








去除率/%Cr(Ⅵ) removal efficiency</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{C}-{C}_{0}=-kt $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">实验条件Experimental conditions</td><td class="table_top_border" align="center" valign="middle">拟合级数Fitting series</td><td class="table_top_border" align="center" valign="middle">动力学方程Kinetic equation</td><td class="table_top_border" align="center" valign="middle">速率常数<i>k</i>Rate constant <i>k</i></td><td class="table_top_border" style="width:8%" align="center" valign="middle"><i>R</i><sup>2</sup></td><td class="table_top_border" align="center" valign="middle">Cr(Ⅵ)去除率/%Cr(Ⅵ) removal efficiency</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{C}-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M1.jpg"><img class="graphic" src="2021120501_M1.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">3.022 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.5253</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=1</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M2.jpg"><img class="graphic" src="2021120501_M2.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0286 h<sup>−1</sup></td><td align="center" valign="middle">0.6589</td><td align="center" valign="middle">79.4</td></tr><tr><td align="center" valign="middle"></td><td align="center" valign="middle">二级</td><td align="center" valign="middle"><inline-formula><tex-math id="M3">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M3.jpg"><img class="graphic" src="2021120501_M3.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0002 L(mg· h)<sup>-1·</sup></td><td align="center" valign="middle">0.7508</td><td align="center" valign="middle"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M4">$ C-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M4.jpg"><img class="graphic" src="2021120501_M4.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">13.436 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.8007</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=3</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M5">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M5.jpg"><img class="graphic" src="2021120501_M5.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.1765 h<sup>−1</sup></td><td align="center" valign="middle">0.9358</td><td align="center" valign="middle">99.9</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">二级</td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M6">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M6.jpg"><img class="graphic" src="2021120501_M6.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.0012 L(mg· h)<sup>-1·</sup></td><td class="table_bottom_border" align="center" valign="middle">0.9459</td><td class="table_bottom_border" align="center" valign="middle"></td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td class="table_top_border2" align="center" valign="middle">3.022 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.5253</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=1</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">实验条件Experimental conditions</td><td class="table_top_border" align="center" valign="middle">拟合级数Fitting series</td><td class="table_top_border" align="center" valign="middle">动力学方程Kinetic equation</td><td class="table_top_border" align="center" valign="middle">速率常数<i>k</i>Rate constant <i>k</i></td><td class="table_top_border" style="width:8%" align="center" valign="middle"><i>R</i><sup>2</sup></td><td class="table_top_border" align="center" valign="middle">Cr(Ⅵ)去除率/%Cr(Ⅵ) removal efficiency</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{C}-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M1.jpg"><img class="graphic" src="2021120501_M1.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">3.022 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.5253</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=1</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M2.jpg"><img class="graphic" src="2021120501_M2.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0286 h<sup>−1</sup></td><td align="center" valign="middle">0.6589</td><td align="center" valign="middle">79.4</td></tr><tr><td align="center" valign="middle"></td><td align="center" valign="middle">二级</td><td align="center" valign="middle"><inline-formula><tex-math id="M3">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M3.jpg"><img class="graphic" src="2021120501_M3.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0002 L(mg· h)<sup>-1·</sup></td><td align="center" valign="middle">0.7508</td><td align="center" valign="middle"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M4">$ C-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M4.jpg"><img class="graphic" src="2021120501_M4.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">13.436 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.8007</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=3</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M5">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M5.jpg"><img class="graphic" src="2021120501_M5.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.1765 h<sup>−1</sup></td><td align="center" valign="middle">0.9358</td><td align="center" valign="middle">99.9</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">二级</td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M6">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M6.jpg"><img class="graphic" src="2021120501_M6.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.0012 L(mg· h)<sup>-1·</sup></td><td class="table_bottom_border" align="center" valign="middle">0.9459</td><td class="table_bottom_border" align="center" valign="middle"></td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td align="center" valign="middle">0.0286 h<sup>−1</sup></td><td align="center" valign="middle">0.6589</td><td align="center" valign="middle">79.4</td></tr><tr><td align="center" valign="middle"></td><td align="center" valign="middle">二级</td><td align="center" valign="middle"><inline-formula><tex-math id="M3">$ 1/C-1/{C}_{0}=kt $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">实验条件Experimental conditions</td><td class="table_top_border" align="center" valign="middle">拟合级数Fitting series</td><td class="table_top_border" align="center" valign="middle">动力学方程Kinetic equation</td><td class="table_top_border" align="center" valign="middle">速率常数<i>k</i>Rate constant <i>k</i></td><td class="table_top_border" style="width:8%" align="center" valign="middle"><i>R</i><sup>2</sup></td><td class="table_top_border" align="center" valign="middle">Cr(Ⅵ)去除率/%Cr(Ⅵ) removal efficiency</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{C}-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M1.jpg"><img class="graphic" src="2021120501_M1.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">3.022 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.5253</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=1</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M2.jpg"><img class="graphic" src="2021120501_M2.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0286 h<sup>−1</sup></td><td align="center" valign="middle">0.6589</td><td align="center" valign="middle">79.4</td></tr><tr><td align="center" valign="middle"></td><td align="center" valign="middle">二级</td><td align="center" valign="middle"><inline-formula><tex-math id="M3">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M3.jpg"><img class="graphic" src="2021120501_M3.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0002 L(mg· h)<sup>-1·</sup></td><td align="center" valign="middle">0.7508</td><td align="center" valign="middle"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M4">$ C-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M4.jpg"><img class="graphic" src="2021120501_M4.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">13.436 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.8007</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=3</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M5">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M5.jpg"><img class="graphic" src="2021120501_M5.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.1765 h<sup>−1</sup></td><td align="center" valign="middle">0.9358</td><td align="center" valign="middle">99.9</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">二级</td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M6">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M6.jpg"><img class="graphic" src="2021120501_M6.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.0012 L(mg· h)<sup>-1·</sup></td><td class="table_bottom_border" align="center" valign="middle">0.9459</td><td class="table_bottom_border" align="center" valign="middle"></td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td align="center" valign="middle">0.0002 L(mg· h)<sup>-1·</sup></td><td align="center" valign="middle">0.7508</td><td align="center" valign="middle"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M4">$ C-{C}_{0}=-kt $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">实验条件Experimental conditions</td><td class="table_top_border" align="center" valign="middle">拟合级数Fitting series</td><td class="table_top_border" align="center" valign="middle">动力学方程Kinetic equation</td><td class="table_top_border" align="center" valign="middle">速率常数<i>k</i>Rate constant <i>k</i></td><td class="table_top_border" style="width:8%" align="center" valign="middle"><i>R</i><sup>2</sup></td><td class="table_top_border" align="center" valign="middle">Cr(Ⅵ)去除率/%Cr(Ⅵ) removal efficiency</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{C}-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M1.jpg"><img class="graphic" src="2021120501_M1.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">3.022 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.5253</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=1</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M2.jpg"><img class="graphic" src="2021120501_M2.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0286 h<sup>−1</sup></td><td align="center" valign="middle">0.6589</td><td align="center" valign="middle">79.4</td></tr><tr><td align="center" valign="middle"></td><td align="center" valign="middle">二级</td><td align="center" valign="middle"><inline-formula><tex-math id="M3">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M3.jpg"><img class="graphic" src="2021120501_M3.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0002 L(mg· h)<sup>-1·</sup></td><td align="center" valign="middle">0.7508</td><td align="center" valign="middle"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M4">$ C-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M4.jpg"><img class="graphic" src="2021120501_M4.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">13.436 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.8007</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=3</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M5">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M5.jpg"><img class="graphic" src="2021120501_M5.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.1765 h<sup>−1</sup></td><td align="center" valign="middle">0.9358</td><td align="center" valign="middle">99.9</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">二级</td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M6">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M6.jpg"><img class="graphic" src="2021120501_M6.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.0012 L(mg· h)<sup>-1·</sup></td><td class="table_bottom_border" align="center" valign="middle">0.9459</td><td class="table_bottom_border" align="center" valign="middle"></td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td class="table_top_border2" align="center" valign="middle">13.436 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.8007</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=3</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M5">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">实验条件Experimental conditions</td><td class="table_top_border" align="center" valign="middle">拟合级数Fitting series</td><td class="table_top_border" align="center" valign="middle">动力学方程Kinetic equation</td><td class="table_top_border" align="center" valign="middle">速率常数<i>k</i>Rate constant <i>k</i></td><td class="table_top_border" style="width:8%" align="center" valign="middle"><i>R</i><sup>2</sup></td><td class="table_top_border" align="center" valign="middle">Cr(Ⅵ)去除率/%Cr(Ⅵ) removal efficiency</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{C}-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M1.jpg"><img class="graphic" src="2021120501_M1.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">3.022 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.5253</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=1</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M2.jpg"><img class="graphic" src="2021120501_M2.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0286 h<sup>−1</sup></td><td align="center" valign="middle">0.6589</td><td align="center" valign="middle">79.4</td></tr><tr><td align="center" valign="middle"></td><td align="center" valign="middle">二级</td><td align="center" valign="middle"><inline-formula><tex-math id="M3">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M3.jpg"><img class="graphic" src="2021120501_M3.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0002 L(mg· h)<sup>-1·</sup></td><td align="center" valign="middle">0.7508</td><td align="center" valign="middle"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M4">$ C-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M4.jpg"><img class="graphic" src="2021120501_M4.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">13.436 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.8007</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=3</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M5">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M5.jpg"><img class="graphic" src="2021120501_M5.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.1765 h<sup>−1</sup></td><td align="center" valign="middle">0.9358</td><td align="center" valign="middle">99.9</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">二级</td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M6">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M6.jpg"><img class="graphic" src="2021120501_M6.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.0012 L(mg· h)<sup>-1·</sup></td><td class="table_bottom_border" align="center" valign="middle">0.9459</td><td class="table_bottom_border" align="center" valign="middle"></td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td align="center" valign="middle">0.1765 h<sup>−1</sup></td><td align="center" valign="middle">0.9358</td><td align="center" valign="middle">99.9</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">二级</td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M6">$ 1/C-1/{C}_{0}=kt $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">实验条件Experimental conditions</td><td class="table_top_border" align="center" valign="middle">拟合级数Fitting series</td><td class="table_top_border" align="center" valign="middle">动力学方程Kinetic equation</td><td class="table_top_border" align="center" valign="middle">速率常数<i>k</i>Rate constant <i>k</i></td><td class="table_top_border" style="width:8%" align="center" valign="middle"><i>R</i><sup>2</sup></td><td class="table_top_border" align="center" valign="middle">Cr(Ⅵ)去除率/%Cr(Ⅵ) removal efficiency</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M1">$ \mathrm{C}-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M1.jpg"><img class="graphic" src="2021120501_M1.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">3.022 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.5253</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=1</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M2">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M2.jpg"><img class="graphic" src="2021120501_M2.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0286 h<sup>−1</sup></td><td align="center" valign="middle">0.6589</td><td align="center" valign="middle">79.4</td></tr><tr><td align="center" valign="middle"></td><td align="center" valign="middle">二级</td><td align="center" valign="middle"><inline-formula><tex-math id="M3">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M3.jpg"><img class="graphic" src="2021120501_M3.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.0002 L(mg· h)<sup>-1·</sup></td><td align="center" valign="middle">0.7508</td><td align="center" valign="middle"></td></tr><tr><td class="table_top_border2" align="center" valign="middle"></td><td class="table_top_border2" align="center" valign="middle">零级</td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M4">$ C-{C}_{0}=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M4.jpg"><img class="graphic" src="2021120501_M4.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">13.436 mg·(L· h)<sup>−1</sup></td><td class="table_top_border2" align="center" valign="middle">0.8007</td><td class="table_top_border2" align="center" valign="middle"></td></tr><tr><td align="center" valign="middle">S/Fe=3</td><td align="center" valign="middle">一级</td><td align="center" valign="middle"><inline-formula><tex-math id="M5">$ \mathrm{l}\mathrm{n}(C/{C}_{0})=-kt $</tex-math><alternatives><img class="graphic" src="2021120501_M5.jpg"><img class="graphic" src="2021120501_M5.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.1765 h<sup>−1</sup></td><td align="center" valign="middle">0.9358</td><td align="center" valign="middle">99.9</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"></td><td class="table_bottom_border" align="center" valign="middle">二级</td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M6">$ 1/C-1/{C}_{0}=kt $</tex-math><alternatives><img class="graphic" src="2021120501_M6.jpg"><img class="graphic" src="2021120501_M6.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.0012 L(mg· h)<sup>-1·</sup></td><td class="table_bottom_border" align="center" valign="middle">0.9459</td><td class="table_bottom_border" align="center" valign="middle"></td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.0012 L(mg· h)<sup>-1·</sup></td><td class="table_bottom_border" align="center" valign="middle">0.9459</td><td class="table_bottom_border" align="center" valign="middle"></td></tr></tbody>
</table></div></foreignObject></svg>)
-
硫化亚铁(FeS)常被用于六价铬(Cr(Ⅵ))污染水体的治理修复,主要通过吸附、还原、和共沉淀作用等去除水体中的Cr(Ⅵ). FeS材料的颗粒尺寸、比表面积、形状、存在形态、稳定性等因素对其Cr(Ⅵ)的去除性能有显著影响[1-4]. 与天然FeS矿物相比,人工合成的FeS颗粒具有更大的比表面积和更高的反应活性;采用羧甲基纤维素、海藻酸钠等稳定剂对其进行表面修饰后,可以有效防止颗粒的聚集,使得FeS活性更稳定,对Cr(Ⅵ)的去除性能可得到显著提升[5-7].
FeS的形貌对Cr(Ⅵ)的去除性能可能产生较大影响. Li等[8]使用合成的梭形FeS颗粒修复铬污染土壤,当FeS/Cr(Ⅵ)物质的量比=1.5:1时,在3 d内可以去除98%的Cr(Ⅵ),Cr(Ⅵ)浓度由1407 mg·kg−1降低至16 mg·kg−1. Liu等[7]采用椭球型纳米FeS处理水溶液Cr(Ⅵ),在pH=5.6时去除性能为683 mg Cr(Ⅵ)/g FeS,去除率高达92.48%;Wang等[9]使用棒状FeS去除水溶液中Cr(Ⅵ),当FeS/Cr(Ⅵ)物质的量比=4时,可在30 min内完全去除水溶液中Cr(Ⅵ).
利用溶剂热法、均相沉淀法、NaBH4还原法、生物法等方法[8, 10-12]制备的FeS颗粒形貌差异较大. Sines等[13]采用溶剂热法在Fe2+/S2-物质的量比为5:3.12、200 ℃下反应4 h后,合成了FeS纳米片,平均厚度约为30 nm. Li等[8]在氮气保护下将等量的S2-溶液逐滴加入至Fe2+-CMC溶液中制备了梭形的FeS,平均长度为400 nm,中间直径约为100 nm. Kim等[10]采用改性的NaBH4还原法将S2O42-溶液以3:1的体积比加入至Fe3+溶液中合成了球形FeS. Xiong等[14]制备了微球状FeS纳米颗粒,并通过批处理试验表明FeS纳米颗粒可以有效地固定沉积物中的Hg,当FeS/Hg物质的量比为26.5时,Hg浸出浓度降低97%,毒性浸出率降低99%. 程千文[12]利用希瓦氏菌在代谢过程中将Fe3+和S2O32-还原为Fe2+和S2- 后,合成了球状FeS,粒径范围在20—100 nm.
目前,水热法已被应用于合成形貌多样的硫化物材料,反应时间、反应温度、溶剂、反应物的物质的量比等合成条件会对产物形貌产生显著影响[15]. Gorai等[16]研究表明在乙二胺和水比例在100:0—0:100范围内,随着乙二胺和水的比例减小,趋于球形的铜离子与硫脲的配合物逐渐形成,使Cu1-xSx形貌从树枝状转变为棒状最后成为球状. 用水热法合成Bi2S3微/纳米材料时,当反应时间从10 min延长到6 h时,合成的Bi2S3形貌从纳米带束转变为完整的、均匀的花状结构[17]. Kar和Chaudhuri[17]证实了在反应温度为150—230 ℃范围内,升高温度可使FeS2由纳米线转变为纳米片; Wu等[18]发现,当S/Zn物质的量比分别为1:1、2:1时,ZnS形貌从片状转变为球形. 但是,当前对影响水热法合成FeS颗粒形貌的主要影响因素仍不清楚[19].
本研究旨在观测识别水热法控制FeS形貌的影响因素,并以水溶液中六价铬Cr(Ⅵ)作为目标污染物,探究不同形貌的FeS对Cr(Ⅵ)的去除性能. 具体包括:(1)分析溶剂比、反应温度、反应时间和铁硫物质的量比等因素对人工合成FeS颗粒形貌的影响;(2)探究具有棒状和片状等形貌的人工合成FeS颗粒对Cr(Ⅵ)的去除性能.
-
主要试剂:氯化铁(FeCl3·6H2O)、L-半胱氨酸(C3H7NO2S)、乙二胺(EN)、无水乙醇(C2H5OH)、重铬酸钾(K2Cr2O7)、吗啉乙磺酸(MES)、氢氧化钠(NaOH)、丙酮(C3H6O),以上试剂均为分析纯.
主要仪器:马弗炉(SX2-2.5-10NP,上海一恒科学仪器有限公司)、回旋振荡器(THZ-98C,上海一恒科学仪器有限公司)、冷冻干燥机(SCIENTZ-18N,宁波新芝生物科技有限公司)、台式低速离心机(TD5A,金坛市科析仪器有限公司)、真空干燥箱(DZF-6010,重庆东悦仪器有限公司)、高压反应釜(100 mL,西安常仪仪器设备有限公司)、紫外可见分光光度计(T6,北京普析通用仪器有限公司)、超纯水机(UPT-Ⅱ-10T,成都超纯科技有限公司).
-
采用Min等[19]报道的FeS微粒的水热合成方法,称取L-半胱氨酸(C3H7NO2S)溶于35 mL乙二胺溶液中,称取氯化铁(FeCl3·6H2O)溶于35 mL乙二胺溶液中,用磁力搅拌混合30 min后移入100 mL聚四氟乙烯内衬的高压反应釜中,置入马弗炉中加热至180、190、200 ℃,反应 4、8、24 h,而后将高压反应釜从马弗炉中转移出,等待其冷却至室温,将高压反应釜中的混合溶液转移至聚丙烯离心管中,在5000 r·min−1条件下离心10 min后,弃去上清液并向离心管中加入无氧水,振荡离心管使固体重新悬浮后继续离心,洗涤3次后用无水乙醇重复上述步骤3次. 将洗净的FeS固体置入真空干燥箱,于50 ℃干燥8 h后研磨过200目标准筛,充氮气后于4 ℃密封保存.
-
在250 mL锥形瓶中加入K2Cr2O7溶液,用MES和NaOH 溶液将pH调节至5.5;溶液体积100 mL,Cr(Ⅵ)初始浓度为170 mg·L−1,溶液中投加0.5 g·L−1的FeS;采用氮气吹脱保持无氧条件,密封后置于温度为25 ℃、振荡速率为200 r·min−1的恒温振荡器内持续振荡,每组实验设有两个平行样,待反应一定时间间隔后移取上清液,采用0.45 um滤膜过滤,测定滤液中的Cr(Ⅵ)浓度.
-
采用《水质 六价铬的测定 二苯碳酰二肼分光光度法》(GB7464—87)测定水溶液中Cr(Ⅵ)浓度. 采用D/max 2500PC X-射线粉末衍射仪(XRD)(日本理学)对样品进行测试,分析衍射图谱. 测试为Cu靶,扫描为连续扫描,扫描范围为 10°—90°,扫描速度为4( °)·min−1,步长为0.02°,管压和管流分别为40.0 kV和150.0 mA;采用JEOL公司的JSM-7800F场发射扫描电镜对样品进行形貌分析.
-
Fe(Ⅲ)被乙二胺还原后生成[Fe(EN)2]2+配合物,该配合物可取代L-半胱氨酸分子中—COOH上的质子,并形成HSCH2(NH2)COO-Fe-(OH)2,其性质不稳定,极易分解形成FeS晶核,晶核逐渐生长为FeS微粒[19-20]. HSCH2CH(NH2)COO-Fe-(OH)2是FeS合成的真实前驱体. 反应方程式见式(1—3):
图1展示了合成样品的SEM-EDS和XRD谱图. 图1a、b可以看出,在合成的样品中检测到了Fe、S元素,且二者质量比分别为58.5%和41.5%,说明合成样品由Fe、S元素组成. 图1c中,S/Fe物质的量比为1和3合成的样品在2θ=17.64°均出现了尖锐的衍射峰,可以对应FeS(JCPDS 15-0037)在001晶面处的特征衍射峰,由此可进一步确定所合成的物质为FeS晶体,与Sines[13]和Thomas[11]等用水热法合成的FeS的表征结果一致.
-
图2展示了用不同水/乙二胺(EN)溶剂体积比时合成FeS的SEM图像. 随着乙二胺(EN)比例的增大,合成的FeS颗粒形主要呈现片状,部分镶嵌组合在一起. 当水/EN=1:1和1:2时,单个片状紧密聚集并形成分层结构,厚度约为1—3 μm;水/EN=1:3时,FeS转变为片状镶嵌的形貌,厚度减小,均小于1 μm;当溶剂为纯EN时合成的FeS呈现分散的薄片,大小不均匀,而且分散度更大. 片状FeS是L-半胱氨酸分解后释放H2S气体促使球状FeS分裂而成[21-22]. 在FeS晶核生长过程中由于乙二胺吸附在晶体表面,晶体的团聚会被阻止[20]. 随着乙二胺浓度的增大,乙二胺对Fe元素的络合能力逐渐加强,通过双齿双核络合成键作用使FeS晶体片相互聚集. 但是,过量乙二胺的存在会使络合成键方式由双齿双核转变为双齿单核,FeS晶体片又恢复到了分散的状态[16, 23].
-
L-半胱氨酸的分解温度小于400 ℃[24],由图3可知,当温度为180 ℃时,FeS呈现一些不定形的碎片;当温度为190 ℃和200 ℃时,FeS微粒的均一性更好,主要呈现为片状;当反应温度为180 ℃时,L-半胱氨酸未能完全分解,在乙二胺中的溶解度不高,参与反应的S离子数量不足,无法完全还原Fe(Ⅲ),因此FeS的纯度不高;当温度升高到200 ℃时,出现了多边形薄片,边界也更加明显,薄片厚度约<1 μm,说明升高温度有利于晶体生长,同时边界的明显化也有助于活性位点的充分暴露[25]. 但温度过高也不利于FeS微粒的形貌的均一性[20],温度高于200 ℃后,溶于乙二胺的L-半胱氨酸由于相转变发生过度分解,此时溶液从均质状态转变为非均质状态,L-半胱氨酸存在浓度梯度,导致FeS晶体以不同速率生长,从而使FeS微粒呈现不规则的形貌. 因此,200 ℃条件更有利于水热合成FeS微粒的形貌保持均一性.
-
由图4可知,反应4 h后FeS微粒生长为花形的片状镶嵌结构;随着反应时间的延长,薄片继续生长,反应8 h时FeS晶体生长为薄片更大、更加松散的花状镶嵌结构. 图3(c)表明,反应24 h后FeS晶体薄片结构更大,原本镶嵌的薄片结构散开,呈现出独立的片状结构. 这一过程可由晶体生长理论来解释[22]:反应初期,FeS晶核会聚集为球形以降低表面能,而L-半胱氨酸在高温下分解释放H2S气体促使球状FeS分裂成片状FeS,随着反应时间的延长,片状FeS不断长大、拉伸,自发形成更大的薄片. Wu[19]通过水热法用乙酸锌和L-半胱氨酸合成ZnS的研究发现当反应时间为4 h时,产物由40 nm的纳米ZnS和平均粒径为400 nm的球形ZnS组成;随着反应时间延长至10 h,产物转变为平均粒径为420 nm的球形ZnS;进一步延长时间至24 h,ZnS形貌没有发生变化,粒径不断增大,约为570 nm. 因此,随着反应时间的延长,合成产物会自发生长、尺寸持续增大.
-
图5展示了S/Fe物质的量比对FeS微粒表观形貌的影响. 随着S投加量的增大,FeS微粒形貌由片状逐步变为棒状. 当S/Fe=1时,FeS微粒的形貌呈现花状和片状;当S/Fe=2时,出现了多种形貌的FeS微粒,主要由少量的棒状和球状颗粒构成;当S/Fe=3和S/Fe=4时,FeS微粒形貌均为棒状;但是S/Fe=4时棒条更平整,粗细也更加均匀,直径大致在300—500 nm之间. 可以表明,采用水热法合成FeS微粒时S/Fe物质的量比对产物的形貌影响较大,增大S源的浓度有利于FeS由片状向棒状转变. 这与Zhang等[26]用水热法合成Bi2S3的研究发现保持一致,其在以水为溶剂、温度150 ℃时将L-半胱氨酸含量从0.15 g增大到0.35 g,固体颗粒形貌从纳米薄片转变为纳米棒. 同时,乙二胺是一种具有较强极性和螯合能力的表面活性剂,可以控制FeS不同晶面的生长速率[11,20],由图1可知,本研究合成的FeS晶体微粒具有沿着[001]方向优先生长的取向,使FeS定向生长为棒状[23].从而,通过控制S/Fe物质的量比,采用水热法可合成具有不同形貌的FeS微粒,同时在水热合成过程中提高反应物的投加量,可抑制产物的继续生长,有效的控制晶体形貌[19].
-
为了研究FeS形貌对Cr(Ⅵ)的去除性能的影响,分别选取S/Fe=1、S/Fe=3代表片状和棒状两种典型形貌的FeS微粒进行水溶液中Cr(Ⅵ)去除试验. 图6展示了水溶液中Cr(Ⅵ)的去除动力学,反应180 h后,S/Fe=3条件下合成的FeS微粒对Cr(Ⅵ)去除率可达到99.9%,Cr(Ⅵ)的浓度接近0 mg L-1;S/Fe=1条件下合成的FeS微粒对Cr(Ⅵ)去除率为79.4%,Cr(Ⅵ)的浓度为35 mg·L−1. 因此,S/Fe=1和S/Fe=3时合成的FeS微粒对Cr(Ⅵ)的去除率存在显著性差异.
由表1可知,S/Fe=1和S/Fe=3条件下合成的FeS对Cr(Ⅵ)的去除过程较好地符合拟二级反应动力学,反应速率常数分别是0.0002 L·(mg·h)−1、0.0012 L·(mg·h)−1. 因此,当溶液pH=5.5时,S/Fe=3条件下Cr(Ⅵ)的去除率和去除速率常数均大于S/Fe=1条件.
随着S/Fe物质的量比从1增大至3,合成的FeS与Cr(Ⅵ)的反应速率增大,主要是由于L-半胱氨酸提供S源的同时也提高了反应物的浓度,从而提高了FeS微粒对Cr(Ⅵ)的去除速率[16]. 同时,由于棒状FeS微粒具有更大的比表面积,可提供更大的接触面积和更多的活性位点[9],从而可使反应快速进行.
-
(1)利用水热法合成FeS微粒分别具有棒状和片状等两种典型形貌.
(2)水/乙二胺体积比、反应温度、反应时间对水热合成FeS微粒形貌的影响不显著,均为片状;S/Fe物质的量比对FeS矿物形貌具有显著影响,随着S/Fe物质的量比的增大,FeS的颗粒从片状转变为棒状. 当S/Fe =3和S/Fe =4时,FeS颗粒均呈棒状,粒径可达200—300 nm.
(3)具有棒状的水热合成FeS微粒对水溶液中Cr(Ⅵ)去除率更高. 棒状FeS微粒在180 h内可去除水溶液中99.9%的Cr(Ⅵ),而片状FeS微粒对Cr(Ⅵ)的去除率仅为79.4%.
硫化亚铁微粒的水热法合成及其对水溶液中Cr(Ⅵ)的去除性能
Hydrothermal synthesis of ferrous sulfide particles and their performance for the removal of Cr(Ⅵ) from aqueous solution
-
摘要: 借助扫描电镜(SEM)、X射线衍射(XRD)等手段,对水热合成的硫化亚铁(FeS)微粒进行特性表征,探究了水/乙二胺体积比、反应时间、反应温度、S/Fe物质的量比等因素对FeS微粒微观形貌的影响;通过批实验观测了具有不同形貌的FeS微粒对水溶液中Cr(Ⅵ)的去除性能. 结果表明,在S/Fe物质的量比=1—4时,随着S/Fe物质的量比的增大,FeS形貌由片状逐渐转变为棒状;水/乙二胺体积比、反应温度和反应时间对FeS的形貌的影响不显著,均呈片状. FeS微粒可有效去除水溶液中的Cr(Ⅵ),当溶液pH=5.5时,0.5 g·L-1的片状和棒状FeS对初始浓度为170 mg·L-1的水溶液中Cr(Ⅵ)的去除率分别为79.4%和99.9%.Abstract: Ferrous sulfide (FeS) particles were prepared via a hydrothermal synthesis method, and then characterized by scanning electron microscopy and X-ray diffraction. The effects of water-to-ethylenediamine volume ratio, reaction time, reaction temperature and S: Fe molar ratio on the micromorphology of FeS particles were investigated. The removal of aqueous Cr(Ⅵ) using FeS particles with various morphologies was determined through batch tests. The results showed that the morphology of FeS particles gradually changed from flake-shaped to rod-shaped, as the S: Fe molar ratio increased from 1 to 4. The water-to-ethylenediamine volume ratio, reaction temperature, and reaction time had insignificant effects on the morphology of FeS particles, which remained flake-shaped. The removal efficiencies of aqueous Cr(Ⅵ) using 0.5 g·L-1 flake-shaped and rod-shaped FeS at a pH of 5.5 and initial Cr(Ⅵ) concentration of 170 mg·L-1 were 79.4% and 99.9%, respectively.
-
Key words:
- hydrothermal synthesis /
- FeS particles /
- hexavalent chromium /
- influencing factors.
-
硫化亚铁(FeS)常被用于六价铬(Cr(Ⅵ))污染水体的治理修复,主要通过吸附、还原、和共沉淀作用等去除水体中的Cr(Ⅵ). FeS材料的颗粒尺寸、比表面积、形状、存在形态、稳定性等因素对其Cr(Ⅵ)的去除性能有显著影响[1-4]. 与天然FeS矿物相比,人工合成的FeS颗粒具有更大的比表面积和更高的反应活性;采用羧甲基纤维素、海藻酸钠等稳定剂对其进行表面修饰后,可以有效防止颗粒的聚集,使得FeS活性更稳定,对Cr(Ⅵ)的去除性能可得到显著提升[5-7].
FeS的形貌对Cr(Ⅵ)的去除性能可能产生较大影响. Li等[8]使用合成的梭形FeS颗粒修复铬污染土壤,当FeS/Cr(Ⅵ)物质的量比=1.5:1时,在3 d内可以去除98%的Cr(Ⅵ),Cr(Ⅵ)浓度由1407 mg·kg−1降低至16 mg·kg−1. Liu等[7]采用椭球型纳米FeS处理水溶液Cr(Ⅵ),在pH=5.6时去除性能为683 mg Cr(Ⅵ)/g FeS,去除率高达92.48%;Wang等[9]使用棒状FeS去除水溶液中Cr(Ⅵ),当FeS/Cr(Ⅵ)物质的量比=4时,可在30 min内完全去除水溶液中Cr(Ⅵ).
利用溶剂热法、均相沉淀法、NaBH4还原法、生物法等方法[8, 10-12]制备的FeS颗粒形貌差异较大. Sines等[13]采用溶剂热法在Fe2+/S2-物质的量比为5:3.12、200 ℃下反应4 h后,合成了FeS纳米片,平均厚度约为30 nm. Li等[8]在氮气保护下将等量的S2-溶液逐滴加入至Fe2+-CMC溶液中制备了梭形的FeS,平均长度为400 nm,中间直径约为100 nm. Kim等[10]采用改性的NaBH4还原法将S2O42-溶液以3:1的体积比加入至Fe3+溶液中合成了球形FeS. Xiong等[14]制备了微球状FeS纳米颗粒,并通过批处理试验表明FeS纳米颗粒可以有效地固定沉积物中的Hg,当FeS/Hg物质的量比为26.5时,Hg浸出浓度降低97%,毒性浸出率降低99%. 程千文[12]利用希瓦氏菌在代谢过程中将Fe3+和S2O32-还原为Fe2+和S2- 后,合成了球状FeS,粒径范围在20—100 nm.
目前,水热法已被应用于合成形貌多样的硫化物材料,反应时间、反应温度、溶剂、反应物的物质的量比等合成条件会对产物形貌产生显著影响[15]. Gorai等[16]研究表明在乙二胺和水比例在100:0—0:100范围内,随着乙二胺和水的比例减小,趋于球形的铜离子与硫脲的配合物逐渐形成,使Cu1-xSx形貌从树枝状转变为棒状最后成为球状. 用水热法合成Bi2S3微/纳米材料时,当反应时间从10 min延长到6 h时,合成的Bi2S3形貌从纳米带束转变为完整的、均匀的花状结构[17]. Kar和Chaudhuri[17]证实了在反应温度为150—230 ℃范围内,升高温度可使FeS2由纳米线转变为纳米片; Wu等[18]发现,当S/Zn物质的量比分别为1:1、2:1时,ZnS形貌从片状转变为球形. 但是,当前对影响水热法合成FeS颗粒形貌的主要影响因素仍不清楚[19].
本研究旨在观测识别水热法控制FeS形貌的影响因素,并以水溶液中六价铬Cr(Ⅵ)作为目标污染物,探究不同形貌的FeS对Cr(Ⅵ)的去除性能. 具体包括:(1)分析溶剂比、反应温度、反应时间和铁硫物质的量比等因素对人工合成FeS颗粒形貌的影响;(2)探究具有棒状和片状等形貌的人工合成FeS颗粒对Cr(Ⅵ)的去除性能.
1. 材料与方法(Materials and methods)
1.1 试剂与仪器
主要试剂:氯化铁(FeCl3·6H2O)、L-半胱氨酸(C3H7NO2S)、乙二胺(EN)、无水乙醇(C2H5OH)、重铬酸钾(K2Cr2O7)、吗啉乙磺酸(MES)、氢氧化钠(NaOH)、丙酮(C3H6O),以上试剂均为分析纯.
主要仪器:马弗炉(SX2-2.5-10NP,上海一恒科学仪器有限公司)、回旋振荡器(THZ-98C,上海一恒科学仪器有限公司)、冷冻干燥机(SCIENTZ-18N,宁波新芝生物科技有限公司)、台式低速离心机(TD5A,金坛市科析仪器有限公司)、真空干燥箱(DZF-6010,重庆东悦仪器有限公司)、高压反应釜(100 mL,西安常仪仪器设备有限公司)、紫外可见分光光度计(T6,北京普析通用仪器有限公司)、超纯水机(UPT-Ⅱ-10T,成都超纯科技有限公司).
1.2 FeS颗粒的水热合成
采用Min等[19]报道的FeS微粒的水热合成方法,称取L-半胱氨酸(C3H7NO2S)溶于35 mL乙二胺溶液中,称取氯化铁(FeCl3·6H2O)溶于35 mL乙二胺溶液中,用磁力搅拌混合30 min后移入100 mL聚四氟乙烯内衬的高压反应釜中,置入马弗炉中加热至180、190、200 ℃,反应 4、8、24 h,而后将高压反应釜从马弗炉中转移出,等待其冷却至室温,将高压反应釜中的混合溶液转移至聚丙烯离心管中,在5000 r·min−1条件下离心10 min后,弃去上清液并向离心管中加入无氧水,振荡离心管使固体重新悬浮后继续离心,洗涤3次后用无水乙醇重复上述步骤3次. 将洗净的FeS固体置入真空干燥箱,于50 ℃干燥8 h后研磨过200目标准筛,充氮气后于4 ℃密封保存.
1.3 水溶液中Cr(Ⅵ)的去除
在250 mL锥形瓶中加入K2Cr2O7溶液,用MES和NaOH 溶液将pH调节至5.5;溶液体积100 mL,Cr(Ⅵ)初始浓度为170 mg·L−1,溶液中投加0.5 g·L−1的FeS;采用氮气吹脱保持无氧条件,密封后置于温度为25 ℃、振荡速率为200 r·min−1的恒温振荡器内持续振荡,每组实验设有两个平行样,待反应一定时间间隔后移取上清液,采用0.45 um滤膜过滤,测定滤液中的Cr(Ⅵ)浓度.
1.4 测试与表征
采用《水质 六价铬的测定 二苯碳酰二肼分光光度法》(GB7464—87)测定水溶液中Cr(Ⅵ)浓度. 采用D/max 2500PC X-射线粉末衍射仪(XRD)(日本理学)对样品进行测试,分析衍射图谱. 测试为Cu靶,扫描为连续扫描,扫描范围为 10°—90°,扫描速度为4( °)·min−1,步长为0.02°,管压和管流分别为40.0 kV和150.0 mA;采用JEOL公司的JSM-7800F场发射扫描电镜对样品进行形貌分析.
2. 结果与讨论(Results and discussion)
2.1 FeS晶体生成机制
Fe(Ⅲ)被乙二胺还原后生成[Fe(EN)2]2+配合物,该配合物可取代L-半胱氨酸分子中—COOH上的质子,并形成HSCH2(NH2)COO-Fe-(OH)2,其性质不稳定,极易分解形成FeS晶核,晶核逐渐生长为FeS微粒[19-20]. HSCH2CH(NH2)COO-Fe-(OH)2是FeS合成的真实前驱体. 反应方程式见式(1—3):
Fe3++EN→[Fe(EN)2]2+ (1) [Fe(EN)2]2++L-cysteine→HSCH2CH(NH2)COO-Fe-(OH)2 (2) HSCH2CH(NH2)COO-Fe-(OH)2→FeS(s) (3) 图1展示了合成样品的SEM-EDS和XRD谱图. 图1a、b可以看出,在合成的样品中检测到了Fe、S元素,且二者质量比分别为58.5%和41.5%,说明合成样品由Fe、S元素组成. 图1c中,S/Fe物质的量比为1和3合成的样品在2θ=17.64°均出现了尖锐的衍射峰,可以对应FeS(JCPDS 15-0037)在001晶面处的特征衍射峰,由此可进一步确定所合成的物质为FeS晶体,与Sines[13]和Thomas[11]等用水热法合成的FeS的表征结果一致.
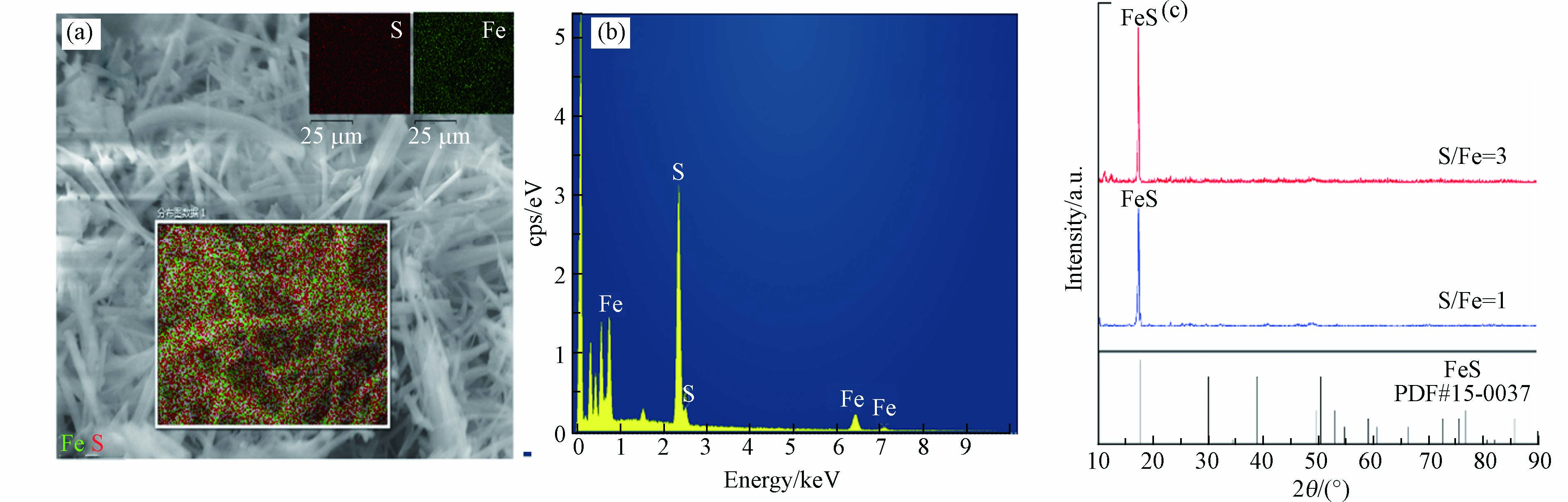 图 1 FeS微粒的(a) SEM图像、(b) EDS图和(c) XRD谱(纯EN Pure EN,反应温度 Reaction temperature 200 ℃,反应时间Reaction time 4 h)Figure 1. XRD patterns and SEM-EDS images of FeS by (a) SEM analysis,(b) EDS analysis,(c) XRD analysis
图 1 FeS微粒的(a) SEM图像、(b) EDS图和(c) XRD谱(纯EN Pure EN,反应温度 Reaction temperature 200 ℃,反应时间Reaction time 4 h)Figure 1. XRD patterns and SEM-EDS images of FeS by (a) SEM analysis,(b) EDS analysis,(c) XRD analysis2.2 人工合成FeS颗粒形貌的影响因素
2.2.1 水/乙二胺(EN)体积比
图2展示了用不同水/乙二胺(EN)溶剂体积比时合成FeS的SEM图像. 随着乙二胺(EN)比例的增大,合成的FeS颗粒形主要呈现片状,部分镶嵌组合在一起. 当水/EN=1:1和1:2时,单个片状紧密聚集并形成分层结构,厚度约为1—3 μm;水/EN=1:3时,FeS转变为片状镶嵌的形貌,厚度减小,均小于1 μm;当溶剂为纯EN时合成的FeS呈现分散的薄片,大小不均匀,而且分散度更大. 片状FeS是L-半胱氨酸分解后释放H2S气体促使球状FeS分裂而成[21-22]. 在FeS晶核生长过程中由于乙二胺吸附在晶体表面,晶体的团聚会被阻止[20]. 随着乙二胺浓度的增大,乙二胺对Fe元素的络合能力逐渐加强,通过双齿双核络合成键作用使FeS晶体片相互聚集. 但是,过量乙二胺的存在会使络合成键方式由双齿双核转变为双齿单核,FeS晶体片又恢复到了分散的状态[16, 23].
 图 2 不同溶剂体积比条件下合成FeS微粒的SEM图Figure 2. SEM images of the FeS synthesized by (a) water/EN =1:1,(b) water/EN =1:2,(c) water/EN =1:3,(d) pure EN(a) 水/EN =1:1,(b) 水/EN =1:2,(c) 水/EN =1:3,(d) 纯EN(S/Fe =1,反应温度Reaction Temperature 200 ℃,反应时间Reaction time 24 h)
图 2 不同溶剂体积比条件下合成FeS微粒的SEM图Figure 2. SEM images of the FeS synthesized by (a) water/EN =1:1,(b) water/EN =1:2,(c) water/EN =1:3,(d) pure EN(a) 水/EN =1:1,(b) 水/EN =1:2,(c) 水/EN =1:3,(d) 纯EN(S/Fe =1,反应温度Reaction Temperature 200 ℃,反应时间Reaction time 24 h)2.2.2 反应温度
L-半胱氨酸的分解温度小于400 ℃[24],由图3可知,当温度为180 ℃时,FeS呈现一些不定形的碎片;当温度为190 ℃和200 ℃时,FeS微粒的均一性更好,主要呈现为片状;当反应温度为180 ℃时,L-半胱氨酸未能完全分解,在乙二胺中的溶解度不高,参与反应的S离子数量不足,无法完全还原Fe(Ⅲ),因此FeS的纯度不高;当温度升高到200 ℃时,出现了多边形薄片,边界也更加明显,薄片厚度约<1 μm,说明升高温度有利于晶体生长,同时边界的明显化也有助于活性位点的充分暴露[25]. 但温度过高也不利于FeS微粒的形貌的均一性[20],温度高于200 ℃后,溶于乙二胺的L-半胱氨酸由于相转变发生过度分解,此时溶液从均质状态转变为非均质状态,L-半胱氨酸存在浓度梯度,导致FeS晶体以不同速率生长,从而使FeS微粒呈现不规则的形貌. 因此,200 ℃条件更有利于水热合成FeS微粒的形貌保持均一性.
 图 3 不同反应温度条件下合成FeS微粒的SEM图Figure 3. SEM images of FeS under (a) 180 ℃,(b) 190 ℃,(c) 200 ℃(a) 180 ℃,(b) 190 ℃,(c) 200 ℃(纯EN Pure EN,S/Fe=1,反应时间Reaction time 24 h)
图 3 不同反应温度条件下合成FeS微粒的SEM图Figure 3. SEM images of FeS under (a) 180 ℃,(b) 190 ℃,(c) 200 ℃(a) 180 ℃,(b) 190 ℃,(c) 200 ℃(纯EN Pure EN,S/Fe=1,反应时间Reaction time 24 h)2.2.3 反应时间
由图4可知,反应4 h后FeS微粒生长为花形的片状镶嵌结构;随着反应时间的延长,薄片继续生长,反应8 h时FeS晶体生长为薄片更大、更加松散的花状镶嵌结构. 图3(c)表明,反应24 h后FeS晶体薄片结构更大,原本镶嵌的薄片结构散开,呈现出独立的片状结构. 这一过程可由晶体生长理论来解释[22]:反应初期,FeS晶核会聚集为球形以降低表面能,而L-半胱氨酸在高温下分解释放H2S气体促使球状FeS分裂成片状FeS,随着反应时间的延长,片状FeS不断长大、拉伸,自发形成更大的薄片. Wu[19]通过水热法用乙酸锌和L-半胱氨酸合成ZnS的研究发现当反应时间为4 h时,产物由40 nm的纳米ZnS和平均粒径为400 nm的球形ZnS组成;随着反应时间延长至10 h,产物转变为平均粒径为420 nm的球形ZnS;进一步延长时间至24 h,ZnS形貌没有发生变化,粒径不断增大,约为570 nm. 因此,随着反应时间的延长,合成产物会自发生长、尺寸持续增大.
 图 4 不同反应时间条件下合成FeS微粒的SEM图Figure 4. SEM images of FeS under different react time (a) 4 h,(b) 8 h,(c) 24 h(a) 4 h,(b) 8 h,(c) 24 h (纯EN Pure EN,S/Fe =1,反应温度Reaction Temperature 200 ℃)
图 4 不同反应时间条件下合成FeS微粒的SEM图Figure 4. SEM images of FeS under different react time (a) 4 h,(b) 8 h,(c) 24 h(a) 4 h,(b) 8 h,(c) 24 h (纯EN Pure EN,S/Fe =1,反应温度Reaction Temperature 200 ℃)2.2.4 S/Fe物质的量比
图5展示了S/Fe物质的量比对FeS微粒表观形貌的影响. 随着S投加量的增大,FeS微粒形貌由片状逐步变为棒状. 当S/Fe=1时,FeS微粒的形貌呈现花状和片状;当S/Fe=2时,出现了多种形貌的FeS微粒,主要由少量的棒状和球状颗粒构成;当S/Fe=3和S/Fe=4时,FeS微粒形貌均为棒状;但是S/Fe=4时棒条更平整,粗细也更加均匀,直径大致在300—500 nm之间. 可以表明,采用水热法合成FeS微粒时S/Fe物质的量比对产物的形貌影响较大,增大S源的浓度有利于FeS由片状向棒状转变. 这与Zhang等[26]用水热法合成Bi2S3的研究发现保持一致,其在以水为溶剂、温度150 ℃时将L-半胱氨酸含量从0.15 g增大到0.35 g,固体颗粒形貌从纳米薄片转变为纳米棒. 同时,乙二胺是一种具有较强极性和螯合能力的表面活性剂,可以控制FeS不同晶面的生长速率[11,20],由图1可知,本研究合成的FeS晶体微粒具有沿着[001]方向优先生长的取向,使FeS定向生长为棒状[23].从而,通过控制S/Fe物质的量比,采用水热法可合成具有不同形貌的FeS微粒,同时在水热合成过程中提高反应物的投加量,可抑制产物的继续生长,有效的控制晶体形貌[19].
 图 5 不同S/Fe比合成FeS微粒的SEM图Figure 5. SEM images of FeS by (a) S/Fe =1,(b) S/Fe =2,(c) S/Fe =3,(d) S/Fe =4(a) S/Fe =1,(b) S/Fe =2,(c) S/Fe =3,(d) S/Fe =4
图 5 不同S/Fe比合成FeS微粒的SEM图Figure 5. SEM images of FeS by (a) S/Fe =1,(b) S/Fe =2,(c) S/Fe =3,(d) S/Fe =4(a) S/Fe =1,(b) S/Fe =2,(c) S/Fe =3,(d) S/Fe =42.3 人工合成FeS微粒对水溶液中Cr(Ⅵ)的去除性能
为了研究FeS形貌对Cr(Ⅵ)的去除性能的影响,分别选取S/Fe=1、S/Fe=3代表片状和棒状两种典型形貌的FeS微粒进行水溶液中Cr(Ⅵ)去除试验. 图6展示了水溶液中Cr(Ⅵ)的去除动力学,反应180 h后,S/Fe=3条件下合成的FeS微粒对Cr(Ⅵ)去除率可达到99.9%,Cr(Ⅵ)的浓度接近0 mg L-1;S/Fe=1条件下合成的FeS微粒对Cr(Ⅵ)去除率为79.4%,Cr(Ⅵ)的浓度为35 mg·L−1. 因此,S/Fe=1和S/Fe=3时合成的FeS微粒对Cr(Ⅵ)的去除率存在显著性差异.
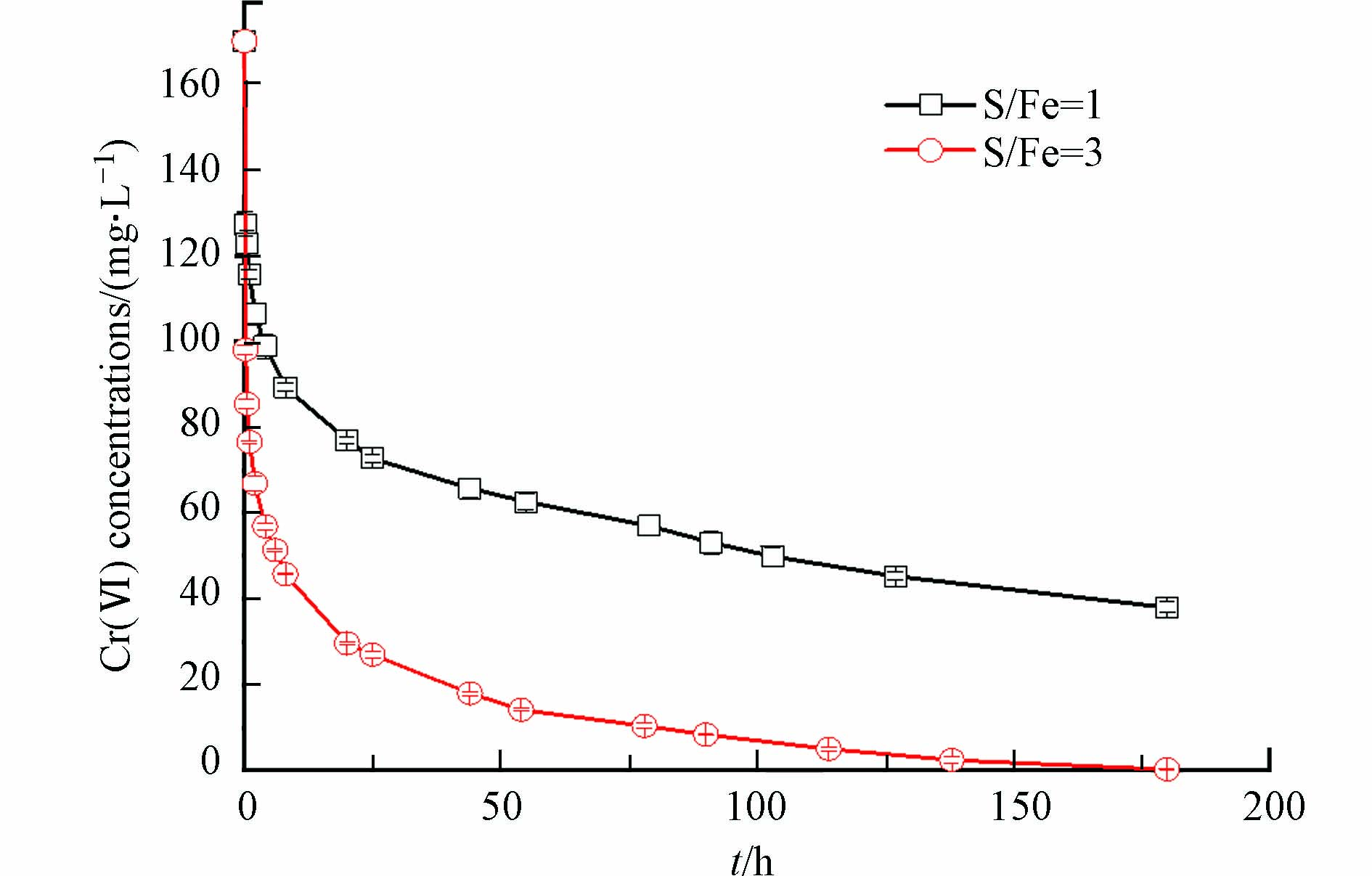 图 6 水热合成FeS微粒去除水溶液中Cr(Ⅵ)的性能Figure 6. The removal performance of Cr(Ⅵ) in aqueous solution by hydrothermal synthesized FeS particles
图 6 水热合成FeS微粒去除水溶液中Cr(Ⅵ)的性能Figure 6. The removal performance of Cr(Ⅵ) in aqueous solution by hydrothermal synthesized FeS particles由表1可知,S/Fe=1和S/Fe=3条件下合成的FeS对Cr(Ⅵ)的去除过程较好地符合拟二级反应动力学,反应速率常数分别是0.0002 L·(mg·h)−1、0.0012 L·(mg·h)−1. 因此,当溶液pH=5.5时,S/Fe=3条件下Cr(Ⅵ)的去除率和去除速率常数均大于S/Fe=1条件.
表 1 人工合成FeS对水溶液中Cr(Ⅵ)的去除性能Table 1. The removal performances of Cr(Ⅵ) in aqueous solution by hydrothermal synthesized FeS particles(pH = 5.5, Cr(Ⅵ)0 = 170 mg·L−1 , FeS投加量 = 0.5 g·L−1, 200 r·min−1)实验条件Experimental conditions 拟合级数Fitting series 动力学方程Kinetic equation 速率常数kRate constant k R2 Cr(Ⅵ)去除率/%Cr(Ⅵ) removal efficiency 零级 C−C0=−kt 3.022 mg·(L· h)−1 0.5253 S/Fe=1 一级 ln(C/C0)=−kt 0.0286 h−1 0.6589 79.4 二级 1/C−1/C0=kt 0.0002 L(mg· h)-1· 0.7508 零级 C−C0=−kt 13.436 mg·(L· h)−1 0.8007 S/Fe=3 一级 ln(C/C0)=−kt 0.1765 h−1 0.9358 99.9 二级 1/C−1/C0=kt 0.0012 L(mg· h)-1· 0.9459 | Show Table DownLoad:
CSV
DownLoad:
CSV
随着S/Fe物质的量比从1增大至3,合成的FeS与Cr(Ⅵ)的反应速率增大,主要是由于L-半胱氨酸提供S源的同时也提高了反应物的浓度,从而提高了FeS微粒对Cr(Ⅵ)的去除速率[16]. 同时,由于棒状FeS微粒具有更大的比表面积,可提供更大的接触面积和更多的活性位点[9],从而可使反应快速进行.
3. 结论(Conclusion)
(1)利用水热法合成FeS微粒分别具有棒状和片状等两种典型形貌.
(2)水/乙二胺体积比、反应温度、反应时间对水热合成FeS微粒形貌的影响不显著,均为片状;S/Fe物质的量比对FeS矿物形貌具有显著影响,随着S/Fe物质的量比的增大,FeS的颗粒从片状转变为棒状. 当S/Fe =3和S/Fe =4时,FeS颗粒均呈棒状,粒径可达200—300 nm.
(3)具有棒状的水热合成FeS微粒对水溶液中Cr(Ⅵ)去除率更高. 棒状FeS微粒在180 h内可去除水溶液中99.9%的Cr(Ⅵ),而片状FeS微粒对Cr(Ⅵ)的去除率仅为79.4%.
-
图 1 FeS微粒的(a) SEM图像、(b) EDS图和(c) XRD谱(纯EN Pure EN,反应温度 Reaction temperature 200 ℃,反应时间Reaction time 4 h)
Figure 1. XRD patterns and SEM-EDS images of FeS by (a) SEM analysis,(b) EDS analysis,(c) XRD analysis
图 2 不同溶剂体积比条件下合成FeS微粒的SEM图
Figure 2. SEM images of the FeS synthesized by (a) water/EN =1:1,(b) water/EN =1:2,(c) water/EN =1:3,(d) pure EN
图 3 不同反应温度条件下合成FeS微粒的SEM图
Figure 3. SEM images of FeS under (a) 180 ℃,(b) 190 ℃,(c) 200 ℃
图 4 不同反应时间条件下合成FeS微粒的SEM图
Figure 4. SEM images of FeS under different react time (a) 4 h,(b) 8 h,(c) 24 h
图 5 不同S/Fe比合成FeS微粒的SEM图
Figure 5. SEM images of FeS by (a) S/Fe =1,(b) S/Fe =2,(c) S/Fe =3,(d) S/Fe =4
图 6 水热合成FeS微粒去除水溶液中Cr(Ⅵ)的性能
Figure 6. The removal performance of Cr(Ⅵ) in aqueous solution by hydrothermal synthesized FeS particles
表 1 人工合成FeS对水溶液中Cr(Ⅵ)的去除性能
Table 1. The removal performances of Cr(Ⅵ) in aqueous solution by hydrothermal synthesized FeS particles(pH = 5.5, Cr(Ⅵ)0 = 170 mg·L−1 , FeS投加量 = 0.5 g·L−1, 200 r·min−1)
实验条件Experimental conditions 拟合级数Fitting series 动力学方程Kinetic equation 速率常数kRate constant k R2 Cr(Ⅵ)去除率/%Cr(Ⅵ) removal efficiency 零级 C−C0=−kt 3.022 mg·(L· h)−1 0.5253 S/Fe=1 一级 ln(C/C0)=−kt 0.0286 h−1 0.6589 79.4 二级 1/C−1/C0=kt 0.0002 L(mg· h)-1· 0.7508 零级 C−C0=−kt 13.436 mg·(L· h)−1 0.8007 S/Fe=3 一级 ln(C/C0)=−kt 0.1765 h−1 0.9358 99.9 二级 1/C−1/C0=kt 0.0012 L(mg· h)-1· 0.9459
下载: 导出CSV
-
[1] YANG Y, ZHANG Y H, WANG G Y, et al. Adsorption and reduction of Cr(Ⅵ) by a novel nanoscale FeS/chitosan/biochar composite from aqueous solution [J]. Journal of Environmental Chemical Engineering, 2021, 9(4): 105407. doi: 10.1016/j.jece.2021.105407 [2] WU J, WANG X B, ZENG R J. Reactivity enhancement of iron sulfide nanoparticles stabilized by sodium alginate: Taking Cr (Ⅵ) removal as an example [J]. Journal of Hazardous Materials, 2017, 333: 275-284. doi: 10.1016/j.jhazmat.2017.03.023 [3] GONG Y Y, GAI L S, TANG J C, et al. Reduction of Cr(Ⅵ) in simulated groundwater by FeS-coated iron magnetic nanoparticles [J]. Science of the Total Environment, 2017, 595: 743-751. doi: 10.1016/j.scitotenv.2017.03.282 [4] LYU H H, TANG J C, HUANG Y, et al. Removal of hexavalent chromium from aqueous solutions by a novel biochar supported nanoscale iron sulfide composite [J]. Chemical Engineering Journal, 2017, 322: 516-524. doi: 10.1016/j.cej.2017.04.058 [5] ZHANG H, PENG L, CHEN A W, et al. Chitosan-stabilized FeS magnetic composites for chromium removal: Characterization, performance, mechanism, and stability [J]. Carbohydrate Polymers, 2019, 214: 276-285. doi: 10.1016/j.carbpol.2019.03.056 [6] WANG T, LIU Y Y, WANG J J, et al. In-situ remediation of hexavalent chromium contaminated groundwater and saturated soil using stabilized iron sulfide nanoparticles [J]. Journal of Environmental Management, 2019, 231: 679-686. [7] LIU Y Y, XIAO W Y, WANG J J, et al. Optimized synthesis of FeS nanoparticles with a high Cr(Ⅵ) removal capability [J]. Journal of Nanomaterials, 2016, 2016: 7817296. [8] LI Y J, WANG W Y, ZHOU L Q, et al. Remediation of hexavalent chromium spiked soil by using synthesized iron sulfide particles [J]. Chemosphere, 2017, 169: 131-138. doi: 10.1016/j.chemosphere.2016.11.060 [9] WANG X B, LIU J, ZHAO D L, et al. Preparation of CMC-stabilized FeS nanoparticles and their enhanced performance for Cr(Ⅵ) removal[J]. Advanced Materials Research, 2011, 287/288/289/290: 96-99. [10] KIM E J, KIM J H, AZAD A M, et al. Facile synthesis and characterization of Fe/FeS nanoparticles for environmental applications [J]. ACS Applied Materials & Interfaces, 2011, 3(5): 1457-1462. [11] THOMAS M P, ULLAH A, PHAM R H, et al. Morphology control in the hydrothermal synthesis of FeS nanoplatelets [J]. Crystal Growth & Design, 2020, 20(9): 5728-5735. [12] 程千文. 高效还原六价铬的生物纳米杂合体系的构建及其还原机制研究[D]. 镇江: 江苏大学, 2019. CHENG Q W. Development of bio-nano hybrid system for efficient reduction of hexavalent chromium[D]. Zhenjiang: Jiangsu University, 2019(in Chinese).
[13] SINES I T, VAUGHN II D, MISRA R, et al. Synthesis of tetragonal mackinawite-type FeS nanosheets by solvothermal crystallization [J]. Journal of Solid State Chemistry, 2012, 196: 17-20. doi: 10.1016/j.jssc.2012.07.056 [14] XIONG Z, HE F, ZHAO D Y, et al. Immobilization of mercury in sediment using stabilized iron sulfide nanoparticles [J]. Water Research, 2009, 43(20): 5171-5179. doi: 10.1016/j.watres.2009.08.018 [15] ZHU Y J, LIU T, MA W, et al. Biomolecule-assisted hydrothermal synthesis and electrochemical properties of copper sulfide hollow spheres [J]. Chemistry Letters, 2015, 44(10): 1321-1323. doi: 10.1246/cl.150448 [16] GORAI S, GANGULI D, CHAUDHURI S. Shape selective solvothermal synthesis of copper sulphides: Role of ethylenediamine-water solvent system [J]. Materials Science and Engineering:B, 2005, 116(2): 221-225. doi: 10.1016/j.mseb.2004.10.003 [17] 李艳平, 方鑫, 王逸伦, 等. 水热法制备不同形貌的Bi2S3微纳米材料 [J]. 实验技术与管理, 2017, 34(9): 47-51,55. LI Y P, FANG X, WANG Y L, et al. Preparation of Bi2S3 micro/nano materials with various morphologies by hydrothermal method [J]. Experimental Technology and Management, 2017, 34(9): 47-51,55(in Chinese).
[18] WU X, LI K W, WANG H. Facile synthesis of ZnS nanostructured spheres and their photocatalytic properties [J]. Journal of Alloys and Compounds, 2009, 487(1/2): 537-544. [19] MIN Y L, CHEN Y C, ZHAO Y G. A small biomolecule-assisted synthesis of iron sulfide nanostructures and magnetic properties [J]. Solid State Sciences, 2009, 11(2): 451-455. doi: 10.1016/j.solidstatesciences.2008.07.005 [20] XIONG S L, XI B J, XU D C, et al. L-cysteine-assisted tunable synthesis of PbS of various morphologies [J]. Journal of Physical Chemistry C, 2007, 111(45): 16761-16767. doi: 10.1021/jp075096z [21] NATH M, CHOUDHURY A, KUNDU A, et al. Synthesis and characterization of magnetic iron sulfide nanowires [J]. Advanced Materials, 2003, 15(24): 2098-2101. doi: 10.1002/adma.200306042 [22] 荣华, 王春刚, 周明. 用作锂离子电池负极的FeS2微球的制备及性能 [J]. 高等学校化学学报, 2020, 41(3): 447-455. doi: 10.7503/cjcu20190545 RONG H, WANG C G, ZHOU M. Synthesis and electrochemical performance of FeS2 microspheres as an anode for Li-ion batteries [J]. Chemical Journal of Chinese Universities, 2020, 41(3): 447-455(in Chinese). doi: 10.7503/cjcu20190545
[23] KAR S, CHAUDHURI S. Solvothermal synthesis of nanocrystalline FeS2 with different morphologies [J]. Chemical Physics Letters, 2004, 398(1/2/3): 22-26. [24] DEMAZEAU G. Solvothermal reactions: An original route for the synthesis of novel materials [J]. Journal of Materials Science, 2008, 43(7): 2104-2114. doi: 10.1007/s10853-007-2024-9 [25] 蔡炜, 韩倩雯, 宫业辉, 等. 水热温度对纳米Cr2O3/CeO2复合氧化物低温催化氧化NO性能的影响 [J]. 环境化学, 2021, 40(10): 3226-3235. doi: 10.7524/j.issn.0254-6108.2020061304 CAI W, HAN Q W, GONG Y H, et al. Effect of different hydrothermal temperatures on catalytic performance for NO oxidation at low temperature over nanostructured Cr2O3/CeO2 [J]. Environmental Chemistry, 2021, 40(10): 3226-3235(in Chinese). doi: 10.7524/j.issn.0254-6108.2020061304
[26] ZHANG B, YE X C, HOU W Y, et al. Biomolecule-assisted synthesis and electrochemical hydrogen storage of Bi2S3 flowerlike patterns with well-aligned nanorods [J]. The Journal of Physical Chemistry B, 2006, 110(18): 8978-8985. doi: 10.1021/jp060769j 期刊类型引用(0)
其他类型引用(1)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 4015
- HTML全文浏览数: 4015
- PDF下载数: 121
- 施引文献: 1
