







-
氯苯(chlorobenzene,CB)是最简单的氯芳烃,自19世纪合成以来,即大量用于生产DDT,至今,氯苯依然是年产量超过100万磅的高产量化学品[1]. 环境中的氯苯大多来源于人类的工业活动,据报道,美国氯苯类化合物的环境排放量可达到每年980吨[2]. 氯苯在自然界中的降解速度较慢,具有很强的生物积累性和生物毒性,有研究显示氯苯除了对中枢神经系统和呼吸系统有影响之外,还可造成肾脏和肝脏的损伤[3].
目前已经有很多研究者关注到氯苯的无害化处理问题,传统的氯苯处理方法主要包括吸附法、生物降解法和化学氧化法. 这些方法大多具有二次污染、效率低、选择性差等特点. 基于单过硫酸盐化合物(PMS)的高级氧化技术因其高氧化效率在降解氯代有机污染物的过程中表现出了优异的性能. 许多研究结果表明,钴氧化物(CoO、CoO2、Co2O3、CoO(OH)、Co3O4)具有活化PMS的良好能力,但单钴氧化物的比表面积非常低,以团聚,导致活性位有限,显著抑制其催化活性[4]. 有研究表明通过将钴氧化物分散在多孔材料的孔道中,可以将活性金属限域在特定孔结构中,从而使活性金属实现高度分散,这种方法可以极大提高钴基材料的催化活性[5]. SBA-15具有较高的比表面积、稳定的结构和有序的孔径,是一种良好的催化剂载体. 由于金属盐与模板剂之间的强相互作用,通过固相研磨法将金属盐与未去除模板的SBA-15充分混合之后,经过焙烧可以得到高金属分散度的催化剂. 因此,在本研究中,采用固相研磨法合成催化剂CoOx@SBA-15,并对其活化PMS降解氯苯的性能进行测试,并进一步探究反应中的各种因素对反应活性的影响机制及反应体系的主要活性物种.
-
试剂:P123(Sigma-Aldrich,99%),正硅酸四乙酯(国药,AR),六水合硝酸钴(阿拉丁,99%),氯苯(麦克林,AR),单过硫酸盐化合物(Sigma-Aldrich,99.9%),2,2,6,6-四甲基哌啶(Sigma-Aldrich,AR),5,5-二甲基-1-吡咯啉-N-氧化物(百灵威,AR),甲醇(TEDIA,HPLC),盐酸(国药,AR),实验中所用水均为去离子水.
-
以SBA-15为载体利用固相研磨法制备限域型CoOx@SBA-15[6]:按文献报道方法,以正硅酸四乙酯(TEOS)为硅源,三嵌段共聚化合物P123为模板剂合成介孔氧化硅SBA-15[7];在室温条件下将一定量的Co(NO3)2·6H2O与1 g未去除模板剂的SBA-15在研钵中混合并研磨1 h得到CoOx@SBA-15;将所得的混合物置于马弗炉中,以2 °C·min−1 的升温速率升温至500 °C,并保持5 h,焙烧所得产物标记为CoOx(X)@SBA-15,其中X是钴的负载量(以质量分数计).
采用传统浸渍法制备CoOx/SBA-15和CoOx/SiO2催化剂. 首先将SBA-15置于马弗炉中焙烧,以1 °C·min−1的升温速率升温到550 °C,并保持6 h,目的是碳化并去除SBA-15中的模板剂;将购得的SiO2置于马弗炉中焙烧,以2 °C·min−1的升温速率升温到300 °C,并保持4 h,目的是去除其中可能存在的杂质;随后将一定量的Co(NO3)2溶液与1 g载体在室温条件下混合搅拌2 h以上,并在90 °C水浴中蒸干,在100 °C烘箱中干燥过夜,干燥后所得材料标记为CoOx(X)/Y,其中X是负载量(以质量分数计),Y是载体.
催化剂透射电镜分析(TEM)在日本JEOL公司,JEM-200CX型透射电子显微镜上检测;X射线衍射分析(XRD):采用日本Rigaku公司D/max-rA型X射线衍射仪,Cu 靶(Kα1,λ=0.154056 nm,扫描速度6(°)· min−1),操作条件:40 kV、30 mA,扫描范围:10°—80°;催化剂中Co含量采用原子吸收光谱(AAS,美国Thermo公司)测定;催化剂比表面积、孔径孔容采用比表面积测定仪(ASAP 2020,美国Micromeritics公司)分析;催化剂的在不同温度的还原状态采用泛泰公司生产的Finesorb-3010程序升温化学吸附仪进行测定.
-
氯苯的降解实验在250 mL三口烧瓶中进行,温度保持在(25±0.5)℃,具体操作方法如下:储备液用去离子水稀释至反应所需浓度,三口烧瓶中溶液总体积为200 mL,随后将一定量的催化剂(5—40 mg)分散在溶液中,搅拌1 h以达到吸附平衡并保证催化剂充分分散. 加入一定量的PMS储备液开始反应,在反应开始后的固定时间(1、3、5、10、20、30、50、70、90、120 min)取出反应溶液,并通过0.22 μm PTFE过滤器(Anpel)进行过滤. 将过滤后的1 mL反应溶液转移到装有0.5 mL甲醇的2 mL棕色液相小瓶中以清除残留的自由基.
使用配备有C-18色谱柱(ZORBAX Eclipse XDB-C18)的高效液相色谱仪(HPLC,1220 Infinity II)检测滤液中的氯苯. 仪器条件:流动相包括纯水和甲醇(30/70,V/V),流速为0.8 mL·min−1. 紫外检测波长为223 nm,柱温30 ℃.
-
催化剂活化PMS产生的自由基采用德国Bruker BioSpin有限公司生产的顺磁共振波谱仪(Electron Paramagnetic Resonance Spectrometer,EMX PLUS(PPMS))检测. 具体方法如下:称取适量催化剂分散在去离子水中,涡旋振荡30 s确保催化剂充分分散,取一定量PMS溶液加入到催化剂的溶液中,并使其充分混匀;在2.5 mL尖头离心管中加入1 mL所得混合溶液和100 µL 1 mol·L−1的自由基捕获剂(5,5-二甲基吡咯啉氧化物(DMPO)或2,2,6,6-四甲基-4-哌啶(TEMP))储备液,自由基捕获剂需溶解在pH = 7.4的磷酸缓冲液中,涡旋振荡30 s;将混合后的溶液转移至EPR样品管中,进行EPR分析. EPR操作参数:中心场为348.0 mT,扫描宽度为20 mT,微波频率为9.77 GHz,调制频率为100 GHz,能量为20 mW.
-
图1(a)是焙烧去除模板后的SBA-15、固相研磨法合成的催化剂CoOx@SBA-15以及浸渍法合成的催化剂CoOx/SBA-15的小角XRD图谱,小角XRD图谱可以用于分析材料的孔结构的有序度. 从图1(a)中可以看出,去除模板后的SBA-15在2θ为0.75°到2°范围内有3个明显的特征衍射峰,分别位于0.88°、1.52°和1.76°,对应于于SBA-15的(100)、(110)和(200)衍射面,该结果表明合成的SBA-15具有高度的二维六方介孔结构和p6mm对称性[7]. 此外,催化剂CoOx@SBA-15和CoOx/SBA-15的图谱中也呈现着3个明显的特征衍射峰,这表明在500 ℃焙烧后载体SBA-15的介孔结构并没有被破坏.
催化剂的广角XRD图谱如图1(b)所示. 从图1可以看出,浸渍法合成的CoOx/SiO2催化剂在2θ 为36.56°、55.3°、59.96°处均有明显的特征衍射峰,此处衍射峰可归属于尖晶石Co3O4的(311)、(422)、(511)晶面,这表明焙烧过程中Co(NO3)2在载体SiO2形成了较大的Co3O4微晶,这是因为金属和载体之间的相互作用较弱[8]. 此外在该材料的XRD图谱上还可以观察到CoO(2θ = 42.9°)和金属Co(2θ = 44.2°)的特征衍射峰,这是因为在焙烧过程中产生的微量C、N会还原部分Co3O4. 从CoOx@SBA-15和CoOx/SBA-15的广角XRD图谱中可以看出,2种材料与载体SBA-15一样均在2θ = 22°附近有一处较宽的衍射峰,可归属于无定型SiO2的特征峰[9]. 其中催化剂CoOx/SBA-15可以观察到一处微弱的CoO(2θ = 42.9°)特征衍射峰,表明位于SBA-15上的Co(NO3)2焙烧时部分形成了CoO分散在SBA-15表面或孔道中,而在CoOx@SBA-15中未观察到明显的衍射峰,这表明金属在CoOx@SBA-15中高度分散.
去模板后的载体SBA-15以及催化剂CoOx@SBA-15和CoOx/SBA-15的N2吸附-脱附等温线和孔径分布如图2所示,各材料的结构参数汇总在表1. 从图2(a)中可以看出,SBA-15的N2吸附-脱附等温线在P/P0 0.64—0.85之间出现了明显的H1型回滞环,且曲线为典型的IV型等温线,验证了实验中合成的SBA-15具有有序的介孔结构[10]. CoOx@SBA-15和CoOx/SBA-15的等温线形状与SBA-15类似,分别在P/P0 0.50—0.85和0.49—0.82之间存在H1型回滞环,这说明经过焙烧后的催化剂依旧具有和SBA-15一样的有序介孔结构. 但材料的氮气吸附量呈现出CoOx@SBA-15 < CoOx/SBA-15 < SBA-15的趋势,这是由于固相研磨法合成的催化剂中金属会在焙烧过程中更多地被限域在SBA-15的孔道中,从而影响材料的吸附量. 除此之外,如图2(b)所示,催化剂CoOx@SBA-15和CoOx/SBA-15和载体SBA-15的孔径均集中分布在4—10 nm之间,最可几孔径分别为6.38 nm、6.33 nm和6.49 nm,这表明金属的负载并不会影响SBA-15的中孔结构. 除此之外,从表1中可以看出,CoOx@SBA-15和CoOx/SBA-15的孔容分别为0.68 cm3·g-1和0.82 cm3·g−1,相较于SBA-15的孔容1.10 cm3·g−1有很大降低,孔径和孔容结果也进一步验证了催化剂CoOx@SBA-15上SBA-15对钴氧化物的限域作用.
图3为催化剂CoOx@SBA-15、CoOx/SBA-15和CoOx/SiO2的TEM图. 从图3(a)、(b)可以观察到清晰的孔道结构,这与SBA-15的典型孔道结构相一致[7]. 此外CoOx/SBA-15和CoOx/SiO2的TEM图中可以观察到明显的金属颗粒,这是因为传统浸渍法合成的材料易在载体表面形成团聚而呈现出较大的金属颗粒. 对比图3(a)可以发现,利用固相研磨法合成的材料CoOx@SBA-15中金属颗粒借由焙烧过程中和模板剂P123的相互作用高度分散在SBA-15的孔道之中,而未见明显的金属颗粒.
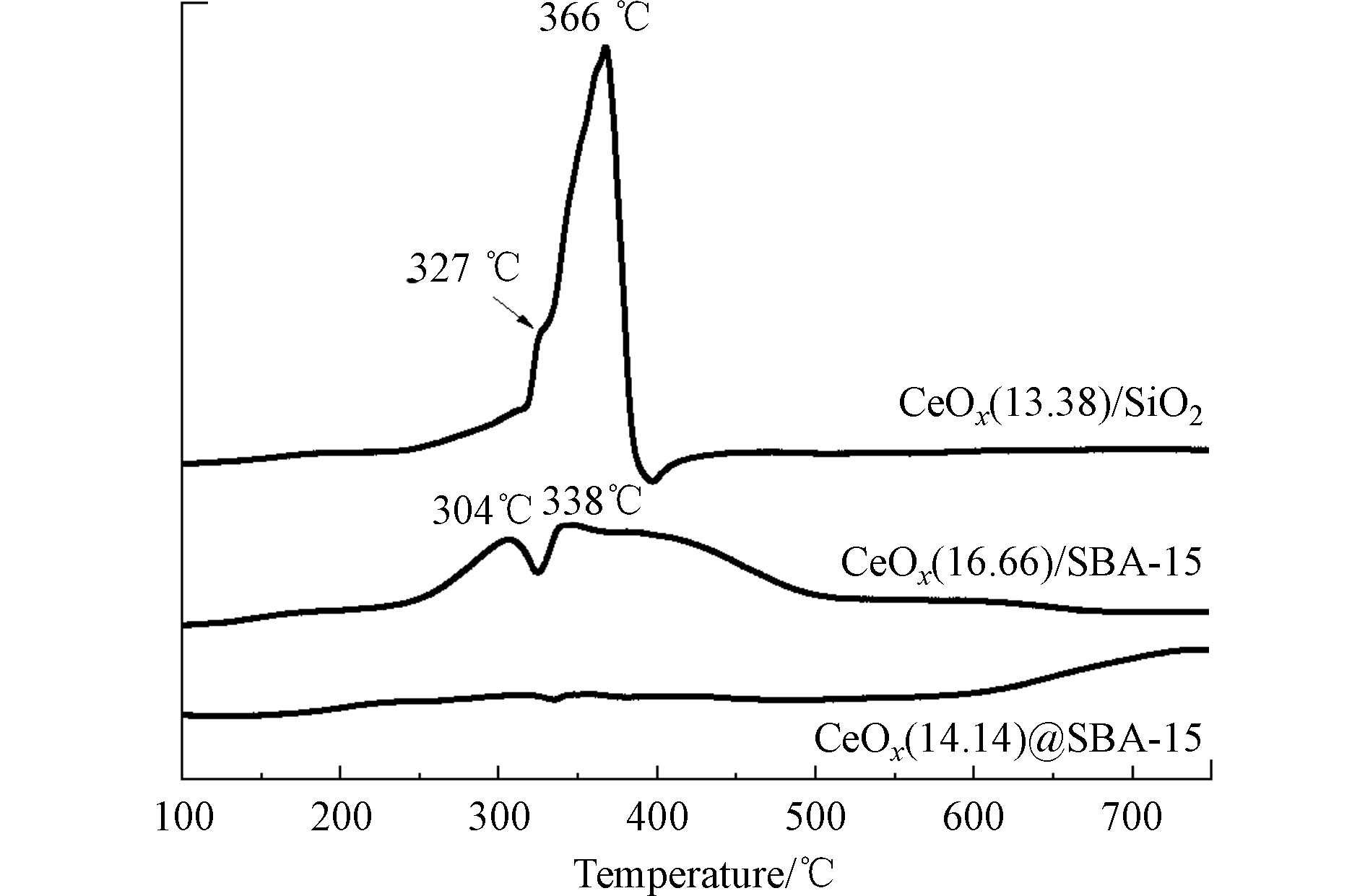
为了进一步探究催化剂中的CoOx和载体之间的相互作用,对3种催化剂进行了氢气程序升温还原实验,所得结果如图4所示. 从图4可以看出,CoOx/SBA-15和CoOx/SiO2的TPR曲线中均可观察到2个明显的连续还原峰,其中CoOx/SiO2的还原峰出现在327°C和366°C附近,分别对应于Co3O4还原为CoO和CoO还原为金属Co两个过程[11],且第二个还原峰的强度较高,这是因为负载在SiO2上的Co3O4的颗粒粒径较大,因而CoO的还原程度较高[12]. CoOx/SBA-15的2个还原峰出现在304°C和338°C附近,且300—500°C之间的还原峰强度较低、范围较广,这归因于SBA-15的介孔结构. 虽然同为浸渍法合成,但相较于CoOx/SiO2,CoOx/SBA-15上的CoOx分散度更高、颗粒更小. 相对地,CoOx@SBA-15的TPR曲线在100—600°C 范围内未观察到明显的还原峰,仅在700°C后出现了强度较弱的还原峰,该还原峰归属于Co2+与SBA-15强相互作用形成的高分散的硅酸钴类物质的还原峰[12],表明CoOx@SBA-15中CoOx为高分散,与TEM和XRD结果一致.
-
氯苯的初始浓度为0.18 mmol·L−1,催化剂的投加量为50 mg·L−1,PMS浓度为5 mmol·L−1时,分别以CoOx(14.14)@SBA-15、CoOx(16.66)/SBA-15和CoOx(13.38)/SiO2为催化剂在室温条件下进行氯苯的降解实验,所得结果如图5所示. 结果显示,3种催化剂均在活化PMS催化降解氯苯的反应中表现出较强的活性,反应120 min后,氯苯在CoOx(14.14)@SBA-15、CoOx(16.66)/SBA-15和CoOx(13.38)/SiO2 的3种催化剂上去除率分别为100%、89.3%和84.7%. 结合催化剂表征来看,SBA-15的介孔结构使得的催化活性CoOx(16.66)/SBA-15高于CoOx(13.38)/SiO2,具体来说,SBA-15的均匀孔结构有利于活性位点的分散同时还可以促进污染物在催化剂上的扩散,提高氯苯的降解效率. 而CoOx(14.14)@SBA-15的催化活性高于CoOx(16.66)/SBA-15是因为采用固相研磨法合成催化剂的过程中Co2+与模板剂P123互作用,使得CoOx高度分散在SBA-15的孔道中,形成对活性位点的限域作用[13]. 限域在孔道中的活性位点能够更高效地与PMS接触,提高PMS的活化效率,进而提高反应活性.
-
氯苯的初始浓度为0.18 mmol·L−1,PMS浓度为5 mmol·L−1时,以CoOx(14.14)@SBA-15为催化剂探究催化剂投加量对CoOx@SBA-15催化PMS降解氯苯的影响,所得结果如图6所示. 催化剂投加量为50 mg·L−1和100 mg·L−1时,氯苯分别在70 min和30 min时实现完全降解,而当催化剂投加量为25 mg·L−1时,氯苯在120 min时的去除率仅为93.2%,这表明氯苯的降解速率随着催化剂投加量的增加而增大. 通过计算催化剂在最初3 min内的催化活性可以验证反应过程是否受降解中间体的竞争性吸附影响[14],催化活性计算结果如图6(b)所示. 从图6可以看出,不同催化剂投加量时的氯苯降解反应的初活性基本相同,因此该反应不受催化剂传质阻力影响.
-
氯苯的初始浓度为0.18 mmol·L−1,催化剂的投加量为50 mmol·L−1,PMS浓度为5 mmol·L−1时,分别以不同负载量的CoOx@SBA-15为催化剂在室温条件下进行氯苯的降解实验,所得结果如图7所示. 从图7可以看出,当负载量为14.14%和26.02%时,CoOx@SBA-15可以在70 min内实现氯苯的完全降解,而当催化剂的负载量为8.14%时,反应120 min后氯苯的去除率仅为83%,氯苯的降解速率随着催化剂负载量的增加而增大. 图7(b)中展示的是不同负载量的CoOx@SBA-15催化剂的反应初活性,随着催化剂负载量的增加,催化反应在3 min内的初活性从0.368增长到2.297 mmol·L−1gCat−1min−1,这是因为负载量的增加使得催化反应的主要活性位点CoOx增多.
-
氯苯的初始浓度为0.18 mmol·L−1,催化剂的投加量为50 mg·L−1时,在其他条件不变的情况下,改变每次降解实验中氯苯和PMS的初始浓度,以进一步探究PMS在CoOx(14.14)@SBA-15上的活化机制,所得结果如图8、9所示. 从图8中可以看出,当PMS的浓度固定为5 mmol·L−1,氯苯的初始浓度从0.1 mmol·L−1增加至0.4 mmol·L−1时,氯苯的降解速率随之加快,反应初活性也从1.512 mmol·L−1gCat−1min−1增至3.185 mmol·L−1gCat−1min−1,这是因为随着氯苯初始浓度的增加,氯苯在CoOx(14.1)@SBA-15表面的吸附量不断增加,从而促进了氯苯的降解. 从图9中可以看出,当氯苯的初始浓度固定为0.1 mmol·L−1时,随着PMS浓度从2.5增加到10 mmol·L−1,反应初活性从1.429 mmol·L−1gCat−1min−1增至2.460 mmol·L−1gCat−1min−1,这说明PMS在催化剂表面的吸附量也会影响氯苯的降解反应.
为了进一步探究反应物在催化剂表面的吸附对反应的影响,使用Langmuir-Hinshelwood(L-H)模型对实验数据进行拟合,Langmuir吸附方程为:
其中,θ、b、c分别代表吸附的反应物覆盖在催化剂表面的分数,吸附常数和反应物的浓度. 而在该反应中,可能吸附在催化剂表面的反应物有氯苯和PMS,因此假设氯苯和PMS的初始浓度分别为
c1 和c2 ,当氯苯的初始浓度c1 固定时,θ的计算公式为:式中,
r1 为氯苯的初始降解速率,k为反应速率常数,b1 和b2 分别为氯苯和PMS的吸附常数. 从式4可以看出,当c2 固定时,1/r1 与1/c1 成正比. 同理,当PMS浓度c2 固定时,1/r2与1/c2 也呈正比.L-H模型假设反应速率与吸附在催化剂表面的反应物浓度成正比,进而证明反应物的吸附是催化反应的速率控制步骤. 因此将
1/r1 与1/c1 、1/r2 与1/c2 分别进行拟合,拟合结果如图8(b)和图9(b)的插图所示. 从图中可以看出,当氯苯的初始浓度改变时,1/r1 与1/c1 的线性关系良好(R2 > 0.98),同样的,当PMS的浓度改变时,1/r2 与1/c2 也呈现了良好的线性关系(R2 > 0.98),这表明氯苯和PMS在催化剂表面的吸附均在氯苯的降解过程中起着重要的作用,是反应的速率控制步骤. -
氯苯的初始浓度为0.18 mmol·L−1,PMS浓度为5 mmol·L−1,催化剂的投加量为50 mg·L−1 时,以CoOx(14.14)@SBA-15为催化剂探究反应体系初始pH氯苯降解的影响,所得结果如图10所示. 从图10可看出,当反应体系初始pH为6.0和10.0时,氯苯在70 min时可以实现完全降解,这表明CoOx@SBA-15催化剂在中性和碱性条件下都具有较高的催化活性. 然后当反应体系初始pH为3.0时,120 min时氯苯的去除率仅为54.3%,这表明酸性条件会抑制催化剂活化PMS降解氯苯,这是因为酸性条件下,H+易于HSO5-结合形成氢键,影响PMS的活化[15].
-
采用EPR技术检测反应体系中的活性自由基,具体方法见1.4,所得结果如图11所示. 从图11(a)可以看出,CoOx@SBA-15+PMS的EPR光谱图中未见明显的DMPO·-OH和DMPO·-
SO−4 特征峰,这表明该体系中没有产生SO·−4 和·OH,但在该体系的光谱中出现了强度为1:2:1:2:1:2:1的7条分裂峰,这类分裂峰可归属于DMPO-X,形成原因为DMPO受单原子直接氧化,类似的EPR信号曾出现在Co2+-PMS体系中[16]. 除此之外,在CoOx@SBA-15+PMS+CB、PMS+CB、DMPO体系的EPR光谱图中还出现了3个等强度的分裂缝,此处可归属于DMPO的分裂峰[17]. CoOx@SBA-15/PMS/CB催化体系的DMPO-EPR结果表明,PMS在催化剂CoOx@SBA-15上的活化存在非自由基过程,PMS会与CoOx@SBA-15表面的活性位点结合形成亚稳态复合物,这种复合物具有强氧化活性,从而促进氯苯的氧化降解. 这也再次验证了,在氯苯的降解过程中,PMS在催化剂表面的吸附是反应的关键步骤. 为了鉴定反应体系中可能存在的其他活性自由基,使用TEMP(2,2,6,6-四甲基-4 哌啶)作为单线态氧(1O2)的捕获剂,所得结果如图11(b)所示,TEMP-EPR光谱图中出现了3个等强度的分裂峰,归属于TEMP-1O2,这表明CoOx@SBA-15/PMS/CB催化体系中存在单线态氧(1O2).为了进一步验证反应体系中存在的自由基种类,在反应体系中加入一定量自由基猝灭剂[18],其中甲醇(Methanol)用于猝灭
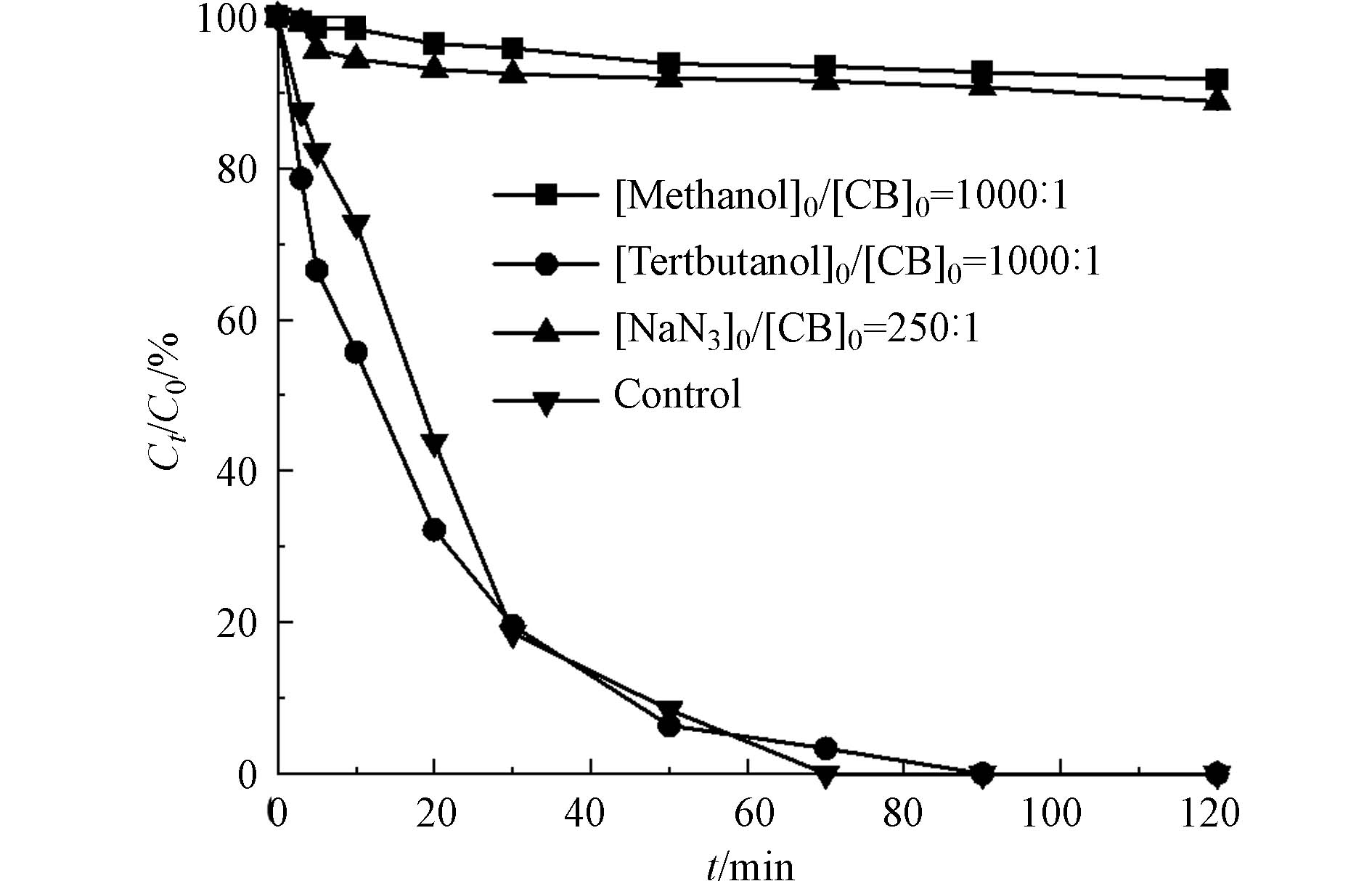
SO·−4 和·OH,叔丁醇(tert-butanol)用于猝灭·OH,叠氮化钠(NaN3)则用于猝灭1O2,实验结果如图12所示. 与Control组相比,当反应体系中存在甲醇和叠氮化钠时,氯苯在120 min时的去除率从100%分别下降到9.3%和10.2%. 而当反应体系中存在叔丁醇时,氯苯的降解反应未被明显抑制,虽然降解速率有所下降但在90 min时可以实现完全降解. 因此反应体系中的主要活性物种是SO·−4 和1O2,·OH的作用较小. -
(1)相较于传统浸渍法合成的催化剂CoOx/SBA-15和CoOx/SiO2,固相研磨法合成的催化剂CoOx@SBA-15的活性位点颗粒更小、分散度更高,因而在氯苯的降解实验中具有更高的催化活性.
(2) CoOx@SBA-15催化活化PMS降解氯苯的反应符合Langmuir-Hinshelwood模型,表明PMS和氯苯在催化剂表面的吸附是该反应的关键步骤.
(3)酸性条件下,H+易于HSO5-结合形成氢键,不利于PMS的活化,进而影响氯苯的降解效率.
(4)自由基抑制试验表明CoOx@SBA-15/PMS/CB催化体系中的主要活性物种为
SO·−4 、·OH和1O2.
SBA-15限域的CoOx催化过硫酸盐降解水中氯苯的机制研究
CoOx confined in SBA-15 as the highly effective catalyst to activate peroxymonosulfate for chlorobenzene degradation
-
摘要: 以中孔SBA-15为载体采用固相研磨法合成限域型催化剂CoOx@SBA-15,采用传统浸渍法合成负载型催化剂CoOx/SBA-15和CoOx/SiO2作为对比. 利用X射线衍射(XRD)、透射电子显微镜测试(TEM)等表征技术对催化剂的结构和组成进行分析,并对催化剂活化PMS降解氯苯的性能进行了研究. 结果表明,CoOx@SBA-15具有更高的催化活性,氯苯的降解反应受氯苯和PMS在催化剂表面吸附控制,CoOx@SBA-15/PMS/CB催化体系中的主要活性物种为
SO·−4 和1O2.-
关键词:
- CoOx@SBA-15 /
- 氯苯降解 /
- PMS活化 /
- 限域催化剂.
Abstract: Supported Co catalysts on SBA-15 were prepared using a solid-state reaction between Co(NO3)2 and organic template-occluded SBA-15. The catalysts were characterized by X-ray diffraction (XRD), N2 adsorption-desorption, H2 temperature-programmed reduction and Transmission Electron Microscope (TEM). Catalytic activities of the catalysts were investigated by activating PMS for chlorobenzene degradation. The results showed that CoOx@SBA-15 had a higher activity than other catalysts. The degradation of chlorobenzene on catalyst followed the Langmuir-Hinshelwood model, reflecting that the activation of adsorbed PMS was the rate controlling step. EPR results showed that the main active species involved in the catalytic system CoOx@SBA-15/PMS/CB wereSO·−4 and 1O2.-
Key words:
- PMS activation /
- chlorobenzene degradation /
- CoOx@SBA-15 /
- confinement atalysts
-
清洁水和卫生设备供应不足是全球性最大的挑战之一,特别是在中低收入国家和地区[1]。据报道,世界上有21亿人不能或难以获得清洁安全的供水[2—3]。氯化和臭氧化是最为广泛使用的化学消毒方法[4—5]。它们能够有效地杀死有害微生物,但仍存在一些问题。例如,氯化处理会导致致癌消毒副产物(disinfection byproducts , DBPs)的形成,甚至会引发军团杆菌等耐氯病原体的生长,以及在处理后的水中产生不良的气味[6—7]。臭氧化同样会产生有害的DBPs,在大规模臭氧生产、储存和运输过程中还体现出急性毒性和腐蚀性特征[8—10]。相对来讲,煮沸是一种有效常用且不会产生DBPs的家庭水处理方法[11—12],但由于其需要大量额外供能而不适于大规模水消毒。此外,与煮沸相比,使用免费阳光的SODIS技术更加具有可持续性。根据光热催化材料的存在与否,将SODIS分为光热催化杀菌和紫外线杀菌。紫外线杀菌是利用UVC和UVB(200—280 nm)来破坏DNA,形成胸腺嘧啶二聚体来阻断繁殖并灭活微生物[11]。然而,紫外线在太阳光谱中占比极低(约4 %),导致对水的消毒效率低下,尤其是对病毒。早期研究表明,需要超过30 h的太阳光照射,才能灭活99.9%的噬菌体MS2[13]。相比之下,光热催化杀菌主要通过光热催化材料产生热量和活性氧物种(ROSs)来协同灭菌,更加具有广谱灭菌性,包括对VBNC(viable but non-culturable)细菌以及病毒都有高灭活效率[14—15]。优良的光热催化材料对紫外光、可见光甚至红外光都能产生响应,从而充分利用太阳能。因此,光热催化消毒法在实际水杀菌,特别是在终端(point of use,POU)水处理中展现出强大的应用潜力。
1. 光热催化杀菌纳米材料(Photothermal disinfection by nanostructures)
常用的光热材料包括(1)通过局域表面等离子体共振效应(SPR)来转换光热的纳米金属及其化合物,如金、银、铂、镍和铜等;(2)直接吸收光子热量的碳材料如炭黑,碳量子点、碳纳米管,石墨烯等;(3)具有红外响应光催化效应的半导体材料如窄隙半导体(CuS、黑磷))、重掺杂半导体(WO3–x、MoO3–x) 等。这些光热材料具有高消光系数ε和高光热转换效率η,能够有效吸收光辐射电磁波且不让其发散,并将其快速转换为热量,因此利于实现太阳能高效利用。
1.1 等离子体NPs(nanoparticles)
贵金属(如Au、Ag和Pt)是应用最广泛的等离子体纳米颗粒,表现出良好的光热催化杀菌性能。贵金属的光热活性在很大程度上取决于其形态、颗粒大小、颗粒间排列和周围环境[16—18]。以金纳米颗粒为例,虽然比表面积和活性位点数会随着颗粒的减小而增加,但较小的颗粒直径可能会使较多Au原子被覆盖,从而导致SPR(surface plasmonic resonance)强度下降。根据之前的研究,表现出最有效的光热催化杀菌效果的Au NPs的最佳直径为2—40 nm [16];但当Au颗粒被控制为粒径小于2 nm的Au团簇时,SPR几乎可以忽略[17]。在高温和近红外辐照下,Au NPs或Au纳米棒会形成较大的团聚体,导致比表面积减小和催化活性降低[18]。Au的形状也会显著影响其光热催化性能。Loeb等制备了Au纳米立方体(nanocubes,NCs)和纳米棒(nanorods,NRs),并比较了它们的光热催化杀菌性能[1]。Au NRs(25 μmol·L−1)能分别杀灭约5.6×106、5.5×106、1.61×106 CFU·mL−1的K-12大肠杆菌、MS2噬菌体和PR772噬菌体,而Au NCs在相同条件下对上述微生物率仅灭活约4.1×106、2.0×106、0.51×106 CFU·mL−1。结果表明,Au NRs在光热催化杀菌过程中表现出更高的潜力,而Au NRs具有高生物相容性和低细胞毒性。
考虑到纯贵金属纳米颗粒的光稳定性低、易在近红外辐射下聚集等缺点,随后设计和制备了贵金属基复合材料来解决上述问题。Zhao等制备了负载Au NRs的多隔室介孔二氧化硅NPs(mesoporous silica , MMSN@AuNR),发现其具有超高的光稳定性和优异的光热催化活性[19]。MMSN@AuNR能在808 nm近红外光照射下快速杀死细胞,并在11次照射启动/关闭的循环后保持高灭菌效率。MMSN@AuNR比纯Au NRs具有更高的稳定性,这主要是由于MMSN的保护能有效抑制Au NRs在近红外光下的团聚。
为降低材料成本,采用廉价的非贵金属如镍(Ni)和铜(Cu)作为替代等离子体材料。例如,He等开发了Ni-TiO2异质结构,并在该系统中观察到SPR介导的载流子转移[20]。在可见光照射下,Ni通过等离子体激发产生热电子和热空穴。然后热载流子从Ni转移到TiO2,占据氧空位,产生Ti3+,并固定在TiO2的表面氧上。Ni NRs负载的氧化石墨烯(Ni/RGO)表现出高效的光热转换,在氙灯(850 mW·cm−2)照射400 s内将水从25 ℃加热到50 ℃以上[21]。虽然贵金属纳米颗粒表现出了强光热转换能力,但其高成本限制了其大规模应用。因此,更经济的廉价金属或非贵金属光热催化剂在抗菌应用中受到关注。
1.2 碳NPs
宽而强的光吸收能力使碳纳米颗粒能够进行高效光热催化反应。碳纳米颗粒,如碳黑、碳纳米管、碳纤维和纳米氧化石墨烯等,具有完整的紫外-可见-近红外吸收,已被广泛开发并应用于杀菌[1]、产生蒸汽[22]和有机物聚合[23]。与金属基材料相比,碳纳米颗粒作为光热催化剂除了具有广谱吸收特性外,还具有成本低、光腐蚀少、无金属释出等优点。
碳黑优异的光热转换性能已被广泛报道。Han等证明了碳黑粉末及其纳米流体在200 nm到2500 nm的宽波长范围内表现出良好的吸收[24]。在光照射下,碳黑纳米流体的温度在42 min内从24.4 ℃上升到38.4 ℃,而纯水的温度仅上升到31.2 °C,表明了碳黑良好的光热转换能力。Loeb等人工作中[1]表明,在日光照射(AM 1.5G)时长分别为60 min和100 min的条件下,碳黑纳米颗粒对大肠杆菌几乎无杀灭效果,对噬菌体MS2有轻微杀灭效果。与纯碳黑和Au相比,其复合膜材料对噬菌体PR722的光热催化灭活作用增强。
碳纳米管(carbon Nanotubes = CNTs),由于其大表面积、优秀的光学性能(如高效光热转换和广谱吸收)和高光热导率,已成为一种很有前途的抗菌材料。将碳纳米管与等离子体材料复合已被证明是提高光热效率的有效策略。在模拟日光照射(AM 1.5)下,将等离子体Ni NPs嵌入N掺杂CNTs的表面温度在2 min内迅速上升至56.8 ℃,展现了有效的光热转换能力[25]。Ag修饰的多壁碳纳米管(MWCNTs)表现出更高的导热性和光热活性,在670 nm照射下实现了细胞的有效光热消融[26]。Sun等报道了一种Au纳米颗粒/羧基功能化的碳纳米管(AuNP/CNT-COOH)[27]具有优异的光热转换能力。在852 nm激光的照射下,这种碳纳米管基材料可以将水从约20 ℃加热至75 ℃。
氧化石墨烯纳米复合材料具有强烈的近红外光吸收、光催化活性和“纳米刀”效应,可实现有效光热催化杀菌。值得注意的是,纯氧化石墨烯表现出有限的光热转换效率,在近红外照射8 min后温度只有小幅升高[28]。因此,人们制造了不同的氧化石墨烯基复合材料,并将其应用于水消毒。氨基化的氧化石墨烯(GO-NH2)纳米片可以通过静电引力轻易吸附细菌细胞,并表现出显著增强的光热催化抗菌性能[29]。如图1,在白光照射(159 mW·cm−2)下,GO-NH2浓度为0.10 mg·mL−1和0.25 mg·mL−1时,水的温度分别从20.5 ℃快速上升至55.5 ℃和81.4 ℃。GO-NH2纳米片对金黄色葡萄球菌和大肠杆菌的光热催化抗菌活性分别提高了16倍和32倍。此外,通过扫描电镜观察发现GO-NH2纳米片的锐利边缘所产生的“纳米刀”效应在细菌失活中起着关键作用。
 图 1 (a) 在光照射下不同GO-NH2的升温曲线;(b) GO-NH2作用 2min前后金黄色葡萄球菌和大肠杆菌的图像;(c)不同浓度的GO-NH2对金黄色葡萄球菌和大肠杆菌的灭活[29]Figure 1. (a) Heating curves of GO-NH2 with different catalyst concentrations irradiated by white light (159 mW·cm-2). (b) SEM images of (A, B) S. aureus and (C, D) E. coli before and after interaction with GO-NH2 for 2 min. (c) Growth inhibition of S. aureus and E. coli after the photothermal treatment by GO-NH2.
图 1 (a) 在光照射下不同GO-NH2的升温曲线;(b) GO-NH2作用 2min前后金黄色葡萄球菌和大肠杆菌的图像;(c)不同浓度的GO-NH2对金黄色葡萄球菌和大肠杆菌的灭活[29]Figure 1. (a) Heating curves of GO-NH2 with different catalyst concentrations irradiated by white light (159 mW·cm-2). (b) SEM images of (A, B) S. aureus and (C, D) E. coli before and after interaction with GO-NH2 for 2 min. (c) Growth inhibition of S. aureus and E. coli after the photothermal treatment by GO-NH2.1.3 缺陷型光催化剂
在光催化剂中制造缺陷结构(也称为空位),通过空位可以缩小能带隙、促进电荷转移和/或引起局部SPR效应,从而可以使宽带隙半导体产生近红外光诱导的光热催化性能。例如,被广泛报道的存在氧缺陷的WO3-x [30],In2O3-x [31—32]、ZrO2-x [33]和MoO3-x [34—35]等光催化剂,不仅在可见光到近红外光区域表现出可调谐的光吸收,而且可以通过调控颗粒尺寸和氧缺陷的比例[36]来进一步增强其光热催化性能。然而,吸附在空位上的O2和H2O会导致氧缺陷光催化剂被氧化,故存在化学不稳定性。构建缺陷型复合材料被认为是提高稳定性和光催化活性的有效策略[37]。例如,Zhang等通过一锅水热法制备了WO3-x/C纳米片[30],其中氧空位和碳涂层的存在显著延长了可见到红外光区域的光吸收带。除了提高光催化性能外,碳涂层还促进了电荷载流子的分离,从而提高了光热催化效率。在Zhao等[38]的另一项研究中,半金属Bi与有氧缺陷的BiO1-xI结合,形成Bi/BiO1-xI复合材料,具有光热协同催化消毒能力。Bi和氧空位不仅在600—1400 nm范围内引起了表面等离子体效应,而且还显著增加了光生电子和空穴的生成量。机理研究表明,活性物种(1O2、h+和·O2−)与热协同作用可有效灭活细菌。
1.4 窄带隙光催化剂
窄带隙半导体,如磷系催化剂、MoS2、Bi2S3和CuS等,表现出很强的近红外吸收,也有有用作光热催化剂的潜力。磷是一种地球富含的非金属元素,有3种同素异形体,即红、黑、白磷。其中红磷(red Phosphorus ,RP)和黑磷(black Phosphorus,BP)可作为光催化剂或光热催化剂来实现光催化和/或光热消毒。BP和RP都是无毒的,具有生物相容性,但RP比BP更具成本效益[39]。Zhang等评估了在不同照射波长下Ti-RP/GO薄膜的光热灭菌效果[40]。在模拟日光照射下,Ti-RP/GO膜在20 min内迅速灭活99.9%金黄色葡萄球菌和大肠杆菌(1×107 CFU·mL−1)。Li等将BP纳米片作为POU末端水消毒系统中的光热催化剂[41]。在该体系中,壳聚糖水凝胶与黑色BP纳米片逐层叠加形成了三明治式过滤器。基于BP纳米片的过滤器表现出优异的近红外光驱动的光热特性,能够实现高杀菌温度(> 140 ℃),导致粘附的枯草芽孢杆菌和大肠杆菌完全失活。其他含硫半导体如MoS2织物的表面温度迅速上升到77 ℃左右,并伴随着ROS例如·O2−的产生。结果表明,MoS2织物对革兰氏阴性大肠杆菌和革兰氏阳性金黄色葡萄球菌(细胞密度= 1×106 CFU·mL−1)均有有效的灭活效果,3 h内的抑菌效率分别为58%和60%左右。这些窄带隙半导体不仅可以作为光热剂直接灭活细菌,还可以作为释放热敏性药物的载体进行间接抗菌处理。
1.5 MOFs(Metal organic Frameworks)
MOFs是一类新兴的多孔固体催化剂,含有与有机配体配位的金属离子/团簇。它们作为抗菌材料时主要是利用生物毒性金属离子的释放[42]。此外,MOFs通常具有较宽的带隙,例如,MOF-5的带隙为3.4 eV [43-44], ZIF-8为3.87 eV[45],使得它们不适合宽光谱响应和光热转换。然而,考虑到金属离子或有机配体的合理调节赋予了MOFs在分子水平上的高设计性,MOFs展示出用于抗菌处理的光热催化剂的潜力。Wang等发现通过在空气中200 ℃下对ZIF-8 NPs进行简单的热处理,会改变ZIF-8中配体结构 (例如, 生成了—N=C=O键),进而造成ZIF-8 MOF的光吸收从紫外到可见和近红外区域的显著延长[45]。此研究证明了宽带隙MOFs作为光热催化剂的可行性。此外,一些MOFs,比如PCN-224(Eg= 1.81 eV)[46],IRMOF-M2a(Eg= 1.5 eV)[43],和Sr-MOF(Eg= 2.3 eV)[43],表现出窄带隙和宽光谱吸收的性质,也可以用于光热催化剂。Wu等通过将Cu2+引入卟啉环的核心,开发了一种Cu掺杂的PCN-224 MOF [47],能够高效光热催化灭菌。一方面,掺杂的Cu2+促进了载流子的转移,从而促进了ROSs的生成,例如1O2等的生成。另一方面,由于d-d跃迁,Cu2+在660 nm处表现出了额外的吸光,并增强了光热转换。由于协同作用,在660 nm光照射(0.4 W·cm−2)下20 min内,Cu掺杂的PCN-224对金黄色葡萄球菌的抗菌效果达到99.71%。表1为近几年报道的光热催化剂及其细菌杀菌性能。
表 1 最近报道的纳米结构的光热细菌失活性能的比较Table 1. Comparison of the photothermal bacterial inactivation by the recently reported nanostructures催化剂Catalysts 辐照(强度)Irradiation(intensity) 催化剂浓度/(mg·mL−1)Catalyst concentration 光热杀菌性能Photothermal disinfection performance 参考文献References Au纳米棒 模拟日光 4.93×10−3 100 min内,分别灭活5.6-lg CFU·mL−1、5.5-lg CFU·mL−1和1.6-lg CFU·mL−1 左右的大肠杆菌K-12、MS2噬菌体和PR772噬菌体 [1] Ni/rGO 808 nm 激光(2 W·cm−2) 0.025 8 min内,对2×106 CFU·mL−1的大肠杆菌和枯草芽孢杆菌分别达到99.6%和99.5%的灭活率 [28] GO-NH2 白光(0.159 W·cm−2) 0.032 10 min内,对107 CFU·mL−1大肠杆菌和金黄色葡萄球菌的灭活率超过90% [29] RP 模拟日光(0.2 W·cm−2) 0.2 20 min内,对5×106 CFU·mL−1金黄色葡萄球菌的灭活率达到99.98% [39] Ti-RP/GO 模拟日光(0.2 W·cm−2) N.A. 15 min内,对107 CFU·mL−1的大肠杆菌的灭活率达到99.91% [40] WO3-x/C 带有700 nm截止滤光片的氙灯(0.2 W·cm−2) 1 40 min内,灭活了1.2×107 CFU·mL−1的大肠杆菌 [30] 碳化ZIF-8 808 nm 激光(3 W·cm−2) 0.16 10 min内,对107 CFU·mL−1的金黄色葡萄球菌的灭活率达到80%左右 [43] PB-PCN-224 600 nm LED(0.3 W·cm−2) 1 15 min内,对1×107 CFU·mL−1的金黄色葡萄球菌的灭活率达到99.84% [47] | Show Table DownLoad:
CSV
DownLoad:
CSV
2. 光热催化杀菌机制(Photothermal disinfection mechanism)
2.1 高温和ROSs对微生物的攻击
如前所述,在光照射下,光热催化材料会通过光热转换产生局部高温和/或通过光催化和热催化生成ROSs进行协同作用, 如图2 [14]。在光热催化材料界面会形成局部热场而升温至约50 ℃以上[48-49]。光热催化材料表面的高温会导致蛋白质变性,导致微生物一旦接触到材料表面就会迅速失活。局部热场会扩散到周围环境,导致体相及水溶液温度升高。当细菌暴露于亚沸温度溶液(55—60 ℃)时,细胞膜上的蛋白质和脂质将被破坏[50]。随后,酶、核酸和其他胞内成分随着照射时间的延长而失活,这与巴氏杀菌相似。同时,生成的ROSs攻击细胞会诱导微生物产生氧化应激以致生理系统紊乱,进而导致细胞膜破裂、胞内成分(如蛋白质、核酸、K+等)的泄漏氧化以及细胞的最终死亡。
 图 2 Ag/MnO2光热催化杀菌机理图[14]Figure 2. Scheme of photothermalcatalytic inactivation over Ag/MnO2.
图 2 Ag/MnO2光热催化杀菌机理图[14]Figure 2. Scheme of photothermalcatalytic inactivation over Ag/MnO2.2.2 细菌细胞的破坏和表征技术
(1)细胞膜的损伤
细菌细胞膜主要由脂质、蛋白质和少量碳水化合物组成。它是细菌抵御外界攻击和环境变化的第一层保护层。在光照射下暴露于光热催化剂时,细胞膜的脂质双分子层会受到热和ROSs的攻击。ROSs与细胞膜不饱和脂肪酸之间的反应引发了随后的链式反应,导致脂质过氧化。ROSs和脂质过氧化产物都会对细菌细胞造成损伤。用MDA检测试剂盒测定细胞膜氧化情况。此外,与底物运输、特异性识别和呼吸相关的膜蛋白对细菌代谢至关重要[19]。在光热处理中,ROSs和局域热场会引起胞内氨基酸氧化和蛋白质变性。如果目标病原微生物是病毒(如MS2),ROSs和局域热场则会破坏蛋白质衣壳并导致抗原性降低[51]。
光热催化杀菌可通过两条途径增加细胞膜的渗透性:①通过脂质过氧化破坏细胞膜的微观结构和降低细胞膜的流动性;②通过ROSs和局域热场灭活在细胞呼吸和跨膜运输中起重要作用的膜蛋白和ATP酶[52]。首先,细菌膜通透性的增加可以破坏钠钾(Na+-K+)泵,导致K+离子等小分子的释放。因此,释放的K+的量被用于测量细胞膜渗透性的变化。此外,利用邻硝基苯-β-D-吡楠半乳糖苷(ONPG)结合比色法可以测定细胞质膜的穿透性[14]。8-苯胺基-1-萘磺酸(ANS) 会与外膜结合发出荧光,也可用于检测外膜的通透性。
此外,利用扫描电镜(SEM)、透射电镜(TEM)和原子力显微镜(AFM)观察细菌细胞膜的完整性和形态变化。在光热催化处理之前,大肠杆菌和金黄色葡萄球菌保持光滑的表面和完整的微观结构。在光热系统中照射10 min后,细胞膜发生严重变形和皱缩,出现凸出和凹坑的变形。光热催化处理10 min后,在细胞膜上观察到一些孔洞。透射电镜提供了细菌样品的高分辨率成像,并显示了细胞膜和胞内成分的变化:在光热催化处理下大肠杆菌细胞膜的功能紊乱并受损,导致细胞质分离和胞内组分渗漏[53].
(2)胞内成份的释放和氧化
在破坏细胞膜的形态和改变其通透性后,进一步检测胞内组分在ROSs和热攻击下的变化,以更好地了解杀菌机制。细胞膜通透性的增加和破坏使ROSs得以加速通过。荧光探针法可用于检测细胞内ROS水平,其中2',7'-二氯二氢荧光素二乙酸酯是检测·OH和H2O2的常用荧光探针。羟苯基荧光素(HPF)和二氢乙啶(HE)也可分别作为·OH和·O2−的荧光探针[54—55]。
通过谷胱甘肽(GSH)、超氧化物酶(SOD)、过氧化氢酶(CAT)以及ATP的量可以分析细菌受到攻击时的自卫能力。GSH不仅是H2O2和·O2−的清除剂,而且还能产生分解ROSs的酶[56-57]。此外,GSH能稳定酶活性,维持细胞内氧化平衡,阻止血红蛋白被氧化。SOD则通过与·O2−特异性反应而参与细菌的自卫系统。CAT在H2O2的防御系统中起着重要作用。用对应的检测试剂盒通过分光光度法测定SOD、CAT、GSH的含量。基本能量载体ATP的合成与细胞代谢活性直接相关。ATP含量用ATP检测试剂盒监测,通过测量636 nm处的吸光度来定量分析[58]。值得注意的是,在光热催化杀菌初期,细胞会产生更多的GSH、SOD和CAT来保护自己免受氧化,并且合成上述抗氧化物质需要更多的能量,ATP水平呈上升趋势。但是,随着处理时间的延长,ROSs和局域热场的持续攻击会使细菌代谢紊乱。最终所有的抗氧化物质和ATP都会被分解。
细菌包膜的破坏也导致细胞内成分的释放,如K+、核酸和蛋白质等。释放的蛋白质可以通过二喹啉甲酸(BCA)法监测,因为蛋白质的肽键结构可以在碱性条件下将Cu2+转换为Cu+,然后BCA可以与Cu+反应形成紫色化合物, 可以通过分光光度计在562 nm处定量分析。然后利用2D电泳进行定性分析可深入了解蛋白质的释放和氧化,还可以用分光光度计测定释放的核酸浓度,有关DNA/RNA的特征吸收峰位于260 nm附近[40]。进一步采用三维荧光激发-发射矩阵技术,通过分析溶解有机物的变化来研究生物分子的破坏。此外,利用单细胞的傅立叶变换红外吸收光谱和拉曼显微光谱还可以分析细胞内成分结构的演化[51-53]。
(3)核酸的损伤
为了更好地理解ROS和局域热场对细菌核酸的损伤,进行了DNA琼脂糖凝胶电泳和转录组分析。前者是用Ezup柱式细菌基因组DNA抽提试剂盒提取染色体DNA,然后用DNA琼脂糖凝胶电泳验证。此外,转录组学研究中的样品制备和数据分析也比电泳法复杂。一般情况下,提取总RNA、片段化处理mRNA、合成cDNA、末端修复、添加单核苷酸后,选择样品进行琼脂糖凝胶电泳、PCR扩增,然后定量定性分析[54]。通过这种方法可以确定参与各种正常生理活动(如代谢活动、氧化应激反应和细胞呼吸过程等)的基因表达的变化,为细菌失活机制提供了更深入的见解[54-55]。总有机碳(TOC)的测定也可以表示细菌矿化程度[56]。
3. 展望(Perspectives)
本文总结了光热催化消毒的研究进展,显示出了实际应用的巨大潜力。然而,光热催化消毒技术仍面临挑战,需要采取进一步的策略来降低成本,提高效率。为了实现光热催化剂的实际应用,较高的材料和运行成本在一定程度上限制了大规模的光热应用。使用低成本和可持续的材料,如生物质碳,非贵金属等离子体NPs和丰富的自然资源(例如,矿物),更适于大规模水消毒处理。虽然可以利用各种方法来分析光热催化灭菌,但对光热催化过程中生物分子变化的深入认识还有待进一步评价。此外,由于天然水或污水中TOC和浊度高、各种微生物的共存、pH值不理想等原因,其杀菌效果是完全不同的;需要进一步设计和优化光热反应器如采用间歇式和连续流式反应器。总之,光热催化法有望成为环境修复(包括但不限于水消毒)的一种有效策略。
-
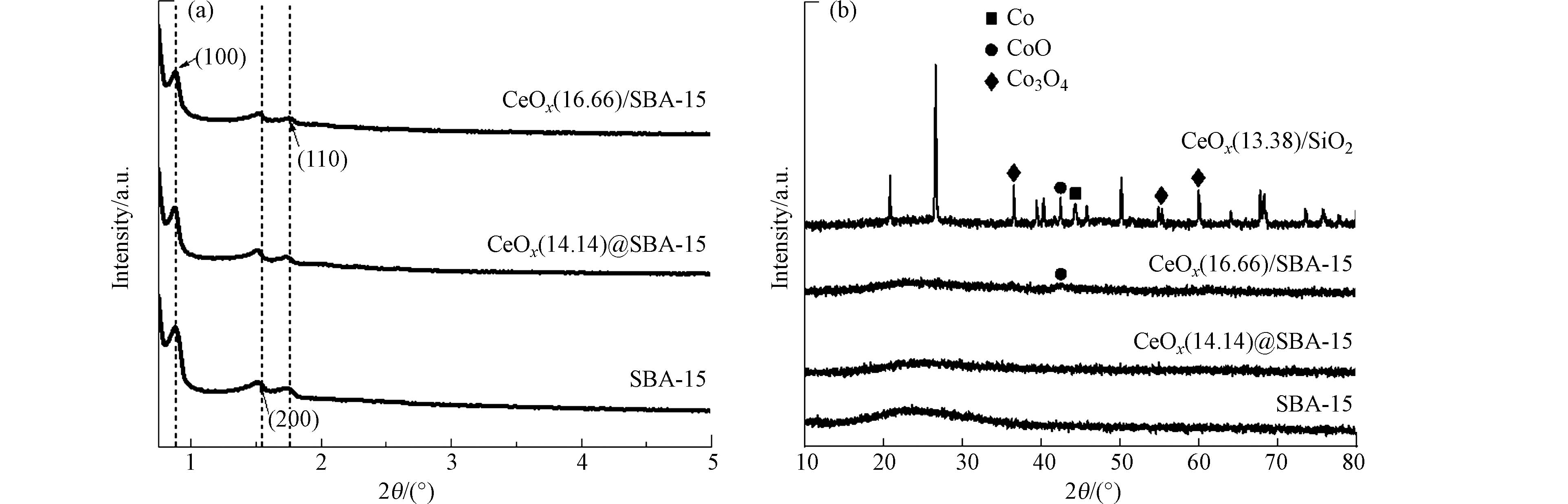
图 1 催化剂的(a)SAXRS谱图和(b)WAXRD谱图
Figure 1. (a) SAXRS patterns and (b) WAXRD patterns of catalysts
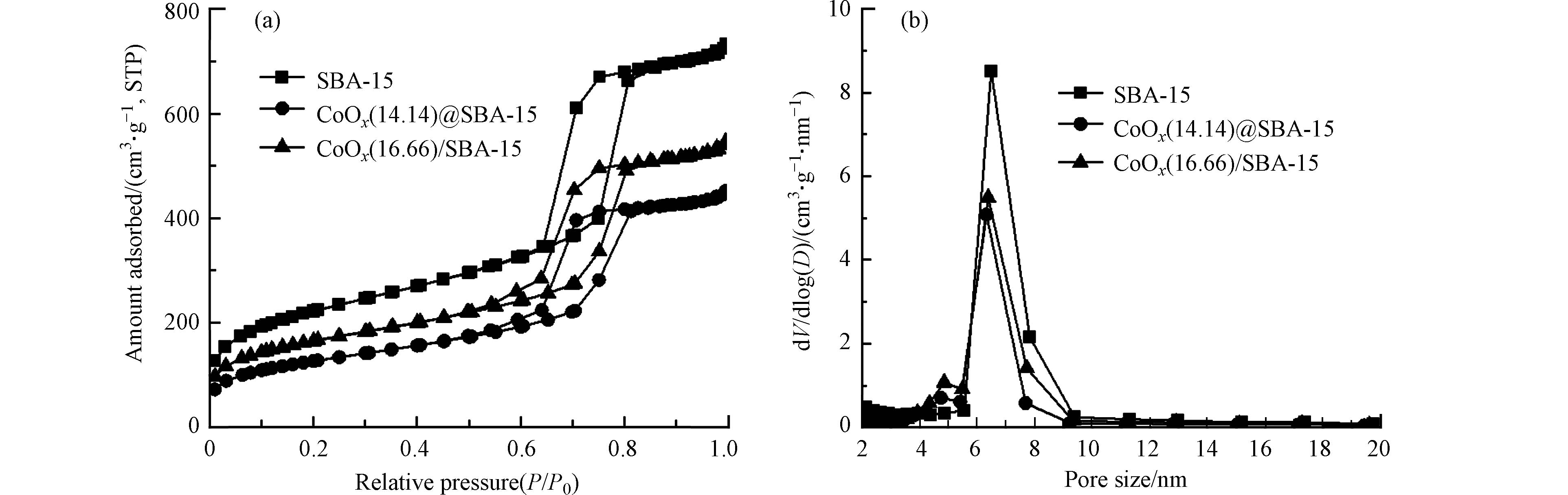
图 2 (a)催化剂的N2吸附-脱附等温线及(b)孔径分布
Figure 2. (a) N2 adsorption-desorption curves and (b) pore size distributions of samples
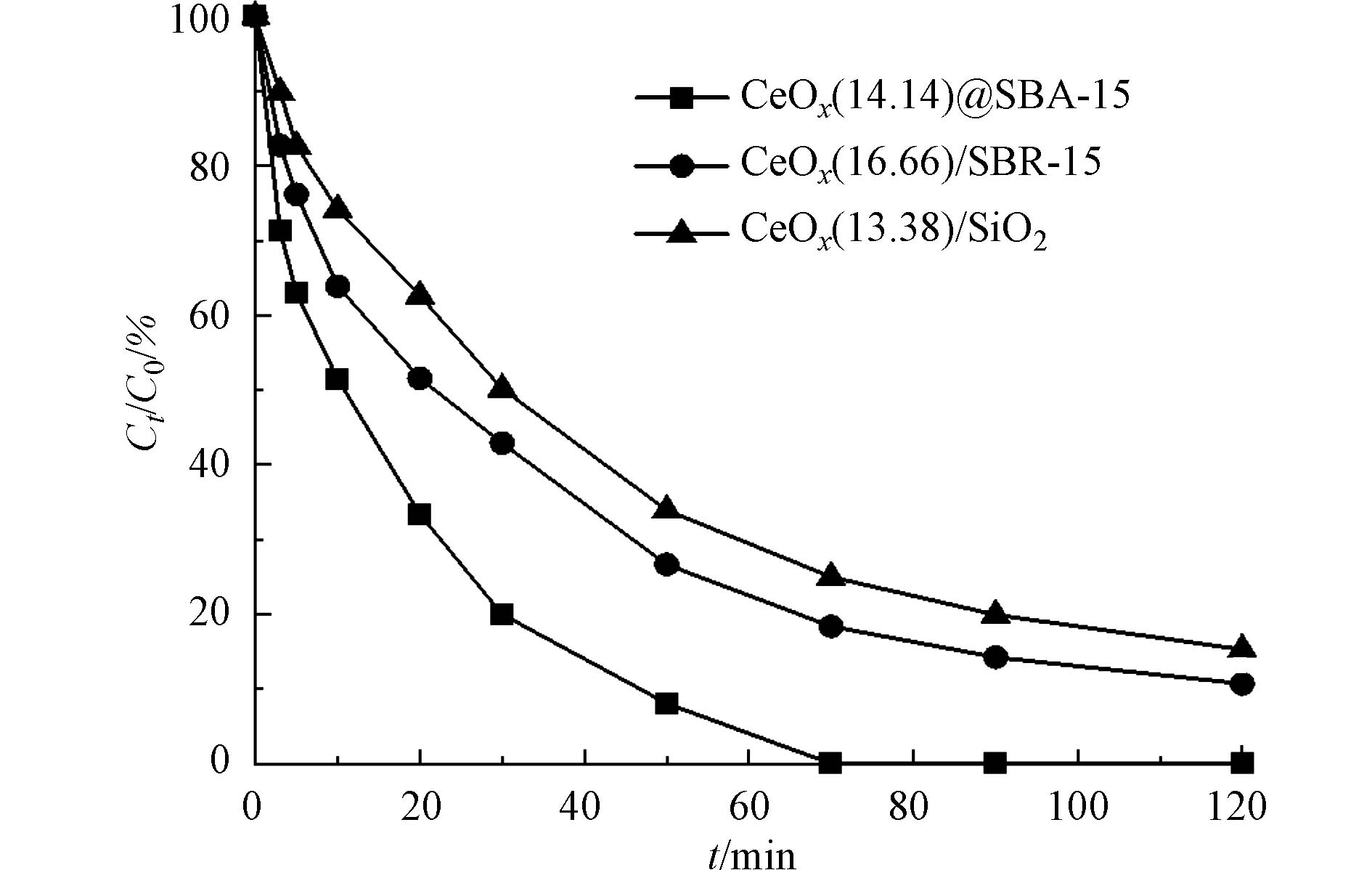
图 5 不同催化剂条件下氯苯的去除率对比
Figure 5. comparison of catalytic activities for CB degradation using different catalysts
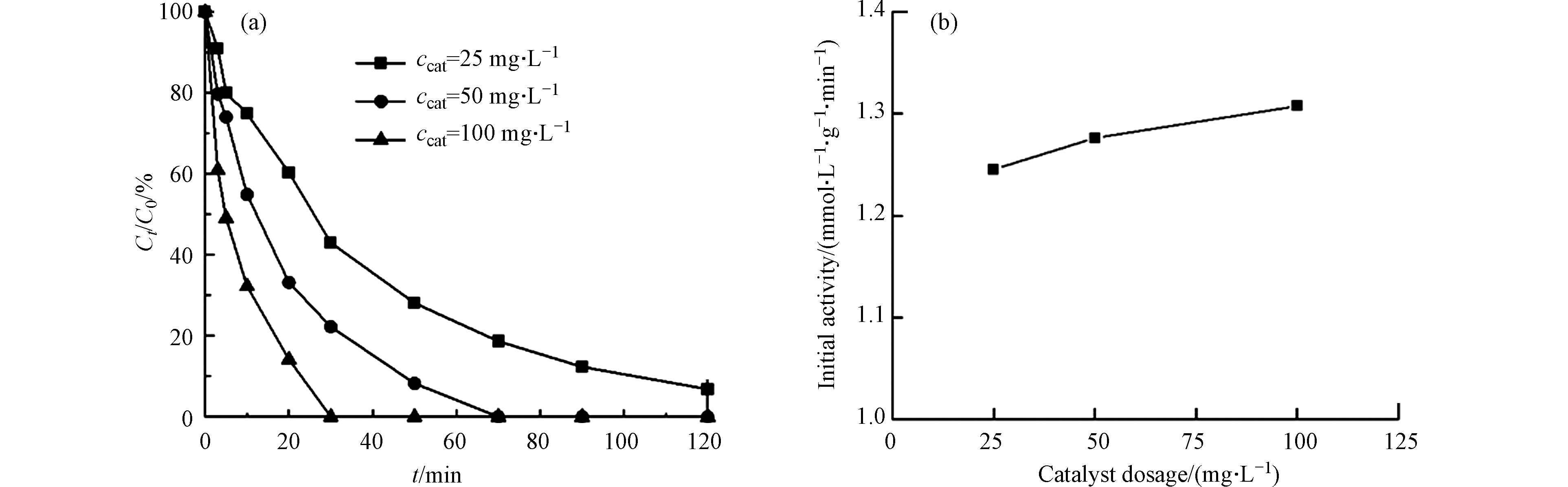
图 6 (a)催化剂投加量对CoOx@SBA-15催化PMS降解氯苯的影响;(b)在最初3 min内的相应初始活性
Figure 6. (a) Effect of catalyst dosage on CB degradation by PMS over CoOx@SBA-15 and (b) the corresponding initial activity within initial 3 min
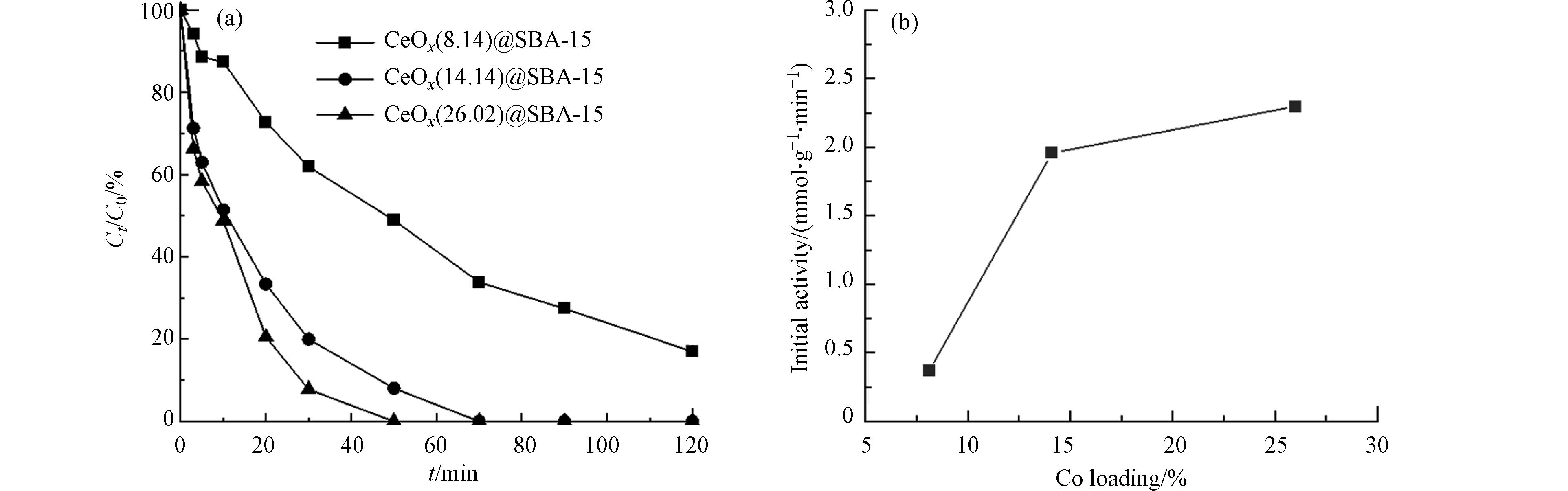
图 7 (a)催化剂负载量对CoOx@SBA-15催化PMS降解氯苯的影响;(b)在最初3 min内的相应初始活性
Figure 7. (a) Effect of loading amount on CB degradation by PMS over CoOx@SBA-15 and (b) the corresponding initial activity within initial 3 min
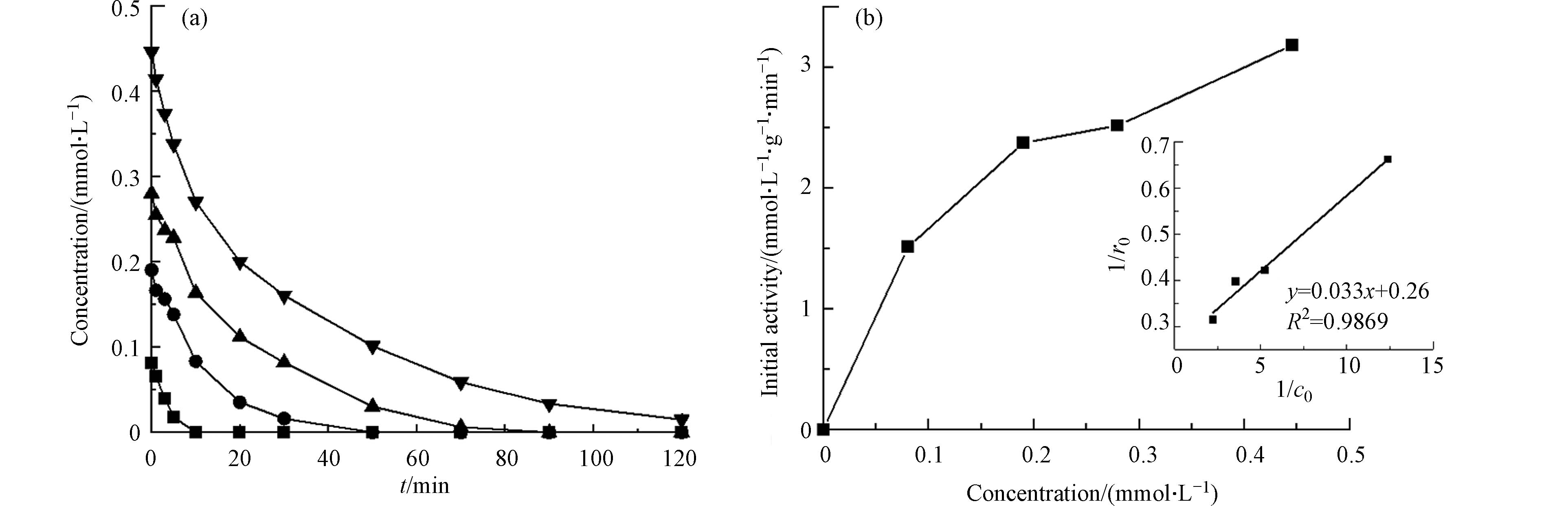
图 8 氯苯的初始浓度对CoOx@SBA-15催化PMS降解氯苯的影响
Figure 8. Effect of initial CB concentrations on CB degradation by PMS over CoOx@SBA-15

图 9 PMS的初始浓度对CoOx@SBA-15催化PMS降解氯苯的影响
Figure 9. Effect of initial PMS concentrations on CB degradation by PMS over CoOx@SBA-15
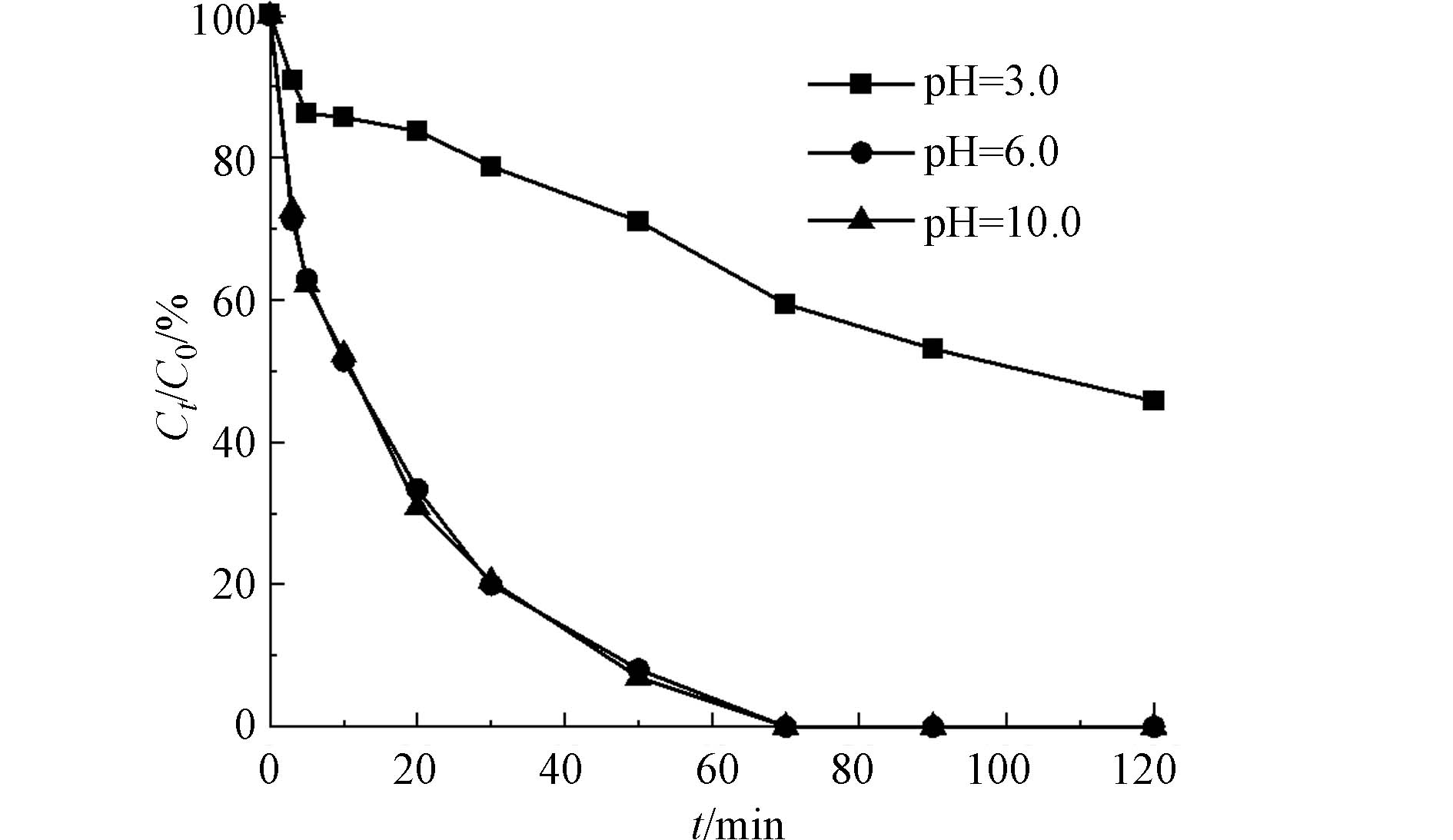
图 10 pH对CoOx@SBA-15催化PMS降解氯苯的影响
Figure 10. Effect of pH on CB degradation by PMS over CoOx@SBA-15
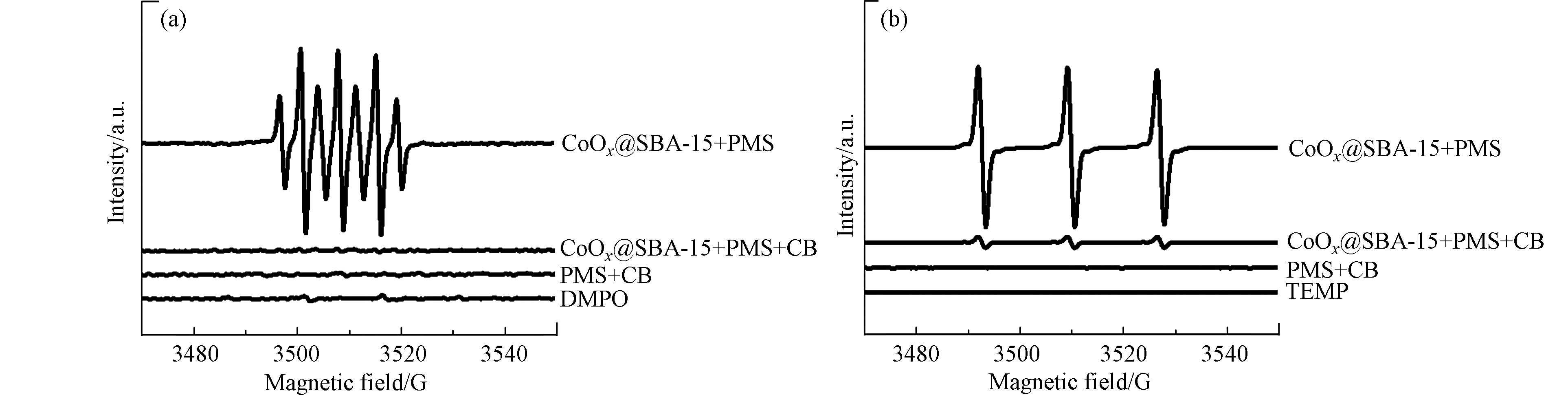
图 11 使用(a)DMPO和(b)TEMP作为自由基自旋捕获剂的CoOx@SBA-15-PMS系统中的EPR光谱
Figure 11. EPR spectra in CoOx@SBA-15-PMS system using (a) DMPO and (b) TEMP
表 1 SBA-15及钴基催化剂的结构参数
Table 1. Structural parameters of SBA-15 and Cobalt-based catalyst
样品 Sample SBETa/(m2·g−1) Smicrob/(m2·g−1) Vtc/(cm3·g−1) Vmicrob/(cm3·g−1) DBJHd/nm SBA-15 744 75 1.10 0.051 6.68 CoOx(14.14)@SBA-15 430 38 0.68 0.024 6.70 CoOx(16.66)/SBA-15 552 69 0.82 0.045 6.80 a calculated by Brunauer-Emmett-Teller (BET) method;b calculated by t-plot method;c obtained at P/P0=0.995;d from BJH method.
下载: 导出CSV
-
[1] KOWALSKY E S, JENNINGS A A. Worldwide regulatory guidance values for chlorinated benzene surface soil contamination [J]. Journal of Environmental Engineering, 2012, 138(11): 1085-1105. doi: 10.1061/(ASCE)EE.1943-7870.0000574 [2] MALCOLM H. Chlorobenzenes other than hexachlorobenzene: Environmental aspects [Z]. 2004 [3] National Center for Biotechnology Information. Pubchem compound summary for CID 7964, Chlorobenzene [Z]. 2022. [4] LIU X H, YI R, ZHANG N, et al. Cobalt hydroxide nanosheets and their thermal decomposition to cobalt oxide nanorings [J]. Chemistry - an Asian Journal, 2008, 3(4): 732-738. doi: 10.1002/asia.200700264 [5] WANG Y , WU Z , SHI L , et al. Rapid functionalization of mesoporous materials: Directly dispersing metal oxides into as-prepared SBA-15 occluded with template [J]. Advanced Materials, 2005, 17(3): 323-327. doi: 10.1002/adma.200400860 [6] NING X, LU Y Y, FU H Y, et al. Template-mediated Ni(II) dispersion in mesoporous SiO2 for preparation of highly dispersed Ni catalysts: Influence of template type [J]. ACS Applied Materials & Interfaces, 2017, 9(22): 19335-19344. [7] ZHAO D, FENG J, HUO Q, et al. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores [J]. Science, 1998, 279(5350): 548-552. doi: 10.1126/science.279.5350.548 [8] PARK K S, SARAVANAN K, PARK S J, et al. Effects of CO2 on the deactivation behaviors of Co/Al2O3 and Co/SiO2 in CO hydrogenation to hydrocarbons [J]. Catalysis Science & Technology, 2017, 7(18): 4079-4091. [9] ZHAN H J, XIA Q H, LU X H, et al. Selective epoxidation of styrene with air catalyzed by CoOx and CoOx/SiO2 without any reductant [J]. Catalysis Communications, 2007, 8(10): 1472-1478. doi: 10.1016/j.catcom.2006.12.026 [10] LIU H M, LI Y M, WU H, et al. Effects of β-cyclodextrin modification on properties of Ni/SBA-15 and its catalytic performance in carbon dioxide reforming of methane [J]. Catalysis Communications, 2012, 28: 168-173. doi: 10.1016/j.catcom.2012.08.035 [11] VISWANATHAN B, GOPALAKRISHNAN R. Effect of support and promoter in Fischer-Tropsch cobalt catalysts [J]. Journal of Catalysis, 1986, 99(2): 342-348. doi: 10.1016/0021-9517(86)90359-3 [12] ZHANG X Y, GUAN Y Y, XIONG Y, et al. Nitrous oxide decomposition over Co/SBA-15 catalysts: The influence of metal loading, cobalt precursor, and solvent effect [J]. Materials Research Innovations, 2016, 20(7): 487-494. doi: 10.1179/1433075X15Y.0000000062 [13] CHEN W, FAN Z L, PAN X L, et al. Effect of confinement in carbon nanotubes on the activity of Fischer-Tropsch iron catalyst [J]. Journal of the American Chemical Society, 2008, 130(29): 9414-9419. doi: 10.1021/ja8008192 [14] ILINITCH O M, CUPERUS F P, NOSOVA L V, et al. Catalytic membrane in reduction of aqueous nitrates: Operational principles and catalytic performance [J]. Catalysis Today, 2000, 56(1/2/3): 137-145. [15] TAN C Q, GAO N Y, DENG Y, et al. Radical induced degradation of acetaminophen with Fe3O4 magnetic nanoparticles as heterogeneous activator of peroxymonosulfate [J]. Journal of Hazardous Materials, 2014, 276: 452-460. doi: 10.1016/j.jhazmat.2014.05.068 [16] HUANG Y F, HUANG Y H. Behavioral evidence of the dominant radicals and intermediates involved in Bisphenol A degradation using an efficient Co2+/PMS oxidation process [J]. Journal of Hazardous Materials, 2009, 167(1/2/3): 418-426. [17] CANO M, QUINTANA J, JULIÁ L, et al. Trapping of cyclopropenyl radicals by 5, 5-dimethyl-1-pyrroline-N-oxide [J]. The Journal of Organic Chemistry, 1999, 64(14): 5096-5099. doi: 10.1021/jo9900026 [18] QIN X, FANG S W, ZHAO L, et al. Cobalt super-microparticles anchored on nitrogen-doped graphene for aniline oxidation based on sulfate radicals [J]. Science of the Total Environment, 2017, 601/602: 99-108. doi: 10.1016/j.scitotenv.2017.05.198 期刊类型引用(1)
1. 闫兴雨,王艳,王善虎,莫原野,许伟,刘阳,孟宪荣,施维林. 金属-硅核壳结构诱导强化nZVI@SiO_2/PDS体系降解地下水氯苯. 环境科学学报. 2024(02): 125-135 .  百度学术
百度学术
其他类型引用(1)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 2138
- HTML全文浏览数: 2138
- PDF下载数: 95
- 施引文献: 2
























