


-
铁氧化物广泛的存在于自然界中,常见的有针铁矿、水铁矿、纤铁矿等. 铁氧化物因比表面积大及氧化活性高等特点,影响重金属元素(如Cu、Pb和Zn等)、有机污染物和营养元素(如C、N、P和S等)的迁移、转化和归趋[1]. 因此,自然环境中铁氧化物的形成过程在环境化学和土壤学等领域备受关注. 铁氧化物可由Fe(Ⅱ)氧化或其他铁氧化物转化形成[2-5]. 自然界中的铁氧化物与锰氧化物常交结伴生,它们常以铁锰结核或包覆物的形式存在[6-9]. 锰氧化物是环境中重要的氧化剂,它们直接氧化Fe(Ⅱ)的过程被认为是铁锰结核重要形成途径之一[10]. 通常,Fe(Ⅱ)与四价锰氧化物的反应过程如式(1):
四价锰氧化物被还原,Fe(Ⅱ)被氧化形成铁氧化物. 但Fe(Ⅱ)氧化速率及产物类型受锰氧化物晶体结构影响[11-16]. 例如,在水钠锰矿氧化FeCl2-NH4OH反应中,Fe(Ⅱ)的氧化产物为针铁矿与四方纤铁矿;软锰矿、锰钾矿和黑锰矿氧化Fe(Ⅱ)产物则为纤铁矿、四方纤铁矿和磁铁矿[11-12];六方锰矿(γ-MnO2)与Fe(Ⅱ)氧化产物为MnFe2O4和水铁矿. 因此,不同反应体系中,生成产物及反应机制存在明显差异. 锰氧化物氧化Fe(Ⅱ)速率受pH影响[17-18]. pH < 4.0时,水钠锰矿能快速氧化Fe(Ⅱ),而pH > 4.0时反应速率大幅降低[17]. 现有研究多关注高价态锰氧化物对Fe(Ⅱ)的氧化过程. 黑锰矿由于结构中含有大量的Mn(Ⅲ),既可以在酸性条件下歧化生成高价锰氧化物,又易在碱性条件下发生氧化,因此在环境中难以长期大量稳定存在. 其与Fe(Ⅱ)的相互作用,特别是溶解氧参与的氧化过程有助于认识表生环境铁锰氧化物的形成与共生机理.
本文以典型低价锰氧化物黑锰矿为例,考察其对溶解氧氧化Fe(Ⅱ)过程的影响,采用XRD、FTIR、TEM和XAFS表征过程中间产物以明确黑锰矿氧化Fe(Ⅱ)机理,考察Fe(Ⅱ)浓度、pH和溶解氧对Fe(Ⅲ)氧化物形成过程的影响.
-
借鉴Mckenzie[19]报道的方法合成黑锰矿. 具体过程为:用除氧的超纯水配置等体积的0.2 mol·L−1 MnSO4和0.5 mol·L−1 NaOH溶液,将MnSO4溶液与NaOH溶液利用磁力搅拌混合,并不断通入氮气. 将反应生成Mn(OH)2沉淀用去氧超纯水清洗至pH ≤ 8.5. 用超纯水将沉淀物分散至烧杯中,于室温下通入空气氧化5 h,所得固相产物用超纯水洗涤至上清液电导小于20 μS·cm−1.
合成针铁矿具体过程[1]:将180 mL 5 mol·L−1 NaOH与100 mL 1 mol·L−1 Fe(NO3)3溶液混合于2 L 烧杯,加入超纯水至体积为2.0 L,随后密封并置于70 ℃烘箱老化72 h. 随后用超纯水将终产物洗涤至电导小于20 μS·cm−1. 所得黑锰矿和针铁矿固体经40 ℃真空干燥24 h后研磨过0.165 mm标准筛备用. 利用草酸与KMnO4的氧化还原反应,滴定测得黑锰矿锰平均氧化度为2.69[19].
-
在空气氛围中,将黑锰矿与不同浓度FeSO4在50 mL离心管中混合,黑锰矿浓度为0.5 g·L−1, Fe(Ⅱ)浓度设定为0—10 mmol·L−1,背景电解质为20 mmol·L−1 NaNO3. 溶液总体积为20 mL,反应体系控制在恒温摇床(25 ℃)中进行. 反应过程中用0.1/1.0 mol·L−1 HNO3和0.01/0.1 mol·L−1 NaOH调节pH值为3.0和5.0. 反应完之后在手套箱中用0.22 μm水系滤膜过滤,滤液保留待测. 为降低空气中氧气的影响,溶液配制及材料准备等实验操作均在氮气氛围的手套箱中进行. 配置溶液均采用除氧超纯水(DDW, 18 MΩ·cm),其余实验操作均与上述相同.
考虑到实验的可操作性,使用五口反应瓶研究Fe(Ⅱ)氧化动力学过程. 在500 mL去氧超纯水中加入0.25 g黑锰矿和20 mol·L−1 NaNO3,并持续磁力搅拌,反应过程控制室温持续通入空气,流速保持为气泡持续鼓出. 用浓度为0.1/1.0 mol·L−1 HNO3和0.01/0.1 mol·L−1 NaOH溶液12 h内持续调节pH恒定为5.0. 在不同反应时间点取悬浊液过滤待测. 为考察溶解氧对反应的影响,反应过程中持续鼓入高纯氮气(99.99%)和空气进行实验,利用溶解氧仪测得氮气氛围溶解氧含量接近0,其余实验条件保持一致.
此外,为明确反应过程中的中间产物对Fe(Ⅱ)氧化的影响,在8.0 mmol·L−1 Fe(Ⅱ)溶液中,加入0.86 g·L−1针铁矿和6.5 mmol·L−1 MnSO4考察其对Fe(Ⅱ)氧化的影响.
-
反应过程中过滤所得样品用X-射线衍射仪(XRD,Bruker D8 ,Germany, Cu Kα(λ=0.15406 nm))表征产物组分. 用傅里叶变换红外光谱仪(FTIR,Bruker VERTEX 70,Germany)表征产物的官能团,制样过程采用KBr压片,扫描范围为4000—400 cm−1. 用透射电镜(TEM,Hitachi H-7650,Japan)观察样品的微观形貌. 不同反应阶段的取出的滤液中Fe(Ⅱ)浓度的测定采用邻菲啰啉比色法,释放的Mn(Ⅱ)浓度的测定采用原子吸收(AA240FS),精度为0.01 mg·L−1. XAFS吸收光谱用于分析产物的平均锰氧化度和铁氧化物组分. 在北京同步辐射(1W2B)收集X-射线吸收谱. 采集的Mn K边能量范围为6340—7327 eV,Mn箔用作能量校正(6539 eV). Fe K边能量范围为6953—7884 eV,Fe箔作能量校正(7112 eV). 用Athena软件进行数据处理. 使用Combo方法对黑锰矿及其转化样品归一化的Mn K边X射线吸收近边结构光谱(X-ray absorption near edge structure, XANES)进行最小二乘法线性拟合,以分析锰氧化物中不同价态Mn含量和平均氧化度[20].
-
图1为pH 5.0下,不同初始浓度Fe(Ⅱ)(0—10 mmol)与黑锰矿(0.5 g·L−1)在空气和氮气氛围下相互作用24 h后溶液中Mn(Ⅱ)和Fe(Ⅱ)浓度. 在氮气和空气条件下,随初始Fe(Ⅱ)浓度升高,体系中溶解释放的Mn(Ⅱ)浓度先增加后保持平衡. 当初始Fe(Ⅱ)浓度 < 5.0 mmol·L−1时,测得生成的Mn(Ⅱ)与消耗的Fe(Ⅱ)比例接近1:1. 依据电荷守恒,若Fe(Ⅱ)完全被黑锰矿氧化,生成的Mn(Ⅱ)与消耗的Fe(Ⅱ)比例为3:1,表明有过量的溶解态Fe(Ⅱ)被析出,可能是溶解态的Fe(Ⅱ)被黑锰矿吸附所致. 这与Postma报道的水钠锰矿和Fe(Ⅱ)反应也观察到过量的Fe(Ⅱ)消耗反应规律一致[15]. 当Fe(Ⅱ)浓度增至 ≥ 5.0 mmol·L−1时,溶液中释放的Mn(Ⅱ)浓度小于消耗的Fe(Ⅱ)浓度,黑锰矿对Fe(Ⅱ)吸附达到饱和,溶液中有剩余的溶解态Fe(Ⅱ). 与氮气氛围结果比较发现,空气体系下消耗的Fe(Ⅱ)量明显增加,也即溶解氧参与了Fe(Ⅱ)的氧化过程.
由图1结果可见,当初始Fe(Ⅱ)浓度较低时,Fe(Ⅱ)可被完全吸附和氧化. 为此设定Fe(Ⅱ)初始浓度为8 mmol·L−1研究有溶解氧参与的Fe(Ⅱ)氧化过程与机理. 考察了pH 5.0下、氮气和空气体系中0.5 g·L−1黑锰矿与8.0 mmol·L−1 Fe(Ⅱ)反应不同时间点溶解的Fe(Ⅱ)和Mn(Ⅱ)浓度(图2). 反应开始2 h内,氮气和空气氛围下Mn(Ⅱ)浓度均快速升高后保持平衡(图2a). N2氛围下,在反应开始1 h内Fe(Ⅱ)浓度快速降低,随后趋于平衡,而空气氛围下Fe(Ⅱ)浓度在反应过程中持续降低(图2b). 对比反应开始1h内,空气氛围下消耗的Fe(Ⅱ)速率与释放的Mn(Ⅱ)量均低于氮气氛围,表明溶解氧降低了Fe(Ⅱ)被氧化的速率.
在水锰矿氧化S2−反应过程中也观察到氧气的存在使得锰氧化物氧化S2−速率降低[21]. 在本研究中,由于黑锰矿与溶解氧均可参与Fe(Ⅱ)的氧化,但溶解氧氧化Fe(Ⅱ)的能力弱于黑锰矿,因此空气氛围下氧气竞争吸附在黑锰矿表面导致Fe(Ⅱ)氧化速率下降. 当释放的Mn(Ⅱ)浓度不再增加时,Fe(Ⅱ)仍可在空气氛围下被氧化,而氮气体系下Fe(Ⅱ)氧化没有明显变化.
这些结果表明,溶解氧虽降低Fe(Ⅱ)氧化速率,但可促进Fe(Ⅱ)被氧化的总量. 而Fe(Ⅱ)在单一的溶解氧氛围下的氧化速率几乎可以忽略(图2c),因此Fe(Ⅱ)的进一步氧化可能是中间产物在溶解氧作用下催化氧化的结果.
研究表明,FeCl2溶液在pH 5.0的开放体系中反应可生成纤铁矿,而锰钾矿和黑锰矿存在时Fe(Ⅱ)的氧化产物则分别为四方纤铁矿和磁铁矿[12]. 为阐明黑锰矿氧化Fe(Ⅱ)的机理,用XRD和FTIR表征了0.5 g·L−1黑锰矿和8.0 mmol·L−1 Fe(Ⅱ)在空气条件下不同时间点的固相产物(图3). 黑锰矿特征峰随着反应进行逐渐减弱,Fe(Ⅱ)被氧化主要形成了针铁矿和纤铁矿(图3a). 图3b为不同时间段产物的FTIR图谱,在3430 cm−1和1634 cm−1处为H2O中—OH的伸缩振动吸收峰[22]. 626、520、424 cm−1为黑锰矿中Mn—O的伸缩振动峰,随反应的进行其峰强逐渐减弱[22]. 880 cm−1为Fe(OH)3(s)中Fe—O—H伸缩振动,610 cm−1为针铁矿的特征吸收峰,1116、1054、454 cm−1为纤铁矿的特征吸收峰[23-24].
FTIR分析结果进一步表明,pH 5.0下Fe(Ⅱ)最终氧化产物为针铁矿和纤铁矿的混合物. 研究表明,在MnO2氧化Fe(Ⅱ)与As(Ⅲ)共存的体系中,Fe(Ⅱ)与As(Ⅲ)存在竞争氧化,Fe(Ⅱ)被氧化后可在锰氧化物表面形成了施氏矿物层,这降低了As(Ⅲ)的氧化速率[25]. 施氏矿物是砷酸盐和亚砷酸盐的重要寄主矿物[26],因此溶液中的As(Ⅲ)可以通过施氏矿物涂层并到达MnO2表面进行氧化. 而本工作的研究结果表明,当Fe(Ⅱ)被黑锰矿氧化形成铁氧化物包覆的黑锰矿后,氮气氛围下Fe(Ⅱ)氧化被抑制,即Fe(Ⅱ)并不能穿过铁氧化物包覆层继续被氧化. 但空气氛围下Fe(Ⅱ)可被继续氧化,而单一的Fe(Ⅱ)在溶解氧环境被氧化的速率可以忽略. 因此,反应后期Fe(Ⅱ)的氧化主要是溶解氧存在下形成铁氧化物的催化氧化作用导致.
锰平均氧化度常被用于判断锰氧化物氧化程度. 黑锰矿在氧化Fe(Ⅱ)过程中,结构中的Mn(Ⅲ)被还原伴随着Mn(Ⅱ)的释放. 利用XAS表征了pH 5.0条件下,黑锰矿氧化Fe(Ⅱ) 0.5 h和6 h后固相产物Mn的平均价态(图4a). 采用Combo方法分析产物中Mn(Ⅱ、Ⅲ、Ⅳ)的相对含量及平均价态,拟合结果见表1. 当黑锰矿与Fe(Ⅱ)在pH 5.0条件下反应0.5 h后,锰氧化度为2.91,反应6 h后降为2.71,Mn(Ⅲ)的相对含量从75%降至69%,Mn(Ⅱ)相对含量从17%增加至30%,表明黑锰矿只能被部分还原,与体系中形成铁氧化物包覆层后Mn(Ⅱ)的浓度不再增加一致.
利用XRD难以鉴定Fe(Ⅱ)在氧化过程中形成的弱晶态铁氧化物. 因此采用Fe K边EXAFS光谱的线性拟合确定pH 5.0下可能的中间产物(图4b). 拟合过程中所用标样包括针铁矿、纤铁矿和水铁矿. 结果表明黑锰矿氧化Fe(Ⅱ)的中间产物主要为水铁矿及少量纤铁矿和针铁矿. 反0.5 h后产物中水铁矿、纤铁矿和针铁矿的相对含量分别为94.4%、1.2%和4.4%;反应6 h后相对含量分别为96.2%、0%和3.8%. 也即反应后期(6 h)水铁矿的相对含量略有增加. 有研究发现在水钠锰矿氧化Fe(Ⅱ)反应体系中,Fe(Ⅱ)的前期氧化产物为弱晶质铁氧化物,随后转化形成针铁矿和纤铁矿[11]. 也有研究发现Fe(Ⅱ)在弱晶质铁氧化物表面被氧化会生成与原始铁氧化物不同晶相的产物[4,27],因此,本工作中反应前期生成的水铁矿只是针铁矿和纤铁矿形成的前驱体. 反应后期Fe(Ⅱ)进一步被催化氧化,促进了水铁矿向针铁矿和纤铁矿的转化.
图5为pH 5.0空气氛围下0.5 g·L−1黑锰矿和8.0 mmol·L−1 Fe(Ⅱ)在0、1 h、6 h、12 h的透射电镜图片. 黑锰矿的初始形貌为方块状,粒径约50—100 nm. 反应进行1 h后,黑锰矿表面生成少量薄片状颗粒物. 在6 h和12 h时黑锰矿表面形成絮状颗粒物,同时片状颗粒物长大,表明黑锰矿逐渐被还原,生成薄片状针铁矿. XAS和TEM分析结果进一步表明,pH 5.0下Fe(Ⅱ)氧化的中间产物主要为水铁矿,终产物为针铁矿和纤铁矿的混合物.
以上结果表明黑锰矿与Fe(Ⅱ)的氧化体系中,Fe(Ⅱ)被氧化生成水铁矿随后转化成针铁矿和纤铁矿,同时黑锰矿还原释放Mn(Ⅱ)到溶液,总的反应式如下:
当Fe(Ⅱ)不足量时,Fe(Ⅱ)被完全吸附和氧化,过量的Fe(Ⅱ)消耗是溶解态的Fe(Ⅱ)被吸附所致的. 吸附的Fe(Ⅱ)一般可由0.4 mol·L−1 HCl提取[28-29],但黑锰矿在低pH下易溶解并发生歧化反应. 此外,当初始Fe(Ⅱ)浓度较低时,氮气和空气氛围下消耗的Fe(Ⅱ)没有差异,均可被完全吸附或氧化. Fe(Ⅱ)足量时,在反应后期Mn(Ⅱ)浓度平衡后,只有空气氛围下Fe(Ⅱ)才可进一步被溶解氧氧化. 因此本工作中没有单独区分吸附和溶解态Fe(Ⅱ),统一归为消耗的Fe(Ⅱ).
在pH 5.0的空气氛围下,8 mmol·L−1 Fe(Ⅱ)被空气氧化的速率极其缓慢,1 d内的氧化量几乎可忽略. 然而黑锰矿存在的反应体系中,反应前期黑锰矿快速Fe(Ⅱ)氧化,反应后期当体系中有溶解氧存在时,Fe(Ⅱ)才能被进一步缓慢氧化. 在Fe(Ⅱ)与黑锰矿反应后期,Fe(Ⅱ)的氧化产物主要为针铁矿及还原的Mn(Ⅱ). 而反应前期形成水铁矿由于结构不稳定被转化为针铁矿和纤铁矿,故水铁矿是针铁矿形成的前驱体. 为此进一步考察了体系中生成的主要固相产物针铁矿及黑锰矿被还原形成的Mn(Ⅱ)对Fe(Ⅱ)氧化过程的影响,图6a所示为Mn(Ⅱ)和Fe(Ⅱ)共存体系中不同时间段Fe(Ⅱ)和Mn(Ⅱ)浓度. 反应12 h后,Mn(Ⅱ)和Fe(Ⅱ)浓度均没有变化,也即Mn(Ⅱ)对Fe(Ⅱ)的氧化过程无明显影响. 在氮气氛围下,加入针铁矿后对Fe(Ⅱ)的氧化无影响(图6b). 而空气氛围下,随反应进行Fe(Ⅱ)浓度明显降低. 反应12 h后,Fe(Ⅱ)浓度由8.0 mmol·L−1降为6.2 mmol·L−1,这表明生成的针铁矿可在溶解氧作用下催化氧化Fe(Ⅱ),进而促进Fe(Ⅱ)进一步氧化.
-
pH显著影响Fe2+的氧化[17]. 图7a—c为低pH(pH 3.0)环境下,0.5 g·L−1黑锰矿与不同浓度Fe(Ⅱ)在空气和氮气氛围下相互作用24 h后溶液中Mn(Ⅱ)、Fe(Ⅱ)浓度和空气与氮气消耗的Fe(Ⅱ)浓度差.
未加入Fe(Ⅱ)时,溶液中就有较高浓度的Mn(Ⅱ),表明黑锰矿发生了溶解. 随初始Fe(Ⅱ)浓度升高,体系中溶解释放的Mn(Ⅱ)浓度先逐渐增加后保持平衡. 且空气氛围下消耗的溶解态Fe(Ⅱ)量大于氮气氛围,即低pH环境溶解氧促进了Fe(Ⅱ)氧化.
图8为pH 3.0下,0.5 g·L−1黑锰矿与8.0 mmol·L−1 Fe(Ⅱ)体系中不同时间点溶解的Fe(Ⅱ)和Mn(Ⅱ)浓度. 随反应进行,溶解的Mn(Ⅱ)浓度逐渐升高且12 h仍未平衡,Fe(Ⅱ)浓度逐渐降低且空气和氮气氛围下Fe(Ⅱ)氧化速率基本一致. 这些结果表明低pH条件下,溶解氧并未影响黑锰矿氧化Fe(Ⅱ)速率,但促进了Fe(Ⅱ)被氧化的总量. 与pH 5.0相比,空气氛围pH 3.0体系消耗的溶解态Fe(Ⅱ)量更少,即pH升高有利于Fe(Ⅱ)氧化. 这与水钠锰矿氧化Fe(Ⅱ)的速率规律不同[29]. 水钠锰矿在pH 3.0和pH 5.0均较稳定,依据能斯特方程可知酸性条件下更有利于Fe(Ⅱ)氧化. 此外,在pH < 4.0时水钠锰矿氧化Fe(Ⅱ)速度快,反应几分钟内就有75%的Fe(Ⅱ)被氧化;当pH > 4.0时,Fe(Ⅱ)初始氧化速率较快. 随着反应的进行,水钠锰矿氧化Fe(Ⅱ)形成的FeOOH沉淀覆盖了锰氧八面体上的活性位点. 因此Fe(Ⅱ)的氧化速率表现出随pH升高而降系[17,30]. 而在黑锰矿与Fe(Ⅱ)反应体系中,pH 3.0时黑锰矿不稳定,易发生歧化反应,生成的产物氧化能力随之减弱. 此外随着反应进行,Fe(Ⅱ)被氧化后在黑锰矿表面形成了铁氧化物包覆层,抑制了Fe(Ⅱ)进一步氧化. 体系中溶解氧在反应后期参与了Fe(Ⅱ)氧化. pH可显著影响Fe(Ⅱ)的氧化速率,pH升高使得Fe(Ⅱ)水解加剧生成Fe(OH)+和Fe(OH)2等,它们与溶解氧反应活性要比游离态Fe(Ⅱ)高几个数量级[31-32]. 因此溶解氧存在时高pH下黑锰矿更能快速氧化Fe(Ⅱ). 另外,pH升高后黑锰矿表面负电荷增加,更多Fe(Ⅱ)吸附在黑锰矿表面,提升了氧化速率.
为明确pH对黑锰矿氧化Fe(Ⅱ)产物的影响,利用XRD表征空气氛围下0.5 g·L−1黑锰矿与不同初始浓度Fe(Ⅱ)在pH 3.0和5.0下反应24 h后的固相产物(图9).
先前研究表明在pH 3.0时,初始的黑锰矿会歧化成δ-MnO2,随后转化成γ-MnO2或是β-MnO2[33]. 且Fe(Ⅱ)浓度增加,黑锰矿(JCPDS No. 14-0644)的特征峰逐渐减弱,即使与8.0 mmol·L−1初始Fe(Ⅱ)反应,仍能观察到黑锰矿的晶体结构. Fe(Ⅱ)氧化生成的针铁矿的结晶度随初始Fe(Ⅱ)浓度逐渐增强(图9a). 当pH为5.0时,未加入Fe(Ⅱ)时黑锰矿晶体结构稳定. 随初始Fe(Ⅱ)浓度升高,黑锰矿的特征峰逐渐减弱. 黑锰矿与8.0 mmol·L−1 Fe(Ⅱ)反应的产物主要为针铁矿(α-FeOOH, JCPDS card No. 81-0464)和少量纤铁矿(γ-FeOOH, JCPDS card No. 74-1877)混合物.
-
环境中广泛存在高吸附和氧化活性的铁锰氧化物,它们影响着重金属和有机物的迁移转化[34-35]. 本研究结果也表明,在偏酸性环境Fe(Ⅱ)可被黑锰矿氧化,并在其表面形成包覆层抑制了Fe(Ⅱ)的进一步氧化,同时释放Mn(Ⅱ). 这可能会阻碍一些酸性矿山废水和海水孔隙水环境里有毒元素如Cd(Ⅱ)、Sb(Ⅲ)的吸附固定,增加它们的潜在环境风险[25,33]. 有研究发现β-MnO2氧化Fe(Ⅱ)的反应过程中,Fe(Ⅱ)多次加入后仍可被锰氧化物氧化,且形成的复合物具有较高的氧化活性[36]. 在我们的研究中也发现生成的铁氧化物包覆层具有催化效应,在溶解氧作用下可促进Fe(Ⅱ)的进一步氧化,这可能会影响环境中的一些变价有毒(类)重金属元素如Se(Ⅳ)、Cr(Ⅲ)、As(Ⅲ)及有机物的迁移和毒性[37]. 因此,明确环境中常见的黑锰矿与Fe(Ⅱ)的反应机理及影响因素有助于认识重金属元素和微量元素的循环过程.
-
(1)在偏酸性环境中Fe(Ⅱ)可被黑锰矿氧化,并在其表面形成了以针铁矿为主和少量纤铁矿组成的包覆层,阻碍了Fe(Ⅱ)的进一步氧化.
(2)形成的铁氧化物包覆层具有催化效应,可在溶解氧作用下催化氧化Fe(Ⅱ),促进Fe(Ⅱ)的进一步及铁氧化物生成.
(3)pH 影响Fe(Ⅱ)的氧化速率和产物,Fe(Ⅱ)的氧化速率随pH 升高而升高. 且低pH环境有利于针铁矿的形成.
黑锰矿催化氧化Fe(Ⅱ)生成铁氧化物过程及影响因素
Catalytic oxidation process of Fe(Ⅱ) to iron oxides by hausmannite in the presence of dissolved oxygen and its influence factors
-
摘要: 自然界中的锰氧化物常与铁氧化物交结伴生,其形成和转化过程相互影响. Mn(Ⅳ)氧化物与Fe(Ⅱ)反应过程已有较多研究,然而有氧环境中低价锰氧化物氧化Fe(Ⅱ)生成铁氧化物的过程尚缺乏系统研究. 本工作以黑锰矿为例,研究了开放体系中Fe(Ⅱ)在低价氧化锰矿物表面的氧化行为,分析了Fe(Ⅱ)浓度、溶解氧以及pH对Fe(Ⅲ)氧化物形成的影响. 结果表明,黑锰矿在氧化Fe(Ⅱ)形成针铁矿和纤铁矿的同时,自身被部分还原释放Mn(Ⅱ). 当反应体系pH值为3.0时,溶解氧氧化Fe(Ⅱ)作用弱,主要为黑锰矿氧化Fe(Ⅱ)并在其表面生成针铁矿. 当反应体系pH值升高至5.0,黑锰矿催化加速了溶解氧对低浓度(<5.0 mmol·L−1)Fe(Ⅱ)的氧化并生成水铁矿,随后转化成纤铁矿和针铁矿;Fe(Ⅱ)浓度升高(>5.0 mmol·L−1),反应初期Fe(Ⅱ)直接被黑锰矿氧化,形成了以针铁矿和少量纤铁矿组成的包覆层,并导致氧化速率减弱,而溶解氧在Fe(Ⅱ)后期氧化过程中发挥了主导作用. 这些结果丰富了表生环境铁锰氧化物的形成与共生机理,为土壤矿物形成演化提供了基础数据.Abstract: The formation and transformation processes of manganese oxides are usually associated with iron oxides in soils. The reaction of Fe (Ⅱ) with Mn(Ⅳ) oxides has attracted much attention. However, the oxidation process of dissolved Fe(Ⅱ) to iron oxides on the surface of low-valent manganese oxides still lacks systematic research in aerobic environments. In this work, the oxidation behavior of Fe(Ⅱ) on the surface of a representative low-valent manganese oxide mineral, hausmannite, was investigated in an open system, and the effects of Fe(Ⅱ) concentration, pH and dissolved oxygen on the oxidation processes and kinetics were also analyzed. The results showed that hausmannite was reduced to Mn(Ⅱ) and Fe(Ⅱ) was oxidized with the formation goethite and lepidocrocite. At pH 3.0, the oxidation of Fe(Ⅱ) was mainly mediated by hausmannite to form goethite, and the presence of dissolved oxygen had no significant effect on the increase of Fe(Ⅱ) oxidation rate. When pH was increased to 5.0, the catalytic oxidation of Fe(Ⅱ) of low-concentration (< 5.0 mmol·L−1) occurred by hausmannite in the presence of dissolved oxygen, and only ferrihydrite was formed as intermediate, which was subsequently transformed to goethite and lepidocrocite. When the concentration of Fe(Ⅱ) was increased to above 5.0 mmol·L−1, Fe(Ⅱ) was directly oxidized by hausmannite to generate a coating composed of goethite and a small amount of lepidocrocite at the initial stage, resulting in a decrease in the oxidation rate and then dissolved oxygen played a major role in the oxidation of Fe(Ⅱ) oxidation. These results have a better understanding about the formation and symbiosis mechanism of iron and manganese oxides in supergene environments, and provide the basis data on formation and evolution of soil minerals.
-
Key words:
- manganese oxide /
- iron oxide /
- Fe(Ⅱ) /
- catalytic oxidation /
- dissolved oxygen
-
在众多的污水处理方法中,活性污泥法受到人们的广泛关注,活性污泥法作为重要的处理污水方法之一,具有很多优势. 但是随着国内外对污水治理的日益重视和城市污水处理厂的不断建设,大量的剩余污泥作为活性污泥法处理污水的副产物排出[1]. 污泥因其含水率高、含有大量病原体和微生物等有害生物、重金属及有机物含量高等特点,容易对环境造成二次污染[2],污泥的有效处理处置是亟待解决的重要问题. 污泥脱水是常规的污泥处理方法,在污泥脱水之前需要经过一定的调理使其满足后续脱水要求,所以,选择合适的污泥调理方法对改善污泥脱水性能尤为重要.
过氧化钙(CaO2)作为一种热稳定性好的环境友好型材料,被广泛应用于农业种植、水产养殖、食品保存、医疗以及环境领域[3]. CaO2具有高能的过氧化物共价键,当CaO2与水接触时,能够缓慢释放过氧化氢(H2O2),同时还会生成羟基自由基、过氧化氢自由基等具有强氧化性的自由基(反应式见式(1—5))[4]. 近年来,因其具有稳定的氧化性,CaO2在污泥处理方面的应用成为一个新的研究热点. Wang 等研究发现,通过CaO2预处理污泥后,难降解有机物可以转化为可生物降解,促进污泥中可生物降解基质的水解和分解代谢,进而增强污泥厌氧消化效果[5]. 有研究表明,CaO2可以破解污泥EPS结构,释放污泥中的束缚水[6]. Wang等的研究表明,通过联合CaO2和微波预处理污泥,预处理后污泥的CST值相较于原泥下降52% [7]. 通过热处理与CaO2联合调理,可以提升污泥脱水性能[8].
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) stringUtils.convertMath(!{formula.content}) (5) 除了直接使用CaO2对目标物进行氧化,对CaO2进行活化也是一种常用的技术[8]. 有研究认为,通过微波活化CaO2,能促进CaO2产生更多的HO·和·O2-[7]. 通过过渡金属(Fe2+/Fe3+和Ag+)活化CaO2分解是常用的活化方法[9]. 利用Fe2+活化CaO2可以形成类芬顿反应,但如果不进行pH调节, Fe2+易于被氧化成Fe3+,限制了芬顿反应的效率. 有研究指出,利用含铁矿物对H2O2进行活化可以克服这一缺陷[10]. 黄铁矿(FeS2)是一种常见的脉石矿物,与矿床中的有价矿物伴生,可通过常规浮选方法轻松处理[11]. 最近有研究发现,利用黄铁矿活化CaO2降解磺胺,相比常规的芬顿反应,磺胺的氧化效率从30%提升至80%,(主要反应见式(6—9))[12]. Zhou等研究表明利用黄铁矿活化CaO2处理邻苯二甲酸二乙酯(DEP),78%的DEP在24 h内被降解[13]. 这些结果说明,通过黄铁矿活化CaO2能有效促进HO·产生,但目前尚未发现关于利用黄铁矿活化过氧化钙调理污泥的研究,其对污泥脱水性能的影响及机理尚未清晰,因此本研究利用黄铁矿-CaO2作为一种新型的芬顿法对污泥进行调理,以期达到破解EPS从而释放结合水的效果,并通过EPS性质及污泥絮体性质变化探究其对污泥脱水性能的影响机理.
stringUtils.convertMath(!{formula.content}) (6) stringUtils.convertMath(!{formula.content}) (7) stringUtils.convertMath(!{formula.content}) (8) stringUtils.convertMath(!{formula.content}) (9) 本研究对不同污泥样品进行EPS的提取,并对提取出来的EPS样品进行含量测定、三维荧光光谱检测,以表征调理前后污泥EPS性质变化. 同时对不同污泥样品的粒径分布进行检测,探究调理方法对污泥絮体团聚性能变化的影响.
1. 材料与方法(Materials and methods)
1.1 实验材料
本研究中污泥取自于广州市某污水处理厂二沉池,污泥取至实验室后,先过20目筛,去除大颗粒杂质和毛发,之后置于冰箱在4 ℃下保存. CaO2采购于上海麦克林生化科技有限公司. 黄铁矿采购于佛山市大昌顺材料科技有限公司,黄铁矿在使用之前对其进行研磨,并过100目筛,利用0.1 mol·L−1HNO3 洗去表面杂质及氧化层,干燥后备用[14].
1.2 实验方法
1.2.1 污泥脱水性能实验
为了探究不同调理条件对污泥脱水性能的影响,本研究对黄铁矿单独调理、CaO2单独调理以及两者复合调理污泥进行实验室规模的污泥脱水性能实验,250 mL的烧杯作为污泥调理容器,在调理容器中加入100 mL污泥样品进行实验. 利用重量法对污泥总固体(TS)进行测定[15]. 在黄铁矿单独调理实验中,设置6组不同黄铁矿调理剂量实验组,各组黄铁矿投加量分别为0、1、2、4、6 g·L−1. CaO2单独调理实验中,设置6组不同CaO2调理剂量实验组,各组CaO2投加量分别为10、30、50、80、100 mg·g−1 TS. 为了研究单独调理与复合调理以及不同复合调理方法之间的污泥脱水性能变化,设置了两组复合调理实验,第一组:CaO2投加量30 mg·g−1 TS,黄铁矿投加剂量1 g·L−1,第二组:CaO2投加量100 mg·g−1 TS,黄铁矿投加剂量1 g·L−1. 将单独调理和复合调理的实验组分别设置为A30、A100和B30、B100. 其中,A30为30 mg·g−1 TS CaO2单独调理,B30为30 mg·g−1 TS CaO2 +1 g·L−1黄铁矿复合调理,A100为100 mg·g−1 TS CaO2单独调理,B100为100 mg·g−1 TS CaO2 +1 g·L−1黄铁矿复合调理.
1.3 分析方法
1.3.1 污泥脱水性能
本研究中利用毛细吸水时间(CST)作为评价污泥脱水性能的指标. CST利用CST测定仪进行测定(HDFC-10A),利用测定后CST数据进行标准化CST(SCST)计算[16],计算公式如下:
stringUtils.convertMath(!{formula.content}) 其中,CSTa为调理后污泥样品的CST值,CST0为原泥的CST值.
1.3.2 EPS提取及其分析
在本研究中,EPS根据其存在形态分类为溶解性EPS(S-EPS)、松散束缚EPS(LB-EPS)和紧密束缚EPS(TB-EPS)[17],本研究采用一种改进的热提取方式对EPS进行提取,具体方法参照文献[18]. EPS中的多糖含量利用硫酸-蒽酮法测定,蛋白质含量利用福林酚法进行测定[19].
1.3.3 三维荧光光谱(3D-EEM)测定方法
本研究中利用荧光光谱仪(Hitachi F-4600)对提取出的EPS进行3D-EEM的测定,光谱数据的发射波长(Em)以及激发波长(Ex)范围从220 nm到450 nm,采集间隔为10 nm. 光谱数据的利用5 nm的发射和激发狭缝带宽以及1500 nm·min−1的扫描速度进行收集.
1.3.4 污泥絮体粒径测定方法
本研究利用激光粒度仪(Mastersize 3000)对污泥絮体粒径分布及絮体粒径D50和D90值的测定. 其中,D50与D90分别定义为颗粒直径的第50和第90百分位数[20].
2. 结果与讨论(Results and discussion)
2.1 污泥脱水性能变化
由图1可见,单独投加CaO2之后,污泥SCST值随着CaO2的投加量的增加呈现先下降再上升的趋势,单独投加CaO2,投加量为30 mg·g−1 TS的实验组SCST值最低为0.61. 在投加剂量不高于80 mg·g−1 TS时,CaO2单独调理有利于提升脱水性能,但当CaO2投加量增加至100 mg·g−1 TS时,SCST值增加至1.39,说明过量的CaO2不仅不会提升污泥脱水性能,反而会使得原污泥脱水性能下降. 随着黄铁矿投加量增加,黄铁矿单独调理的SCST值也表现出先下降再上升的,最优黄铁矿单独调理剂量为1 g·L−1,SCST值为0.70. 但当投加量继续增加时,黄铁矿单独调理对污泥脱水性能的提升效果变弱,在投加量为6 g·L−1的单独调理下,SCST值为0.92,污泥脱水性能提升不明显. 这说明过量的过氧化钙投加,带来过强的氧化性能,会使得污泥的脱水性能下降,这一趋势与Chen等的研究结果相似,过强的氧化性可能会导致过量的EPS释放,降低污泥脱水性能[6]. 但在CaO2投加量为30 mg·g−1 TS复合调理时,虽然氧化性能更强,但污泥有更佳的脱水性能,SCST值下降至0.55,这说明利用黄铁矿活化过氧化钙对污泥进行复合调理能有效提升污泥的脱水性能.
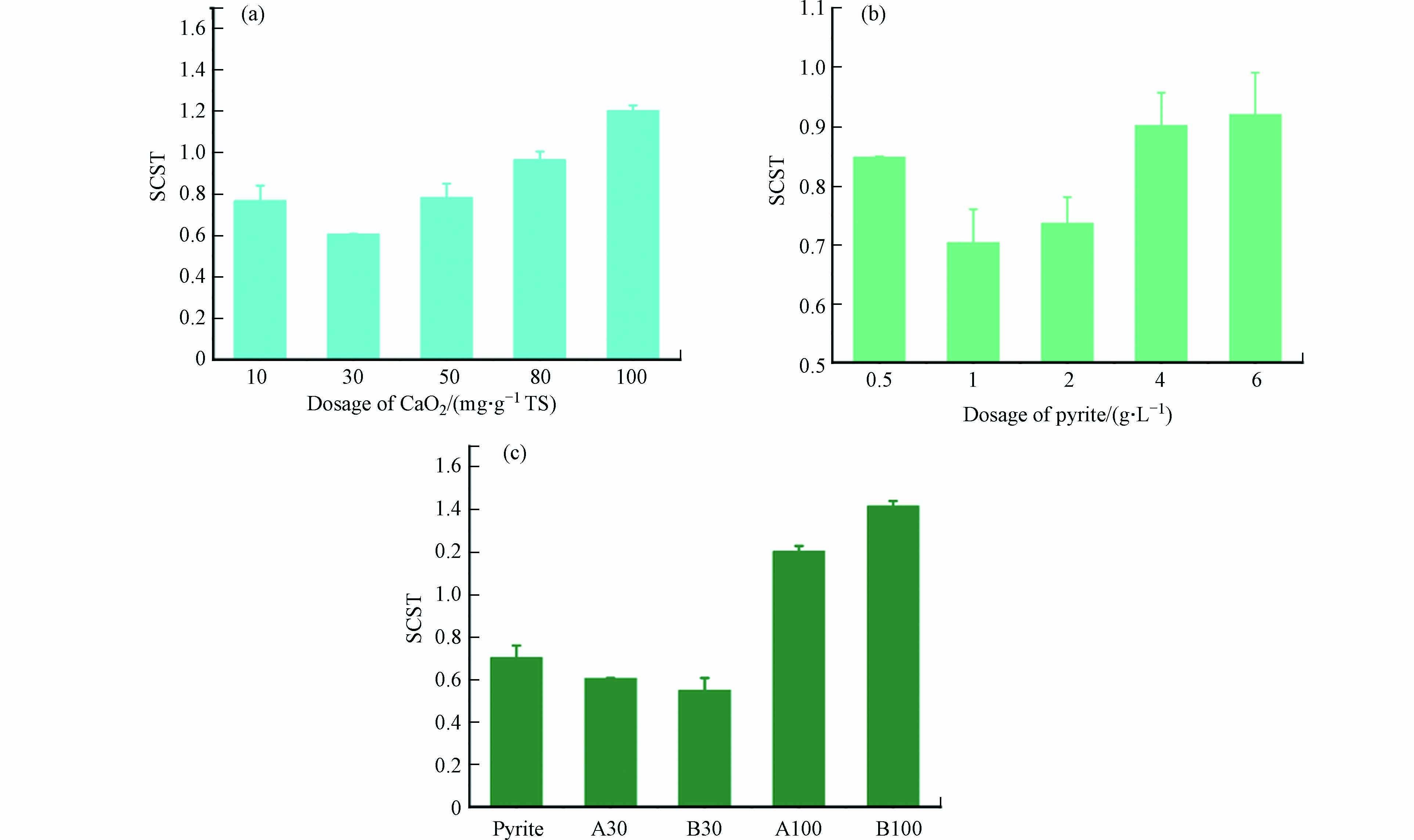 图 1 过氧化钙(a)、黄铁矿(b) 单独调理和复合调理(c)污泥样品的SCST值Figure 1. SCST values of calcium peroxide (a), pyrite (b) single conditioning and composite conditioning (c) sludge samples
图 1 过氧化钙(a)、黄铁矿(b) 单独调理和复合调理(c)污泥样品的SCST值Figure 1. SCST values of calcium peroxide (a), pyrite (b) single conditioning and composite conditioning (c) sludge samples2.2 EPS含量变化
不同结构的EPS对剩余污泥的脱水性能影响程度可能不同,Dai等认为S-EPS中有机物含量较高或LB-EPS中有机物含量较低,具有较好的脱水性能[21]. He等指出污泥脱水性与S-EPS中有机物浓度呈正相关,而与LB-EPS中生物聚合物含量呈负相关[22]. 剩余污泥脱水性能除了和EPS的组成结构有关,还与EPS的组成成分相关,Wei等研究发现,污泥脱水性能与EPS中蛋白质含量呈负相关性[23],而且蛋白质含量是决定污泥脱水性能的关键因素[24],为了进一步探究污泥调理过程中污泥性质的变化,本研究对提取出的EPS样品进行蛋白质和多糖含量的测定. CaO2调理后污泥EPS结构发生明显的变化(图2a),在30 mg·g−1 TS的CaO2投加量下,S-EPS蛋白质含量略有下降,而内层EPS(LB-EPS、TB-EPS)蛋白质含量增加,相较于单独调理,CaO2/黄铁矿复合调理由于其更强的氧化性能,在CaO2投加量为30 mg·g−1 TS时的复合调理污泥样品中,内层EPS蛋白质含量增加幅度更大. 当CaO2投加量增加至100 mg·g−1 TS后,所有层EPS中蛋白质含量均增加,与低CaO2投加量相似,复合调理因其更强的氧化性,内部EPS含量较单独调理增加更多. 调理后污泥的总EPS(T-EPS)蛋白质含量均增加,高剂量CaO2导致更多的蛋白质释放,而复合调理对蛋白质含量的提升高于单独调理.
 图 2 原泥以及调理后污泥EPS中蛋白质含量(a),多糖(b)蛋白质-多糖含量比率(c)变化Figure 2. The concentration of protein (a), polysaccharide (b) and the ratio of protein to polysaccharide (c) in EPS of raw sludge and conditioned sludge
图 2 原泥以及调理后污泥EPS中蛋白质含量(a),多糖(b)蛋白质-多糖含量比率(c)变化Figure 2. The concentration of protein (a), polysaccharide (b) and the ratio of protein to polysaccharide (c) in EPS of raw sludge and conditioned sludge调理前后EPS多糖含量的变化见图2b,随着CaO2投加量增加,内外层EPS多糖含量均增加. 值得注意的是,高CaO2投加剂量的复合调理样品中,S-EPS和LB-EPS的多糖含量较单独调理均下降. T-EPS中多糖的变化趋势与蛋白质不同,T-EPS中多糖含量随着氧化性能的增强表现出先增加后下降的趋势,这可能是低CaO2剂量调理下,EPS结构被破解,内层EPS释放至外层. 但在高剂量CaO2的复合调理下,多糖类物质可能被分解为更小的有机分子或直接被矿化,导致T-EPS中多糖含量下降.
有研究认为,LB-EPS中蛋白质/多糖比率(PN/PS)与脱水性有负相关性[25]. 本实验中,B30样品LB-EPS的PN/PS最小(图2c),且无论高剂量或低剂量,在同一剂量下复合调理得到的LB-EPS样品,其PN/PS值均小于单独调理. 但当用高剂量过氧化钙对污泥进行调理后,LB-EPS中的PN/PS上升,污泥脱水性能下降. 但本实验发现,高剂量的过氧化钙调理后虽然PN/PS上升,但仍然低于原泥,这与脱水性能变化不一致,这是因为污泥脱水性能的变化影响十分复杂,并不能只靠EPS中的PN/PS进行指示.
从EPS含量变化可以看出,使用CaO2单独调理以及CaO2/黄铁矿复合调理都可以改变EPS原有结构,破解EPS结构. 在同一CaO2投加量下,复合调理得到的EPS破解效果更加明显. 结合污泥脱水结果分析,污泥调理方法在一定范围内对EPS结构进行破解,可能有利于污泥脱水性能的提升,但对EPS结构的过度破解可能会使得大量有机质的释放,进而使得污泥脱水性能下降.
2.3 3D-EEM
为了更深入地了解调理前后以及各调理方法对各层EPS的性质以及其含量的影响,本研究利用三维荧光光谱对各层EPS的有机成分进行表征,各样品EPS的三维荧光光谱见图3. 本研究中EPS的荧光光谱峰主要有两个,分别为A峰(Em/Ex:340 nm/225 nm)和B峰(Em/Ex:350 nm/280 nm). 根据Wen等提出的三维荧光光谱分区方法,A峰位于区域Ⅱ,归类为芳香类蛋白物质,B峰位于区域Ⅳ,归类为色氨酸和类蛋白物质[26].
 图 3 原泥(a)、A30(b)、B30(c)、A100(d)、B100(e)EPS样品三维荧光光谱图Figure 3. 3D-EEM spectra of EPS samples of raw sludge(a); A30(b); B30(c); A100(d); B100(e)
图 3 原泥(a)、A30(b)、B30(c)、A100(d)、B100(e)EPS样品三维荧光光谱图Figure 3. 3D-EEM spectra of EPS samples of raw sludge(a); A30(b); B30(c); A100(d); B100(e)A峰在原泥S-EPS中强度较低,但经过调理后,A峰强度上升,芳香类蛋白含量增加. 在A100中,S-EPS中的A峰出现最强的荧光强度,说明在此调理方法下内层EPS和胞内的芳香类蛋白向外释放,聚集在外层EPS中. 但经过氧化性更强的B100调理后,A峰强度下降,这可能是由于芳香类蛋白的分解导致含量下降. S-EPS中B峰的荧光强度在A30和B30调理下均下降,当CaO2投加量增加后,S-EPS的B峰强度增加,S-EPS中B峰最强峰强度出现在B100调理下. 原泥中LB-EPS中A峰和B峰强度稍强于S-EPS,经过预处理后污泥LB-EPS中A、B峰强度增加,且两峰强度的增加幅度明显大于S-EPS. 不同调理方法对LB-EPS的荧光光谱图影响与S-EPS相似,A、B峰在B100调理下均出现最强荧光强度. 原泥TB-EPS中的芳香类蛋白和色氨酸含量明显高于S-EPS和LB-EPS,这一结果与EPS含量一致. 不同调理手段下B峰强度在TB-EPS中的变化与在S-EPS、LB-EPS中的变化相似,B峰在A100调理下出现最大荧光强度,随后下降. 但与 S-EPS、LB-EPS 变化趋势不一致的是,TB-EPS 中 A 峰的最大荧光强度出现在 B30 调理下, 这一结果说明,芳香类蛋白比色氨酸更易于从胞内和内层 EPS 释放至胞外和外层 EPS.
荧光峰强度变化趋势可以说明,在一定条件下,随着调理方法的氧化性的增强,EPS中物质被分解,EPS结构破解程度增加,胞内物质向TB-EPS转移,同时TB-EPS中的物质向外层的LB-EPS和S-EPS转移. 当调理方法氧化性能过强,各层EPS中物质被分解甚至矿化,导致各层EPS中荧光峰强度下降,同时还发现,各层EPS中不同物质对于不同调理方法的变化趋势并不完全相同.
2.4 调理方法对絮体粒径的影响
由图4a可以看出,经过调理后的污泥絮体粒径分布曲线均向左移动,同时图4b中看到原泥有最大的D90以及D50值,调理后污泥的D50以及D90均有明显的下降,说明调理后污泥的絮体粒径下降. 这是由于强氧化性的调理方法将EPS结构破解后,会使得污泥絮体分解,形成尺寸更小的絮体[27]. 随着调理方法的氧化性能增强,污泥的粒径分布曲线左移程度越大,且有更小的D50和D90值,可以认为氧化性能越强的调理方法能够更高效、更彻底地破坏原有污泥絮体结构,使得原有稳定的大颗粒絮体失稳进而形成众多小尺寸的絮体. 这一现象与Ling等研究结果一致,通过对污泥絮体的破解,可以有效地释放束缚水,提升污泥脱水性能[28]. 在本研究中,在同一CaO2投加量下,复合调理后的污泥样品相较于单独调理后的污泥样品有更小的粒径,这也再次说明本研究中复合调理有更高效的EPS破解性能,但高剂量的过氧化钙投加量可能会过度破解絮体结构,过度破解絮体使得絮体粒径下降可能会增加小颗粒污泥对过滤介质的堵塞作用,降低污泥的脱水性能[29].
 图 4 原泥以及调理后污泥样品的絮体粒径分布(a)和粒径 D50、D90 变化(b)Figure 4. Floc particle size change distribution (a) and particle size D50, D90 change (b) of raw sludge and conditioned sludge samples
图 4 原泥以及调理后污泥样品的絮体粒径分布(a)和粒径 D50、D90 变化(b)Figure 4. Floc particle size change distribution (a) and particle size D50, D90 change (b) of raw sludge and conditioned sludge samples3. 结论(Conclusion)
本研究提出一种利用黄铁矿活化CaO2的污泥调理技术,结果表明,单独利用CaO2或者黄铁矿对污泥进行调理,随着CaO2或黄铁矿投加量的增加,污泥脱水性能呈现先上升后下降的趋势,在30 mg·g−1 TS CaO2和1 g·L−1黄铁矿的投加量下分别得到过氧化钙和黄铁矿的最优单独调理效果,同时发现,当CaO2和黄铁矿投加量为30 mg·g−1 TS和1g L−1时,复合调理后的污泥样品脱水性能优于单独调理. 但实现污泥脱水性能的提升需要对调理药剂投加量进行控制,过多的药剂投加可能会带来污泥脱水性能的下降.
-

图 1 pH 5.0体系,空气(a)和氮气(b)条件下,0.5 g·L−1黑锰矿与不同初始浓度Fe(Ⅱ)反应24 h后溶液中Fe(Ⅱ)、Mn(Ⅱ)、消耗的Fe(Ⅱ)及空气与氮气消耗的Fe(Ⅱ)浓度差(c)
Figure 1. Concentrations of dissolved Fe(Ⅱ), dissolved Mn(Ⅱ), consumed Fe(Ⅱ) in the systems of 0.5 g·L−1 hausmannite and Fe(Ⅱ) with different initial concentrations in air (a), N2 (b) and the consumtion difference of Fe(Ⅱ) (c) by air and N2 at pH 5.0 for 24 h
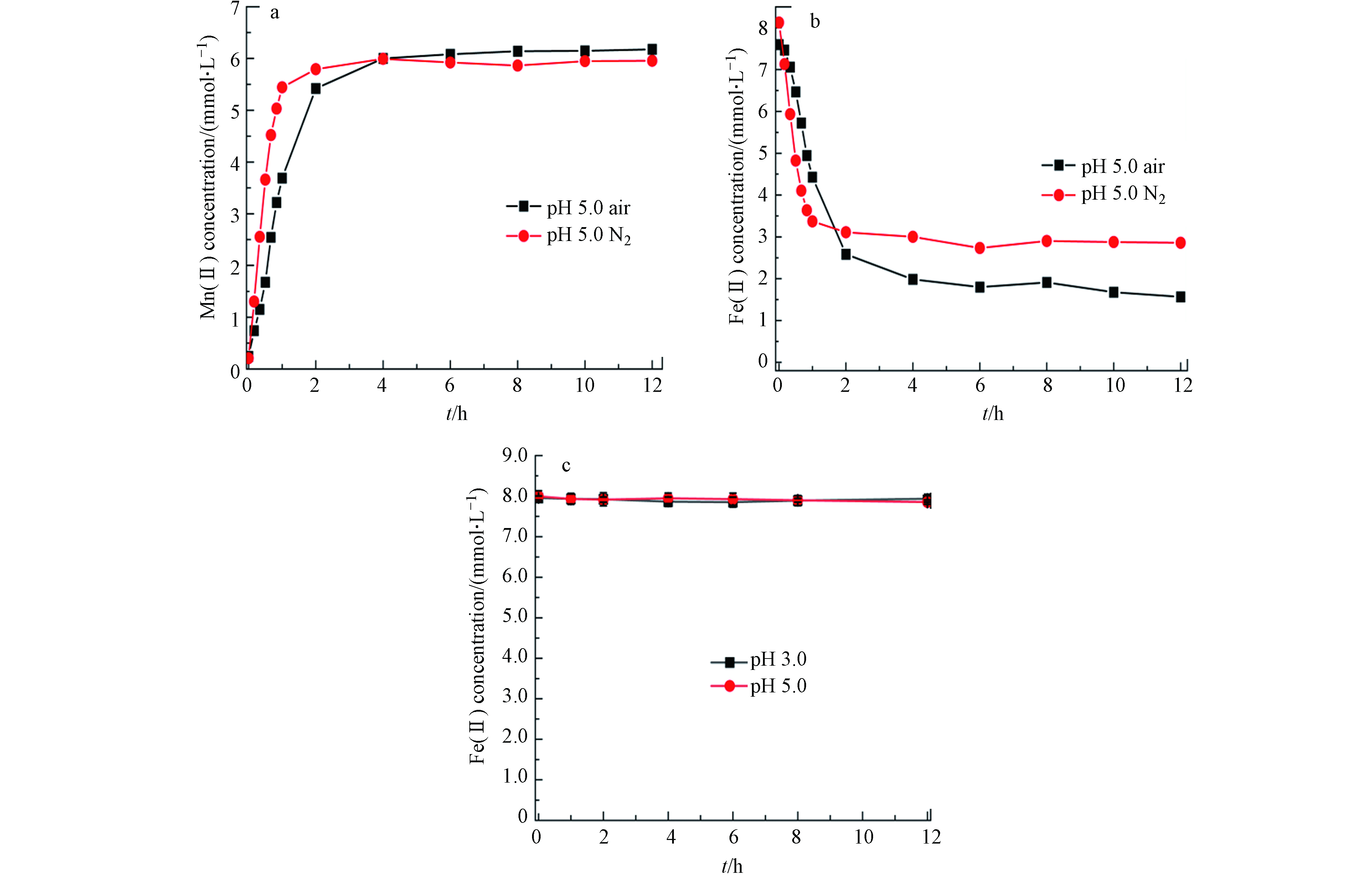
图 2 pH 5.0氮气和空气氛围下,0.5 g·L−1黑锰矿与8.0 mmol·L−1 Fe(Ⅱ)体系中不同时间溶解的Mn(Ⅱ) (a),Fe(Ⅱ) (b) 和8.0 mmol·L−1 Fe(Ⅱ)在不同pH,空气氛围下氧化12h后的Fe(Ⅱ) (c)浓度
Figure 2. Concentrations of dissolved Mn(Ⅱ) and Fe(Ⅱ) in the reaction system of 0.5 g·L−1 hausmannite and 8.0 mmol·L−1 Fe(Ⅱ) in air and nitrogen atmosphere at pH 5.0 (a, b) and Fe(Ⅱ) concentration in the reaction system of 8.0 mmol·L−1 Fe(Ⅱ) in air

图 3 pH 5.0空气氛围下,0.5 g·L−1黑锰矿和8.0 mmol·L−1 Fe(Ⅱ)在不同时间反应产物的XRD(a)和FTIR(b)谱图
Figure 3. XRD patterns (a) and FTIR spectra (b) of the products collected in the reaction system of 0.5 g·L−1 hausmannite and 8.0 mmol·L−1 Fe(Ⅱ) in air at pH 5.0
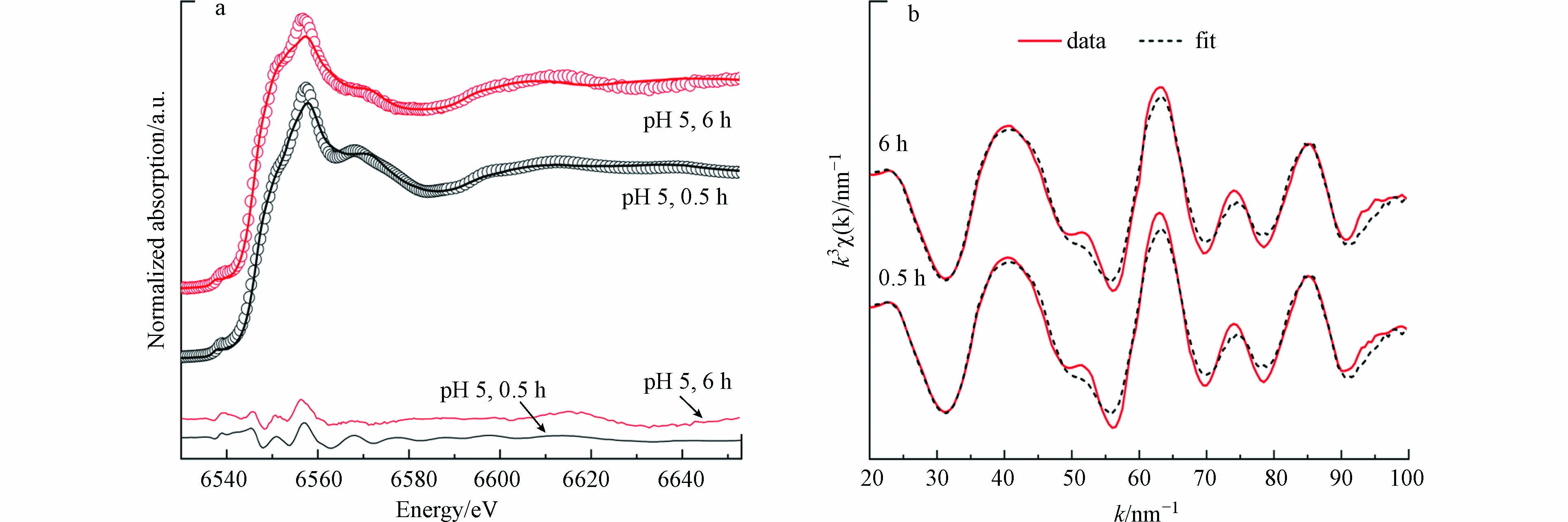
图 4 用Combo法和线性拟合方法对0.5 g·L−1黑锰矿和8.0 mmol·L−1 Fe(Ⅱ)在pH 5.0的空气氛围下产物的Mn K边XANES谱图(a)和Fe K边 EXAFS 谱图(b)拟合(图4a中圆圈为实验谱,实线为拟合谱,下方实线为残差谱)
Figure 4. Normalized Mn K-edge XANES (a) and the linear combination fitting of Fe K-edge k3-weighted EXAFS (b) spectra of the solid products collected in the rection system of 0.5 g·L−1 hausmannite and 8.0 mmol·L−1 Fe(Ⅱ) in air at pH 5.0

图 5 pH 5.0空气氛围下,0.5 g·L−1黑锰矿和8.0 mmol·L−1 Fe(Ⅱ) 反应 0 h(a)、1 h(b)、6 h(c)、12 h(d)产物的TEM照片
Figure 5. TEM images of the solid products collected in the reaction system of 0.5 g·L−1 hausmannite and 8.0 mmol·L−1 Fe(Ⅱ) after 0 h (a), 1 h (b), 6 h (c), 12 h (d) at pH 5.0 in air atmosphere
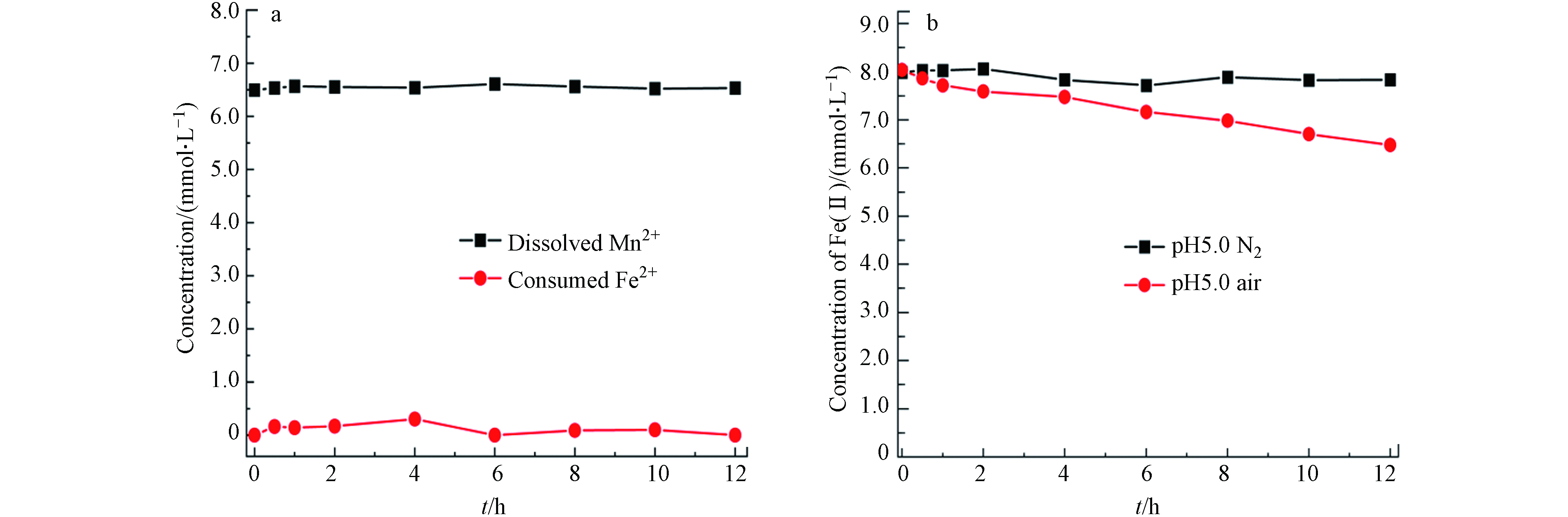
图 6 pH 5.0空气氛围下,6.5 mmol·L−1 Mn(Ⅱ) 和8.0 mmol·L−1 Fe(Ⅱ)(a)和pH 5.0空气、氮气氛围下0.86 g·L−1 针铁矿和8.0 mmol·L−1 Fe(Ⅱ)(b)体系中不同时间点Mn(Ⅱ)和Fe(Ⅱ) 的浓度
Figure 6. Concentrations of dissolved Fe(Ⅱ) and Mn(Ⅱ) in the aqueous system of 6.5 mmol·L−1 Mn(Ⅱ) and 8.0 mmol·L−1 Fe(Ⅱ) (a) and the concentration of dissolved Fe(Ⅱ) in the system of 0.86 g·L−1 goethite and 8.0 mmol·L−1 Fe(Ⅱ) (b) at pH 5.0 in air
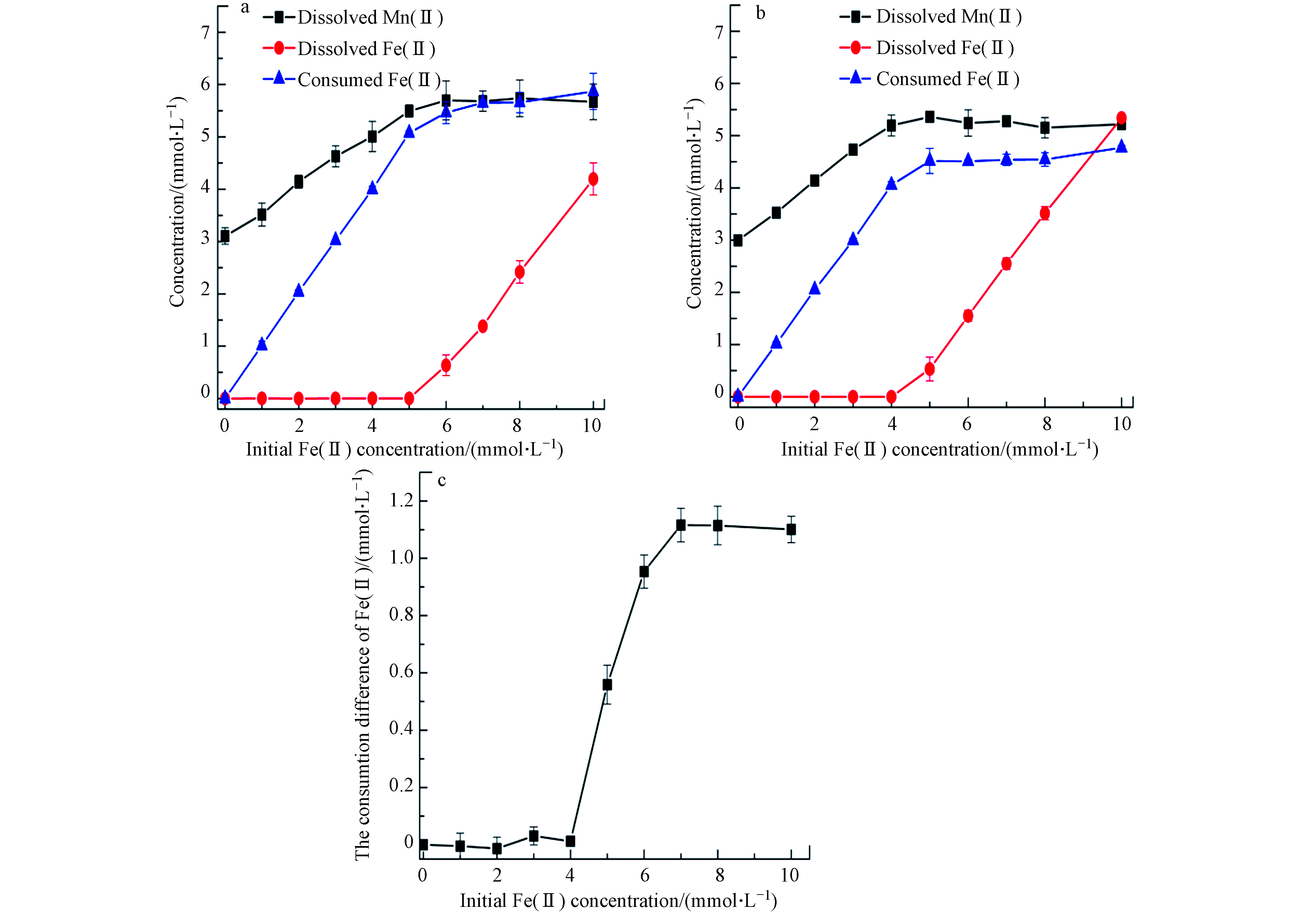
图 7 pH 3体系,空气(a)和氮气(b)条件下, 0.5 g·L−1黑锰矿与不同初始浓度Fe(Ⅱ)反应24 h后溶液中Fe(Ⅱ)、Mn(Ⅱ)消耗的Fe(Ⅱ)浓度及空气与氮气消耗的Fe(Ⅱ)浓度差(c)
Figure 7. Concentrations of dissolved Fe(Ⅱ), dissolved Mn(Ⅱ), consumed Fe(Ⅱ) in the systems of 0.5 g·L−1 hausmannite and Fe(Ⅱ) with different initial concentrations in air (a), N2(b) and the consumtion difference of Fe(Ⅱ) by air and N2 (c) at pH 3.0 for 24 h
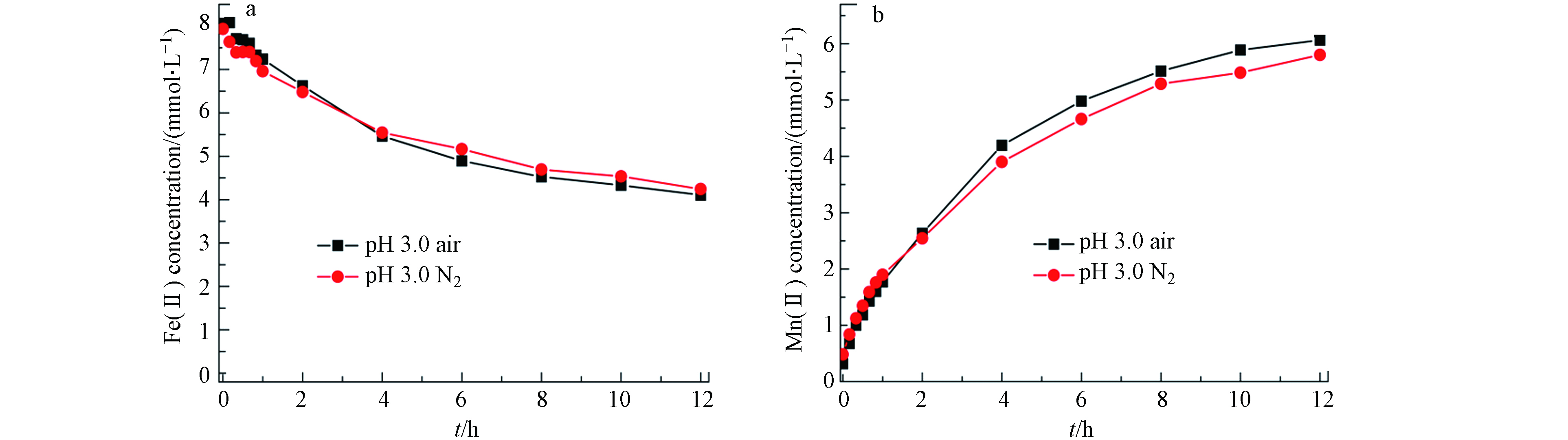
图 8 pH 3体系,0.5 g·L−1黑锰矿与8 mmol·L−1 Fe(Ⅱ)在空气与氮气体系中不同时间溶解的Fe(Ⅱ) (a)和Mn(Ⅱ)浓度(b)
Figure 8. Concentrations of Fe(Ⅱ) (a) and Mn(Ⅱ) (b) in the systems of 0.5 g·L−1 hausmannite and 8.0 mmol·L−1 Fe(Ⅱ) in air and nitrogen atmosphere at pH 3.0
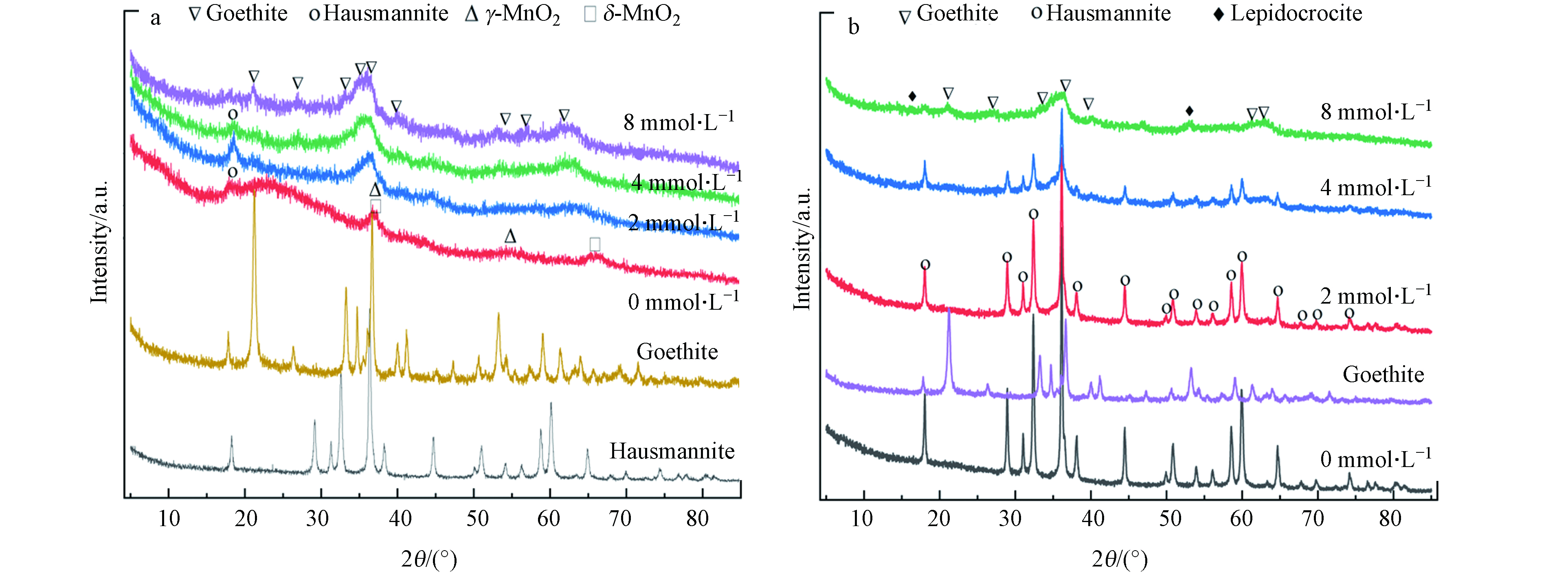
图 9 空气氛围下, pH 3.0(a), pH5.0(b)体系中0.5 g·L−1黑锰矿与不同初始浓度Fe(Ⅱ)反应24 h固相产物XRD谱图
Figure 9. XRD patterns of the solid products collected in the systems of 0.5 g·L−1 hausmannite and Fe(Ⅱ) with different initial concentration at pH 3.0 (a) and pH5.0 (b) for 24 hs in air atmosphere
表 1 pH 5.0空气氛围下,0.5 g·L−1黑锰矿和8.0 mmol·L−1 Fe(Ⅱ)反应产物的Mn(Ⅱ、Ⅲ、Ⅳ)的相对含量及平均锰氧化度
Table 1. Mn AOS and relative content of Mn(Ⅱ, Ⅲ, Ⅳ) of the solid products collected from the reaction of 0.5 g·L−1 hausmannite and 8.0 mmol·L−1 Fe(Ⅱ) for 0.5 and 6 h in air atmosphere at pH 5.0
反应时间/h Reaction time Mn(Ⅱ) Mn(Ⅲ) Mn(Ⅳ) Mn AOS 0 0.20 0.73 0.07 2.87 0.5 0.17 0.75 0.08 2.91 6 0.30 0.69 0.01 2.71  下载: 导出CSV
下载: 导出CSV
-
[1] 童蕾, 曾梦玲, 李民敬, 等. 铁锰氧化物对地下水环境中金霉素的降解 [J]. 环境化学, 2016, 35(5): 917-924. doi: 10.7524/j.issn.0254-6108.2016.05.2015120102 TONG L, ZENG M L, LI M J, et al. Degradation of chlorotetracycline by iron and manganese oxides under simulated groundwater environment [J]. Environmental Chemistry, 2016, 35(5): 917-924(in Chinese). doi: 10.7524/j.issn.0254-6108.2016.05.2015120102
[2] 胡敏, 李芳柏. 土壤微生物铁循环及其环境意义 [J]. 土壤学报, 2014, 51(4): 683-698. HU M, LI F B. Soil microbe mediated iron cycling and its environmental implication [J]. Acta Pedologica Sinica, 2014, 51(4): 683-698(in Chinese).
[3] 罗瑶, 李珊, 刘立虎, 等. 水锰矿与Fe2+的相互作用与转化过程 [J]. 岩石矿物学杂志, 2016, 35(4): 703-711. doi: 10.3969/j.issn.1000-6524.2016.04.010 LUO Y, LI S, LIU L H, et al. Interaction and transformation processes of manganite and Fe2+ [J]. Acta Petrologica et Mineralogica, 2016, 35(4): 703-711(in Chinese). doi: 10.3969/j.issn.1000-6524.2016.04.010
[4] LUO Y, LIU L H, QIAO W C, et al. Facile crystal-structure-controlled synthesis of iron oxides for adsorbents and anode materials of lithium batteries [J]. Materials Chemistry and Physics, 2016, 170: 239-245. doi: 10.1016/j.matchemphys.2015.12.044 [5] PEDERSEN H D, POSTMA D, JAKOBSEN R, et al. Fast transformation of iron oxyhydroxides by the catalytic action of aqueous Fe(II) [J]. Geochimica et Cosmochimica Acta, 2005, 69(16): 3967-3977. doi: 10.1016/j.gca.2005.03.016 [6] HOCHELLA M F J, LOWER S K, MAURICE P A, et al. Nanominerals, mineral nanoparticles, and Earth systems [J]. Science, 2008, 319(5870): 1631-1635. doi: 10.1126/science.1141134 [7] PALUMBO B, BELLANCA A, NERI R, et al. Trace metal partitioning in Fe-Mn nodules from Sicilian soils, Italy [J]. Chemical Geology, 2001, 173(4): 257-269. doi: 10.1016/S0009-2541(00)00284-9 [8] TEBO B M, BARGAR J R, CLEMENT B G, et al. BIOGENIC MANGANESE OXIDES: Properties and mechanisms of formation [J]. Annual Review of Earth and Planetary Sciences, 2004, 32: 287-328. doi: 10.1146/annurev.earth.32.101802.120213 [9] 鲁安怀, 卢晓英, 任子平, 等. 天然铁锰氧化物及氢氧化物环境矿物学研究 [J]. 地学前缘, 2000, 7(2): 473-483. doi: 10.3321/j.issn:1005-2321.2000.02.015 LU A H, LU X Y, REN Z P, et al. New advances in environmental mineralogy of natural oxides and hydroxides of iron and manganese [J]. Earth Science Frontiers, 2000, 7(2): 473-483(in Chinese). doi: 10.3321/j.issn:1005-2321.2000.02.015
[10] JUNTA J L, HOCHELLA M F. Manganese (II) oxidation at mineral surfaces: A microscopic and spectroscopic study [J]. Geochimica et Cosmochimica Acta, 1994, 58(22): 4985-4999. doi: 10.1016/0016-7037(94)90226-7 [11] GAO T Y, SHEN Y G, JIA Z H, et al. Interaction mechanisms and kinetics of ferrous ion and hexagonal birnessite in aqueous systems [J]. Geochemical Transactions, 2015, 16(1): 16. doi: 10.1186/s12932-015-0031-3 [12] KRISHNAMURTI G S R, HUANG P M. Influence of manganese oxide minerals on the formation of iron Oxides [J]. Clays and Clay Minerals, 1988, 36(5): 467-475. doi: 10.1346/CCMN.1988.0360513 [13] VILLINSKI J E, O'DAY P A, CORLEY T L, et al. In situ spectroscopic and solution analyses of the reductive dissolution of MnO2 by Fe(II) [J]. Environmental Science & Technology, 2001, 35(6): 1157-1163. [14] NESBITT H W, CANNING G W, BANCROFT G M. XPS study of reductive dissolution of 7Å-birnessite by H3AsO3, with constraints on reaction mechanism [J]. Geochimica et Cosmochimica Acta, 1998, 62(12): 2097-2110. doi: 10.1016/S0016-7037(98)00146-X [15] POSTMA D, APPELO C A J. Reduction of Mn-oxides by ferrous iron in a flow system: Column experiment and reactive transport modeling [J]. Geochimica et Cosmochimica Acta, 2000, 64(7): 1237-1247. doi: 10.1016/S0016-7037(99)00356-7 [16] SIEBECKER M, MADISON A S, LUTHER G W. Reduction kinetics of polymeric (soluble) manganese (IV) oxide (MnO2) by ferrous iron (Fe2+) [J]. Aquatic Geochemistry, 2015, 21(2/3/4): 143-158. [17] POSTMA D. Concentration of Mn and separation from Fe in sediments-I. Kinetics and stoichiometry of the reaction between birnessite and dissolved Fe(II) at 10 ℃ [J]. Geochimica et Cosmochimica Acta, 1985, 49(4): 1023-1033. doi: 10.1016/0016-7037(85)90316-3 [18] MYERS C R, NEALSON K H. Microbial reduction of manganese oxides: Interactions with iron and sulfur [J]. Geochimica et Cosmochimica Acta, 1988, 52(11): 2727-2732. doi: 10.1016/0016-7037(88)90041-5 [19] MCKENZIE R M. The synthesis of birnessite, cryptomelane, and some other oxides and hydroxides of manganese [J]. Mineralogical Magazine, 1971, 38(296): 493-502. doi: 10.1180/minmag.1971.038.296.12 [20] LUO Y, TAN W F, SUIB S L, et al. Dissolution and phase transformation processes of hausmannite in acidic aqueous systems under anoxic conditions [J]. Chemical Geology, 2018, 487: 54-62. doi: 10.1016/j.chemgeo.2018.04.016 [21] 罗瑶, 李珊, 谭文峰, 等. 水锰矿氧化水溶性硫化物过程及其影响因素 [J]. 环境科学, 2016, 37(4): 1539-1545. doi: 10.13227/j.hjkx.2016.04.045 LUO Y, LI S, TAN W F, et al. Oxidation process of dissolvable sulfide by manganite and its influencing factors [J]. Environmental Science, 2016, 37(4): 1539-1545(in Chinese). doi: 10.13227/j.hjkx.2016.04.045
[22] GAO T Y, SHI Y, LIU F, et al. Oxidation process of dissolvable sulfide by synthesized todorokite in aqueous systems [J]. Journal of Hazardous Materials, 2015, 290: 106-116. doi: 10.1016/j.jhazmat.2015.02.018 [23] KIRILLOV S A, ALEKSANDROVA V S, LISNYCHA T V, et al. Oxidation of synthetic hausmannite (Mn3O4) to manganite (MnOOH) [J]. Journal of Molecular Structure, 2009, 928(1/2/3): 89-94. [24] RAHIMI S, MOATTARI R M, RAJABI L, et al. Iron oxide/hydroxide (α, γ-FeOOH) nanoparticles as high potential adsorbents for lead removal from polluted aquatic media [J]. Journal of Industrial and Engineering Chemistry, 2015, 23: 33-43. doi: 10.1016/j.jiec.2014.07.039 [25] HAN X, LI Y L, GU J D. Oxidation of As(Ⅲ) by MnO2 in the absence and presence of Fe(Ⅱ) under acidic conditions [J]. Geochimica et Cosmochimica Acta, 2011, 75(2): 368-379. doi: 10.1016/j.gca.2010.10.010 [26] RICHMOND W R, LOAN M, MORTON J, et al. Arsenic removal from aqueous solution via ferrihydrite crystallization control [J]. Environmental Science & Technology, 2004, 38(8): 2368-2372. [27] CHIRIŢĂ P, DESCOSTES M, SCHLEGEL M L. Oxidation of FeS by oxygen-bearing acidic solutions [J]. Journal of Colloid and Interface Science, 2008, 321(1): 84-95. doi: 10.1016/j.jcis.2008.01.024 [28] REDDY T R, FRIERDICH A J, BEARD B L, et al. The effect of pH on stable iron isotope exchange and fractionation between aqueous Fe(Ⅱ) and goethite [J]. Chemical Geology, 2015, 397: 118-127. doi: 10.1016/j.chemgeo.2015.01.018 [29] 刘承帅, 李芳柏, 陈曼佳, 等. Fe(Ⅱ)催化水铁矿晶相转变过程中Pb的吸附与固定 [J]. 化学学报, 2017, 75(6): 621-628. doi: 10.6023/A17030093 LIU C S, LI F B, CHEN M J, et al. Adsorption and stabilization of lead during Fe(Ⅱ)-catalyzed phase transformation of ferrihydrite [J]. Acta Chimica Sinica, 2017, 75(6): 621-628(in Chinese). doi: 10.6023/A17030093
[30] SCHOTT J, BERNER R A. X-ray photoelectron studies of the mechanism of iron silicate dissolution during weathering [J]. Geochimica et Cosmochimica Acta, 1983, 47(12): 2233-2240. doi: 10.1016/0016-7037(83)90046-7 [31] ROSSO K M, MORGAN J J. Outer-sphere electron transfer kinetics of metal ion oxidation by molecular oxygen [J]. Geochimica et Cosmochimica Acta, 2002, 66(24): 4223-4233. doi: 10.1016/S0016-7037(02)01040-2 [32] KING D W. Role of carbonate speciation on the oxidation rate of Fe(II) in aquatic systems [J]. Environmental Science & Technology, 1998, 32(19): 2997-3003. [33] 李颖, 顾雪元. 土壤中锰氧化物的形态及其化学提取方法综述 [J]. 环境化学, 2022, 41(1): 9-21. doi: 10.7524/j.issn.0254-6108.2021061603 LI Y, GU X Y. Soil manganese oxides and its extraction methods: A review [J]. Environmental Chemistry, 2022, 41(1): 9-21(in Chinese). doi: 10.7524/j.issn.0254-6108.2021061603
[34] SUN Q, CUI P X, FAN T T, et al. Effects of Fe(Ⅱ) on Cd(Ⅱ) immobilization by Mn(Ⅲ)-rich δ-MnO2 [J]. Chemical Engineering Journal, 2018, 353: 167-175. doi: 10.1016/j.cej.2018.07.120 [35] 刘立虎, 樊萍, 孙学成, 等. 电化学驱动水钠锰矿高效吸附去除混合重金属离子 [J]. 环境化学, 2022, 41(2): 740-748. LIU L H, FAN P, SUN X C, et al. High-efficiency adsorption removal for multiple heavy metal ions using birnessite under electrochemical drive [J]. Environmental Chemistry, 2022, 41(2): 740-748(in Chinese).
[36] SCHAEFER M V, HANDLER R M, SCHERER M M. Fe(Ⅱ) reduction of pyrolusite (β-MnO2) and secondary mineral evolution [J]. Geochemical Transactions, 2017, 18: 7. doi: 10.1186/s12932-017-0045-0 [37] RADY O, LIU L H, YANG X, et al. Adsorption and catalytic oxidation of arsenite on Fe-Mn nodules in the presence of oxygen [J]. Chemosphere, 2020, 259: 127503. doi: 10.1016/j.chemosphere.2020.127503 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4338
- HTML全文浏览数: 4338
- PDF下载数: 111
- 施引文献: 0
