吸附苯表现出良好的吸附性能;Bahri等[15]采用等离子体协同MIL-101、MIL-53材料降解甲苯,发现MOFs材料的加入可以提高甲苯的降解效果、降低副产物O3的生成. 因此,研制适合DBD体系的高性能MOFs催化剂,提高两者的协同作用效果,对于推动该技术工业化应用具有重要的意义.
MOF-74由二价过渡金属(硝酸盐、醋酸盐)和配体2,5-二羟基-对苯二甲酸合成(DOT),具有一维六角形孔洞结构和高密度“开放”金属位点,具有高比表面积、有机配体和金属离子可调性剂以及很好的稳定性,具有优异的电化学性能及光催化特性,在吸附分离、光电催化产氢、化学传感、烟气脱硝等领域受到广泛关注[16-18]. 例如,Chen等[16]采用微波辅助法合成的Ni-MOF-74对CO2吸附性能好:Feng等[8]将MOF-74(Mn-Co-Ni)催化剂与NTP协同应用于甲苯降解,相同条件下甲苯去除率提高了42.9%,并有效控制了副产物O3生成. 目前,将MOF-74材料与DBD协同降解VOCs还鲜有报道[8],催化剂引入DBD后在等离子体内较低温度下O2是否参与吸附活化为O-和O2-等参与催化反应的机理还不明晰.
本研究采用溶剂热法合成制备了Mn基MOF-74(Mn-MOF-74)材料,并通过改变配体为1, 4-苯二甲酸(TPA)制备了Mn-TPA-DMF多孔材料,将两种催化剂引入DBD等离子体降解甲苯气体. 采用XRD、FTIR、SEM、BET和XPS等表征技术对催化剂的结构进行分析,对比了两种Mn基MOFs材料加入DBD后甲苯的降解效果、副产物的形成,推测了DBD催化降解甲苯的反应机理.
1. 实验部分(Experimental section)
1.1 材料
四水合硝酸锰(Mn(NO3)2·4H2O)、2,5-二羟基对苯二甲酸、1,4-苯二甲酸(TPA)、N,N-二甲基甲酰胺(DMF)、乙醇和丙醇等,所有试剂均为分析纯,不需经进一步纯化可以直接使用.
1.2 催化剂的制备
采用溶剂热合成法制备催化剂. 准确称取2.84 mL Mn(NO3)2·4H2O和0.66 g DOT,溶解在15:1:1(V:V:V)的DMF-乙醇-水混合物中,反应混合物超声处理5 min,然后转移到高压釜中,放置在120 ℃热空气烘箱内24 h. 然后取出高压釜在室温下冷却,将获得的棕色晶体用乙醇洗涤后去离子水洗涤多次,然后将样品置于60 ℃烘箱中干燥,制备Mn-MOF-74催化剂.
另外,称取0.92 g Mn(NO3)2·4H2O 和0.90 g TPA,同时溶解在50 mL DMF中,超声处理10 min后,将所得溶液转移到高压釜中,在120 ℃ 的热空气烘箱中保持24 h. 将反应混合物冷却至室温后,得到浅白色结晶化合物,用DMF、甲醇和水洗涤多次,获得的产物在130 ℃下真空干燥,制备Mn-TPA-DMF催化剂.
1.3 催化剂的表征
使用X射线衍射仪(XRD,PANalytical,荷兰),获得制备的催化剂的晶体结构. 催化剂样品的形态通过来扫描电子显微镜(SEM,ZESS,德国的Gemini 300)表征,相应的元素分布通过能量色散X射线光谱仪(EDS)分析系统获得. 使用氮气吸附-脱附实验测定样品的孔隙信息,BET和BJH方程中的吸附数据计算催化剂的比表面积、孔径分布和孔体积. X射线光电子能谱(XPS)通过具有Al-Kα辐射源的Thermo Scientific K α谱仪测量,用C1s为284.8 eV的数据校准测试的元素的结合能数据.
1.4 催化剂的性能评价
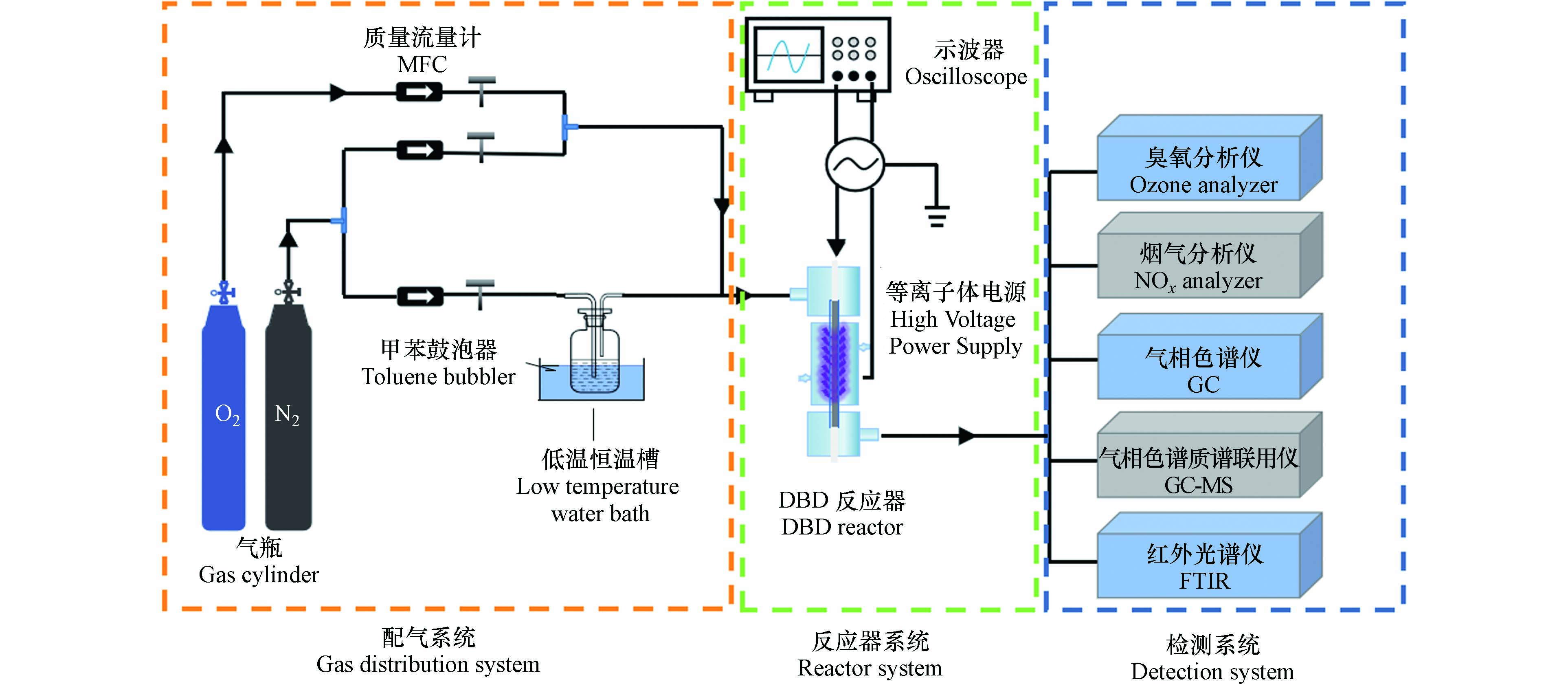
DBD催化降解甲苯气体的工艺流程如图1所示. 该系统由三部分组成:甲苯气体发生器系统、DBD等离子体反应器系统和废气检测系统. 氮气通过装有液体甲苯的气体鼓泡器发生器来生成一定浓度的含甲苯气体. 使用3个质量流量控制器(MFC)分别控制氮气、氧气的流量,得到所需的甲苯初始浓度. 实验时,甲苯初始浓度1368 mg·m−3,气体流量1 L·min−1,氧含量4%(体积分数).
DBD反应器为50 mm放电长度、3 mm放电间隙的同轴结构(图1),采用石英管作为介质层,使用循环水作为接地电极,内管放置1根不锈钢棒作为高压电极. 高压电源为南京苏曼等离子体科技有限公司生产的CTP-2000 K实验电源,输出频率范围为5—20 kHz. DBD催化实验时,将0.3 g催化剂(40—60目)与1.5 g石英砂混合,装入DBD反应器中. 使用高压探头和电流探头测定电压和电流,并通过数字示波器(Tektronix TBS 1000 C)记录放电波形,通过利萨如(V-Q Lissajous)方法计算DBD放电功率P(方程1),借助方程(2)计算能量密度(SED). 本文气体流量(Q)为1 L·min−1,通过调节放电电压来实现SED的变化.
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) 式中,f为放电频率(9.6 kHz),C为电容(0.47 μF),A为利萨如图面积,P为放电功率(W),Q为气体流量
(L⋅min−1) 降解前后的甲苯、CO和CO2的浓度均使用气相色谱仪(GC9790型,浙江福立分析仪器有限公司)在线测定,其中CO和CO2通过Ni转化炉转化为甲烷后由FID检测器测定. O3和NOx(NO2和NO)的浓度分别用O3分析仪(2B Model 106-M,美国)和烟气分析仪(MGA 6-plus,德国)测定. 采用气相色谱-质谱联用仪(GC-MS)对尾气中的有机副产物进行了分析. 尾气用正己烷吸收20 min,再超声1 h. 样品进样至GC-MS中,通过自动进样器进行分析. 甲苯降解率(η)、矿化度(Md)、CO2选择性(
SCO2 stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) stringUtils.convertMath(!{formula.content}) (5) stringUtils.convertMath(!{formula.content}) (6) 式中,Cin和Cout分别为甲苯进口和出口的浓度(mg·m−3),M为甲苯的分子量(g·mol−1),[CO2]和[CO]分别为出口CO2和CO的浓度(mol·m−3),Ey为反应器的能量效率(g·kWh−1).
2. 结果与讨论(Results and discussion)
2.1 催化剂的表征分析
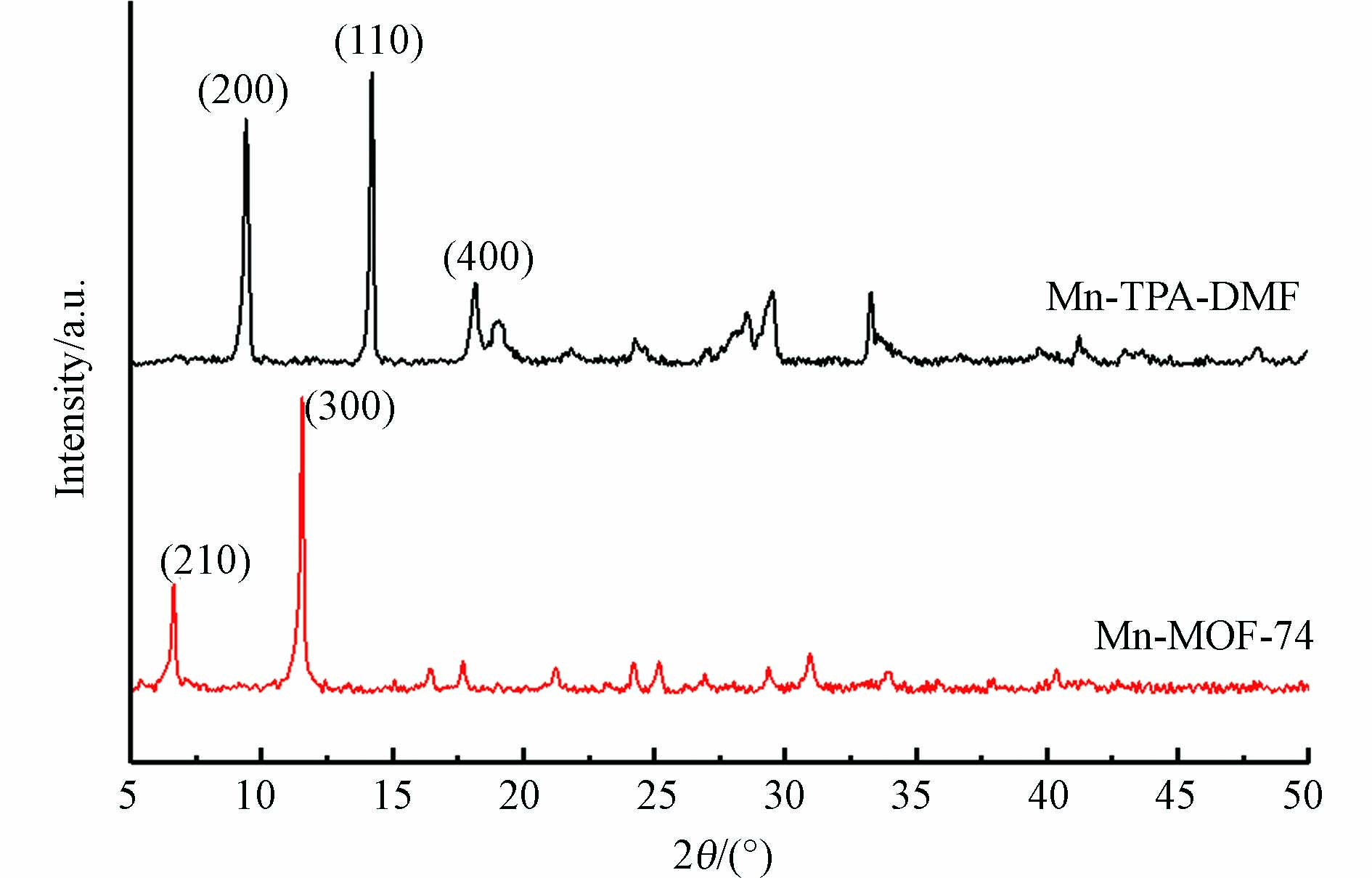
如图2所示,为了确定材料的晶体结构,对制备的样品进行了XRD表征. Mn-MOF-74材料在2θ=6.8°和11.8°附近有两个很强的衍射峰,分别对应于Mn-MOF-74的(210)和(300)晶面[18],其他位置的衍射峰都相对较弱. Mn-TPA-DMF材料在2θ=9.41°、14.22°、18.17°附近的3个衍射峰分别对应材料的(200)、(110)和(400)晶面[19]. 本研究中合成的Mn-MOF-74和Mn-TPA-DMF的XRD谱图的峰位置、强度及顺序与先前文献报道的相一致[20-21],表明材料合成成功.
图3(a)为Mn-MOF-74使用前后的FTIR图. 3422 cm−1附近的特征峰为OH或H2O的伸缩振动吸收峰,表明水或其他溶剂分子存在于Mn-MOF-74通道中[22]. 1567 cm−1和1401 cm−1附近的峰为MOF-74中COO-的不对称和对称伸缩振动峰[23],1240 cm−1附近的峰为C—N键振动吸收峰,说明溶剂DMF吸附在催化剂表面或者孔道内[24],1196 cm−1处的特征峰为C—O单键伸缩振动峰,886 cm−1和806 cm−1处的特征峰为苯环C—H键面内和面外摇摆振动峰,574 cm−1处的特征峰归属于Mn—O伸缩振动峰[25],表明Mn-MOF-74的成功合成.
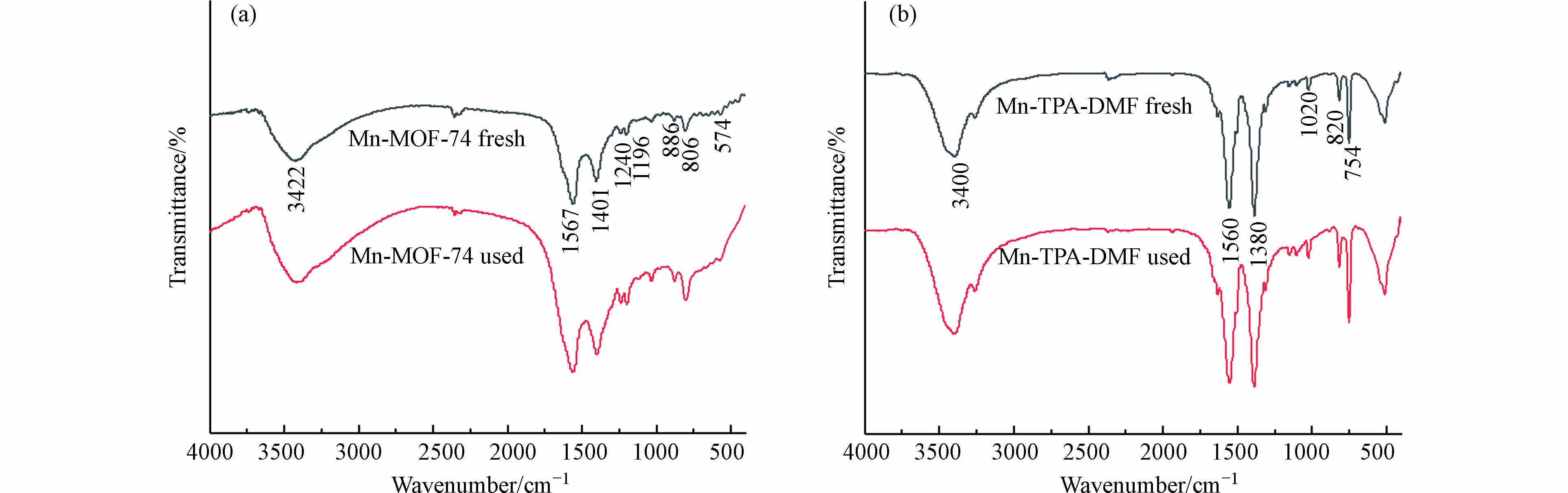 图 3 Mn-MOF-74和Mn-TPA-DMF反应前后的红外图谱Figure 3. FTIR spectra of Mn-MOF-74 and Mn-TPA-DMF before and after catalysis reaction
图 3 Mn-MOF-74和Mn-TPA-DMF反应前后的红外图谱Figure 3. FTIR spectra of Mn-MOF-74 and Mn-TPA-DMF before and after catalysis reaction图3(b)为Mn-TPA-DMF使用前后(fresh 和 used表示)的FTIR图,具有与Mn-MOF-74相似的特征峰,但特征峰向低频(波数)轻微偏移,这可能是由于Mn与2,5-二羟基对苯二甲酸和1,4-苯二甲酸两种不同的有机配体结合引起的. 此外,通过反应前后的红外图谱对比可知,Mn-MOF-74和Mn-TPA-DMF在反应前后没有发生结构上的改变,表明其具有良好的稳定性,适合实际工业应用.
本研究通过SEM分析了催化剂的形貌结构,如图4所示. Mn-MOF-74和Mn-TPA-DMF均呈现出不规则的二维片状结构,表面上有细小的块状颗粒附着. 此外,催化剂的元素映射图表明了Mn、O、C元素的存在,且呈均匀分布.
 图 4 (a—c)Mn-MOF-74和(d—f)Mn-TPA-DMF的SEM和元素映射Figure 4. SEM image and element mapping of (a—c) Mn-MOF-74 and (d—f) Mn-TPA-DMF
图 4 (a—c)Mn-MOF-74和(d—f)Mn-TPA-DMF的SEM和元素映射Figure 4. SEM image and element mapping of (a—c) Mn-MOF-74 and (d—f) Mn-TPA-DMF图5为催化剂的N2吸附-脱附曲线,根据曲线信息计算出的BET比表面积和孔隙体积如表1所示. Mn-MOF-74和Mn-TPA-DMF的N2吸附-脱附曲线均为IV型,并在高的压力下表现出脱附滞后,形成回滞环,说明两种材料都是含有中孔(2—50 nm)结构的材料. 与Mn-TPA-DMF相比(比表面积和孔体积分别为29.123 m2·g−1和0.02421 cm3·g−1),Mn-MOF-74呈现更高的比表面积及孔体积,分别为83.368 m2·g−1和0.17630 cm3·g−1,本文合成的Mn-MOF-74的比表面积高于相似的方法合成的Mg-MOF-74催化剂(18.04 m2·g−1)[26],可以提供更多的表面活性位点.
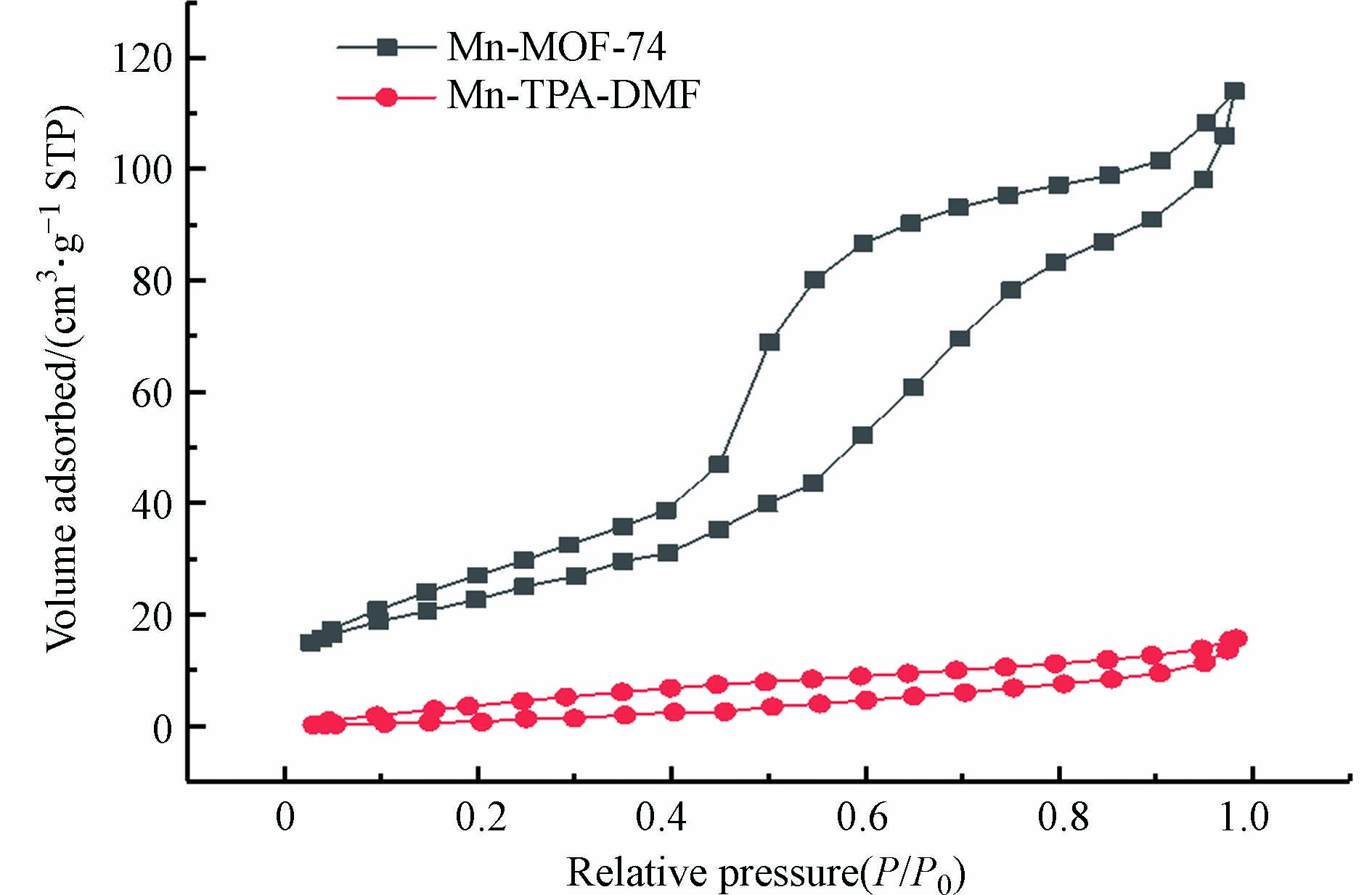 图 5 Mn-MOF-74和Mn-TPA-DMF的氮气吸附-脱附曲线Figure 5. Nitrogen adsorption and desorption curves of Mn-MOF-74 and Mn-TPA-DMF表 1 Mn-MOF-74和Mn-TPA-DMF的比表面积和孔容分析Table 1. BET surface area and pore volume analysis of Mn-MOF-74 and Mn-TPA-DMF
图 5 Mn-MOF-74和Mn-TPA-DMF的氮气吸附-脱附曲线Figure 5. Nitrogen adsorption and desorption curves of Mn-MOF-74 and Mn-TPA-DMF表 1 Mn-MOF-74和Mn-TPA-DMF的比表面积和孔容分析Table 1. BET surface area and pore volume analysis of Mn-MOF-74 and Mn-TPA-DMF催化剂 Catalyst 比表面积/(m2·g−1) Specific surface area 孔容/(cm3·g−1) Pore volume 平均孔径/nm Average diameter Mn-MOF-74 83.368 0.17630 8.460 Mn-TPA-DMF 29.123 0.02421 3.326 | Show Table DownLoad:
CSV
DownLoad:
CSV
图6所示为Mn-MOF-74和Mn-TPA-DMF在甲苯催化反应前后的XPS分析. 图6(a)的全谱分析中显示有Mn、C、N和O元素的存在,表明MOFs材料的成功合成,N元素的存在,说明DMF分子也参与了Mn-MOF-74的配位[27]. 图6(b)为Mn-MOF-74的Mn 2p XPS光谱,位于640.7 eV和652.0 eV的峰分别归因于Mn 2p3/2和Mn 2p1/2的特征峰. 将Mn 2p3/2进行分峰拟合,在结合能为639.3、640.7 、644.7 eV处的峰分别归属于Mn2+、Mn3+和Mn4+[10, 28]. 以上结果表明,Mn以多价态存在,反应过程中可以相互转换,有利于催化反应的进行. 气相中的O2、O3和电子能够借助Mn2+、Mn3+、Mn4+之间的表面转换,被吸附和转移到催化剂表面的甲苯和中间产物上,产生活性粒子(OH、·O等),从而将甲苯进一步矿化为CO2. 如表2所示,通过Mn 2p光谱的定量分析可知,两种Mn基催化剂反应前Mn3+占比都较高,Mn3+的存在能够增加氧空穴,有利于将O2和O3激活为活性氧,进而参与反应;反应后Mn3+占比变化不大,说明Mn3+一直参与甲苯的氧化反应,比较稳定. Mn3+略有下降的原因可能为反应后氧被激活,消耗了部分Mn3+. 在多价态的锰中,高价态的锰可以氧化甲苯,低价态的锰可以通过等离子体催化过程中臭氧的活化而被再氧化. 此外,位于656.0 eV附近的峰归因于Mn 2p特征峰的卫星峰. 反应后Mn的特征峰的强度变低且位置向高结合能的位置轻微偏移,这可能是由于催化剂在放电过程中参与甲苯氧化反应引起的.
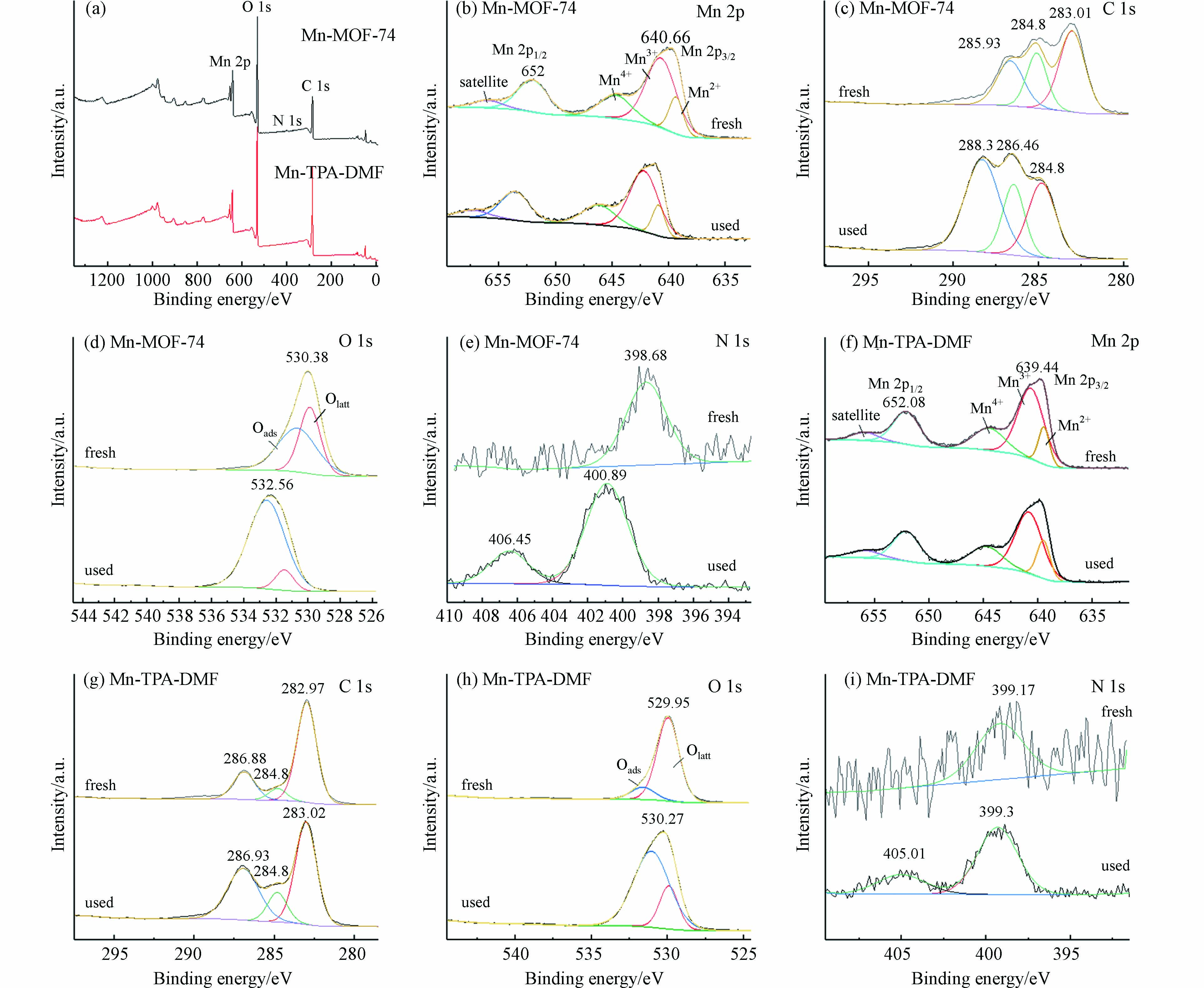 图 6 Mn-MOF-74和Mn-TPA-DMF催化反应前后的XPS分析Figure 6. XPS analysis of Mn-MOF-74 and Mn-TPA-DMF before and after catalytic reaction(a)全谱分析,(b-e)Mn-MOF-74的Mn 2p, C 1s, O 1s, N 1s XPS光谱,(f-i)Mn-TPA-DMF的Mn 2p, C 1s, O 1s, N 1s XPS光谱(a) Full spectrum analysis, and (b-e) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-Muf-74, and(f-i) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-TPA-DMF表 2 Mn-MOF-74和Mn-TPA-DMF各元素价态的组成Table 2. Valence composition of the elements of Mn-MOF-74 and Mn-TPA-DMF
图 6 Mn-MOF-74和Mn-TPA-DMF催化反应前后的XPS分析Figure 6. XPS analysis of Mn-MOF-74 and Mn-TPA-DMF before and after catalytic reaction(a)全谱分析,(b-e)Mn-MOF-74的Mn 2p, C 1s, O 1s, N 1s XPS光谱,(f-i)Mn-TPA-DMF的Mn 2p, C 1s, O 1s, N 1s XPS光谱(a) Full spectrum analysis, and (b-e) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-Muf-74, and(f-i) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-TPA-DMF表 2 Mn-MOF-74和Mn-TPA-DMF各元素价态的组成Table 2. Valence composition of the elements of Mn-MOF-74 and Mn-TPA-DMF催化剂 Catalyst 表面元素比Mn3+/(∑Mnn+) Surface element ratio Mn3+/(∑Mnn+) Oads/Olatt Before After Before After Mn-MOF-74 0.63 0.62 1.23 8.60 Mn-TPA-DMF 0.62 0.60 0.16 6.28 | Show TableDownLoad:
CSV
图6(c)为Mn-MOF-74的C 1s XPS谱,在283.01 eV、285.1 eV和286.65 eV处分峰拟合为3个峰,分别对应于C—C,C—O 和O—C=O[10],反应后特征峰的位置向高结合能的位置轻微偏移,这可能是由于等离子放电或与甲苯及中间产物的结合.
图6(d)为Mn-MOF-74的O 1s XPS光谱,通过分峰拟合可以分为两个特征峰. 位于约529.9 eV处的峰可归因于材料中的晶格氧(O2-)(标记为Olatt),位于约530.7 eV处的峰则归因于表面吸附氧[10, 29]. O2在催化剂上吸附后通常会经过以下变化:吸附氧Oads→O2-(ads)→O-(ads)→Olatt. 一般认为,源自氧空位的Oads比Olatt具有更高的迁移率,并且在VOCs催化氧化过程中比Olatt更有效[30, 31]. 通过表2可知,反应前吸附氧(Oads)的含量比较少,反应后Oads含量明显升高,说明反应过程中催化剂能活化氧物种产生大量Oads,使Oads参与了甲苯的氧化反应. 其中,Mn-MOF-74催化剂显示出较高的Oads/Olatt摩尔比,这表明在Mn-MOF-74催化剂表面上有较高含量的Oads,即表面氧空位密度越高,O2分子越容易在催化剂表面吸附和活化,催化性能越好. 图6(d)表明,发生DBD催化反应后Oads的占比显著增加,表明Mn-MOF-74可以活化氧物种参与甲苯的氧化反应.
图6(e)为Mn-MOF-74的N 1s XPS光谱,在398.7 eV 的峰归属于N(C)3,是由残留DMF引起的. 反应后406.5 eV处出现了1个特征峰,是由等离子体电荷效应引起,说明有少量NOx副产物产生[32].
图6(f-i)为Mn-TPA-DMF的Mn 2p, C 1s, O 1s, N 1s XPS光谱,均呈现和Mn-MOF-74相似的特征峰,从图中也可以分析得出,Mn和表面活性氧物种参与了甲苯的氧化反应.
2.2 DBD催化降解甲苯的效果分析
如图7(a)显示了甲苯在不同DBD催化体系中的降解行为. 由图7可知,甲苯的降解效率随SED的增加也就是放电电压的升高而增加,DBD-催化体系比单独DBD体系具有更高的甲苯去除效率. SED为605.97 J·L−1时,DBD、DBD+Mn-TPA-DMF和DBD+Mn-MOF-74的3个体系中甲苯去除率分别为74.67%、91.23%和94.91%. 能量密度越高,产生的电流脉冲的数量和强度越大,形成的微放电越多,从而通过高能电子和大量活性物质改善了VOCs的降解效果[33-34]. 换句话说,SED升高,增大了高能电子的绝对数量及与气体分子的碰撞几率,导致体系中活性粒子数增加,同时可能诱导催化剂表面发生反应,改变催化剂的特性和微孔放电特性. 此外,两种催化剂相比,Mn-MOF-74比Mn-TPA-DMF表现出更好的催化协同性能. 从表2中可以看出,Mn-TPA-DMF和Mn-MOF-74的金属元素的价态及含量差别不大,但与Mn-TPA-DMF相比,Mn-MOF-74呈现更高的比表面积及孔体积. 结合催化活性测试结果即Mn-MOF-74与等离子体结合时甲苯降解效果更好,说明比表面积和孔体积是影响催化剂活性的主要因素,这与以往的文献报道一致[35].
 图 7 DBD催化体系中不同SED甲苯的去除效率和能量效率Figure 7. (a) Toluene removal efficiency and (b) energy efficiency under different SED in DBD-catalytic systems
图 7 DBD催化体系中不同SED甲苯的去除效率和能量效率Figure 7. (a) Toluene removal efficiency and (b) energy efficiency under different SED in DBD-catalytic systems能量效率可以定义为单位电耗降解甲苯的量,可以说明等离子体催化过程的能耗. 图7(b)可以观察到,能量效率都随着能量密度的升高而降低,说明从经济的角度能量密度越小越好. 与单独DBD相比,相同的SED下DBD催化能量效率有所提高,说明能耗降低了. 当SED为263.68 J·L−1时,单独使用DBD系统的能量效率值为7.43 g·kWh−1,DBD+Mn-MOF-74的能量效率值为11.22 g·kWh−1,说明等离子体与催化剂之间可能存在协同效应. 因此,DBD与催化剂的组合可以提高能量效率,有效降低甲苯降解的能耗,提高DBD系统的经济性.
为了进一步评价DBD-催化体系的性能,进一步研究了CO/CO2浓度、CO2选择性和甲苯矿化度,如图8所示. 如图8(a)所示,在所有情况下,COx浓度都随SED的增加而升高. 此外,DBD催化时生成的CO和CO2的浓度与单独DBD降解时浓度相比显著增加. 当SED为605.87 J·L−1时,DBD+Mn-MOF-74中CO2和CO的浓度分别为1375 mg·m−3和1019.13 mg·m−3,DBD单独体系中为785.71 mg·m−3和712.75 mg·m−3,CO2和CO生成量为单独DBD的175%和143%.
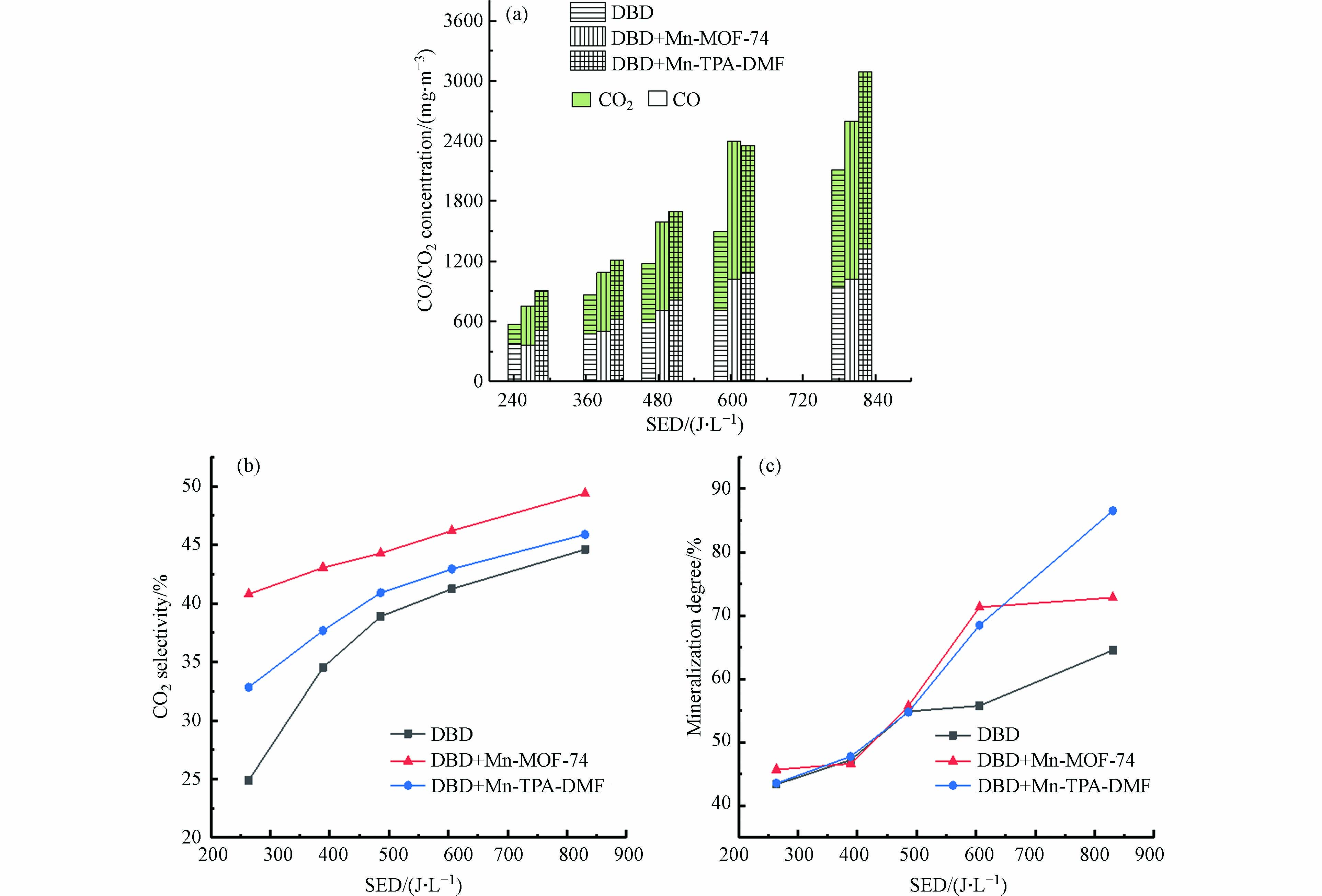 图 8 DBD催化体系(a) CO和CO2浓度(b) CO2选择性和(c)矿化度随SED的变化Figure 8. Changes in (a) CO and CO2 concentration, (b) CO2 selectivity and (c) mineralization degree with SED under DBD catalyst systems
图 8 DBD催化体系(a) CO和CO2浓度(b) CO2选择性和(c)矿化度随SED的变化Figure 8. Changes in (a) CO and CO2 concentration, (b) CO2 selectivity and (c) mineralization degree with SED under DBD catalyst systems图8(b)显示DBD+Mn-TPA-DMF、DBD+Mn-MOF-74和单独DBD体系的CO2选择性随着能量密度的增加而增加. DBD+Mn-MOF-74体系的CO2选择性在能量密度为263.68 J·L−1时为40.79%,比单独DBD体系(24.89%)提高了15.9%.
如图8(c)所示,随着SED的增加,DBD单独体系和DBD-催化剂体系的矿化度都增加. 当能量密度为605.87 J·L−1时,DBD+Mn-TPA-DMF和DBD+Mn-MOF-74体系的矿化率分别为68.48%和71.31%,比单独DBD体系(55.78%)分别提高了12.7%和15.53%. 当SED为830.57 J·L−1时,DBD+Mn-TPA-DMF体系甲苯的矿化度达到最高值86.53%,表明DBD+Mn-TPA-DMF在较高的SED下更有利于甲苯彻底降解为CO2.
NOx和O3是DBD降解VOCs的主要副产物. 图9显示了单独DBD和DBD催化系统中副产物O3和NOx浓度. 在图9(a)中,O3浓度随着SED的增加而降低,这可能是由于O3参与了COx和NOx等其他活性物种的生成[36]. 外加Mn基MOFs催化材料后,DBD降解甲苯气体中副产物O3浓度显著下降,例如,当SED为388.75 J·L−1时,单独DBD系统中O3浓度约为390.64 mg·m−3,而在DBD+Mn-MOF-74和DBD+Mn-TPA-DMF体系中O3浓度分别降至264.64 mg·m−3和303.64 mg·m−3,O3浓度的降低可归因于O3在催化剂表面分解成O2并形成活性原子氧物质(O* )[37]. 就挥发性有机化合物氧化的反应性而言,O* 是比O3更具化学活性的物质,因此,将O3分解为原子氧的能力是等离子体催化过程中挥发性有机化合物降解的重要因素[38]. 此外,Mn2+/Mn3+和Mn4+/Mn3+的氧化还原循环也可以加速表面氧物种的释放,有利于甲苯的氧化[39]. 与Mn-TPA-DMF催化剂相比,Mn-MOF-74催化剂具有较大的比表面积,更有利于O3的吸附和分解,在催化剂表面形成更多的活性氧,提高了催化剂的去除效率和CO2选择性[40].
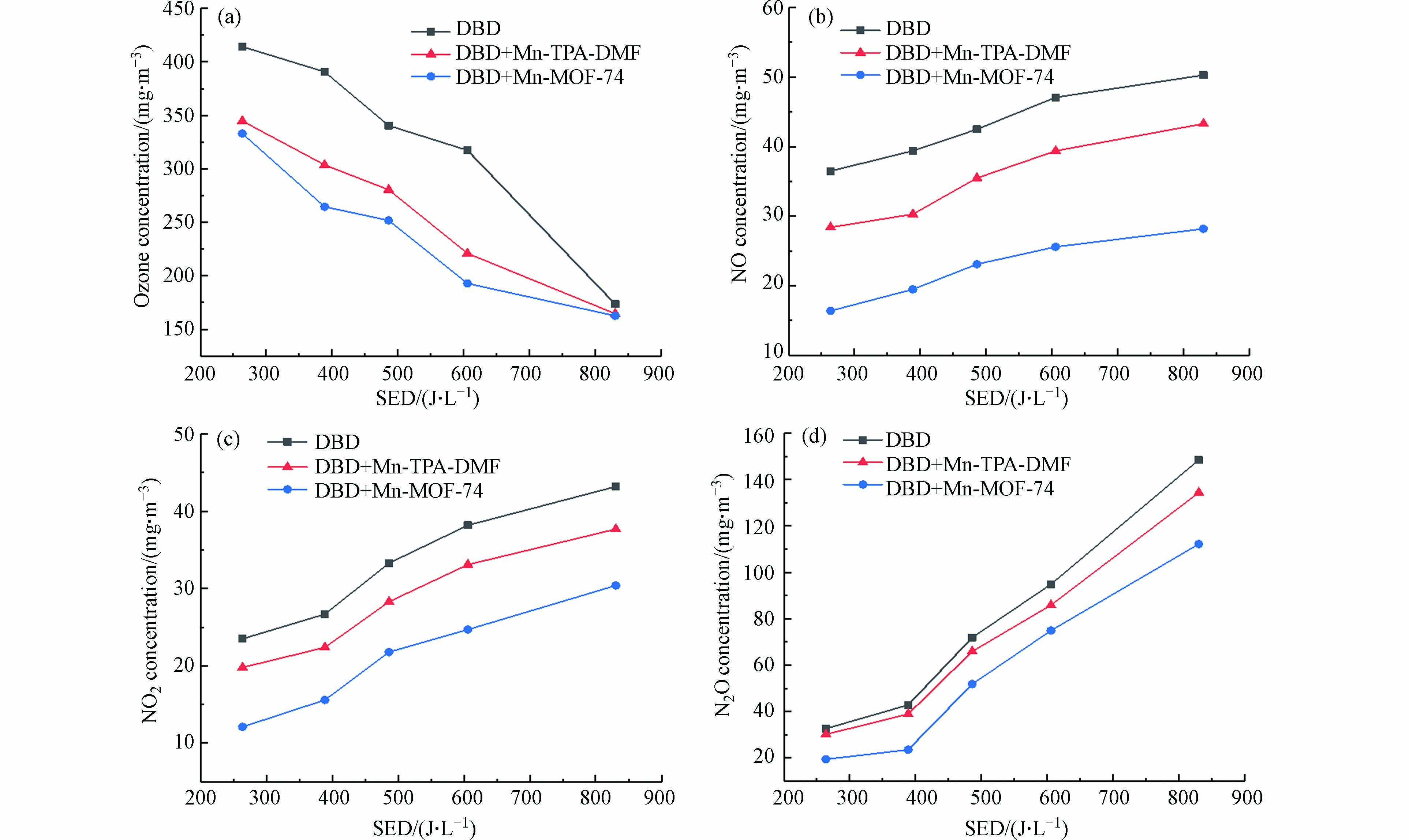 图 9 DBD、DBD+Mn-MOFs催化降解甲苯(a)O3浓度和(b)、(c)、(d) NOx浓度随能量密度的变化Figure 9. DBD and catalyst added (a) O3 concentration and (b) , (c) , (d) NOx concentration as a function of energy density
图 9 DBD、DBD+Mn-MOFs催化降解甲苯(a)O3浓度和(b)、(c)、(d) NOx浓度随能量密度的变化Figure 9. DBD and catalyst added (a) O3 concentration and (b) , (c) , (d) NOx concentration as a function of energy density采用烟气分析仪检测了DBD和DBD-催化体系中3种NOx(NO、N2O和NO2)的浓度,得到NOx生成浓度与SED的关系(图9 b-d). 如图9b-d所示,NO、NO2和N2O的浓度均随SED的增大而增大,与单独DBD系统相比,DBD-催化体系中生成的NOx浓度明显降低,且Mn-MOF-74催化剂的NOx生成量低于Mn-TPA-DMF,表明Mn-MOF-74催化剂更有利于抑制NOx的生成. 例如,当SED为263.68 J·L−1时,单独DBD系统产生的NO2浓度为26.7 mg·m−3,DBD+Mn-TPA-DMF和DBD+Mn-MOF-74体系分别降至22.4 mg·m−3和12.1 mg·m−3. 通过上述结果可知,催化剂可以提高反应的选择性,进而抑制副产物NOx等的生成. 其生成量降低的原因在于Mn基催化剂的引入有利于O3在催化剂的表面生成氧气和表面活性氧,生成的活性氧进而参与甲苯的降解反应,避免了与N2生成NOx. 此外,催化剂的加入还有利于高能电子传输,并降低NOx的解离能进而可以使部分NOx解离为N2[41- 42].
2.3 DBD/Mn-MOFs催化降解甲苯的机理分析
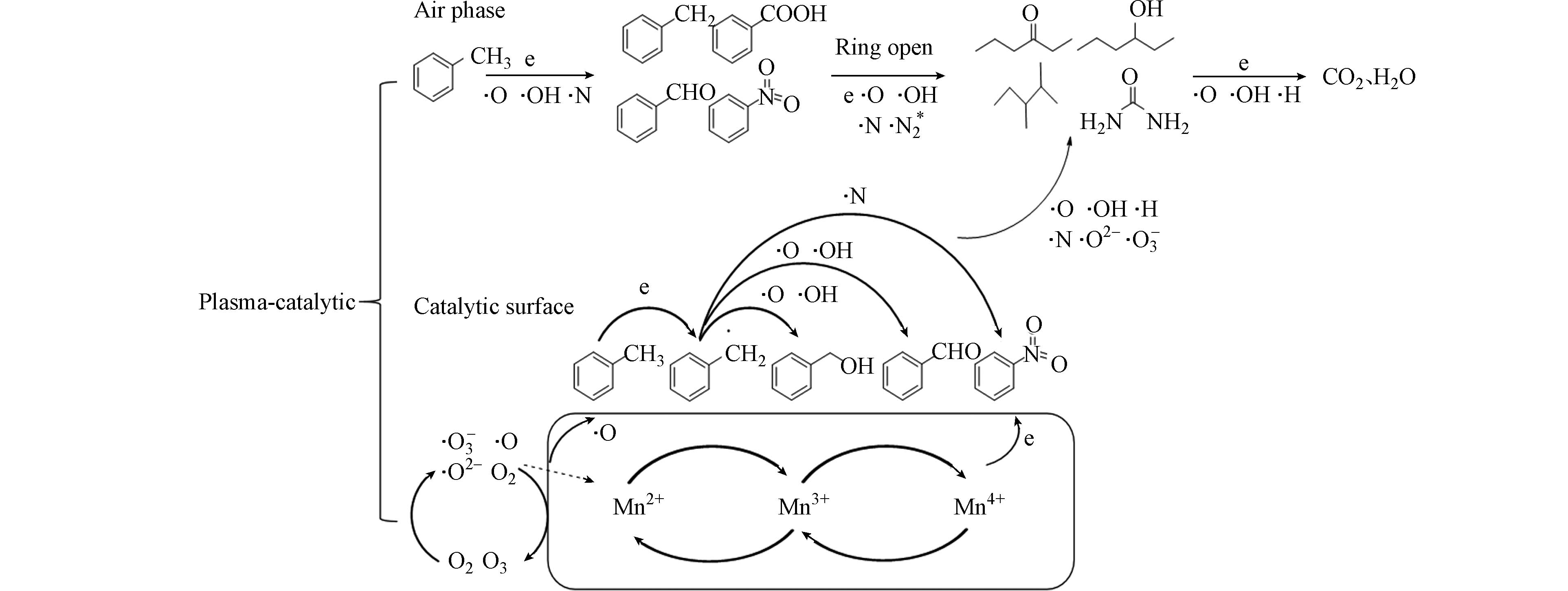
采用气相色谱-质谱联用技术(GC-MS)对DBD及DBD复合催化剂的有机副产物进行了分析. 将反应气体用正己烷吸收20 min,然后将吸收液注入GC-MS仪器进行检测. 结合降解前后材料表征结果及甲苯降解效果,推测DBD/Mn-MOFs催化降解甲苯的机理如下图:由图10可知,等离子体Mn-MOFs催化降解甲苯主要包括3个路径. 等离子体电场内具有丰富的高能电子,高能电子轰击甲苯分子导致甲苯降解是一个主要途径;高能电子与空气中的O2、N2等产生活性粒子(·OH, N, N2*, ·O, N2*, O2- ),活性粒子降解甲苯是第二条途径;再次,催化剂表面的反应取决于甲苯及中间产物的化学吸附、吸附氧的数量及Mn2+、Mn3+、Mn4+之间的转换. 气相中的O2、O3和电子能够借助Mn2+、Mn3+、Mn4+之间的表面转换,被吸附和转移到催化剂表面的甲苯和中间产物上,进一步产生活性粒子(OH、·O等),从而将甲苯进一步矿化为CO2. XPS结果显示催化反应后催化剂的Oads的占比显著增加,也进一步证实Mn基MOFs表面氧空位作为表面缺陷成为催化反应的活性位点,导致O3在催化剂表面分解产生更多的表明氧物种(O和O2),促进甲苯和中间产物的深度氧化[10,31]. 当然,Mn基MOFs催化剂的引入,也会使得等离子体中表面局部场强增大,改变了等离子体放电特性,对降解也会产生影响.
3. 结论(Conclusion)
本文制备了Mn-MOF-74和Mn-TPA-DMF两种Mn基MOFs催化材料,放置在DBD等离子体反应器对甲苯进行催化降解实验. 通过材料表征、降解效果分析等,探索了DBD催化降解机理,结论如下:
1) 与单独DBD相比,DBD与Mn基MOFs催化剂联用显著提高了甲苯的去除率、CO2选择性、矿化度和能量效率,并显著抑制了O3和NOx副产物的生成. DBD+Mn-MOF-74催化降解甲苯效果最好,当SED为605.87 J·L−1时,甲苯降解率达94.91%、CO2选择性达46.2%、矿化度达71.3%,与单独DBD相比,副产物O3和NO生成量下降到50%左右.
2) Mn-MOF-74与Mn-TPA-DMF催化剂相比,表现出更优异的催化活性,这个与Mn-MOF-74具有更高的比表面积、更多的Oads含量、更强的氧化还原性能相关.
3) XPS材料表征表明,Mn基修饰的MOFs材料中Mn作为电荷转移媒介,促进了电荷的循环转移. 通过DBD催化反应前后的催化剂的FTIR图谱可知,Mn-MOF-74和Mn-TPA-DMF在反应前后没有发生结构上的改变,说明催化剂性能稳定.
-
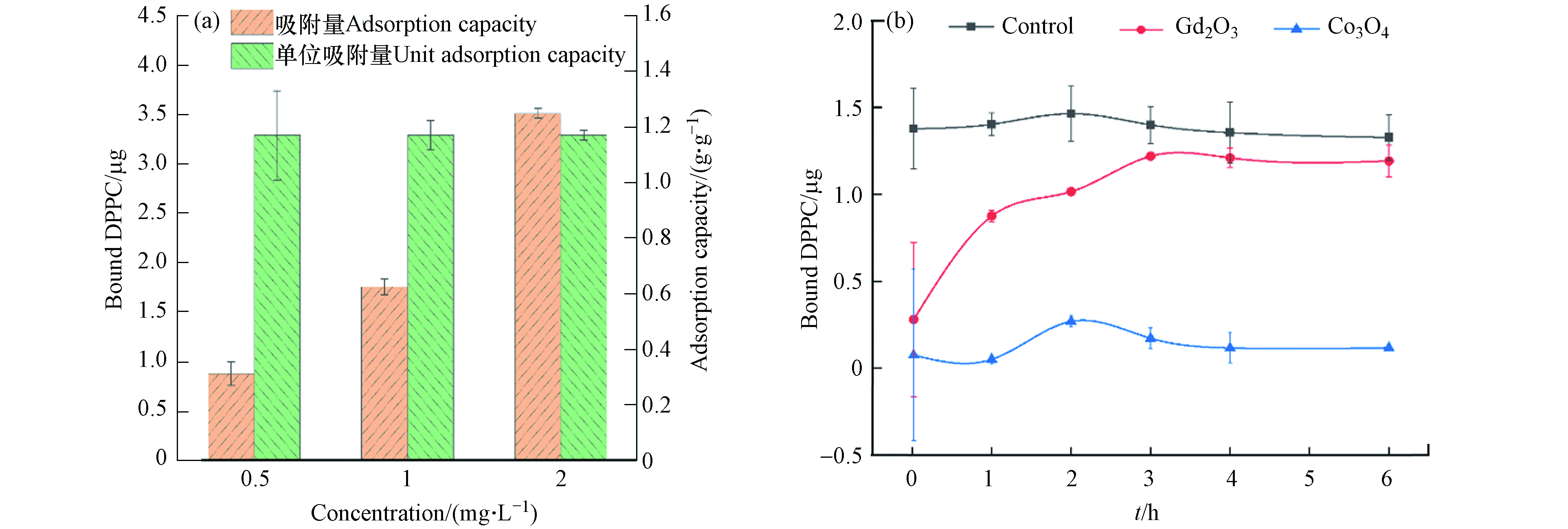
图 1 不同浓度Gd2O3对DPPC的吸附量(a)Gd2O3和Co3O4对DPPC的吸附动力学曲线(b)
Figure 1. Adsorption capacity of Gd2O3 with different concentrations for DPPC (a) And Adsorption kinetics of Gd2O3 and Co3O4 (b)
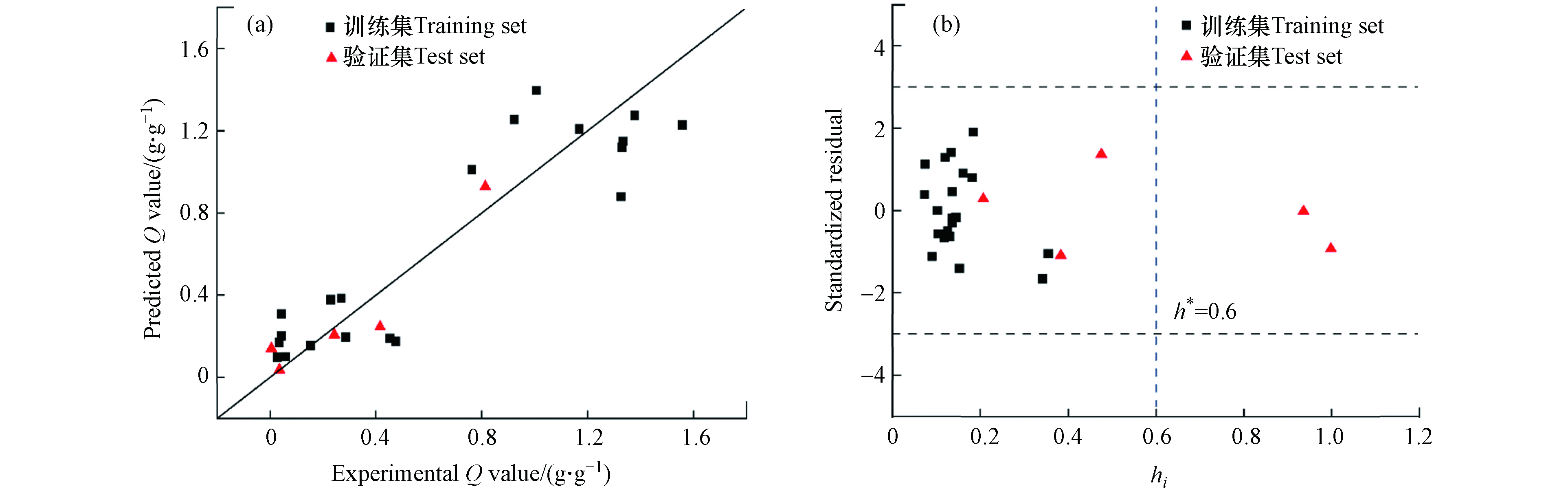
图 3 MeONPs对DPPC吸附量实测值与模型预测值的对比图(a)和PLS预测模型应用域表征的Williams 图(b)
Figure 3. Plot of experimental versus predicted Q values (a) And Williams plot of the application domain of the PLS model (b)
-
[1] BASANT N, GUPTA S. Multi-target QSTR modeling for simultaneous prediction of multiple toxicity endpoints of nano-metal oxides [J]. Nanotoxicology, 2017, 11(3): 339-350. doi: 10.1080/17435390.2017.1302612 [2] KIM T, HYEON T. Applications of inorganic nanoparticles as therapeutic agents [J]. Nanotechnology, 2014, 25(1): 012001. doi: 10.1088/0957-4484/25/1/012001 [3] LI Y, YU S L, YUAN T Z, et al. Rational design of metal oxide nanocomposite anodes for advanced lithium ion batteries [J]. Journal of Power Sources, 2015, 282: 1-8. doi: 10.1016/j.jpowsour.2015.02.016 [4] ZHAO Z H, TIAN J, SANG Y H, et al. Structure, synthesis, and applications of TiO2 nanobelts [J]. Advanced Materials, 2015, 27(16): 2557-2582. doi: 10.1002/adma.201405589 [5] KAPRALOV A A, FENG W H, AMOSCATO A A, et al. Adsorption of surfactant lipids by single-walled carbon nanotubes in mouse lung upon pharyngeal aspiration [J]. ACS Nano, 2012, 6(5): 4147-4156. doi: 10.1021/nn300626q [6] ZHAO Q, LI Y J, CHAI X L, et al. Interaction of pulmonary surfactant with silica and polycyclic aromatic hydrocarbons: Implications for respiratory health [J]. Chemosphere, 2019, 222: 603-610. doi: 10.1016/j.chemosphere.2019.02.002 [7] KIM K H, KABIR E, KABIR S. A review on the human health impact of airborne particulate matter [J]. Environment International, 2015, 74: 136-143. doi: 10.1016/j.envint.2014.10.005 [8] RAESCH S S, TENZER S, STORCK W, et al. Proteomic and lipidomic analysis of nanoparticle Corona upon contact with lung surfactant reveals differences in protein, but not lipid composition [J]. ACS Nano, 2015, 9(12): 11872-11885. doi: 10.1021/acsnano.5b04215 [9] MONOPOLI M P, ÅBERG C, SALVATI A, et al. Biomolecular coronas provide the biological identity of nanosized materials [J]. Nature Nanotechnology, 2012, 7(12): 779-786. doi: 10.1038/nnano.2012.207 [10] PARRA E, PÉREZ-GIL J. Composition, structure and mechanical properties define performance of pulmonary surfactant membranes and films [J]. Chemistry and Physics of Lipids, 2015, 185: 153-175. doi: 10.1016/j.chemphyslip.2014.09.002 [11] GUAGLIARDO R, PÉREZ-GIL J, de SMEDT S, et al. Pulmonary surfactant and drug delivery: Focusing on the role of surfactant proteins [J]. Journal of Controlled Release, 2018, 291: 116-126. doi: 10.1016/j.jconrel.2018.10.012 [12] THORLEY A J, RUENRAROENGSAK P, POTTER T E, et al. Critical determinants of uptake and translocation of nanoparticles by the human pulmonary alveolar epithelium [J]. ACS Nano, 2014, 8(11): 11778-11789. doi: 10.1021/nn505399e [13] YU Q L, WANG H G, PENG Q, et al. Different toxicity of anatase and rutile TiO2 nanoparticles on macrophages: Involvement of difference in affinity to proteins and phospholipids [J]. Journal of Hazardous Materials, 2017, 335: 125-134. doi: 10.1016/j.jhazmat.2017.04.026 [14] KONDURU N V, DAMIANI F, STOILOVA-MCPHIE S, et al. Nanoparticle wettability influences nanoparticle-phospholipid interactions [J]. Langmuir, 2018, 34(22): 6454-6461. doi: 10.1021/acs.langmuir.7b03741 [15] LUO Z, LI S X, XU Y, et al. Extracting pulmonary surfactants to form inverse micelles on suspended graphene nanosheets [J]. Environmental Science:Nano, 2018, 5(1): 130-140. doi: 10.1039/C7EN00843K [16] LUO Z, LI S X, XU Y, et al. The role of nanoparticle shape in translocation across the pulmonary surfactant layer revealed by molecular dynamics simulations [J]. Environmental Science:Nano, 2018, 5(8): 1921-1932. doi: 10.1039/C8EN00521D [17] CHHODEN T, CLAUSEN P A, LARSEN S T, et al. Interactions between nanoparticles and lung surfactant investigated by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry [J]. Rapid Communications in Mass Spectrometry, 2015, 29(11): 1080-1086. doi: 10.1002/rcm.7199 [18] WANG F, LIU J W. Liposome supported metal oxide nanoparticles: Interaction mechanism, light controlled content release, and intracellular delivery [J]. Small, 2014, 10(19): 3927-3931. doi: 10.1002/smll.201400850 [19] ZHANG X, PANDIAKUMAR A K, HAMERS R J, et al. Quantification of lipid Corona formation on colloidal nanoparticles from lipid vesicles [J]. Analytical Chemistry, 2018, 90(24): 14387-14394. doi: 10.1021/acs.analchem.8b03911 [20] BYLESJÖ M, RANTALAINEN M, CLOAREC O, et al. OPLS discriminant analysis: Combining the strengths of PLS-DA and SIMCA classification [J]. Journal of Chemometrics, 2006, 20(8/9/10): 341-351. [21] SCHÜÜRMANN G, EBERT R U, CHEN J W, et al. External validation and prediction employing the predictive squared correlation coefficient test set activity mean vs training set activity mean [J]. Journal of Chemical Information and Modeling, 2008, 48(11): 2140-2145. doi: 10.1021/ci800253u [22] FATEMI M, GHORBANNEZHAD Z. Estimation of the volume of distribution of some pharmacologically important compounds from their structural descriptors [J]. Journal of the Serbian Chemical Society, 2011, 76(7): 1003-1014. doi: 10.2298/JSC101104091F [23] HUANG Y, LI X H, XU S J, et al. Quantitative structure-activity relationship models for predicting inflammatory potential of metal oxide nanoparticles [J]. Environmental Health Perspectives, 2020, 128(6): 67010. doi: 10.1289/EHP6508 [24] GRAMATICA P. Principles of QSAR models validation: Internal and external [J]. QSAR & Combinatorial Science, 2007, 26(5): 694-701. [25] 祁晓娟, 李雪花, 黄杨, 等. 预测农药植物角质层-水分配系数的LSER模型 [J]. 农药学学报, 2020, 22(2): 249-255. doi: 10.16801/j.issn.1008-7303.2020.0053 QI X J, LI X H, HUANG Y, et al. LSER model for predicting cuticle-water partition coefficients of pesticides [J]. Chinese Journal of Pesticide Science, 2020, 22(2): 249-255(in Chinese). doi: 10.16801/j.issn.1008-7303.2020.0053
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 1446
- HTML全文浏览数: 1446
- PDF下载数: 105
- 施引文献: 0










