







-
由于干旱和半干旱地区地表水资源稀缺且具有间歇性,地下水往往是支撑中国西北煤矿地区用水需求的主要水源[1]。然而,煤炭开采与水资源管理之间很难达到平衡。此外,煤层上覆多层含水层的存在使得这种情况更加复杂。一方面,采矿可能会以各种方式影响地下水质量,这取决于水文地质环境和相邻含水层的化学性质[2-3]。另一方面,多层含水层以下的深部煤层,在开采过程中工作面或巷道附近可能发生突水,会对开采构成威胁[4-5]。为了支持矿山水资源的可持续利用和突水预测,有必要阐明地下水的来源、循环和水-岩作用对水化学演化过程的影响[6-7]。
近年来,人们对地下水系统的水化学演化进行了广泛的研究,其中水-岩作用是地下水系统水化学演化的主要机制。简单的散点图,多元统计分析,同位素等方法已被应用于调查地下水系统的水化学过程[8-10]。此外,不同离子之间的相关性同样为获取水化学演化过程中水岩相互作用信息提供了有效途径[11-12]。然而,这些方法仅利用部分现有数据定性描述可能的水化学演化过程,难以量化这些过程中各组分的形式和物质转移量。相对于这些定性方法,水化学反向模拟是在多种理论和技术方法基础上发展起来的定量方法[13]。重要的是,反演模型可以计算各种组分的形式、地下水系统中的物质转移量和水-岩平衡状态,从而揭示地下水系统中的水化学过程[14]。
本文选取的研究区为我国西北地区的大海则井田,通过水化学图、离子比例分析、饱和指数等方法探讨了多层含水层地下水的水化学特征和演化规律,并且通过水化学反向模拟的方法定量解释了控制不同含水层地下水化学演化的水文地球化学过程。
-
大海则井田位于中国陕西省榆林市西北方向50 km处,井田南北长14.7 km,东西宽12—22 km,占地面积280.03 km2。研究区的水文地质补勘工作于2013年12月完成。目前,井田的巷道钻打和基础设施建设工作已经完成,正式采煤工作计划于2021年年底开始。研究区位于毛乌素沙漠南缘,沙漠覆盖率达70%以上。主要地貌类型为沙漠洼地和沙丘,黄土梁地貌仅见于东南部地区。研究区地形较为平坦,最高海拔为1327 m,最低海拔为1210 m。
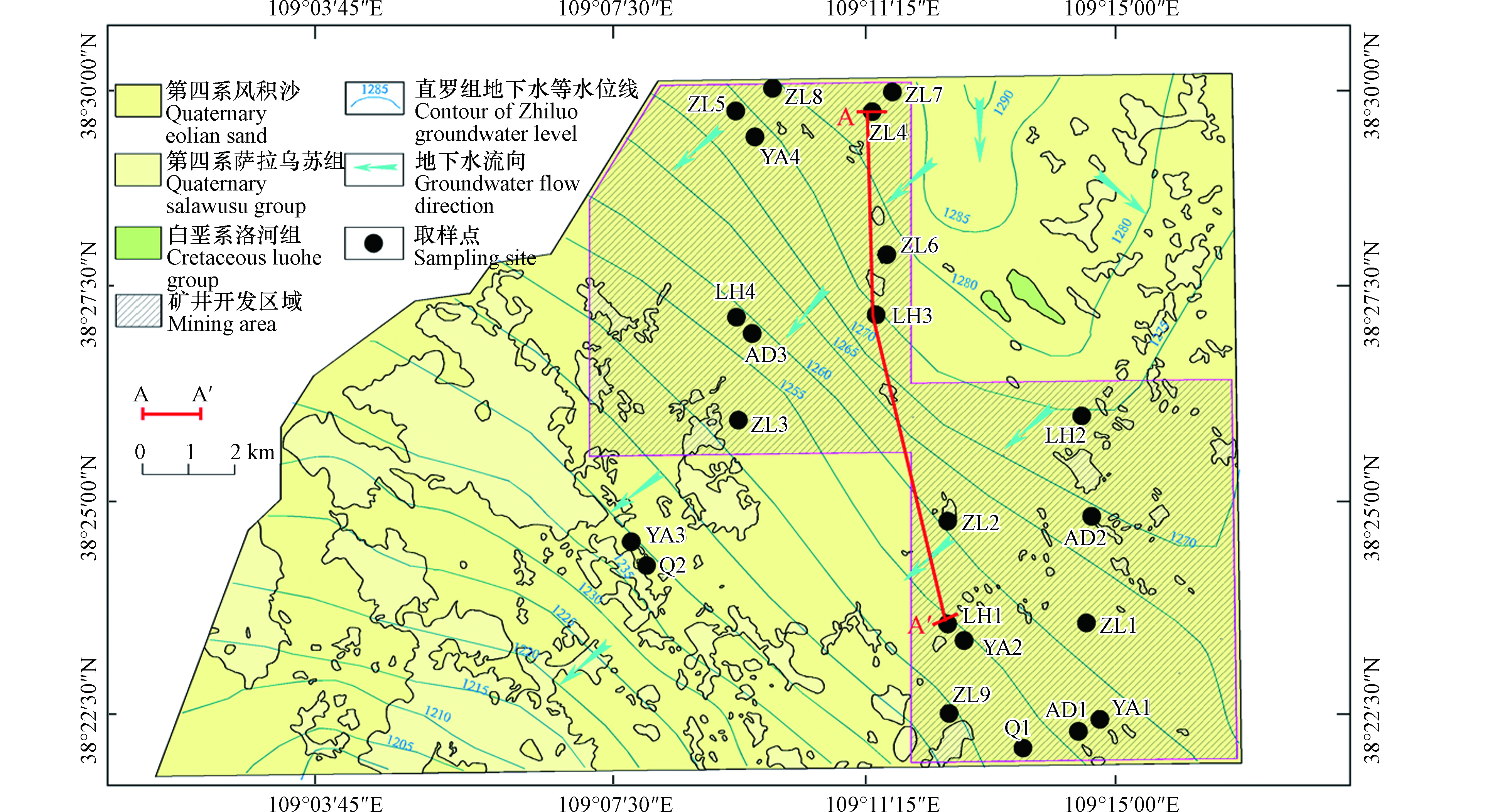
研究区位于鄂尔多斯盆地中部(图1),地表大部分被第四系风积沙覆盖,区内无大断层,也无岩浆活动。井田内发育有多个含水层,其中,第四系松散潜水含水层(单位涌水量q = 0.143—0.466 L·(s·m)−1)富水性中等-强;白垩系下统洛河组孔隙-裂隙含水层(单位涌水量q = 0.1—0.276 L·(s·m)−1)富水性中等;侏罗系中统安定组裂隙含水层(q = 0.003—0.02 L·(s·m)−1)富水性弱;侏罗系中统直罗组裂隙含水层(q = 0.033—0.271 L·(s·m)−1)富水性弱-中等;侏罗系中统延安组裂隙含水层(q = 0.0004—0.012 L·(s·m)−1)富水性极弱。研究区多层含水层系统地下水主要受降水补给,地下水流动主要受地形控制,流动方向由东北向西南。因此,多层含水层的地下水具有相同的补给面积和流向(图2)。此外,侧向径流排泄是地下水的主要排泄方式。
-
2011年10月,在大海则井田共采集地下水样24组(图1),其中包括第四系地下水样2组(Q1—Q2),洛河组地下水样4组(LH1—LH4),安定组地下水样3组(AD1—AD3),直罗组地下水样9组(ZL1—ZL9),延安组地下水样4组(YA1—YA4)以及矿井水水样2组(M1—M2)。
地下水样品的测试主要由陕西省核工业地质调查院进行的。结果如表1所示,现场参数(pH值、水温)通过便携式多参数监测仪进行测试。主要阳离子(Ca2+、Mg2+、Na++K+)采用电感耦合等离子体发射光谱仪(ICP-OES)测定,阴离子(SO42–、Cl–)采用离子色谱仪(ICS-1100)测定,HCO3–用HCl标准溶液(0.025 mol·L–1)滴定,可溶性SiO2用硅钼黄分光光度法测定。总溶解固体(TDS)是通过计算得到。除HCO3–外,阳离子和阴离子的精密度为0.01 mg·L–1;HCO3–精密度为0.60 mg·L–1。大部分样品电荷平衡误差小于5%,只有少数样品的电荷平衡误差较大,但仍小于10%,在合理范围内。
-
多层含水层地下水描述性统计分析结果见表2。第四系、洛河组和安定组地下水中阴阳离子以HCO3−和Ca2+为主,矿化度较低(TDS < 1000 mg·L–1)。直罗组和延安组地下水中阴阳离子以SO42−、Na++K+和Ca2+为主,为中等矿化度的微咸水(1000 mg·L–1 < TDS < 3000 mg·L–1)。然而,矿井水以SO42−和Na++K+为主,表现为高矿化度的咸水(TDS > 5000 mg·L–1)。《国家地下水质量标准》(GB/T 14848—2017)包含TDS等93项指标,将地下水质量标准分为5个等级。以TDS为依据,除第四系、洛河组、安定组和延安组地下水的YA4(428.1 mg·L–1)外,其他水样均超过Ⅲ型标准(500 mg·L–1 ≤ TDS ≤ 1000 mg·L–1),甚至部分地下水样属于Ⅴ型标准(TDS > 2000 mg·L–1)。说明安定组含水层以下的深层含水层的地下水不适宜饮用,但经适当处理后可用于工农业生产。
-
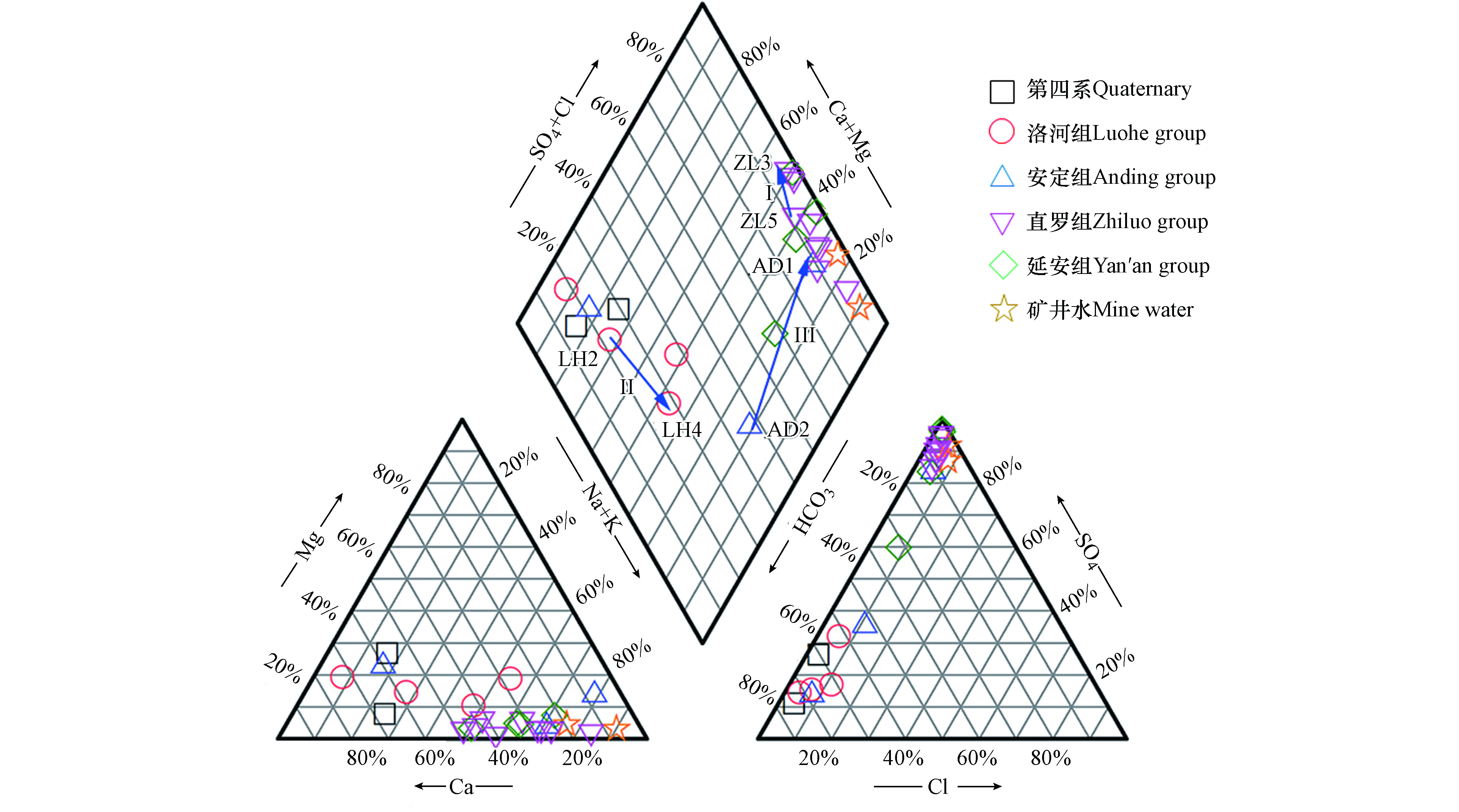
如图3所示,Piper图中多层含水层地下水样品分散:第四系地下水以HCO3·SO4-Ca和HCO3-Ca·Mg型为主;洛河组地下水类型为HCO3-Ca·Na+K、HCO3·SO4-Na+K·Ca和HCO3-Ca型;安定组地下水类型为SO4-Na+K·Ca、HCO3-Ca和HCO3·SO4-Na+K型;直罗组地下水类型为SO4-Ca·Na+K、SO4-Na+K和SO4-Na+K·Ca型;延安组地下水类型为SO4-Na+K·Ca和SO4·HCO3-Na+K型;矿井水的水化学类型为SO4-Na+K型,与直罗组地下水类似,说明直罗组地下水和矿井水之间存在水力联系,是煤矿开采过程中潜在的突水水源。
此外,Piper图可以用于表示地下水的水化学演化[15]。根据水文地质条件可知,多层含水层具有相同的补给区域和地下水流向(图1),这有助于研究不同含水层水化学演化的异同。此次研究主要调查了直罗组含水层、洛河组含水层和安定组含水层的水化学演化规律(图3)。
第一个演化路径(箭头Ⅰ:ZL5→ZL3)代表直罗组地下水的水化学演化,水化学类型从SO4-Na+K·Ca型变为SO4-Ca·Na+K,TDS沿着地下水流动方向明显增加。总体上,主要离子的变化规律是Ca2+、Na++K+、Mg2+和SO42−含量增加,HCO3−含量降低,而Cl−含量基本保持不变。其中,Ca2+和Mg2+同步增加,说明可能存在白云石的溶解(方程1);Ca2+和SO42−同步增加,这可能是由于石膏溶解导致(方程2),但SO42−增长幅度明显大于Ca2+,说明可能受到其他水岩作用的影响。除了石膏的溶解,黄铁矿的氧化可能是煤矿地下水中SO42−的另一个潜在来源(FeS2 + 7/2O2 + H2O = Fe2+ + 2SO42− + 2H+)[16]。但是研究区地下水总铁含量较低,且pH值呈中性(表1)。因此,SO42−的主要来源是石膏的溶解,而不是黄铁矿的氧化。由此可知,SO42−浓度逐渐高于Ca2+浓度是由于Ca2+的减少,而不是SO42−的增加,说明可能受到阳离子交换作用的影响(方程3)。此外,沿着流动路径Na++K+含量明显增大,而Cl−保持相对恒定,进一步验证了阳离子交换作用。
虽然从ZL5到ZL3的直罗组地下水中SiO2含量略有增加(表1),说明硅铝酸盐的溶解可能是Na+和/或K+增大的潜在来源(方程4—5)。但是地下水样品的可溶性SiO2含量较低,且pH值呈中性,因此硅铝酸盐的溶解不是Na+和/或K+的主要来源[17]。相比之下,HCO3−浓度沿着流动路径明显减小,而且大部分直罗组水样点方解石的饱和指数均大于零(表1),说明方解石呈过饱和状态,有可能沿流动路径析出沉淀。综上所述,直罗组地下水的演化路径(ZL5→ZL3)可能发生的水化学过程包括:白云石、石膏和少量硅铝酸盐的溶解,阳离子交换作用和方解石沉淀。
第二条演化路径(箭头Ⅱ:LH2→LH4)代表了洛河组地下水由HCO3-Ca·Na+K型向HCO3-Na+K·Ca型的水化学演化。水化学类型转变的实质是Na++K+、Mg2+、Cl−、SO42−和HCO3−含量增加,Ca2+含量减少。Na++K+和Cl−的增加表明岩盐的溶解,而Na++K+增加幅度明显大于Cl−,说明可能存在阳离子交换作用。Mg2+和SO42−的增加分别来自白云石和石膏的溶解作用。因此,第二条演化路径的主要水岩作用包括岩盐、白云岩和石膏的溶解并伴随阳离子交换。
第三条演化路径(箭头Ⅲ:AD2→AD1)代表安定组地下水由HCO3·SO4-Na+K型转化为SO4-Na+K·Ca型的演化过程。沿流动路径主要离子的变化是Na++K+、Ca2+、Cl−和SO42−含量增加,HCO3−含量下降,Mg2+含量略有下降。由表1可知,安定组地下水样方解石和白云石的饱和指数均大于零,说明方解石和白云石处于饱和/过饱和状态。因此,演化过程中HCO3−浓度的减小可能由方解石和白云石析出沉淀导致 [18]。此外,Ca2+与SO42−的浓度同步增大,说明Ca2+的主要来源是石膏的溶解。对于Na++K+和Cl−,二者浓度均发生了增大,说明了岩盐溶解的存在。但是,Na++K+增大幅度明显大于Cl−,说明可能受到了阳离子交换的影响。此外,硅铝酸盐的溶解(SiO2略有增加)和方解石和/或白云石的沉淀可能是导致Na++K+增加和Ca2+减少的其他原因。因此,第三条演化路径的水化学过程包括岩盐、石膏和硅铝酸盐的溶解,方解石和白云石的沉淀,并伴随阳离子交换作用。
-
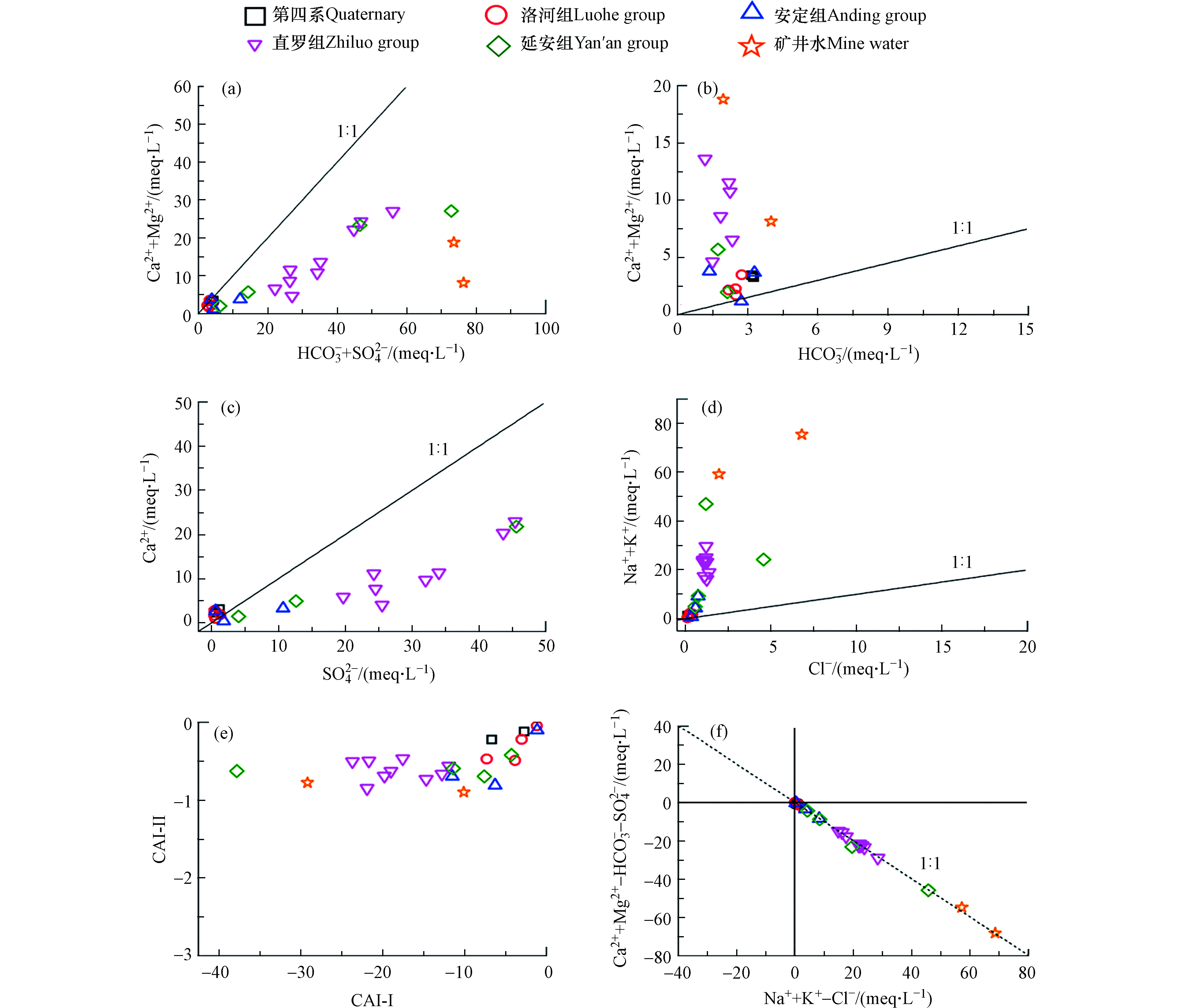
不同来源或条件下形成的地下水中离子的比值关系存在明显差异,因此常用不同离子的比值来识别地下水的来源或水化学演化过程[19]。(Ca + Mg)/(SO4 + HCO3)和(Ca + Mg)/HCO3值可以表明碳酸盐(方解石、白云石等)和硫酸盐(石膏)的溶解或沉淀对地下水化学成分的贡献[20]。第四系、洛河组和安定组地下水的(Ca + Mg)/(SO4 + HCO3),(Ca + Mg)/HCO3和Ca/SO4值均接近于1:1线(图4a—c),说明存在碳酸盐岩的溶解和石膏的溶解。直罗组、延安组地下水和矿井水的(Ca + Mg)/(SO4 + HCO3)低于1:1线,(Ca + Mg)/HCO3值高于1:1线,一方面说明石膏的溶解是Ca2+的主要来源,另一方面说明可能受到了碳酸盐沉淀的影响。此外,由图4c可知,直罗组、延安组地下水和矿井水的SO42–含量是要明显高于Ca2+的,这说明石膏的溶解过程还有可能受到了阳离子交换或者碳酸盐沉淀的影响。
如果盐岩和钾盐的溶解是Na+和K+的唯一来源,那么(Na + K)/Cl的值应是1。然而,在大多数情况下,Na+和K+可能有其他来源(如硅酸盐风化和阳离子交换),从而导致(Na + K)/Cl大于1[21]。如图4d所示,第四系、洛河组和安定组地下水的(Na + K)/Cl值接近于1,说明Na+和K+主要来源于岩盐和钾盐的溶解。相比之下,直罗组、延安组地下水和矿井水的水样点的分布明显高于1:1线,即(Na + K)/Cl >> 1,表明存在Na+和K+的其他来源,如硅铝酸盐的溶解。而水样中的SiO2含量证明了该趋势的可能性(表1),与上述规律一致。此外,阳离子交换作用是造成Na++K+过量的常见原因。通常可利用地下水的Schoeller指数表征地下水化学演化过程中阳离子交换的强度(方程6—7)。如果 Schoeller 指数得到是负的,说明 Na+、K+从含水层矿物中析出,Ca2+、Mg2+取代了它们的位置;如果指数是正的,说明发生了相反的交换[22]。如图4e可知,研究区水样点的Schoeller 指数均为负值,且研究区的含水层中富含黏土矿物[23],含有丰富的Na+和K+,因此,可能发生了含水层矿物中 Na+和K+取代水溶液中 Ca2+和Mg2+的阳离子交换作用。
根据(Na++K+–Cl–)与(Ca2++Mg2+–HCO3––SO42–)关系同样可以判断阳离子交换是否为主导的水文地球化学作用[24]。(Na++K+–Cl–)表示除去岩盐和钾盐溶解后剩余的 Na++K+,(Ca2++Mg2+–HCO3––SO42–)表示除去碳酸盐矿物和石膏溶解后剩余的(Ca2++Mg2+)。若样点落在第Ⅳ 象限内,说明样点中 Na+、K+除了盐岩和钾盐溶解有额外的增加,而碳酸盐矿物和石膏溶解产生的Ca2+、Mg2+有减少,可能是由于正向阳离子交换反应释放了Na+、K+,减少了水中的 Ca2+、Mg2+;若样点落在第Ⅱ 象限,说明样点中 Ca2+、Mg2+除了碳酸盐矿物和石膏溶解有额外的增加,而岩盐和钾盐溶解产生的 Na+、K+有减少,可能是含水介质中的 Ca2+、Mg2+离子与水中 Na+、K+离子发生反向阳离子交换反应。从图4f可知,几乎所有地下水样点在斜率为-1的线附近分布,并沿右侧分布,表明地下水中 Na+增多而 Ca2++Mg2+减少,表明正向阳离子交换作用在研究区广泛发生,与Schoeller指数得出的结论一致。
-
水化学反向模拟能够定量解释水化学成分的变化和水化学演化的主要水岩反应[25]。本研究采用NETPATH程序对上述水化学演化过程进行了定量模拟和定性分析。为了验证和量化这些路径中的水化学过程,本研究选择前文分析提到的直罗组含水层的ZL5→ZL3、洛河组含水层的LH2→LH4和安定组含水层的AD2→AD1作为模拟路径。
-
根据前文的分析,可能的矿物相包括岩盐、石膏、白云石、方解石和硅铝酸盐(如钠长石)。同时,阳离子交换是本研究中必不可少的水化学过程,也应考虑到模型中。根据3个模拟路径不同组分的饱和指数变化可知(表3),3条路径中岩盐、石膏均发生溶解,方解石趋于沉淀,直罗组和洛河组地下水中白云石均发生溶解,而安定组地下水中的白云石趋于沉淀。除了可能矿物相,约束变量是在质量平衡模型中要考虑的水化学元素[26]。根据水化学测试结果,设定Na++K+、Ca2+、Mg2+、HCO3–、SO42–、Cl–和SiO2为模拟约束变量。
-
由表4可知,直罗组地下水的演化路径(ZL5→ZL3)主要反应包括石膏、白云岩、岩盐、钠长石和CO2的溶解,方解石的沉淀和阳离子交换。直罗组含水层与含煤地层延安组含水层相邻(图2),所以当延安组含水层进行巷道掘进工作时,直罗组含水层极容易生成导水裂隙。所以,CO2可以通过掘进产生的裂隙进入地下水,从而促进白云石和钠长石的溶解(方程1和5)。岩盐的溶解和阳离子交换作用产生Na++K+,使得Na++K+不断进入地下水。Ca2+主要受到石膏和白云石的溶解、方解石的沉淀和阳离子交换的影响。虽然阳离子交换可以导致Ca2+的降低,但由于石膏的大量溶解,使得Ca2+的浓度仍呈现出持续升高的趋势。由于Ca2+的增加,地下水水化学类型由ZL5的SO4-Na+K·Ca型转化为ZL3的SO4-Ca·Na+K型。
沿着演化路径LH2→LH4,洛河组地下水的水化学演化经历了石膏、白云岩、岩盐和CO2的溶解;方解石的沉淀与阳离子交换。根据地层剖面图可知(图2),洛河组含水层与第四系含水层相邻,洛河组地下水为潜水,属于开放系统。CO2可以直接进入地下水从而加速白云石的溶解(方程1),另外,阳离子交换作用和岩盐的溶解可以不断产生Na+,且导致Ca2+的减少。虽然白云石和石膏的溶解会产生新的Ca2+,但是方解石的沉淀和阳离子交换的程度更大,从而使得Ca2+总浓度减小。总体上,由于Na+的增加和Ca2+的减少,导致演化路径上水化学类型由LH2的HCO3-Ca·Na+K型转化为LH4的HCO3-Na+K·Ca型。
安定组地下水演化路径(AD2→AD1)的主要水化学过程包括石膏、岩盐和钠长石的溶解;方解石、白云石的沉淀与阳离子交换作用。少量的岩盐、钠长石的溶解和阳离子交换作用使得Na+不断进入地下水。而石膏的溶解、方解石、白云石的沉淀和阳离子交换是控制Ca2+的水化学过程。虽然碳酸盐岩(方解石和白云石)的沉淀和阳离子交换可以降低Ca2+浓度,但石膏的大量溶解导致Ca2+总浓度的增加。同时,石膏的溶解、方解石和白云石的沉淀会导致SO42–的升高和HCO3–的降低。最终,由于Ca2+、SO42–的增加和HCO3–的减少,AD2的水化学类型从HCO3·SO4-Na+K型转化为AD1的SO4-Na+K·Ca型。上述3种演化路径的水化学过程可由下式表示(单位:mmol·L–1):
综上所述,利用水化学反向模拟可以识别出3个不同含水层的水化学演化路径,并且与定性分析的结果相符。研究发现,不同含水层的演化路径中水岩作用和物质转移量都不同,从而导致水化学类型的差异。根据研究区水文地质背景,由于无明显断层,含水层之间较弱的水力联系是造成多层含水层系统水化学类型和水化学演化差异的主要原因。虽然目前相邻含水层之间的水力联系相对较差,但后续的煤矿开采会产生导水裂隙,进而促进相邻含水层之间的水力联系。因此,需要对开采过程中和开采后的水化学演化进行连续监测,预防高矿化度矿井水对主要供水含水层的污染。
-
(1)研究区多层含水层地下水的主要水化学类型为:第四系地下水为HCO3·SO4-Ca型和HCO3-Ca·Mg型;洛河组地下水为HCO3-Ca·Na+K、HCO3·SO4-Na+K·Ca和HCO3-Ca型;安定组地下水为SO4-Na+K·Ca、HCO3-Ca和HCO3·SO4-Na+K型;直罗组地下水为SO4-Ca·Na+K、SO4-Na+K和SO4-Na+K·Ca型;延安组地下水为SO4-Na+K·Ca和SO4·HCO3-Na+K型;矿井水水化学类型以SO4-Na+K型为主,与直罗组地下水类似,说明直罗组地下水是潜在的突水水源,应予以特别重视。
(2)主要离子关系和水化学模拟结果表明,不同矿物的溶解和沉淀以及阳离子交换作用是控制水化学类型的主要因素。然而,来自不同含水层的演化途径表现出了完全不同的水化学过程,这是研究区缺乏断层,相邻含水层之间的水力联系相对较差所导致。
大海则井田多层含水层系统水化学特征及演化规律
Hydrochemical characteristics and evolution of groundwater in multi-layer aquifer system in the Dahaize Coalfield
-
摘要: 研究多层含水层系统地下水化学特征及演化规律对于矿区水资源保护和突水灾害防治具有重要意义。本文运用Piper图解法、离子比值法和反向地球化学模拟研究了我国西北地区大海则井田多层含水层的水化学过程和水质特征。结果表明,不同含水层地下水的水化学类型差异明显,主要表现为浅层地下水水呈以HCO3型为主的淡水,而深层地下水呈以SO4型为主的微咸水和咸水。其中,矿井水和直罗组地下水的水化学类型接近,说明直罗组地下水是矿井水的主要水源,也是潜在的突水水源。此外,通过离子关系和水化学模拟得出,不同矿物的溶解和沉淀以及阳离子交换作用是导致不同含水层地下水水化学类型差异性特征的主要因素。Abstract: It is of great significance for water resources protection and water inrush prevention to study hydrochemical characteristics and evolution of coalfield groundwater in multi-layer aquifer system. The groundwater in Dahaize Coalfield in Northwest China was stored in typical multi-layer aquifers. To get better understanding of its hydrochemical characteristics, hydrogeological field investigations and tests were carried out in the Coalfield. The Piper diagram, ion ratios and inverse geochemical simulation were applied to identify the hydrochemical process and water quality in the multi-layer aquifer. The results showed that the hydrochemical types of the groundwater in the different layers of the aquifer system were significant different. The groundwater in the shallow layers was fresh water with a dominated hydrochemical type of HCO3. However, it was brackish or salty water in the deep layers with a dominated hydrochemical type of SO4. Meanwhile, the hydrochemical type of the mine water was approximately the same with that of the groundwater in Zhiluo group. This indicated that the groundwater in Zhiluo group could be the main source of the mine drainage and water inrush of the Coalfield. As a result of this, together with the ion relationships and hydrochemical simulation results, the dissolution and precipitation of the minerals and cation exchange could be the main factors that influenced the Coalfield hydrochemical types of the groundwater in the different layers of the aquifer system.
-
Key words:
- multi-layer aquifer /
- hydrochemical evolution /
- inverse modeling /
- Dahaize Coalfield
-
抗生素在医疗、畜牧、养殖等行业广泛应用。由于不正当地使用和无组织排放,致使其大量流入环境介质中,近年来,国内外在水环境[1]、土壤[2]、动植物组织[3]等环境介质中均有抗生素的检出报道[4-5]。畜禽养殖业中抗生素的不合理使用引发了一系列潜在危害,主要体现在产生抗药基因、破坏生物体内的微环境、人畜共患病增多等方面。动物源性食品安全成为了重大问题,为了保证人类健康,世界卫生组织(WHO)和联合国粮食及农业组织(FAO)曾共同建立了农药和兽药的最大残留限量(MRL)标准来对其进行控制。
检测食品中微量或痕量级别抗生素最常采用仪器法,有高效液相色谱法(HPLC)[6]、高效液相色谱-串联质谱法(HPLC-MS/MS)[7-9]、液相色谱-紫外-质谱法(LC-UV-MS)[10]、液相色谱-荧光检测方法[11]等。仪器检测法的灵敏度高,检测数据准确,但由于其前处理复杂、耗时长等劣势,限制了其在原位快速检测中的应用。生物检测技术因其高灵敏度、高选择性被广泛用于动物源性食品中的抗生素残留量检测研究。基于抗体的生物免疫检测技术最为成熟,研究应用也比较广泛。而稳定性好、适用范围广的核酸适配体与响应快、操作简便的传感器相结合成为了生物检测研究的热门发展方向。基于此,本文综述了以抗体和核酸适配体为生物识别元件的生物检测技术特点及其在动物源性食品中抗生素残留量检测的研究进展,并在归纳总结生物检测技术检测抗生素的研究基础上,预测抗生素检测技术未来的发展趋势。
1. 基于抗体的免疫检测技术(Antibody-based immunoassay technology)
免疫分析技术是基于抗体、抗原之间特异性结合的一种生物化学技术,被广泛应用于环境和食品行业中药物残留等有害物质的定性和定量。
1.1 酶联免疫吸附分析法(ELISA)
ELISA是通过被酶标记的抗体对抗生素的竞争,结合到固相包被抗原上,洗涤后加入酶底物,固定在载体(酶标板)上的酶催化底物出现显色反应,最后通过分光光度法(酶标仪)对待测物质进行定量分析。近年来,双斑点法、直接(图1A)和间接性竞争ELISA在检测抗生素残留研究方面皆有开发。
 图 1 抗生素的免疫测定原理Figure 1. Principles of the immunoassays for the detection of antibiotics.(A)ELISA(B)RIA(C)FIA(D)CLIA(A)酶联免疫吸附分析法(B)放射免疫分析法(C)荧光免疫分析法(D)化学发光免疫分析法
图 1 抗生素的免疫测定原理Figure 1. Principles of the immunoassays for the detection of antibiotics.(A)ELISA(B)RIA(C)FIA(D)CLIA(A)酶联免疫吸附分析法(B)放射免疫分析法(C)荧光免疫分析法(D)化学发光免疫分析法直接性ELISA和间接性ELISA有着不同的优势,Ashu等[12]比较了两种ELISA测定抗生素的灵敏性,找到了一种特异性强、灵敏度高的多克隆抗体As172,采用直接竞争ELISA法建立了用于饲料中氟喹诺酮类抗生素的检测方法,其中该抗体对恩诺沙星的检测能力达20 ng·g−1,对8种氟喹诺酮类抗生素有高达42%的交叉反应率,可实现多种抗生素残留物的同时测定。为了使ELISA更加简便且节省抗原,Wei等[13]制备出6H3D5单克隆抗体,研究出了一种双斑点ELISA可同时检测卡那霉素(KANA)和链霉素,该方法的固相载体为硝酸纤维素膜(NCM),经酶促反应后从视觉上可观测到显色情况,其检测限分别为0.09 ng·mL−1和1.37 ng·mL−1。
酶联免疫的方法操作简单,普适性强,但在其方法的特异性和灵敏度上,对抗体的要求比较高,因此,采用其他标记物的新方法也被研究。
1.2 放射免疫分析法(RIA)
RIA的检测原理为被放射性同位素标记的抗原与待测抗原竞争性结合有限的特异性抗体的位点(图1B),洗涤后加入闪烁液(示踪剂),经Charm闪烁计数器分析后,闪烁计数值越高,样品中待测物质浓度越低,常用于抗生素的定性与半定量检测。
Meyer等[14]在购置的检测限为1 μg·L−1左右的Charm Ⅱ RIA的基础上加以优化,用于检测样品中残留的四环素类抗生素。结果表明,该RIA的灵敏度低于传统的LC/MS法,有待于开发灵敏度更高的RIA。为了拓宽RIA的适用范围,Yang和Carlson[15]将样品先经固相萃取(SPE)技术纯化后,再对四环素和磺酰胺进行半定量分析,该方法的检测限低至0.05 μg·L−1。Al-Mazeedi等[16]利用Charm Ⅱ RIA筛选出阳性和疑似阳性样品,而后通过LC/MS-MS进行确认,完成了1517份乳制品中四环素残留的调查。
虽然放射免疫分析法无法对待测样品进行准确定量,但它适用于大批量样品筛查,针对RIA的假阳性结果,可对样品进一步浓缩纯化来改善解决。不过由于该方法中使用到的放射性物质对人类健康和生态环境有一定的危害,现如今已多被ELISA所取代。
1.3 荧光免疫分析法(FIA)
荧光免疫分析通常采用荧光物质进行标记(图1C),有荧光色素、镧系螯合物以及荧光酶等化合物。FIA具有高特异性、高灵敏度、检测速度快等特点,但由于一般的荧光标记物产生荧光测定本底值较高。为了减少误差,开发出了时间分辨荧光免疫分析法(TRFIA),利用了镧系螯合物的量子产率高、荧光寿命长等特性,通过时间分辨技术来测定荧光强度。
Le等[17-18]利用Sm3+和Eu3+标记不同的抗体,将双标记TRFIA用于检测金霉素和强力霉素、磺胺二甲嘧啶和磺胺喹喔啉,其检测限在0.02—0.04 ng·mL−1。Ashuo等[19]利用生物素和链霉亲和素的高亲和度制备出了Eu3+微球-mAb荧光探针,建立了一种基于TRFIA的侧向免疫测定法。该试纸条可通过扫描QR码获取相应信息,搭配荧光读取器,可实现10 min左右完成牛奶样品中3种抗生素的实时检测,检测限分别为0.10、0.06、0.27 ng·mL−1。一些自身具备荧光特性的纳米材料(如上转换纳米颗粒(UCNPs))以及不具荧光特性的磁性纳米粒子(MNPs)也被广泛应用于检测环境样品中的抗生素残留(表1)。
FIA是一种可靠且强大的筛选方法,其信号强度大,但量子点的最终荧光发射易受干扰,所以需对样品进行脱色,以达最佳效果。
1.4 化学发光免疫分析法(CLIA)
CLIA是将高灵敏度的化学发光技术和高特异性的免疫反应结合起来的一种操作简便、特异性强、灵敏度高、分析速度快、线性范围宽的超微量检测技术。它通过添加氧化剂或酶底物,使体系中化学发光物质被氧化至不稳定的激发态中间体,而后回到稳定的基态并发射光子,利用发光信号测量仪进行定量。CLIA有两大类型,一种是发光物质直接对抗体进行标记(图1D),加入发光启动剂即可;另一种是采用酶(如HRP)标记抗体,然后加入相应的发光酶底物(如鲁米诺Luminol)。
Li等[20]制备出了一种能够同时识别32种磺酰胺的mAb,采用化学发光直接竞争酶免疫测定法(CL-dcELISA)对鸡肉进行检测。Yu等[21]采用CL-dcELISA,以单链可变片段(scFvs)代替抗体提高了与FQs的交叉反应性(CR),用于检测鱼虾类20种氟喹诺酮(FQs),其中诺氟沙星(NOR)的检出限为0.017 μg·kg−1,线性范围在0.04—1.08 μg·kg−1。不少学者将微流控芯片技术和纳米材料引入CLIA用于检测动物源性食品中抗生素的残留(表1),显著减少了试剂和样品用量,增强了发光系统。
限制CLIA的有化合物的溶解度以及闪光的即时测量等因素,但是该方法的灵敏度远高于ELISA、FIA等,反应时间优于TRFIA,在免疫检测市场上,有着无可替代的地位。
表 1 基于不同纳米材料用于检测动物源性食品抗生素残留的免疫技术Table 1. Immunoassay based on different nanomaterials for the screening of antibiotics in animal-derived foods检测类型 Assay mode 纳米材料Nanomaterials 抗生素 Antibiotics 样品 Sample 检测限/ (ng·mL−1)LOD 回收率/% Recovery 参考文献 Reference FIA MNPs 环丙沙星 牛奶 8 90—100 [28] QDs 链霉素 牛奶 0.005 80.21—108.3 [29] 四环素 80.5—109.2 [29] 青霉素 82.4—101.4 [29] NaYF4∶Yb, Er 诺氟沙星 牛奶 0.01 82.37—132.22 [30] CLIA MNPs 氯霉素 食品 2 × [31] AuNPs 虾、蜂蜜 3.3 × [32] SiO2 虾、蜂蜜 3.3 × 83.7—115.1 [33] EIA MOFs/AuPt 马杜霉素 鸡蛋 0.045 96.4—106 [34] PAMAM-Au 诺氟沙星 牛奶、鸡蛋、猪肉 0.3837 91.6—106.1 [35] PEDOT/MWCNT 瘦肉精 牛肉 4.66 85—111 [36] | Show Table DownLoad:
CSV
DownLoad:
CSV
1.5 胶体金免疫层析法(CGIA)
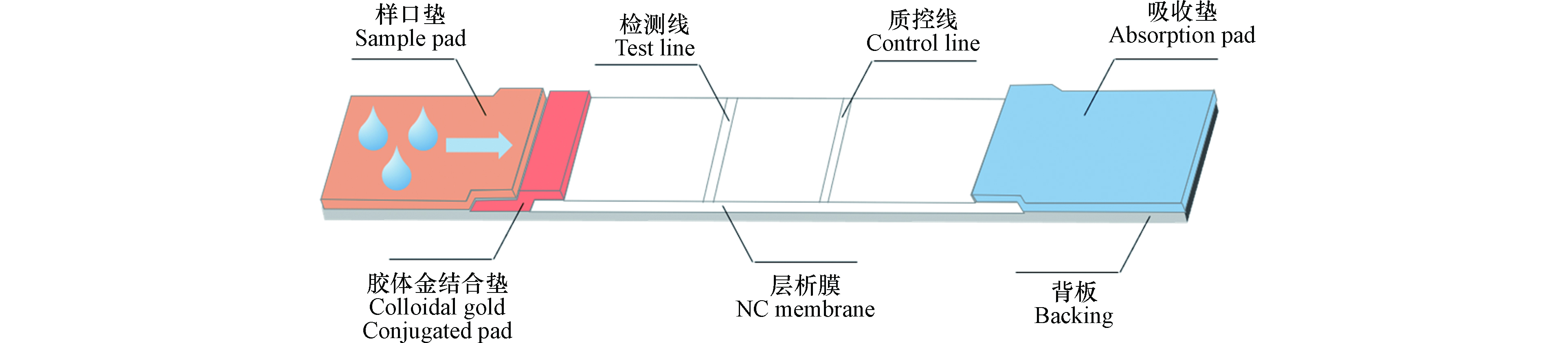
胶体金通常是氯金酸(HAuCl4)在还原剂的作用下,聚合形成粒径均匀的金粒子,粒子间的静电作用使其形成了稳定的胶体状态。由于AuNPs表面的强吸附作用,可以很好地同蛋白质结合进行标记。当大量标记物聚集时,视觉上呈红色,在不使用仪器的情况下即可通过肉眼比色来完成检测物的定性与半定量。CGIA是在硝酸纤维素膜上的检测线和质控线内固定好特异性抗原或抗体,样品加在样品垫,借助毛细作用向另一端流动(图2),到达T或C线相应的抗体或抗原被截留,实现分离,整个检测过程在10 min左右。
Luo等[22]使用对马波沙星(MBF)有高灵敏度的M4E3 mAb开发了一种基于视觉的CGIA,检测限在1 ng·mL−1,还可灵敏地检测6种氟喹诺酮类似物,交叉率超过20%。Guo等[23]开发了一种用于筛选鸡胸肉中卡巴氧(CBX)和喹赛多(CYA)残留物的CGIA。该方法以(E)-2-(hydr甲基)喹喔啉1,4-二氧化物作半抗原制备了mAb,在最佳条件下,视觉检测限达8 μg·kg−1,利用条带扫描仪定量检出限为2.92 μg·kg−1、2.68 μg·kg−1。Xu等[24]则建立了一种无需进行样品前处理的CGIA,用于检测牛奶中的氯霉素(CAP)。与SERS技术结合,可极大提高检测的灵敏度,对新霉素的检测限低至0.216 pg·mL−1,证实该方法具有较高的重现性和稳定性[25]。
CGIA无法进行多种抗生素的同时检测,但基于CGIA开发的试纸条与微流控芯片检测技术结合,经小型仪器扫描将色度值转化为电信号,在8 min内即可完成现场快速检测,具有良好的稳定性和准确性[26]。
1.6 电化学免疫分析法(EIA)
电化学免疫传感器(EIS)是由生物感受器和换能器组成的一种小型分析设备。其原理为生物受体识别目标分析物会产生特定的生物信号,此信号通过换能器转换,成为可测量的电信号,分析物的浓度与电信号存在相对应的关系。利用高灵敏度、低成本的EIS可对痕量待测物完成准确定量。根据检测的信号类型不同,通常将EIS分为电流型、电容型、电位型和电导型4类。
不同的纳米材料可不同程度地提升传感器的电化学性能(表1)。纳米酶是一种具有酶活性的纳米材料,其特点是成本低,稳定性强,在EIS的信号放大方面起着极其重要的作用。Xiao等[27]将二维的Cu-TCPP(Fe)用作纳米酶修饰于电极上,基于Cu2+和聚乙烯亚胺(PEI)的强亲和力致使酶活性降低,进而引起电化学电流下降的原理,开发了一种新型电化学免疫传感器,用于测定磺酰胺,检测限达0.395 ng·mL−1。但电极传感器的制造需耗费大量时间,可以通过批量生产来用于现场检测。
抗体作为识别元件时,生物蛋白的特异性受制诸多因素制约,如pH、温度、存放时间等。而核酸适配体作为一种“化学抗体”,它具有良好的稳定性、可修饰性以及宽适用范围等特性,因而近年来被广泛应用于动物源性食品中抗生素的残留检测。
2. 基于核酸适配体的生物检测技术(Aptamer-based bioassay technology)
核酸适配体是通过SELEX,即指数富集的配体系统进化技术筛选出的单链DNA或RNA寡核苷酸序列。适配体有着复杂多样的二级结构,能够形成凸环、发夹、G-四联体等立体结构,这些结构特性使之被广泛应用于生物传感器的开发。它可通过范德华力、氢键、静电相互作用与靶分子相结合[37-38],具有高度的亲和性和特异性。相对于抗体来说,适配体通过化学合成,减少了批次间的差异,缩短了生产时间,成本相对较低;良好的热稳定性,使其更易储存;经过化学修饰和标记后仍具有良好的特异性;适用范围更广泛,从离子到细胞的任何靶点,包括非免疫或有毒的靶点[39-40] 。由于适配体的优良特性,目前在分析动物源性食品中抗生素的残留检测应用中有着良好的前景。与基于抗体的酶联免疫吸附法相比,酶联适体法具有更高的灵敏度和回收率。一些基于适配体的生物传感器,如电化学适配体传感器、光学适配体传感器等[41],不断被设计开发应用。适配体的可扩增性使得利用PCR技术实现信号放大成为可能。
2.1 基于核酸适配体的生物传感器抗生素检测技术
核酸适配体生物传感器(aptamer biosensor,简称AB)是一种由生物识别元件和换能器组成的装置,其生物识别元件通常以核酸适配体为主。适配体具有易修饰性,与纳米材料结合可增强传感系统的灵敏度,提高传感器的性能。AB法通常会引入一定的物质用于放大信号系统,进一步提升传感器的灵敏度。当目标分析物接触识别元件时,若有目标物的存在,适配体则会适应型折叠形成特殊的空间结构与目标物特异性结合,换能器将结合瞬间的信号转换为物理信号,最后经电子系统放大并显示。对物理信号进行分析,就可以完成对于样品中目标物的定性与定量。
2.1.1 光学核酸适配体传感器
光学生物传感器的换能器通常利用光的分光、荧光、折射等性质来对目标物进行定量检测。
光学核酸适配体传感器中最常采用的是基于比色的检测,目前开发的比色核酸适配体传感器大都基于胶体的测定,最常用的就是AuNPs,由于AuNPs具有独特的光学性质和高消光系数,研究者们利用这些优势开发出来许多应用于抗生素检测的AB。也有一些研究者建立了一种酶联适体测定法[42-43],通过测定吸光度来完成检测抗生素,具有较宽的线性检测范围和较低的检测限。
较短的单链DNA(ssDNA)其表面密度更高,与载体有更强的亲和力、更好的动力学。Wu等[44]基于ssDNA这一特性,利用长链(CAP适配体)、短链(TET适配体)偶联于AuNPs表面造成密度差异这一原理引发的AuNPs溶液颜色变化,开发了一种无标记超灵敏比色适体传感器。Abedalwafa等[45]和 Liu等[46]都开发了一种便携式比色传感器条用于动物源性食品中KANA的检测。前者是通过谷氨酸(GA)接枝到乙酸纤维素(CA)来制备带有羧基的电纺纳米纤维膜(NFM),将KANA适体标记在NFM上,适体与cDNA@AuNPs偶联,以此获得比色生物传感器条,当KANA存在时,传感条由粉色变为白色,检测限低至2.5 nmol·L−1;后者是基于AuNPs-apt作探针和AgNPs-cDNA作为信号放大元件开发了检测KANA的试纸条传感器,通过仪器扫描可实现1—30 nmol·L−1的线性检测范围和0.0778 nmol·L−1的检测限。Emrani等[47]设计了一种荧光猝灭和比色适配体传感器,传感器基于AuNPs、双链DNA(dsDNA)和FAM标记的cDNA(互补链DNA)。当链霉素不存在时,溶液由红变蓝,有强烈的荧光;反之,溶液呈红色,荧光被AuNPs猝灭。THMS(三螺旋结构)的结构比dsDNA更加坚固,能够保护AuNPs免受盐诱导的聚集,利用这一特点,Ramezani等[48]设计了一种比色适配体传感器,对四环素的检测限为266 pmol·L−1。
另一种最常采用的检测信号就是荧光强度,荧光信号一般是由荧光染料或是具有荧光性质的纳米材料发出的,适体与靶标特异性结合后,一些物质可通过荧光能量共振转移对荧光进行猝灭或使荧光信号得以释放,辅以一定手段对荧光信号进行放大,通过测定即可完成分析物的定量。
MNPs、UCNPs[49-50]、GO[51]等纳米材料和噻唑橙[52]等荧光染料常被用于光学核酸适配体传感器,灵敏、快速的检测抗生素。 Yang等[53]使用典型的MB-SELEX技术进行磺胺二甲嘧啶(SMZ)适体的选择,并引入裸羧基MB和SMR-MB用于反选择,提高SMZ适体的特异性。筛选得到的SA07适体用于开发化学发光适配体传感器,与27种磺酰胺类抗生素几乎无交叉反应性,检测限为0.92 ng·mL−1。Liu等[54]开发了一种比例荧光传感器,其中适配体标记上荧光染料,锆卟啉MOF(PCN-222)作为荧光猝灭剂,PCN-222与apt通过π-π堆积、静电等相互作用吸附结合,由FRET发生荧光猝灭。该传感器可在26 min内完成残留于牛奶和虾中CAP的检测,检测范围0.1 pg·mL−1—10 ng·mL−1,检测限低至0.08 pg·mL−1。Rouhbakhsh等[55]使用成本低的液晶(LC)设计了一种无标记向列型LC适体传感器,其原理是利用靶标诱导适体构象转换进而改变LC的垂直取向,可通过偏振光学显微镜计算出图像的灰度强度来定量牛奶中四环素的浓度。
表面等离子共振(SPR)的量子力学现象是SPR生物传感器的基本原理,平行于入射平面的偏振光撞击导电薄膜表面,在两种介质间产生的表面等离子激元被完全反射,当适配体结合分析物时,薄膜的折射率及共振角发生改变,反射光强度随结合分析物的质量呈比例变化。SPR生物传感器对于无标记、未经预处理的浑浊的样品也可完成高灵敏、高通量的检测。Wang等[56]在金膜表面引入了DNA四面体以纳米级距离定向的方式固定四环素适配体,提升了对TC的捕获效率,从而提高了SPR适体传感器的灵敏度,在蜂蜜样品中TC的回收率可达80.2%—114.3%,检测限为0.0069 μg·kg−1,较未引入DNA四面体的适体传感器低至10倍。
表面增强拉曼散射(SERS)是一种高度灵敏的光学测量方法,它基于光在纳米级粗糙贵金属表面附近原子或分子上的增强的非弹性散射(拉曼散射)提供信号。SERS适体传感器有着极高的灵敏度,成本低、响应速度快等优势,使用各种纳米材料能够不同程度的提升SERS适体传感器的灵敏度。Nguyen等[57]开发了一种基于GO/AuNPs的SERS传感器用于检测牛奶中的KANA,该纳米复合材料有较高的热稳定性和低热辐射,降低了微流体检测系统中的闪烁效应,获得了1—100 nmol·L−1的线性范围,0.75 nmol·L−1的检测限。Jiang等[58]通过监测4-巯基苯甲酸(4-MBA)的特征峰强度变化来定量牛奶中KANA的浓度,4-MBA修饰于Au@AgNPs表面,适体加入时削弱了4-MBA的拉曼强度,当KANA存在时,恢复了对4-MBA的表面增强作用。Meng等在适配体连接的两种大小的AuNPs之间构建了拉曼热点,当鱼粉中存在OTC时,系统拉曼强度增加,其检测限低至4.35
× 2.1.2 电化学核酸适配体传感器
固定于电极上的适配体与目标分析物特异性结合,这一反应会被电极表面的电活性物质所识别,从而引起可测量的电性能,如电流、电势、电容,发生变化,由此可完成目标分析物灵敏的定量检测。为了增强电化学性能,经常采用导电纳米材料或化学物质(如β-CD(环糊精)[59])来修饰电极,以放大信号,提高传感器的灵敏度。
Chen等[60]基于场效应晶体管(FET)开发了一种可集成的小型电子传感器,用于检测妥布霉素。还原氧化石墨烯(rGO)纳米片用作通道材料,适体RNA作为探针偶联于rGO,并采用了MCH和PBA对未结合适体的位点进行封闭,降低特异性结合引起的误差,传感器可在5 s内做出反应,0.3 nmol·L−1的检测限。有人在笔状石墨电极(PGE)上依次固定了rGO和AuNPs,巯基化适配体偶联于AuNPs表面,组建了电化学适体传感器,成功应用于肉类及牛奶样品中磺胺二甲氧嘧啶[61]和四环素[62]的检测,在最佳条件下,检测范围宽至1×10−16—1×10−6 mol·L−1,检测限低至3×10−17 mol·L−1。石墨相氮化碳(g-C3N4)是一种2D半导体纳米材料,具有独特的光学、电子、机械和化学特性,广泛应用于生物传感器。Li等[63]将g-C3N4修饰于钒酸铋(BiVO4)电极表面,g-C3N4通过π-π共轭吸附固定DNA适配体,开发出了一种通用的光电化学(PEC)传感器,改变DNA适体即可实现对于抗生素的灵敏性检测。有人将适体探针和微芯片电泳(MCE)平台相结合,根据apt-分析物和dsDNA产生的峰位置及强度不同,对牛奶及鱼肉中的抗生素进行定量检测[64]。Huang等[65]将四环素的短适体(Apt40)和长适体(Apt76)混合物固定在金丝网印刷电极表面上,使二者有良好的空间序列可与含TC的蜂蜜样品有较大的接触面积,识别能力远高于使用单个适体,检测限为0.0073 ng·mL−1,96.45%—114.6%良好的回收率。
基于核酸适配体的传感器有着高灵敏度、强选择性、响应速度快、成本低等特点,微量样品即可完成检测,被广泛应用于动物源性食品中抗生素的残留检测。而核酸适配体本身作为一段核苷酸序列是可以在体外利用聚合酶链式反应(PCR)技术进行扩增的,利用这一特性,不少学者开发出了基于适配体的新型抗生素生物检测方法。
2.2 基于聚合酶链式反应(PCR)扩增信号的抗生素测定技术
PCR技术能够实现在体外快速扩增目的DNA序列,早期应用于转基因和克隆技术,现被成功引入到环境监测领域。核酸适配体或互补寡核苷酸链通过标记生物素和链霉亲和素来与纳米磁珠载体进行结合,在外磁场作用下,可高效分离目标扩增DNA,减少后续PCR中的非特异性扩增,提高方法的特异性。
Tao等[66]将抗体和DNA结合设计了一种用于检测牛奶中CAP的方法,把报告基因DNA连接在CAP抗体上,而后通过EDC/NHS交联法将其固定于磁珠表面,以此获得免疫磁珠。牛奶样品中的CAP结合到一部分免疫磁珠后,剩余的免疫磁珠被固定于表面有CAP的PCR管中,经洗涤后对报告基因进行实时荧光定量PCR(qPCR),样品中CAP的浓度与阈值循环(Ct)值呈正比,有0.0008 μg·L−1的检测限,回收率在75%—155%之间。与免疫磁分离(IMS)相比,适体磁捕获(AMC)的捕获效率随着靶标数量减少而提高[67],与PCR信号放大技术的成功联用,实现了微量样品的高灵敏检测。Duan等[68]将互补链固定于磁珠上,适体与互补链杂交,当氯霉素(CAP)加入后,适体从磁珠上解离,收集含有适体的上清液进行qPCR,在最佳实验条件下,基于AMC的qPCR检测限为0.1 ng·mL−1,有0.1—20 ng·mL−1的线性检测范围。在AMC和PCR的基础上,Zhou等[69]结合微芯片电泳(MCE)阵列开发了一种可同时检测牛奶和鱼肉中KANA和CAP的传感器。适体固定于磁性金粒子上,与其互补链杂交,当待测物存在时,互补链被解离释放至上清液,将互补链与内标链共同扩增20个循环,产物通过MCE确定不同位置的电流强度,检测限分别达0.0025 nmol·L−1和0.006 nmol·L−1。除了单独利用PCR增强检测信号外,Ma等[70]设计了一种基于PCR和链霉亲和素(SA)双重放大的荧光偏振(FP)适体传感器,最佳条件下,对CAP的线性检测范围宽至0.001—200 nmol·L−1。适体和CAP孵育一段时间后,利用氧化石墨烯(GO)除去未结合适体,适体-CAP复合物作为模板,加入FAM标记的正向引物和生物素标记的反向引物后进行PCR,在产物中加入SA以增大分子量,该方法中FP与CAP的浓度呈正比。
qPCR是用于基因序列快速测定、精准定量的重要技术。通过加入各种类型的荧光标记物,在扩增过程中,荧光信号随着PCR产物的增加而增强,从而完成分析物的高灵敏度测定。
3. 结论与展望(Conclusion and perspective)
综上所述,在对低浓度抗生素检测方法的研究中,基于抗体的免疫检测技术是高通量、高灵敏度、强特异性分析方法中不可或缺的一项,其应用领域及技术已经相当成熟。但RIA由于放射性元素危害人体健康,限制了其发展,逐渐被其他免疫技术所取代。基于CGIA的免疫试纸条技术可在十几分钟内完成样品的半定量测定,非常适宜于对抗生素的快速筛查,而荧光、化学发光和电化学免疫检测技术是提高检测技术灵敏度的良好选择,其最低检测限可达pg·mL−1,线性范围跨越2个数量级。与抗体识别元件相比更为稳定的适配体具有特异性强、热稳定性、成本低、易于修饰等特点,以其为识别元件的生物传感器检测方法研究发展迅速,广泛应用于动物源性食品中抗生素的检测。随着适配体筛选技术不断改进,越来越多的高特异性适配体被选出,进一步扩大了适配体识别元件的应用范围。PCR技术能够指数级地扩增模板信号,加上后续对产物进行凝胶电泳来定量或是加入荧光基团或染料可以进行全过程荧光定量,可实现对动物源性食品中痕量抗生素的检测。
未来动物源性食品中抗生素的检测技术发展趋向于简便、高灵敏、高通量的快速原位检测。对于新的生物检测技术的开发,可从以下几个方面着手:
(1)提高技术的选择性能:传统的生物抗体识别可被性能更加优越的核酸适配体所替代,因此,筛选出高特异性适配体以及提升适配体与靶物质的亲和度,提升技术抗扰能力是未来建立单一靶物质测定的研究方向之一。
(2)降低技术的操作成本:目前食源性食品中抗生素的残留趋向于一大类抗生素乃至多种类抗生素残留,因此建立能够同时对多种抗生素产生反应的检测方法,可以提高其在实际样品中的实用性。例如可将不同抗生素的核酸适配体同时固定于丝网印刷电极或磁性粒子上。
(3)研发微型智能化技术:计算机技术和微电子技术发展迅猛,设计开发适合原位快速检测的便携技术,借助计算机网络实现数据的实时在线监测分析,改善在市场应用的可行性。
-
表 1 研究区水化学数据
Table 1. Hydrochemical data from the study area
编号ID 浓度(mg·L−1) 平衡误差/%Balance error TDS/(mg·L−1) 水温/℃Temperature pH SiO2/(mg·L−1) 总铁/(mg·L−1)Total Fe SI Na++K+ Ca2+ Mg2+ Cl– HCO3– SO42– 方解石Calcite 白云石Dolomite 石膏Gypsum Q1 26.4 61.10 4.30 5.30 195.3 57.60 0.29 252.35 14 7.5 10.7 <0.08 –0.04 –1.04 –1.83 Q2 14.5 44.04 12.6 6.04 199.2 20.37 0.45 197.15 13 7.47 7.2 0 –0.2 –0.76 –2.39 LH1 6.81 54.65 8.8 4.92 168.13 23.46 10.58 182.71 14 7.81 10. 6 0.17 0.18 –0.24 –2.24 LH2 18.54 33.95 5.2 6.89 133.98 21 2.76 152.57 15 8.2 10. 6 0.06 0.29 –0.02 –2.44 LH3 47.8 36.1 5.5 8.9 158.6 64.8 5.72 242.4 10 7.7 14.3 <0.08 –0.21 –1.12 –1.97 LH4 44.68 19.95 8.28 14.27 153.51 28.81 3.04 192.75 18 7.7 5 0.1 –0.34 –0.8 –2.55 AD1 211.78 65.41 6 26.02 83.52 513.15 1.07 864.12 13 8.57 14.08 0.18 0.46 0.07 –1.08 AD2 98.81 7.88 9 20.82 164.67 87.26 5.50 306.11 14 8.73 10.6 0.8 0.18 0.62 –2.53 AD3 17.5 53.1 12.2 12.4 210.5 28.8 3.96 229.25 16 7.8 5.4 0.4 0.27 0.12 –2.19 ZL1 539.27 405.5 22 36.43 66.12 2095.57 –0.63 3131.83 16 7.63 15.8 0.04 0.03 –0.99 –0.09 ZL2 537.81 226.46 27.24 41.33 110.56 1631.79 1.71 2519.91 19 7.08 18 0.08 –0.46 –1.58 –0.38 ZL3 535.70 457.88 17.00 44.40 87.00 2180.13 –1.25 3278.61 17 7.73 17.6 0.79 –0.32 –1.8 –0.05 ZL4 436.10 153.30 10.9 48.90 112.90 1176.70 –0.71 1882.35 17 7.9 17 <0.08 0.22 –0.46 –0.58 ZL5 371.80 222.40 4.9 44.30 134.20 1164.70 –0.04 1875.2 19 7.8 15.2 0 0.39 –0.61 –0.44 ZL6 574.80 192.40 13.40 42.50 137.30 1537.00 0.63 2428.75 22 8.5 — 0.17 0.98 1.1 –0.47 ZL7 554.00 80.20 7.30 37.20 91.50 1224.80 2.16 1949.25 23 8.77 — 0.06 0.73 0.73 –0.86 ZL8 398.00 116.20 8.50 39.00 143.40 946.20 2.67 1579.6 18 8.58 — <0.08 0.9 0.92 –0.75 ZL9 682.00 468.90 42.60 42.50 88.50 2617.60 –0.98 3897.85 18 8.49 — 0.1 1 1.22 –0.01 YA1 1086.17 473.24 40.80 42.80 71.34 3442.00 –0.09 5120.68 15 8.52 7.04 0.18 0.84 0.84 0.04 YA2 556.71 435.11 18.8 35.42 55.68 2190.12 –7.29 3264 17 7.7 10.6 0.8 0.05 –1.01 –0.06 YA3 215.94 98.78 8.91 26.67 106.14 605.88 –0.94 1009.25 15 8.18 10.6 0.4 0.37 –0.09 –0.89 YA4 112.6 29.1 6.1 20.2 131.2 194.5 1.29 428.1 13 7.8 4.8 0.04 –0.32 –1.14 –1.68 M1 1368.17 304.8 42.65 161.5 119.6 3436.9 3.18 5373.82 18 7.78 — 0.08 0.21 –0.17 –0.16 M2 1734.4 110.2 31.6 241.1 244.1 3472.6 0.52 5711.95 18 8.22 11.8 0.79 0.5 0.71 –0.6
下载: 导出CSV
表 2 不同含水层水化学组分统计(mg·L−1)
Table 2. Statistics of hydrochemical compositions in different aquifers (mg·L−1)
含水层Aquifer 统计量Statistics Na++K+ Ca2+ Mg2+ Cl− HCO3− SO42− TDS 第四系Quaternary 最大值 26.40 61.10 12.60 6.04 199.20 57.60 263.34 最小值 14.50 44.04 4.30 5.30 195.30 20.37 186.16 第四系Quaternary 平均值 20.45 52.57 8.45 5.67 197.25 38.99 224.75 标准差 5.95 8.53 4.15 0.37 1.95 18.62 38.59 洛河组Luohegroup 最大值 47.80 54.65 8.80 14.27 168.13 64.80 242.40 最小值 6.81 19.95 5.20 4.92 133.98 21.00 152.57 平均值 29.46 36.16 6.95 8.75 153.56 34.52 192.61 标准差 17.32 12.34 1.61 3.49 12.46 17.71 32.33 安定组Andinggroup 最大值 211.78 65.41 12.20 26.02 210.50 513.15 864.12 最小值 17.50 7.88 6.00 12.40 83.52 28.80 229.25 平均值 109.36 42.13 9.07 19.75 153.63 209.74 466.86 标准差 79.66 24.73 2.53 5.61 52.68 215.87 282.70 直罗组Zhiluo group 最大值 682.00 468.90 42.60 48.90 143.40 2617.60 3897.85 最小值 371.80 80.20 4.90 36.43 66.12 946.20 1579.60 平均值 514.39 258.14 17.09 41.32 107.94 1619.39 2504.29 标准差 91.25 139.62 11.29 3.71 25.11 532.93 736.86 延安组Yan’an group 最大值 1086.17 473.24 40.80 42.80 131.20 3442.00 5120.68 最小值 112.60 29.10 6.10 20.20 55.68 194.50 428.10 平均值 492.86 259.06 18.65 31.27 91.09 1608.13 2455.51 标准差 379.93 197.13 13.63 8.57 29.49 1294.67 1868.08 矿井水Mine water 最大值 1734.40 304.80 42.65 241.10 244.10 3472.60 5711.95 最小值 1368.17 110.20 31.60 161.50 119.60 3436.90 5373.82 平均值 1551.29 207.50 37.13 201.30 181.85 3454.75 5542.89 标准差 183.12 97.30 5.53 39.80 62.25 17.85 169.07
下载: 导出CSV
表 3 不同演化路径的主要矿物饱和指数
Table 3. Main mineral saturation index in different evolution paths
样品编号Sample ID 路径ⅠPath I 路径II Path II 路径III Path III ZL5 ZL3 LH2 LH4 AD2 AD1 SI(石膏Gypsum) −0.44 −0.05 −2.44 −2.55 −2.53 −1.08 SI(方解石Calcite) 0.39 0.32 0.29 0.34 0.18 0.46 SI(白云石Dolomite) −0.61 −1.8 −0.02 −0.8 0.62 0.07 SI(岩盐Halite) −6.43 −6.31 −8.42 −7.74 −7.23 −6.84 SI(CO2) −2.77 −2.25 −3.14 −2.56 −3.6 −3.77 SI(钠长石Albite) −0.68 −0.28 −2.69 −2.79 −2.78 −1.33
下载: 导出CSV
表 4 矿物转移量(mmol·L−1)
Table 4. Mineral transfer amount (mmol·L−1)
路径Path ZL5→ZL3 LH2→LH4 AD2→AD1 白云石Dolomite 0.48 0.13 –0.12 方解石Calcite –1.79 –0.09 –1.08 石膏Gypsum 10.62 0.08 4.44 CO2 0.06 0.15 — 岩盐Halite 0.005 0.2 0.13 钠长石Albite 0.26 — 1.18 阳离子交换Cation exchange 6.86 0.94 3.6 注:正值表示矿物发生溶解,进入地下水;负值表示矿物在地下水中沉淀析出离开地下水. Note: positive value indicate mineral dissolution and enter groundwater; negative value indicate mineral precipitation and leave groundwater.
下载: 导出CSV
-
[1] 柳凤霞, 史紫薇, 钱会, 等. 银川地区地下水水化学特征演化规律及水质评价 [J]. 环境化学, 2019, 38(9): 2055-2066. doi: 10.7524/j.issn.0254-6108.2019043003 LIU F X, SHI Z W, QIAN H, et al. Evolution of groundwater hydrochemical characteristics and water quality evaluation in Yinchuan area [J]. Environmental Chemistry, 2019, 38(9): 2055-2066(in Chinese). doi: 10.7524/j.issn.0254-6108.2019043003
[2] LIU P, HOTH N, DREBENSTEDT C, et al. Hydro-geochemical paths of multi-layer groundwater system in coal mining regions—Using multivariate statistics and geochemical modeling approaches [J]. Science of the Total Environment, 2017, 601/602: 1-14. doi: 10.1016/j.scitotenv.2017.05.146 [3] 吴耀国, 沈照理, 钟佐, 等. 淄博煤矿区矿井水的化学形成及其模拟 [J]. 环境科学学报, 2000, 20(4): 401-405. doi: 10.3321/j.issn:0253-2468.2000.04.004 WU Y G, SHEN Z L, ZHONG Z S, et al. Chemical origin of mine drainage and its simulation for Zibo coal mining district [J]. Acta Scientiae Circumstantiae, 2000, 20(4): 401-405(in Chinese). doi: 10.3321/j.issn:0253-2468.2000.04.004
[4] QIAO X J, LI G M, LI M, et al. Influence of coal mining on regional Karst groundwater system: A case study in West Mountain area of Taiyuan City, Northern China [J]. Environmental Earth Sciences, 2011, 64(6): 1525-1535. doi: 10.1007/s12665-010-0586-3 [5] SINGH A K, MAHATO M K, NEOGI B, et al. Hydrogeochemistry, elemental flux, and quality assessment of mine water in the pootkee-balihari mining area, jharia coalfield, India [J]. Mine Water and the Environment, 2011, 30(3): 197-207. doi: 10.1007/s10230-011-0143-7 [6] QU S, WANG G C, SHI Z M, et al. Using stable isotopes (δD, δ18O, δ34S and 87Sr/86Sr) to identify sources of water in abandoned mines in the Fengfeng coal mining district, Northern China [J]. Hydrogeology Journal, 2018, 26(5): 1443-1453. doi: 10.1007/s10040-018-1803-5 [7] 孙芳强, 侯光才, 窦妍, 等. 鄂尔多斯盆地白垩系地下水循环特征的水化学证据: 以查布水源地为例 [J]. 吉林大学学报(地球科学版), 2009, 39(2): 269-275,293. SUN F Q, HOU G C, DOU Y, et al. Hydrogeochemistry evidence of groundwater circulation features in Ordos Cretaceous basin—A case study in chabu well field [J]. Journal of Jilin University (Earth Science Edition), 2009, 39(2): 269-275,293(in Chinese).
[8] 陈晨, 高宗军, 李伟, 等. 泰莱盆地地下水化学特征及其控制因素 [J]. 环境化学, 2019, 38(6): 1339-1347. doi: 10.7524/j.issn.0254-6108.2018090504 CHEN C, GAO Z J, LI W, et al. Characteristics and possible factors of hydrochemistry in the groundwater in Tailai basin [J]. Environmental Chemistry, 2019, 38(6): 1339-1347(in Chinese). doi: 10.7524/j.issn.0254-6108.2018090504
[9] 韩佳君, 周训, 姜长龙, 等. 柴达木盆地西部地下卤水水化学特征及其起源演化 [J]. 现代地质, 2013, 27(6): 1454-1464. doi: 10.3969/j.issn.1000-8527.2013.06.025 HAN J J, ZHOU X, JIANG C L, et al. Hydrochemical characteristics, origin and evolution of the subsurface brines in western Qaidam basin [J]. Geoscience, 2013, 27(6): 1454-1464(in Chinese). doi: 10.3969/j.issn.1000-8527.2013.06.025
[10] 华琨, 李洲, 李志. 黄土区长武塬地下水水化学特征及控制因素分析 [J]. 环境化学, 2020, 39(8): 2065-2073. doi: 10.7524/j.issn.0254-6108.2019052703 HUA K, LI Z, LI Z. The hydrochemical characteristics and controlling factors of groundwater in the Changwu loess tableland [J]. Environmental Chemistry, 2020, 39(8): 2065-2073(in Chinese). doi: 10.7524/j.issn.0254-6108.2019052703
[11] ANDRÉ L, FRANCESCHI M, POUCHAN P, et al. Using geochemical data and modelling to enhance the understanding of groundwater flow in a regional deep aquifer, Aquitaine Basin, south-west of France [J]. Journal of Hydrology, 2005, 305(1/2/3/4): 40-62. [12] 林永生, 裴建国, 杜毓超, 等. 基于多元统计方法的岩溶地下水化学特征及影响因素分析 [J]. 环境化学, 2016, 35(11): 2394-2401. doi: 10.7524/j.issn.0254-6108.2016.11.2016032801 LIN Y S, PEI J G, DU Y C, et al. Hydrochemical characteristics of Karst groundwater and their influencing factors based on multiple statistical analysis [J]. Environmental Chemistry, 2016, 35(11): 2394-2401(in Chinese). doi: 10.7524/j.issn.0254-6108.2016.11.2016032801
[13] SOUMYA B S, SEKHAR M, RIOTTE J, et al. Inverse models to analyze the spatiotemporal variations of chemical weathering fluxes in a granito-gneissic watershed: Mule Hole, South India [J]. Geoderma, 2011, 165(1): 12-24. doi: 10.1016/j.geoderma.2011.06.015 [14] SHARIF M U, DAVIS R K, STEELE K F, et al. Inverse geochemical modeling of groundwater evolution with emphasis on arsenic in the Mississippi River Valley alluvial aquifer, Arkansas (USA) [J]. Journal of Hydrology, 2008, 350(1/2): 41-55. [15] 张丽, 陈永金, 刘加珍, 等. 东平湖水化学特征及成因分析 [J]. 环境化学, 2021, 40(5): 1490-1502. doi: 10.7524/j.issn.0254-6108.2019122502 ZHANG L, CHEN Y J, LIU J Z, et al. Analysis on hydrochemical characteristics and causes of Dongping Lake [J]. Environmental Chemistry, 2021, 40(5): 1490-1502(in Chinese). doi: 10.7524/j.issn.0254-6108.2019122502
[16] GAMMONS C H, BROWN A, POULSON S R, et al. Using stable isotopes (S, O) of sulfate to track local contamination of the Madison Karst aquifer, Montana, from abandoned coal mine drainage [J]. Applied Geochemistry, 2013, 31: 228-238. doi: 10.1016/j.apgeochem.2013.01.008 [17] HAN Y, WANG G C, CRAVOTTA C A III, et al. Hydrogeochemical evolution of Ordovician limestone groundwater in Yanzhou, North China [J]. Hydrological Processes, 2013, 27(16): 2247-2257. doi: 10.1002/hyp.9297 [18] HUANG X J, WANG G C, LIANG X Y, et al. Hydrochemical and stable isotope (δD and δ18O) characteristics of groundwater and hydrogeochemical processes in the ningtiaota coalfield, northwest China [J]. Mine Water and the Environment, 2018, 37(1): 119-136. doi: 10.1007/s10230-017-0477-x [19] LI P Y, WU J H, TIAN R, et al. Geochemistry, hydraulic connectivity and quality appraisal of multilayered groundwater in the hongdunzi coal mine, northwest China [J]. Mine Water and the Environment, 2018, 37(2): 222-237. doi: 10.1007/s10230-017-0507-8 [20] 沈照理. 水文地球化学基础[M]. 北京: 地质出版社, 1993. SHEN Z L. Fundamental hydrogeochemistry [M]. Beijing: Geological Publishing House, 1993(in Chinese).
[21] EDMUNDS W M, GUENDOUZ A H, MAMOU A, et al. Groundwater evolution in the continental intercalaire aquifer of southern Algeria and Tunisia: Trace element and isotopic indicators [J]. Applied Geochemistry, 2003, 18(6): 805-822. doi: 10.1016/S0883-2927(02)00189-0 [22] 栾风娇, 周金龙, 贾瑞亮, 等. 新疆巴里坤-伊吾盆地地下水水化学特征及成因 [J]. 环境化学, 2017, 36(2): 380-389. doi: 10.7524/j.issn.0254-6108.2017.02.2016062001 LUAN F J, ZHOU J L, JIA R L, et al. Hydrochemical characteristicsand formation mechanism of groundwater in plain areas of Barkol-Yiwu Basin, Xinjiang [J]. Environmental Chemistry, 2017, 36(2): 380-389(in Chinese). doi: 10.7524/j.issn.0254-6108.2017.02.2016062001
[23] 马庆伟, 杨晨光. 陕北地区砂岩的技术指标特性 [J]. 筑路机械与施工机械化, 2019, 36(2): 83-86. doi: 10.3969/j.issn.1000-033X.2019.02.015 MA Q W, YANG C G. Technical characteristics of sandstone in northern Shaanxi [J]. Road Machinery & Construction Mechanization, 2019, 36(2): 83-86(in Chinese). doi: 10.3969/j.issn.1000-033X.2019.02.015
[24] QU S, SHI Z M, LIANG X Y, et al. Multiple factors control groundwater chemistry and quality of multi-layer groundwater system in Northwest China coalfield—Using self-organizing maps (SOM) [J]. Journal of Geochemical Exploration, 2021, 227: 106795. doi: 10.1016/j.gexplo.2021.106795 [25] 刘瑞平, 徐友宁, 亢文婷. 基于phreeqci和netpath联合反演水文地球化学过程: 以小秦岭太峪水库为例 [J]. 西北地质, 2019, 52(1): 239-243. LIU R P, XU Y N, KANG W T. Based on phreeqci and netpath joint inversion hydrology geochemistry process: Example from the Xiaoqinling Tianyu reservoir [J]. Northwestern Geology, 2019, 52(1): 239-243(in Chinese).
[26] GASTMANS D, HUTCHEON I, MENEGÁRIO A A, et al. Geochemical evolution of groundwater in a basaltic aquifer based on chemical and stable isotopic data: Case study from the Northeastern portion of Serra Geral Aquifer, São Paulo state (Brazil) [J]. Journal of Hydrology, 2016, 535: 598-611. doi: 10.1016/j.jhydrol.2016.02.016 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3678
- HTML全文浏览数: 3678
- PDF下载数: 101
- 施引文献: 0
