




-
药品和个人护理用品(PPCPs)作为一种新型有机微污染物,近年来受到了人们的广泛关注[1-3]. 其中,三氯生(triclosan,TCS)是一种多环芳烃类广谱抗菌剂,被广泛用于各类生活用品(如肥皂、清洁剂、牙膏、洗发水等)中[4]. 当前,随着TCS产品的普及,其在生产和使用过程中已不可避免地进入自然水体. 研究表明,全球范围地表水中的TCS浓度已高达ng·L−1至μg·L−1级别.TCS易通过生物富集和生物放大作用对摄入它的水生物种和哺乳动物造成内分泌干扰和毒性损伤,且能够诱导抗性基因的产生和传播,对人体健康和生态系统的潜在威胁不容忽视. 早在2016年,美国食品和药物管理局已开始禁止含TCS抗菌皂和沐浴露的销售,欧盟也于同年通过了禁用含TCS杀菌液的议案. 然而,我国对TCS的管控相对滞后[5].
传统的污水生物处理工艺对TCS的去除效率不佳,且在其处理过程中易产生危害性更大的亲脂性甲基衍生物[6]. 吸附技术处理水中TCS具有能耗低、反应过程不生成有毒物质的优点,但处理费用昂贵,并存在材料再生过程的二次污染隐患[7]. 相比之下,高级氧化技术因其高效、矿化率高、二次污染小等优势成为该领域研究的热点. 目前,国内对TCS废水的高级氧化处理已有较多报道,如臭氧氧化[8]、紫外/过氧化氢[9]、紫外/过硫酸盐[10]以及光催化氧化[11-12]等技术已被证实可有效削减水中的TCS.
过氧乙酸(peracetic acid,PAA)通常作为一种高效漂白、除菌剂被广泛用于食品加工、医疗制药、水产养殖等领域中,具有耐储存、pH依赖性低、氧化能力强、不产生有害消毒副产物、对人体危害小、成本低等特点[13]. 与过氧化氢相比,PAA是一种具有潜力的活性自由基前驱体,其氧化还原电位高达1.96 V[14],但过氧键能(38 kcal·mol−1)却远低于过氧化氢(51 kcal·mol−1)[15],理论上更易被活化.近两年,已有研究报道紫外光、过渡金属(Fe2+、Co2+、Mn2+)可在广泛pH条件下活化PAA,并获得丰度较高的活性物种(如羟基自由基HO·、有机自由基CH3C(=O)O·、CH3C(=O)OO·等)[16],将此类技术用于水中TCS去除的研究还未见报道.
为探讨PAA高级氧化体系去除水体中TCS的可行性,本研究采用价格低廉、来源广泛且二次污染较小的过渡金属离子Fe2+与PAA构建均相氧化体系(Fe2+/PAA),考察反应初始pH、PAA投加量、共存阴阳离子和天然有机物(NOM)对TCS去除效率的影响. 此外,通过自由基选择清除试验鉴别体系中的主要自由基物种,并利用超高效液相色谱-四极杆飞行时间质谱仪(UPLC-Q-TOF-MS)对反应过程中产生的中间产物进行分析,进而推断TCS在Fe2+/PAA体系中的转化途径.
-
试剂:TCS(HPLC级,纯度≥99%)、PAA溶液(15%纯度,H2O2/PAA物质的量比=1.4)、甲醇(纯度≥99.8%)、叔丁醇(纯度≥99.8%);腐殖酸、NaOH、NaHCO3、NaCl、FeSO4、CuSO4、FeCl3、H2SO4、Na2S2O3均为分析纯。
仪器:高精密电子分析天平(FA224),恒温水浴振荡器(HH-2),电热鼓风干燥箱(101-3AB),pH计(雷磁PHS-3C),液相色谱仪(HPLC,Ultimate 3000),UPLC-Q-TOF-MS型液质联用仪(ACQUITY UPLC,Quattro Premier XE,Waters),超纯水机(UPHW-H-90T)。
-
实验在置于恒温磁力搅拌水浴槽的烧杯(250 mL)中进行,实验温度控制为(25±2)oC. TCS标准储备液配制时,采用0.5 mol·L−1NaOH溶液作为助溶剂,以500 r·min−1的速率搅拌12 h使TCS充分溶解,H2SO4将溶液pH调至7.0后定容至1 L. PAA母液稀释50倍作为使用液,其有效浓度为0.039 mol·L−1. 实验时取一定量TCS标准储备液配制成浓度为1 μmol·L−1的使用液,取100 mL于水浴槽中的烧杯,其后根据设定的PAA投加量加入PAA使用液,采用浓度为1 mol·L−1的H2SO4和NaOH调节pH(作为初始pH),再投加FeSO4溶液后,反应即开始计时. 在指定的时间取出1 mL样品置于含有0.5 mLNa2S2O3溶液(浓度为0.1 mol·L−1)的液相小瓶中并迅速测试. 除产物分析实验,所有实验均至少重复3次,结果取平均值.
-
采用HPLC和UPLC-Q-TOF-MS对TCS及其降解产物进行分析[17]. HPLC条件:色谱柱为Waters C18柱(4.6 mm×150 mm,5 μm),流动相为0.1%冰乙酸/甲醇=23/77(V/V),流速为1.2 mL·min−1,检测波长为221 nm,柱温为30 ℃,进样体积为20 μL. UPLC-Q-TOF-MS条件:色谱柱为C18柱(2.1 mm×100 mm,1.7 μm),载气为高纯氦气,90 kPa;流动相为A(0.1%的甲酸)和B(乙腈)组成,采用梯度洗脱的方式,首先B为10%,持续0.2 min;然后0.2—4.5 min,B线性上升到95%,保持0.5 min;最后5.0—7.0 min,B下降至10%,流速为0.5 mL·min−1. 进样体积为5 μL. 采用电喷雾电离(ESI),在负离子模式下进行扫描,扫描范围在m/z 50—800. 溶液pH值利用pH计(雷磁PHS-3C)进行测定.
-
研究表明,Fe2+活化PAA可产生HO·和多种有机自由基(如CH3·、CH3O2·、CH3C(=O)O·和CH3C(=O)OO·等),其反应如式(1)—(6)所示[18]. 其中,HO·、CH3C(=O)O·和CH3C(=O)OO·具有强氧化性,已被证实能够有效去除双氯芬酸、磺胺甲恶唑、扑热息痛等PPCPs污染物[18-21]. CH3·易和O2反应快速产生CH3O2·,但CH3O2·氧化性极弱,难与有机物发生反应[18]. 鉴于此,为探讨Fe2+/PAA体系对TCS的降解效率及机理,本研究在相同条件下进行了4组TCS降解对照实验.
由图1可知,TCS在4个体系中的降解均遵循准一级反应动力学.其中,TCS可被PAA直接氧化去除,反应速率常数为0.04 min−1. 相比之下,Fe2+/PAA体系氧化性明显更强,仅5 min即可去除90.7%的TCS,反应速率常数高达0.46 min−1,该结果表明自由基氧化是Fe2+/PAA体系去除TCS的主要途径. 为进一步探讨Fe2+/PAA体系中自由基对TCS降解的贡献,本研究采用TBA和MeOH分别对体系中自由基进行清除.其中,TBA是一种良好的HO·淬灭剂(k=7.6×108 L·(mol·s)−1)[18];而MeOH不仅可以有效淬灭HO·,两者的二级反应速率常数为9.7×108 L·(mol·s)−1,且已被证实可以有效清除CH3C(=O)O·和CH3C(=O)OO·[22].
如图1所示,当TBA存在时,Fe2+/PAA对TCS的降解明显受到抑制,反应速率常数降至0.05 min−1;MeOH的加入使TCS的降解速率进一步减至0.04 min−1,与PAA单独氧化TCS的速率相近. 上述结果证明,HO·是Fe2+/PAA体系中的主要活性物种,但有机自由基(主要是CH3C(=O)O·和CH3C(=O)OO·)对TCS降解的贡献也不容忽视.
-
图2为Fe2+/PAA体系中初始pH值对TCS降解的影响. 在pH 3.5—11.0的范围内,TCS在20 min时的去除率随pH值的升高由94.9%降低至61.9%. 其原因可能是在酸性条件下,Fe2+与体系中的H2O2发生Fenton反应,该过程与PAA的活化协同进行,高效产生了HO·;溶液pH值上升至5.0后,Fenton反应受到抑制,HO·产量下降,但PAA活化过程仍可为体系提供足够的自由基;溶液pH进一步升至8.5时,因PAA的pKa值为8.2,此时部分PAA以去质子化形态存在,易发生自解离反应(式(7))[23],同时Fe2+因水解生成氢氧化物而失去活化能力,抑制了自由基的产生;最后,当溶液pH达到11.0时,PAA自解离作用增强,且消耗体系内H2O2(式(8)),使体系氧化性进一步下降[18].
-
如图3所示,PAA投加量由1增至10 mmol·L−1时,TCS降解速率常数由0.46 min−1提升至0.74 min−1. 其原因是随着PAA溶液投加量的提升,体系中自由基前驱体PAA和H2O2的浓度相应升高,能够被活化产生更多自由基. 虽然H2O2也是HO·捕获剂,两者的二级反应速率常数高达2.7×107 L·(mol·s)−1[24],但随着体系内自由基稳态浓度的大幅提高,它和PAA对HO·的清除作用并未明显影响TCS的降解.
鉴于PAA投加量在1至5 mmol·L−1范围内,TCS均可在20 min内得到高效去除,综合考虑药剂成本和二次污染隐患,本研究选取1 mmol·L−1作为PAA最佳投加量.
-
水体中天然有机物是影响高级氧化效率的重要因素. 研究表明,它们的影响机制包括吸附、增强电子传递和自由基捕集等[25]. 在NOM中,腐殖质含量约占50%,而其中溶解态的腐殖酸(HA)组分广泛存在于自然水体中[26]. 鉴于此,本研究采用HA作为典型NOM,探讨其对Fe2+/PAA体系降解TCS的影响. 如图4可知,HA的存在会抑制TCS的降解,且随其浓度的增加,抑制作用越明显.当HA浓度达到10 mg·L−1时,TCS在20 min时的去除率降至81%. 研究表明,NOM与HO·的二级反应速率常数为2.23×108 L·(mol C·s)−1 [27]. 因此,体系中的NOM可能与TCS共同竞争HO·,从而降低了TCS的去除效率.
-
HCO3−和Cl−是广泛存在于自然水体中的阴离子. 由于它们可与HO·发生反应,在高级氧化体系中其可能会对目标污染物的降解产生影响[24,28-29]. 因此,探讨它们对Fe2+/PAA体系的影响对于该技术在处理实际水体中TCS的应用具有重要意义. HCO3−是一种常见的HO·淬灭剂,其与HO·反应(k=8.5×106 L·(mol·s)−1)生成碳酸根自由基(CO3•−)(氧化还原电位为1.59 V)(见式(9))[24]. 此外,HCO3−的水解反应导致溶液中OH−增多,当其浓度为20 mmol·L−1时,溶液初始pH值升至7.8. 由2.2节所述,该初始pH值不利于TCS的降解. 然而,本研究并未发现HCO3−对Fe2+/PAA体系有明显抑制作用,随其浓度由0增至20 mmol·L−1,TCS在20 min时的去除率仅下降了8%,如图5(a)所示.
其原因是体系产生的CO3•−也可进攻TCS中富含电子的酚类结构,虽然已有研究表明该反应的二级速率常数仅为4.2×107 L·(mol·s)−1[30],远低于TCS与HO·的反应速率常(k=5.4×109 L·(mol·s)−1)[31],但由于体系内CO3•−稳态浓度较高,仍能满足TCS的有效去除. 图5(b)为Cl−对TCS降解的影响.Cl−同样可与HO·发生淬灭反应,其二级反应速率常数为(4.3 ± 0.4)×109 L·(mol·s)−1[32],然而,TCS的去除并未受到Cl−的明显干扰. 其原因可能是酸性氛围中Cl−与HO·反应生成的ClOH•−又进一步转化为Cl·和Cl2•−等氯衍生物种(见式(10)—(12)). Cl·和Cl2•−的氧化还原电位分别高达2.4 V和2.0 V,它们可能也参与了TCS的氧化[29].
-
Fe3+和Cu2+是类Fenton反应的常用催化剂[33,34],它们的存在可能对Fe2+/PAA体系降解TCS造成影响. 通常,Fe3+在pH 2.5—4.0的范围内可催化H2O2产生Fe2+和HO2·,额外产生的Fe2+可以同时活化PAA和H2O2产生更多自由基,故理论上Fe3+应该促进TCS的降解. 然而,如图6(a)所示,体系内Fe3+反而轻微抑制了TCS的降解,当Fe3+浓度由0增至10 mg·L−1时,TCS在20 min时的去除率降低9%. 其原因可能是TCS首先与Fe3+发生了络合反应[35],而该产物对自由基反应的敏感性较低. 如图6(b)所示,Cu2+对TCS的降解几乎没有影响,表明在pH值为3.5的条件下,Cu2+可能无法有效催化PAA和H2O2产生自由基,该结果与Peng等研究Cu2+/H2O2体系降解TCS得出的结论类似[36].
-
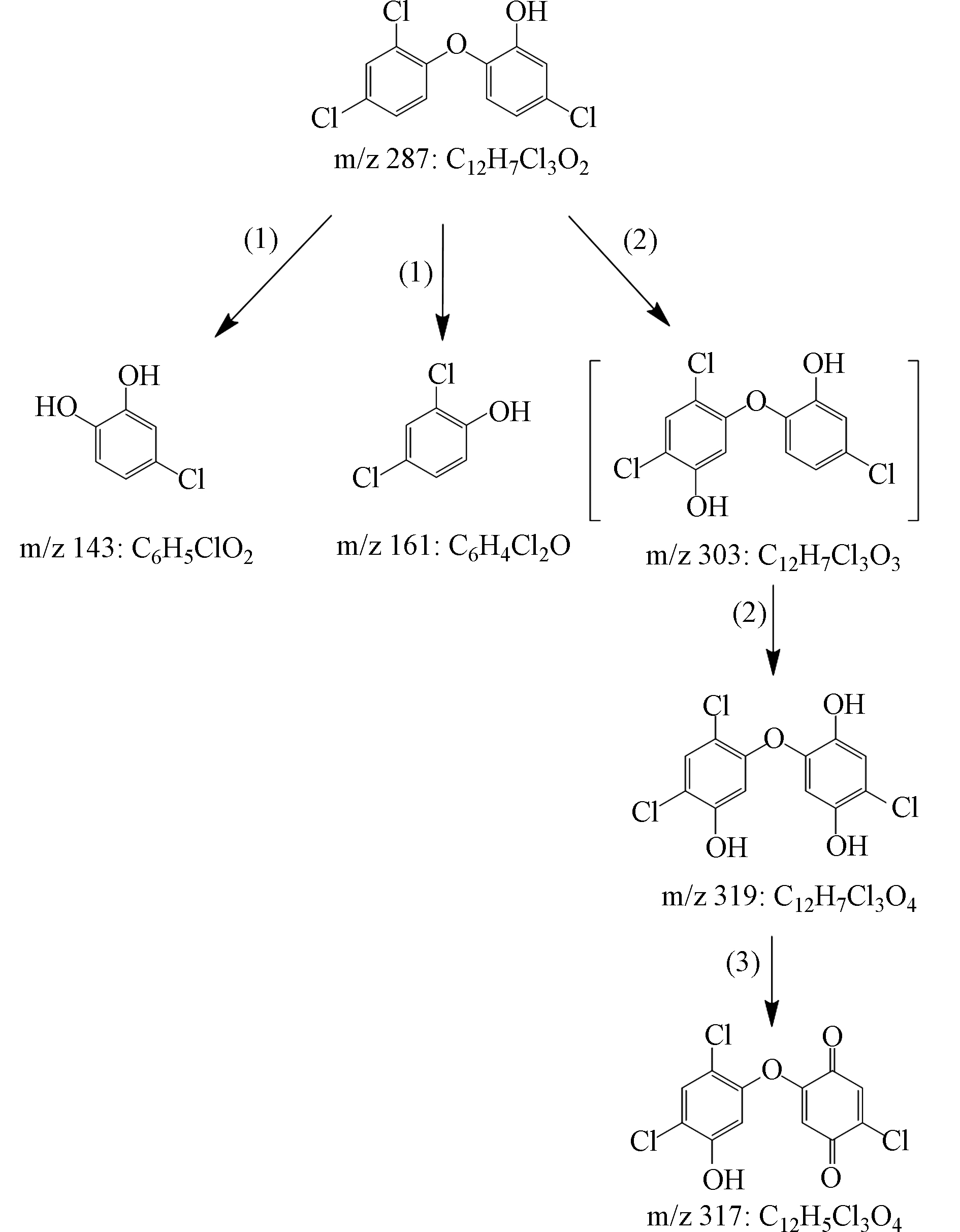
基于TCS降解过程中检出的4种主要产物(表1),提出了TCS在Fe2+/PAA体系中的降解途径,分别为:(1)醚键断裂、(2)羟基化和(3)脱氢,如图7所示. 醚键断裂和羟基化是高级氧化技术降解TCS的两种常见途径,已被大量研究证实[37-38]. 其中,TCS醚键断裂主要是由于HO·对醚键的进攻,导致m/z 143和m/z 161的产生;而羟基化主要通过HO·加成至TCS不饱和键实现[38],生成m/z 303. 虽然本文未检测到该物质,但它的后续羟基化产物m/z 319被检出,因而不能忽视其存在的可能. 最后,m/z 319通过脱氢反应生成m/z 317.
-
(1)Fe2+/PAA体系能够高效去除水中TCS,在TCS浓度为1 μmol·L−1,初始pH值为3.5、Fe2+投加量为10 μmol·L−1、PAA投加量为1 mmol·L−1、反应温度为25 oC、反应时间为20 min的条件下,TCS去除率高达94.9%.
(2)HO·和有机活性物种(主要为CH3C(=O)O·和CH3C(=O)OO·)均参与了TCS的降解,但以HO·对TCS的氧化降解为主.
(3)水中NOM因自由基竞争明显抑制了TCS在Fe2+/PAA体系中的降解;而共存阴阳离子对TCS的降解影响较小.
(4)TCS在Fe2+/PAA体系中的转化途径包含醚键断裂、羟基化和脱氢.
亚铁联合过氧乙酸降解水中的三氯生
Degradation of triclosan by Fe2+-peracetic acid system in water
-
摘要: 研究了Fe2+/过氧乙酸(Fe2+/PAA)高级氧化体系对水中三氯生(TCS)的降解.考察了初始pH、PAA投加量、常见阴阳离子(HCO3−,Cl-,Fe3+,Cu2+)和天然有机物(NOM)对TCS降解的影响,探讨了TCS在该体系中的降解机理和转化途径. 结果表明,TCS在Fe2+/PAA体系中的降解符合准一级反应动力学,其反应速率常数为0.46 min−1. 在TCS浓度为1 μmol·L−1,初始pH值为3.5、Fe2+投加量为10 μmol·L−1、PAA投加量为1 mmol·L−1、反应温度为25 oC、反应时间为20 min的条件下,TCS去除率高达95%.水中的NOM通过自由基竞争抑制TCS的降解,而共存阴阳离子的影响较小. HO•氧化是TCS降解的主要途径,但有机自由基对其的氧化作用亦不容忽视. 基于TCS降解过程中检出的4种产物,提出了它的转化途径,包含醚键断裂、羟基化和脱氢.Abstract: The degradation of triclosan(TCS)by activated peracetic acid(PAA)using Fe2+ was investigated in this study. The effects of operational parameters such as initial pH, PAA dosage and common water matrix on the degradation of TCS were investigated. Finally, the degradation mechanism and transformation pathway of TCS were studied. Results indicated that the TCS degradation by Fe2+/PAA followed the pseudo first-order kinetic model(k = 0.46 min−1). 95% of TCS was removed in 20 min under the conditions of TCS initial concentration of 1 μmol L−1, inital pH of 3.5, Fe2+ dosage of 10 μmol L−1, PAA dosage of 1 mmol L−1, temperature of 25 oC and reaction time of 20 min. The presence of NOM inhibited the TCS degradation through radical competition, while the co-existing inorganic anions and metal cations had no obvious influence on the removal of TCS. The oxidation of TCS by HO• was the major pathway during its degradation, while the organic radicals might also be the important active species. According to four identified intermediates, the transformation pathway of TCS by Fe2+/PAA was proposed including ether bond cleavage, hydroxylation and dehydrogenation.
-
Key words:
- ferrous ion /
- peracetic acid /
- advanced oxidation /
- triclosan
-

图 4 NOM对TCS去除效果的影响实验条件:[PAA]0 = 1 mmol·L-1,[Fe2+]0 = 10 μmol·L-1,[TCS]0 = 1 μmol·L-1,初始 pH = 3.5,T = 25 oC
Figure 4. Effect of NOM on TCS removal

图 5 阴离子HCO3-(a),Cl-(b)对TCS去除效果的影响
Figure 5. Effect of HCO3-(a)and Cl-(b)on TCS removal

图 6 阳离子Fe3+(a),Cu2+(b)对TCS去除效果的影响
Figure 6. Effect of Fe3+(a)and Cu2+(b)on TCS removal
表 1 TCS在Fe2+/PAA体系中的降解产物
Table 1. Degradation products of TCS in Fe2+/PAA system
序号Serial number 质荷比(m/z)Mass to charge ratio 分子式Formula 结构式Structure TCS 287 C12H7Cl3O2 Mass to charge ratio</td><td class="table_top_border" align="center" valign="middle">分子式Formula</td><td class="table_top_border" align="center" valign="middle">结构式Structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">TCS</td><td class="table_top_border2" align="center" valign="middle">287</td><td class="table_top_border2" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>2</sub></td><td class="table_top_border2" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.87cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">序号Serial number</td><td class="table_top_border" align="center" valign="middle">质荷比(m/z)Mass to charge ratio</td><td class="table_top_border" align="center" valign="middle">分子式Formula</td><td class="table_top_border" align="center" valign="middle">结构式Structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">TCS</td><td class="table_top_border2" align="center" valign="middle">287</td><td class="table_top_border2" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>2</sub></td><td class="table_top_border2" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.87cm"><img class="graphic" src="2021051402-b1-1.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">1</td><td align="center" valign="middle">143</td><td align="center" valign="middle">C<sub>6</sub>H<sub>5</sub>ClO<sub>2</sub></td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-2.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">2</td><td align="center" valign="middle">161</td><td align="center" valign="middle">C<sub>6</sub>H<sub>4</sub>Cl<sub>2</sub>O</td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-3.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">3</td><td align="center" valign="middle">317</td><td align="center" valign="middle">C<sub>12</sub>H<sub>5</sub>Cl<sub>3</sub>O<sub>4</sub></td><td align="center" valign="middle"><styled-content style="width:2.67cm"><img class="graphic" src="2021051402-b1-4.jpg"></styled-content></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">4</td><td class="table_bottom_border" align="center" valign="middle">319</td><td class="table_bottom_border" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>4</sub></td><td class="table_bottom_border" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.8cm"><img class="graphic" src="2021051402-b1-5.jpg"></styled-content></td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td></tr><tr><td align="center" valign="middle">1</td><td align="center" valign="middle">143</td><td align="center" valign="middle">C<sub>6</sub>H<sub>5</sub>ClO<sub>2</sub></td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">序号Serial number</td><td class="table_top_border" align="center" valign="middle">质荷比(m/z)Mass to charge ratio</td><td class="table_top_border" align="center" valign="middle">分子式Formula</td><td class="table_top_border" align="center" valign="middle">结构式Structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">TCS</td><td class="table_top_border2" align="center" valign="middle">287</td><td class="table_top_border2" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>2</sub></td><td class="table_top_border2" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.87cm"><img class="graphic" src="2021051402-b1-1.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">1</td><td align="center" valign="middle">143</td><td align="center" valign="middle">C<sub>6</sub>H<sub>5</sub>ClO<sub>2</sub></td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-2.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">2</td><td align="center" valign="middle">161</td><td align="center" valign="middle">C<sub>6</sub>H<sub>4</sub>Cl<sub>2</sub>O</td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-3.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">3</td><td align="center" valign="middle">317</td><td align="center" valign="middle">C<sub>12</sub>H<sub>5</sub>Cl<sub>3</sub>O<sub>4</sub></td><td align="center" valign="middle"><styled-content style="width:2.67cm"><img class="graphic" src="2021051402-b1-4.jpg"></styled-content></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">4</td><td class="table_bottom_border" align="center" valign="middle">319</td><td class="table_bottom_border" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>4</sub></td><td class="table_bottom_border" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.8cm"><img class="graphic" src="2021051402-b1-5.jpg"></styled-content></td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td></tr><tr><td align="center" valign="middle">2</td><td align="center" valign="middle">161</td><td align="center" valign="middle">C<sub>6</sub>H<sub>4</sub>Cl<sub>2</sub>O</td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">序号Serial number</td><td class="table_top_border" align="center" valign="middle">质荷比(m/z)Mass to charge ratio</td><td class="table_top_border" align="center" valign="middle">分子式Formula</td><td class="table_top_border" align="center" valign="middle">结构式Structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">TCS</td><td class="table_top_border2" align="center" valign="middle">287</td><td class="table_top_border2" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>2</sub></td><td class="table_top_border2" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.87cm"><img class="graphic" src="2021051402-b1-1.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">1</td><td align="center" valign="middle">143</td><td align="center" valign="middle">C<sub>6</sub>H<sub>5</sub>ClO<sub>2</sub></td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-2.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">2</td><td align="center" valign="middle">161</td><td align="center" valign="middle">C<sub>6</sub>H<sub>4</sub>Cl<sub>2</sub>O</td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-3.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">3</td><td align="center" valign="middle">317</td><td align="center" valign="middle">C<sub>12</sub>H<sub>5</sub>Cl<sub>3</sub>O<sub>4</sub></td><td align="center" valign="middle"><styled-content style="width:2.67cm"><img class="graphic" src="2021051402-b1-4.jpg"></styled-content></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">4</td><td class="table_bottom_border" align="center" valign="middle">319</td><td class="table_bottom_border" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>4</sub></td><td class="table_bottom_border" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.8cm"><img class="graphic" src="2021051402-b1-5.jpg"></styled-content></td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td></tr><tr><td align="center" valign="middle">3</td><td align="center" valign="middle">317</td><td align="center" valign="middle">C<sub>12</sub>H<sub>5</sub>Cl<sub>3</sub>O<sub>4</sub></td><td align="center" valign="middle"><styled-content style="width:2.67cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">序号Serial number</td><td class="table_top_border" align="center" valign="middle">质荷比(m/z)Mass to charge ratio</td><td class="table_top_border" align="center" valign="middle">分子式Formula</td><td class="table_top_border" align="center" valign="middle">结构式Structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">TCS</td><td class="table_top_border2" align="center" valign="middle">287</td><td class="table_top_border2" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>2</sub></td><td class="table_top_border2" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.87cm"><img class="graphic" src="2021051402-b1-1.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">1</td><td align="center" valign="middle">143</td><td align="center" valign="middle">C<sub>6</sub>H<sub>5</sub>ClO<sub>2</sub></td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-2.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">2</td><td align="center" valign="middle">161</td><td align="center" valign="middle">C<sub>6</sub>H<sub>4</sub>Cl<sub>2</sub>O</td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-3.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">3</td><td align="center" valign="middle">317</td><td align="center" valign="middle">C<sub>12</sub>H<sub>5</sub>Cl<sub>3</sub>O<sub>4</sub></td><td align="center" valign="middle"><styled-content style="width:2.67cm"><img class="graphic" src="2021051402-b1-4.jpg"></styled-content></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">4</td><td class="table_bottom_border" align="center" valign="middle">319</td><td class="table_bottom_border" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>4</sub></td><td class="table_bottom_border" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.8cm"><img class="graphic" src="2021051402-b1-5.jpg"></styled-content></td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">4</td><td class="table_bottom_border" align="center" valign="middle">319</td><td class="table_bottom_border" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>4</sub></td><td class="table_bottom_border" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.8cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">序号Serial number</td><td class="table_top_border" align="center" valign="middle">质荷比(m/z)Mass to charge ratio</td><td class="table_top_border" align="center" valign="middle">分子式Formula</td><td class="table_top_border" align="center" valign="middle">结构式Structure</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">TCS</td><td class="table_top_border2" align="center" valign="middle">287</td><td class="table_top_border2" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>2</sub></td><td class="table_top_border2" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.87cm"><img class="graphic" src="2021051402-b1-1.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">1</td><td align="center" valign="middle">143</td><td align="center" valign="middle">C<sub>6</sub>H<sub>5</sub>ClO<sub>2</sub></td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-2.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">2</td><td align="center" valign="middle">161</td><td align="center" valign="middle">C<sub>6</sub>H<sub>4</sub>Cl<sub>2</sub>O</td><td align="center" valign="middle"><styled-content style="width:1.82cm"><img class="graphic" src="2021051402-b1-3.jpg"></styled-content></td></tr><tr><td align="center" valign="middle">3</td><td align="center" valign="middle">317</td><td align="center" valign="middle">C<sub>12</sub>H<sub>5</sub>Cl<sub>3</sub>O<sub>4</sub></td><td align="center" valign="middle"><styled-content style="width:2.67cm"><img class="graphic" src="2021051402-b1-4.jpg"></styled-content></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">4</td><td class="table_bottom_border" align="center" valign="middle">319</td><td class="table_bottom_border" align="center" valign="middle">C<sub>12</sub>H<sub>7</sub>Cl<sub>3</sub>O<sub>4</sub></td><td class="table_bottom_border" style="padding-bottom:2pt;" align="center" valign="middle"><styled-content style="width:2.8cm"><img class="graphic" src="2021051402-b1-5.jpg"></styled-content></td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td></tr></tbody>
</table></div></foreignObject></svg>)
1 143 C6H5ClO2 2 161 C6H4Cl2O 3 317 C12H5Cl3O4 4 319 C12H7Cl3O4  下载: 导出CSV
下载: 导出CSV
-
[1] 余绵梓, 袁啸, 李适宇, 等. 典型PPCPs在河流沉积物中的吸附特性 [J]. 中国环境科学, 2019, 39(4): 1724-1733. doi: 10.3969/j.issn.1000-6923.2019.04.046 YU M Z, YUAN X, LI S Y, et al. Experimental and modeling study of sorption characteristics of selected PPCPs onto river sediments [J]. China Environmental Science, 2019, 39(4): 1724-1733(in Chinese). doi: 10.3969/j.issn.1000-6923.2019.04.046
[2] 王建龙. 废水中药品及个人护理用品(PPCPs)的去除技术研究进展 [J]. 四川师范大学学报(自然科学版), 2020, 43(2): 143-172,140. WANG J L. Removal of pharmaceuticals and personal care products(PPCPs) from wastewater: A review [J]. Journal of Sichuan Normal University (Natural Science), 2020, 43(2): 143-172,140(in Chinese).
[3] WANG J L, WANG S Z. Removal of pharmaceuticals and personal care products (PPCPs) from wastewater: A review [J]. Journal of Environmental Management, 2016, 182: 620-640. doi: 10.1016/j.jenvman.2016.07.049 [4] DANN A B, HONTELA A. Triclosan: environmental exposure, toxicity and mechanisms of action [J]. Journal of Applied Toxicology, 2011, 31(4): 285-311. doi: 10.1002/jat.1660 [5] LU Y C, MAO J H, ZHANG W, et al. A novel strategy for selective removal and rapid collection of triclosan from aquatic environment using magnetic molecularly imprinted nano−polymers [J]. Chemosphere, 2020, 238: 124640. doi: 10.1016/j.chemosphere.2019.124640 [6] YING G G, YU X Y, KOOKANA R S. Biological degradation of triclocarban and triclosan in a soil under aerobic and anaerobic conditions and comparison with environmental fate modelling [J]. Environmental Pollution, 2007, 150(3): 300-305. doi: 10.1016/j.envpol.2007.02.013 [7] 戴步峰, 燕强, 贺超, 等. 抗生素去除技术在饮用水处理领域的研究进展 [J]. 应用化工, 2020, 49(8): 2091-2095,2100. doi: 10.3969/j.issn.1671-3206.2020.08.048 DAI B F, YAN Q, HE C, et al. Advances in antibiotic removal techniques in the field of drinking water treatment [J]. Applied Chemical Industry, 2020, 49(8): 2091-2095,2100(in Chinese). doi: 10.3969/j.issn.1671-3206.2020.08.048
[8] SUAREZ S, DODD M C, OMIL F, et al. Kinetics of triclosan oxidation by aqueous ozone and consequent loss of antibacterial activity: Relevance to municipal wastewater ozonation [J]. Water Research, 2007, 41(12): 2481-2490. doi: 10.1016/j.watres.2007.02.049 [9] 罗从伟, 马军, 江进, 等. UV/H2O2降解三氯生动力学及反应机理 [J]. 哈尔滨工业大学学报, 2017, 49(2): 26-31. doi: 10.11918/j.issn.0367-6234.2017.02.005 LUO C W, MA J, JIANG J, et al. Degradation of triclosan by UV/H2O2: Kinetics and reaction mechanism [J]. Journal of Harbin Institute of Technology, 2017, 49(2): 26-31(in Chinese). doi: 10.11918/j.issn.0367-6234.2017.02.005
[10] GAO J, LUO C W, GAN L, et al. A comparative study of UV/H2O2 and UV/PDS for the degradation of micro-pollutants: Kinetics and effect of water matrix [J]. Environmental Science and Pollution Research, 2020, 27(19): 24531-24541. doi: 10.1007/s11356-020-08794-1 [11] AZARPIRA H, SADANI M, ABTAHI M, et al. Photo-catalytic degradation of triclosan with UV/iodide/ZnO process: Performance, kinetic, degradation pathway, energy consumption and toxicology [J]. Journal of Photochemistry and Photobiology A:Chemistry, 2019, 371: 423-432. doi: 10.1016/j.jphotochem.2018.10.041 [12] ABA-GUEVARA C G, MEDINA-RAMÍREZ I E, HERNÁNDEZ-RAMÍREZ A, et al. Comparison of two synthesis methods on the preparation of Fe, N-Co-doped TiO2 materials for degradation of pharmaceutical compounds under visible light [J]. Ceramics International, 2017, 43(6): 5068-5079. doi: 10.1016/j.ceramint.2017.01.018 [13] CHHETRI R K, THORNBERG D, BERNER J, et al. Chemical disinfection of combined sewer overflow waters using performic acid or peracetic acids [J]. Science of the Total Environment, 2014, 490: 1065-1072. doi: 10.1016/j.scitotenv.2014.05.079 [14] CAI M Q, SUN P Z, ZHANG L Q, et al. UV/peracetic acid for degradation of pharmaceuticals and reactive species evaluation [J]. Environmental Science & Technology, 2017, 51(24): 14217-14224. [15] BIANCHINI R, CALUCCI L, CARETTI C, et al. An EPR study on wastewater disinfection by peracetic acid, hydrogen peroxide and UV irradiation [J]. Annali Di Chimica, 2002, 92(9): 783-793. [16] 田丹, 吴玮, 沈芷璇, 等. Co(Ⅱ)活化过氧乙酸降解有机染料研究 [J]. 环境科学学报, 2018, 38(10): 4023-4031. TIAN D, WU W, SHEN Z X, et al. Degradation of organic dyes with peracetic acid activated by Co(Ⅱ) [J]. Acta Scientiae Circumstantiae, 2018, 38(10): 4023-4031(in Chinese).
[17] CHEN J, QU R J, PAN X X, et al. Oxidative degradation of triclosan by potassium permanganate: Kinetics, degradation products, reaction mechanism, and toxicity evaluation [J]. Water Research, 2016, 103: 215-223. doi: 10.1016/j.watres.2016.07.041 [18] WANG S X, WANG H B, LIU Y Q, et al. Effective degradation of sulfamethoxazole with Fe2+-zeolite/peracetic acid [J]. Separation and Purification Technology, 2020, 233: 115973. doi: 10.1016/j.seppur.2019.115973 [19] WANG Z P, WANG J W, XIONG B, et al. Application of cobalt/peracetic acid to degrade sulfamethoxazole at neutral condition: Efficiency and mechanisms[J]. Environmental Science & Technology, 2019: acs. est. 9b04528. [20] KIM J, ZHANG T Q, LIU W, et al. Advanced oxidation process with peracetic acid and Fe(II) for contaminant degradation [J]. Environmental Science & Technology, 2019, 53(22): 13312-13322. [21] GHANBARI F, GIANNAKIS S, LIN K Y A, et al. Acetaminophen degradation by a synergistic peracetic acid/UVC-LED/Fe(II) advanced oxidation process: Kinetic assessment, process feasibility and mechanistic considerations [J]. Chemosphere, 2021, 263: 128119. doi: 10.1016/j.chemosphere.2020.128119 [22] ZHANG L, LIU Y Q, FU Y S. Degradation kinetics and mechanism of diclofenac by UV/peracetic acid [J]. RSC Advances, 2020, 10(17): 9907-9916. doi: 10.1039/D0RA00363H [23] YUAN Z, NI Y, van HEININGEN A R P. Kinetics of peracetic acid decomposition: Part I: Spontaneous decomposition at typical pulp bleaching conditions [J]. The Canadian Journal of Chemical Engineering, 1997, 75(1): 37-41. doi: 10.1002/cjce.5450750108 [24] SHAH N S, HE X X, KHAN H M, et al. Efficient removal of endosulfan from aqueous solution by UV-C/peroxides: A comparative study [J]. Journal of Hazardous Materials, 2013, 263: 584-592. doi: 10.1016/j.jhazmat.2013.10.019 [25] REZAEI R, MOHSENI M. Impact of natural organic matter on the degradation of 2, 4-dichlorophenoxy acetic acid in a fluidized bed photocatalytic reactor [J]. Chemical Engineering Journal, 2017, 310: 457-463. doi: 10.1016/j.cej.2016.05.086 [26] KANG Y M, KIM M K, ZOH K D. Effect of nitrate, carbonate/bicarbonate, humic acid, and H2O2 on the kinetics and degradation mechanism of Bisphenol-A during UV photolysis [J]. Chemosphere, 2018, 204: 148-155. doi: 10.1016/j.chemosphere.2018.04.015 [27] WESTERHOFF P, MEZYK S P, COOPER W J, et al. Electron pulse radiolysis determination of hydroxyl radical rate constants with Suwannee River fulvic acid and other dissolved organic matter isolates [J]. Environmental Science & Technology, 2007, 41(13): 4640-4646. [28] LIAO C H, KANG S F, WU F A. Hydroxyl radical scavenging role of chloride and bicarbonate ions in the H2O2/UV process [J]. Chemosphere, 2001, 44(5): 1193-1200. doi: 10.1016/S0045-6535(00)00278-2 [29] WANG J L, WANG S Z. Effect of inorganic anions on the performance of advanced oxidation processes for degradation of organic contaminants [J]. Chemical Engineering Journal, 2021, 411: 128392. doi: 10.1016/j.cej.2020.128392 [30] HUANG Y, KONG M H, WESTERMAN D, et al. Effects of HCO3– on degradation of toxic contaminants of emerging concern by UV/NO3– [J]. Environmental Science & Technology, 2018, 52(21): 12697-12707. [31] LATCH D E, PACKER J L, STENDER B L, et al. Aqueous photochemistry OF triclosan: Formation OF 2, 4-DICHLOROPHENOL, 2, 8-DICHLORODIBENZO-p-DIOXIN and oligomerization products [J]. Environmental Toxicology and Chemistry, 2005, 24(3): 517. doi: 10.1897/04-243R.1 [32] KEEN O S, LOVE N G, LINDEN K G. The role of effluent nitrate in trace organic chemical oxidation during UV disinfection [J]. Water Research, 2012, 46(16): 5224-5234. doi: 10.1016/j.watres.2012.06.052 [33] MOFFETT J W, ZIKA R G. Reaction kinetics of hydrogen peroxide with copper and iron in seawater [J]. Environmental Science & Technology, 1987, 21(8): 804-810. [34] LEE C, YOON J. Temperature dependence of hydroxyl radical formation in the hv/Fe3+/H2O2 and Fe3+/H2O2 systems [J]. Chemosphere, 2004, 56(10): 923-934. doi: 10.1016/j.chemosphere.2004.04.047 [35] SUN K, LI S Y, YU J L, et al. Cu2+-assisted laccase from Trametes versicolor enhanced self-polyreaction of triclosan [J]. Chemosphere, 2019, 225: 745-754. doi: 10.1016/j.chemosphere.2019.03.079 [36] PENG J B, LI J H, SHI H H, et al. Oxidation of disinfectants with Cl-substituted structure by a Fenton-like system Cu2+/H2O2 and analysis on their structure-reactivity relationship [J]. Environmental Science and Pollution Research, 2016, 23(2): 1898-1904. doi: 10.1007/s11356-015-5454-y [37] GAO H P, CHEN J B, ZHANG Y L, et al. Sulfate radicals induced degradation of Triclosan in thermally activated persulfate system [J]. Chemical Engineering Journal, 2016, 306: 522-530. doi: 10.1016/j.cej.2016.07.080 [38] PENG J B, ZHANG Y Z, ZHANG C N, et al. Removal of triclosan in a Fenton-like system mediated by graphene oxide: Reaction kinetics and ecotoxicity evaluation [J]. Science of the Total Environment, 2019, 673: 726-733. doi: 10.1016/j.scitotenv.2019.03.354 -

 点击查看大图
点击查看大图

计量
- 文章访问数: 3767
- HTML全文浏览数: 3767
- PDF下载数: 62
- 施引文献: 0
