


-
毛细管液相色谱是一种微型化的液相色谱分离技术[1],毛细管直径范围从几微米至几百微米. 由于其具有柱内体积小(0.01—30 μL)、节约试剂(0.1—10 L·a−1)、所需样品量少以及易与当前使用较为广泛的检测器(如质谱、光学和电化学检测器等)在线联用等诸多优势[2],在痕量样本检测方面具有独特优势[3-4].
毛细管柱的填充方法包括多种:有湿法填充[5](包括上行法和下行法)、干法填充[6]和电泳填充法[7]. 在上世纪70年代,Ishii[8]和Novotny[9]等就开展了一系列的研究工作,致力于填充出高效毛细管柱. 他们填充的毛细管柱内径较大,约为几百微米. 之后Jorgenson和Kennedy等研发了多种填充方法,极大地提高了柱效,并且能够填充超低内径的毛细管柱(低至12 μm)[2,9-14]. 但是上行法装填毛细管柱的过程中,为了防止填料的自然沉降,采用磁子对匀浆持续地搅拌[15]. 这种填充方式带来的弊端就是会使填料破碎,这样会导致填料的硅羟基暴露于色谱环境中,不断地吸附样品,例如碱性物质、大分子蛋白质、核酸等分子等[16],进而影响色谱柱的柱效、分离度下降、色谱柱寿命降低等情况. 因此,毛细管柱的装填过程应格外注意保护填料,避免破碎. 与上行法相比较,下行法无需磁力搅拌装置,可降低破碎率. 但是下行法为了抵抗由于重力引起的填料聚沉,多采用快速填充方式,利用低粘度的有机溶剂进行快速填充[17]. 填料在快速下行过程中,仍然会与管口碰撞而导致破碎.
为解决此类难题,关亚风研发了干法填充技术[6],用于填充大内径色谱柱(例如200—500 μm),填料尺寸为5—10 μm的球形填料. 在过程采用低压填充(低于1 MPa),因此完全避免了填料的破碎,色谱柱的柱效高且性能稳定、惰性好. 但是干法填充技术不适合采用尺寸≤ 3 μm的填料和填充内径低于50 μm的毛细管柱,而匀浆法没有这个限制.
针对以上问题,本文拟解决细内径毛细管填充所面临的填料易破碎、柱效不稳定等问题. 首先,自主设计一款填充装置,装置内部为一款自制的尼龙漏斗,为进一步减小填料碰撞,尼龙的出料口直径与柱内径相同;本文也研制出一款下行式填充方法,采用体积比为9:1丙酮/甲醇作为匀浆溶剂进行缓慢填充,确保在填充过程中填料可均匀分散,提高柱效. 本实验填充的毛细管柱内径为25—250 μm, 有效长度为100 mm和150 mm.
-
石英毛细管(外径365 μm,内径分别为25、50、75、100、200、250 μm);填料为大连化学物理研究所赠与,填料表面修饰C18的硅胶球(填料直径3 μm). 所用匀浆试剂为乙腈、丙酮、甲醇、己烷(色谱纯,Merck)和曲拉通X-100(Triton X-100,美仑生物). 4种测试柱效的试剂分别为硝基苯、萘、菲和芘(分析纯,阿拉丁),用乙腈/水(80:20)配成0.25 g·L−1的测试混合样品. 筛板采用正硅酸乙酯(TEOS)、盐酸(1.2 mol·L−1)以及5 μm的硅胶填料制作.
-
岛津液相色谱泵;Jasco CE-975多波长紫外检测器(日本),设置波长254 nm;采用色谱3010工作站采集数据和计算实际柱效;倒置显微镜IX30;场发射扫描电子显微镜(FESEM)型号JSM-7800F(日本).
-
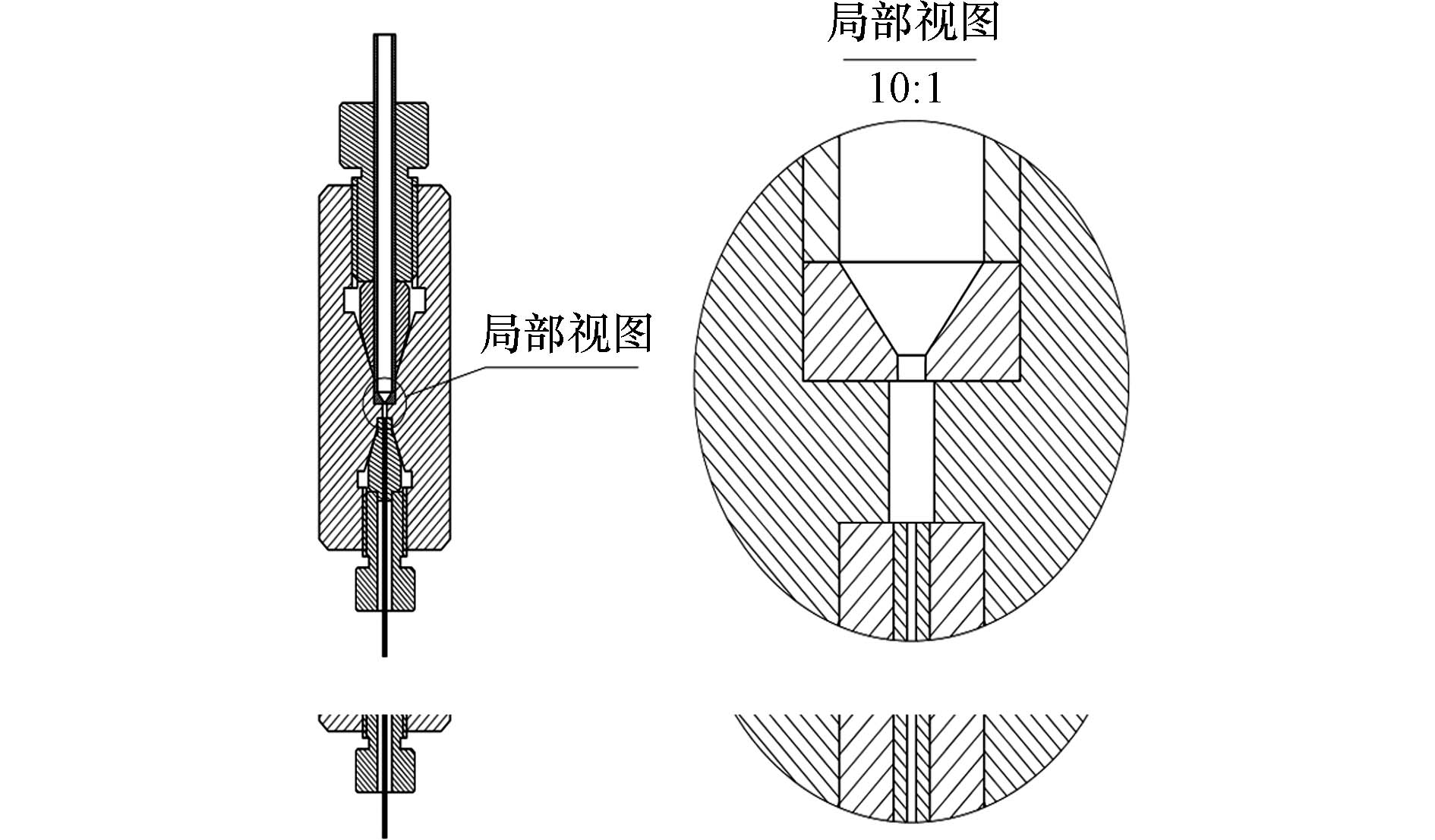
本实验研发一款填充装置,该装置为一个不锈钢罐,罐的内部添加一个尼龙漏斗,罐的上部连接液相色谱泵,罐的下部与毛细管柱连接,并采用PEEK管进行密封(外径1/16英寸,内径0.4 mm). 该填充装置的设计图如图1所示.
-
为测定填料的粒径分布,采用场发射扫描电子显微镜(field emission scanning electron microscopy, FESEM)观测填料的粒径和形貌,在照片中选取约143颗填料进行粒径分析. 形貌见图2(A),填料均呈现均匀的圆形且较完整,其粒径分布统计图见2(B). 经Gwyddion软件分析,粒径平均为3.68 μm (RSD=5.56%).
-
分别采用甲醇、丙酮、己烷、体积比为9:1丙酮/甲醇以及体积比为99:1水/Triton X-100作为匀浆溶剂,配置浓度为5 mg·mL−1和10 mg·mL−1的匀浆,将匀浆在超声仪中放置10 min后,移取10 μL匀浆置于显微镜下观察其匀浆状态;然后将上述匀浆重新超声5 min测定填料下沉时间.
-
在填充毛细管柱之前,首先制作毛细管柱的筛板. 制作的简要步骤如下:取115 μL HCl(1.2 mol·L−1),加入200 μL的TEOS后进行超声混匀,然后迅速加入5 μm的硅胶填料,两者的混合比例约为100 mL:30 mg,迅速进行超声2 min. 随后匀浆填料虹吸至毛细管内,注意筛板的长度约为几毫米,若太长则会影响色谱柱的通透性. 虹吸之后放入60℃烘箱,加热34 h,生成耐高压的筛板[18].
本实验采用3.68 μm的C18硅胶色谱填料,填充不同内径的毛细管柱. 在填充之前,经过实验优化,确定了5—30 mg·mL−1为最佳浓度,因为浓度大于30 mg·mL−1则会导致填料迅速沉降,堵塞管口,造成填充失败;而浓度低于5 mg·mL−1,则会导致填料浓度过低,柱中出现柱塌陷和孔隙率较大的情况,进而导致柱效低的情况,因此,综合以上情况来看,5—30 mg·mL−1为最佳浓度. 将上述匀浆经超声处理10 min[19],快速放入匀浆罐进行填充. 色谱泵中溶剂为纯水,采用高压填充,流速为0.1 mL·min−1. 由于采用恒流模式,在填充过程中,柱压会不断升高,当填充压力为35 MPa时,关闭流动相,最后,在20 MPa下过夜冲洗(甲醇和水各6 h).
-
为了最小化柱外效应,采用柱上检测方式(On-Column Detection),采用4种常见的苯系物硝基苯、萘、菲和芘为分析物,测定实际的柱效和分离度,分析流动相为体积比为80:20的ACN/H2O,改变测试流速并绘制h-V曲线. 评价完毕后取出填料并在电镜下观测其形貌.
-
由于上行法所采用的磁力搅拌会增加填料破碎率[20],因此,本文研发下行法填充毛细管. 设计装置如图1所示. 装置内部有一个尼龙漏斗,其出口内径与柱内径相同减少填充过程中的冲击. 色谱柱评价后,填料在电镜下的照片如图3所示,经高压冲洗和测试后,填料仍可保持完整.
-
填料的分散状态会影响填充色谱柱的性能[20-21]. 填料表面的电荷会导致填料彼此吸引,出现大块的粘结现象,填充过程中容易堵塞毛细管口,造成填充失败. 并且经过研究发现,相互“粘结”的填料在填充过程中容易导致柱子填充不紧实,而造成较大的“死体积”,进而导致低柱效. 因此,匀浆溶剂的选择十分重要,需要充分的分散填料. 本实验选择体积比为99:1水/Triton X-100、丙酮、体积比为9:1丙酮/甲醇、己烷和甲醇等作为匀浆溶剂,测定填料的分散状态,相比之下,图4(A)中体积比为99:1水/Triton X-100和图4(C)中体积比为9:1丙酮/甲醇的分散能力相对较好,可作为C18硅胶填料的匀浆溶剂.
由于重力的作用,填料会自然沉降,导致柱内填料的均匀度变差. 因此,需要在填料完全沉降之前完成色谱柱填充. 经测定,填料沉降时间见表1,并且填料浓度越高,沉降速度越快. 填料在体积比为99:1水/Triton X-100中的沉降较慢,在甲醇中沉降最快,在丙酮、体积比为9:1丙酮/甲醇和己烷中的沉降速度相当.
-
基于以上测定结果,最终以体积比为99:1水/Triton X-100、体积比为9:1丙酮/甲醇和己烷作为匀浆溶剂,填充内径为50 μm(i.d.)的毛细管柱并测定柱效,见表2. 结果表明, 采用体积比为9:1丙酮/甲醇填充的柱子性能最优. 为了去除溶解残留,柱子填充后分别采用甲醇和水冲洗6 h. 图5为25 μm(i.d.)毛细管柱在折合流速(V=2.4)下的色谱图,4种物质的调整保留值(k′)分别为0.60、1.62、2.97、4.68(此色谱图中的信噪比可达200). 其最小折合塔板高仅为2.25(k′=4.68),非常接近理论值(h=2). 图6为内径为25 μm(i.d.)毛细管柱的Van Deemter曲线(分析物为芘).
-
为研究柱效与柱内径的关系,本文制备了6种规格的毛细管柱. 6种色谱柱匀浆浓度的变化范围是5—30 mg·mL−1,除此之外,其他填充条件均相同,实验条件见1.5节,实验结果见表3,柱内径越细,柱效越高.
-
Van Deemter方程(式1)可以较直观的反映出色谱柱的性能.
图7为不同内径毛细管色谱柱的Van Deemter 方程中A、B、C三个系数随内径的变化情况. 在Van Deemter方程中的系数中,A(涡流扩散项的系数)与柱内径呈现出良好的线性关系,随着柱内径减小,A数值变小. 原因是:色谱柱内具有靠近管壁较疏松的“管壁区”与靠近中心区域较紧密的“核心区”,两个区域的相比不同,分析物在两个区域的保留时间将产生差异;内径越细,两个区域的差异越小,由此所造成的峰展宽也会随之减小[22]. 直至颗粒与柱直径之比降低到6以下,核心区域将消失[23],分析物在柱内的移动速度趋于一致. 因此,柱内径越细,色谱柱内部的均匀程度越高,涡流扩散(A)项越小.
B(纵向扩散项的系数)与毛细管的内径无明显线性关系,从Van Deemter 方程(式1)分析可知,纵向扩散项(B项)与色谱柱中填料的填充因子(γ≈0.6)和溶质在流动相中的扩散系数有关(DM≈10−6 — 10−5cm2·s−1),与柱内径无关. 从图7(C)可知,B项数值不随内径的改变而呈现线性变化,其原因是不同的色谱柱的填充密度与填充的均匀程度略有差异,与填充方式有关,与柱内径无关.
C(传质阻力项的系数)也不随内径的改变而呈现线性变化. 因为C项为固定相传质阻力和流动相传质阻力的加和,取决于分析物在流动相中的扩散系数和填料粒径及固定相膜厚度等因素,由于不同内径的毛细管柱所用的固定相和流动相均相同,因此,各个毛细管柱的C项相差不大.
-
填充细内径毛细管柱是一个精细且复杂的过程. 常规毛细管内径约为(200—500 μm),管径较大,填充较容易,可以采用干法填充和湿法填充;而细内径毛细管由于内径较小(仅约25—150 μm),在填充过程中容易导致填料堵塞管口而造成填充失败,并且由于填充过程需要较高压力(20—40 MPa),应该格外注意安全,仅能使用湿法填充. 其柱效会受到填充溶剂、填充压力和填料质量等因素的影响. 实际上,随着色谱柱内径和颗粒直径的减小,填充的难度会急剧增加. 本文采用自行设计的填充装置以下行式匀浆法填充细内径毛细管液相色谱柱,降低了填充时填料的破碎率. 采用体积比为9:1丙酮/甲醇作为匀浆溶剂进行缓慢填充,在填充过程中填料在匀浆溶剂中可保持均匀分散的状态,获得高柱效.
本实验研究了内径与Van Deemter方程中A, B, C项的系数及柱效的关系. 在细内径毛细管柱(25—250 μm)中,随着内径降低,毛细管色谱柱的柱效呈现明显增加的趋势. 在柱内径25 μm时,最小折合塔板高度仅为2.25(k′=4.68),柱效高达148200 m−1. 在应用方面,因为细内径毛细管柱对于样品的稀释效应较小,其对于痕量样品分析具有极大的优势,有望应用于环境、生物样品中痕量成分的高效分离分析.
下行式匀浆法填充细内径毛细管液相色谱柱
Preparation of small-diameter capillary liquid chromatography columns using downward packing method
-
摘要: 本文采用下行式匀浆法填充内径为25—250 μm,有效长度为100 mm和150 mm的毛细管液相色谱柱,填料为3 μm C18修饰的多孔硅球. 经实验条件优化,匀浆溶剂是体积比为9 : 1丙酮/甲醇,匀浆浓度的变化范围为5—30 mg·mL−1. 在此条件下,色谱柱的柱效随柱内径减小而提高. 通过比较Van Deemter方程中A、B和C项系数与毛细管内径之间的关系发现,随着柱内径减小,A项的系数呈线性减小的趋势,B和C项的系数与内径无明显关系. 在最佳流速下,内径为25 μm毛细管柱的折合塔板高度低至2.25,接近理论塔板高度.
-
关键词:
- 下行法 /
- 毛细管柱 /
- 柱效 /
- Van Deemter方程.
Abstract: We developed a downward packing method for preparing the efficient capillary column with inner diameters (i.d.) from 25—250 µm in the effective length of 100—150 mm. The columns were packed by 3 μm of C18 modified silica particles with 5—30 mg·mL−1 slurries in acetone/methanol (9:1, V/V). In this condition, the column efficiency was increased as the diameter of the column decreased. By comparing the coefficients of A, B and C term in the Van Deemter equation with the inner diameters of the capillary columns, we found the coefficient of A term decreased linearly with decreasing the i.d. of the column. However, there is no linear relationship between the coefficients of the B and C terms with the i.d. of the column. At the optimum reduced velocity, the hmin (the minimum plate height) of the 25 μm i.d. column was as low as 2.25, which is very close to the theoretical plate (HETP).-
Key words:
- downward packing method /
- capillary columns /
- column efficiency /
- Van Deemter equation.
-
抗生素是一类由微生物(包括细菌、真菌、放线菌属)分泌或化学合成等途径产生的复杂分子,可以抑制微生物活性,提高人和动物的抵抗力,因而在医疗和养殖行业被广泛应用[1]. 滥用现象导致的抗生素耐药性问题,已经使其成为备受关注的一类新型污染物. 抗生素在环境中极为隐蔽、检出难度大、扩散性强且极易被生物积累,对生态系统稳定性和人体健康造成潜在威胁[2].
与人类医疗行业使用的抗生素类型不同,四环素类、磺胺类和喹诺酮类药物在畜禽养殖中被广泛使用[3]. 多数兽用抗生素在畜禽体内生物利用率较低,未完全代谢的抗生素极易随动物排泄物进入环境[4]. 因此,畜禽粪便被认为是环境抗生素的主要污染源之一. 进入土壤中的抗生素,一部分在生物因素(如土壤微生物)和非生物因素(如土壤组分)的作用下发生吸附或降解反应,另一部分则会持续对土壤微生物施加选择压力,改变土壤微生物的活性和群落组成,甚至会促进环境中耐药菌的传播和抗生素抗性基因的污染. 此外,抗生素不仅会通过多种途径迁移到动植物体内,还会伴随地表径流以及淋溶作用迁移到地表水和地下水中,扩大污染范围(图1)[5 − 6]. 因此,土壤环境中抗生素的污染现状及其生态风险逐渐成为研究热点.
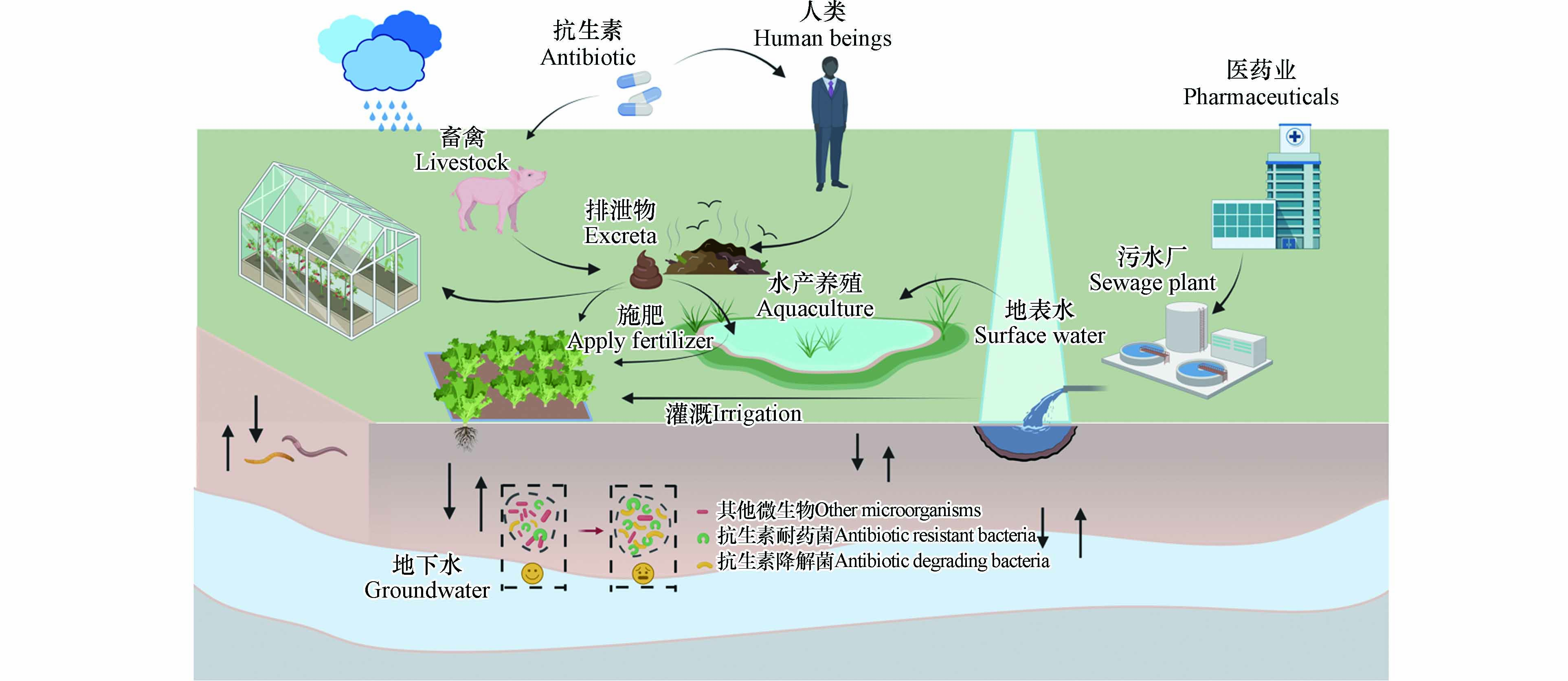 图 1 环境中抗生素的来源及生态危害Figure 1. Sources of antibiotics in the environment and ecological hazards
图 1 环境中抗生素的来源及生态危害Figure 1. Sources of antibiotics in the environment and ecological hazards近年来,作为化肥的替代品,以畜禽粪便为重要来源的有机肥成为了农业生产必不可少的营养来源. 与其他类型的农用地相比,菜地往往因作物生长周期短且经济效益高而轮作频繁,对有机肥的消耗量也远高于其他类型作物[7],因此菜地较粮食作物地更可能受到畜禽粪便污染,也更易出现抗生素残留. 调查显示,相对于常规菜地,施用有机肥的菜地土壤中抗生素残留量更高,部分地区抗生素残留量达到mg·kg−1级别[6, 8]. 尽管学者对抗生素污染的来源、现状和归趋已经有了深入研究成果,但是各类抗生素在菜地系统中的污染现状调查仍然存在技术壁垒等客观限制. 抗生素在环境中具有一定的内在生物活性,能够对生物和人类健康产生毒性作用. 目前关于抗生素的潜在生态危害的相关研究较少,其产生生态毒性效应机制尚不明确. 文章综述了菜地系统中抗生素的污染情况、影响因素和可能造成的生态效应,重点分析了抗生素对土壤生物的毒性作用机制,以期为减轻环境抗生素污染提供理论支撑和实践指导.
1. 菜地土壤抗生素污染特征及其影响因素(Characteristics and influencing factors of antibiotic contamination in vegetable soil)
1.1 土壤中抗生素的空间分布特征
我国菜地土壤受到不同程度抗生素污染,具体污染状况与区域位置密切相关,总体呈现“北高南低,东高西低”的特征(表1). 截至2021年底,我国菜地土壤抗生素的残留量范围在4.59—2101.4 μg·kg−1之间,平均残留量为186.98 μg·kg−1. 其中,西北和华中部分区域的抗生素污染相对严重,平均含量分别高达243.20 μg·kg−1和724.13 μg·kg−1,而东部沿海典型污染带主要分布在黄淮海和长江三角洲地区,该地区抗生素平均总含量达166.32 μg·kg−1. 我国菜地土壤抗生素含量的空间差异可能与当地抗生素的使用情况有关. Zhang等[9]对我国抗生素使用情况调研显示,华东(38800 t)>华北(27900 t)>华中(21100 t)>西南(18300 t)>华南(9030 t)>东北(6070 t)>西北(2360 t). 然而,西北地区抗生素的使用量和土壤抗生素残留量呈现负相关关系,这极可能与区域农户的施肥习惯有关[10]. 此外,研究区域中四环素类抗生素的检出量最高,其次是喹诺酮类和磺胺类抗生素(表1). 可能原因如下:(1)四环素类在畜禽养殖中使用量最大[11]且具有高Koc值[12];(2)喹诺酮类抗生素在土壤中的吸附能力较低,即使畜禽养殖对其消耗量较大,其在土壤中的残留水平仍低于四环素类抗生素[13];(3)磺胺类抗生素较低的pKa值会导致去质子化物种的比例增加进而降低对土壤的吸附[12]. 然而,由于缺少合适的测定方法,复杂基质中的低浓度水平抗生素难以被检出,因此难以全面评估土壤抗生素污染情况. 此外,我国对菜地土壤抗生素污染的研究多集中在人口稠密、经济发达地区,如京津冀、长三角和珠三角地区,对西北地区的关注有限,有关部门理应给予警惕.
表 1 我国菜地表层土壤抗生素检出浓度Table 1. Antibiotic concentrations in the surface soil of vegetable fields in China地理区域Geographic area 省份Provinces 调查地点Investigation sites 抗生素含量/(μg·kg−1)Antibiotic content 参考文献References ∑TCs ∑SAs ∑QNs SUM 华南地区South China 广东省Guangdong Province 珠三角The Pearl River Delta Area 242.6 321.4 1537.4 2101.4 [3] 84.8 9 — 93.8 [32] 佛山市Foshan 4.24 — 17.83 22.07 中山市Zhongshan 14 — — 14 东莞市Dongguan 15.4 0.96 50.23 66.59 广州市Guangzhou 27.48 1.53 55.81 84.82 [33] 37.1 96.2 — 133.3 [34] — — 48.85 48.85 [35] 西南地区Southwest China 云南省Yunnan Province 昆明市Kunming 19.9 0.2 15.2 35.3 [23] 重庆市Chongqing 重庆Chongqing 79.81 — — 79.81 [19] 9.36 2.564 29.83 41.754 [27] 贵州省Guizhou Province 贵阳市Guiyang 5.07 0.62 5.28 10.97 [36] 1.52 0.62 5.28 7.42 [15] 三峡库区the Three Gorges Reservoir 53.3 99.2 75.9 228.4 [37] 华东地区East China 安徽省Anhui Province 安庆市Anqing 31.9 — — 31.9 [38] 合肥市Hefei — 4.59 — 4.59 [39] 上海市Shanghai 上海Shanghai 17.1 7.6 62.5 87.2 [23] 江苏省Jiangsu Province 徐州市Xuzhou 460.8 6.6 54.1 521.5 南京市Nanjing 64.5 10.2 38.5 113.2 黄淮海平原Huang-Huai-Hai Plain 24.01 0.03 16.32 40.36 [40] 浙江省Zhejiang Province 宁波市Ningbo 41.43 0.07 8.97 50.47 [41] 长三角Yangtze River Delta 27.03 3.25 42.67 72.95 [42] 山东省Shandong Province 135.664 32.866 — 168.53 [43] 274 3.91 73.05 350.96 [44] 华中地区Central China 河南省Henan Province 723.42 0.61 0.1 724.13 [45] 华北地区North China 北京Beijing 北京Beijing 103.58 13.41 7.35 124.34 [46] 102 1.1 86 189.1 [47] 河北省Hebei Province 石家庄市Shijiazhuang — — 170.775 170.775 [48] 西北地区Northwest China 宁夏省Ningxia Province 银川市Yinchuan 462.24 — — 462.24 [10] 陕西省Shaanxi Province 杨凌市Yangling 12.77 1.14 51.76 65.67 [49] 甘肃省Gansu Province — 201.676 — 201.676 [50] 东北地区Northeast China 黑龙江省Heilongjiang Province 哈尔滨市Harbin 181.74 — — 181.74 [51] 河北、河南、四川和江苏省Hebei, Henan, Sichuan and Jiangsu Province 82.75 2.61 12.78 98.14 [52] — ,未检测;TCs,四环素类;SAs,磺胺类;QNs,喹诺酮类. — , non-detection; TCs, tetracyclines; SAs, sulfonamides; QNs, quinolones. | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 土壤中抗生素污染特征的影响因素
如上所述,我国菜地土壤普遍存在抗生素污染且污染水平在地理空间上差异较大. 土壤作为抗生素富集的一种典型环境载体,其中的抗生素残留量还与很多因素有关,主要包括以下4种.
1.2.1 输入水平
农业生产中灌溉和施肥是大部分土壤中抗生素的主要来源. 污水和粪肥中抗生素含量与土壤抗生素总含量表现显著正相关关系. 厉文辉等[14]对凉水河两岸污灌区和井水灌溉区农田土壤样品进行采集检测,发现污灌区土壤抗生素残留量高于井水灌溉区,这意味着抗生素会从灌溉水转移到农田土壤中. 考虑到不同来源灌溉水污染水平的差异,Pan等[12]比较了用鱼塘水和生活污水灌溉后土壤中抗生素污染情况,发现鱼塘水灌溉的土壤中抗生素污染水平较高,这警示相关部门需要加强对水产养殖业的药品管控. 此外,距灌溉水源越近,土壤中抗生素检出率和残留量越高,反之越低[15]. 在Zhao等[16]的研究中,施用鸡、猪、牛等3类粪肥后土壤抗生素残留水平高低与这3类粪肥中抗生素含量高低一致,说明粪肥施用可显著增加土壤抗生素含量,且土壤抗生素残留水平与输入抗生素量息息相关. 重复性施肥可增加抗生素在土壤中的累积,不过也有学者研究发现,在施用粪肥的前期,土壤中抗生素的浓度会有所降低. 例如徐秋桐等[17]发现,在粪肥施加前期(第8天)施用1%有机肥处理组比未添加有机肥处理组4种抗生素降解率分别高出了12.5%、13.5%、24.8%和12.0%,这与Zhang等[18]的研究结论基本一致. 原因可能是有机肥提高了土壤中某些微生物的活性,从而加速了抗生素的生物降解.
1.2.2 种植条件
种植条件对菜地土壤中抗生素残留水平也存在一定影响. 一般来说,温室菜地中抗生素浓度要高于露天菜地,可能是在温室种植模式下,蔬菜的轮作更加频繁,粪肥的施用频率和总量更高,导致土壤中更多抗生素残留. 但彭秋等[19]和罗凯等[20]的调查发现,大棚菜地土壤抗生素浓度也可能会低于露天菜地. 这可能是该露天菜地靠近污染源,接触了由大气颗粒物携带的抗生素类污染物[21],也可能是露天菜地常以附近地表水作为灌溉水源,而灌溉水源受到了抗生素污染[20]. 其次,相较于露天环境,大棚的高温高湿条件有利于土壤微生物对抗生素的降解[22]. 种植年限与土壤抗生素残留水平存在密切关系. 在Zhang等[23]研究中,四环素类和氟喹诺酮类残留量随种植年限呈(中期)6年至10年<(长期)10年以上<(短期)5年以下的变化趋势,但土壤抗生素残留量的增长率却随种植年限延长而降低,这是由于进入土壤后抗生素的消散速率最初被抑制而后恢复,导致高含量的四环素类和喹诺酮类抗生素出现在短期种植的土壤中. 该现象提醒了今后研究还需关注抗生素代谢产物在土壤中的污染机制. 值得注意的是,磺胺类抗生素呈现出相反的情况. 在长期种植年限的土壤中往往能检测出更高含量的磺胺类[23],Fang等[24]研究也得到类似的结论. 从抗生素本身性质来说,一方面磺胺类抗生素的水溶性较四环素类和氟喹诺酮类更高,因而不易被土壤颗粒吸附. 另一方面,磺胺类抗生素对土壤有机质表现出高亲和力,这减缓了它在土壤中的迁移. 上述结论进一步证实了抗生素在土壤中的动态持久性.
1.2.3 土壤条件
理化性质和颗粒组成等土壤条件制约了抗生素在土壤中的移动. 土壤pH通过改变抗生素的电荷状态间接干扰抗生素的吸附. 多数抗生素(如四环素类和喹诺酮类等)的吸附能力随pH上升呈下降趋势,而磺胺类的吸附能力与pH有强烈的正相关性[25 − 26]. 抗生素进入土壤就会与土壤固相紧密结合,不难推断土壤抗生素含量与土壤有机质呈显著正相关关系[27]. 而有机质多分布在土壤黏粒上,意味着土壤黏粒比重越高土壤抗生素含量越高. 但也出现了不一样的研究结论,即在粘土比例较低的土壤中四环素类抗生素含量反而最高[28],这可能是在多因素的作用下土壤有机质含量下降了[29 − 30]. 因此可以认为土壤性质与抗生素的环境行为密切相关.
1.2.4 其他
关于气候条件对土壤中抗生素分布特征的研究较少. Hu等[6]报道,与夏季相比,冬季菜地土壤抗生素的残留更多,不难推测在一定条件下南方土壤抗生素残留水平要低于北方. 究其原因,适宜的水热条件极大帮助了土壤微生物对抗生素的降解[31],而低温低湿环境不利于抗生素在土壤中的降解和迁移. 此外,蔬菜种类的差异也是影响土壤抗生素含量的重要因素,比如磺胺类抗生素在土壤中表现为根茎类蔬菜土壤(289 μg·kg−1)>瓜果类蔬菜土壤(143 μg·kg−1)>叶菜类蔬菜土壤(98.1 μg·kg−1)[22];四环素类抗生素表现为叶菜类蔬菜土壤(77.4 μg·kg−1)>瓜果类蔬菜土壤(67.8 μg·kg−1)>茄果类蔬菜土壤(54.1 μg·kg−1)>豆类蔬菜土壤(47.7 μg·kg−1)[19],喹诺酮类抗生素总含量表现为果类蔬菜(44.8 μg·kg−1)>根茎类蔬菜(37.0 μg·kg−1)>叶菜类蔬菜(32.1 μg·kg−1)[8].
综上所述,菜地抗生素赋存差异性是多因素共同作用的结果. 其中,输入水平直接决定了土壤中抗生素的污染水平,而抗生素性质、种植条件、土壤条件和气候条件等在一定程度上影响着抗生素在土壤中含量的变化. 如吸附能力强的抗生素更易在土壤中累积,重复的施肥可以增加土壤中抗生素含量,而高温高湿环境有利于抗生素的降解.
2. 蔬菜中抗生素的累积迁移特征(Accumulation and migration characteristics of antibiotics in vegetables)
暴露在受抗生素污染的土壤中时,蔬菜可能会吸收抗生素并在体内不断累积. 某些抗生素甚至能够在较低浓度下被蔬菜吸收并累积,出现蔬菜体内抗生素浓度高于土壤中的现象. 如Hu等[6]发现林可霉素在土壤中的检出量低于在蔬菜中的检出量,Migliore等[53]发现,恩诺沙星在4种果蔬中的含量远高于培养基中的水平. 以上事实均表明,蔬菜对抗生素具有极强的生物累积性,而且实际环境下蔬菜对抗生素的累积系数远低于实验室模拟值,因此可以推断在更严重的污染条件下,该累积系数会增大.
蔬菜对抗生素的吸收按照吸收方式分为主动吸收和被动吸收. 主动吸收主要受蒸腾作用影响,需要消耗能量,而被动运输不需要其他辅助. 有研究比较了田间试验和盆栽试验发现,前者四环素类抗生素含量显著高于后者[54]. 这是由于田间开放条件下的蔬菜蒸腾作用更高,更有利于蔬菜对抗生素的主动吸收. 有研究发现磺胺多辛、土霉素和林可霉素在蔬菜中含量均很高,意味着这些抗生素的理化性质(如水溶性和半衰期)几乎不影响蔬菜对其的吸收,说明了蔬菜对这些抗生素的吸收是被动吸收[6]. 目前认为,蔬菜对有机污染物的吸收以被动吸收为主,并伴有一定程度的主动吸收.
蔬菜对抗生素的吸收和累积受多种因素影响,主要包括蔬菜种类、抗生素性质以及其他环境因子. 不同种类蔬菜中抗生素的残留水平差异很大,一般为叶菜类>果蔬类>块茎类[55],但是Li等[56]调查了蔬菜可食用部分氟喹诺酮的累积情况,发现茄果类>叶菜类. 这可能是不同类型蔬菜对抗生素的吸收能力不同,当然也不能忽略其他影响因素的作用,如土壤性质可以通过控制抗生素的生物可利用度来影响蔬菜对抗生素的吸收[56 − 58]. 抗生素性质似乎是影响抗生素吸收过程的主要因素. 一般认为,强土壤吸附性、高疏水性的四环素类抗生素的生物可利用度较低,而较弱土壤吸附性、较高水溶性的磺胺类和喹诺酮类抗生素生物可利用度较高[59]. 因此可以推测,磺胺类和喹诺酮类更容易被蔬菜吸收并累积. 然而,在某些实际环境中,蔬菜对这3类抗生素的吸收情况不符合上述结论. 如图2所示,前4种蔬菜中四环素类含量显著高于磺胺类和喹诺酮类,这与土壤中不同种类抗生素含量的变化一致[6],说明土壤中抗生素污染水平也是一个重要的影响因素. 此外,抗生素降解、土壤结合和浸出也限制了蔬菜吸收抗生素[2].
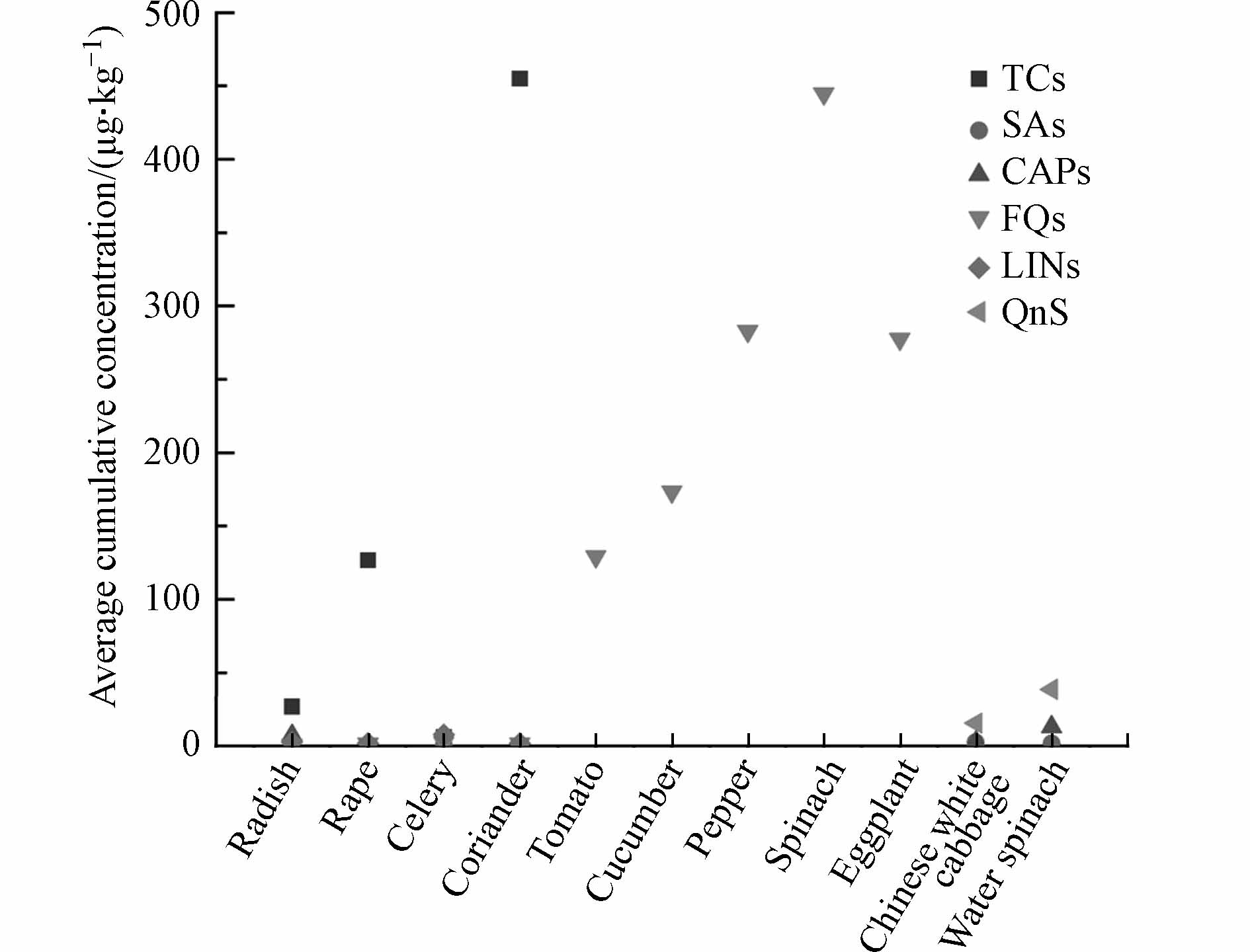 图 2 抗生素在不同种类蔬菜的累积情况Figure 2. Accumulation of antibiotics in different types of vegetables
图 2 抗生素在不同种类蔬菜的累积情况Figure 2. Accumulation of antibiotics in different types of vegetables表2中抗生素在叶中含量要大于茎和根,但是Migliore等[60]发现,根中抗生素含量高于叶. 这是由于抗生素在蔬菜体内的转运受蒸腾作用驱动,而蒸腾水量的差异最终会影响抗生素在根和叶中含量的差异[61]. 此外,抗生素种类也是影响其在蔬菜组织中分布水平差异的重要因素. 抗生素从植物根部向地上部分的转运能力常用易位因子测定. 大多数四环素类抗生素、喹诺酮类抗生素和林可霉素易位因子>1,但磺胺类抗生素和大环内酯类抗生素易位因子<1[62]. 这就造成了前者在植物体内均能被检出,而后者很难在植物地上部分被检出. 值得注意的是,大环内酯类药物尺寸较大,多数情况下很难在植物中被检测到[63]. 抗生素在蔬菜内的分布随不同生长阶段变化. 如采集收获前15 d蔬菜样品,发现抗生素在叶菜类根部含量高于叶片,而对成熟期采集的蔬菜样品检测得到相反的结论[6].
蔬菜Types of vegetables 抗生素Types of antibiotics 抗生素含量/(μg·kg−1)Antibiotic content 根Root 茎Stem 叶Leaves 萝卜Radish TCs 8.3 22.1 24.4—76.4 SAs 0.1—0.4 0.2—0.5 0.2—0.6 CAPs ND. 1.4—3.4 8—30 QNs ND. 0.5 0.8—1.6 LINs 0.9—3.1 1.5—3.9 1.4—5.4 油菜Rape TCs ND. — 61.1—192.1 SAs 0.1—0.5 — 0.5—1.4 CAPs ND. — 0.7 FQs ND. — 0.7—2.1 LINs 0.5—3.5 — 0.7—3.2 芹菜Celery TCs — 1.0—2.4 15.7 SAs — 0.1—0.3 0.3—0.7 FQs — 0.5—1.9 2.8—4.7 LINs — 1.2—5.1 5—20 香菜Coriander TCs 128.2—690.1 — 133.9—867.6 SAs 0.1—0.5 — 0.5—1.3 FQs ND. — 0.7—3.5 LINs 0.4—2.4 — 0.8—3.8 小白菜Chinese white cabbage TCs ND. — 5.5 QNs 5.9 — 9.6 CAPs ND. — 2.6 SAs 0.8 — ND. 空心菜Water spinach TCs 4.8 — 6.3 QNs 16.9 — 21.8 CAPs 3 — 10.1 SAs 1.7 — ND. —,未检测;ND.,未检测出;TCs,四环素类;SAs,磺胺类;CAPs,氯霉素类;QNs,喹诺酮类;LINs,林可酰胺类;FQs,氟喹诺酮类. —, non-detection; ND., not detected; TCs, tetracyclines; SAs, sulfonamides; CAPs, chloramphenicols; QNs, quinolones; Lins, lincolamide; FQs, fluoroquinolones. | Show TableDownLoad:
CSV
抗生素的理化性质以及蔬菜种类、生长阶段和蒸腾速率是导致其在蔬菜中含量不同的影响因素. 目前已有学者开展了一些蔬菜可食用部分的人体暴露影响的研究,但是仍然缺乏蔬菜摄食途径下抗生素的人体健康风险的全面评估. 因此,需要进一步研究来评估这些抗生素在食物链中的积累,以确定人类使用的安全浓度.
3. 菜地系统中抗生素的生态毒性效应(Ecotoxicity of antibiotics in vegetable field system)
抗生素具有易被生物积累的特性. 大量研究证实,长期的农业生产不仅会促使抗生素在植物间迁移[57, 64 − 65],还会诱导微生物产生抗生素抗性,严重破坏了土壤微生态系统的稳定性[66 − 67],同时,长期暴露在抗生素污染环境中的动物也会表现出诸多不良反应.
3.1 抗生素诱发植物毒性
抗生素对植物生长的影响具有两面性,一方面,抗生素可以帮助植物抵抗病害、提高植物体内Ca、Mg、K、N等营养元素的含量[68 − 69],另一方面,某些条件下,抗生素的存在反而会诱发植物毒性,并持续作用于植物生长发育的不同阶段. 抗生素对蔬菜生长的影响表现为低浓度促进高浓度抑制[68]. 金彩霞等[70]研究发现,低浓度磺胺嘧啶钠(1 mg·kg−1)对大白菜的芽伸长有一定促进作用. 在0、50、100、5000 μg·kg−1恩诺沙星处理的土壤中,黄瓜、菜豆、萝卜和莴苣生长发生变化,低浓度(0—50 μg·kg−1)条件下,根长、下胚轴长、子叶和叶片长度及数量均受到刺激作用,随着恩诺沙星浓度提高,抑制作用逐渐明显[53]. 在种子萌发时期,抗生素的毒性作用影响不大[71],这可能是抗生素难以穿过种皮,因而对胚根生长的抑制作用不大[72]. 从这点来看,为缓解抗生素对种子的毒性作用,未来可以利用现代生物技术培育适宜质地的种皮,以达到减少抗生素渗透的目的[73]. 抗生素对植物的潜在毒性作用常通过干扰蚯蚓和微生物介导的土壤速效养分间接抑制植物生产力[74]. 在抗生素的直接或间接作用下,农业系统可持续性将受到严重威胁.
与其他污染物类似的是,抗生素干预了蔬菜光合作用等许多重要的生理过程. 如四环素类、氟喹诺酮类和大环内酯类等抗生素会影响蔬菜的叶绿体和线粒体蛋白质合成[75 − 76],环丙沙星和头孢菌素会降低气孔导度[77]. 这意味着与光合作用有关的叶绿素和类胡萝卜素的减少和叶片气孔导度的降低,会导致蔬菜光合作用速率降低,从而影响蔬菜的生长[78]. 从根本来看,抗生素的毒性作用机理是破坏遗传物质或限制酶作用途径. 喹诺酮类抗生素能够抑制核酸合成或代谢过程,主要通过改变DNA拓扑异构酶Ⅱ活性、抑制酶的切割和链接,进而对真核DNA复制造成不同程度的影响并抑制了叶绿素的转录,最终影响植物生长[53, 75]. 磺胺类抗生素作用机制是改变能量代谢过程,其具有类似氨基苯甲酸结构,能竞争性作用于叶酸合成途径中的相关酶,阻止叶酸合成,最终干扰根系伸长、木质素和光呼吸作用[79 − 80]. 四环素类和大环内酯类抗生素对植物生长的抑制作用表现在能够显著抑制蛋白质生物合成或诱导染色体变异,而β-内酰胺会影响低等植物的质体分裂[75]. 目前很多有关植物毒性的研究都在实验室环境中进行,其设计的抗生素浓度并不一定会在土壤中出现,因此在实际土壤环境中这些抗生素是否仍会表现出毒性作用有待商榷. 如红扁豆在含较低浓度抗生素营养液中根伸长减缓、植物干重降低,而在相应浓度土壤基质的红扁豆则没有出现植物毒性效应[69].
3.2 抗生素对土壤动物新陈代谢和身体机能的影响
抗生素对土壤动物的毒性作用的相关研究并不多,且各方观点不一. Baguer等[81]发现蚯蚓、线虫和弹尾虫在3000—5000 mg·kg−1土霉素和泰乐菌素胁迫下,其生长并未受到严重影响,说明这两种抗生素不太可能对土壤动物构成直接风险. 然而,一些研究认为,抗生素对动物的毒性具有明显的剂量-效应关系. 如在1.0—2.0 mg·kg−1浓度抗生素胁迫下,蚯蚓的生长和呼吸作用受抑制、繁殖率下降、回避反应增强,且随浓度提高,蚯蚓的反应越明显[82]. 除了蚯蚓,抗生素也在破坏着跳虫和白符䖴的健康机能[83 − 84]. 暴露于10 mg·kg−1诺氟沙星和土霉素中,跳虫出现明显的体重下降[83]. 与对照组相比,当白符䖴暴露在浓度为1000 mg·kg−1的诺氟沙星环境下,其繁殖数和成虫体长分别减少34.4%和9%[84].
抗生素对土壤动物的毒性作用首先表现在对DNA的破坏性. 抗生素代谢产物的自由基和碱基位点等会直接导致土壤动物细胞DNA断裂,造成细胞DNA损伤[85]. DNA损伤程度与抗生素剂量呈显著正相关关系,即使是最低暴露剂量(0.3 mg·kg−1)仍然会造成严重DNA损伤[85]. 其次,抗生素还会引起土壤动物酶活性的变化. 在抗生素的暴露下,动物会出现氧化应激反应,导致其脂质过氧化,最终促进脂质过氧化产物丙二醛的形成并诱导过氧化氢酶、过氧化物酶和超氧化物歧化酶的表达[84]. 如Eisenia foetida蚯蚓在环丙沙星(0—51.2 mg·kg−1)污染土壤中发生了蛋白质羟基化造成的氧化损伤[86]. 为了抵消抗生素产生的负面影响,动物肠道菌群物种组成发生改变,身体机能迅速反应形成一道强硬的防御系统. 动物体内产生的抗氧化酶、抗氧化剂和蛋白水解系统等可以极大缓解细胞的氧化损伤,在一定程度上保护了机体[87]. 此外,动物肠道会将已经发生改变的肠道菌群输送到土壤中,进而引起周边土壤菌群发生改变,形成微生物菌群对抗生素的共代谢模式,最终促成对土壤抗生素的削减. 但是目前关于肠道内生菌对抗生素的降解机制依旧未知.
3.3 抗生素影响土壤微生物群落结构和生态功能
土壤微生物是土壤生态系统的关键组成成分,它们在促进土壤有机物分解、提高土壤肥力和增进作物产量的过程中扮演者重要角色[88]. 而外源输入的抗生素会对土壤生态系统产生一系列的影响[89 − 90],造成土壤微生物群落特征的改变[91],干扰微生物对碳源利用[92],且不利于微生物硝化和反硝化等过程[93].
3.3.1 改变土壤微生物群落特征
多数抗生素为抑菌药物,将其添加到土壤中会显著降低土壤细菌数量、改变群落结构并促使群落演替. Hammesfahr等[94]发现,4 d后暴露在磺胺嘧啶中的细菌/真菌比值从70%减少到57%. 磺胺嘧啶还对土壤氨氧化微生物群落分布和活性有显著影响[95 − 96]. 在磺胺嘧啶处理的土壤中,氨氧化细菌丰度会显著下降,而氨氧化古细菌基本稳定[97],Radl等[98]也得到类似的结论. 但另一项研究得到了不同的结论,在施加粪肥土壤中,具有生物有效性的磺胺嘧啶对全程硝化菌活性的抑制作用最强,其次是氨氧化古细菌,对氨氧化细菌的影响可以忽略不记[99]. 部分抗生素还会对革兰氏阳性菌和革兰氏阴性菌起到选择作用. 如当添加高浓度四环素8 d后,革兰氏阳性菌和革兰氏阴性菌的比例会下降,这可能是环境中耐四环素细菌多为革兰氏阴性菌,而磺胺类抗生素则不受此影响[94,100]. 在添加抗生素的处理中,相较于有益菌群,潜在致病菌群的丰度更高[101],即使是将生物质堆肥处理后仍可能增加土壤中病原菌丰度[102]. 但抗生素对真菌的影响仍然不清楚.
3.3.2 影响碳源利用效率和酶活性
进一步研究表明,抗生素会限制微生物对碳源的正常利用,进而干扰其生长繁殖过程. 有研究检测了施加100 mg·kg−1磺胺甲恶唑水稻土经21 d培育后水稻土中微生物对碳源的利用情况,结果显示,微生物对碳源的利用受到抑制且其Shannon指数降低[103]. 但抗生素也可以作为微生物可利用的碳源,改变其呼吸强度. 有研究显示,最初添加磺胺嘧啶和金霉素处理组的土壤呼吸活性均受到抑制,随抗生素添加频率的增加,土壤呼吸活性逐渐提高[24]. 抗生素对土壤微生物的呼吸作用的影响较复杂,如低浓度的抗生素有促进作用,环境浓度抗生素却无显著作用[104 − 105]. 这些影响一般比较短暂,可能是由于抗生素生物可利用性会随时间推移而降低,最终导致了抗生素对微生物呼吸作用受限[106]. 同样,抗生素的种类和暴露时间也影响了呼吸作用[107]. 在抗生素的胁迫下,土壤微生物的活性出现明显的差异,其活性可能增强也可能受抑制[108 − 109]. 如较低浓度四环素对土壤脱氢酶和磷酸酶活性有明显的抑制效果[110],而微生物在较高浓度土霉素中表现出较低的生物活性[111]. 一个可能的原因是,土壤中的酶一般是由真菌分泌产生,而某些抗生素能够促进真菌繁殖造成土壤酶活性的增加[101]. 反之,酶活性受抑制可能是土壤微生物不能够抵抗抗生素的选择压力,出现了生长停缓或死亡等情况.
3.3.3 干扰硝化反硝化过程
此外,抗生素对微生物的影响还表现在干扰土壤的硝化、反硝化和产甲烷化等过程,最终阻碍了土壤养分循环. 抗生素能够刺激土壤微观世界中氮循环,主要体现在对土壤微生物的硝化和反硝化作用所造成的影响[112]. Ma等[113]观察到高浓度的土霉素(30 mg·kg−1)和磺胺嘧啶(100 mg·kg−1)抑制了土壤微生物的硝化作用. 然而,部分研究发现仅最低浓度的环丙沙星和诺氟沙星(1 mg·kg−1)会刺激土壤微生物的硝化作用[114 − 115]. 低浓度的磺胺甲恶唑、磺胺嘧啶、庆大霉素、甲基盐霉素(500 μg·kg−1)还会抑制反硝化过程,但是更低浓度(<1 μg·kg−1)会促进该过程[116]. 抗生素还被证实可以干扰土壤中铁的转化率和产甲烷化过程. 如超过10 mg·kg−1磺胺甲恶唑和土霉素暴露会强烈抑制三价铁的还原[117],而500 μg·kg−1磺胺甲恶唑可以显著刺激土壤微生物产甲烷化过程[118]. 以上事实证明了抗生素在一定程度上不利于地球化学循环.
4. 结果与展望(Results and perspective)
综上所述,我国菜地系统存在不同程度的抗生素污染,且与输入水平、土壤性质、抗生素种类以及气候条件等息息相关. 抗生素从畜牧业向农业的流动过程不仅对土壤生物产生直接毒性作用,还能干扰土壤速效养分造成植物减产,提高致病菌丰度,并对动、植物生长构成严重威胁. 为维护农业的可持续性,相关领域目前已经开展一系列研究,但仍存在一定局限性,未来研究可以关注以下方向:
(1)抗生素在全国尺度上的长期追踪调查目前还未实现,这不利于全面评估我国土壤抗生素污染现状. 因此,未来可以进一步增加对不同区域的调查研究,尤其是人口密度和经济发展水平较低的西北地区,以期为制定相关政策法规提供科学依据.
(2)学者们已认识到抗生素的分子结构是决定抗生素作用机制的关键因素,抗生素的转化和降解速率很大程度上取决于抗生素的结构. 因此,未来研究可以关注如何利用分子手段等高新技术改造相应的官能团,以保障抗生素本身功效的同时安全高效的实现抗生素降解为研究目标.
(3)为全方面控制农田系统中抗生素污染现状,准确定量土壤中抗生素及其降解产物是极为必要的. 对于复杂环境基质中痕量分析,建议尝试更先进的样品前处理方法以及更高性能的数据采集和分析平台. 吸附、转化和降解是土壤中抗生素主要的环境行为,除抗生素自身性质外,这些过程还受到很多环境因素影响. 因此,未来研究可侧重于土壤性质、水热条件等对抗生素的作用机制. 对于参与土壤中抗生素降解的微生物,其降解能力的遗传效率同样值得研究. 掌握调控微生物降解抗生素的关键功能基因技术,尝试将该基因技术运用到培育具备高降解特性的功能性降解菌,这对未来土壤抗生素污染的修复具有重要意义.
(4)目前对于评估抗生素生态风险的方法并不统一,用于毒性试验的物种仍然局限,抗生素对生态环境造成的风险可能被低估了. SSD和风险熵值法是常见的环境风险评估方法,但易受太多不同因素影响而不可靠. 因此,应进一步扩充多营养级多物种的毒性数据,构建更科学的抗生素生态风险评估体系.
-

图 2 (A)扫描电镜下色谱填料的形貌(×3000),(B)色谱填料粒径分布图
Figure 2. (A)FESEM photograph of packing material(×3000), (B) size distribution of the packing material

图 3 色谱填料经过高压填充过程后的扫描电镜图(×3000)
Figure 3. FESEM photograph of chromatographic packing materials after high pressure packing(×3000)
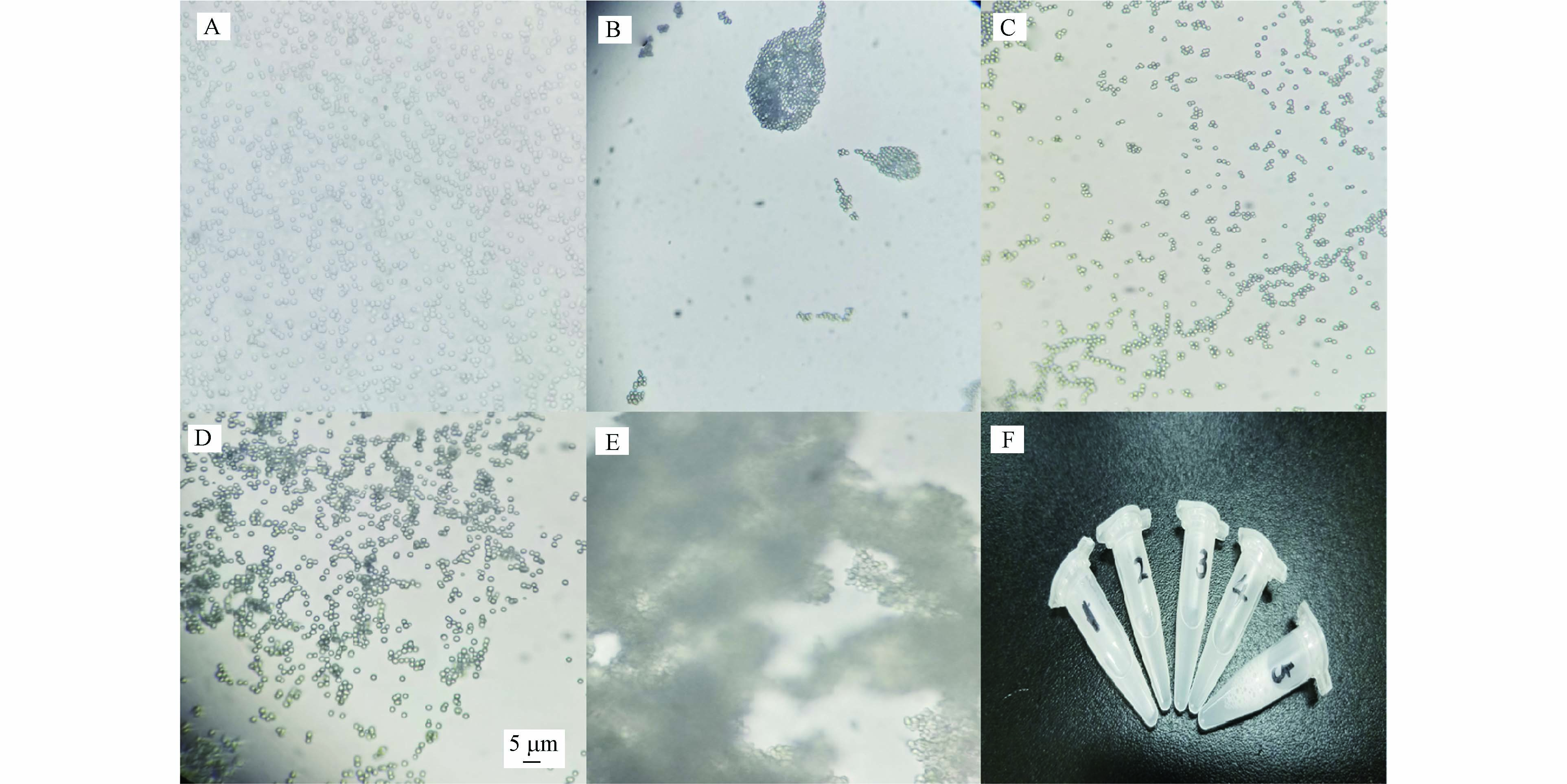
图 4 色谱填料在5种溶剂中的分散情况(×200)
Figure 4. Photograph of the dispersion of five solvents for the chromatographic packing materials(×200)
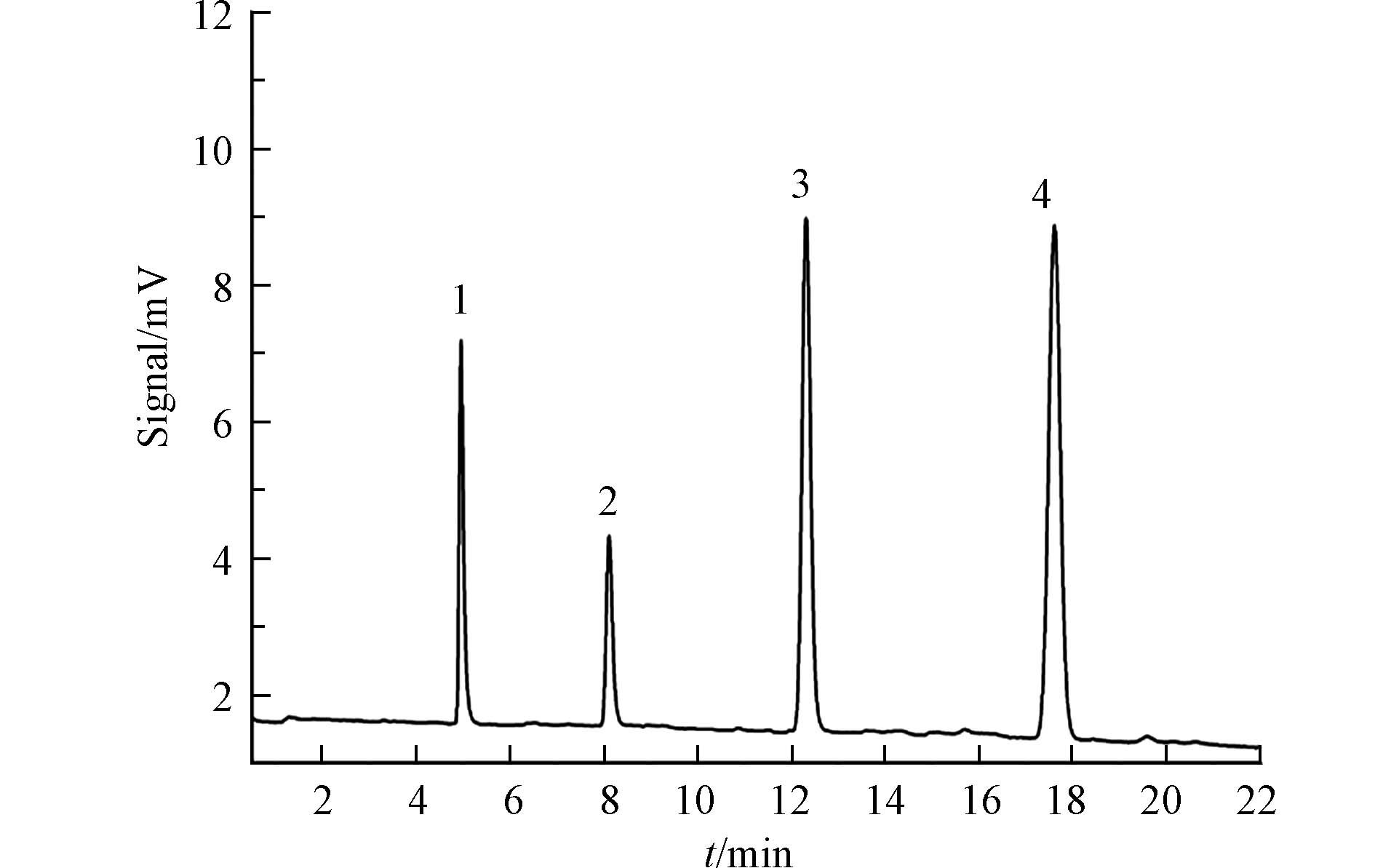
图 5 25 μm i.d.毛细管柱在V=2.4下的色谱图
Figure 5. Chromatogram of a capillary column of 25 μm i.d. at V=2.4

图 6 Van Deemter 曲线(芘;25 μm i.d.)
Figure 6. The Van Deemter curve of 25 μm i.d. capillary column (pyrene)
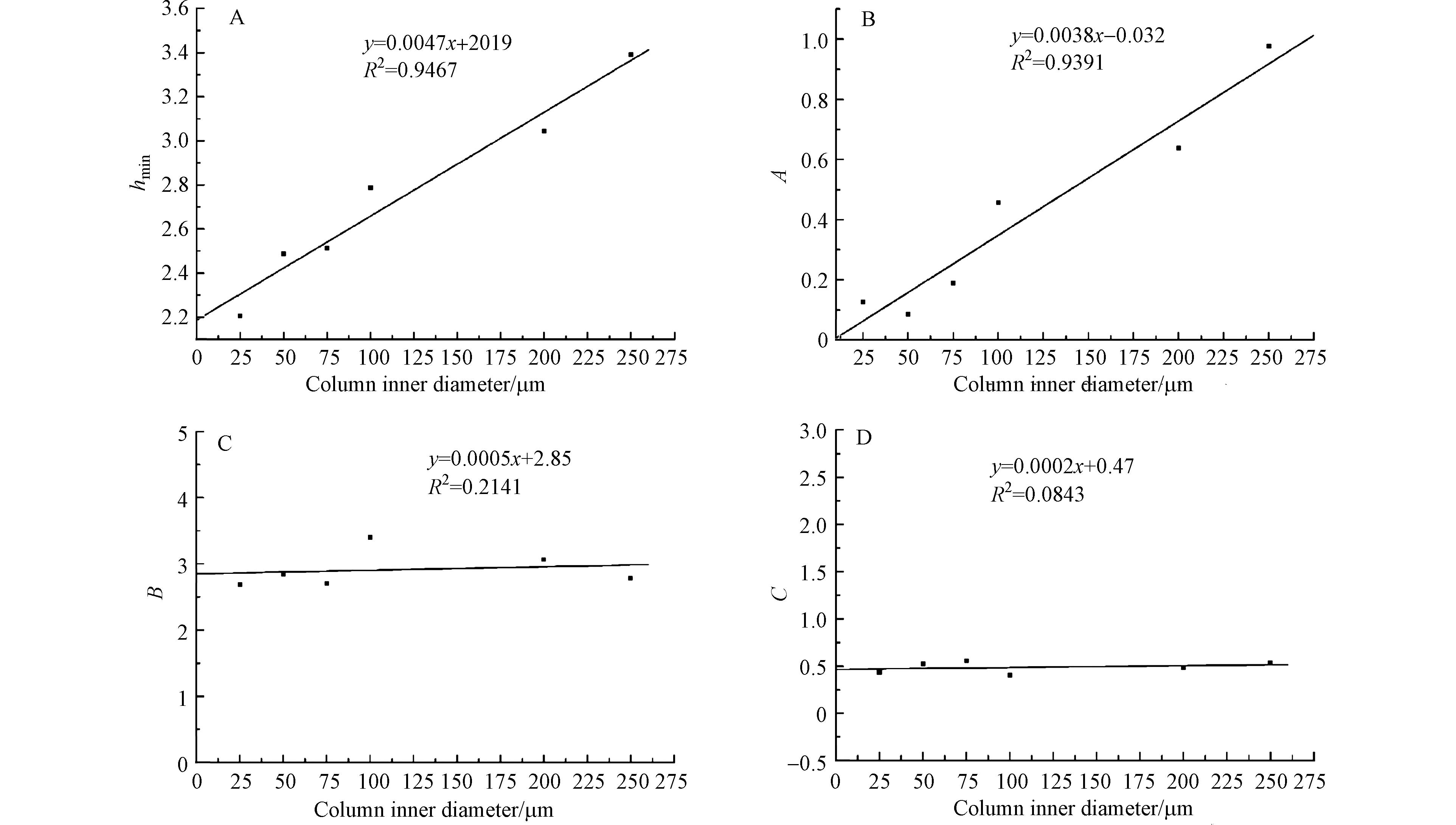
图 7 (A)色谱柱最小折合塔板高度及(B)A项、(C)B项和(D)C项系数随柱内径的变化
Figure 7. (A) The values of the h min, (B) A, (C) B and (D) C coefficient vary with the inner diameter of the columns.
表 1 不同浓度下色谱填料在溶剂中的沉降时间
Table 1. Sedimentation time of chromatography packing materials in solvents at different concentrations
匀浆溶剂 Slurry solvents 沉降时间/ min Sedimentation time 甲醇 (30 mg·mL−1) 2.5 丙酮 (30 mg·mL−1) 12 己烷 (30 mg·mL−1) 14 丙酮/甲醇(9:1,V/V)(30 mg·mL−1) 8 水/Triton X-100(99:1,V/V)(30 mg·mL−1) 67 甲醇 (10 mg·mL−1) 3 丙酮 (10 mg·mL−1) 14 己烷 (10 mg·mL−1) 16 丙酮/甲醇(9:1,V/V)(10 mg·mL−1) 10 水/Triton X-100(99:1,V/V)(10 mg·mL−1) 70 甲醇 (5 mg·mL−1) 5 丙酮 (5 mg·mL−1) 16 己烷 (5 mg·mL−1) 18 丙酮/甲醇(9:1,V/V)(5 mg·mL−1) 12 水/Triton X-100(99:1,V/V)(5 mg·mL−1) 75
下载: 导出CSV
表 2 不同匀浆溶剂填充下毛细管柱的柱效及柱压比较(50 μm i.d.)
Table 2. Comparison of the column efficiency of capillary column packed by different slurry solvents (50 μm i.d.)
匀浆溶剂 Solvents 理论塔板数/mN 折合塔板高度 Reduced h min 压力* / MPa Pressure 水/Triton X-100(99:1,V/V) 98340 2.76 2.6 丙酮/甲醇(9:1,V/V) 131970 2.06 4.8 己烷 114380 2.38 4.7 *压力为最佳流速下的柱压(MPa)(The column pressure is detected at the optimal flow rate)
下载: 导出CSV
表 3 不同内径毛细管柱的柱效比较
Table 3. Comparison of column efficiency of different inner diameter capillary columns
柱内径/μm Column inner diameter 匀浆浓度/(mg·mL−1) Slurry concentration 柱效N/ m−1 折合塔板高度 Reduce h min 25 5 148200 1.83 50 10 134000 2.03 75 15 132700 2.05 100 20 119600 2.27 200 25 109500 2.48 250 30 98300 2.76
下载: 导出CSV
-
[1] JORGENSON J W. Capillary liquid chromatography at ultrahigh pressures [J]. Annual Review of Analytical Chemistry (Palo Alto, Calif. ), 2010, 3: 129-150. doi: 10.1146/annurev.anchem.1.031207.113014 [2] MACNAIR J E, LEWIS K C, JORGENSON J W. Ultrahigh-pressure reversed-phase liquid chromatography in packed capillary columns [J]. Analytical Chemistry, 1997, 69(6): 983-989. doi: 10.1021/ac961094r [3] NOVOTNY M V. Development of capillary liquid chromatography: A personal perspective [J]. Journal of Chromatography A, 2017, 1523: 3-16. doi: 10.1016/j.chroma.2017.06.042 [4] WILSON S R, VEHUS T, BERG H S, et al. Nano-LC in proteomics: Recent advances and approaches [J]. Bioanalysis, 2015, 7(14): 1799-1815. doi: 10.4155/bio.15.92 [5] BORRA C, HAN S M, NOVOTNY M. Quantitative analytical aspects of reversed-phase liquid chromatography with slurry-packed capillary columns [J]. Journal of Chromatography A, 1987, 385: 75-85. doi: 10.1016/S0021-9673(01)94623-0 [6] GUAN Y F, ZHOU L M, SHANG Z H. Dry-packed capillary columns for micro HPLC [J]. Journal of High Resolution Chromatography, 1992, 15(7): 434-436. doi: 10.1002/jhrc.1240150706 [7] JORGENSON J W, LUKACS K D. High-resolution separations based on electrophoresis and electroosmosis [J]. Journal of Chromatography A, 1981, 218: 209-216. doi: 10.1016/S0021-9673(00)82057-9 [8] ISHII D, ASAI K, HIBI K, et al. A study of micro-high-performance liquid chromatography [J]. Journal of Chromatography A, 1977, 144(2): 157-168. doi: 10.1016/S0021-9673(00)99351-8 [9] GLUCKMAN J C, HIROSE A, MCGUFFIN V L, et al. Performance evaluation of slurry-packed capillary columns for liquid chromatography [J]. Chromatographia, 1983, 17(6): 303-309. doi: 10.1007/BF02270662 [10] GODINHO J M, REISING A E, TALLAREK U, et al. Implementation of high slurry concentration and sonication to pack high-efficiency, meter-long capillary ultrahigh pressure liquid chromatography columns [J]. Journal of Chromatography A, 2016, 1462: 165-169. doi: 10.1016/j.chroma.2016.08.002 [11] MELLORS J S, JORGENSON J W. Use of 1.5-microm porous ethyl-bridged hybrid particles as a stationary-phase support for reversed-phase ultrahigh-pressure liquid chromatography [J]. Analytical Chemistry, 2004, 76(18): 5441-5450. doi: 10.1021/ac049643d [12] REISING A E, GODINHO J M, JORGENSON J W, et al. Bed morphological features associated with an optimal slurry concentration for reproducible preparation of efficient capillary ultrahigh pressure liquid chromatography columns [J]. Journal of Chromatography A, 2017, 1504: 71-82. doi: 10.1016/j.chroma.2017.05.007 [13] BLUE L E, JORGENSON J W. 1.1 μm Superficially porous particles for liquid chromatography [J]. Journal of Chromatography A, 2015, 1380: 71-80. doi: 10.1016/j.chroma.2014.12.055 [14] REISING A E, GODINHO J M, BERNZEN J, et al. Axial heterogeneities in capillary ultrahigh pressure liquid chromatography columns: Chromatographic and bed morphological characterization [J]. Journal of Chromatography A, 2018, 1569: 44-52. doi: 10.1016/j.chroma.2018.07.037 [15] TREADWAY J W, WYNDHAM K D, JORGENSON J W. Highly efficient capillary columns packed with superficially porous particles via sequential column packing [J]. Journal of Chromatography A, 2015, 1422: 345-349. doi: 10.1016/j.chroma.2015.10.013 [16] QI Y X, WU D P, WEI J Y, et al. Selective extraction of low molecular weight proteins by mesoporous silica particles with modified internal and external surfaces [J]. Analytical and Bioanalytical Chemistry, 2010, 398(4): 1715-1722. doi: 10.1007/s00216-010-4081-1 [17] 唐意红, 朱道乾, 关亚风. 不锈钢宽口径填充毛细管液相色谱柱的制备及评价 [J]. 分析化学, 2001, 29(10): 1228-1232. doi: 10.3321/j.issn:0253-3820.2001.10.030 TANG Y H, ZHU D Q, GUAN Y F. Preparation and evaluation of stainless-steel wide-bore packed capillary high performance liquid chromatographic columns [J]. Chinese Journal of Analytieal Chemistry, 2001, 29(10): 1228-1232(in Chinese). doi: 10.3321/j.issn:0253-3820.2001.10.030
[18] DENG N, HE Y Z, WANG L, et al. Reversed-phase electrochromatography with a monolithic microcolumn prepared in a 2.2-mm-inner diameter fused-silica tube [J]. Analytical Chemistry, 2005, 77(17): 5622-5627. doi: 10.1021/ac050589q [19] BRUNS S, FRANKLIN E G, GRINIAS J P, et al. Slurry concentration effects on the bed morphology and separation efficiency of capillaries packed with sub-2 μm particles [J]. Journal of Chromatography A, 2013, 1318: 189-197. doi: 10.1016/j.chroma.2013.10.017 [20] KARLSSON K E, NOVOTNY M. Separation efficiency of slurry-packed liquid chromatography microcolumns with very small inner diameters [J]. Analytical Chemistry, 1988, 60(17): 1662-1665. doi: 10.1021/ac00168a006 [21] VISSERS J P C. Recent developments in microcolumn liquid chromatography [J]. Journal of Chromatography A, 1999, 856(1/2): 117-143. [22] HSIEH S, JORGENSON J W. Preparation and evaluation of slurry-packed liquid chromatography microcolumns with inner diameters from 12 to 33 microns [J]. Analytical Chemistry, 1996, 68(7): 1212-1217. doi: 10.1021/ac950682m [23] KNOX J H, PARCHER J F. Effect of the column to particle diameter ratio on the dispersion of unsorbed solutes in chromatography [J]. Analytical Chemistry, 1969, 41(12): 1599-1606. doi: 10.1021/ac60281a009 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2258
- HTML全文浏览数: 2258
- PDF下载数: 90
- 施引文献: 0
