


-
木质纤维素是一种来源丰富、分布广泛的可再生资源,可用来生产多种生物基产品(如燃料乙醇、乳酸等)[1-2]. 由于目前全球面临食品短缺、能源危机和环境污染等问题,将木质纤维素转化为生物基化学品和清洁燃料的研究一直备受关注[3-5]. 然而,由于木质纤维素的生物顽抗性,微生物和酶很难将其降解利用. 预处理能够将这种生物质顽抗性破坏,增大纤维素的孔隙率和比表面积,有效降低纤维素的结晶度,提高酶解时酶对纤维素底物的可及性,提高木质纤维素生物质的转化率. 因此合适的预处理方法,是木质纤维素生物质高值转化为发酵底物——糖(葡萄糖、木糖等)的重要步骤. 目前国内外主要使用物理技术(机械破碎[6]、微波[7]、超声[8])、化学技术(酸、碱水解[9-10]、有机溶剂、离子液体预处理[11-12])、生物技术(真菌处理[13]、生物酶法[14])和物理化学技术(蒸汽爆破、氨纤维爆破、水热预处理[15-17])等预处理技术,去除木质纤维素原料中的木质素和少量半纤维素,增加纤维素孔隙度以增强酶对纤维素的可及性,提高酶水解和糖转化的效率.
水热预处理是处理木质纤维素生物质的常用方法,它具有环境友好,操作成本低的优势[18]. 水热预处理利用热水作为唯一的试剂并且在相对较低的温度(150—230 ℃)和压力下进行,被认为是一种经济有效的生物质预处理技术[19]. 碱预处理也是一种常见的生物质预处理方法,通常使用NaOH来处理木质纤维素. 碱法预处理能够打破木质素和碳水化合物之间的酯键,溶解木质素和部分半纤维素,使得木质纤维素内表面积增加,降低纤维素结晶度和聚合度,大大提高了原料的酶解得率[20]. 本实验室在前人[21]的基础上,建立了一种新型的预处理方式,乙二胺(EDA / Ethanediamine)预处理. EDA预处理可广泛去除木质纤维素中的木质素和乙酰基,并将I型或II型结晶纤维素转变为III型或异形体结构,导致酶解得率增加. EDA预处理过程中不需要添加水或其他溶剂,是一种“干”法到“干”法的过程,避免了预处理后的固液分离. 另外,由于EDA良好的挥发性,可以在预处理后轻易地回收利用[22]. 因此,EDA预处理是一种非常具有前景的预处理方式.
本文以我国丰富的稻草(RS / Rice straw)为原料,研究了水热、NaOH、EDA的 3种预处理方式对其化学组成和物理化学结构特性的影响,以探讨影响木质纤维素底物酶解转化的关键因素,为稻草等木质纤维素原料的生物转化提供研究基础.
-
本研究所用稻草来自天津市宝坻县,原料自然风干,剪成2—3 cm小段,用微型植物试验粉碎机粉碎,过40—80目筛,装进密封袋于干燥阴凉处保存. 用于成分分析的稻草原料在空气中干燥至水分低于10%.
-
水热预处理 称取20 g 稻草,以固液比1:10加入蒸馏水,搅拌使其充分混匀,反应温度为150—180 ℃,反应时间为10—30 min. 反应结束后待反应体系温度降至50 ℃以下时,将所得的预处理物料用蒸馏水进行漂洗除杂处理,用纱布分离液体和固体部分. 过滤后将所得物料于60 ℃烘干,得到预处理的稻草,于密封袋中保存,用于后续实验分析.
NaOH预处理 首先将指定量的NaOH(0.9—3.6 g)与200 mL 水混合均匀,随后加入20 g 稻草搅拌使其充分混匀,反应时间为10—30 min,反应温度为90—150 ℃. 反应结束后待反应体系温度降至50 ℃ 以下时,将所得的预处理物料用蒸馏水进行漂洗除杂处理,用纱布分离液体和固体部分. 过滤后将所得物料于60 ℃ 烘干,得到预处理的稻草,于密封袋中保存,用于后续实验分析.
EDA预处理 称取20 g 稻草,EDA用量为10—30 mL,搅拌使其充分混匀,反应时间为10—30 min,反应温度为90—150 ℃. 反应结束后待反应体系温度降至50 ℃ 以下时,将所得的预处理物料用蒸馏水进行漂洗除杂处理,用纱布分离液体和固体部分. 过滤后将所得物料于60 ℃ 烘干,得到预处理的稻草,于密封袋中保存,用于后续实验分析.
-
反应体系的总体积为20 mL,固含量为1%葡聚糖,纤维素酶和半纤维素酶的用量为27 mg纤维素酶 (CTec2)和15 mg 半纤维素酶(HTec2)每克葡聚糖. 加入1 mL 1 mol·L−1 柠檬酸钠缓冲液和0.2 mL 20 g·L−1 叠氮化钠用以维持体系的 pH(4.8)和防止微生物污染. 酶解反应条件为50 ℃ 和 200 r·min−1,反应72 h 后,终止酶解反应,将酶解液取出12000 r·min−1离心取上清,用0.22 μm 的水相注射器滤膜过滤酶解液后通过高效液相色谱法测定糖浓度.
-
纤维素原料的化学成分分析方法主要参照美国国家能源部可再生能源实验室(NREL)制定的实验室分析操作规程(LAP)的标准方法进行分析[23].
利用高效液相色谱法(high performance liquid chromatography,HPLC)测定样品水解液中的单糖浓度. HPLC分析条件为:色谱柱为Biorad Aminex HPX-87H色谱柱,进样量10 μL,流动相为5 mmol·L−1 H2SO4,流速0.6 mL·min−1,柱温控制在65 ℃,示差折光检测器温度 50 ℃. 纤维素和半纤维素的主要成分是葡聚糖和木聚糖,得到的葡萄糖和木糖的产量分别基于 0.9 g 葡聚糖/1.0 g 葡萄糖和 0.88 g 木聚糖/1.0 g木糖的比率换算[23]. 纤维素和半纤维素回收率的计算公式(1)、(2):
其中,Cg、Cx分别表示水解液中葡萄糖和木糖浓度,单位为 g·L−1. 生物质干重的单位为g.
酸不溶性木质素(AIL)回收率的计算方法见公式(3):
其中,AIL%表示酸不溶性木质素的含量,m1、m2、m3、m4分别表示滤纸和固体的质量(g)、滤纸的质量(g)、灰分质量(g)和生物质干重(g).
酶解得率的计算方法见公式(4)、(5):
其中,Cg、Cx分别表示酶解液中葡萄糖和木糖浓度,单位为 g·L−1. 酶解添加量即固含量为1%葡聚糖的样品质量,单位为 g. 木糖含量为样品中半纤维素所占的比值,单位为%.
-
扫描电子显微镜(SEM) 预处理前后稻草的表面微观结构变化通过电子扫描显微镜测定. 测定前,干燥的样品需要喷金. 测定时,扫面电压控制在 5 kV到 15 kV 之间.
傅立叶转换红外光谱(FT-IR)分析用傅立叶红外光谱测定预处理及酶解前后纤维素底物化学成分的变化,红外光谱的仪器分辨率为4 cm−1,波数范围4000—400 cm−1,扫描次数16次,样品处理采用KBr压片法,2 mg 样品与300 mg KBr混合,压片,使样品粒径降低,分散均匀.
X射线衍射仪分析 用X射线衍射方法分析预处理前后及酶解过程中纤维素底物的纤维素结晶度的变化规律,在 2θ =10°—40°范围内扫描,变化速率为2(°)·min−1. 结晶度计算见公式(6):
式中,I002为002衍射晶面2θ=22.8°的强度,Iam为2θ=18.0°散射峰的强度.
-
之前研究表明,水热预处理能够去除玉米秸秆、小麦秸秆和甘蔗中80%左右的半纤维素[24-26]. 水热预处理对稻草化学成分的影响如图1所示,图中的固体回收率为预处理前后样品的质量比值. 由于预处理去除了部分木质素组分,分解了部分纤维素和半纤维素(葡聚糖和木聚糖,下同),即可通过样品质量的变化来体现出预处理效果.
随着预处理温度和处理时间的提高,稻草固体回收率降低,说明水热预处理去除了稻草中的组分,且去除能力随着预处理强度的增加而增加. 其中半纤维素去除的量最多,这表明半纤维素容易在水热预处理中发生水解. 180 ℃ 条件下,当预处理时间从10 min 延长到30 min 时,稻草的半纤维素回收率从70.4%降到39.7%;而在相同的时间条件下(30 min),当预处理温度从150 ℃ 升高到180℃时,固体回收率从83.2%降到63.9%. 之前研究表明,木质纤维素原料中半纤维素的乙酰基团容易在高温环境下脱落形成乙酸,从而进一步促进预处理的过程[27]. 随着预处理条件从150 ℃,10 min 增强到180 ℃,30 min,木质素回收率下降明显,从67.5%降至33.4%,说明木质素、半纤维素的去除会随着水热预处理强度的增强而增强. 随着预处理强度的增加,纤维素的回收率一直维持在80%—84%之间,在较低预处理强度下能够去除80.7%的纤维素,而较高的预处理强度下纤维素的回收率没有明显增加. 这可能是由于纤维素中的非结晶区容易在水热条件下降解,而结晶区相对比较稳定,非结晶区去除之后继续增强预处理强度纤维素的结晶区仍然得以保留. 然而,水热预处理过程中,80%—84%的纤维素回收率,不利于对稻草中有效组分纤维素的利用.
-
虽然水热预处理能够有效去除半纤维素和木质素,但是其纤维素组分损失较多,表明水热预处理对木质素的选择去除能力较差. 因此,本研究考察了NaOH预处理对稻草组分的影响(图2). 可以看出,与水热预处理相似,随着NaOH预处理强度的提高,稻草的固体回收率逐渐降低,说明稻草组分得到了一定程度的去除. 当NaOH用量由0.9 g 增加到3.6 g 时(120 ℃,20 min),预处理后稻草中的木质素回收率由62.6%降低至36.5%,半纤维素回收率也随之降低(89.9%—72.5%). 随着预处理时间的延长,木质素回收率也逐渐降低. 对比水热预处理,在相同条件下(150 ℃,20 min),NaOH预处理的固体回收率(71.4%)低于水热预处理(84.8%),且木质素回收率明显减少(64.9%—37.3%),这表明NaOH可以有效破坏木质素结构,使木质素间的醚键断裂,降解为小分子的片段并溶出[28].
-
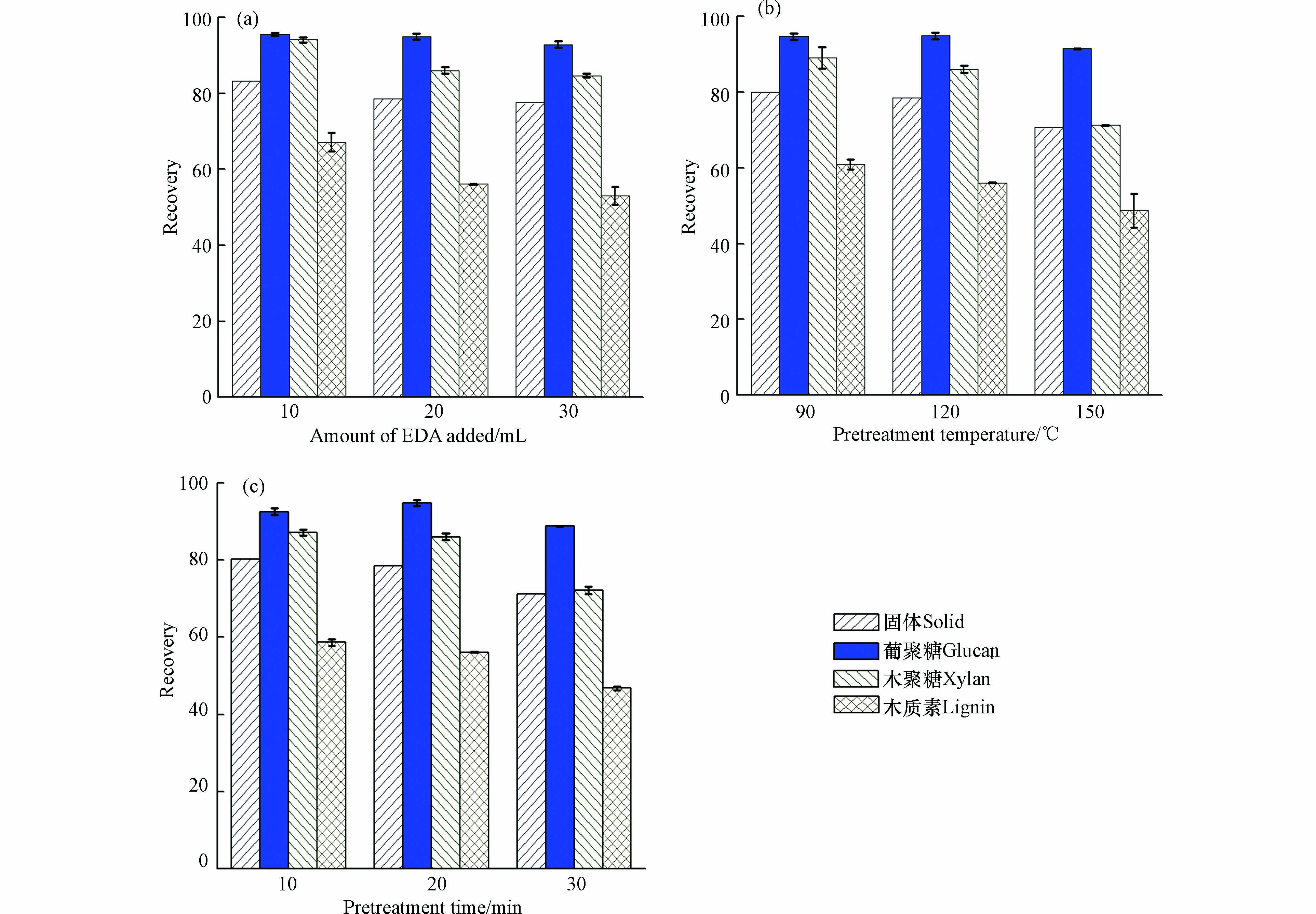
之前的研究表明, EAD预处理中温度和添加量会影响木质素的去除和木质纤维素的酶解率[29]. 因此,本研究主要考察了EDA用量、反应温度和反应时间对稻草组分的影响(图3). 结果表明,EDA预处理与NaOH预处理对木质纤维素组分变化的影响相近,随着预处理强度的增加,木质素回收率逐渐下降,固体回收率逐渐降低. 例如,当预处理温度和时间分别为120 ℃ 和20 min 时,随着EDA用量(10—30 mL)的增加,半纤维素回收率和木质素回收率分别从94.1%和67.2%降为84.7%和53.1%. 在相同预处理条件下(150 ℃,20 min),EDA预处理的半纤维素回收率(71.2%)明显低于NaOH预处理的半纤维素回收率(80.9%),EDA预处理的木质素回收率(48.8%)明显低于水热预处理的木质素回收率(64.9%). 这表明EDA预处理去除木质素和半纤维素的能力较强,可以在较低的温度条件下进行操作.
-
酶解得率是对预处理效果进行评价的重要指标,通过酶解后的葡萄糖和木糖浓度来衡量. 对经过水热预处理的稻草进行酶解,结果如图4所示. 未经预处理的稻草酶解得率只有29.5%和6.6%(葡萄糖和木糖,下同),而水热预处理在150 ℃、30 min 的条件下,酶解得率提高到39.4%和15.4%. 增加预处理强度,酶解得率显著增加,当温度升到180 ℃时,酶解得率达到了73.8%和50.4%,最高预处理强度下(180 ℃,30 min),对比未经预处理的稻草,其酶解得率提高了181.7%和609.9%. 由此可以看到,随着水热预处理强度的增加,稻草的酶解得率得到了明显提高.
-
NaOH预处理对稻草的酶解影响如图5. 从图5可以看出,NaOH添加量和反应温度对稻草酶解得率影响显著,与未经预处理的稻草酶解得率相比,随着NaOH添加量和温度的增加,NaOH预处理酶解得率提升较为显著. 由图5可看出,当NaOH添加量由0.9 g 增加到3.6 g 时(120 ℃,20 min),纤维素和半纤维素的酶解得率由43.8%和28.2%提高到82.4%和65.0%;当温度由90 ℃ 升到150 ℃(1.8 g,20 min),酶解得率由62.0%和43.6%提高到81.6%和61.0%;而反应时间的变化对酶解得率影响不是很明显,随着时间延长,酶解得率波动不大,当时间为由10 min 延长到30 min 时(120 ℃,1.8 g),酶解得率由68.6%和51.7%变为73.9%和58.6%. 对比水热预处理,在相同的与处理温度和时间条件下(150 ℃,20 min),NaOH预处理稻草的酶解得率有所提升,说明碱预处理能够有效提高酶解得率. 主要原因可能是由于碱性预处理能够去除大量木质素,使得木质纤维素孔隙率和纤维素比表面积增加,增加了纤维素酶与底物的可及性,减少了酶的无效吸附[30].
-
对经过不同EDA预处理条件下的稻草进行酶解糖化,结果如图6所示. 由图6可以明显看出,EDA预处理后的稻草酶解得率,明显都高于NaOH和水热预处理. EDA添加量、反应温度和时间均对稻草酶解得率影响明显. 当EDA添加量由10 mL 增加到30 mL 时(120 ℃,20 min),纤维素和半纤维素酶解得率由74.7%和53.8%提高到99.7%和68.2%;当预处理温度由90℃升高到150 ℃ 时(20 min,20 mL),酶解得率由83.4%和63.2%提高到99.8%和84.7%;当反应时间由10 min 延长到30 min 时(120 ℃,20 mL),酶解得率由86.5%和63.7%提高到99.3%和79.6%. 对比水热和NaOH预处理,EDA预处理对稻草酶解得率的提高非常显著,当EDA预处理强度足够大时,纤维素的酶解得率几乎达到100%,可能是因为EAD预处理大量去除了木质纤维素组分中的半纤维素和木质素,极大地增加了酶解可及性,从而显著提高木质纤维素的酶解得率[30].
-
预处理前后稻草的表观形态如图7所示. 未经过预处理的稻草(图7a)表面平整较为光滑,纤维排列规则有序,物理结构致密. 经过水热预处理后的稻草(图7b),其细胞壁表面明显粗糙,结构变得较为疏松. 稻草出现片状和卷曲的纤维,可能是由于从细胞壁上去除了半纤维素,木质素熔化并随后重新沉积所致[31-32]. 经过NaOH预处理后的稻草(图7c),其物理结构出现一定程度破坏,出现了不同大小的空隙,内部纤维外露,纤维束之间的连接变得松散. 通过扫描电子显微镜观察,用NaOH溶液预处理的生物质其孔隙度增加,从而增加了生物质表面积[33].
经过EDA预处理后的稻草(图7d),结构发生了非常明显的变化,致密的物理结构消失不见,结构变得极其松散且充满裂缝,大量纤维束外露,可能是EDA预处理将纤维束之间的连接成分大量溶解造成的. 这些预处理方法基本都是使稻草致密的网络结构变得松散,暴露出纤维素,增加酶与底物的可及性,提高酶解得率.
-
预处理前后稻草的傅里叶红外光谱图如图8所示. 1737 cm−1 和1247 cm−1 分别与半纤维素中的C=O和C-O的伸缩振动有关,1737 cm−1 处的吸收峰对应于半纤维素中的乙酰基和木质素与碳水化合物连接酯键的非共轭羰基C=O伸缩振动,1515 cm−1 处的吸收峰与木质素相关,来源于木质素芳香骨架的变形振动. 对比未预处理稻草,水热预处理稻草的1737 cm−1 吸收峰明显减弱,而NaOH和EDA预处理的稻草1737 cm−1 吸收峰完全消失,这些现象表明,与半纤维素相连接的乙酰基和木质素因为酯键的断裂而脱除. 水热预处理的稻草在1515 cm−1 处的吸收峰有所增强,主要是因为这种预处理方法去除了大量半纤维素,导致木质素的相对含量升高;而NaOH和EDA预处理的稻草在1515 cm−1 处吸收峰明显降低,说明除去了大量的木质素,这与之前的成分分析结果相一致. 之前有研究表明,草本植物中的木质素主要是由对香豆酸和阿魏酸通过酯键与半纤维素连接的[34]. 因此,断裂酯键是预处理去除木质素的重要原因[35].
-
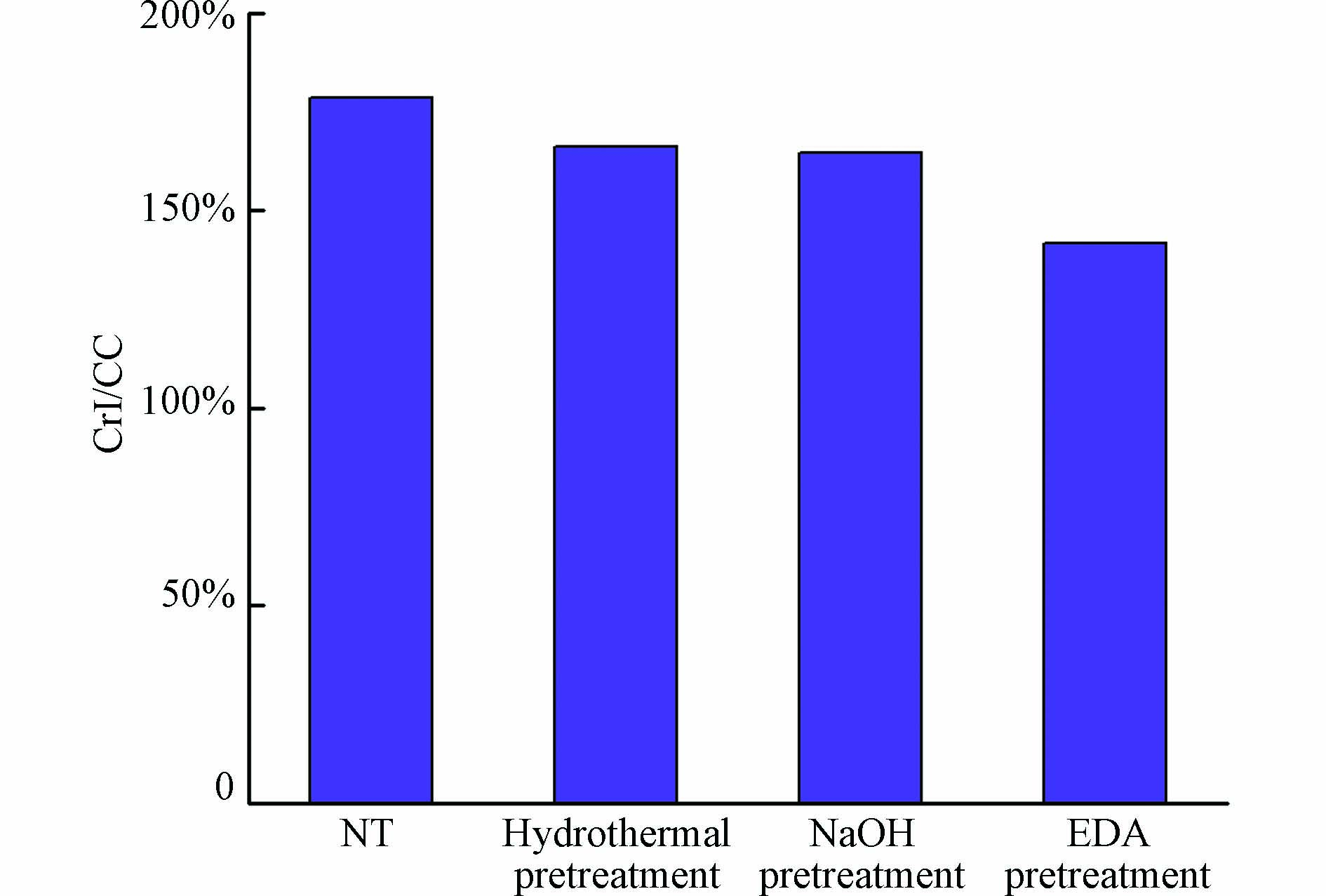
预处理后稻草的相对结晶度如图9所示. 由图9可以看出,未预处理的稻草相对结晶度为178.5%,经过不同预处理后相对结晶度都有所降低,效果最显著的为EDA预处理,相对结晶度降为141.6%. 因此,EDA预处理方法降低稻草的相对结晶度最为有效.
-
水热预处理、NaOH预处理和EDA预处理方法均能不同程度地影响稻草的物理形态和化学成分,从而提高稻草的酶解得率. 水热预处理能够有效去除稻草中大部分半纤维素,但其木质素去除能力较差,对酶解得率的提高并不显著. NaOH预处理和EDA预处理对稻草的物理形态改变较大,对木质素和半纤维素的选择性去除能力较好,稻草的酶解得率得到了明显的提高. FT-IR结果表明,因为碱预处理破坏了木质素和半纤维素相连接的酯键,从而提高了木质素去除能力. 相比于水热预处理和NaOH预处理,EDA预处理能够有效降低纤维素的相对结晶度,显著提高纤维素的酶解得率. 与NaOH预处理和EDA预处理相比,水热预处理需要更高的温度,会极大增加预处理的经济成本. NaOH预处理和EDA预处理温度较低,但其化学试剂本身会腐蚀预处理设备且难以回收,会造成环境污染,预处理后的样品仍需调节pH后才能进行下一步微生物发酵,这会给木质纤维素高值转化增加额外的经济成本.
不同预处理方法对稻草化学组分和酶解得率的影响
Effects of different pretreatment methods on chemical components and enzymatic hydrolysis accessibility of rice straw
-
摘要: 稻草是我国产量极大的木质纤维素代表,将其转化为生物基产品具有重要的意义. 因此研究采用水热、NaOH、乙二胺(EDA / Ethanediamine)预处理方式对稻草进行预处理,以打破木质纤维素的生物顽抗性,研究稻草在不同预处理过程中主要组分的迁移规律及酶解得率;通过扫描电镜(SEM)、傅里叶红外光谱(FT-IR)和X射线衍射(XRD)等分析方法对预处理前后的底物进行微观结构分析,研究其物理形态和化学结构的变化规律. 结果表明,水热预处理的选择性去除能力较差,在去除半纤维素的同时会导致较多纤维素组分的损失;NaOH和EDA预处理能够有效去除木质素组分,同时较好的保留纤维素和半纤维素组分;3种预处理方法都能够促进稻草的酶解得率. 其中,以20 mL乙二胺负载量,在150 ℃条件下油浴20 min预处理的稻草酶解得率得到了显著提高(26.2%—99.8%);预处理后,稻草致密的物理结构变得疏松,大量纤维束外露,增加了纤维素与酶的可及性,与半纤维素相连接的乙酰基和木质素因为酯键的断裂而脱除,与木质素相关的吸收峰在碱性预处理中减弱,而在水热预处理中增强,表明碱性预处理能够降低稻草的相对结晶度,同时能够有效去除木质素.Abstract: Rice straw is the representative of lignocellulose with large yield in China, and it is of great significance to transform it into bio-based products. Therefore, hydrothermal pretreatment, NaOH pretreatment and ethylenediamine pretreatment were used to pretreat rice straw to break the biological resistance of lignocellulose, and to study the migration law of main components and enzymatic hydrolysis rate of rice straw in different pretreatment processes. Scanning electron microscopy (SEM), Fourier transform infrared spectroscopy (FT-IR) and X-ray diffraction (XRD) were used to analyze the microstructure of the substrates before and after pretreatment, and to study the change of their physical morphology and chemical structure. The results show that the selective removal ability of hydrothermal pretreatment is poor, and the removal of hemicellulose will lead to the loss of more cellulose components.The pretreatment of NaOH and EDA could effectively remove the lignin components and retain the cellulose and hemicellulose components. All three pretreatment methods could improve the yield of rice straw. The enzymatic hydrolysis rate of rice straw was significantly increased (26.2%—99.8%) with 20 mL ethanediamine (EDA) loading and oil bath for 20 min at 150 ℃. After pretreatment, straw dense physical structure become loose, a large number of fiber bundle exposed, increased accessibility, cellulose and enzymes of the acetylation of connected to hemicellulose and lignin removal by ester bond rupture, the absorption peak associated with lignin in alkaline pretreatment is abate, and enhanced in the hydrothermal pretreatment, show that alkali pretreatment can reduce the relative crystallinity of straw, At the same time, it can effectively remove lignin.
-
Key words:
- lignocellulose /
- pretreatment /
- enzymatic hydrolysis yield /
- microstructure.
-
多环芳烃 (polycyclic aromatic hydrocarbons,PAHs)是一类由2个及2个以上苯环组成的有机污染物,原型及其衍生物达400多种[1]。美国环保局(USEPA)将16种对人体健康危害较大的PAHs列入了优先控制污染物名单[2],其中苯并[a]芘(BaP)被确定为强致癌物质。
多环芳烃广泛分布在各种环境介质中。由于具有疏水性和亲脂性,PAHs在水中的溶解性较差,主要被悬浮颗粒物吸附,并可随悬浮颗粒物沉降至沉积物,沉积物中的PAHs经过解吸和再悬浮作用重新进入水体,成为新的污染源,同时通过生物积累和生物放大对生态系统和人体健康构成潜在危害。随着研究工作的广泛开展,不同类型地表水和沉积物中PAHs研究取得较大进展。我国对水体和沉积物中PAHs的研究主要集中在水库[3-4]、河流[5-7]、湖泊[8-10]、江海[11]及地下河[12]等,积累了大量数据;浅层地下水中PAHs的研究较少,主要集中在江苏[13]、河北[14]、安徽[15]和河南[16];然而在我国广大农村地区,沟塘数量众多且分布广泛。2016—2018年在淮河流域5个区县调查显示,49.7%(1272/2559)的农村居民报告住宅周边有沟塘,而且报告沟塘水体质量较差,24.3%(622/2559)的沟塘有异味。我国尚未开展农村地区沟塘水和沉积物中PAHs水平及其对周边浅层地下水影响的研究。
本文选取地处河南省西平县的5个沟塘水、3个沉积物和21户居民家中的浅层地下水作为研究对象,在2016年8月采集浅层地下水、沟塘水及沉积物样品,测定16种PAHs(各多环芳烃化合物的缩写详见表1)的含量,分析PAHs的空间分布特征和组分特征,评价农村地区沟塘水对其周边浅层地下水的影响,评估沟塘水和沉积物的生态风险及浅层地下水的人群健康风险。
表 1 浅层地下水、沟塘水及沉积物中PAHs含量Table 1. Concentration of PAHs in the surface sediment, surrounding shallow groundwater and ditch pond water化合物Compounds 环数Rings TEF 浅层地下水Shallow groundwater(N=21) 沟塘水Ditch pond water(N=5) 沉积物Surface sediment(N=3) 检出率/%Detection ration 平均值/(ng·L−1)Average concentration 范围/(ng·L−1)Concentration range 检出率/%Detection ration 平均值/(ng·L−1)Average concentration 范围/(ng·L−1)Concentration range 检出率/%Detection ration 平均值/(ng·kg−1)Average concentration 范围/(ng·kg−1)Concentration range 奈 (Nap) 2环 0.001 95.2 3.17 ND.—10.2 60.0 3.88 N.D.—13.6 100 8.49 1.07—12.2 苊烯(Acy) 3环 0.001 76.2 4.73 ND.—39.2 0.00 N.D. ND. 100 5.57 1.36—12.2 苊(Ace) 3环 0.001 95.2 14.3 ND.—83.7 100 5.56 0.299—25.6 100 12. 6 10.3—15.1 芴(Flu) 3环 0.001 85.7 9.42 ND.—55.9 100 2.06 0.245—5.39 66.7 3.13 ND.—5.59 菲(Phe) 3环 0.001 100 51.1 0.61—349 100 45.7 7.09—180 66.7 13.7 ND.—22.3 蒽(Ant) 3环 0.01 85.7 13.1 ND.—101 40.0 11.4 ND.—54.9 100 2.77 0.167—4.81 荧蒽(Fl) 4环 0.001 100 40.6 1.39—243 100 82.7 2.59—390 100 28.2 1.63—74.7 芘(Pyr) 4环 0.001 100 40.5 0.782—356 100 48.4 1.35—228 100 23.1 0.0940—61.9 苯并(a)蒽(BaA) 4环 0.1 100 14.5 0.706—122 100 25.0 0.710—115 100 7.70 1.53—18.8 䓛(Chr) 4环 0.01 100 15.9 0.601—115 60 28.9 ND.—136. 100 20.9 7.74—39.0 苯并(b)荧蒽(BbF) 5环 0.1 100 3.93 0.547—27.8 100 11.1 0.825—50.5 66.7 14.4 ND.—37.5 苯并(k)荧蒽(BkF) 5环 0.1 100 1.34 0.203—7.74 100 5.18 0.295—23.8 100 7.84 1.74—19.0 苯并芘(BaP) 5环 1 100 2.72 0.164—17.3 100 11.1 0.0420—55.0 100 16.0 2.18—41.4 二苯并[a,h]蒽(DBA) 5环 1 100 0.83 0.111—5.33 100 11.2 0.274—50.2 100 55.3 0.624—161 苯并[g,h,i]芘(BP) 6环 0.01 100 1.81 0.168—10.2 80.0 17.2 ND.—85.1 100 25.4 0.0520—67.8 茚并[1,2,3-cd]芘(InP) 6环 0.1 100 0.81 0.320—2.87 100 7.30 0.633—28.3 100 10.9 2.27—26.2 ∑7PAHs 40.0 3.36—298 99.7 2.84—414 133 21.9—191 ∑16PAHs 219 8.39—1234 317 15.4—1372 256 101—458 TEQ(BaP)7 5.77 0.727—39.8 27.5 0.568—83.1 75.6 4.01—167 TEQ(BaP)16 6.09 0.763—41.8 28.0 0.589—85.3 76.0 4.16—167 注:1) 粗体为国际癌症研究机构(IARC)划定的7种致癌性PAHs;2) ND.表示未检出;3) TEF表示毒性当量。 Note: 1) In bold means the seven carcinogenic PAHs as defined by the International Agency for Research on Cancer (IARC); 2) ND.means no detected; 3) TEF means toxic equivalent. | Show Table DownLoad:
CSV
DownLoad:
CSV
1. 材料与方法(Materials and methods)
1.1 研究区域的基本情况
研究区域位于河南省西平县的滞洪区内,浅层地下水埋藏深度为30—45 m,含水层中含水介质导水性差,地下水径流缓慢,径流方向由西北和西南向东南。
1.2 沟塘水质及周边环境状况
选择5个常年有水、周围居住人群较为密集的沟塘为研究对象,沟塘水体面积约为100—200 m2. 沟塘水体均呈现重度浑浊并伴有较强的嗅味,表面漂浮大量绿藻和生活垃圾(塑料、餐盒、秸秆、农药瓶等);调查的21户居民中有13户(占61.9%)报告会将生活垃圾和生活污水直接倾倒或排放到附近沟塘里,有9户(占42.9%)在沟塘里或周围养殖禽畜,其中2家直接将养殖废水排放到沟塘中。
1.3 点位布设与样品采集
在拟定沟塘和距离1500 m范围内采集水样,浅层地下水的采集以沟塘为原点向南或东南方向布设,在拟定点位附近的住户家中采样,沟塘水和沉积物的采集点位尽量布设在靠近沟塘中心部位,点位布设参见图1。共采集浅层地下水样21个、沟塘水样5个、沉积物3个,浅层地下水的平均井深为34 m(10.0—50.0 m)。采样方法参照《地表水和污水监测技术规范》(HJ/T91—2002)、《地下水环境监测技术规范》(HJ/T164—2004)和《土壤环境监测技术规范》(HJ/T166—2004)。采样器和盛水容器用稀盐酸 (优级纯)清洗,采样前用目标水样清洗3次。测量记录pH值和温度等基本信息后,用2.5L棕色广口瓶采集水样2瓶。以纯净水作为现场采样和样品运输保存的空白样。
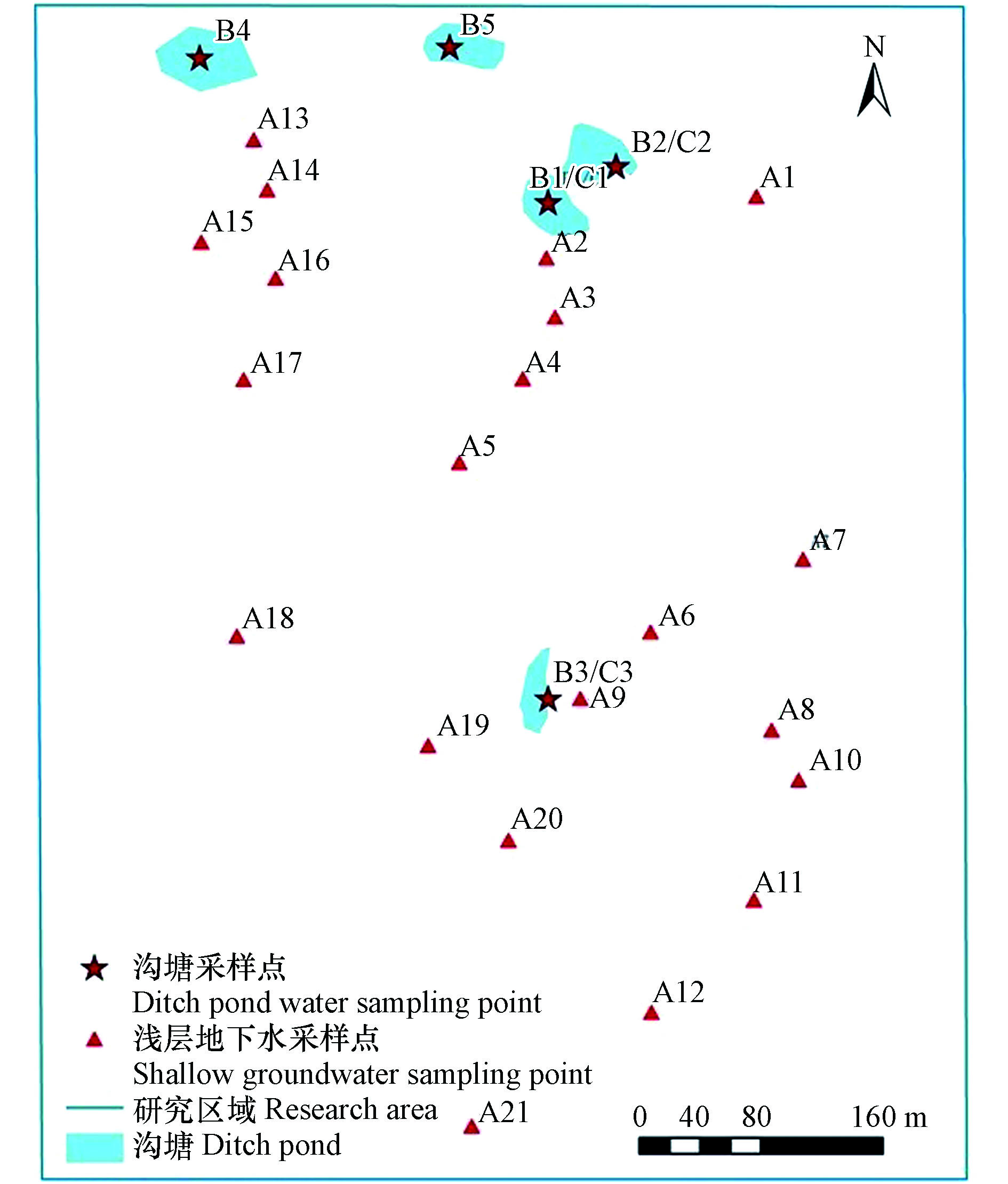 图 1 浅层地下水、沟塘水及沉积物采样示意图Figure 1. Sampling sites of in the surface sediment, surrounding shallow groundwater and ditch pond water
图 1 浅层地下水、沟塘水及沉积物采样示意图Figure 1. Sampling sites of in the surface sediment, surrounding shallow groundwater and ditch pond water1.4 主要仪器和试剂
Waters alliance e2695高效液相色谱仪,配备Waters 2998光电二极管阵列检测器、Waters 2475多波长荧光检测器和四元泵(美国Waters公司);Waters PAH C18色谱柱,4.6 mm×250 mm×5.0 μm(Part No. 186001265,美国Waters公司);Mettler Toledo XS205十万分之一分析天平;IKA MS3 basic旋涡振荡器(德国IKA);KQ-250DV超声仪(昆山市超声仪器有限公司);N-EVAPTM112氮吹浓缩仪(美国Organomation Associates公司);0.22 μm有机系针筒式微孔滤膜过滤器(天津津腾公司);50 mL聚丙烯离心管。
16种多环芳烃混合标准溶液(100 μg·mL−1溶于甲醇,美国Chem Service公司),存放于-20 ℃冰箱中。乙腈(色谱纯,德国Merck公司)。实验用水为Milli-Q Plus超纯水制备系统(美国Millipore公司)临用现制的超纯水(电阻率>18 MΩ·cm)。氮气(纯度大于99.999%)。
1.5 样品前处理及测定
1.5.1 底泥样品
底泥样品在通风柜中自然干燥后磨至80目以下,干燥、阴凉密封保存。称取5 g样品于50 mL离心管(聚丙烯材质)中,加入8 mL乙腈,震荡摇匀。30 ℃下超声30 min后,4000 r·min−1离心2 min. 提取上清液至15 mL的离心管(聚丙烯材质),再用5 mL乙腈萃取残余淤泥,震荡摇匀,同样的条件下超声、离心,重复2次,提取液移到同一离心管中,氮吹浓缩至0.5 mL以下,再用乙腈定容至1.5 mL. 用振荡器将定容后的提取液摇匀,过0.22 μm的滤膜,存于2 mL的色谱瓶中,−20 ℃冷冻保存待测。
1.5.2 沟塘水和浅层地下水样品
取1000 mL沟塘水或浅层地下水样品缓慢加入经二氯甲烷、甲醇活化的C18 SPE小柱过滤,小柱4℃冰箱保存。用13 mL甲醇洗脱,经氮吹浓缩至0.5 mL以下,加入3 mL乙腈,氮吹浓缩至0.5 mL以下,再精确定容至0.5 mL,存于2 mL的色谱瓶中,−20 ℃冷冻保存待测。样品实行平行操作测定。
1.5.3 HPLC分析
采用乙腈-水梯度洗脱,进样量为20 μL,以色谱保留时间和各通道响应信号的一致程度定性,采用外标法峰面积定量。分别进行高、低浓度的加标回收试验,相对标准偏差(RSD) 为5.2%—8.6%。试验结果满足样品分析质量控制的要求,表明分析方法准确可靠。
1.6 数据处理方法
采用SPSS 10.0进行数据的处理及相关统计分析.
2. 结果与讨论(Results and discussion)
2.1 浅层地下水、沟塘水及沉积物中PAHs污染水平
浅层地下水、沟塘水及沉积物中PAHs含量、TEQ(BaP)(Toxic Equivalent Quantity, BaP毒性当量浓度)如表1所示。
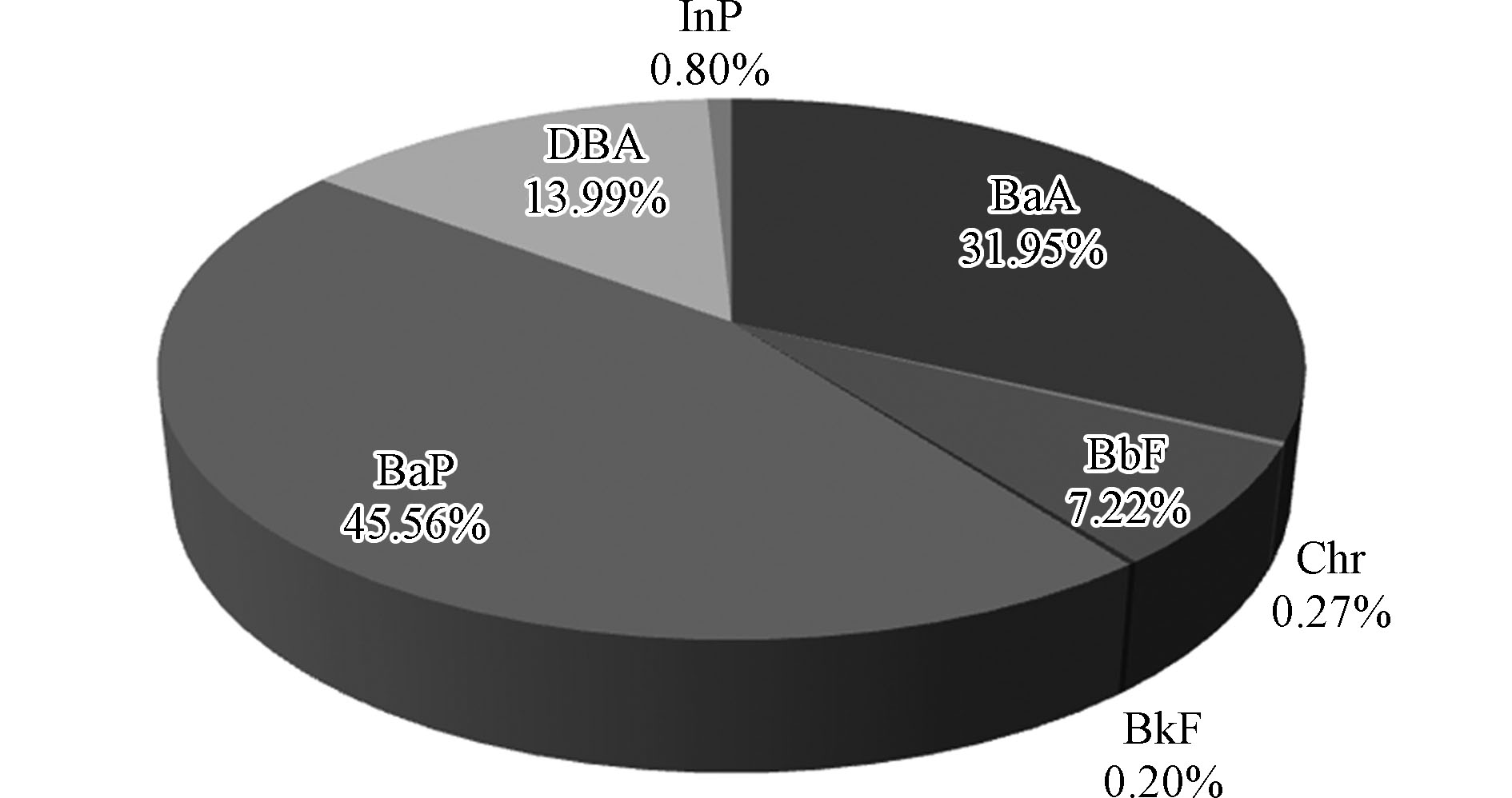
沉积物中PAHs单体的检出率均高于浅层地下水和沟塘水。浅层地下水中,∑16PAHs(16种PAHs总量)为219 ng·L−1(8.39—1234 ng·L−1),其中Phe、Fl和Pyr对∑16PAHs的贡献大,合计占比为60.4%,w(Bap)(Bap的质量分数)为2.72 ng·L−1 (0.164—17.3 ng·L−1),其中1个点位高于我国地下水环境质量标准(GB /T 14848—2017)中Ⅲ类水质规定的限值(10 ng·L−1),超标倍数为0.733倍,与国内其他区域的研究结果相比较发现,该研究中浅层地下水中∑16PAHs和w(Bap)较低;沟塘水中,∑16PAHs为317 ng·L−1 (15.4—1372 ng·L−1),同浅层地下水一样,Phe、Fl和Pyr对∑16PAHs的贡献大,合计占比为55.8%,w(Bap)为11.1 ng·L−1 (0.0420—55.0 ng·L−1),其中1个样品高于我国地表水环境质量标准(GB 3838—2002)限值(2.8 ng·L−1),超标倍数为18.6倍;沉积物中,∑16PAHs为256 ng·kg−1 (101—458 ng·kg−1),其中DBA、Fl、BP、Pyr和Chr对∑16PAHs的贡献大,合计占比为59.7%,w(Bap)为16.0 ng·kg−1 (2.18—41.4 ng·kg−1)。
国际癌症研究机构(IARC)划定7种致癌性PAHs[17]。本研究浅层地下水中,这7种PAHs的质量分数(∑7PAHs)为40.0 ng·L−1 (3.36—298 ng·L−1),占∑16PAHs的18.3%;沟塘水的∑7PAHs为99.7 ng·L−1 (2.84—414 ng·L−1),占∑16PAHs的31.5%.;沉积物的∑7PAHs为133 ng·kg−1 (21.9—191 ng·kg−1),占∑16PAHs的52.0%。
采用毒性当量因子计算浅层地下水、沟塘水及沉积物中PAHs的TEQ(BaP),结果显示,浅层地下水中,TEQ(Bap)7为5.77 ng·L−1 (0.727—39.8 ng·L−1),TEQ(Bap)16为6.09 ng·L−1 (0.763—41.8 ng·L−1);沟塘水中,TEQ(Bap)7为27.5 ng·L−1 (0.568—83.1 ng·L−1),TEQ(Bap)16为28.0 ng·L−1 (0.589—85.3 ng·L−1);沉积物中,TEQ(Bap)7为75.6 ng·kg−1 (4.01—167 ng·kg−1),TEQ(Bap)16为76.0 ng·kg−1 (4.16—167 ng·kg−1). BaP含量、TEQ(BaP)含量、∑7PAHs占比依次为:沉积物>沟塘水>浅层地下水。
2.2 浅层地下水中PAHs空间分布特征
图2为沟塘1南向浅层地下水中PAHs单体的情况. 可以明显看出,距离沟塘越近,浅层地下水中PAHs含量越高,其中Fl的相关系数为0.9464,BaP的相关系数为0.7359,∑16PAHs的相关系数为0.9011,TEQ(BaP)16的相关系数为0.9541,表明沟塘水质对浅层地下水中PAHs的影响较大。
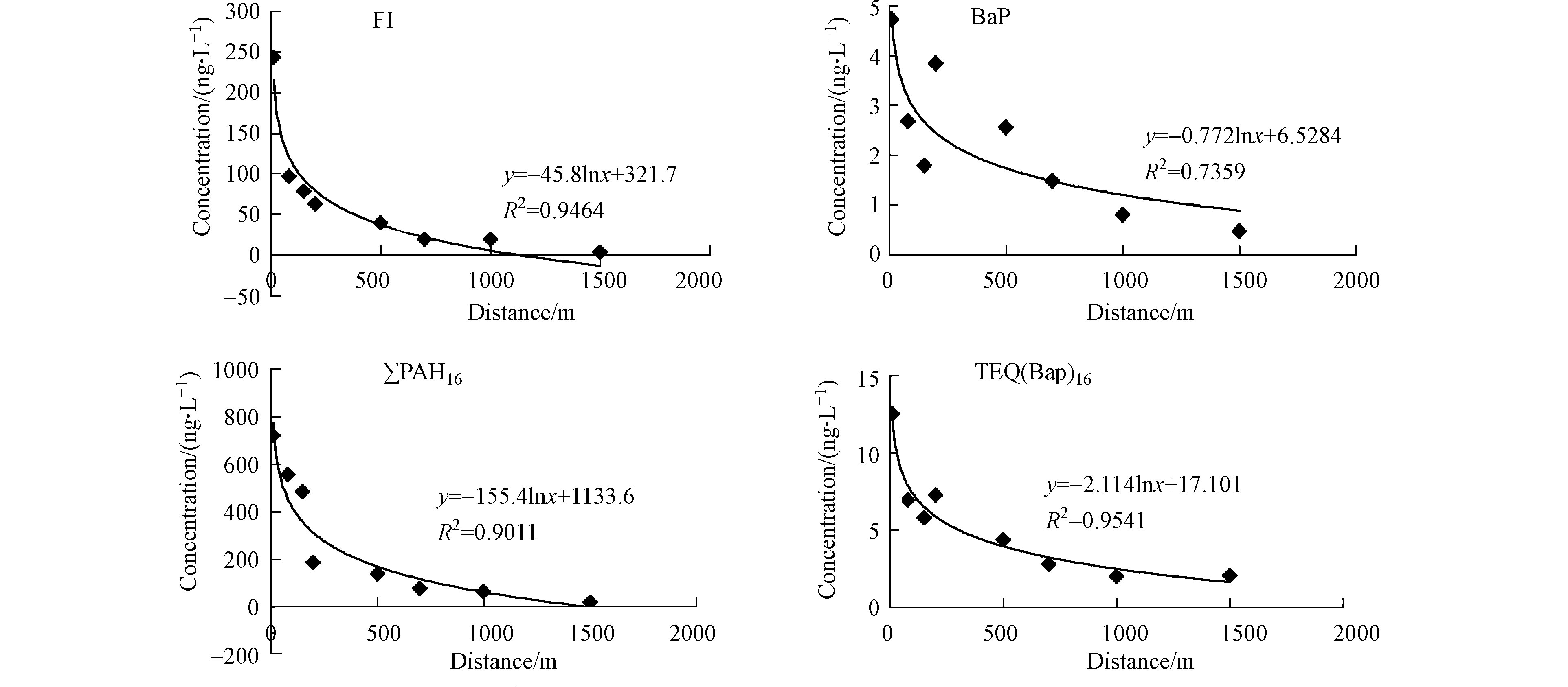 图 2 浅层地下水中PAHs水平与其距沟塘1距离的散点图Figure 2. The scatter plots of PAHs in the shallow groundwater and their distances to the ditch pond1
图 2 浅层地下水中PAHs水平与其距沟塘1距离的散点图Figure 2. The scatter plots of PAHs in the shallow groundwater and their distances to the ditch pond12.3 浅层地下水、沟塘水及沉积物中PAHs的组分特征
环境中PAHs的来源可分为天然和人为两种,绝大部分环境中的PAHs都与人类的生产生活紧密相关[18],其中,人为来源主要包括未经燃烧的煤和石油类产品(如石油挥发和泄漏、公路建设材料等)和各种不充分燃烧(如机动车尾气的排放、工业炼焦、电解铝、炼油、火力发电、煤炭、秸秆与薪材燃烧和吸烟等)。不同分子量的PAHs在土壤中的分布与其来源密切相关[19]。高环(4环及以上)PAHs主要来源于煤和石油类等化石燃料的高温燃烧,低环(2—3环)PAHs主要来源于有机物的低温转化和石油产品的泄露[20-22]。因此通过不同环数PAHs的分析比较可以解析其来源。该研究将16种PAHs单体分为2—3环、4环、5—6环3组进行分析比较(见图3)。浅层地下水PAHs组分中,2—3环PAHs占38.42%(9.63%—79.27%),4环PAHs占51.78%(17.36%—79.80%),5—6环PAHs占9.80%(1.60%—28.32%);沟塘水中2—3环PAHs占45.09%(18.55%—68.12%),4环PAHs占35.80%(21.63%—63.40%),5—6环PAHs占19.11%(8.31%—42.29%);沉积物中2—3环PAHs占25.74%(7.69%—54.80%),4环PAHs占25.57%(9.78%—42.45%),5—6环PAHs占48.69%(20.73%—82.53%)。从整体来看,浅层地下水中PAHs以4环居多,沟塘水中PAHs以2—3环居多,沉积物中PAHs以5—6环居多. 5—6环PAHs占比依次为沉积物>沟塘水>浅层地下水,说明PAHs在环境介质之间迁移分配的过程中,具有致癌性的5—6环PAHs化合物更容易存蓄在沉积物中。在采样过程中观察到从事家庭养殖活动的沟塘占40.00%,PAHs亲脂疏水特性使其可以在生物体内大量富集,通过食物链最终进入人体,从而对人体健康产生影响,因此需要对沟塘PAHs污染开展防治工作,特别需要对日积月累堆积的含高环的致癌性PAHs较多的沉积物加以治理。
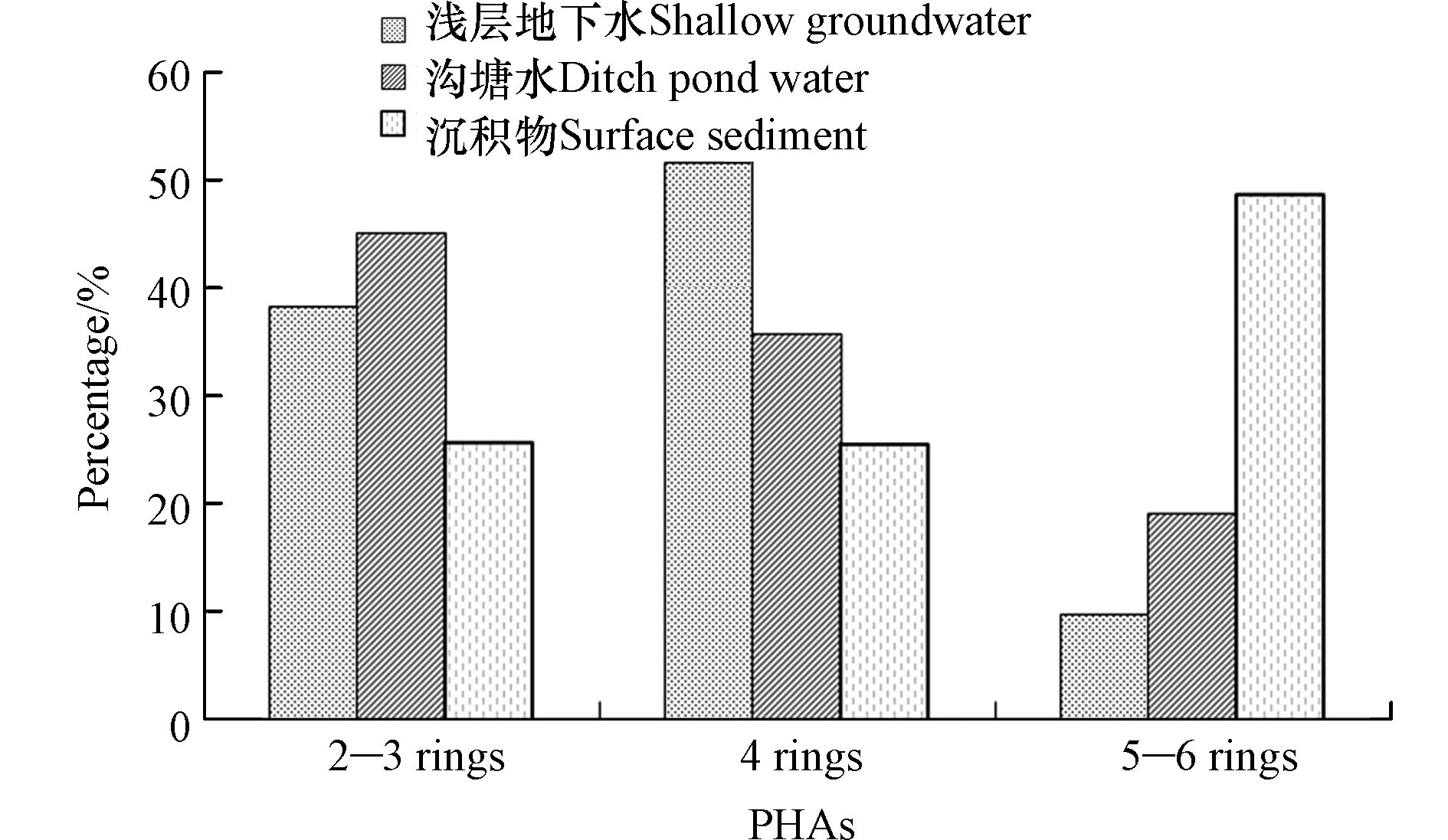 图 3 浅层地下水、沟塘水及沉积物中不同环数PAHs组成Figure 3. The composition of PAHs with different rings in the surface sediment, surrounding shallow groundwater and ditch pond water
图 3 浅层地下水、沟塘水及沉积物中不同环数PAHs组成Figure 3. The composition of PAHs with different rings in the surface sediment, surrounding shallow groundwater and ditch pond water2.4 浅层地下水、沟塘水及沉积物中PAHs的来源解析
Ant/(Ant+Phe)和Fl/(Fl+Pyr)的比值可用来判断环境中PAHs污染物的来源[23-25]。研究表明[25],当Ant/(Ant+Phe)比值小于0.10为石油源,大于0.10为燃烧源;Fl/(Fl+Pyr)比值小于0.40,表明PAHs主要来自石油源,大于0.50表明PAHs主要是生物质和煤炭的燃烧源,介于0.40—0.50则是石油燃烧源。图4为浅层地下水、沟塘水及沉积物中PAHs的Ant/(Ant+Phe)和Fl/(Fl+Pyr)分子比率图,浅层地下水中Ant/(Ant+Phe)比率为0.16(0.00—0.48),沟塘水为0.07(0.00—0.23),沉积物为0.40(0.01—1.00);浅层地下水中Fl/(Fl+Pyr)比率为0.59(0.15—0.64),沟塘水为0.64(0.60—0.66),沉积物为0.68(0.54—0.95)。除了浅层地下水一个点位,其余所有采样点的Fl/(Fl+Pyr)值都大于0.5,说明污染主要来源于燃烧,并以生物质和煤炭燃烧为主。浅层地下水、沟塘水及沉积物中PAHs有相似的来源,在采样过程中发现有40.00%的沟塘中有纳污现象,当地居民用燃烧煤炭和生物质来做饭和取暖,直接将底灰倾倒在沟塘内,或者直接在沟塘边燃烧秸杆和枯枝树叶等生物质,这些可能与浅层地下水、沟塘水及沉积物中PAHs的存在有密切关系。
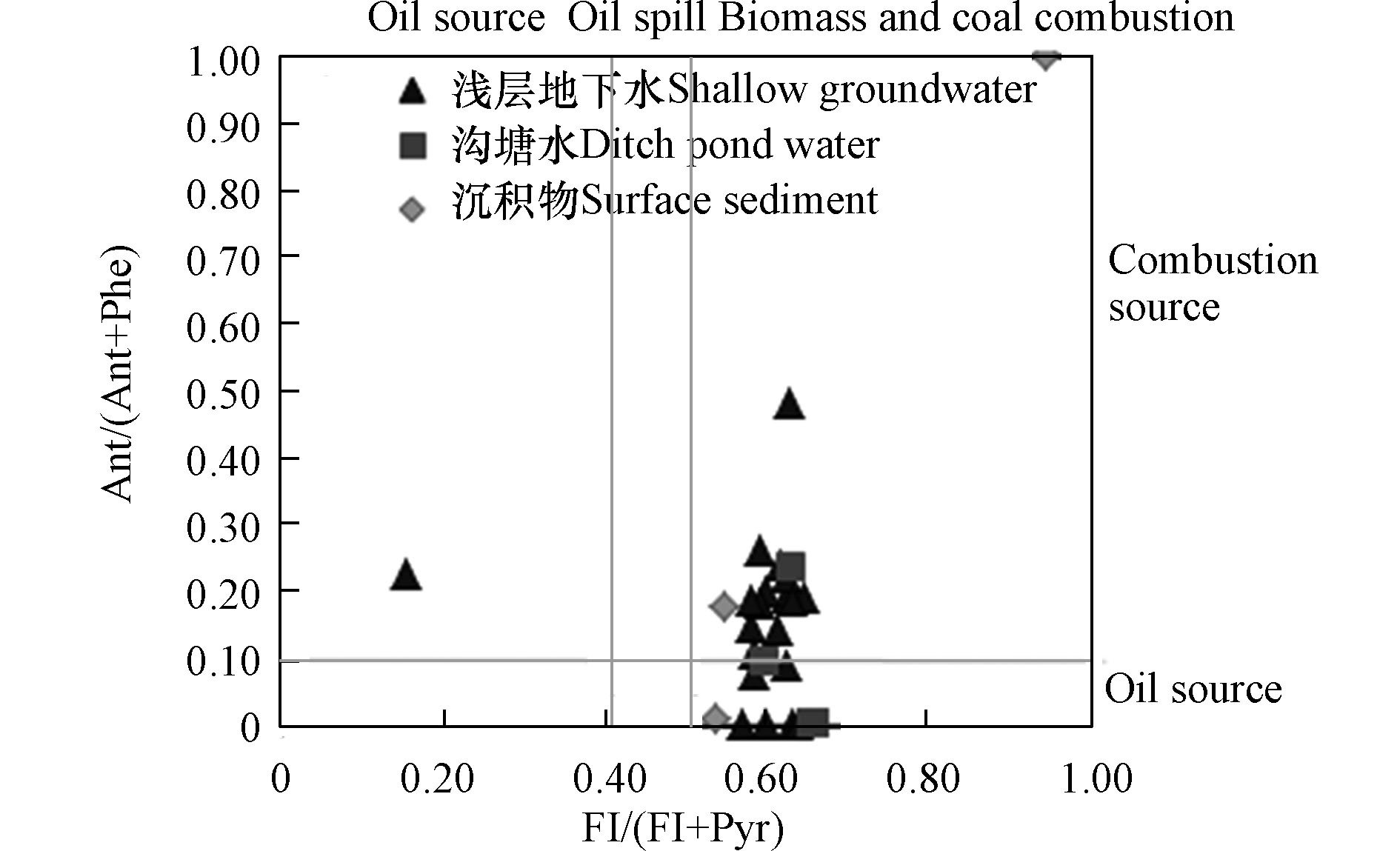 图 4 浅层地下水、沟塘水及沉积物中PAHs异构体比值源解析Figure 4. Cross plot for the isomeric rations of Ant/(Ant+Phe)vs. Fl/(Fl+Pyr)in the surrounding shallow groundwater,ditch pond water and surface sediment
图 4 浅层地下水、沟塘水及沉积物中PAHs异构体比值源解析Figure 4. Cross plot for the isomeric rations of Ant/(Ant+Phe)vs. Fl/(Fl+Pyr)in the surrounding shallow groundwater,ditch pond water and surface sediment2.5 浅层地下水健康风险评价
浅层地下水曾经广泛作为农村地区的饮用水,目前仍有局部区域人群以其作为饮用水。因此,在健康风险评估时仅考虑经口摄入的暴露途径,健康风险评估方法参考美国国家科学院(NAS)提出的四步法[26],采用US EPA模型计算当地人群的浅层地下水PAHs经口暴露剂量,公式如下:
ADD=C×IR×EF××EDBW×AT (1) 式中,ADD为PAHs的日平均暴露剂量[mg·(kg·d)−1];C为浅层地下水中PAHs的浓度(mg·L−1);IR为日均饮水摄入量(L·d−1);EF为暴露频率(d·a−1),本文中为365 d·a−1;ED为暴露持续时间(a),成人的ED为30 a[26];BW为人群体重(kg);AT为平均接触时间(d),对于致癌风险AT为:70 a×365 d·a−1,对于非致癌风险,AT为ED×365 d·a−1。IR和BW分别参照河南省数据[27-29]。
非致癌健康风险和致癌风险的计算公式(2)和(3)如下:
HQ=ADDRfD (2) RI=ADD×SF (3) 式中,HQ为发生某种特定有害健康效应而造成的危险度;RI为人群患癌终身超额危险度;RfD为PAHs经口暴露的非致癌毒性参考剂量[mg·(kg·d)−1];SF为PAHs经口暴露的致癌斜率系数,[mg·(kg·d)−1]−1。RfD、SF和关键效应来源于美国EPA的风险评估信息系统(RAIS)[30]和综合风险信息系统(IRIS)[31]。
9 种非致癌PAHs的暴露剂量的P95非致癌风险(HQ)在6.1×10−4—1.9×10−2之间,远小于1,见图5,表明饮用此浅层地下水摄入的9种PAHs的非致癌风险为可接受水平。
 图 5 不同人群经饮水入PAHs的非致癌健康风险(HQ)和致癌健康风险(RI)Figure 5. Non-carcinogenic health risks (HQ) and carcinogenic health risks (R) of PAHs through drinking water for different subpopulations
图 5 不同人群经饮水入PAHs的非致癌健康风险(HQ)和致癌健康风险(RI)Figure 5. Non-carcinogenic health risks (HQ) and carcinogenic health risks (R) of PAHs through drinking water for different subpopulationsPAHs的P95致癌风险(RI)在1.8×10−8—7.0×10−6之间,如果从出生就开始饮用该浅层地下水,在6岁时RI为1.1×10−6,且女孩高于男孩,超出一般可接受的致癌风险水平(1×10−6),主要产生致癌风险的污染物为BaP和BaA(图6)。2018年,我们开展的问卷调查中,西平县有23.5%的农村居民饮用浅层地下水,以浅层地下水作为饮用水的健康风险亟需关注。
污染物的暴露途径一般包括经口摄入(饮水)、呼吸和皮肤的3种. 本文只讨论了16种PAHs单体经饮水途径的健康风险,没有考虑经污染的水产品或农副产品摄入所致健康风险,也没有考虑另外两种途径(呼吸和皮肤)的暴露;而且健康风险评估仅涉及9种非致癌和7种致癌的PAHs,因此可能低估其非致癌和致癌健康风险。
2.6 沟塘水生态风险评价
本研究中沟塘水中BaP含量变化范围为0.042—54.974 ng·L−1,均值为11.12 ng·L−1。参照国家《地表水环境质量标准》(GB 3838—2002),20.00%沟塘水体中BaP含量高于标准限值(2.8 ng·L−1)。然而,上述标准仅能对BaP的生态风险作出评价,未能充分考虑16种PAHs单体的生态风险。为全面了解沟塘内水体中∑PAHs的综合毒性,采用风险商值法[16] (risk quotient,RQ),综合评价沟塘水中PAHs的生态风险,结果见表2。沟塘水中BaA和BbF的RQMPCs≥1,表明沟塘水中这些单体为高风险,另外12种多环芳烃的RQMPCs<1,而RQNCs≥1,表明上覆水中Ace、Flu、Phe、Ant、Fl、Pyr、Chr、BkF、BaP、DBA、BP和InP均为中等风险。B4沟塘水中有6种PAHs单体(Fl、Pyr、BaA、BbF、BaP和BP)的RQMPCs≥1,且RQΣ PAHs(MPCs)≥1,RQΣPAHs(NCs)≥800,表明B4沟塘水中多环芳烃处于高风险水平,应当重点关注该类污染物. 在采样过程中发现,该沟塘主要用于纳污,需立即采取必要措施,B4沟塘水中BaA对生态风险RQMPCs的贡献最大,达到40.69%,虽然B4沟塘水中Fl的浓度最高,但是其对生态风险RQMPCs的贡献较小。B3沟塘水中有12种PAHs单体(Ace、Flu、Phe、Ant、Fl、Pyr、BaA、Chr、BbF、BkF、DBA和BP)的RQNCs≥1,DBA的RQMPCs≥1,且RQΣPAHs(MPCs)≥1,RQΣPAHs(NCs)<800,表明B3沟塘水中PAHs处于中等风险2水平,应予以关注。在采样过程中也发现,该沟塘主要用于纳污,需考虑控制和修复措施。B3沟塘水中DBA和BaA对生态风险RQMPCs的贡献最大,分别达到36.89%和23.60%。
表 2 风险评价参数及评价结果Table 2. Risk evaluation parameters and resultsPAHs 经口摄入非致癌参考剂量RfDo/(mg·(kg·d)−1) 经口摄入致癌斜率因子SFo/ (kg·d ·mg−1) 沟塘水Ditch pond water 沉积物Surface sediment 中国标准China standard EPA标准EPA standard 水体Water body 评价结果Evaluation result 质量基准法阈值/(ng·g−1)Quality reference method threshold 评价结果Evaluation result 质量标准法阈值/(ng·g−1)Quality reference method threshold 评价结果Evaluation result NCs MPCs RQNCs RQMPCs ERL ERM RCF>1 REL TEL OEL PEL FEL 区间Range 点位Point position 奈(Nap) 4.0×10−2 — — — 12.0 1200 0.324 0.00324 160 2100 17 35 120 390 1200 苊烯(Acy) 6.0×10−2 — — 1.2×106 0.7 70 0 0 16 500 3.3 5.9 30 130 340 TEL-OEL C3 苊(Ace) 6.0×10−2 — — — 0.7 70 7.94 0.0794 44 640 3.7 6.7 21 89 940 TEL-OEL C1、C2、C3 芴(Flu) 4.0×10−2 — — 1.3×106 0.7 70 2.94 0.0294 19 540 10 21 61 140 1200 菲(Phe) 3.0×10−2 — — — 3.0 300 15.2 0.152 240 1500 25 42 130 520 1100 蒽(Ant) 3.0×10−1 — — 9.6×106 0.7 70 16.2 0.162 85.3 1100 16 47 110 240 1100 荧蒽(Fl) 4.0×10−2 — — 3×105 3.0 300 27.6 0.276 600 5100 47 110 450 2400 4900 REL-TEL C3 芘(Pyr) 3.0×10−2 — — 9.6×105 0.7 70 69.2 0.692 665 2600 29 53 230 880 1500 TEL-OEL C3 苯并(a)蒽(BaA) — 7.3×10−1 — 4.4 0.1 10 250 2.50 261 1600 14 32 120 390 760 REL-TEL C3 䓛(Chr) — 7.3×10−3 — 4.4 3.4 340 8.49 0.0849 384 2800 26 57 240 860 1600 REL-TEL C3 苯并(b)荧蒽(BbF) — 7.3×10−1 — 4.4 0.1 10 110 1.10 N.A. N.A. — — — — — 苯并(k)荧蒽(BkF) — 7.3×10−2 — 4.4 0.4 40 12.9 0.129 N.A. N.A. — — — — — 苯并芘(BaP) — 7.3 2.8 4.4 0.5 50 22.2 0.222 430 1600 11 32 150 780 3200 TEL-OEL C3 二苯并[a,h]蒽(DBA) — 7.3 — 4.4 0.5 50 22.4 0.224 63.4 260 C1 3.3 6.2 43 140 200 REL-FEL C1、C3 苯并[g,h,i]芘(BP) 3.0×10−2 — — — 0.3 30 57.2 0.572 — — — — — — — 茚并[1,2,3-cd]芘(InP) — 7.3×10−1 — 4.4 0.4 40 18.3 0.183 — — — — — — — ∑PAHs — — — — — — 641 6.41 — — — — — — — 1) “—”表示无数据;2) NCs表示沟塘水中PAHs最低风险标准值,MPCs表示沟塘水中PAHs最高风险标准值. 1) “—” means no data;2) NCs means minimum risk standard value of PAHs in ditch pond water, MPCs means maximum risk standard value of PAHs in ditch pond water. | Show TableDownLoad:
CSV
2.7 沉积物生态风险评价
本研究采用质量基准法[32]和质量标准法[33]评估沉积物中PAHs潜在生态风险,评价结果见表2。质量基准法分为效应低值(ERL)对生物体毒副作用发生的风险几率<10%和对生物体毒副作用发生的风险几率>50%的效应中值(ERM),相对污染系数RCF是对沉积物中PAHs污染的定量表征,RCF为沉积物中PAHs浓度与效益低值的比值;质量标准法分为5个阈值REL、TEL、OEL、PEL和FEL,分别表示罕见效应、临界效应、偶然效应、可能效应以及频繁效应的浓度阈值,将16种PAHs任一种超过最高限值的点位给标注出。
质量基准法评价结果显示,沟塘沉积物中只有DBA在C1超出ERL,介于ERL—ERM之间,证明沟塘沉积物中PAHs潜在生态风险发生几率不大。但在沟塘沉积物中无最低安全阈值的致癌PAHs单体BkF和InP在沟塘沉积物中检出率均高达100%,无最低安全阈值的PAHs只要存在于环境中就会对生物体产生毒副作用。
质量标准法评价结果显示,C2介于TEL和OEL之间,对生物的不良影响概率较低;C1、C3介于REL和FEL之间,且PAHs含量也较高,需要引起关注,并对沉积物毒性风险进行评估,对PAHs污染采取治理措施。沟塘沉积物中检出的PAHs浓度效应值都低于FEL值,表明沟塘对生物体潜在风险不高,但C1和C2沟塘从事家庭养殖活动,PAHs势必会通过食物链进入人体,影响人体健康,因此该区域需要重点开展清淤工作,清淤后的底泥需要妥善处置,以免引起二次污染。结合质量基准法和质量标准法可以看出,沟塘沉积物中最易对生物造成危害的PAHs单体化合物是DBA以及没有最低安全限值的BbF和BkF,对生物的不良影响概率最高处是C1.
3. 结论(Conclusion)
(1)本研究中BaP含量、TEQ(BaP)含量、∑7PAHs、高环PAHs占比依次为:沉积物>沟塘水>浅层地下水。浅层地下水、沟塘水及沉积物中PAHs有相似的来源,主要源于燃烧,并以生物质和煤炭燃烧为主;沟塘水对浅层地下水中PAHs的影响较大,采样点距离沟塘越近,浅层地下水中PAHs含量越高。
(2)经饮水摄入浅层地下水中7种致癌PAHs的P95致癌风险在1.8×10−8—7.0×10−6之间,在6岁时为1.1×10−6,且女孩高于男孩,超出一般可接受的致癌风险水平(1×10−6),主要产生致癌风险的污染物为BaP和BaA,以浅层地下水作为饮用水的健康风险亟需关注;P95非致癌风险在6.1×10−4—1.9×10−2之间,HQ均小于1,为可接受水平。
(3)纳污的4号沟塘水中PAHs处于高生态风险水平,对生态风险贡献最大的污染物是BaA,达40.69%,需立即采取必要措施。同样纳污的3号沟塘水中PAHs处于中等风险2水平,对生态风险贡献最大的污染物是DBA和BaA,分别达到了36.89%和23.60%。
(4)沟塘沉积物中各PAHs化合物的浓度只有C1超过效应区间中值(ERM),所有沉积物中PAHs均超过了TEL,说明沟塘沉积物的PAHs污染已经具有一定程度的“临界效应”,需要采取相应的措施进行污染控制和削减,尤其是C1沟塘需要立即停止养殖活动,采取清淤治理。
-
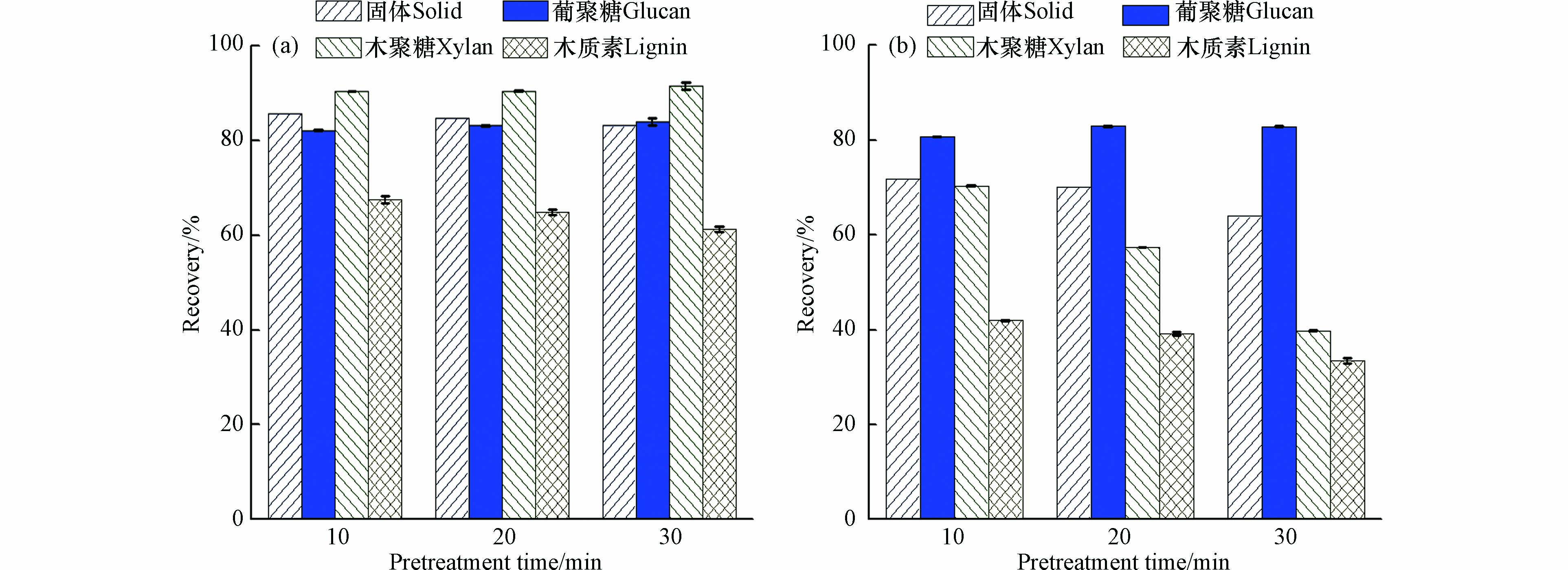
图 1 不同温度条件下水热预处理对稻草成分的影响
Figure 1. Effect of hydrothermal pretreatment at different temperatures on composition of rice straw
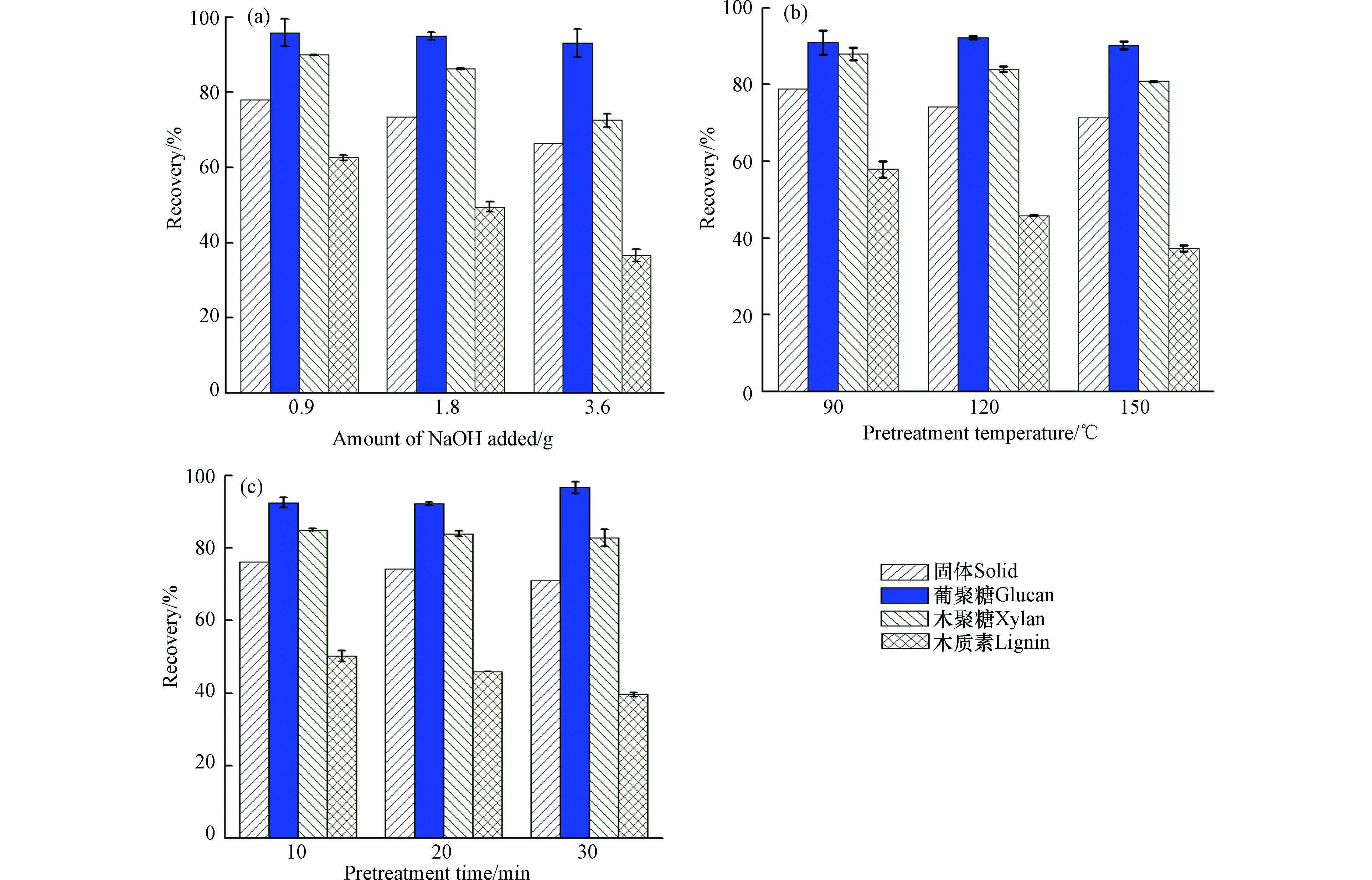
图 2 NaOH预处理对稻草成分的影响
Figure 2. Effect of NaOH pretreatment on the composition of rice straw
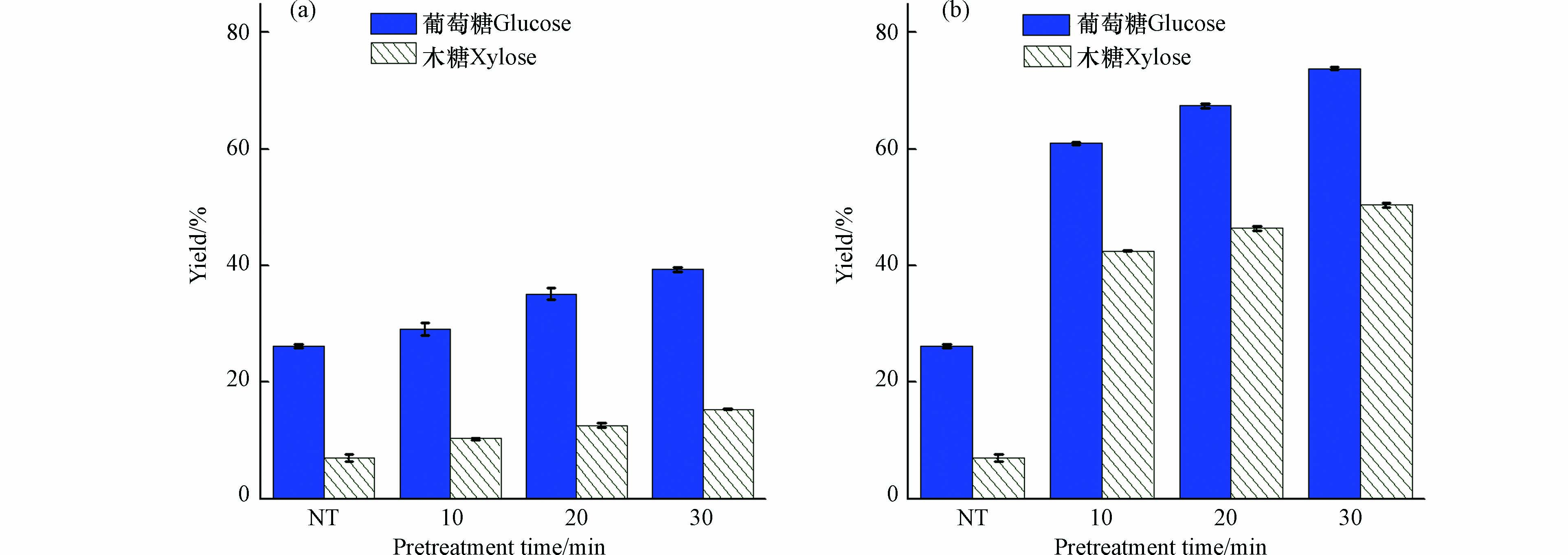
图 4 水热预处理对稻草酶解得率的影响
Figure 4. Effect of hydrothermal pretreatment on enzymatic hydrolysis yield of rice straw
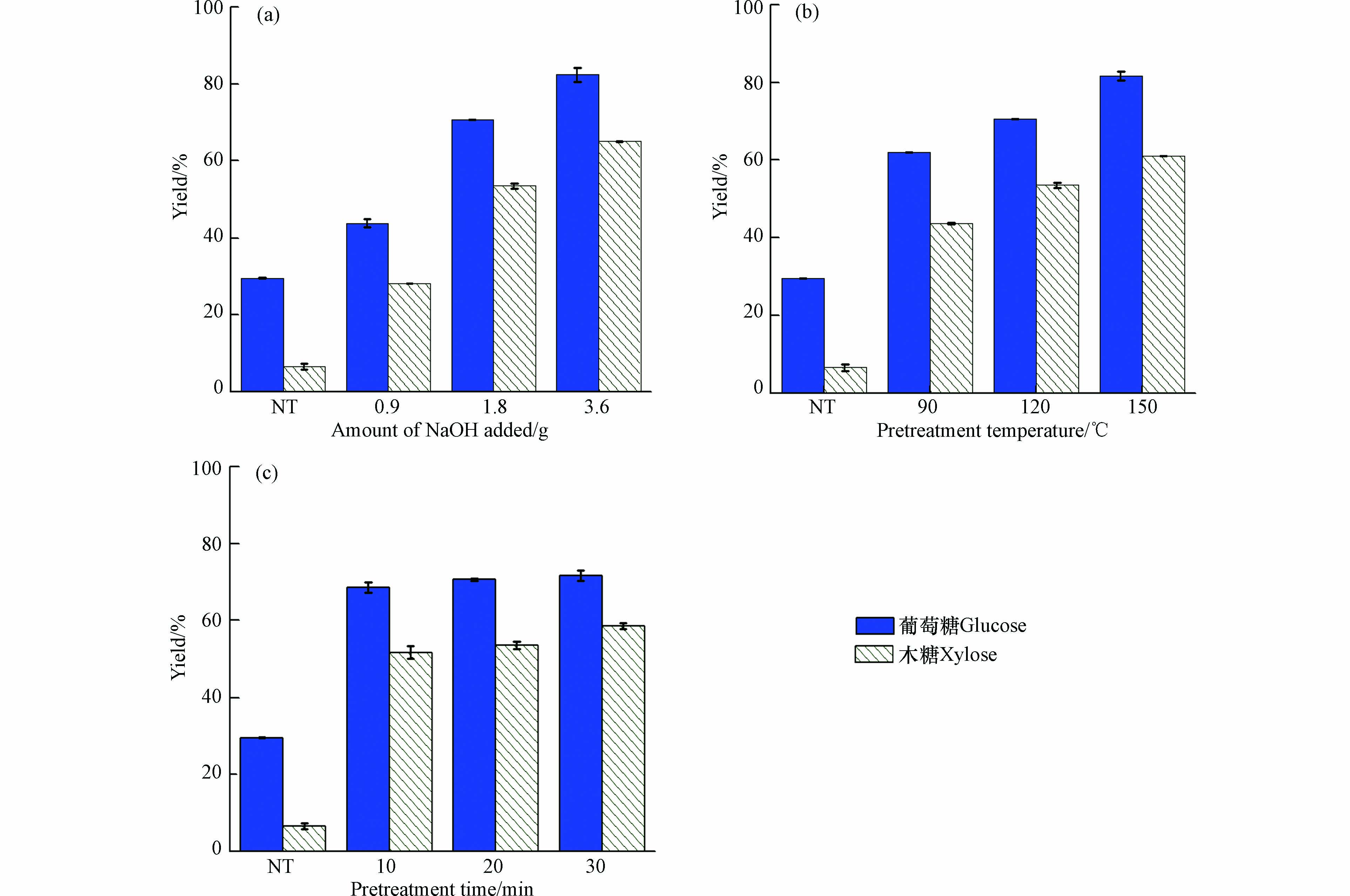
图 5 NaOH预处理对稻草酶解得率的影响
Figure 5. Effect of NaOH pretreatment on enzymatic hydrolysis yield of rice straw
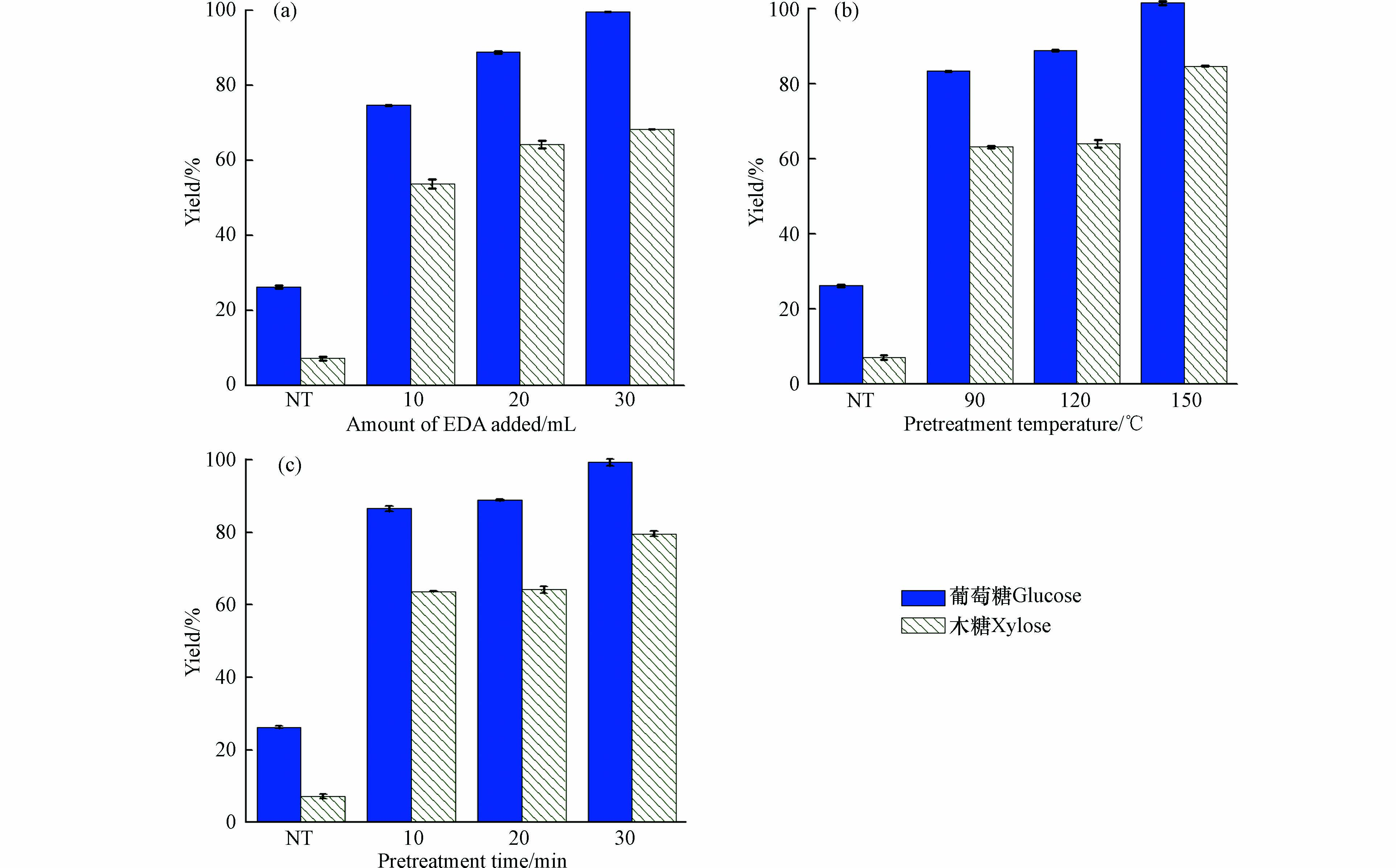
图 6 EDA预处理对稻草酶解得率的影响
Figure 6. Effect of EDA pretreatment on enzymatic hydrolysis yield of rice straw

图 7 不同预处理前后稻草扫描电镜图
Figure 7. SEM images of rice straw before and after different pretreatment

图 8 预处理前后稻草的傅里叶红外光谱图
Figure 8. FT-IR spectra of rice straw before and after pretreatment
-
[1] 岳国君. 纤维素乙醇工程概论[M]. 北京: 化学工业出版社, 2015. YUE G J. An introduction to cellulosic ethanol engineering[M]. Beijing: Chemical Industry Press, 2015(in Chinese).
[2] CHEN H Z, LIU Z H. Steam explosion and its combinatorial pretreatment refining technology of plant biomass to bio-based products [J]. Biotechnology Journal, 2015, 10(6): 866-885. doi: 10.1002/biot.201400705 [3] 朱振兴, 颜涌捷, 亓伟, 等. 铁炭曝气微电解预处理纤维素发酵废水 [J]. 环境化学, 2008, 27(6): 779-781. doi: 10.3321/j.issn:0254-6108.2008.06.016 ZHU Z X, YAN Y J, QI W, et al. Pretreatment of cellulose zymolytic wastewater with iron-carbon aeration microelectrolysis method [J]. Environmental Chemistry, 2008, 27(6): 779-781(in Chinese). doi: 10.3321/j.issn:0254-6108.2008.06.016
[4] LI W C, LI X, ZHU J Q, et al. Improving xylose utilization and ethanol production from dry dilute acid pretreated corn stover by two-step and fed-batch fermentation [J]. Energy, 2018, 157: 877-885. doi: 10.1016/j.energy.2018.06.002 [5] HARUN R, DANQUAH M K. Enzymatic hydrolysis of microalgal biomass for bioethanol production [J]. Chemical Engineering Journal, 2011, 168(3): 1079-1084. doi: 10.1016/j.cej.2011.01.088 [6] MUSSATTO S I. Preface[M]//Biomass Fractionation Technologies for a Lignocellulosic Feedstock Based Biorefinery. Amsterdam: Elsevier, 2016: xxiii-xxv. [7] HOANG A T, NIŽETIĆ S, ONG H C, et al. Insight into the recent advances of microwave pretreatment technologies for the conversion of lignocellulosic biomass into sustainable biofuel [J]. Chemosphere, 2021, 281: 130878. doi: 10.1016/j.chemosphere.2021.130878 [8] da SILVA A R G, TORRES ORTEGA C E, RONG B G. Techno-economic analysis of different pretreatment processes for lignocellulosic-based bioethanol production [J]. Bioresource Technology, 2016, 218: 561-570. doi: 10.1016/j.biortech.2016.07.007 [9] ZHAO R, ZHANG Z Y, ZHANG R Q, et al. Methane production from rice straw pretreated by a mixture of acetic-propionic acid [J]. Bioresource Technology, 2010, 101(3): 990-994. doi: 10.1016/j.biortech.2009.09.020 [10] JIANG D P, GE X M, ZHANG T, et al. Effect of alkaline pretreatment on photo-fermentative hydrogen production from giant reed: Comparison of NaOH and Ca(OH)2 [J]. Bioresource Technology, 2020, 304: 123001. doi: 10.1016/j.biortech.2020.123001 [11] PARK Y C, KIM T H, KIM J S. Effect of organosolv pretreatment on mechanically pretreated biomass by use of concentrated ethanol as the solvent [J]. Biotechnology and Bioprocess Engineering, 2017, 22(4): 431-439. doi: 10.1007/s12257-017-0088-1 [12] ZHANG J X, ZHANG X, YANG M K, et al. Transforming lignocellulosic biomass into biofuels enabled by ionic liquid pretreatment [J]. Bioresource Technology, 2021, 322: 124522. doi: 10.1016/j.biortech.2020.124522 [13] SHAH T A, LEE C C, ORTS W J, et al. Biological pretreatment of rice straw by ligninolytic Bacillus sp. strains for enhancing biogas production [J]. Environmental Progress & Sustainable Energy, 2019, 38(3): e13036. [14] UÇKUN KIRAN E, TRZCINSKI A P, LIU Y. Enhancing the hydrolysis and methane production potential of mixed food waste by an effective enzymatic pretreatment [J]. Bioresource Technology, 2015, 183: 47-52. doi: 10.1016/j.biortech.2015.02.033 [15] 宋永民, 陈洪章. 汽爆秸秆高温固态发酵沼气的研究 [J]. 环境工程学报, 2008, 2(11): 1564-1570. SONG Y M, CHEN H Z. Study on biogas production by thermophilic solid-state fermentation from steam exploded corn stalk [J]. Chinese Journal of Environmental Engineering, 2008, 2(11): 1564-1570(in Chinese).
[16] BEHERA S, ARORA R, NANDHAGOPAL N, et al. Importance of chemical pretreatment for bioconversion of lignocellulosic biomass [J]. Renewable and Sustainable Energy Reviews, 2014, 36: 91-106. doi: 10.1016/j.rser.2014.04.047 [17] SILVA-FERNANDES T, DUARTE L C, CARVALHEIRO F, et al. Hydrothermal pretreatment of several lignocellulosic mixtures containing wheat straw and two hardwood residues available in Southern Europe [J]. Bioresource Technology, 2015, 183: 213-220. doi: 10.1016/j.biortech.2015.01.059 [18] NITSOS C K, MATIS K A, TRIANTAFYLLIDIS K S. Optimization of hydrothermal pretreatment of lignocellulosic biomass in the bioethanol production process [J]. ChemSusChem, 2013, 6(1): 110-122. doi: 10.1002/cssc.201200546 [19] SARKER T R, PATTNAIK F, NANDA S, et al. Hydrothermal pretreatment technologies for lignocellulosic biomass: A review of steam explosion and subcritical water hydrolysis [J]. Chemosphere, 2021, 284: 131372. doi: 10.1016/j.chemosphere.2021.131372 [20] YUAN Z Y, WEN Y B, KAPU N S. Ethanol production from bamboo using mild alkaline pre-extraction followed by alkaline hydrogen peroxide pretreatment [J]. Bioresource Technology, 2018, 247: 242-249. doi: 10.1016/j.biortech.2017.09.080 [21] QIN L, ZHAO X, LI W C, et al. Process analysis and optimization of simultaneous saccharification and co-fermentation of ethylenediamine-pretreated corn stover for ethanol production [J]. Biotechnology for Biofuels, 2018, 11: 118. doi: 10.1186/s13068-018-1118-8 [22] LI W C, ZHANG S J, ZHANG T Z, et al. Bacterial cellulose production from ethylenediamine pretreated Caragana korshinskii kom [J]. Industrial Crops and Products, 2021, 164: 113340. doi: 10.1016/j.indcrop.2021.113340 [23] SLUITER A, HAMES B, RUIZ R, et al. Determination of Structural Carbohydrates and Lignin in Biomass [J]. National Renewable Energy Laboratory Technol, 2008, 4: 456-505. [24] PÉREZ J A, BALLESTEROS I, BALLESTEROS M, et al. Optimizing Liquid Hot Water pretreatment conditions to enhance sugar recovery from wheat straw for fuel-ethanol production [J]. Fuel, 2008, 87(17/18): 3640-3647. [25] MOSIER N, HENDRICKSON R, HO N, et al. Optimization of pH controlled liquid hot water pretreatment of corn stover [J]. Bioresource Technology, 2005, 96(18): 1986-1993. doi: 10.1016/j.biortech.2005.01.013 [26] LASER M, SCHULMAN D, ALLEN S G, et al. A comparison of liquid hot water and steam pretreatments of sugar cane bagasse for bioconversion to ethanol [J]. Bioresource Technology, 2002, 81(1): 33-44. doi: 10.1016/S0960-8524(01)00103-1 [27] CHEN L J, LI J B, LU M S, et al. Integrated chemical and multi-scale structural analyses for the processes of acid pretreatment and enzymatic hydrolysis of corn stover [J]. Carbohydrate Polymers, 2016, 141: 1-9. doi: 10.1016/j.carbpol.2015.12.079 [28] FENG R Z, ZAIDI A A, LI Q Y, et al. NaOH-urea pretreatment for biogas enhancement from algal biomass anaerobic [J]. Journal of Renewable and Sustainable Energy, 2021, 13(3): 033102. doi: 10.1063/5.0048341 [29] QIN L, LI W C, ZHU J Q, et al. Ethylenediamine pretreatment changes cellulose allomorph and lignin structure of lignocellulose at ambient pressure [J]. Biotechnology for Biofuels, 2015, 8: 174. doi: 10.1186/s13068-015-0359-z [30] BALI G, MENG X Z, DENEFF J I, et al. The effect of alkaline pretreatment methods on cellulose structure and accessibility [J]. ChemSusChem, 2015, 8(2): 275-279. doi: 10.1002/cssc.201402752 [31] KŁOSOWSKI G, MIKULSKI D. Impact of lignocellulose pretreatment by-products on S. cerevisiae strain ethanol red metabolism during aerobic and an-aerobic growth [J]. Molecules, 2021, 26(4): 806. doi: 10.3390/molecules26040806 [32] DIEN B S, SARATH G, PEDERSEN J F, et al. Improved sugar conversion and ethanol yield for forage Sorghum (Sorghum bicolor L. moench) lines with reduced lignin contents [J]. BioEnergy Research, 2009, 2(3): 153-164. doi: 10.1007/s12155-009-9041-2 [33] BINOD P, JANU K U, SINDHU R, et al. Hydrolysis of lignocellulosic biomass for bioethanol production[M]//Biofuels. Amsterdam: Elsevier, 2011: 229-250. [34] LOURENÇON T V, HANSEL F A, da SILVA T A, et al. Hardwood and softwood kraft lignins fractionation by simple sequential acid precipitation [J]. Separation and Purification Technology, 2015, 154: 82-88. doi: 10.1016/j.seppur.2015.09.015 [35] BIAN H Y, CHEN L H, GLEISNER R, et al. Producing wood-based nanomaterials by rapid fractionation of wood at 80 ℃ using a recyclable acid hydrotrope [J]. Green Chemistry, 2017, 19(14): 3370-3379. doi: 10.1039/C7GC00669A 期刊类型引用(1)
1. 王泓,晏文菁,杨芸芸,马一可,李东妮,李俊贤,华煜,戴晓虎. 不同水热预处理方法对水稻秸秆组成结构及酶解特性的影响. 环境工程. 2024(09): 285-291 .  百度学术
百度学术
其他类型引用(3)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 4343
- HTML全文浏览数: 4343
- PDF下载数: 119
- 施引文献: 4
