


-
改革开放以来,中国工业飞速发展,表面处理作为现代工业的一个基础行业更是发展迅猛。表面处理中的基础工艺化学镀和电镀,因其能增强镀件耐磨性、耐腐蚀性、硬度和光泽等在许多领域得到广泛应用[1-3]。镀镍废水是化学镀镍或电镀镍工艺各操作单元汇总的工业废水,其中主要污染物包括重金属镍离子,且因络合剂的存在,镍离子拥有较好的溶解性和稳定性。传统的重金属废水处理方法如化学沉淀法、离子交换法和吸附法去除络合态重金属污染物的效果不佳[4-6]。针对络合态重金属的处理,目前普遍采用的方法是先氧化破络释放出重金属离子,再通过沉淀、絮凝、吸附等传统方法分离去除[7-8].
已有许多学者将高级氧化技术应用于电镀废水的处理中,以实现重金属络合物的氧化破络。这些处理体系主要为UV/H2O2、UV/Chlorine、Fenton及类Fenton反应[8-12]。为避免上述反应过程需外源引入氧化剂、处理成本高等问题,近些年逐渐发展出电化学高级氧化技术[13-14],这些电化学技术主要聚焦于电催化阳极的研发及不同工艺之间的耦合实现污染物的高效去除[15-17],而关于电化学体系中重金属络合物的降解规律、机理解析及重金属是否会参与络合物矿化过程鲜有研究。已有研究报道了Cu-EDTA在UV/Chlorine过程中络合态铜能够介导氯自由基的生成从而形成自催化效果,加快反应速率[18]。在电化学膜过滤体系处理Cu-EDTA过程中也发现了类似的自催化降解现象,络合物态铜能够催化H2O2分解生成羟基自由基从而形成自催化破络效果[19]。也有研究报道Ni(Ⅱ)可以催化活性物种生成,Ni(Ⅱ)/过一硫酸盐体系中生成硫酸根自由基和单线态氧能有效降解柠檬酸镍络合物[20];Ni(Ⅱ)物种如Ni(OH)2作为阳极材料通电后能转为高价Ni物种(NiOOH),后者能有效降解甲醛[21]。Ni-EDTA作为镀镍废水中典型污染物,其在电化学氧化体系中的降解规律还未有详细的报道,镍作为一种变价金属是否会形成自催化效果也未知。
本研究旨在构建以混合金属氧化物(MMO)为阳极的电化学氧化体系处理Ni-EDTA,其核心是当电解质溶液中存在一定浓度的Cl−时,MMO阳极产生的活性氯与小分子配体络合态Ni(Ⅱ)反应生成次生活性物种,促进污染物的降解。以MMO为阳极,不锈钢为阴极建立的电化学体系用于Ni-EDTA破络研究,通过检测Ni-EDTA在不同电解质溶液和电流下的降解规律,探讨相关反应过程与机理,明确Ni2+在Ni-EDTA降解过程中起到的作用,为电化学技术用于镀镍废水处理提供理论借鉴。
-
乙二胺四乙酸二钠盐(EDTA-2Na)、六水合硫酸镍(NiSO4·6H2O)、氯化钠(NaCl)、硫酸钠(Na2SO4)、氢氧化钠(NaOH)、甲基苯基亚砜(PMSO)、甲基苯基砜(PMSO2)、甲醇(MeOH)、乙腈(CH3CN)、四丁基溴化铵(TBAB)和甲酸钠购自阿拉丁化学试剂有限公司,甲酸、乙酸和硫酸购自广州化学试剂厂,以上试剂均为分析纯。MMO(RuO2-IrO2/Ti)电极购于昌力特种金属有限公司,不锈钢板电极购于中润鸿发有限公司。
LC-20AT高效液相色谱仪(HPLC)、TOC-VCPH总有机碳分析仪,日本岛津公司;pH计,上海仪电科学仪器股份有限公司;GPD-3303S直流电源,台湾固纬电源有限公司;X射线衍射仪(XRD),荷兰帕纳科公司;X射线光电子能谱仪(XPS),赛默飞世尔科技公司。
-
Ni-EDTA电化学降解实验在圆柱形石英制电解槽中进行。电解槽由MMO阳极(材质为RuO2-IrO2镀层和Ti基底)和不锈钢板阴极组成,电极尺寸为30 mm × 30 mm × 2 mm,阴阳两极平行放置,电极之间距离为15 mm。降解实验在恒电流模式下运行,由直流电源控制电流大小。电化学降解实验水溶液总体积为250 mL,含0.68 mmol·L−1 Ni-EDTA,NaCl和Na2SO4作为电解质,浓度见后文实验条件描述。用H2SO4将溶液初始pH值调整为2.5。磁力搅拌器控制转速为350 r·min−1。反应时间180 min,每隔一定的时间间隔取样,液体样品通过0.45 μm水系滤膜过滤处理。所有实验至少进行3次,结果以平均值±标准差表示。
-
Ni-EDTA、PMSO和PMSO2的浓度采用HPLC测定,具体条件为:对于Ni-EDTA,C18反向色谱柱(Thermo, 5 μm×250 mm×4.6 mm),流动相采用甲酸缓冲液(含有5 mmol·L−1 甲酸钠、15 mmol·L−1甲酸和1 mmol·L−1四丁基溴化铵)和乙腈(V/V = 80:20)的混合液,流速为1.0 mL·min−1,进样量为10 μL,色谱柱温度30 ℃,紫外检测波长为210 nm;对于PMSO和PMSO2,流动相采用0.1%乙酸和乙腈(V/V = 75:25)的混合液,流速为1.0 mL·min−1,进样量为10 μL,色谱柱温度30 ℃,PMSO检测波长为230 nm,PMSO2检测波长为215 nm。
-
为了探讨电解质溶液对电化学氧化体系降解Ni-EDTA的影响,分别在溶液有氯离子体系(NaCl 50 mmol·L−1,Na2SO4 25 mmol·L−1)和溶液无氯离子体系(Na2SO4 50 mmol·L−1)两种情形下进行对照实验。如图1(a)所示,在两个体系中Ni-EDTA均有一定程度的降解。在电解质溶液无氯离子体系中,经180 min处理Ni-EDTA去除率为77.8%,这说明MMO阳极能通过直接或者间接氧化作用促使Ni-EDTA降解;在电解质溶液有氯离子体系中,Ni-EDTA降解率明显提升,180 min处理去除率达到96.3%.
这是因为MMO作为一种析氯性能良好的电催化阳极,在溶液存在氯离子情况下能够生成活性氯[22],活性氯的产生促进了Ni-EDTA的降解[23]。TOC去除也呈现出相似的规律,如图1(b)所示,无氯离子体系180 min处理TOC去除率为29.2%,有氯离子体系TOC去除率上升至44.9%,这说明活性氯的产生有利于污染物质的矿化[24-25]。由图1(c)动力学拟合发现,Ni-EDTA在不同体系中降解均符合一级反应动力学,无氯离子体系反应速率常数为0.0081 min−1,有氯离子体系反应速率常数有所增加。经过180 min运行后,阴极上没有观察到明显的Ni沉积(电极颜色无明显变化)。这是因为Ni2+还原成单质Ni的标准电极电位为−0.257 V[26],低于H+还原的标准电极电位0 V,在酸性条件下更容易进行析氢反应;此外Ni-EDTA矿化率不高,Ni2+以小分子有机络合态存在,增加了Ni2+还原难度[27]。
值得注意的是,在有氯离子体系中反应速率可以分为前后两个阶段,0—60 min内反应速率常数为0.0124 min−1,这个阶段反应速率的提升是因为活性氯的贡献;60—180 min反应速率常数上升至0.0216 min−1,这可能是因为随着反应的进行,生成了某种次生活性物种,促进Ni-EDTA的降解。已有研究证明了高级氧化处理Cu-EDTA过程中存在自催化降解的现象,如UV/Chlorine处理Cu-EDTA过程中,Cu(Ⅱ)-Complex可以通过分子内电荷转移生成Cu(I)-Complex,后者可以介导活性氯生成氯自由基从而加快Cu-EDTA的降解[18]。我们推测在电化学氧化体系中Ni也存在自催化效果,后文将进一步论述。电化学体系pH的变化是反映相关反应过程的重要指标,如图1(d)所示,在无氯离子体系中pH几乎维持不变,这是因为当转移的电荷量相同时,阴极发生的析氢反应(式1)所消耗的氢离子与阳极发生的析氧反应(式2)所蓄积的氢离子持平;在有氯离子体系中,pH逐渐升高,180 min后由2.5上升至7.24,这是因为阴极析氢反应不断消耗氢离子而阳极发生的析氯反应(式3)不会直接提供氢离子,只会通过Cl2溶于水产生氢离子(式4)。由于Cl2溶解性有限,且产生的HClO是弱酸不能完全电离,故反应过程pH不断上升。
进一步发现,在溶液中含有氯离子的电化学体系中,180 min反应后溶液中悬浮着大量灰黑色的固体。将沉淀离心分离干燥后进行物理表征,结果如图2所示。
图2(a)为灰黑色固体的XRD图谱,在2θ为18.8°、34.0°、37.3°和60.6°位置的特征衍射峰对应于Ni3O2(OH)4(JCPDS 06-0144)的(003)、(100)(006)和(110)晶面。Ni3O2(OH)4中Ni的价态为+2和+3混合价态,含有较多的三价Ni。图2(b)为灰黑色固体的Ni 2p XPS图谱,经过XPeaks软件拟合后得到两组信号,855.3 eV处信号峰对应于二价Ni,856.9 eV处信号峰对应于三价Ni[28],进一步证实了灰黑色固体中Ni为+2和+3混合价态。这说明Ni-EDTA在电化学体系降解过程中二价Ni会被活性物种氧化至高价态Ni。在反应过程中,前90 min溶液一直是澄清透明的,90 min后溶液中才逐渐出现灰黑色固体悬浮物,这可能是因为反应过程中溶液pH逐渐上升,含高价态Ni的物质在近中性pH条件下才会以固体析出[29]。实际上,含高价态Ni的物种已被证实能够通过氢和氧原子转移途径氧化多种有机物[30-32];并且已有报道称Ni(Ⅱ)络合物与NaClO反应能够生成高价Ni物种[33]。基于此推测,在溶液有氯离子的电化学体系中,Ni-EDTA降解生成的小分子配体络合态Ni(Ⅱ)会被氧化至高价态Ni,高价态Ni具备强氧化能力,能够通过抽氢反应实现EDTA的逐步脱羧,在Ni-EDTA降解动力学上呈现出反应速率常数增大的现象。至于反应90 min之后反应速率才增大,推测高价态Ni物种的生成需要一定浓度的活性氯;其次,活性氯无法直接氧化EDTA络合态Ni(Ⅱ),只有当Ni-EDTA降解成小分子配体络合态Ni(Ⅱ),活性氯才能氧化其至高价态。
为了验证含氯电化学体系中生成的固体确实含有高价Ni,选用PMSO作为指示物。高价金属能够通过氧转移过程将PMSO氧化至PMSO2[34-36],如图3所示,将电化学体系降解Ni-EDTA生成的灰黑色固体分离出来与PMSO混合起来反应,能够检测到有PMSO2的生成,但是损失的PMSO并没有完全转化为PMSO2,PMSO2转化率大约为60%,说明高价Ni固体还能通过除了氧转移外的其他路径氧化PMSO。PMSO2的生成进一步证明了含氯电化学体系降解Ni-EDTA会产生高价Ni物种。
-
电流是电化学反应的驱动力,电流大小是影响电化学反应速率的关键因素。图4(a)为电流密度大小对电化学降解Ni-EDTA的影响,经过180 min反应,Ni-EDTA的降解效率随着电流增大而增大;当电流密度从11.1 mA·cm−2增加到22.2 mA·cm−2时,降解效率有明显的提升,Ni-EDTA去除率从68.5%上升至94.3%,继续增大电流密度至33.3 mA·cm−2时,降解效率几乎没有改变。由上文可知,在无氯离子电化学体系反应180 min后,Ni-EDTA的去除率为77.8%,这说明施加在MMO上的电势能够直接氧化Ni-EDTA,或者MMO表面上生成的活性物种能够氧化Ni-EDTA。Ni-EDTA的降解效率在电流密度11.1 mA·cm−2至22.2 mA·cm−2范围内显著提升原因有两点,一是电流越大阳极氧化Ni-EDTA的能力越强,二是电流越大,MMO产生的活性氯越多。图4(a)内插图不同电流密度下Ni-EDTA降解动力学拟合也反应了相同的规律(拟合结果见表1),电流密度从11.1 mA·cm−2增加到22.2 mA·cm−2时,反应动力学常数明显增大,继续增大电流密度至33.3 mA·cm−2时增长幅度较小。其中,当电流密度为11.1 mA·cm−2时,kobs = 0.0064 min−1,电流密度从16.7 mA·cm−2开始,反应速率明显可以分为前后两个阶段,60 min至180 min阶段的kobs明显要大于前60 min,这符合在前面的推测,反应过程中会生成高价Ni物种促进Ni-EDTA的降解。电流密度为11.1 mA·cm−2时没有出现自催化现象,推测是因为活性氯的浓度较低,也可能是该条件下体系氧化能力较低没有生成小分子配体络合态Ni(Ⅱ)。之前研究发现,氧化剂如活性氯和过二硫酸盐无法氧化EDTA络合态Ni(Ⅱ),但可以直接氧化Ni2+至高价Ni物种,这是因为大分子配体EDTA的空间位阻作用导致氧化剂无法与Ni(Ⅱ)接触[37]。而小分子配体空间位阻作用较弱,活性物质更容易与Ni(Ⅱ)接触从而将其氧化至高价Ni物种。
氯离子浓度对活性氯的产生有影响,也是电化学反应中的关键因素。图4(b)为不同氯离子浓度下电化学降解Ni-EDTA的情况,溶液中氯离子的出现明显加快了Ni-EDTA的降解,氯离子浓度从0增加到25 mmol·L−1时,反应180 min后Ni-EDTA降解效率从75.9%上升至92.7%。当氯离子在25 mmol·L−1至100 mmol·L−1范围内变化时,Ni-EDTA降解效率变化不大。图4(b)内插图降解动力学拟合更能直观看出氯离子浓度对Ni-EDTA降解的影响(拟合结果见表2),当溶液中存在氯离子时,Ni-EDTA反应速率便可分为两个阶段,后一阶段中活性氯氧化小分子配体络合态Ni(Ⅱ),生成高价态Ni物种促进Ni-EDTA降解。
-
为了验证Ni-EDTA降解过程中小分子络合态Ni(Ⅱ)是否起到催化降解的作用,设计了一组实验,研究了相同类型的EDTA络合物在电化学体系中的降解情况。如图5(a)所示,Fe(Ⅲ)-EDTA在含氯离子的溶液中降解效率略高于无氯离子体系,图5(b)Fe(Ⅲ)-EDTA降解动力学拟合更能直观的说明氯离子的存在加快了Fe(Ⅲ)-EDTA降解。值得注意的是,在含氯离子体系中,Fe(Ⅲ)-EDTA反应动力学常数不像Ni-EDTA那样分前后两个阶段,这说明没有其他活性物种生成的话反应速率是不会发生变化的。为了进一步说明小分子配体络合态Ni(Ⅱ)在含氯电化学体系中能够促进重金属络合物的降解,将不同浓度的Ni2+(以NiSO4形式,避免引入小分子有机配体竞争活性物质)加入到电化学体系中以观察其对Fe(Ⅲ)-EDTA降解的影响。Ni2+在水中是以水合离子[Ni(H2O)6]2+形式存在,其配体是H2O,相对于EDTA、NTA等大分子有机配体来说H2O属于小分子配体,Ni2+可以看作是一种小分子配体络合态Ni。图5(c)所示,Ni2+的引入显著加快了Fe(Ⅲ)-EDTA的去除,当加入的Ni2+分别为20、40、60 mg·L−1时,Fe(Ⅲ)-EDTA去除率从30.7%分别上升至62.2%、67.8%和73.7%。图5(d)Fe(Ⅲ)-EDTA降解动力学拟合可以发现(拟合结果见表3),Ni2+加入到反应体系之后,降解动力学常数便可分为前后两个阶段,0至30 min降解动力学常数几乎没有差异,这是因为此时产生的活性氯量较少不足氧化Ni2+生活高价Ni物种,30 min之后,当加入的Ni2+为20、40、60 mg·L−1时,Fe(Ⅲ)-EDTA降解动力学常数分别为0.0059 、0.0069 、0.0081 min−1,远高于没有Ni2+体系的0.0028 min−1。
-
综合上述实验结果,可以推测电化学体系降解Ni-EDTA的原理(如图6所示)。当溶液中没有氯离子时,MMO阳极可产生吸附态羟基自由基,Ni-EDTA在阳极可通过直接氧化或者羟基自由基介导途径分解。当溶液中含有氯离子时,MMO能够将氯离子氧化成活性氯,活性氯一方面能够作用于Ni-EDTA的降解,生成小分子配体络合态Ni(Ⅱ)[38](如Ni-ED2A,Ni-IMDA和Ni-G,图中用NiII complex表示),随着电解时间延长进一步矿化成小分子;另一方面活性氯还能够氧化小分子配体络合态Ni(Ⅱ)生成高价Ni物种,后者能够作用于Ni-EDTA的降解,形成自催化降解的现象。随着电化学反应的进行,溶液pH逐渐升高,+2和+3混合价态Ni物种会以灰黑色固体形式析出。
-
电化学氧化体系能够有效降解Ni-EDTA。当电流密度为27.8 mA·cm−2时,在电解质溶液有氯和无氯情况下,电化学体系处理180 min后Ni-EDTA的去除率分别达到96.3%和77.8%;动力学拟合发现Ni-EDTA降解符合一级反应动力学,其中在有氯情况下会出现自催化降解现象,降解速率常数在0到90 min阶段为0.0124 min−1,90 min到180 min上升至0.0216 min−1。对含氯体系中出现的固体进行XRD、XPS和PMSO探针实验,确定反应过程生成了高价Ni物种。通过研究电化学体系中有无Ni2+对Fe(Ⅲ)-EDTA降解的影响,发现高价Ni物种能够促进金属有机络合物的降解,从而提高反应速率。Ni-EDTA在电化学氧化体系中自催化降解现象的揭示,为金属有机络合物去除机制提供了新见解。
Ni-EDTA在电化学氧化体系中的自催化降解
Autocatalytic degradation of Ni-EDTA in electrochemical oxidation systems
-
摘要: 氧化破络是去除络合态重金属的关键步骤,本文构建了以商用混合金属氧化物(MMO)为阳极的电化学氧化体系用于降解Ni-EDTA。研究发现,当电流密度为27.8 mA·cm−2时,在溶液有氯和无氯情况下,电化学体系处理180 min后Ni-EDTA的去除率分别达到96.3%和77.8%;动力学拟合发现Ni-EDTA降解符合一级反应动力学,其中在有氯情况下随着反应进行降解速率呈现出加快的现象,降解速率常数在0到90 min阶段为0.0124 min−1,90 min到180 min上升至0.0216 min−1。对含氯体系中出现的固体进行X射线衍射(XRD)、X射线光电子能谱(XPS)和苯基甲基亚砜(PMSO)探针实验,确定反应过程生成了高价Ni物种。通过研究电化学体系中有无Ni2+对Fe(Ⅲ)-EDTA降解的影响,发现高价Ni物种能够促进金属有机络合物的降解,从而提高反应速率。Ni-EDTA在电化学氧化体系中自催化降解现象的揭示,为金属有机络合物去除机制提供了新见解。Abstract: Decomplexation by oxidation is a prerequisite step to remove complexed heavy metals from the aqueous phase. Herein, this study established an electrochemical oxidation system with a commercial mixed metal oxide (MMO) anode for degrading Ni-EDTA. When the current density is 27.8 mA·cm−2, the removal efficiencies of Ni-EDTA were 96.3% and 77.8% after 180-min treatment in the presence and absence of chloride, respectively. The abatement of Ni-EDTA obeyed the first-order kinetic law, and the rate constants increased with the operation time from 0.0124 min−1 to 0.0216 min−1 when chlorine was available in the solution. The characterizations of X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) and the methyl phenyl sulfoxide (PMSO) probe tests were conducted on the solids obtained from the chloride-contained system, suggesting the formation of high-valence Ni species. By investigating the effect of Ni2+ on the degradation of Fe(Ⅲ)-EDTA in the electrochemical system, it was revealed that high-valence Ni species can promote the destruction of metal organic complexes, and thus increase the reaction rate. The autocatalytic degradation of Ni-EDTA in the electrochemical oxidation system provides a new insight into the removal mechanism of metal-organic complexes.
-
Key words:
- electro-oxidation /
- active chlorine /
- Ni-EDTA treatment /
- autocatalytic degradation /
- high-valance metal
-
砷作为一种剧毒的类金属,其毒性与砷的价态有很强的相关性:As(Ⅲ) > As(Ⅴ). 研究报告表明,天然水体中的砷浓度范围很广,从小于0.5 μg·L−1到大于5000 μg·L−1,地热水中的砷浓度甚至可达50 mg·L−1,在地下水缺氧环境中,As(Ⅲ)是主要形态[1]. 全球约有9400万人至2.2亿人可能接触到高砷浓度的地下水,其中绝大多数(94%)在亚洲[2]. 美国和中国都参照世界卫生组织规定要求饮用水中砷的浓度不得高于10 μg·L−1. As(Ⅲ)对混凝剂和吸附剂的亲和力较弱,As(Ⅲ)相较于As(Ⅴ)更难去除,将As(Ⅲ)氧化为As(Ⅴ)是提高总砷去除的关键步骤[3]. 通过氯和臭氧等化学氧化手段可以将As(Ⅲ)氧化为As(Ⅴ),但很可能产生有毒副产物;通过UV/Fe[4]、UV/TiO2[5]、UV/碘化物[6]、UV/氢氧化铁[7]等方法可以将As(Ⅲ)氧化为As(Ⅴ),但引入了金属离子,不利于环境安全. H2O2在pH > 9.0时可以氧化As(Ⅲ)[8],且H2O2作为一种温和无害的氧化剂,具有绿色氧化过程,不会产生有害的副产物,但由于H2O2运输困难、不易保存,因此H2O2的原位产生技术受到了广泛关注.
MB+[9]是一种知名的噻嗪光敏剂,被大量用作染色剂、光催化剂、抗氧化剂、防腐剂和单态氧敏剂等使用,能够在光照下产生单线态氧(1O2). 除MB+外,核黄素[10]、血卟啉衍生物[11]、联吡啶钌[12]和亚甲基紫[13]等光敏剂在光照下也能够产生和MB+类似的效应. 由于MB+等噻嗪染料价格低廉,光照下对水体中污染物具有较好的氧化效果[14],受到国内外学者的广泛研究. 乙二胺四乙酸(EDTA)[15]、H2A[16]、亚硫酸盐[17]等还原剂存在下的MB+的光化学现象已有诸多报道. 当不存在还原剂时,MB+通过光敏化过程(反应1 −3)产生1O2[15]. 但还原形式的无色亚甲基蓝(LMB)只有在还原剂存在时通过反应(4)和(5)才产生,其中Rred和Rox分别表示还原剂的还原形式和氧化形式,反应(6)为反应(4)和反应(5)的总式. 此外,还原性的染料自由基(半还原性MB+·)在没有O2存在时是稳定的,在O2存在时,MB+·会失去其未成对电子,产生过氧化氢自由基(HO2·)(反应(7))[18]. 抗坏血酸(H2A)是常见的抗氧化剂,具有较强的还原性,被广泛应用于有机污染物的降解[19-20]. 已有研究表明H2A/MB+在可见光激发下能够产生抗坏血酸自由基(A·−),进而还原分子氧产生H2O2(反应(8)和反应(9))[21]. 因此,在某些还原剂存在时,MB+的光解可能会诱导分子氧的活化. MB+在许多行业和化学实验室中得到了广泛的应用(如著名的“蓝瓶子”实验). MB+排放前需要对其进行处理,将废弃MB+用来处理其它污染物不失为更好的应用.
MB++hv→1MB+∗ (1) 1MB+∗→3MB+∗ (2) 3MB+∗+O2→MB++1O2 (3) 3MB+∗+Rred→MB+⋅+R⋅ox (4) 2MB+⋅→LMB+MB+ (5) 23MB+∗+2Rred→LMB+MB++2R⋅ox (6) MB+⋅+O2→MB++O⋅−2(HO⋅2) (7) 3MB+∗+HA−→MB+⋅+A⋅− (8) A⋅−+O⋅−2(HO⋅2)→DHA+H2O2 (9) 本研究拟探索可见光/MB+/H2A体系氧化水中As(Ⅲ)的可行性与内在机理,考察了光照、pH、H2A浓度、MB+浓度、As(Ⅲ)初始浓度及水中常见阴离子(Cl−、HCO3−、NO3−)和有机质富里酸(FA)对As(Ⅲ)氧化的影响. 通过活性物种识别(自由基清除实验)和溶液光谱变化确定了可见光/MB+/H2A体系氧化As(Ⅲ)的主要活性物种及其生成机理.
1. 实验部分 (Experimental section)
1.1 实验材料
实验中所用的盐酸、硼氢化钾、氢氧化钾、十水硼酸钠、氢氧化钠、叔丁醇(TBA)、碳酸氢钠、抗坏血酸、硝酸钠、氯化钾、磷酸等均购于国药集团化学试剂公司,亚砷酸钠购于西亚试剂有限公司,砷酸氢二钠七水化合物购于阿法埃莎化学有限公司,富里酸购于阿拉丁生化科技股份有限公司,过氧化氢酶(CAT),超氧化物歧化酶(SOD)购于源叶生物科技有限公司,实验中用水均为去离子水,所用药品均为分析纯.
1.2 仪器
ALB−124电子天平秤(赛多利斯仪器公司),R6200氢化物发生−原子荧光光度计(北京博晖创新光电技术股份有限公司),LC−10 ADVP液相泵(日本岛津公司),超纯水机(四川优普科技有限公司),pH 320−S精密数字酸度计(梅特勒−托利多公司),Bante 980溶解氧仪(上海班特仪器公司),801磁力搅拌装置(上海三信公司),UV-1600紫外可见分光光度计(日本岛津公司).
1.3 实验方法与分析方法
在圆柱形开放式夹套反应器(内直径100 mm)中配制500 mL含有5 μmol·L−1 As(Ⅲ)、0.1 mg·L−1 MB+、100 μmol·L−1 H2A、10 mmol·L−1不同类型缓冲液的反应液(醋酸盐缓冲液是pH=4.0、磷酸盐缓冲液是pH=6.0—8.0、硼酸盐缓冲液是pH=9.0—9.5),通过连接外部水循环恒温系统将反应温度控制在(25±1)℃,将LED灯源放置于反应器四周,通过磁力搅拌装置混匀溶液. 打开LED灯并开始计时,分别于0、10、20、30、45、60、90、120 min取4.5 mL反应液,加入0.5 mL 1:1的盐酸以猝灭反应,再进行分析.
采用液相−氢化物发生−原子荧光光度法(LC−HG−AFS)测定As(Ⅲ)和As(Ⅴ)形态的量[22],流动相为磷酸盐(45 mmol·L−1, pH=5.6)和超纯水,流速为1.3 mL·min−1,色谱柱为离子色谱柱(PRP−X100,hamilton,Switzerland),进样体积为100 μL.
本实验所用LED灯是从网上采购. 其最大发射波长为630 nm (图1). 通过紫外分光光度计在200−800 nm范围内分别扫描MB+、劳氏紫(Thionine)、天青B(Azure B)的光谱. MB+、劳氏紫和天青B的可见光区域最大吸收峰分别为660、600、600 nm(图1).
 图 1 LED灯发射光谱及MB+、劳氏紫、天青的3种噻嗪染料染料溶液的紫外可见吸收光谱Figure 1. Emission spectrum of LED lamp and UV-Vis absorption spectra of the solutions of three thiazine dyes (MB+, Thionine and Azure B).
图 1 LED灯发射光谱及MB+、劳氏紫、天青的3种噻嗪染料染料溶液的紫外可见吸收光谱Figure 1. Emission spectrum of LED lamp and UV-Vis absorption spectra of the solutions of three thiazine dyes (MB+, Thionine and Azure B).2. 结果与讨论(Results and discussion)
2.1 pH、H2A浓度、MB+和砷浓度的影响
图2a为As(Ⅲ)在MB+/H2A体系和可见光/MB+/H2A体系中不同pH条件下的氧化. 当pH < 6.0时,As(Ⅲ)在MB+/H2A体系和可见光/MB+/H2A体系中均未发生氧化;随着pH从8.0增加至9.5,MB+/H2A体系和可见光/MB+/H2A体系中As(Ⅲ)的氧化速率分别从0、0.39 μmol·L−1·h−1增加至1.34、3.73 μmol·L−1·h−1. 由此可见,在MB+/H2A体系中加入可见光后,As(Ⅲ)的氧化速率和氧化率均明显提高.
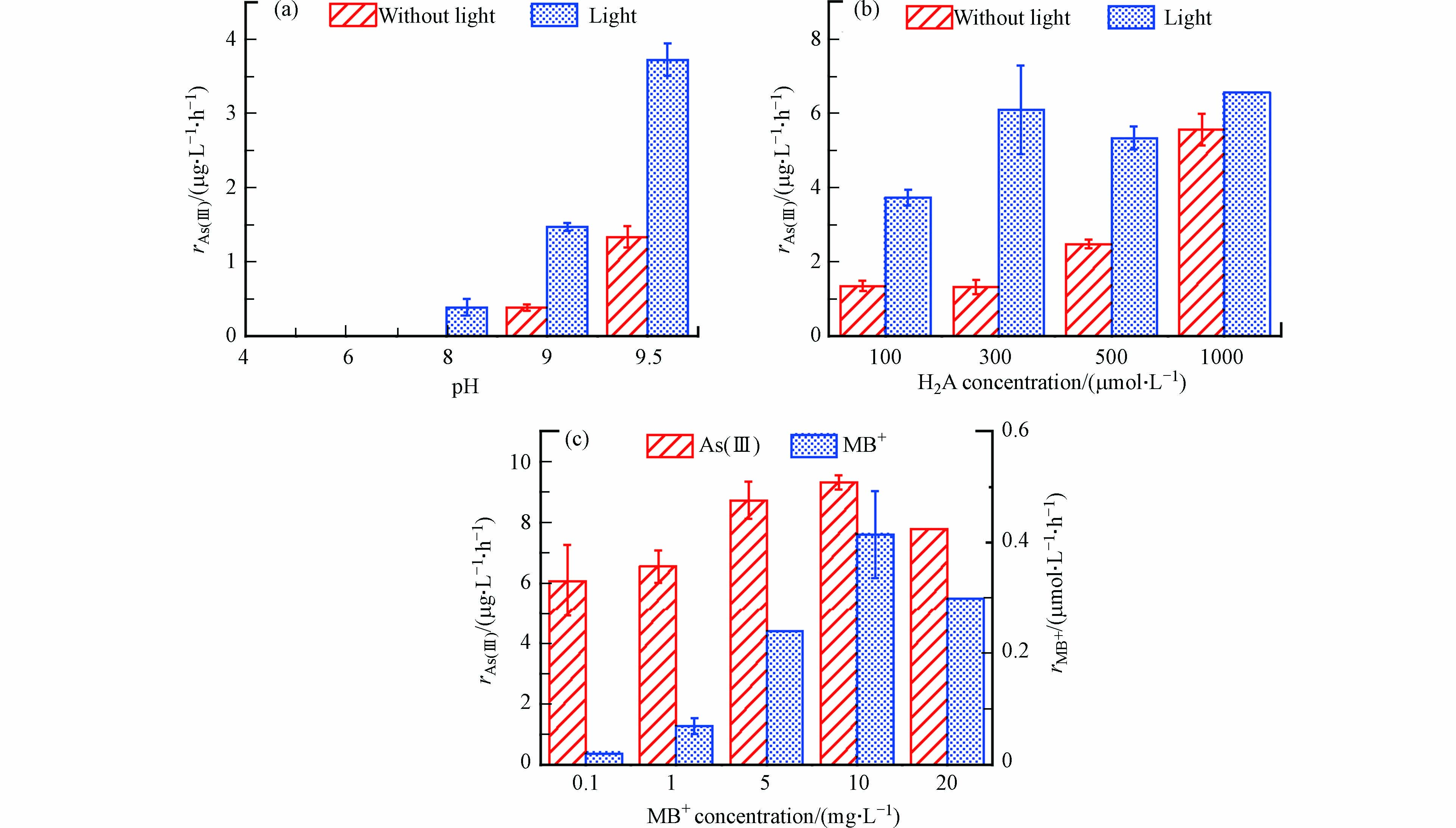 图 2 As(Ⅲ) 氧化的条件实验Figure 2. Effects of solution pH(a), concentration of H2A(b) and MB+(c)on the oxidation of As(Ⅲ).(a)溶液pH值,(b)H2A浓度,(c)MB+浓度
图 2 As(Ⅲ) 氧化的条件实验Figure 2. Effects of solution pH(a), concentration of H2A(b) and MB+(c)on the oxidation of As(Ⅲ).(a)溶液pH值,(b)H2A浓度,(c)MB+浓度pH会影响抗坏血酸和As(Ⅲ)的形态分布与氧化还原电位. 抗坏血酸的两个酸式解离常数分别为4.17、11.57. pH=6—10时,抗坏血酸主要以HA−形态存在,随着pH增加,A2−形态的量会进一步增加,A2−相较于HA−更容易与分子氧反应产生H2O2[8]. As(Ⅲ)的3个酸式解离常数分别为9.22、12.1、13.4,pH > 9.0时As(Ⅲ)主要以解离态存在,解离态的As(Ⅲ)在碱性条件下更容易被H2O2氧化为As(Ⅴ). LMB的3个酸式解离常数分别为1.7、4.5、5.9,因此LMB在溶液中存在四种形态(H3LMB2+、H2LMB+、HLMB、LMB−)[23]. LMB被溶解氧(DO)氧化产生MB+在中性和碱性条件下更快. 考虑到As(Ⅲ)的氧化速率和氧化率,后续反应在pH=9.5下进行.
图2b为H2A浓度对MB+/H2A体系和可见光/MB+/H2A体系中的As(Ⅲ)氧化的影响. As(Ⅲ)在MB+/H2A体系中的氧化速率随着H2A浓度增加而增加,而As(Ⅲ)在可见光/MB+/H2A体系中的氧化速率则随着H2A浓度增加先增加后趋于稳定. H2A投加量的增加一方面会促进活性物种H2O2的产生,另外一方面H2A作为还原剂,过量的H2A则会竞争消耗产生的活性物种. 在Fe(Ⅲ)/H2O2/H2A[24]和Fe(Ⅲ)/PS/H2A[25]体系中,也存在过量的抗坏血酸抑制底物降解的现象. 综上,H2A最佳投加量为300 μmol·L−1.
图2c为MB+浓度对可见光/MB+/H2A体系中的As(Ⅲ)和MB+氧化的影响. 随着MB+浓度从0.1 mg·L−1增加至5 mg·L−1,As(Ⅲ)的氧化速率提高,继续增加MB+浓度至20 mg·L−1,As(Ⅲ)的氧化速率变化不明显. 随着MB+浓度从0.1 mg·L−1增加至10 mg·L−1,MB+的氧化速率从0.02 μmol·L−1·h−1增加至0.42 μmol·L−1·h−1,继续增加MB+浓度至20 mg·L−1,MB+的氧化速率降至0.3 μmol·L−1·h−1. 这是因为随着MB+浓度的增加,MB+吸收光能增多,使得1O2浓度增加,从而有利于反应进行;但MB+达到一定浓度,足以充分使体系中氧分子转变为1O2[26],继续提高MB+浓度并不会对As(Ⅲ)和MB+的氧化产生影响. 综上,MB+最佳投加量为5 mg·L−1.
图3a和图3b分别给出了不同体系中As(Ⅲ)和MB+随时间变化结果. 在可见光/MB+/H2A体系中As(Ⅲ)和MB+的氧化率分别为90%、10%. 图3a显示As(Ⅲ)的氧化率在MB+/H2A体系和可见光/H2A体系中相同,为50%,说明pH 9.5时H2A可以活化分子氧产生H2O2氧化As(Ⅲ). 图3b显示,MB+在MB+/H2A体系中并未发生氧化,说明H2O2并不能氧化MB+. 可见光/MB+体系可以产生1O2,As(Ⅲ)和MB+在可见光/MB+体中氧化率分别为10%和15%,说明1O2对As(Ⅲ)的氧化能力很弱. 而在可见光/MB+/H2A体系中,由于溶液中的H2A会竞争消耗1O2,导致此条件下MB+氧化率低于可见光/MB+体系.
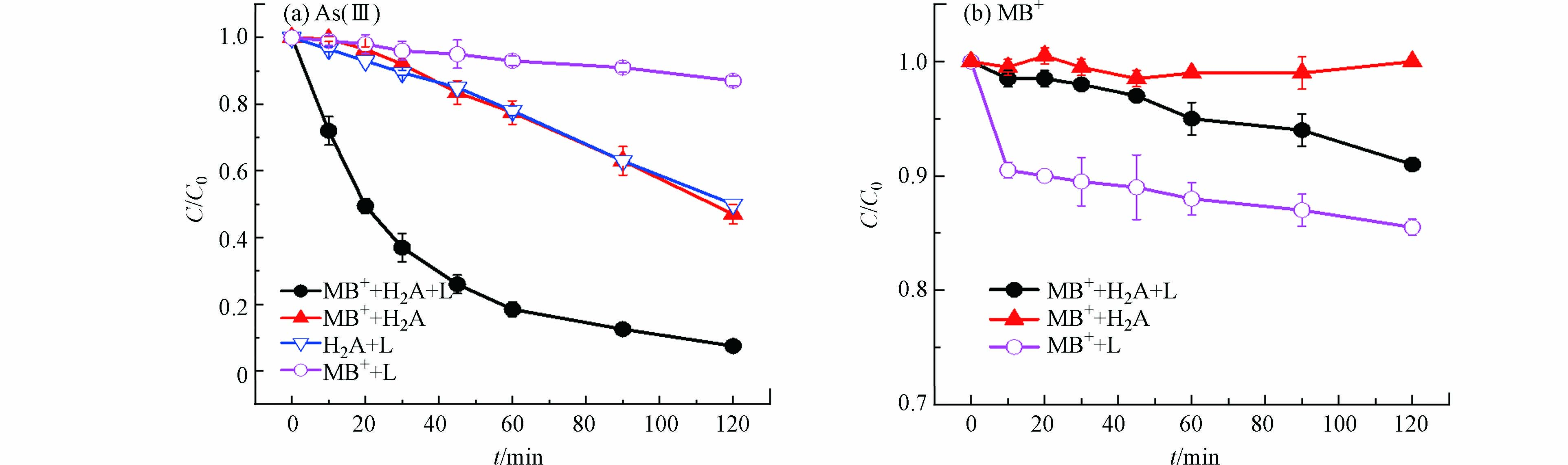 图 3 (a)As(Ⅲ)的氧化与(b)MB+的去除动力学曲线Figure 3. Kinetic curves of As(Ⅲ) oxidation(a) and MB+ removal(b)
图 3 (a)As(Ⅲ)的氧化与(b)MB+的去除动力学曲线Figure 3. Kinetic curves of As(Ⅲ) oxidation(a) and MB+ removal(b)图4a为As(Ⅲ)初始浓度对可见光/MB+/H2A体系氧化As(Ⅲ)的影响. 当As(Ⅲ)浓度从5 μmol·L−1上升到50 μmol·L−1时,As(Ⅲ)的氧化率从90%降低至80%(相应的As(Ⅲ)氧化量分别为2.25 μmol和20 μmol),As(Ⅲ)的氧化率没有显著的改变,但As(Ⅲ)的氧化量显著增加. As(Ⅲ)的初始氧化速率和As(Ⅲ)的浓度成正比(图4b). 这说明可见光/MB+/H2A体系可以用来处理高浓度含砷废水.
 图 4 可见光/MB+/H2A体系中,As(Ⅲ)初始浓度对(a)As(Ⅲ)的氧化与(b)As(Ⅲ)的氧化速率的影响Figure 4. Effect of As(Ⅲ) concentration on oxidation of As(Ⅲ)(a) and oxidation rate of As(Ⅲ)(b) in visible light/ascorbic acid/methylene blue system
图 4 可见光/MB+/H2A体系中,As(Ⅲ)初始浓度对(a)As(Ⅲ)的氧化与(b)As(Ⅲ)的氧化速率的影响Figure 4. Effect of As(Ⅲ) concentration on oxidation of As(Ⅲ)(a) and oxidation rate of As(Ⅲ)(b) in visible light/ascorbic acid/methylene blue system2.2 机理探究与氧气浓度的影响
As(Ⅲ)在可见光/MB+/H2A体系中的氧化可以从MB+的激发、激活的MB+(MB+*)与H2A相互作用以及反应活性物种的产生三个角度来进行说明. MB+是一种最大吸收峰为660 nm的蓝色染料,而LMB为无色且在265 nm处具有最大吸收峰[16]. 当预先用氮气曝气半小时以去除溶液中的DO,可见光照射,可以观察到MB+在660 nm处的最大吸收峰迅速消失(0—5 min),而在265 nm处出现LMB的最大吸收峰(0—120 min)(图5a). 在有氧搅拌条件下可见光照射后,反应液中DO迅速减少,溶液中部分LMB无法氧化,出现LMB的吸收峰(0—10 min),此后溶液复氧(10—120 min), LMB最大吸收峰消失(图5b). 由于实际反应是在空气氛围下进行,与N2氛围相比,MB+下降的速率较慢. 这是因为在O2存在时,MB+首先被H2A还原成LMB,然后LMB被O2氧化成MB+,这个过程非常迅速,所以在O2存在下无法观察到MB+的循环过程. 此外,当可见光照射脱氧的MB+/H2A反应溶液重新暴露于空气中,观察到无色溶液迅速变为蓝色(图6). 这也可以解释上述现象,即LMB很容易被O2氧化[17].
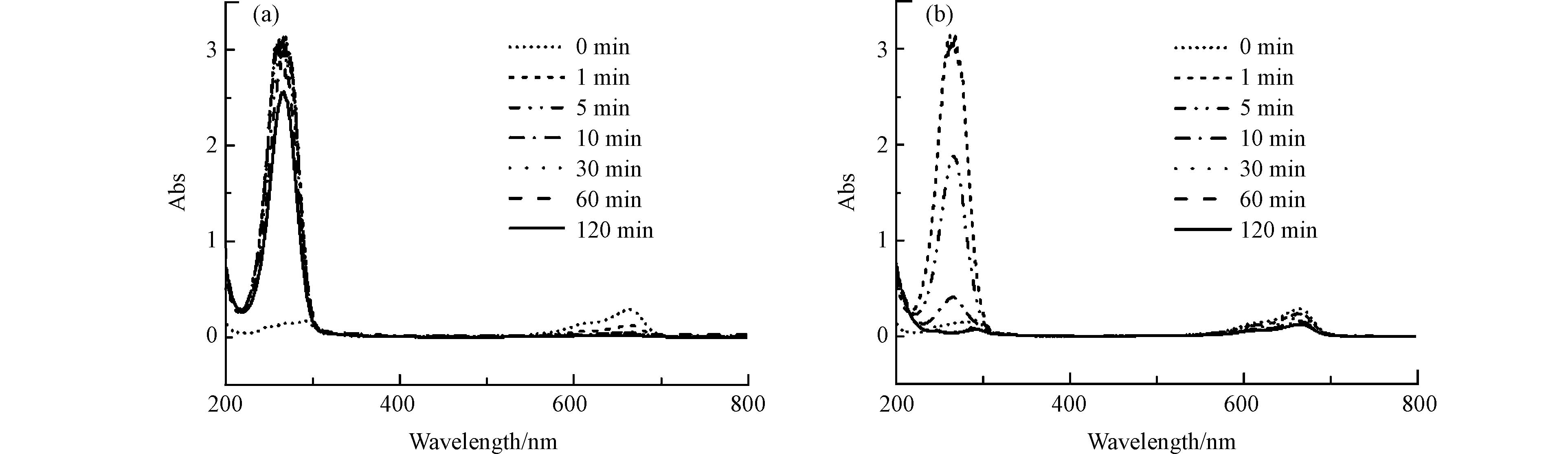 图 5 (a) 氮气条件下和(b)有氧搅拌条件下可见光/MB+/H2A体系中溶液光谱变化Figure 5. Spectral variation of the solution in a visible light/ascorbic acid/methylene blue system under nitrogen conditions(a) and stirring conditions(b)
图 5 (a) 氮气条件下和(b)有氧搅拌条件下可见光/MB+/H2A体系中溶液光谱变化Figure 5. Spectral variation of the solution in a visible light/ascorbic acid/methylene blue system under nitrogen conditions(a) and stirring conditions(b) 图 6 可见光/N2和遮光/O2条件下可见光/MB+/H2A体系中MB+吸光度变化Figure 6. In the visible light/ascorbic acid/methylene blue system under under visible light/N2 and shading/O2 conditions, the absorbance of methylene blue changes.
图 6 可见光/N2和遮光/O2条件下可见光/MB+/H2A体系中MB+吸光度变化Figure 6. In the visible light/ascorbic acid/methylene blue system under under visible light/N2 and shading/O2 conditions, the absorbance of methylene blue changes.为了进一步理解可见光/MB+/H2A体系中的As(Ⅲ)的氧化机理,利用自由基抑制实验来捕获产生的活性物种. SOD和TBA分别作为HO2·和HO·猝灭剂,SOD与HO2·的反应速率常数为1.6×109 L·mol−1·s−1,TBA与HO·的反应速率常数为7.6×108 L·mol−1·s−1. 分别投加100 U·mL−1 SOD和2 mmol·L−1 TBA(图7),均未对As(Ⅲ)的氧化产生明显影响,表明O2·−(HO2·)或HO·不是氧化As(Ⅲ)的主要活性物种. CAT可以快速分解H2O2,因而被用于清除反应中产生的H2O2. 分别投加100 U·mL−1和250 U·mL−1的CAT,反应前30 min,As(Ⅲ)的氧化率由65%下降到25%,此后并未观察到As(Ⅲ)进一步氧化,说明反应30 min后As(Ⅲ)的氧化被完全抑制. 上述结果说明H2O2是氧化As(Ⅲ)的主要活性物种. 如图8所示,pH=9.5时,单独的H2O2能够快速氧化As(Ⅲ).
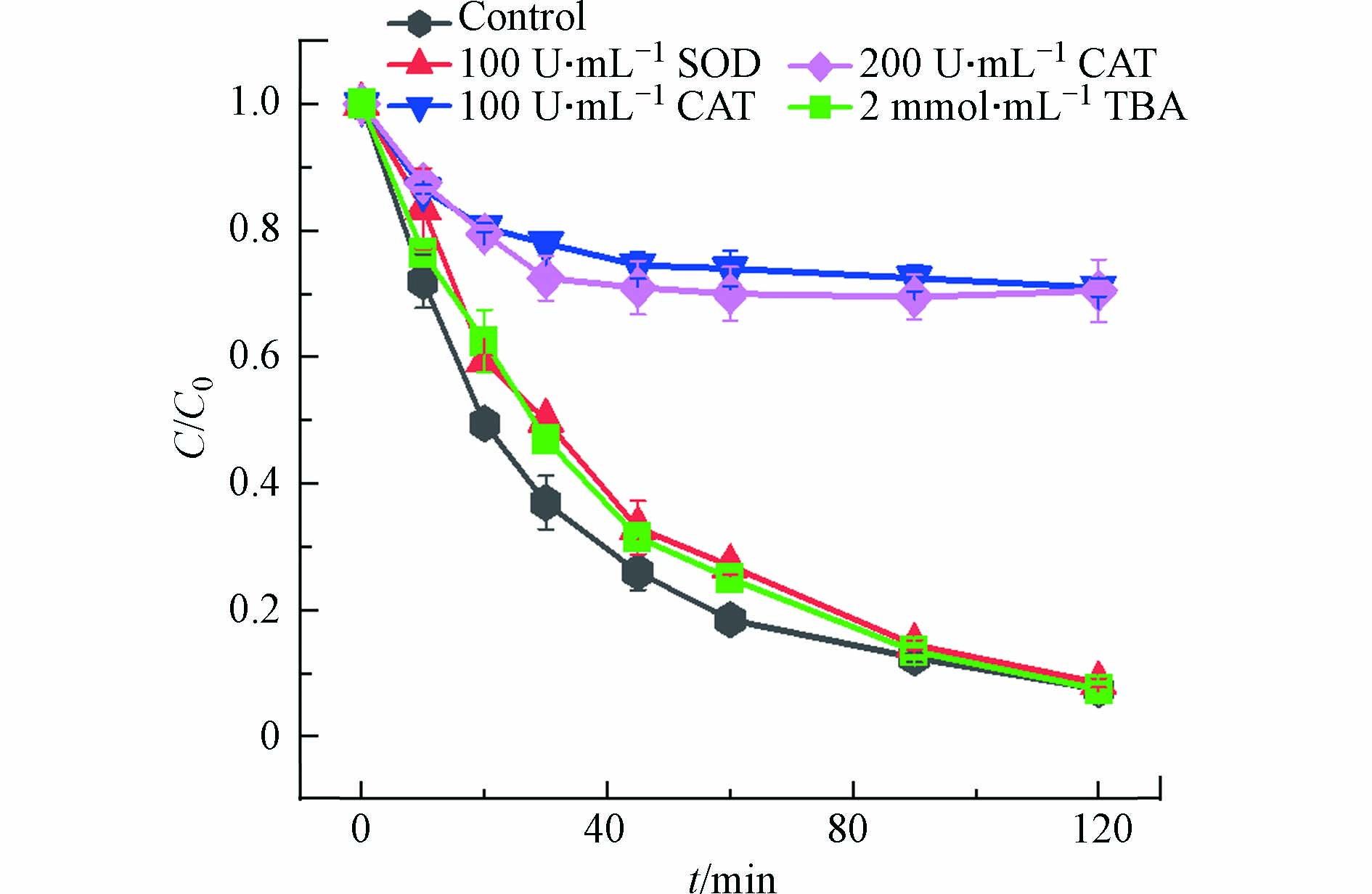 图 7 可见光/MB+/H2A 体系中自由基抑制剂对As(Ⅲ)氧化的影响Figure 7. Effect of free radical inhibitors on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue system
图 7 可见光/MB+/H2A 体系中自由基抑制剂对As(Ⅲ)氧化的影响Figure 7. Effect of free radical inhibitors on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue system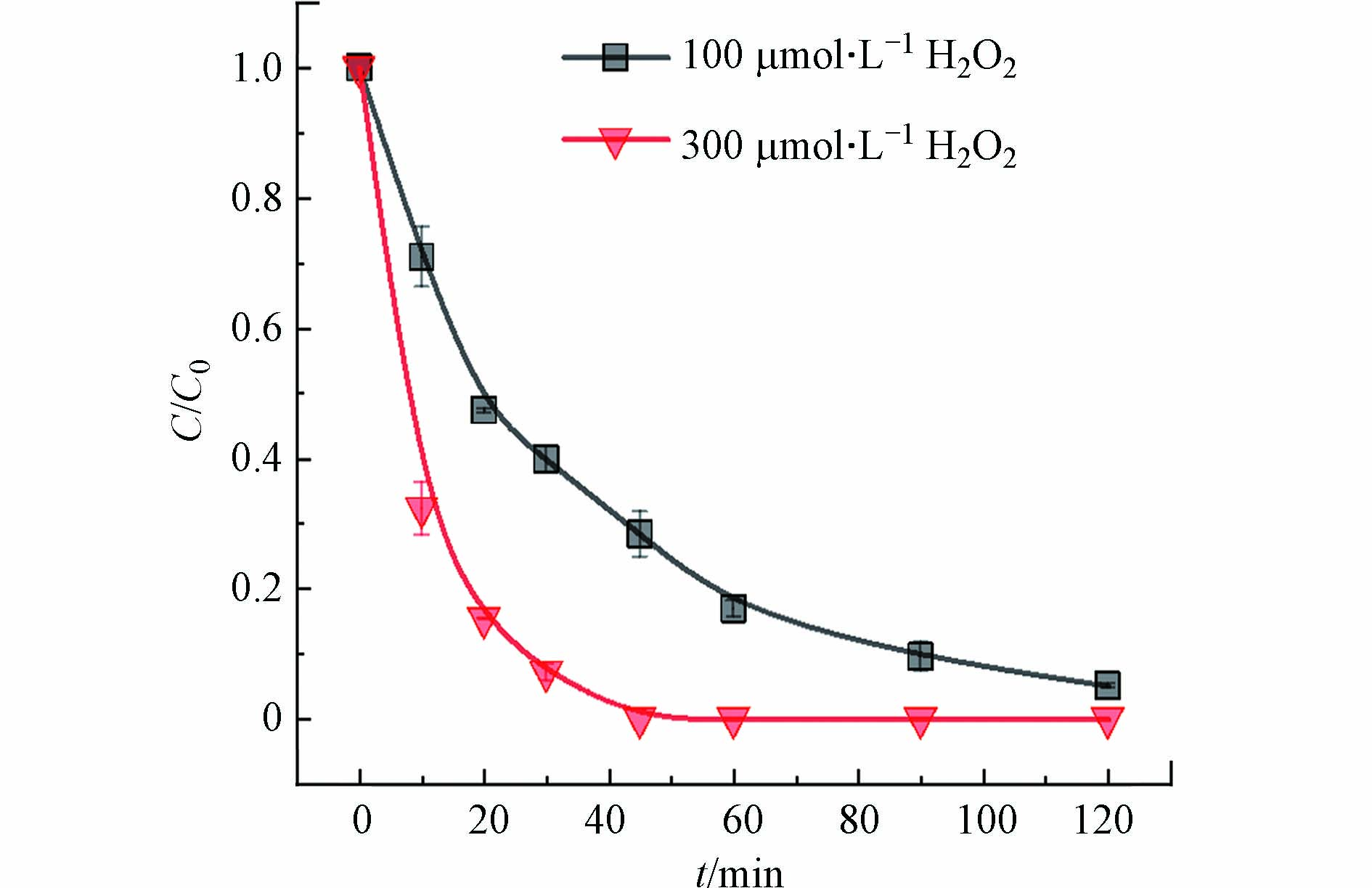 图 8 pH 9.5,H2O2 对As(Ⅲ)氧化的影响Figure 8. Effect of H2O2 on the oxidation of As(Ⅲ) at pH 9.5
图 8 pH 9.5,H2O2 对As(Ⅲ)氧化的影响Figure 8. Effect of H2O2 on the oxidation of As(Ⅲ) at pH 9.5在N2氛围下,As(Ⅲ)在可见光/MB+/H2A体系中不会发生氧化(图9). 说明即便MB+和HA−之间能够电子转移,但是体系中没有分子氧,产生MB+·和A·−也不能进行后续产生H2O2的反应. 这与无氧条件下碱/H2A体系无法产生H2O2的实验结果一致[8]. 而分别通入20% 或100% O2,As(Ⅲ)的氧化效率与搅拌条件下一致,表明过多O2也没有促进体系中As(Ⅲ)的氧化.
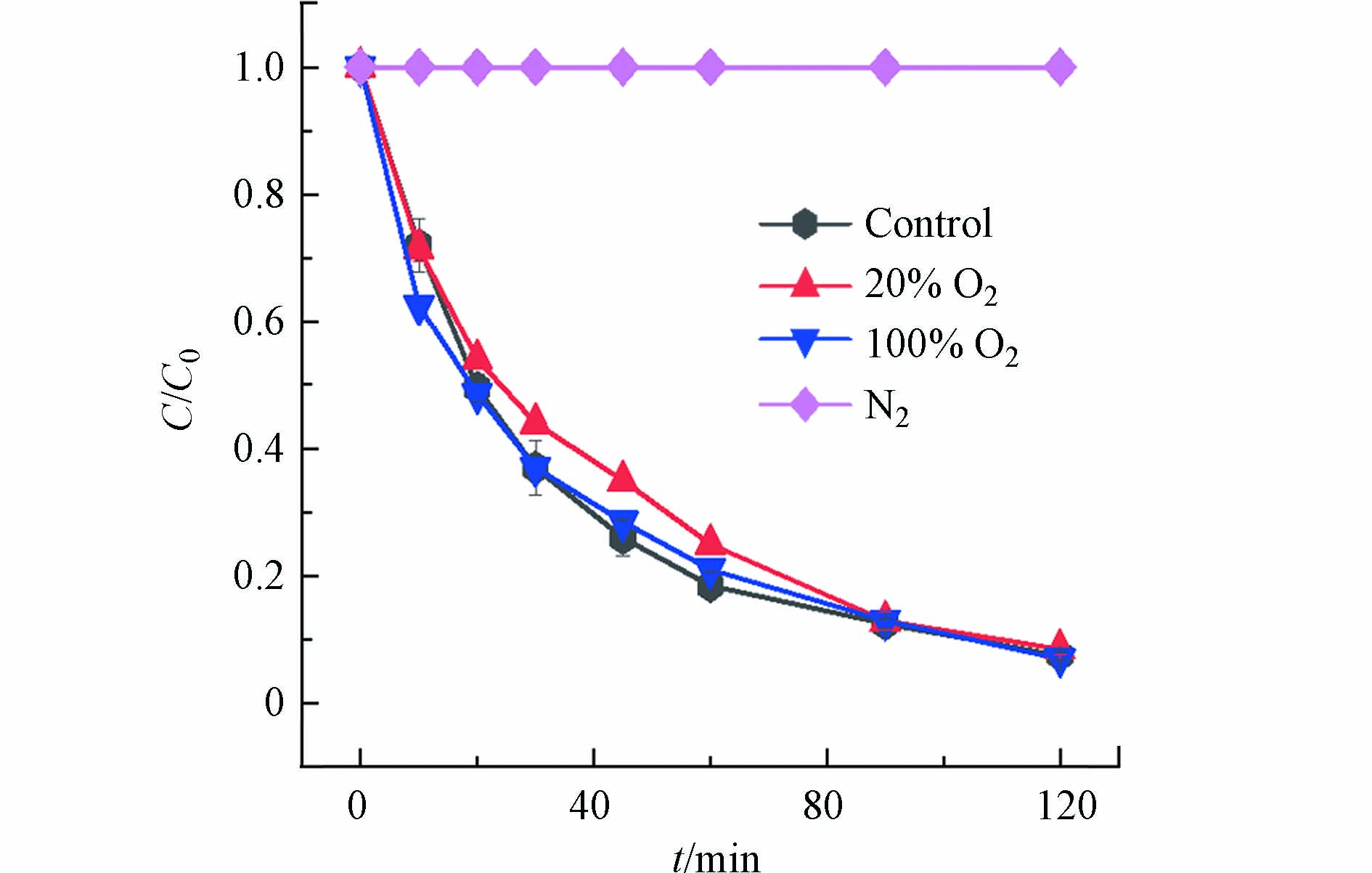 图 9 可见光/MB+/H2A体系中氧气浓度对As(Ⅲ)氧化的影响Figure 9. Effect of oxygen concentration on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.
图 9 可见光/MB+/H2A体系中氧气浓度对As(Ⅲ)氧化的影响Figure 9. Effect of oxygen concentration on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.在可见光/MB+/H2A体系中,活性物种通过以下途径产生(图10):首先,MB+通过可见光激发产生单线态1MB+(公式1),随后1MB+快速转变为三重态(3MB+)(公式2),3MB+和分子氧(3O2)反应产生1O2(公式3);其次,3MB+可以从HA−处得到一个电子生成MB+·和A·−(公式8),MB+·也可以通过歧化反应产生LMB和MB+(公式5),LMB再与O2反应产生MB+;最后,A·−与O2·−(HO2·)反应产生H2O2(公式9). HA−也可以直接与分子氧通过双电子转移产生H2O2,MB+和可见光的引入加速HA−的氧化,从而产生更多的H2O2. 在可见光/MB+/H2A体系中存在H2A可清除产生的1O2, 1O2对As(Ⅲ)的氧化贡献微乎其微,H2O2对As(Ⅲ)的氧化起主要作用.
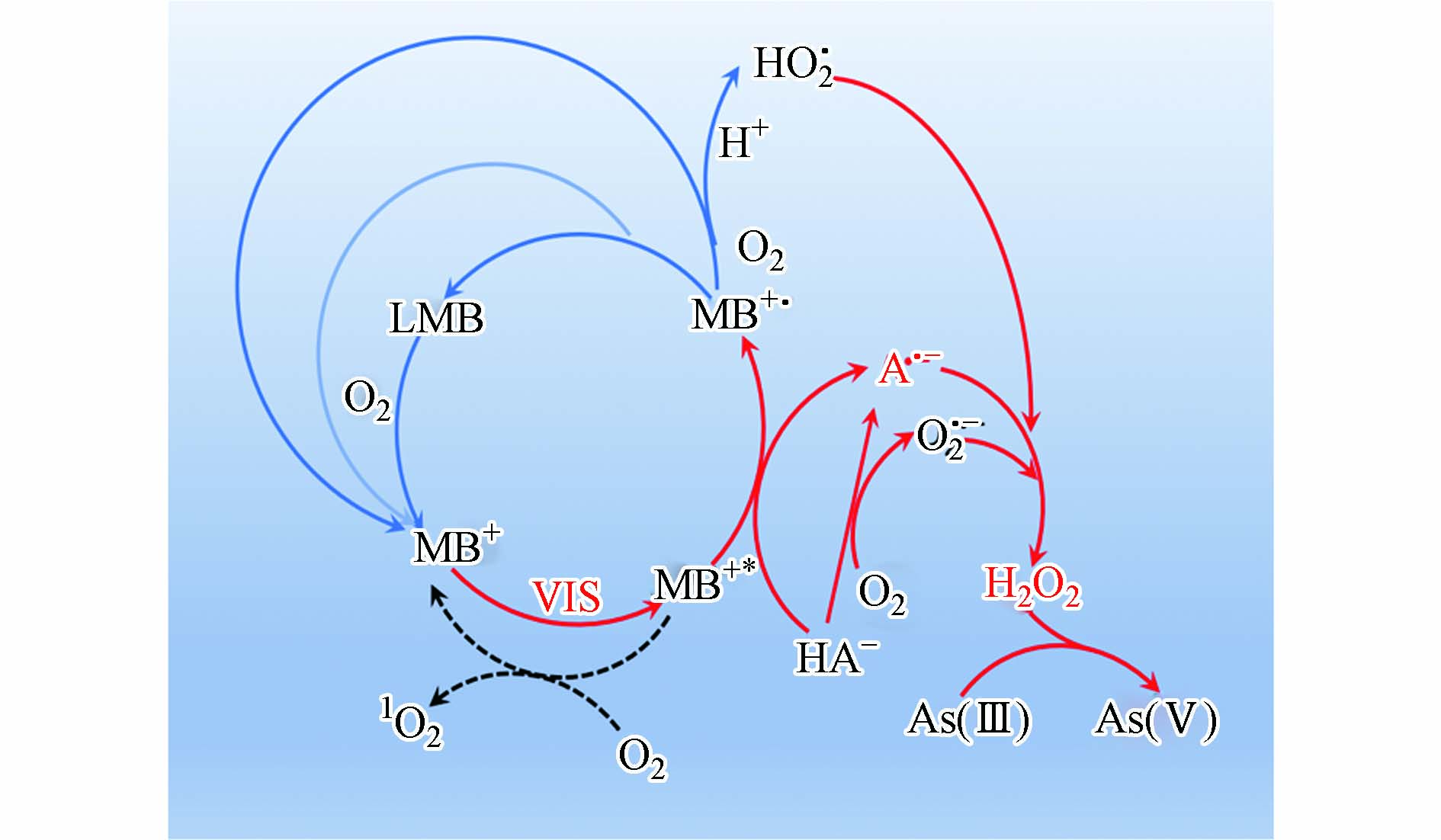 图 10 可见光/MB+/H2A体系中As(Ⅲ)氧化的机理Figure 10. Mechanism diagram of As(Ⅲ) oxidation in a visible light/ascorbic acid/methylene blue system.
图 10 可见光/MB+/H2A体系中As(Ⅲ)氧化的机理Figure 10. Mechanism diagram of As(Ⅲ) oxidation in a visible light/ascorbic acid/methylene blue system.2.3 常见阴离子与有机质的影响
HCO3−、Cl−、NO3−、FA在土壤间隙水和地下水中普遍存在,HCO3−、Cl−、NO3−、FA对As(Ⅲ)氧化的影响如图11所示,HCO3−、Cl−、NO3−对As(Ⅲ)氧化基本无影响,这是因为HCO3−、Cl−、NO3−加入反应液后并不会使MB+的吸收光谱发生变化,因此不会影响可见光/MB+/H2A体系中As(Ⅲ)的氧化,这和碱/H2A体系中环境基质对As(Ⅲ)氧化的影响一致[8]. 但FA对As(Ⅲ)氧化有一定的影响,这是因为FA加入反应液后使得MB+的吸收光谱发生了显著变化,使得MB+在660 nm处的吸收变弱,从而抑制了MB+和HA−之间的电子转移.
 图 11 可见光/MB+/H2A体系中环境基质(HCO3−、Cl−、NO3−、FA)对As(Ⅲ)氧化的影响Figure 11. Effects of environmental matrices (HCO3−、Cl−、NO3−、FAs) on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.
图 11 可见光/MB+/H2A体系中环境基质(HCO3−、Cl−、NO3−、FA)对As(Ⅲ)氧化的影响Figure 11. Effects of environmental matrices (HCO3−、Cl−、NO3−、FAs) on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.2.4 其他两种噻嗪染料的效果
探讨了将其他噻嗪染料用于可见光/H2A体系活化分子氧氧化As(Ⅲ)的可能性. 劳氏紫和天青B也是常见的噻嗪染料,在可见光/H2A/劳氏紫体系和可见光/H2A/天青B体系中分别对测定了As(Ⅲ)和染料的浓度的变化,结果如图12所示. 实验结果表明,劳氏紫和天青B均能促进H2A活化分子氧产生H2O2氧化As(Ⅲ),但促进效果均不如MB+,这是因为相同浓度下MB+有最大的吸光度值,能更好地吸收能量. 噻嗪染料的相似行为可能是由于它们具有相同的官能团和激发状态. 推测其他光敏剂如核黄素[9]和茜素[27]都具有接受电子的能力,同样可以促进H2A活化分子氧产生H2O2氧化水中的As(Ⅲ).
 图 12 可见光/H2A中噻嗪染料天青B 和劳氏紫对As(Ⅲ)的氧化及自身的降解Figure 12. As(Ⅲ) oxidation induced by thiazide dyes Thionine and Azure B, and degradation of the dyes themselves in visible light/ascorbic acid system.
图 12 可见光/H2A中噻嗪染料天青B 和劳氏紫对As(Ⅲ)的氧化及自身的降解Figure 12. As(Ⅲ) oxidation induced by thiazide dyes Thionine and Azure B, and degradation of the dyes themselves in visible light/ascorbic acid system.3. 结论(Conclusion)
本文从可见光/MB+/H2A活化分子氧体系出发,研究了可见光/MB+/H2A体系氧化水中As(Ⅲ)的可行性与内在机理. 其中,可见光光照、pH值、H2A与MB+的投加量对于As(Ⅲ)的氧化过程有着不同影响. 实验结果表明:
(1)可见光光照可以促进MB+/H2A体系氧化As(Ⅲ); pH越高,As(Ⅲ)氧化越快;pH=9.5下,H2A浓度的增加对As(Ⅲ)氧化呈现先促进后稳定的趋势,且O2浓度对于该体系对As(Ⅲ) 的氧化过程的影响较小. 最终得出该体系下As(Ⅲ)氧化的最佳条件:最佳H2A投加量为300 μmol·L−1;最佳MB+投加量为5 mg·L−1.
(2)通过自由基抑制剂分析验证了可见光/MB+/H2A活化分子氧体系氧化As(Ⅲ)的主要活性物种为H2O2, 1O2对As(Ⅲ)的氧化贡献微乎其微.
(3)同MB+一样,在可见光/MB+体系中添加劳氏紫和天青B这两种噻嗪染料也能促进As(Ⅲ)的氧化.
-
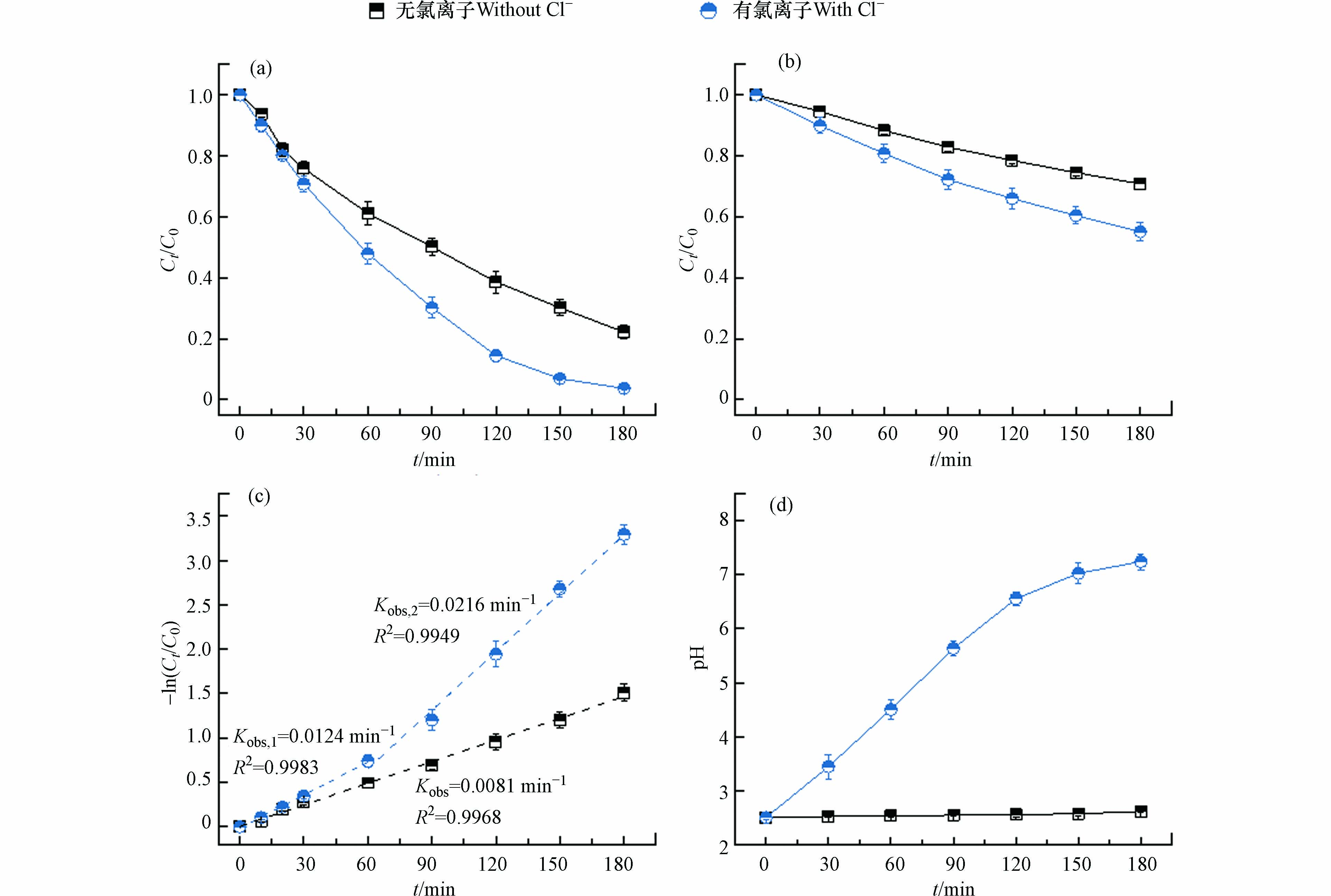
图 1 有无氯情形下电化学体系中(a)Ni-EDTA降解效果;(b)TOC去除效果;(c)Ni-EDTA降解动力学拟合;(d)pH变化.
Figure 1. Comparisons of time-course (a) Ni-EDTA concentration, (b) TOC concentration, (c) kinetic-fitting results, and (d) pH variations in the electrochemical systems in the presence and absence of chloride.
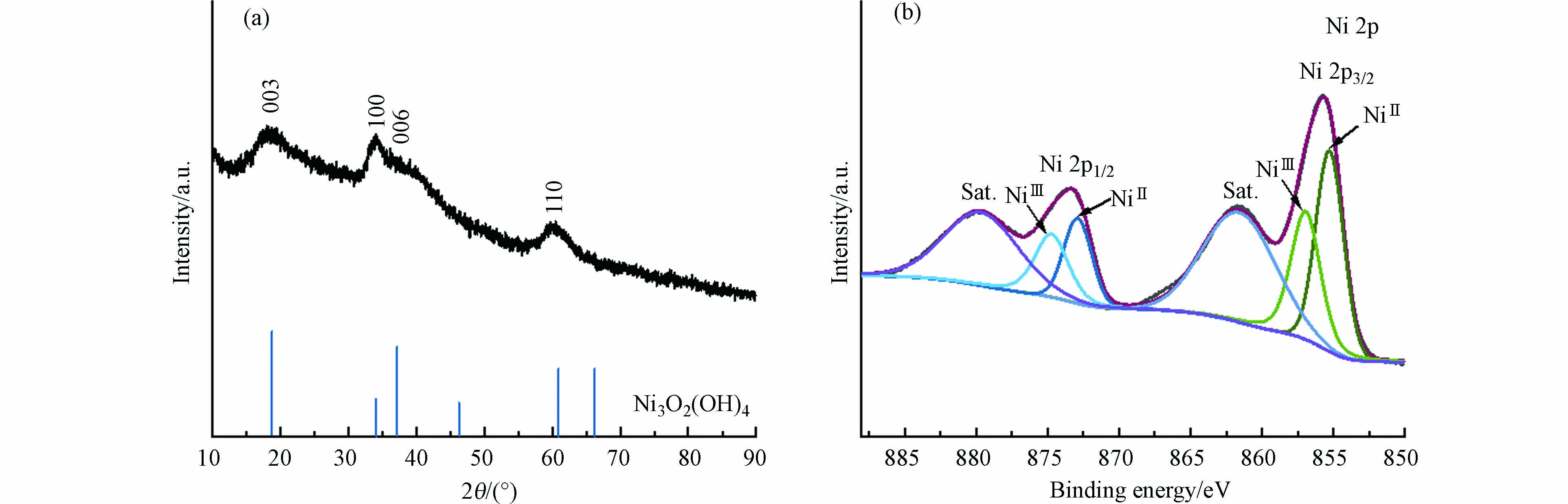
图 2 含氯离子的电化学体系反应后分离出灰黑色固体的表征分析
Figure 2. (a) The XRD pattern and (b) XPS curves of gray black solid obtained from the chloride-containing electrochemical system.
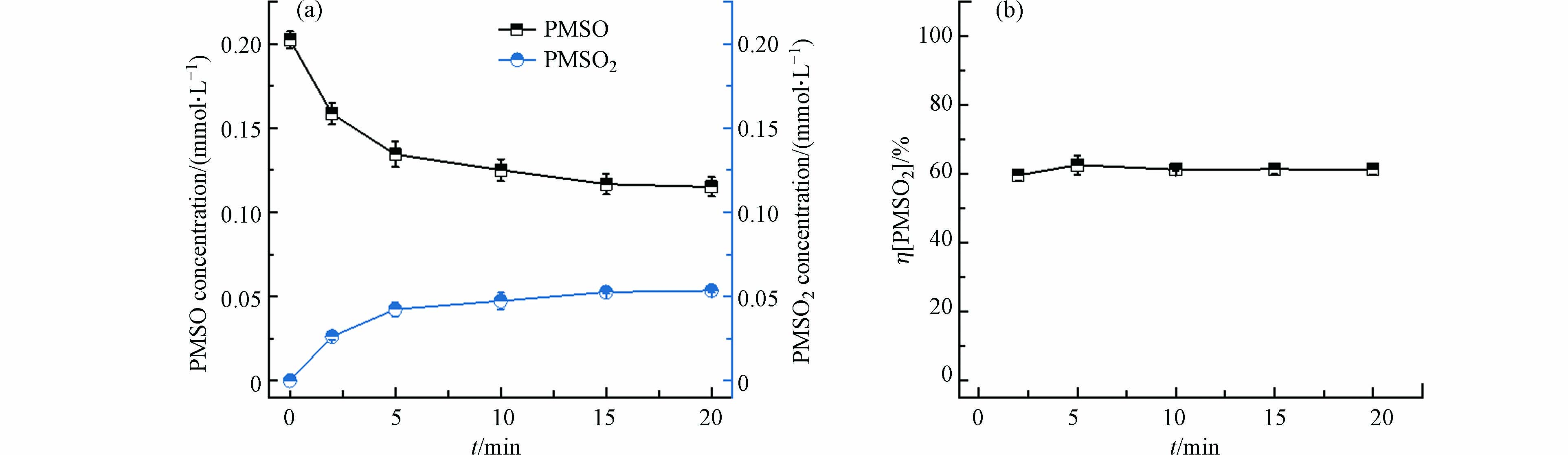
图 3 电化学体系中灰黑色固体氧化PMSO效果
Figure 3. Oxidation of PMSO by the gray black solid obtained from the chloride-containing electrochemical system
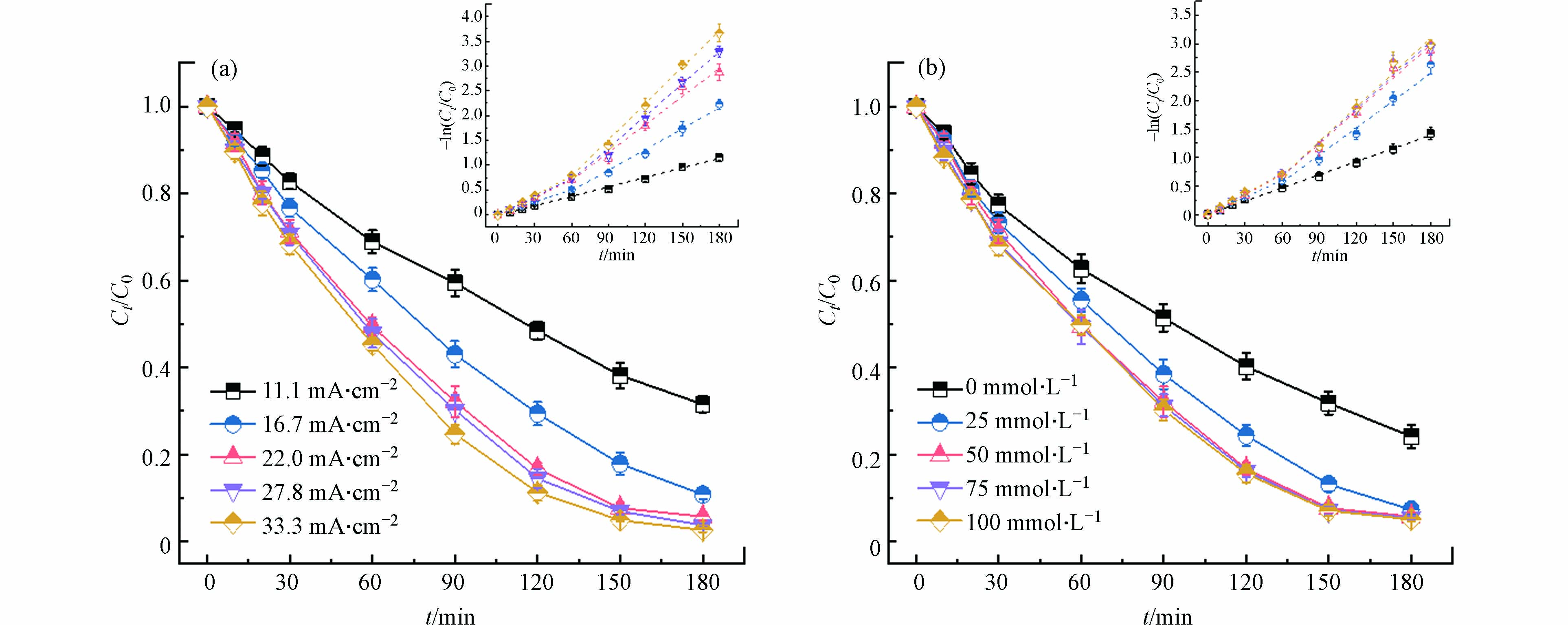
图 4 (a)电流密度和(b)氯离子浓度对Ni-EDTA降解的影响(内插图为Ni-EDTA降解动力学拟合)
Figure 4. Effects of (a) current density and (b) chloride concentration on time-course Ni-EDTA concentration (insert: kinetic-fitting results)

图 5 电化学体系中Fe(Ⅲ)-EDTA(a)降解效果(b)降解动力学拟合;不同Ni2+浓度下Fe(Ⅲ)-EDTA(c)降解效果(d)降解动力学拟合.
Figure 5. Comparisons of time-course (a) Fe-EDTA concentration and (b) kinetic-fitting results in the electrochemical systems. The effects of concentration of added Ni2+ on (c) degradation of Fe(Ⅲ)-EDTA and (d) kinetic-fitting results
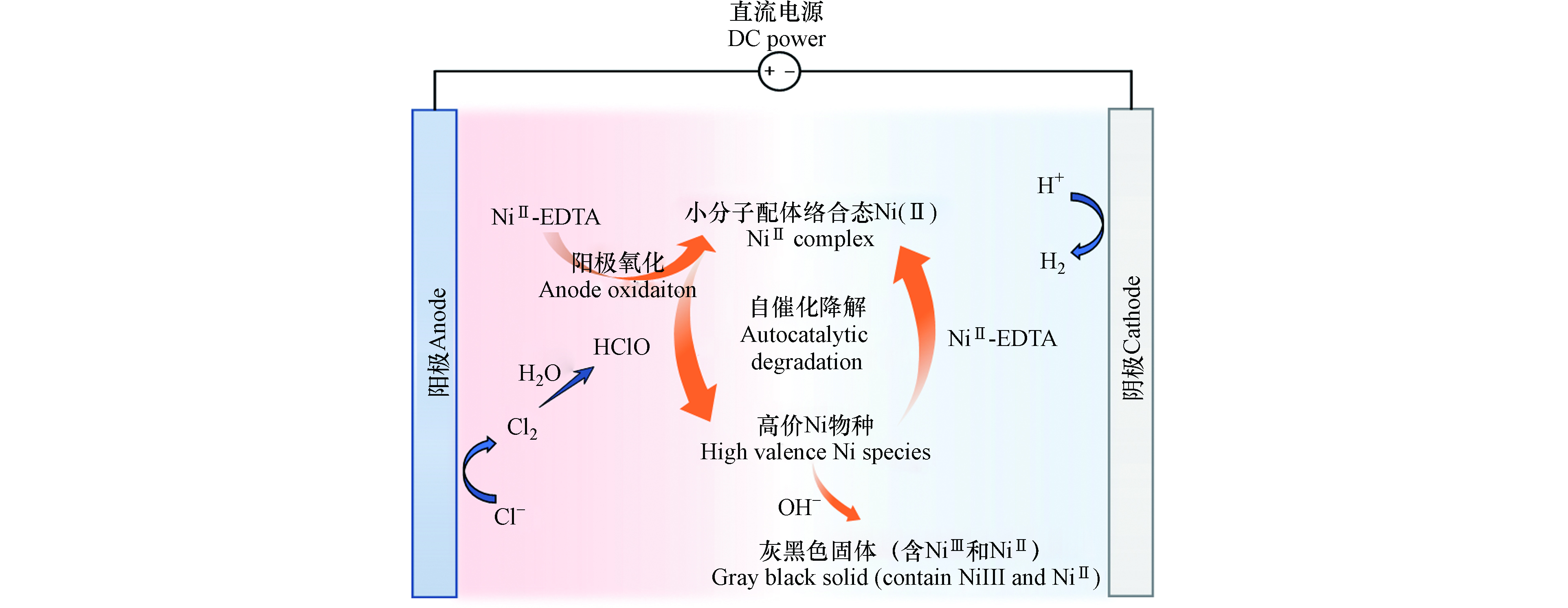
图 6 Ni-EDTA在含氯电化学氧化体系中自催化降解途径示意图.
Figure 6. Schematic representation of the proposed pathways for Ni-EDTA autocatalytic degradation in the chloride-containing electrochemical oxidation system.
表 1 不同电流密度下Ni-EDTA降解一级动力学拟合结果
Table 1. First-order kinetic fitting results of Ni-EDTA degradation under different current density
电流密度/(mA·cm−2)Current density 0—60 min 60—180 min Kobs/min−1 R2 Kobs/min−1 R2 11.1 0.0064 0.9975 0.0064 0.9975 16.7 0.0086 0.9985 0.0139 0.9918 22.2 0.0120 0.9981 0.0189 0.9914 27.8 0.0124 0.9983 0.0216 0.9961 33.3 0.0133 0.9991 0.0244 0.9977  下载: 导出CSV
下载: 导出CSV
表 2 不同氯离子浓度下Ni-EDTA降解一级动力学拟合结果
Table 2. First-order kinetic fitting results of Ni-EDTA degradation under different Cl− concentration
Cl−/( mmol·L−1) 0—60 min 60—180 min Kobs / min−1 R2 Kobs / min−1 R2 0 0.0077 0.9975 0.0077 0.9975 25 0.0100 0.9965 0.0174 0.9868 50 0.0120 0.9981 0.0193 0.9834 75 0.0119 0.9984 0.0197 0.9811 100 0.0117 0.9965 0.0202 0.9865
下载: 导出CSV
表 3 不同镍离子浓度下Fe(Ⅲ)-EDTA降解一级动力学拟合结果
Table 3. First-order kinetic fitting results of Fe(Ⅲ)-EDTA degradation under different Ni2+ concentration
Ni2+/(mmol·L−1) 0—30 min 30—180 min Kobs/min−1 R2 Kobs/min−1 R2 0 0.0028 0.9986 0.0028 0.9986 20 0.0025 0.9859 0.0059 0.9895 40 0.0026 0.9917 0.0069 0.9935 60 0.0032 0.9868 0.0081 0.9949
下载: 导出CSV
-
[1] LIU M H, MENG Y, ZHAO Y, et al. Electropolishing parameters optimization for enhanced performance of nickel coating electroplated on mild steel [J]. Surface and Coatings Technology, 2016, 286: 285-292. doi: 10.1016/j.surfcoat.2015.12.027 [2] LI W L, LI L Y, SUN Q Q, et al. Direct fabrication of high-resolution and high-performance flexible electronics via surface-activation-localized electroless plating [J]. Chemical Engineering Journal, 2021, 416: 127644. doi: 10.1016/j.cej.2020.127644 [3] BULASARA V K, THAKURIA H, UPPALURI R, et al. Combinatorial performance characteristics of agitated nickel hypophosphite electroless plating baths [J]. Journal of Materials Processing Technology, 2011, 211(9): 1488-1499. doi: 10.1016/j.jmatprotec.2011.03.022 [4] LING L L, LIU W J, ZHANG S, et al. Magnesium oxide embedded nitrogen self-doped biochar composites: Fast and high-efficiency adsorption of heavy metals in an aqueous solution [J]. Environmental Science & Technology, 2017, 51(17): 10081-10089. [5] HARGREAVES A J, VALE P, WHELAN J, et al. Impacts of coagulation-flocculation treatment on the size distribution and bioavailability of trace metals (Cu, Pb, Ni, Zn) in municipal wastewater [J]. Water Research, 2018, 128: 120-128. doi: 10.1016/j.watres.2017.10.050 [6] 陈倩, 吴一楠, 蒋天遥, 等. UiO-66(Zr)@多孔陶瓷复合材料的制备及对络合态重金属EDTA-Cu(Ⅱ)的去除 [J]. 环境化学, 2020, 39(3): 677-686. doi: 10.7524/j.issn.0254-6108.2019103108 CHEN Q, WU Y N, JIANG T Y, et al. Synthesis of UiO-66(Zr)@ porous ceramic composite for the removal of EDTA-Cu(Ⅱ) complex [J]. Environmental Chemistry, 2020, 39(3): 677-686(in Chinese). doi: 10.7524/j.issn.0254-6108.2019103108
[7] DU J Q, ZHANG B G, LI J X, et al. Decontamination of heavy metal complexes by advanced oxidation processes: A review [J]. Chinese Chemical Letters, 2020, 31(10): 2575-2582. doi: 10.1016/j.cclet.2020.07.050 [8] ZHU Y, FAN W H, FENG W Y, et al. A critical review on metal complexes removal from water using methods based on Fenton-like reactions: Analysis and comparison of methods and mechanisms [J]. Journal of Hazardous Materials, 2021, 414: 125517. doi: 10.1016/j.jhazmat.2021.125517 [9] 王义, 黄先锋, 郑向勇, 等. UV/氯降解铜络合物的特性与机理 [J]. 环境科学学报, 2019, 39(6): 1763-1771. WANG Y, HUANG X F, ZHENG X Y, et al. Performance and mechanism of Cu(Ⅱ)-organic complexes degradation by UV/chlorine advanced oxidation process [J]. Acta Scientiae Circumstantiae, 2019, 39(6): 1763-1771(in Chinese).
[10] NGUYEN M K, TRAN V S, PHAM T T, et al. Fenton/ozone-based oxidation and coagulation processes for removing metals (Cu, Ni)-EDTA from plating wastewater [J]. Journal of Water Process Engineering, 2021, 39: 101836. doi: 10.1016/j.jwpe.2020.101836 [11] XU Z, SHAN C, XIE B H, et al. Decomplexation of Cu(Ⅱ)-EDTA by UV/persulfate and UV/H2O2: Efficiency and mechanism [J]. Applied Catalysis B:Environmental, 2017, 200: 439-447. doi: 10.1016/j.apcatb.2016.07.023 [12] LAN S Y, XIONG Y, TIAN S H, et al. Enhanced self-catalytic degradation of CuEDTA in the presence of H2O2/UV: Evidence and importance of Cu-peroxide as a photo-active intermediate [J]. Applied Catalysis B:Environmental, 2016, 183: 371-376. doi: 10.1016/j.apcatb.2015.10.030 [13] RONG H Y, ZHANG C Y, SUN Y Y, et al. Electrochemical degradation of Ni-EDTA complexes in electroless plating wastewater using PbO2-Bi electrodes [J]. Chemical Engineering Journal, 2022, 431: 133230. doi: 10.1016/j.cej.2021.133230 [14] SUN Y Y, ZHANG C Y, RONG H Y, et al. Electrochemical Ni-EDTA degradation and Ni removal from electroless plating wastewaters using an innovative Ni-doped PbO2 anode: Optimization and mechanism [J]. Journal of Hazardous Materials, 2022, 424: 127655. doi: 10.1016/j.jhazmat.2021.127655 [15] ZHANG F, WANG W L, XU L, et al. Treatment of Ni-EDTA containing wastewater by electrochemical degradation using Ti3+ self-doped TiO2 nanotube arrays anode [J]. Chemosphere, 2021, 278: 130465. doi: 10.1016/j.chemosphere.2021.130465 [16] ZHAO X, GUO L B, ZHANG B F, et al. Photoelectrocatalytic oxidation of Cu(Ⅱ)–EDTA at the TiO2 electrode and simultaneous recovery of Cu(Ⅱ) by electrodeposition [J]. Environmental Science & Technology, 2013, 47(9): 4480-4488. [17] 杨桂蓉, 魏连雨, 李静, 等. Co-BiVO4薄膜电极光电处理Pb/Cu-EDTA研究 [J]. 环境科学学报, 2014, 34(4): 914-919. YANG G R, WEI L Y, LI J, et al. Photoelectrocatalytic treatment of Pb/Cu-EDTA at Co-BiVO4 film electrode [J]. Acta Scientiae Circumstantiae, 2014, 34(4): 914-919(in Chinese).
[18] HUANG X F, WANG Y, LI X C, et al. Autocatalytic decomplexation of Cu(Ⅱ)–EDTA and simultaneous removal of aqueous Cu(II) by UV/chlorine [J]. Environmental Science & Technology, 2019, 53(4): 2036-2044. [19] LI J Y, MA J X, DAI R B, et al. Self-enhanced decomplexation of Cu-organic complexes and Cu recovery from wastewaters using an electrochemical membrane filtration system [J]. Environmental Science & Technology, 2021, 55(1): 655-664. [20] LU Y, YANG F J, CHEN S Y, et al. Decomplexation of Ni(Ⅱ)-citrate and recovery of nickel from chelated nickel containing electroplating wastewater by peroxymonosulfate with nickel [J]. Separation and Purification Technology, 2022, 283: 120142. doi: 10.1016/j.seppur.2021.120142 [21] TRAFELA Š, ZAVAŠNIK J, ŠTURM S, et al. Controllable voltammetric formation of a structurally disordered NiOOH/Ni(OH)2 redox pair on Ni-nanowire electrodes for enhanced electrocatalytic formaldehyde oxidation [J]. Electrochimica Acta, 2020, 362: 137180. doi: 10.1016/j.electacta.2020.137180 [22] DONG H, YU W L, HOFFMANN M R. Mixed metal oxide electrodes and the chlorine evolution reaction [J]. The Journal of Physical Chemistry C, 2021, 125(38): 20745-20761. doi: 10.1021/acs.jpcc.1c05671 [23] ZENG H B, TIAN S C, LIU H F, et al. Photo-assisted electrolytic decomplexation of Cu-EDTA and Cu recovery enhanced by H2O2 and electro-generated active chlorine [J]. Chemical Engineering Journal, 2016, 301: 371-379. doi: 10.1016/j.cej.2016.04.006 [24] 李勇东, 吴迪, 郑文笑, 等. PbO2/Fe双阳极耦合促进焦化废水除碳脱氮 [J]. 环境化学, 2020, 39(6): 1650-1659. doi: 10.7524/j.issn.0254-6108.2019122801 LI Y D, WU D, ZHENG W X, et al. Enhanced removal of carbon and nitrogen from the coking wastewater via the coupled PbO2/Fe dual-anode electrochemical system [J]. Environmental Chemistry, 2020, 39(6): 1650-1659(in Chinese). doi: 10.7524/j.issn.0254-6108.2019122801
[25] 邱凌峰, 倪尔灵. 电催化氧化阳极制备及其降酚特性 [J]. 环境化学, 2010, 29(6): 1019-1026. QIU L F, NI E L. Preparation of dimensionless stable anode in the electro-catalytic oxidation and its phenoldegrading characteristics [J]. Environmental Chemistry, 2010, 29(6): 1019-1026(in Chinese).
[26] LI C W, YU J H, LIANG Y M, et al. Ni removal from aqueous solutions by chemical reduction: Impact of pH and pe in the presence of citrate [J]. Journal of Hazardous Materials, 2016, 320: 521-528. doi: 10.1016/j.jhazmat.2016.08.030 [27] LI L H, HUANG Z P, FAN X X, et al. Preparation and Characterization of a Pd modified Ti/SnO2-Sb anode and its electrochemical degradation of Ni-EDTA [J]. Electrochimica Acta, 2017, 231: 354-362. doi: 10.1016/j.electacta.2017.02.072 [28] FU Z W, HU J T, HU W L, et al. Quantitative analysis of Ni2+/Ni3+ in Li[NixMnyCoz]O2 cathode materials: Non-linear least-squares fitting of XPS spectra [J]. Applied Surface Science, 2018, 441: 1048-1056. doi: 10.1016/j.apsusc.2018.02.114 [29] HUANG L F, HUTCHISON M J, SANTUCCI R J Jr, et al. Improved electrochemical phase diagrams from theory and experiment: The Ni–water system and its complex compounds [J]. The Journal of Physical Chemistry C, 2017, 121(18): 9782-9789. doi: 10.1021/acs.jpcc.7b02771 [30] CORONA T, DRAKSHARAPU A, PADAMATI S K, et al. Rapid hydrogen and oxygen atom transfer by a high-valent nickel-oxygen species [J]. Journal of the American Chemical Society, 2016, 138(39): 12987-12996. doi: 10.1021/jacs.6b07544 [31] WANG Y R, ZHAO J J, XIONG X Q, et al. Role of Ni2+ ions in TiO2 and Pt/TiO2 photocatalysis for phenol degradation in aqueous suspensions [J]. Applied Catalysis B:Environmental, 2019, 258: 117903. doi: 10.1016/j.apcatb.2019.117903 [32] KIM S, KANG J S, KIM S, et al. Electrochemical regeneration of free chlorine treated nickel oxide catalysts for oxidation of aqueous pollutants [J]. Catalysis Today, 2021, 375: 514-521. doi: 10.1016/j.cattod.2020.03.045 [33] DRAKSHARAPU A, CODOLÀ Z, GÓMEZ L, et al. Spectroscopic analyses on reaction intermediates formed during chlorination of alkanes with NaOCl catalyzed by a nickel complex [J]. Inorganic Chemistry, 2015, 54(22): 10656-10666. doi: 10.1021/acs.inorgchem.5b01463 [34] GAO Y, ZHOU Y, PANG S Y, et al. New insights into the combination of permanganate and bisulfite as a novel advanced oxidation process: Importance of high valent manganese-oxo species and sulfate radical [J]. Environmental Science & Technology, 2019, 53(7): 3689-3696. [35] PESTOVSKY O, BAKAC A. Aqueous ferryl(Ⅳ) ion: Kinetics of oxygen atom transfer to substrates and oxo exchange with solvent water [J]. Inorganic Chemistry, 2006, 45(2): 814-820. doi: 10.1021/ic051868z [36] ZONG Y, GUAN X H, XU J, et al. Unraveling the overlooked involvement of high-valent cobalt-oxo species generated from the cobalt(Ⅱ)-activated peroxymonosulfate process [J]. Environmental Science & Technology, 2020, 54(24): 16231-16239. [37] LIANG S, HU X T, XU H L, et al. Mechanistic insight into the reaction pathway of peroxomonosulfate-initiated decomplexation of EDTA-Ni(Ⅱ) under alkaline conditions: Formation of high-valent Ni intermediate [J]. Applied Catalysis B:Environmental, 2021, 296: 120375. doi: 10.1016/j.apcatb.2021.120375 [38] ZHAO X, GUO L B, HU C Z, et al. Simultaneous destruction of Nickel (Ⅱ)-EDTA with TiO2/Ti film anode and electrodeposition of nickel ions on the cathode [J]. Applied Catalysis B:Environmental, 2014, 144: 478-485. doi: 10.1016/j.apcatb.2013.07.038 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 7077
- HTML全文浏览数: 7077
- PDF下载数: 195
- 施引文献: 0
