


-
随着可用磷资源的减少以及对水环境质量需求的提升,废水中磷的去除和回收工艺越来越受到关注. 从废水中回收磷资源,不仅能缓解磷矿石危机,还能从源头解决磷污染问题,具有广阔的应用前景[1-2]. 目前,常用的废水磷回收技术主要有磷酸铵镁结晶法、羟基磷酸钙结晶法(HAP)、生物富集法、离子交换法以及膜生物反应器等[3-7]. 其中,HAP结晶法在处理不同浓度的含磷废水方面有着广泛应用,尤其适用于强化生物除磷系统中从厌氧沉淀池出水(磷酸盐浓度0—20 mg·L−1)中回收磷[8].
HAP结晶过程包括前体物质的生成和转化,在碱性条件下,溶液中的磷与钙离子反应生成透磷钙石(CaHPO4·2H2O)、无定型磷酸钙(Ca3(PO4)2·xH2O)、磷酸八钙(Ca4H(PO4)3·2.5H2O)、磷酸钙(Ca3(PO4)2)等磷酸钙盐,这些前驱体在不同条件下可以相互转化,最终形成热力学上更加稳定的HAP晶体,实现磷的回收与利用,这一过程受pH值、过饱和度、Ca/P摩尔比、温度、晶种种类及投加量、杂质离子等物理化学因素的影响[9-12]. 在这些影响因素中,杂质离子对磷结晶过程的影响研究较少,尤其是城镇污水中不容忽视的重金属离子. 有研究结果表明,在磷结晶体系中,重金属离子并不是稳定地存在于液相环境中,而是体现出强烈地向磷结晶产物中活跃迁移的特性,进而对磷回收产物的循环利用造成潜在环境风险[13-16],但以往的研究主要集中在磷酸铵镁和透钙磷石结晶体系上,重金属对HAP结晶的相组成和结构的影响研究则较少,也很少考虑HAP结晶的生长和熟化,缺少从诱导时间、结晶速率等结晶动力学层面的评价.
因此,本研究选取了废水体系中常见的3种具有较大生物毒性的重金属离子(Pb2+、Cd2+、Cr3+),分别从结晶动力学、结晶产物组成以及反应终点磷去除率3个方面,探究重金属离子的添加对HAP结晶除磷的影响,以期为高纯度的羟基磷酸钙产物的回收提供理论指导.
-
实验仪器:紫外分光光度计(UV 9100 B)、pH 测定仪(PHSJ-3F,上海仪电科学仪器股份有限公司)、扫描电子显微镜及能谱仪(Zeiss Gemini 300,德国)、X 射线衍射仪(Bruker D8 Advance,德国)、傅里叶变换红外光谱仪(Thermo Scientific Nicolet iS5,美国)、双数显恒温磁力搅拌器(HJ-4B,常州市金坛友联仪器研究所)等.
实验试剂:CaCl2·2H2O、KH2PO4、Cd(NO3)2·4H2O、Cr(NO3)3·9H2O、NaOH等,均为分析纯;硝酸铅溶液(0.01 mol·L−1)购自北京中科二环科技有限公司. 所有溶液均使用去离子水配制.
-
利用KH2PO4配制1 g·L−1(以P计)的磷酸储备液,CaCl2·2H2O配制2.588 g·L−1(以Ca计)的Ca2+储备液(与磷酸储备液的Ca/P摩尔比为2),Cd(NO3)2·4H2O、Cr(NO3)3·9H2O配制重金属浓度为1 g·L−1的重金属储备液. 结合预实验探究重金属对HAP结晶诱导时间影响的初步结果,结晶动力学影响实验中重金属离子投加量为Pb2+(0、5、10、20、25、30、100 mg·L−1)、Cd2+(0、0.5、1、2.5、5、10 mg·L−1)、Cr3+(0、0.1、0.5、1、2、10 mg·L−1);结晶产物特性影响实验中重金属离子投加量为Pb2+(0、1、10、25 mg·L−1)、Cd2+(0、1、10 mg·L−1)、Cr3+(0、0.5、2.5 mg·L−1);磷去除率影响实验中重金属离子投加量为Pb2+(0、5、10、15、20、25 mg·L−1)、Cd2+(0、0.5、1、2.5、10 mg·L−1)、Cr3+(0、0.5、1、1.5、2、2.5 mg·L−1). 设定反应条件总磷TP=20 mg·L−1、Ca/P摩尔比=2.0、初始pH=8.5、水温25 ℃,所有反应于1000 mL锥形瓶中进行,以恒温磁力搅拌器维持反应体系的混合流态(转速为300 r·min−1)和水温,调节反应体系初始pH=8.5±0.05,反应开始后每隔一段时间测定体系中剩余磷浓度,同时对反应体系的pH变化进行连续在线监测(REXDCH 2.0数据采集软件),直至到达HAP熟化期,即反应体系pH和溶液中剩余磷浓度基本保持稳定,最终投加Pb2+组反应时长12 h、Cd2+组反应时长36 h、Cr3+组反应时长52 h. 所得结晶产物经抽滤、烘干、研磨过筛处理后,分别用能谱仪、X射线衍射仪和傅里叶变换红外光谱仪进行材料表征.
-
HAP的成核速率通常难以测量,但成核诱导时间却容易获得. 反应体系中,HAP结晶的成核过程伴随着质子的释放[17],如式(1)所示,故可通过pH变化曲线来区分结晶的各个阶段,共分为诱导期(阶段Ⅰ)、HAP结晶期(阶段Ⅱ)、HAP熟化期(阶段Ⅲ),诱导时间即阶段Ⅰ持续的时长,可通过第一、二阶段曲线切线的交点确定,再根据公式(2)便可求得HAP的成核速率[18-19].
式中,J为成核速率,V为反应体系的体积,ti为诱导时间.
为确保pH监测结果的准确和稳定,每次实验结束后对pH复合电极进行清洗维护,定期进行电极校正,保证同一组反应的pH在线监测结果具有良好的重现性.
-
向HAP结晶体系单独投加不同浓度Pb2+/Cd2+/Cr3+时,溶液pH值变化如图1所示. 结果表明,投加Pb2+、Cd2+或Cr3+并不影响pH曲线呈现以无定型磷酸钙ACP为前驱体生成HAP的三阶段走势,说明3种重金属的投加并没有从本质上改变反应体系中HAP的非经典结晶方式[18],结晶过程仍以介稳态的磷酸钙盐为前体物质,再通过相转化得到最终晶体.
由图1(a)可知,随着Pb2+投加量的增加,HAP结晶速率变化趋势呈两段式,即先增大后减小,在Pb2+投加量为20 mg·L−1时,结晶速率最大为1.290 h−1,并且当Pb2+初始浓度为0—20 mg·L−1时,HAP结晶诱导时间与Pb2+初始浓度呈显著负相关(R2=0.9921);当Pb2+初始浓度为20—100 mg·L−1时,HAP结晶诱导时间与Pb2+初始浓度呈显著正相关(R2=0.9971). 产生这一现象的原因可能是低浓度Pb2+的加入消耗了部分OH-,使得溶液中ACP的生成量减少,剩余钙磷离子浓度相对增多,进而结晶速率有所提高,但高浓度的Pb2+消耗了过多OH-,使得HAP结晶过程被抑制.
由图1(b)可知,Cd2+的存在会明显延长HAP结晶过程的诱导时间,降低结晶速率. 随着Cd2+初始投加量从0 mg·L−1逐步增加到10 mg·L−1,结晶速率由0.2686 h−1降低至0.0374 h−1,诱导时间由3.72 h增加至26.57 h,且诱导时间与Cd2+初始浓度呈显著正相关(R2=0.9658). Cr3+对HAP成核过程存在强烈的抑制作用,如图1(c)所示,随着Cr3+初始投加量从0 mg·L−1逐步增加到2 mg·L−1,结晶速率由0.2686 h−1降低至0.0332 h−1,诱导时间由3.72 h增加至26.57 h,诱导时间与Cr3+初始浓度呈显著正相关(R2=0.9868).
对比3种重金属离子对HAP结晶动力学的影响,当Pb2+、Cd2+和Cr3+投加量从0 mg·L−1增加至100 mg·L−1、10 mg·L−1和2 mg·L−1时,对结晶速率的抑制率最大分别7.07%、86.09%和87.64%,可以得出3种重金属对结晶速率的抑制作用Cr3+>Cd2+>Pb2+.
-
采用能谱仪对样品的元素及其摩尔比进行测试与计算. 从图2中可以看出,产物主要元素均为O、Ca、P、C,但当反应体系Pb2+浓度为25 mg·L−1时,产物中出现了Pb(32.61%)和Cl(0.8%)元素,Ca/P摩尔比也由1.664下降至1.283,表明铅和氯作为掺杂物进入到了羟基磷灰石产物中. 对于羟基磷灰石而言,当溶液中存在金属离子半径与其构晶离子(即Ca2+)半径相似时,容易与Ca2+发生竞争和取代反应[20],故Pb2+可能替代了部分钙离子.
为了解产物的物相结构,对投加Pb前后得到的产物进行XRD测试,结果如图3所示. 1 mg·L−1时产物中的峰主要为HAP的衍射峰,在10—25 mg·L−1时可以观察到其它衍射峰出现(2θ为20.5554°和21.5862°),利用XRD分析软件(MDI Jade 6)对比标准图谱,可以得出产物中含有铅羟基磷灰石(Pb5(PO4)3OH[21]、Ca2.5Pb7.5(PO4)6(OH)2[22])及磷氯铅矿(Pb5(PO4)3Cl)[23]. 同时,Pb的引入使得HAP衍射峰强度降低,晶格面(002)、(211)、(310)、(222)、(004)的相对强度较纯HAP产物要弱,说明含铅晶相生成和HAP共存于产物中,降低HAP的结晶度.
对比投加25 mg·L−1Pb2+前后产物的FT-IR谱图(图4)可以看出,引入Pb之后的产物仍然具有空白组的典型吸收峰. 在565.80 cm−1、602.21 cm−1处出现的是P—O弯曲振动峰,在1036.66 cm−1出现的是P—O伸缩振动峰[24],峰强均明显变弱,查阅文献可知,这可能是因为Pb2+取代Ca2+后使得羟基磷灰石中的磷酸盐振动减弱[21]. 在3445.44 cm−1处出现的是O—H伸缩振动峰,O—H特征峰减弱,这是HAP吸附Pb2+所导致的,吸附后O—H被Pb2+占据,分子内O—H的氢键作用力减弱[25-26].
利用Visual MINTEQ 3.1模拟软件进行结晶体系中含有25 mg·L−1Pb2+时的化学平衡计算,如表1所示,与上述表征分析一致,Pb2+在HAP结晶体系中能与OH-、PO43-生成多种过饱和物质. 同时,模拟结果显示,在Pb2+浓度(0.12066 mmol·L−1)远小于Ca2+浓度(1.2914 mmol·L−1)的条件下,Pb5(PO4)3Cl及Pb5(PO4)3(OH)的SI值远大于HAP的SI值,表明Pb2+通过争夺HAP的构晶离子(OH-和PO43-)的方式能显著抑制HAP结晶.
-
图5为投加10 mg·L−1Cd2+前后产物的EDS元素半定量分析结果,相比空白组产物,出现了Cd元素(11.46%)和极少量的Cl元素(0.13%),Ca/P摩尔比由1.664下降至1.338,说明Cd2+的引入会造成产物纯度的显著下降. 同时,镉组产物(Cd+Ca)/P摩尔比为1.520,小于空白组Ca/P摩尔比1.664,由此可以推断Cd2+与PO43-结合生成了磷酸镉. 利用XRD进行初步物相检测,结果如图6所示,羟基磷灰石为产物里唯一的晶相,未检测出类似于Cd5(PO4)3(OH)和Ca10-xCdx(PO4)6(OH)2之类的Cd2+取代Ca2+所生成的新物质,且除晶格面(210)之外,其他特征峰峰强没有发生明显变化. 其中,晶格面(210)峰强增大,可能是因为反应体系引入Cd2+改变了HAP晶体的择优取向[27]. 同时,镉组产物XRD曲线相比空白组更毛糙,表明投加Cd2+之后生成更多的非晶态物质,这再次证明了Cd2+对产物的纯度造成影响. 对比投加10 mg·L−1Cd2+前后产物的FT-IR谱图(图4),二者吸收峰位置保持一致,且镉组产物没有新的官能团生成,但1036.66 cm−1、565.80 cm−1及602.21 cm−1处的PO43-吸收峰强度减弱,分析可能是因为Cd2+与溶液中PO43-反应生成磷酸盐沉淀于产物中,使得其强度降低. 这与Corami等[28]在研究羟基磷灰石吸附去除溶液中Cd2+时,羟基磷灰石吸附Cd2+前后FTIR光谱的变化类似.
表2是用Visual MINTEQ 3.1软件模拟结晶体系中含有10 mg·L−1 Cd2+时的化学平衡计算结果,除HAP、Ca3(PO4)2、Ca4H(PO4)3·3H2O之外,Cd2+主要形成Cd3(PO4)2沉淀物,与上述分析结果一致. 杨佳妮[13]在研究废水体系磷回收过程中镉对鸟粪石结晶的影响时,得出的结论与本研究相似,低浓度时Cd2+主要以形成含Cd-PO4基团的物质进入产物中,而高浓度时Cd2+由与磷酸根结合逐渐转变为更多的与氢氧根结合而形成含Cd-OH基团物质沉淀于鸟粪石表面,使鸟粪石产量下降.
-
图7为投加2.5 mg·L−1Cr3+前后产物的EDS元素半定量分析结果,虽然反应体系中Cr3+浓度较低,但固相产物中仍有约9.04%的铬元素. 相比空白组产物Ca/P=1.664,2.5 mg·L−1Cr3+组产物Ca/P=1.187,说明少量Cr3+的引入也会造成产物HAP的纯度显著下降,结合2.1节中得出的Cr3+对HAP结晶速率存在强烈的抑制作用,推测该产物中存在较多HAP结晶反应的前体物质,可能包括透磷钙石(DCPD)、无定型磷酸钙(ACP)、磷酸八钙(OCP)、磷酸钙(TCP)等其中的一种或多种磷酸钙盐,它们尚未转化成热力学上更加稳定的HAP结晶[11, 29]. 图8中的XRD图谱表明,铬组产物仍然具备HAP的主要特征峰,且峰位没有偏移,说明HAP仍然是产物里唯一的晶相,但Cr3+的存在改变了晶体衍射峰的相对强度,晶格面(002)、(211)、(222)、(213)的相对强度较空白组要弱,这说明Cr3+的引入降低了HAP的结晶度. 与镉影响类似的是,铬影响下产物的XRD曲线也比空白组的更粗糙,证明投加Cr3+之后也生成更多的非晶态物质,这些物质可能是DCPD、ACP、OCP、TCP等HAP的前驱体,也可能是一些含铬的无定形沉淀,如Cr(OH)3、Cr2O3等[30]. 对比投加2.5 mg·L−1Cr3+前后产物的FT-IR谱图(图4),二者没有显著差异,但有文献提到,引入高浓度Cr3+后,由于在铬的高负荷下产物形成过程中伴随有脱羟基作用,使得约3500 cm−1处的O—H伸缩振动峰强度降低[31].
表3是用Visual MINTEQ 3.1软件模拟结晶体系中含有2.5 mg·L−1 Cr3+时的化学平衡计算结果,可以看到,Cr3+主要以Cr2O3晶体和Cr(OH)3无定形沉淀的形式进入产物中,查阅文献发现,稳定的Cr2O3晶体的形成需要经过长年的陈化[32],因此X射线衍射也检测不出含铬矿物相,这与上述分析结果相符合.
-
向HAP结晶体系分别单独投加不同浓度Pb2+/Cd2+/Cr3+时,反应终点(HAP结晶反应进入熟化期,溶液中剩余磷浓度基本保持稳定)体系除磷率的变化如图9所示,可以看出,随着重金属投加量的增加,3组反应的除磷率均发生显著改变,且变化趋势各不相同. 随着初始Pb2+浓度从0增加到25 mg·L−1,除磷率先降低后升高,在Pb2+浓度在20 mg·L−1附近时,除磷率最低,即说明在低浓度下,Pb2+会抑制体系的磷去除率,而在高浓度下Pb2+对除磷呈反向促进作用;Cd2+对除磷呈促进作用,10 mg·L−1Cd2+时促进率最大为10.61%;Cr3+对除磷则呈抑制作用,2.5 mg·L−1 Cr3+时抑制率最大为13.55%.
结合2.2节的实验结果,分析产生这种现象的原因是:Pb2+在低浓度时通过生成Pb(OH)2抑制HAP结晶,进而导致除磷率下降,而随着Pb2+浓度的升高,开始更多地生成铅羟基磷灰石和磷氯铅矿,弥补了由于HAP减少带来的除磷量的下降;Cd2+与PO43-结合生成Cd3(PO4)2,促进了液相中磷的去除;Cr3+一方面能抑制HAP的成核速率以及前驱体向HAP的转化速率,另一方面Cr3+在生成Cr(OH)3时消耗溶液中OH-,两方面的共同作用导致HAP结晶量大幅下降,除磷率降低.
-
(1)3种重金属(Pb2+、Cd2+、Cr3+)均会降低HAP的结晶速率,抑制幅度Cr3+>Cd2+>Pb2+. 其中,Cr3+和Cd2+的初始浓度与结晶速率呈负相关,两者均会明显延长HAP的诱导时间;Pb2+在低浓度(<20 mg·L−1)时能提高HAP结晶速率,但当其浓度大于20 mg·L−1后,初始浓度与结晶速率呈负相关.
(2)3种重金属均能与HAP的构晶离子(OH-和PO43-)发生反应,进而与HAP共沉淀进入产物中,各自产物类型不同. Pb2+主要生成铅羟基磷灰石(Pb5(PO4)3OH、Ca2.5Pb7.5(PO4)6(OH)2)及磷氯铅矿(Pb5(PO4)3Cl);Cd2+易与PO43-结合生成无定形态物质Cd3(PO4)2赋存于产物中;而Cr3+则易生成Cr2O3和Cr(OH)3.
(3)3种重金属对HAP结晶体系除磷的影响各不相同:Pb2+在低浓度时对除磷有抑制作用,高浓度下则能促进磷的去除;Cd2+对结晶体系除磷呈促进作用,Cr3+则呈稳定的抑制作用.
Pb(Ⅱ)、Cd(Ⅱ)和Cr(Ⅲ)对羟基磷酸钙结晶过程及产物的影响
Effects of Pb(Ⅱ), Cd(Ⅱ) and Cr(Ⅲ) on crystallization process and products of hydroxyapatite
-
摘要: 本文通过批量结晶实验,探究了3种生物毒性较大的典型重金属离子(Pb2+、Cd2+、Cr3+)对羟基磷酸钙(HAP)结晶动力学和反应体系磷去除率的影响,并利用能谱、X射线衍射和傅里叶红外光谱表征手段,结合Visual MINTEQ 3.1模拟软件对结晶产物进行了分析. 实验结果表明,低浓度(<20 mg·L−1)的Pb2+能提高HAP结晶速率,但会抑制磷的去除,当Pb2+浓度大于20 mg·L−1后,能反向促进除磷,且初始浓度与诱导时间呈显著正相关;Cd2+和Cr3+均会降低HAP结晶速率,抑制幅度Cr3+>Cd2+,但Cd2+对结晶体系除磷有促进作用,Cr3+则呈稳定的抑制作用. 对结晶产物的分析结果表明,Pb2+主要生成铅羟基磷灰石(Pb5(PO4)3OH、Ca2.5Pb7.5(PO4)6(OH))及磷氯铅矿(Pb5(PO4)3Cl),Cd2+主要形成Cd3(PO4)2沉淀物,Cr3+的产物主要是Cr2O3和Cr(OH)3.Abstract: Batch crystallization experiments were conducted to investigate the influence of three typical heavy metal ions with relatively high biological toxicity (Pb2+, Cd2+ and Cr3+) on the crystallization kinetics and phosphorus removal rate of hydroxyapatite crystallization (HAP). The phase composition of crystallization products was analyzed by energy dispersive spectroscopy, X-ray diffraction and Fourier transform infrared spectroscopy, combined with Visual MINTEQ 3.1 simulation software. The experimental results indicated that Pb2+ could increase HAP crystallization rate at low concentration (<20 mg·L−1), but inhibit phosphorus removal. When the concentration of Pb2+ was greater than 20 mg·L−1, it could promote phosphorus removal in reverse, and the initial concentration of Pb2+ was significantly positively correlated with induction time. Both Cd2+ and Cr3+ could reduce HAP crystallization rate and the order of inhibitory effect was Cr3+>Cd2+. However, Cd2+ promoted phosphorus removal in crystallization system, while Cr3+ showed stable inhibition. The results of the analysis of the crystallization products showed that Pb2+ mainly formed lead hydroxyapatite (Pb5(PO4)3OH、Ca2.5Pb7.5(PO4)6(OH)) and pyromorphite (Pb5(PO4)3Cl) and Cd2+ mainly formed Cd3(PO4)2 precipitate. The products of Cr3+ were Cr2O3 and Cr(OH)3.
-
Key words:
- hydroxyapatite /
- crystallization /
- heavy metal /
- kinetics /
- product.
-
随着可用磷资源的减少以及对水环境质量需求的提升,废水中磷的去除和回收工艺越来越受到关注. 从废水中回收磷资源,不仅能缓解磷矿石危机,还能从源头解决磷污染问题,具有广阔的应用前景[1-2]. 目前,常用的废水磷回收技术主要有磷酸铵镁结晶法、羟基磷酸钙结晶法(HAP)、生物富集法、离子交换法以及膜生物反应器等[3-7]. 其中,HAP结晶法在处理不同浓度的含磷废水方面有着广泛应用,尤其适用于强化生物除磷系统中从厌氧沉淀池出水(磷酸盐浓度0—20 mg·L−1)中回收磷[8].
HAP结晶过程包括前体物质的生成和转化,在碱性条件下,溶液中的磷与钙离子反应生成透磷钙石(CaHPO4·2H2O)、无定型磷酸钙(Ca3(PO4)2·xH2O)、磷酸八钙(Ca4H(PO4)3·2.5H2O)、磷酸钙(Ca3(PO4)2)等磷酸钙盐,这些前驱体在不同条件下可以相互转化,最终形成热力学上更加稳定的HAP晶体,实现磷的回收与利用,这一过程受pH值、过饱和度、Ca/P摩尔比、温度、晶种种类及投加量、杂质离子等物理化学因素的影响[9-12]. 在这些影响因素中,杂质离子对磷结晶过程的影响研究较少,尤其是城镇污水中不容忽视的重金属离子. 有研究结果表明,在磷结晶体系中,重金属离子并不是稳定地存在于液相环境中,而是体现出强烈地向磷结晶产物中活跃迁移的特性,进而对磷回收产物的循环利用造成潜在环境风险[13-16],但以往的研究主要集中在磷酸铵镁和透钙磷石结晶体系上,重金属对HAP结晶的相组成和结构的影响研究则较少,也很少考虑HAP结晶的生长和熟化,缺少从诱导时间、结晶速率等结晶动力学层面的评价.
因此,本研究选取了废水体系中常见的3种具有较大生物毒性的重金属离子(Pb2+、Cd2+、Cr3+),分别从结晶动力学、结晶产物组成以及反应终点磷去除率3个方面,探究重金属离子的添加对HAP结晶除磷的影响,以期为高纯度的羟基磷酸钙产物的回收提供理论指导.
1. 材料与方法(Materials and methods)
1.1 实验仪器及药品
实验仪器:紫外分光光度计(UV 9100 B)、pH 测定仪(PHSJ-3F,上海仪电科学仪器股份有限公司)、扫描电子显微镜及能谱仪(Zeiss Gemini 300,德国)、X 射线衍射仪(Bruker D8 Advance,德国)、傅里叶变换红外光谱仪(Thermo Scientific Nicolet iS5,美国)、双数显恒温磁力搅拌器(HJ-4B,常州市金坛友联仪器研究所)等.
实验试剂:CaCl2·2H2O、KH2PO4、Cd(NO3)2·4H2O、Cr(NO3)3·9H2O、NaOH等,均为分析纯;硝酸铅溶液(0.01 mol·L−1)购自北京中科二环科技有限公司. 所有溶液均使用去离子水配制.
1.2 实验方法
利用KH2PO4配制1 g·L−1(以P计)的磷酸储备液,CaCl2·2H2O配制2.588 g·L−1(以Ca计)的Ca2+储备液(与磷酸储备液的Ca/P摩尔比为2),Cd(NO3)2·4H2O、Cr(NO3)3·9H2O配制重金属浓度为1 g·L−1的重金属储备液. 结合预实验探究重金属对HAP结晶诱导时间影响的初步结果,结晶动力学影响实验中重金属离子投加量为Pb2+(0、5、10、20、25、30、100 mg·L−1)、Cd2+(0、0.5、1、2.5、5、10 mg·L−1)、Cr3+(0、0.1、0.5、1、2、10 mg·L−1);结晶产物特性影响实验中重金属离子投加量为Pb2+(0、1、10、25 mg·L−1)、Cd2+(0、1、10 mg·L−1)、Cr3+(0、0.5、2.5 mg·L−1);磷去除率影响实验中重金属离子投加量为Pb2+(0、5、10、15、20、25 mg·L−1)、Cd2+(0、0.5、1、2.5、10 mg·L−1)、Cr3+(0、0.5、1、1.5、2、2.5 mg·L−1). 设定反应条件总磷TP=20 mg·L−1、Ca/P摩尔比=2.0、初始pH=8.5、水温25 ℃,所有反应于1000 mL锥形瓶中进行,以恒温磁力搅拌器维持反应体系的混合流态(转速为300 r·min−1)和水温,调节反应体系初始pH=8.5±0.05,反应开始后每隔一段时间测定体系中剩余磷浓度,同时对反应体系的pH变化进行连续在线监测(REXDCH 2.0数据采集软件),直至到达HAP熟化期,即反应体系pH和溶液中剩余磷浓度基本保持稳定,最终投加Pb2+组反应时长12 h、Cd2+组反应时长36 h、Cr3+组反应时长52 h. 所得结晶产物经抽滤、烘干、研磨过筛处理后,分别用能谱仪、X射线衍射仪和傅里叶变换红外光谱仪进行材料表征.
1.3 磷结晶动力学分析
HAP的成核速率通常难以测量,但成核诱导时间却容易获得. 反应体系中,HAP结晶的成核过程伴随着质子的释放[17],如式(1)所示,故可通过pH变化曲线来区分结晶的各个阶段,共分为诱导期(阶段Ⅰ)、HAP结晶期(阶段Ⅱ)、HAP熟化期(阶段Ⅲ),诱导时间即阶段Ⅰ持续的时长,可通过第一、二阶段曲线切线的交点确定,再根据公式(2)便可求得HAP的成核速率[18-19].
3Ca3(PO4)2(s)+Ca2++2H2O→2Ca5(PO4)3(OH)(s)+2H+ (1) J=1/(Vti) (2) 式中,J为成核速率,V为反应体系的体积,ti为诱导时间.
为确保pH监测结果的准确和稳定,每次实验结束后对pH复合电极进行清洗维护,定期进行电极校正,保证同一组反应的pH在线监测结果具有良好的重现性.
2. 结果与讨论(Results and discussion)
2.1 重金属离子对HAP结晶动力学的影响
向HAP结晶体系单独投加不同浓度Pb2+/Cd2+/Cr3+时,溶液pH值变化如图1所示. 结果表明,投加Pb2+、Cd2+或Cr3+并不影响pH曲线呈现以无定型磷酸钙ACP为前驱体生成HAP的三阶段走势,说明3种重金属的投加并没有从本质上改变反应体系中HAP的非经典结晶方式[18],结晶过程仍以介稳态的磷酸钙盐为前体物质,再通过相转化得到最终晶体.
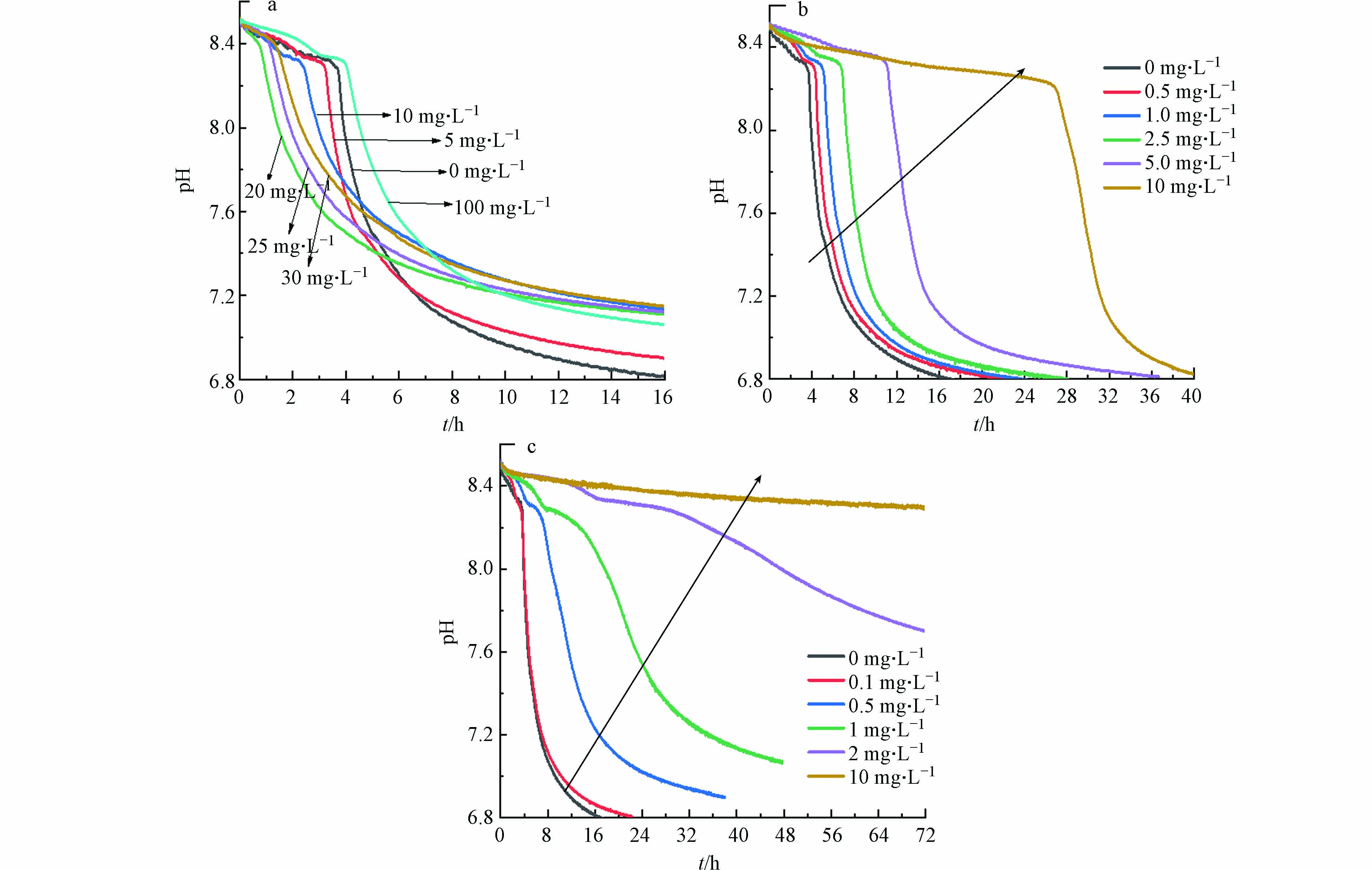 图 1 (a)Pb2+、(b)Cd2+和(c)Cr3+的初始浓度对反应体系pH变化曲线的影响Figure 1. Effect of (a) Pb2+, (b)Cd2+, (c) Cr3+ concentration on the pH curve of reaction system
图 1 (a)Pb2+、(b)Cd2+和(c)Cr3+的初始浓度对反应体系pH变化曲线的影响Figure 1. Effect of (a) Pb2+, (b)Cd2+, (c) Cr3+ concentration on the pH curve of reaction system由图1(a)可知,随着Pb2+投加量的增加,HAP结晶速率变化趋势呈两段式,即先增大后减小,在Pb2+投加量为20 mg·L−1时,结晶速率最大为1.290 h−1,并且当Pb2+初始浓度为0—20 mg·L−1时,HAP结晶诱导时间与Pb2+初始浓度呈显著负相关(R2=0.9921);当Pb2+初始浓度为20—100 mg·L−1时,HAP结晶诱导时间与Pb2+初始浓度呈显著正相关(R2=0.9971). 产生这一现象的原因可能是低浓度Pb2+的加入消耗了部分OH-,使得溶液中ACP的生成量减少,剩余钙磷离子浓度相对增多,进而结晶速率有所提高,但高浓度的Pb2+消耗了过多OH-,使得HAP结晶过程被抑制.
由图1(b)可知,Cd2+的存在会明显延长HAP结晶过程的诱导时间,降低结晶速率. 随着Cd2+初始投加量从0 mg·L−1逐步增加到10 mg·L−1,结晶速率由0.2686 h−1降低至0.0374 h−1,诱导时间由3.72 h增加至26.57 h,且诱导时间与Cd2+初始浓度呈显著正相关(R2=0.9658). Cr3+对HAP成核过程存在强烈的抑制作用,如图1(c)所示,随着Cr3+初始投加量从0 mg·L−1逐步增加到2 mg·L−1,结晶速率由0.2686 h−1降低至0.0332 h−1,诱导时间由3.72 h增加至26.57 h,诱导时间与Cr3+初始浓度呈显著正相关(R2=0.9868).
对比3种重金属离子对HAP结晶动力学的影响,当Pb2+、Cd2+和Cr3+投加量从0 mg·L−1增加至100 mg·L−1、10 mg·L−1和2 mg·L−1时,对结晶速率的抑制率最大分别7.07%、86.09%和87.64%,可以得出3种重金属对结晶速率的抑制作用Cr3+>Cd2+>Pb2+.
2.2 重金属离子对结晶产物特性的影响
2.2.1 Pb对磷回收产物的影响分析
采用能谱仪对样品的元素及其摩尔比进行测试与计算. 从图2中可以看出,产物主要元素均为O、Ca、P、C,但当反应体系Pb2+浓度为25 mg·L−1时,产物中出现了Pb(32.61%)和Cl(0.8%)元素,Ca/P摩尔比也由1.664下降至1.283,表明铅和氯作为掺杂物进入到了羟基磷灰石产物中. 对于羟基磷灰石而言,当溶液中存在金属离子半径与其构晶离子(即Ca2+)半径相似时,容易与Ca2+发生竞争和取代反应[20],故Pb2+可能替代了部分钙离子.
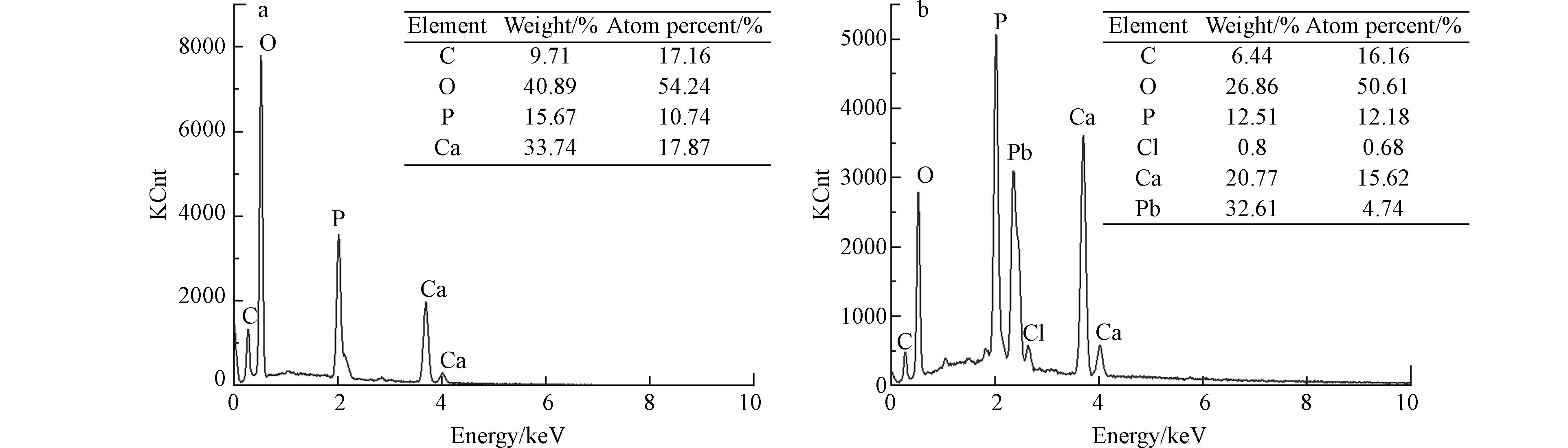 图 2 (a)HAP、(b)加入25 mg·L−1Pb2+产物的EDS谱图Figure 2. EDS spectrums of (a) HAP and (b) product with addition of 25 mg·L−1 Pb2+
图 2 (a)HAP、(b)加入25 mg·L−1Pb2+产物的EDS谱图Figure 2. EDS spectrums of (a) HAP and (b) product with addition of 25 mg·L−1 Pb2+为了解产物的物相结构,对投加Pb前后得到的产物进行XRD测试,结果如图3所示. 1 mg·L−1时产物中的峰主要为HAP的衍射峰,在10—25 mg·L−1时可以观察到其它衍射峰出现(2θ为20.5554°和21.5862°),利用XRD分析软件(MDI Jade 6)对比标准图谱,可以得出产物中含有铅羟基磷灰石(Pb5(PO4)3OH[21]、Ca2.5Pb7.5(PO4)6(OH)2[22])及磷氯铅矿(Pb5(PO4)3Cl)[23]. 同时,Pb的引入使得HAP衍射峰强度降低,晶格面(002)、(211)、(310)、(222)、(004)的相对强度较纯HAP产物要弱,说明含铅晶相生成和HAP共存于产物中,降低HAP的结晶度.
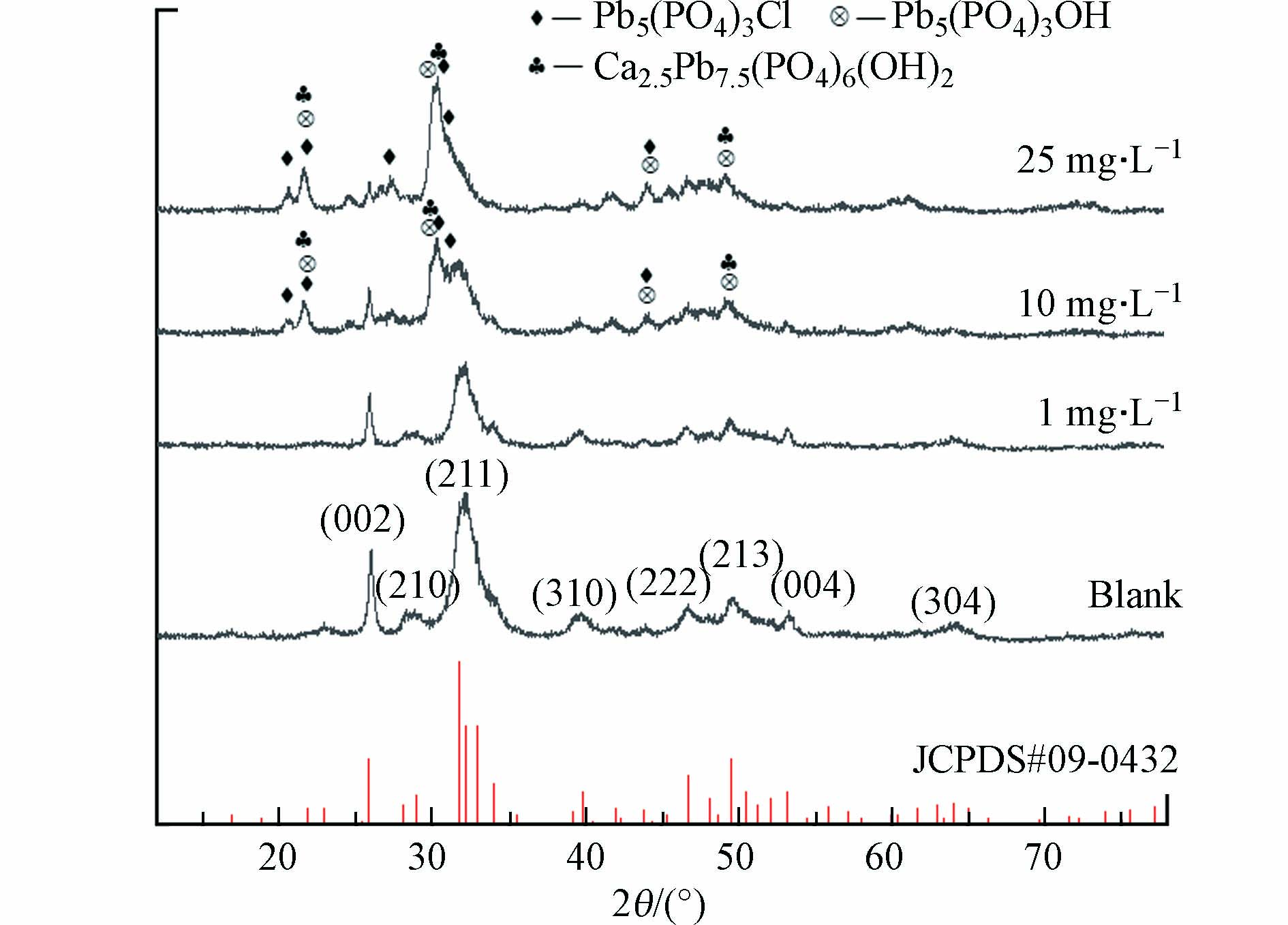 图 3 Pb2+加入前后产物的XRD谱图Figure 3. XRD pattern of product before and after the addition of Pb2+
图 3 Pb2+加入前后产物的XRD谱图Figure 3. XRD pattern of product before and after the addition of Pb2+对比投加25 mg·L−1Pb2+前后产物的FT-IR谱图(图4)可以看出,引入Pb之后的产物仍然具有空白组的典型吸收峰. 在565.80 cm−1、602.21 cm−1处出现的是P—O弯曲振动峰,在1036.66 cm−1出现的是P—O伸缩振动峰[24],峰强均明显变弱,查阅文献可知,这可能是因为Pb2+取代Ca2+后使得羟基磷灰石中的磷酸盐振动减弱[21]. 在3445.44 cm−1处出现的是O—H伸缩振动峰,O—H特征峰减弱,这是HAP吸附Pb2+所导致的,吸附后O—H被Pb2+占据,分子内O—H的氢键作用力减弱[25-26].
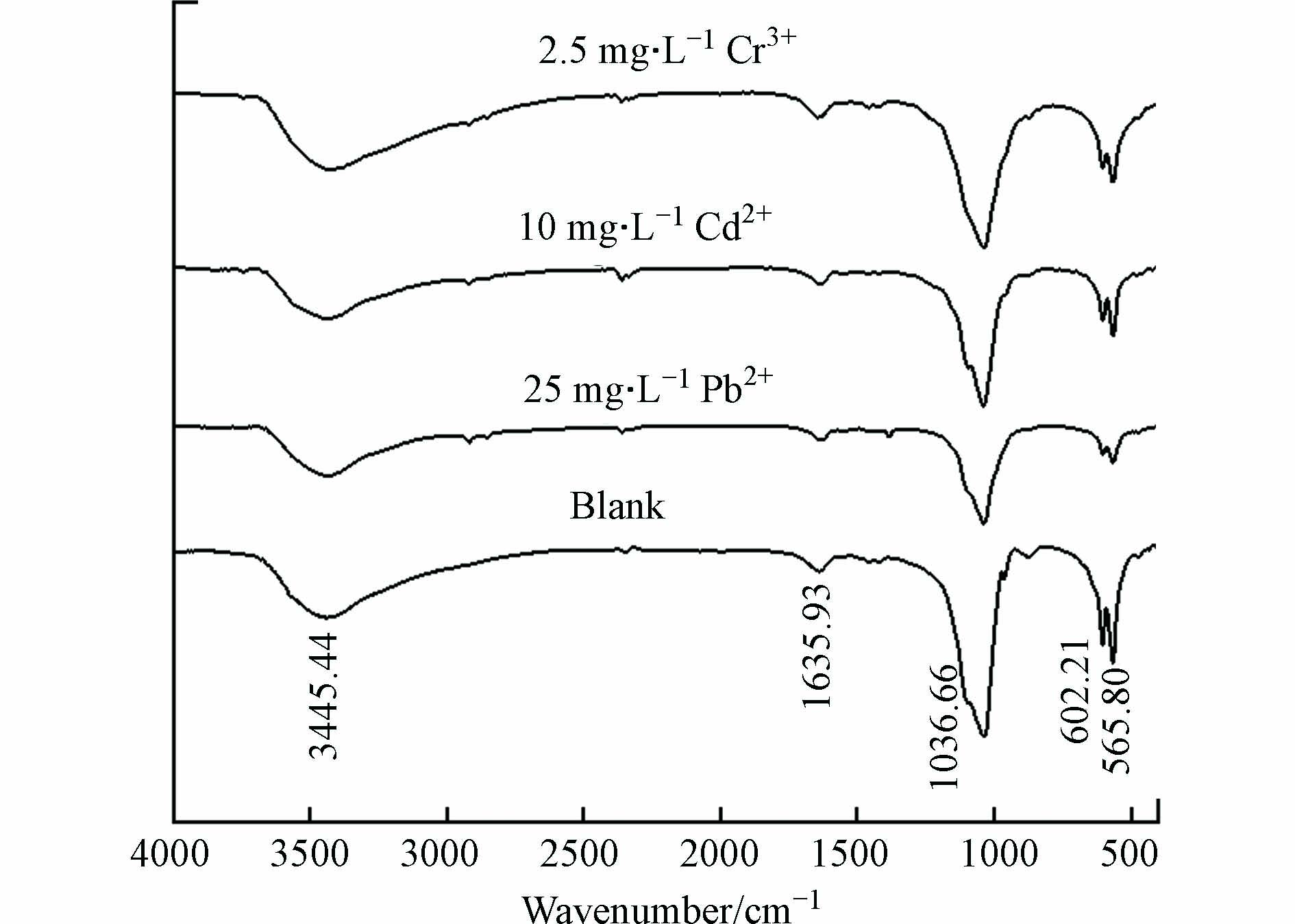 图 4 重金属加入前后产物的FTIR谱图Figure 4. FTIR diagram of product before and after the addition of heavy metals
图 4 重金属加入前后产物的FTIR谱图Figure 4. FTIR diagram of product before and after the addition of heavy metals利用Visual MINTEQ 3.1模拟软件进行结晶体系中含有25 mg·L−1Pb2+时的化学平衡计算,如表1所示,与上述表征分析一致,Pb2+在HAP结晶体系中能与OH-、PO43-生成多种过饱和物质. 同时,模拟结果显示,在Pb2+浓度(0.12066 mmol·L−1)远小于Ca2+浓度(1.2914 mmol·L−1)的条件下,Pb5(PO4)3Cl及Pb5(PO4)3(OH)的SI值远大于HAP的SI值,表明Pb2+通过争夺HAP的构晶离子(OH-和PO43-)的方式能显著抑制HAP结晶.
表 1 结晶体系含25 mg·L−1 Pb2+时主要的过饱和物质及相应的饱和指数SITable 1. Major supersaturated substances and their saturation indexes in the crystal system containing 25 mg·L−1 Pb2+过饱和物质Supersaturated substance 饱和指数Saturation index 过饱和物质Supersaturated substance 饱和指数Saturation index Pb5(PO4)3Cl(c) 34.193 PbHPO4(s) 2.826 Pb5(PO4)3Cl(soil) 30.163 HAP 15.168 Pb5(PO4)3(OH) 23.674 Ca4H(PO4)3·3H2O(s) 4.884 Pb3(PO4)2(s) 13.483 Ca3(PO4)2 (beta) 4.843 Pb2(OH)3Cl(s) 3.907 Ca3(PO4)2 (am2) 4.173 Pb(OH)2(s) 3.761 Ca3(PO4)2 (am1) 1.423 注:SI<0时表示物质在溶液中的浓度未超过其溶解度,不会沉淀;而SI>0时物质将过饱和,出现沉淀. 表中未列出饱和指数<1的物质. Note: SI<0 means that the concentration of the substance in the solution does not exceed its solubility and will not precipitate. SI>0 means that the substance will be supersaturated and will precipitate. Substances with a saturation index less than 1 are not listed in the table. | Show Table DownLoad:
CSV
DownLoad:
CSV
2.2.2 Cd对磷回收产物的影响分析
图5为投加10 mg·L−1Cd2+前后产物的EDS元素半定量分析结果,相比空白组产物,出现了Cd元素(11.46%)和极少量的Cl元素(0.13%),Ca/P摩尔比由1.664下降至1.338,说明Cd2+的引入会造成产物纯度的显著下降. 同时,镉组产物(Cd+Ca)/P摩尔比为1.520,小于空白组Ca/P摩尔比1.664,由此可以推断Cd2+与PO43-结合生成了磷酸镉. 利用XRD进行初步物相检测,结果如图6所示,羟基磷灰石为产物里唯一的晶相,未检测出类似于Cd5(PO4)3(OH)和Ca10-xCdx(PO4)6(OH)2之类的Cd2+取代Ca2+所生成的新物质,且除晶格面(210)之外,其他特征峰峰强没有发生明显变化. 其中,晶格面(210)峰强增大,可能是因为反应体系引入Cd2+改变了HAP晶体的择优取向[27]. 同时,镉组产物XRD曲线相比空白组更毛糙,表明投加Cd2+之后生成更多的非晶态物质,这再次证明了Cd2+对产物的纯度造成影响. 对比投加10 mg·L−1Cd2+前后产物的FT-IR谱图(图4),二者吸收峰位置保持一致,且镉组产物没有新的官能团生成,但1036.66 cm−1、565.80 cm−1及602.21 cm−1处的PO43-吸收峰强度减弱,分析可能是因为Cd2+与溶液中PO43-反应生成磷酸盐沉淀于产物中,使得其强度降低. 这与Corami等[28]在研究羟基磷灰石吸附去除溶液中Cd2+时,羟基磷灰石吸附Cd2+前后FTIR光谱的变化类似.
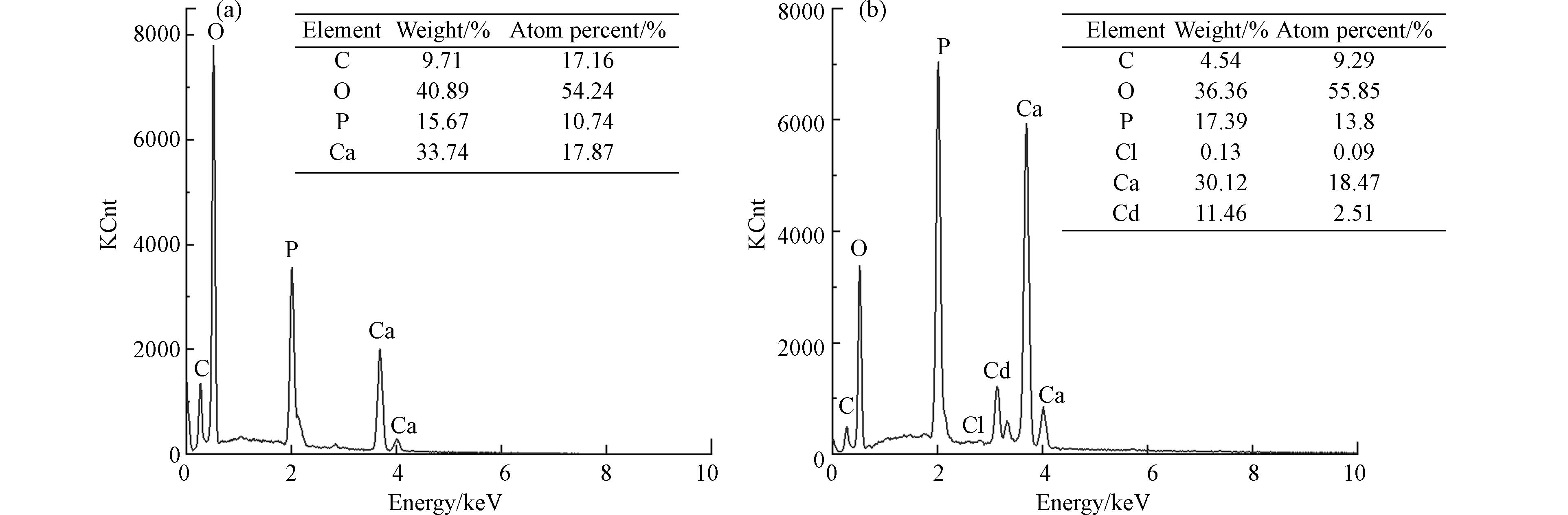 图 5 (a)HAP、(b)加入10 mg·L−1Cd2+产物的EDS谱图Figure 5. EDS spectrums of (a) HAP and (b) product with addition of 10 mg·L−1 Cd2+
图 5 (a)HAP、(b)加入10 mg·L−1Cd2+产物的EDS谱图Figure 5. EDS spectrums of (a) HAP and (b) product with addition of 10 mg·L−1 Cd2+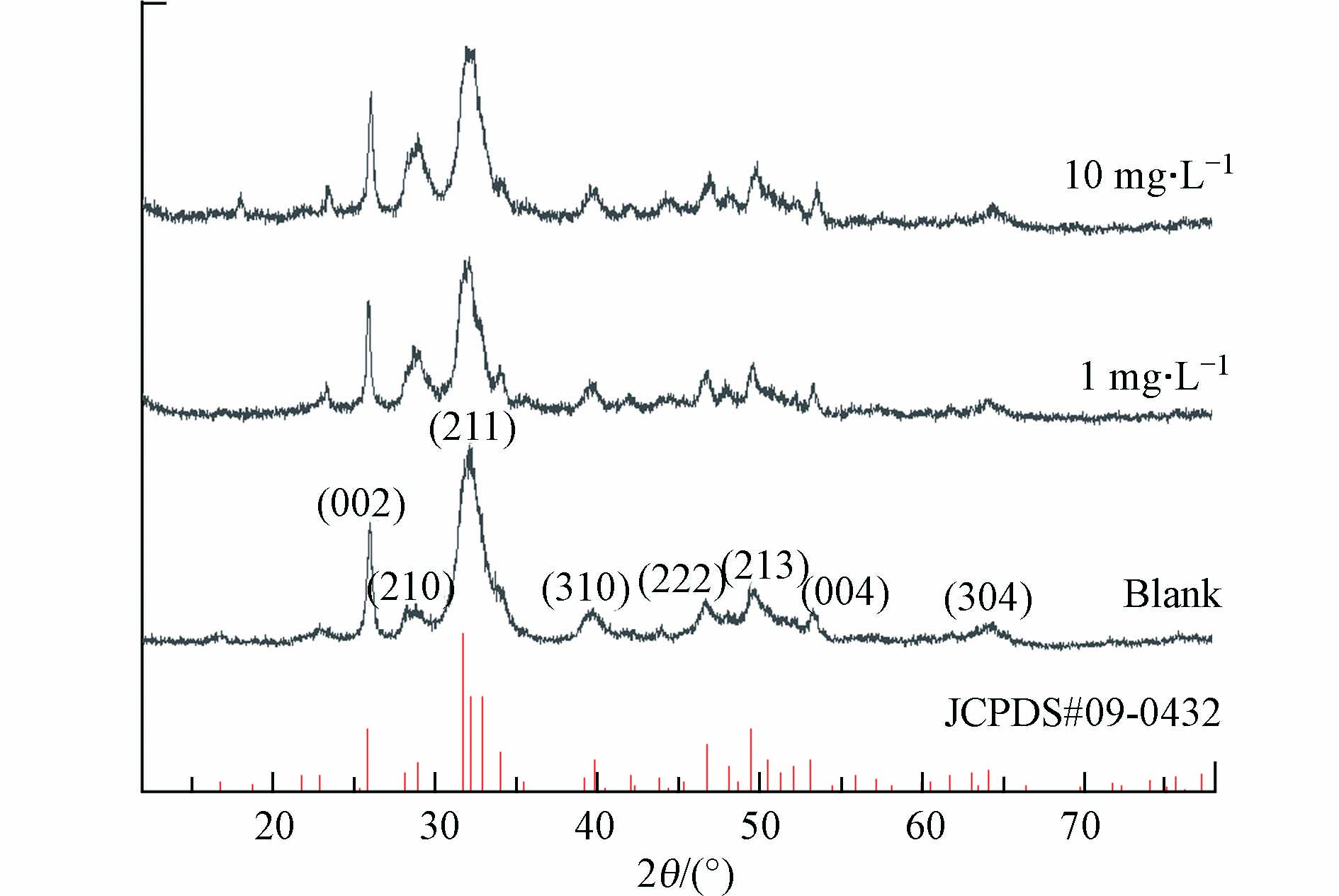 图 6 Cd2+加入前后产物的XRD谱图Figure 6. XRD pattern of product before and after the addition of Cd2+
图 6 Cd2+加入前后产物的XRD谱图Figure 6. XRD pattern of product before and after the addition of Cd2+表2是用Visual MINTEQ 3.1软件模拟结晶体系中含有10 mg·L−1 Cd2+时的化学平衡计算结果,除HAP、Ca3(PO4)2、Ca4H(PO4)3·3H2O之外,Cd2+主要形成Cd3(PO4)2沉淀物,与上述分析结果一致. 杨佳妮[13]在研究废水体系磷回收过程中镉对鸟粪石结晶的影响时,得出的结论与本研究相似,低浓度时Cd2+主要以形成含Cd-PO4基团的物质进入产物中,而高浓度时Cd2+由与磷酸根结合逐渐转变为更多的与氢氧根结合而形成含Cd-OH基团物质沉淀于鸟粪石表面,使鸟粪石产量下降.
表 2 结晶体系含10 mg·L−1 Cd2+时主要的过饱和物质及相应的饱和指数SITable 2. Major supersaturated substances and their saturation indexes in the crystal system containing 10 mg·L−1 Cd2+过饱和物质 Supersaturated substance 饱和指数 Saturation index Cd3(PO4)2 (s) 4.149 Hydroxyapatite 15.109 Ca4H(PO4)3·3H2O (s) 4.82 Ca3(PO4)2 (beta) 4.802 Ca3(PO4)2 (am2) 4.132 Ca3(PO4)2 (am1) 1.382 CaHPO4 (s) 0.263 | Show TableDownLoad:
CSV
2.2.3 Cr对磷回收产物的影响分析
图7为投加2.5 mg·L−1Cr3+前后产物的EDS元素半定量分析结果,虽然反应体系中Cr3+浓度较低,但固相产物中仍有约9.04%的铬元素. 相比空白组产物Ca/P=1.664,2.5 mg·L−1Cr3+组产物Ca/P=1.187,说明少量Cr3+的引入也会造成产物HAP的纯度显著下降,结合2.1节中得出的Cr3+对HAP结晶速率存在强烈的抑制作用,推测该产物中存在较多HAP结晶反应的前体物质,可能包括透磷钙石(DCPD)、无定型磷酸钙(ACP)、磷酸八钙(OCP)、磷酸钙(TCP)等其中的一种或多种磷酸钙盐,它们尚未转化成热力学上更加稳定的HAP结晶[11, 29]. 图8中的XRD图谱表明,铬组产物仍然具备HAP的主要特征峰,且峰位没有偏移,说明HAP仍然是产物里唯一的晶相,但Cr3+的存在改变了晶体衍射峰的相对强度,晶格面(002)、(211)、(222)、(213)的相对强度较空白组要弱,这说明Cr3+的引入降低了HAP的结晶度. 与镉影响类似的是,铬影响下产物的XRD曲线也比空白组的更粗糙,证明投加Cr3+之后也生成更多的非晶态物质,这些物质可能是DCPD、ACP、OCP、TCP等HAP的前驱体,也可能是一些含铬的无定形沉淀,如Cr(OH)3、Cr2O3等[30]. 对比投加2.5 mg·L−1Cr3+前后产物的FT-IR谱图(图4),二者没有显著差异,但有文献提到,引入高浓度Cr3+后,由于在铬的高负荷下产物形成过程中伴随有脱羟基作用,使得约3500 cm−1处的O—H伸缩振动峰强度降低[31].
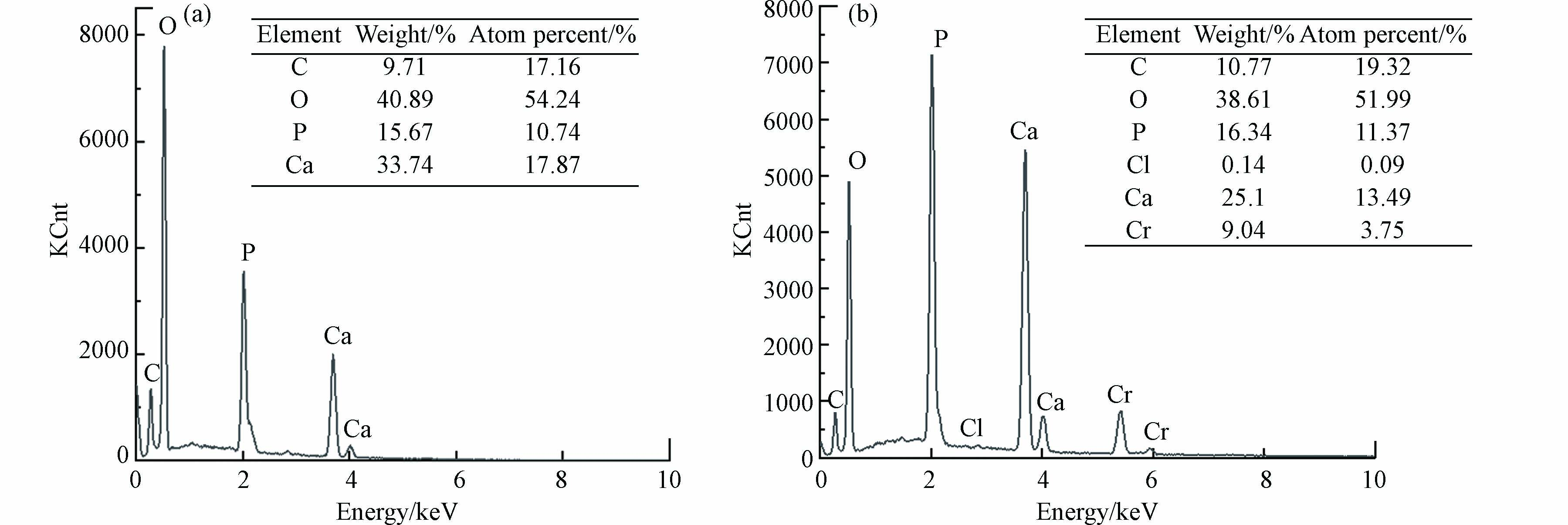 图 7 (a)HAP、(b)加入2.5 mg·L−1Cr3+产物的EDS谱图Figure 7. EDS spectrums of (a) HAP and (b) product with addition of 2.5 mg·L−1 Cr3+
图 7 (a)HAP、(b)加入2.5 mg·L−1Cr3+产物的EDS谱图Figure 7. EDS spectrums of (a) HAP and (b) product with addition of 2.5 mg·L−1 Cr3+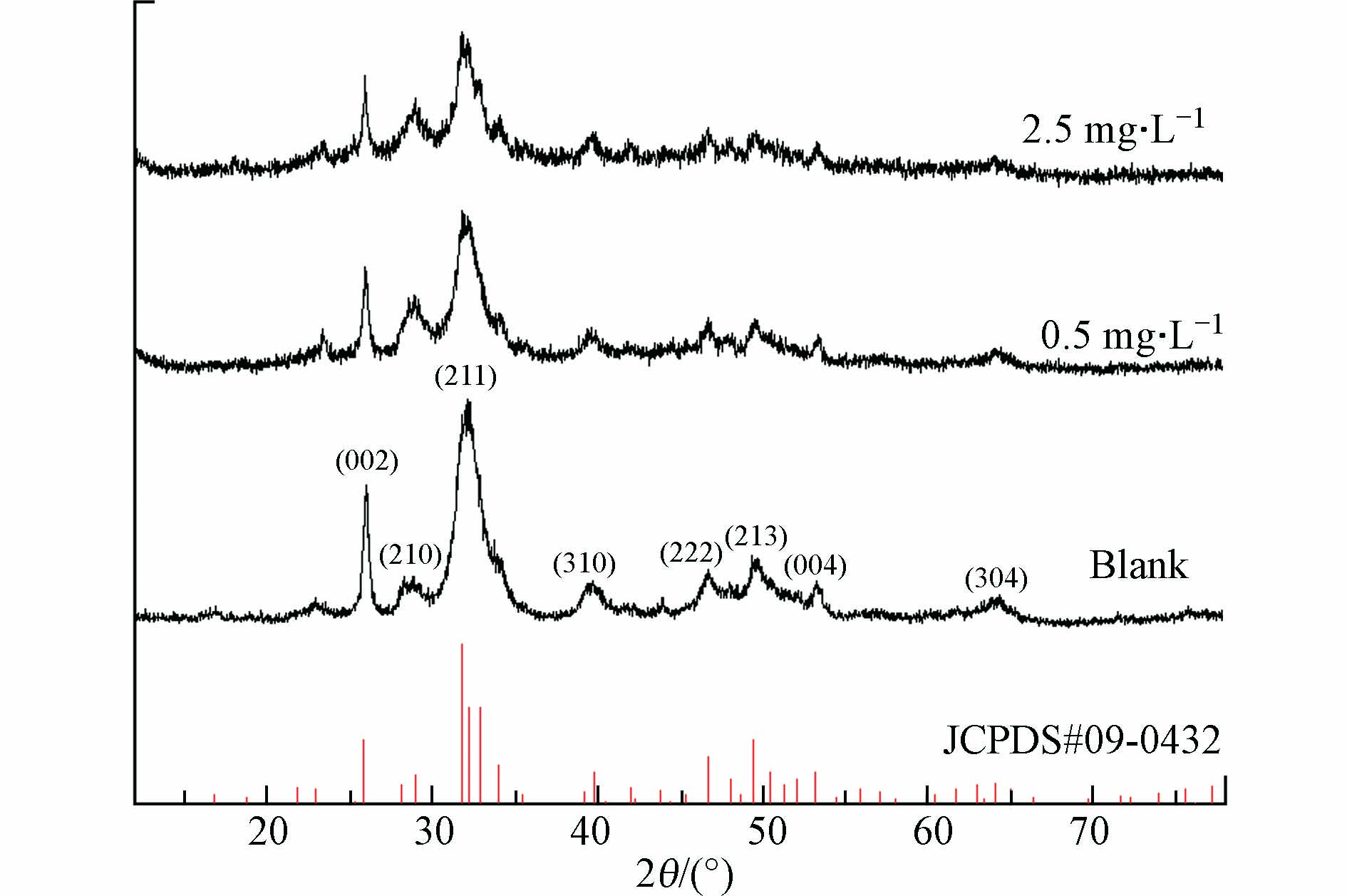 图 8 Cr3+加入前后产物的XRD谱图Figure 8. XRD pattern of product before and after the addition of Cr3+
图 8 Cr3+加入前后产物的XRD谱图Figure 8. XRD pattern of product before and after the addition of Cr3+表3是用Visual MINTEQ 3.1软件模拟结晶体系中含有2.5 mg·L−1 Cr3+时的化学平衡计算结果,可以看到,Cr3+主要以Cr2O3晶体和Cr(OH)3无定形沉淀的形式进入产物中,查阅文献发现,稳定的Cr2O3晶体的形成需要经过长年的陈化[32],因此X射线衍射也检测不出含铬矿物相,这与上述分析结果相符合.
表 3 结晶体系含2.5 mg·L−1 Cr3+时主要的过饱和物质及相应的饱和指数SITable 3. Major supersaturated substances and their saturation indexes in the crystal system containing 2.5 mg·L−1 Cr3+过饱和物质 Supersaturated substance 饱和指数 Saturation index Cr2O3 (c) 5.377 Cr(OH)3 (am) 2.519 Hydroxyapatite 15.181 Ca4H(PO4)3·3H2O (s) 4.896 Ca3(PO4)2 (beta) 4.852 Ca3(PO4)2 (am2) 4.182 Ca3(PO4)2 (am1) 1.432 CaHPO4 (s) 0.29 | Show TableDownLoad:
CSV
2.3 重金属离子对磷去除率的影响
向HAP结晶体系分别单独投加不同浓度Pb2+/Cd2+/Cr3+时,反应终点(HAP结晶反应进入熟化期,溶液中剩余磷浓度基本保持稳定)体系除磷率的变化如图9所示,可以看出,随着重金属投加量的增加,3组反应的除磷率均发生显著改变,且变化趋势各不相同. 随着初始Pb2+浓度从0增加到25 mg·L−1,除磷率先降低后升高,在Pb2+浓度在20 mg·L−1附近时,除磷率最低,即说明在低浓度下,Pb2+会抑制体系的磷去除率,而在高浓度下Pb2+对除磷呈反向促进作用;Cd2+对除磷呈促进作用,10 mg·L−1Cd2+时促进率最大为10.61%;Cr3+对除磷则呈抑制作用,2.5 mg·L−1 Cr3+时抑制率最大为13.55%.
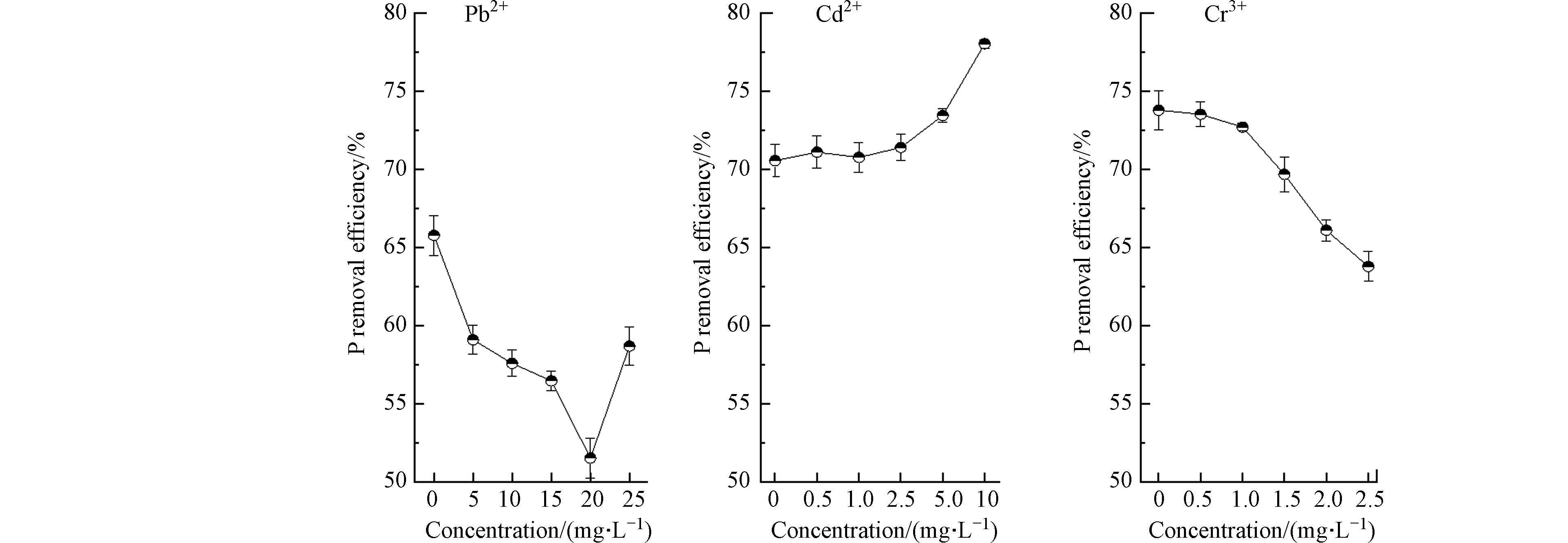 图 9 Pb2+、Cd2+、Cr3+初始浓度对反应体系除磷率的影响Figure 9. Effects of Pb2+,Cd2+,Cr3+ concentration on the phosphorus removal efficiency of reaction system
图 9 Pb2+、Cd2+、Cr3+初始浓度对反应体系除磷率的影响Figure 9. Effects of Pb2+,Cd2+,Cr3+ concentration on the phosphorus removal efficiency of reaction system结合2.2节的实验结果,分析产生这种现象的原因是:Pb2+在低浓度时通过生成Pb(OH)2抑制HAP结晶,进而导致除磷率下降,而随着Pb2+浓度的升高,开始更多地生成铅羟基磷灰石和磷氯铅矿,弥补了由于HAP减少带来的除磷量的下降;Cd2+与PO43-结合生成Cd3(PO4)2,促进了液相中磷的去除;Cr3+一方面能抑制HAP的成核速率以及前驱体向HAP的转化速率,另一方面Cr3+在生成Cr(OH)3时消耗溶液中OH-,两方面的共同作用导致HAP结晶量大幅下降,除磷率降低.
3. 结论(Conclusion)
(1)3种重金属(Pb2+、Cd2+、Cr3+)均会降低HAP的结晶速率,抑制幅度Cr3+>Cd2+>Pb2+. 其中,Cr3+和Cd2+的初始浓度与结晶速率呈负相关,两者均会明显延长HAP的诱导时间;Pb2+在低浓度(<20 mg·L−1)时能提高HAP结晶速率,但当其浓度大于20 mg·L−1后,初始浓度与结晶速率呈负相关.
(2)3种重金属均能与HAP的构晶离子(OH-和PO43-)发生反应,进而与HAP共沉淀进入产物中,各自产物类型不同. Pb2+主要生成铅羟基磷灰石(Pb5(PO4)3OH、Ca2.5Pb7.5(PO4)6(OH)2)及磷氯铅矿(Pb5(PO4)3Cl);Cd2+易与PO43-结合生成无定形态物质Cd3(PO4)2赋存于产物中;而Cr3+则易生成Cr2O3和Cr(OH)3.
(3)3种重金属对HAP结晶体系除磷的影响各不相同:Pb2+在低浓度时对除磷有抑制作用,高浓度下则能促进磷的去除;Cd2+对结晶体系除磷呈促进作用,Cr3+则呈稳定的抑制作用.
-
图 1 (a)Pb2+、(b)Cd2+和(c)Cr3+的初始浓度对反应体系pH变化曲线的影响
Figure 1. Effect of (a) Pb2+, (b)Cd2+, (c) Cr3+ concentration on the pH curve of reaction system
图 2 (a)HAP、(b)加入25 mg·L−1Pb2+产物的EDS谱图
Figure 2. EDS spectrums of (a) HAP and (b) product with addition of 25 mg·L−1 Pb2+
图 3 Pb2+加入前后产物的XRD谱图
Figure 3. XRD pattern of product before and after the addition of Pb2+
图 4 重金属加入前后产物的FTIR谱图
Figure 4. FTIR diagram of product before and after the addition of heavy metals
图 5 (a)HAP、(b)加入10 mg·L−1Cd2+产物的EDS谱图
Figure 5. EDS spectrums of (a) HAP and (b) product with addition of 10 mg·L−1 Cd2+
图 6 Cd2+加入前后产物的XRD谱图
Figure 6. XRD pattern of product before and after the addition of Cd2+
图 7 (a)HAP、(b)加入2.5 mg·L−1Cr3+产物的EDS谱图
Figure 7. EDS spectrums of (a) HAP and (b) product with addition of 2.5 mg·L−1 Cr3+
图 8 Cr3+加入前后产物的XRD谱图
Figure 8. XRD pattern of product before and after the addition of Cr3+
图 9 Pb2+、Cd2+、Cr3+初始浓度对反应体系除磷率的影响
Figure 9. Effects of Pb2+,Cd2+,Cr3+ concentration on the phosphorus removal efficiency of reaction system
表 1 结晶体系含25 mg·L−1 Pb2+时主要的过饱和物质及相应的饱和指数SI
Table 1. Major supersaturated substances and their saturation indexes in the crystal system containing 25 mg·L−1 Pb2+
过饱和物质Supersaturated substance 饱和指数Saturation index 过饱和物质Supersaturated substance 饱和指数Saturation index Pb5(PO4)3Cl(c) 34.193 PbHPO4(s) 2.826 Pb5(PO4)3Cl(soil) 30.163 HAP 15.168 Pb5(PO4)3(OH) 23.674 Ca4H(PO4)3·3H2O(s) 4.884 Pb3(PO4)2(s) 13.483 Ca3(PO4)2 (beta) 4.843 Pb2(OH)3Cl(s) 3.907 Ca3(PO4)2 (am2) 4.173 Pb(OH)2(s) 3.761 Ca3(PO4)2 (am1) 1.423 注:SI<0时表示物质在溶液中的浓度未超过其溶解度,不会沉淀;而SI>0时物质将过饱和,出现沉淀. 表中未列出饱和指数<1的物质. Note: SI<0 means that the concentration of the substance in the solution does not exceed its solubility and will not precipitate. SI>0 means that the substance will be supersaturated and will precipitate. Substances with a saturation index less than 1 are not listed in the table.
下载: 导出CSV
表 2 结晶体系含10 mg·L−1 Cd2+时主要的过饱和物质及相应的饱和指数SI
Table 2. Major supersaturated substances and their saturation indexes in the crystal system containing 10 mg·L−1 Cd2+
过饱和物质 Supersaturated substance 饱和指数 Saturation index Cd3(PO4)2 (s) 4.149 Hydroxyapatite 15.109 Ca4H(PO4)3·3H2O (s) 4.82 Ca3(PO4)2 (beta) 4.802 Ca3(PO4)2 (am2) 4.132 Ca3(PO4)2 (am1) 1.382 CaHPO4 (s) 0.263
下载: 导出CSV
表 3 结晶体系含2.5 mg·L−1 Cr3+时主要的过饱和物质及相应的饱和指数SI
Table 3. Major supersaturated substances and their saturation indexes in the crystal system containing 2.5 mg·L−1 Cr3+
过饱和物质 Supersaturated substance 饱和指数 Saturation index Cr2O3 (c) 5.377 Cr(OH)3 (am) 2.519 Hydroxyapatite 15.181 Ca4H(PO4)3·3H2O (s) 4.896 Ca3(PO4)2 (beta) 4.852 Ca3(PO4)2 (am2) 4.182 Ca3(PO4)2 (am1) 1.432 CaHPO4 (s) 0.29
下载: 导出CSV
-
[1] XUE Q, HE X Y, SACHS S D, et al. The current phosphate recycling situation in China and Germany: A comparative review [J]. Frontiers of Agricultural Science and Engineering, 2019, 6(4): 403. doi: 10.15302/J-FASE-2019287 [2] MAYER B K, BAKER L A, BOYER T H, et al. Total value of phosphorus recovery [J]. Environmental Science & Technology, 2016, 50(13): 6606-6620. [3] 孙雅, 周通, 陈广源, 等. 鸟粪石晶体生长速率关键影响因素的定量分析 [J]. 化工学报, 2021, 72(11): 5831-5839. SUN Y, ZHOU T, CHEN G Y, et al. Quantitative analysis of key factors affecting struvite crystal growth rate [J]. CIESC Journal, 2021, 72(11): 5831-5839(in Chinese).
[4] PERERA M K, ENGLEHARDT J D, DVORAK A C. Technologies for recovering nutrients from wastewater: A critical review [J]. Environmental Engineering Science, 2019, 36(5): 511-529. doi: 10.1089/ees.2018.0436 [5] 钟仁. 废水磷回收过程中鸟粪石结晶对重金属的吸附及共沉淀机制研究[D]. 广州: 广东工业大学, 2021. ZHONG R. Investigation on the adsorption and co-precipitation mechanism of heavy metals on struvite crystals during the process of phosphorus recovery from wastewater[D]. Guangzhou: Guangdong University of Technology, 2021(in Chinese).
[6] YANG W J, SHAN J, PAN Y, et al. A new strategy for obtaining highly concentrated phosphorus recovery solution in biofilm phosphorus recovery process [J]. Journal of Environmental Sciences, 2022, 112: 366-375. doi: 10.1016/j.jes.2021.05.017 [7] LI X, SHEN S T, XU Y Y, et al. Application of membrane separation processes in phosphorus recovery: A review [J]. Science of the Total Environment, 2021, 767: 144346. doi: 10.1016/j.scitotenv.2020.144346 [8] DAI H L, LU X W, PENG Y H, et al. An efficient approach for phosphorus recovery from wastewater using series-coupled air-agitated crystallization reactors [J]. Chemosphere, 2016, 165: 211-220. doi: 10.1016/j.chemosphere.2016.09.001 [9] UCAR S, BJØRNØY S H, BASSETT D C, et al. Formation of hydroxyapatite via transformation of amorphous calcium phosphate in the presence of alginate additives [J]. Crystal Growth & Design, 2019, 19(12): 7077-7087. [10] 金筱英. 基于HAP结晶的污水深度除磷技术与机理研究[D]. 长沙: 长沙理工大学, 2019. JIN X Y. Research on deep phosphorus removal technology and mechanism of wastewater based on HAP crystallization[D]. Changsha: Changsha University of Science & Technology, 2019(in Chinese).
[11] 王铸, 杜兵, 刘寅. 羟基磷酸钙结晶除磷研究进展 [J]. 环境工程, 2015, 33(11): 16-20. doi: 10.13205/j.hjgc.201511004 WANG Z, DU B, LIU Y. Research advances in phosphorus removal by hydroxyapatite crystallization [J]. Environmental Engineering, 2015, 33(11): 16-20(in Chinese). doi: 10.13205/j.hjgc.201511004
[12] XIE B Q, HALTER T J, BORAH B M, et al. Tracking amorphous precursor formation and transformation during induction stages of nucleation [J]. Crystal Growth & Design, 2014, 14(4): 1659-1665. [13] 杨佳妮. 废水体系磷回收过程中重金属对鸟粪石结晶的影响[D]. 广州: 广东工业大学, 2019. YANG J N. Effect of hazardous metals on crystallization of precipitated struvite during phosphorus recovery[D]. Guangzhou: Guangdong University of Technology, 2019(in Chinese).
[14] LUNDAGER MADSEN H E. Influence of foreign metal ions on crystal growth and morphology of brushite (CaHPO4, 2H2O) and its transformation to octacalcium phosphate and apatite [J]. Journal of Crystal Growth, 2008, 310(10): 2602-2612. doi: 10.1016/j.jcrysgro.2008.01.047 [15] DAI H L, TAN X W, ZHU H, et al. Effects of commonly occurring metal ions on hydroxyapatite crystallization for phosphorus recovery from wastewater [J]. Water, 2018, 10(11): 1619. doi: 10.3390/w10111619 [16] TANG C J, LIU Z G, PENG C, et al. New insights into the interaction between heavy metals and struvite: Struvite as platform for heterogeneous nucleation of heavy metal hydroxide [J]. Chemical Engineering Journal, 2019, 365: 60-69. doi: 10.1016/j.cej.2019.02.034 [17] WANG L J, NANCOLLAS G H. Calcium orthophosphates: Crystallization and dissolution [J]. Chemical Reviews, 2008, 108(11): 4628-4669. doi: 10.1021/cr0782574 [18] 蒋淑琴. 无定形磷酸钙为前驱体的羟基磷灰石成核动力学[D]. 杭州: 浙江大学, 2015. JIANG S Q. Amorphous calcium phosphate mediated hydroxyapatite nucleation kinetics[D]. Hangzhou: Zhejiang University, 2015(in Chinese).
[19] CHEN Y, GU W J, PAN H H, et al. Stabilizing amorphous calcium phosphate phase by citrate adsorption [J]. CrystEngComm, 2014, 16(10): 1864-1867. doi: 10.1039/C3CE42274G [20] ZHU X H, LI J, LUO J H, et al. Removal of cadmium (Ⅱ) from aqueous solution by a new adsorbent of fluor-hydroxyapatite composites [J]. Journal of the Taiwan Institute of Chemical Engineers, 2017, 70: 200-208. doi: 10.1016/j.jtice.2016.10.049 [21] PHAM MINH D, TRAN N D, NZIHOU A, et al. Hydroxyapatite gel for the improved removal of Pb2+ ions from aqueous solution [J]. Chemical Engineering Journal, 2013, 232: 128-138. doi: 10.1016/j.cej.2013.07.086 [22] PARK J H, LEE D W, IM S W, et al. Oxidative coupling of methane using non-stoichiometric lead hydroxyapatite catalyst mixtures [J]. Fuel, 2012, 94: 433-439. doi: 10.1016/j.fuel.2011.08.056 [23] OGAWA S, SATO T, KATOH M. Formation of a lead-insoluble phase, pyromorphite, by hydroxyapatite during lead migration through the water-unsaturated soils of different lead mobilities [J]. Environmental Science and Pollution Research, 2018, 25(8): 7662-7671. doi: 10.1007/s11356-017-1093-9 [24] 岳燕丽, 褚伟伟, 卢真真, 等. 绣球花状掺锶碳羟基磷灰石对Pb2+的吸附 [J]. 环境化学, 2017, 36(5): 1131-1139. doi: 10.7524/j.issn.0254-6108.2017.05.2016113002 YUE Y L, CHU W W, LU Z Z, et al. Study on Pb2+ adsorption by Hydrangea-like strontium-doped carbonate hydroxyapatite [J]. Environmental Chemistry, 2017, 36(5): 1131-1139(in Chinese). doi: 10.7524/j.issn.0254-6108.2017.05.2016113002
[25] 何豪, 朱宗强, 刘杰, 等. 镁-钙羟基磷灰石吸附剂对水中Pb2+的去除 [J]. 环境科学, 2019, 40(9): 4081-4090. HE H, ZHU Z Q, LIU J, et al. Removal of Pb2+ from aqueous solution by magnesium-calcium hydroxyapatite adsorbent [J]. Environmental Science, 2019, 40(9): 4081-4090(in Chinese).
[26] 张连科, 王洋, 王维大, 等. 磁性羟基磷灰石/生物炭复合材料的制备及对Pb2+的吸附性能 [J]. 环境科学学报, 2018, 38(11): 4360-4370. ZHANG L K, WANG Y, WANG W D, et al. Preparation of magnetic hydroxyapatite/biochar composite and its adsorption behavior of Pb2+ and recycling performance [J]. Acta Scientiae Circumstantiae, 2018, 38(11): 4360-4370(in Chinese).
[27] 李锦, 张川, 刘伟霞, 等. 溶剂热法制备Cd(OH)2和CdO纳米盘研究性实验 [J]. 物理实验, 2012, 32(1): 1-5. LI J, ZHANG C, LIU W X, et al. Exploring experiments on the preparation of Cd(OH)2 and CdO nanodisks by solvothermal method [J]. Physics Experimentation, 2012, 32(1): 1-5(in Chinese).
[28] CORAMI A, MIGNARDI S, FERRINI V. Cadmium removal from single- and multi-metal (Cd+Pb+Zn+Cu) solutions by sorption on hydroxyapatite [J]. Journal of Colloid and Interface Science, 2008, 317(2): 402-408. doi: 10.1016/j.jcis.2007.09.075 [29] BOWDEN L I, JARVIS A P, YOUNGER P L, et al. Phosphorus removal from waste waters using basic oxygen steel slag [J]. Environmental Science & Technology, 2009, 43(7): 2476-2481. [30] 张曦. 铬污染土壤修复稳定性及Cr(Ⅲ)再氧化研究[D]. 兰州: 兰州交通大学, 2021. ZHANG X. Study on remediation stability of chromium contaminated soil and Cr(Ⅲ) reoxidation[D]. Lanzhou: Lanzhou Jiaotong University. 2021(in Chinese).
[31] SALLAM S M, TOHAMI K M, SALLAM A M, et al. Synthesis and characterization of hydroxyapatite contain chromium [J]. Journal of Biophysical Chemistry, 2012, 3(4): 278-282. doi: 10.4236/jbpc.2012.34033 [32] 酆婧轩, 李芸邑, 师帅, 等. 硫代硫酸钠、磷酸钠联合处理铬渣中的六价铬 [J]. 中国环境科学, 2015, 35(11): 3333-3339. FENG J X, LI Y Y, SHI S, et al. Remediation of Cr6+ in chromite ore processing residue by sodium thiosulfate and sodium phosphate [J]. China Environmental Science, 2015, 35(11): 3333-3339(in Chinese).
期刊类型引用(1)
1. 邱林清,陈煜柠,胡泽昆,韦朝海,邱光磊. 重金属对磷酸钙沉淀回收的影响及其沉积行为. 环境工程. 2025(03): 70-76 .  百度学术
百度学术
其他类型引用(0)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 3299
- HTML全文浏览数: 3299
- PDF下载数: 99
- 施引文献: 1
