























-
有机磷酸酯(organophosphorus esters, OPEs)是一类重要的有机磷阻燃剂(organophosphate flame retardants, OPFRs),主要以磷作为支架,用链烃、芳香烃或卤代烃取代磷酸上的氢原子组成。按性质的差异,OPEs可划分为卤代和非卤代有机磷阻燃剂[1],前者主要是氯代有机磷酸酯(Chlorinated organophosphates, Cl-OPEs),主要包括磷酸三(2-氯乙基)酯(TCEP),磷酸三(2-氯丙基)酯(TCPP)和磷酸三(1,3-二氯-2-丙基)酯(TDCPP)等。20世纪80年代之后,随着溴代阻燃剂在世界范围内的禁用,OPFRs因具有良好的阻燃效果及其低烟、低毒、低卤等特点,得到了广泛应用[2],2015年全球OPFRs消耗量达到68万t[3]。鉴于OPEs是以简单物理添加而非键合的方式添加到材料中,可以通过挥发和表面磨损进入环境,近年来,OPEs在室内灰尘[4-5]、大气[6-7]、水体[8-13] 、土壤[14]、沉积物[11,15]、生物体[12,16]、污泥[11,17]和垃圾填埋场[18]等多种环境介质中频繁检出。尽管确切的人体暴露数据还比较有限,大量利用斑马鱼、大型溞、小鼠等模式生物或借助体外细胞进行的试验表明,OPEs具有多种毒性效应或指向了明显的致毒趋势,如急性毒性[19-22]、生殖与发育毒性[22-30]、神经毒性[24,31-34]、脏器毒性[35]、基因毒性和致突变性[19,36-38]、内分泌干扰性[39-43]和致癌性[20,44]。表1列出了主要OPEs的基本信息。
随着研究的深入,Cl-OPEs作为特殊的一类OPEs越来越受到学界关注。首先,Cl-OPEs是环境赋存最广的一类OPEs,在各介质的检出组成中占据主导地位,其浓度水平为ng·L−1—μg·L−1(水体和垃圾渗滤液),pg·m−3(空气),ng·g−1(土壤、室内灰尘、生物体和沉积物)。例如,TCPP和TCEP被证实是水环境中丰度最高的两种OPEs[8-10],浓度分别可达921 ng·L−1 (TCPP)和268 ng·L−1 (TCEP)[10];以TCPP为主的Cl-OPEs在我国土壤OPEs中占74%以上[14];TCEP在我国城市家庭和大学宿舍室内灰尘中的含量高达208 μg·g−1[5];TCPP在德国北海大气OPEs中的占比高达60%±16%[7],TCPP和TDCPP是韩国石洼人工湖沉积物中的主导OPEs[45],其中TCPP浓度达到2500 ng·g−1。其次,Cl-OPEs具有显著、广泛的生物毒性效应。一方面,在所有OPEs表现出的生物毒性种类中均观察到Cl-OPEs,且只有Cl-OPEs被报道具有致癌性[44];另一方面,研究也证实Cl-OPEs中的TDCPP生物毒性极强,例如TDCPP对虹鳟鱼的96 h-LC50仅为1.1 mg·L−1[20],对斑马鱼仔鱼和胚胎的116 h-LC50仅为7 mg·L−1[19],室内灰尘中的TDCPP可能引起男性激素水平改变和精液质量显著下降[29],其代谢物双(1,3-二氯-2-丙基)磷酸盐(BDCPP)对斑马鱼胚胎是比本体强4个数量级的致畸剂[31]等。另外,Cl-OPEs是最难降解的一类OPEs,由于其对生物和化学降解的抵制倾向,以活性污泥法为基础的常规污水处理技术收效甚微[46],改性沸石[47]和碳纳米管[48]等吸附剂被报道可以有效地吸附水中的Cl-OPEs,但后续处理较为棘手,在此背景下,高级氧化技术(AOPs)在Cl-OPEs降解上得到应用。然而,Cristale等的研究表明TCEP、TDCPP和TCPP在AOPs的应用中也是最顽固的OPFRs[49],例如,UV/H2O2过程中,TCPP、TCEP和TDCPP烷基链上的Cl的存在显著降低了·OH对烷基磷酸酯的反应活性,有机物如腐殖质对UV的辐射吸收抑制了H2O2的消耗,增大了污水处理厂(WWTP)废水中Cl-OPEs的去除难度。
由于Cl-OPEs在环境中检出浓度较高、毒性效应显著且难以生化降解,亟需研究控制Cl-OPEs的替代处理技术。目前,在Cl-OPEs降解领域三种具有代表性的探索方向是水解、微生物降解和光降解。近来,以复合金属/半导体基光催化剂、强自由基发生基质(例如过硫酸盐的活化溶液)和可持续光能体系为优化方向的光催化氧化技术日渐流行。本文综述了Cl-OPEs的3种主要降解途径和机理,其中重点介绍了光降解Cl-OPEs的方法、特点和效果,通过分析比较不同降解途径的优势与不足,对未来Cl-OPEs降解的研究方向和前景提出展望。
-
有机磷物质的水解可以通过两条途径进行:(1) OH−和H2O攻击P原子,该亲电位点较“硬”,反应条件较为苛刻;(2) H2O攻击烷氧基上的α-碳,该亲电位点较“软”,反应较易进行[50]。这两条途径的断键机理如图1所示[51]。
-
鉴于OPEs的基本分子结构(即酯键),磷酸酯与磷酸的非生物水解与它们在水环境中的稳定性密切相关,已有研究证实OPEs的水解具有显著的pH依赖性,而对于含有特征官能团Cl原子的Cl-OPEs,这种依赖性表现为碱性pH有利于水解。早年间,曾有报道称TCPP在海洋固体废物处理场的渗滤液中不会发生非生物降解[52]。近来,Su等将16种OPEs划分为烷基侧链(通式CnH2(n + 1))、氯化烷烃侧链(通式CnHxCly,x+y = 2(n+1))、芳基侧链等类型,分别在pH=7、9、11、13时水解[53],降解动力学表明不同支链类型OPEs的稳定性排序:烷基>含氯烷烃>芳基,3种主要Cl-OPEs即TCEP、TCPP、TDCPP半衰期分别为0.083、11、0.044 d,降解速率常数分别为8.36、0.0621、15.6 d−1,三者均在35 d内被广泛降解。在pH=13的溶液中观察到Cl-OPEs降解速度比烷基OPEs快得多,例如,尽管每个取代基只有一个原子(Cl原子或H原子)的差异,TEP的t1/2,pH=13为16 d,而TCEP的t1/2,pH=13仅为0.083 d。水解产物筛选和质量平衡研究表明Cl-OPEs碱基催化水解的最终产物是二酯而非推测中的单酯,可能是二酯磷酸在碱性条件下被脱质子到相应的共轭碱后性质相当稳定导致的。近来,Wu等通过化合物特异性同位素分析方法(CSIA)表征了OPEs在水生环境中依赖于水解过程的同位素分馏模式[54],发现在较低的pH (pH=2−7)下有显著的碳分馏,但在较高的pH (pH=12)下无明显的碳分馏,在任何水解实验中都没有观察到氢的分馏,表明酸性或中性条件下造成C—O键的断裂,碱性水解则造成P—O键的断裂,进一步区分了两种不同的pH依赖的水解途径。
-
近两年的研究发现矿物催化水解是实际土壤或水生环境Cl-OPEs控制中一个重要的潜在途径。Fang等报道了TDCPP、TCPP、TCEP等8种OPEs在没有和存在金属氧化物矿物情况下的水解[55]。通常Cl-OPEs在中性pH条件的均相溶液中水解缓慢,半衰期t1/2>2年,但在矿物悬浮液中(pH=6,25℃,矿物表面积负载为15 m2·L−1),t1/2可缩短至不到10 d,其中三价铁系氢氧化物和氧化物(α-FeOOH、γ-FeOOH和α-Fe2O3)催化效果最好,铝和硅氧化物(Al2O3和SiO2)催化作用不明显。以代表性的铁系矿物针铁矿为例证实了表面相互作用是促进水解反应的关键,这种相互作用取决于矿物表面和OPFR的结构,进一步阐明了溶解金属离子和矿物表面催化有机磷酸酯水解反应的3种机理,包括表面金属原子与磷酸盐中心直接配位使SN2@P反应更易进行,表面金属原子与酯基直接配位削弱其与磷中心的键合从而促进裂解,以及形成表面Fe(III)羟基物种来提高矿物−水界面上的亲核剂浓度从而促进水解反应。另外,转化产物的高效液相色谱-四极杆飞行时间串联质谱分析(HPLC-Q-TOF-MS)证实酯水解为活性降解途径,单个OPE的kB值是高度可变的,并且与相应的醇离去基的酸离解常数(pKa)密切相关,矿物与OPEs的相互作用是速率控制的,可以通过调控反应速率促进Cl-OPEs的矿物水解。
-
微生物降解方法因能有效避免由化学降解方法产生的二次污染而受到关注。WWTPs中使用的活性污泥法对芳基和非卤代烷基OPFRs有一定的降解效果,但无法降解Cl-OPEs,因为活性污泥菌胶团中的微生物没有能使Cl-OPEs磷酯键断裂的特异性酶。微生物降解法的突出特点是选择性好、降解率高、成本低廉和环境友好,研究的重难点在于筛选出能降解目标化合物的菌种。
近十年来,被报道的可生物降解的OPEs主要是非卤代烷基磷酸酯中的TBP[56-62]和芳香族磷酸酯中的TPhP[63-66],针对Cl-OPEs的生物降解展开研究的主要是日本学者Takahashi领导的课题组,主要目标化合物为TCEP和TDCPP[67-72]。虽然研究规模较小,但该团队在筛选、培养、分离有效菌种,优化降解条件,细胞系统设计,解毒技术开发,基因工程,产物鉴定等方面做了一系列细致而深入的工作。具有降解Cl-OPEs能力的微生物及其降解性能总结见表2。由于TCEP和TDCPP各自的代谢产物2-氯乙醇(2-CE)和1,3-二氯-2-丙醇(1,3-DCP)也具有毒性,仅用目标化合物的去除率不足以完整评估其微生物降解效果,相关研究中特别引入“解毒率”这一概念来量化用微生物方法去除目标化合物以及各自的致毒代谢产物的程度。
2008年,Takahashi等以TCEP或TDCPP为唯一磷源,尝试富集具有降解Cl-OPEs性能的菌种,首次报道了能降解TCEP和TDCPP的微生物[67],降解TCEP和TDCPP的优势菌种Acidovorax-和Sphingomonas-属可分别催化氯醇脱卤和OPEs的磷酸酯键裂解。接着,该团队就TCEP和TDCPP及各自代谢产生的氯醇2-CE和1,3-DCP分别设计了完全解毒的细胞系统[68-69]。一方面,利用TCM1菌株和2-CE降解菌(Xanthobacter autotrophicus菌株GJ10)开发了TCEP的两步反应解毒技术[68],包括TCEP在静息细胞TCM1的作用下24 h内水解成2-CE,然后在生长细胞GJ10的作用下经过144 h降解所有生成的2-CE,联合反应完全解毒9.6 μmol·L−1 TCEP。另一方面,开发了使用菌株TCM1和Arthrobacter sp.株PY1同时降解1,3-DCP而使TDCPP完全解毒的技术[69]。由于TCM1静息细胞对TDCPP脱磷和菌株PY1静息细胞对1,3-DCP脱卤的最佳条件相似,采用两种菌株同时对TDCPP及其代谢物进行降解以缩短反应时间,在30 ℃、pH=9.0、[Tris–H2SO4]=50 mmol·L−1菌株PY1细胞OD660=4.0的情况下,12 h内实现了TDCPP的完全解毒。这两个系统的设计为微生物解毒潜在的有毒持久性有机磷化合物开辟了一条道路。
此外,该团队在基因工程和降解路径推导方面也取得了一些研究成果,降解路径的推导依赖于产物鉴定,主要手段有离子色谱法[57]或衍生化气相色谱法[60]。Abe等从能够降解TCEP和TDCPP的两种菌株Sphingobium sp.TCM1和Sphingobium sp.TDK1中纯化克隆出两种卤代烷基磷水解酶,并分别命名为TDK-HAD和TCM-HAD,这两种磷酸三酯酶成功降解了Cl-OPEs,同时也能广泛降解溴化有机磷(如TDBPP)和三芳基磷酸盐(如TCP和TPhP)[70]。有机磷化合物对微生物细胞生长的利用被认为是由三种类型的磷酸酯酶介导的:磷酸三酯酶(PTE),磷酸二酯酶(PDE)和磷酸单酯酶(PME)。Takahashi等利用TCEP对Sphingobium sp.菌株TCM1进行碱性磷酸酶基因鉴定并推导了TCEP分步降解的路径[72],如图2所示,TCEP在菌株TCM1中的代谢包括全部3种磷酸酯酶,在每一步中,磷酸酯键中的一个被水解生成2-CE,最后一步生成Pi用于细胞生长。
-
Cl-OPEs生物降解的最新进展源于针对混合OPFRs废水的实地生物修复案例。Liu等建立了综合垂直流人工湿地(IVCWs)以研究短期暴露于3种OPFRs(TCrP,TCEP,TCPP)后的环境行为和根际微生物反应[73],揭示了利用IVCWs处理以Cl-OPEs为主的OPFRs混合污染的潜力。研究发现,在处理低碳/氮比废水时,IVCWs对OPFRs废水既表现出良好的TN去除效果((628.13±110.63) mg·m−2·d−1),又具有相当的OPFRs去除效率((48.37%±9.52%)%—(82.28%±7.48%));对比高疏水化合物磷酸三甲苯(TCrP)在土壤、火成岩、沸石和植物组织中的吸附,Cl-OPEs则更容易被吸收并转移到植物体内;OPFRs与根际微生物的丰度、多样性和氮相关的微生物之间存在显著的相互作用;Pseudomonas属和Sphingobium属可能对混合OPFRs具有生物降解功能。
-
高级氧化技术(AOPs)是去除水溶液中难降解有机污染物的有效技术,包括臭氧化、光催化氧化、芬顿氧化、硫酸根自由基氧化等[74],其本质是通过各种途径生成羟基自由基(·OH)和硫酸盐自由基(
⋅SO−4 ),借助活性自由基反应将大分子有机物降解成小分子,最终降解成CO2和H2O。·OH和⋅SO−4 的氧化还原电位分别为1.9—2.7 V和2.5—3.1 V,具体电位取决于溶液pH[75]。在诸多AOPs中,光氧化的降解和解毒潜力越来越受到关注,该方法的吸引力来源于三方面:(a)添加光氧化剂不会进一步污染环境;(b)有希望将太阳光用作光源;(c)是低成本的补救方法,可在现场进行[76]。使用光氧化降解手段处理Cl-OPEs在近十年、尤其是近3年得到了较快的发展。按光源的不同,光氧化降解Cl-OPEs可分为紫外光(UV)降解和可见光(Vis)降解。鉴于Cl-OPEs的降解难度和高能量UV降解的稳定、高效性能,UV是学者开展Cl-OPEs降解研究的首选光源。然而,由于Cl-OPEs侧链烷基缺乏发色团,没有吸收波峰,很难直接光解,通常需要采用基于UV的复合光降解技术。按研究历程有三种复合思路:UV结合生成·OH的氧化剂如H2O2、O3[49,54,75,77-84],UV结合催化剂如TiO2和MOF材料[85-90],UV结合生成
⋅SO−4 的氧化剂如过硫酸盐PS/PMS[83,89,91-93]。UV和这些额外添加的试剂协同生成大量的·OH或⋅SO−4 有效降解Cl-OPEs。能量较低的可见光在应用于Cl-OPEs降解时则往往需要耦合性能更优良的光催化剂,如N、S共掺杂TiO2和优化的MOF材料,或者外加过硫酸盐形成活化体系才能达到不错的降解效果[89,94-95],但其对可见光的利用揭示了构建可持续光能体系的趋势。 -
在含有氧化剂H2O2的UV照射水中能形成大量的·OH,引发了UV/H2O2在全规模的水处理厂降解微污染有机物中的设计和实施[96]。Cl-OPEs在UV-C范围内是较差的光吸收剂,空白实验也表明H2O2不能单独促进化合物的降解,故UV/H2O2是UV和H2O2光解生成·OH氧化降解污染物的协同作用[80]。
多项研究表明,UV/H2O2是降解Cl-OPEs有效、可靠的技术,其光降解性能参数和效率见表3。目前,使用UV/H2O2降解OPEs的研究已经比较体系化,内容包括探究支链结构、光强、pH、底物浓度、氧化剂浓度、天然有机物的竞争性等因素对降解效率的影响,用C/C0、Cl−和
PO3−4 和总有机碳(TOC)量化降解效率,测定二级反应速率kOH以模拟自然水域中的污染物氧化,探究降解前后的生物毒性和生物利用性的变化,以及推导特定目标物的降解路径等。OPEs支链的分子结构显著影响UV/H2O2条件下·OH对目标物的降解速率。Watts等研究了TCEP、TCPP、TBP和TBEP等4种磷酸三酯的二阶反应速率,kOH, TCEP=5.6×108 mol·L−1·s−1,kOH, TCPP=1.98×108 mol·L−1·s−1,kOH, TBP=6.4×109 mol·L−1·s−1,kOH, TBEP=1.03×1010 mol·L−1·s−1,排序为TBEP>TBP>TCEP>TCPP[78],即烷基OPEs的降解速率高于Cl-OPEs,与水解时的规律一致。Cl-OPEs支链上的Cl原子显著降低了H原子的可用性,导致降解速率较慢。另外,随着烷基链的卤化,碳链数和加成物α链上的杂原子(O)也影响了·OH攻击的速率。理论上,TCPP在每个碳链上比TCEP多一个碳,降解速率应高于TCEP,然而实验结果正相反,很可能是受TCPP的α-碳处附加的—CH3的影响。此后,He等在超纯水中,用250 W紫外光照射50 mg·L−1 H2O2降解TCPP[75],动力学计算表明速率常数为0.0035 min−1(R2=0.9871),降解率达到96%,但TOC去除率仅有43.02%,证实了TCPP的矿化难度较大。
UV/H2O2体系中TCEP的去除效果与H2O2的投加量及其氧化特性密切相关。一方面,调控初始H2O2浓度对TCEP的去除非常关键。在5 mg·L−1 TCEP,pH=7碳酸盐缓冲溶液中外加50 mg·L−1 30% H2O2,用低压汞灯(λ=254 nm)照射,降解率达95%以上[77]。但31P核磁共振波谱法对UV/H2O2体系的监测表明,使用高浓度的氧化剂来改善混合OPEs中TCEP的去除可能会导致·OH氧化的抑制,使得TCEP无法完全降解[82]。以反应时间1 h为基本条件考察H2O2用量对UV/H2O2降解143 mg·L−1TCEP的影响[80],发现增加H2O2的用量提高了Cl−和
PO3−4 浓度,5 mmol·L−1 H2O2性价比最高,在最佳条件下,TCEP去除率大于85%,产生的Cl−和PO3−4 、以及总有机碳(TOC)去除率分别达到了86%、94%和97%。另一方面,研究发现H2O2作氧化剂去除Cl-OPEs时自身具有高效低耗的优势。例如,在UV/H2O2和O3氧化的对比研究[49,78,81]中,两者在纯水、腐殖酸(HA)溶液和城市二级废水等不同水基质中都能快速降解OPEs,但UV/H2O2对目标Cl-OPEs的去除率更高,且达到同等氧化效果的能耗更低。需要注意的是,HA和城市二次出水中复杂基质材料的竞争性氧化会通过与目标物竞争紫外光和·OH抑制UV/H2O2对Cl-OPEs的降解。 -
光催化技术及光催化材料的研究是目前最为活跃的研究方向之一。使用光催化技术降解Cl-OPEs的优势在于不需使用昂贵的氧化剂,也不易产生毒性更高的中间产物。
TiO2及其改性产品是最常用和最早实现商品化的光催化剂,具有催化活性高、应用范围广、清洁无二次污染、价格低廉等优点。TiO2的禁带宽度大(3.2 eV),产生的光生电子和空穴氧化还原电势高,且稳定性强,因而在光催化剂向有机半导体基/金属基高分子纵深发展的今天,TiO2仍然被广泛应用于实际工程领域中污染物的去除。UV/TiO2在降解Cl-OPEs上的有效性很早就得到了证实。刘青等采用商品化的TiO2-P25在UV下降解TCPP[85],UV和P25联合作用下12 h降解率达到80%,用高温醇热法进一步合成暴露(001)活性晶面TiO2,与其特征波长365 nm的紫外光联合作用,90 min TCPP降解率超过50%,360 min内完全降解,最优催化加投加量为1 g·L−1。中性和酸性时的降解率要略微大于碱性条件,但不同pH下TCPP在6 h内降解率都达到90%,表明改性催化剂TiO2-001的pH适应性更强,有潜力应用于不同酸碱度TCPP废液的处理。
天然有机物、无机离子和腐殖酸能够显著影响UV/TiO2对Cl-OPEs的降解。有报道称光催化降解TCEP的效率受pH、天然有机物和阴离子影响很大,在实际的水处理过程中,TCEP很难完全矿化[86]。Tang等在使用UV/TiO2光催化降解TCEP、TCPP和TDCPP时重点考察了无机离子和腐殖酸的作用[87],结果表明无机离子如
PO3−4 、SO2−4 、Cl−、NO−3 和HA均能抑制降解,抑制能力排序PO3−4 >SO2−4 >Cl−>NO−3 ,抑制效果随离子浓度的增加而增强,主要的2个抑制机制为:(1)无机离子或HA与TCEP竞争·OH;(2)无机离子和HA抢占TiO2的表面活性位点,发生表面结合氧化,降低了TiO2的催化作用。金属-有机骨架(MOFs)是一类具有超高的比表面积、较高且可调的孔隙率和开放的金属位点的新型多孔材料,作催化剂时具有多相催化后易回收分离、可循环使用的优点。光和MOFs的结合是降解Cl-OPEs的新方向。Hu等制备了禁带宽度为2.41 eV的高纯度规整晶体MIL-101(Fe),结合UV在过硫酸盐体系中降解TCEP[89],合成的MIL-101(Fe)可利用短波可见光(400—520 nm)和紫外光(200—400 nm),被光激活的MIL-101(Fe)使Fe(Ⅲ)转化为Fe(Ⅱ),进而使
S2O2−8 进一步转化为中心点SO−4 ,和·OH共同氧化降解TCEP。尽管420 nm辐照、酸性pH、高温、加入S2O2−8 都能促进TCEP的降解,该体系的实验室最高去除率仅停留在(3 h)80%—85%,小于UV/PS体系达到的最高去除率(280 nm, 3 h)95%—100%,表明MOF材料在去除TCEP上的性能表现不够理想,但420 nm/MIL101(Fe)/PS体系比均相UV/PS体系在实际水基质中去除TCEP方面表现更佳,意味着它将是一种潜在的水体污染去除技术。显然,基于MOFs的多相光催化在实际水体中的稳定性、可用性等方面的应用存在很大的挑战。 -
活化过硫酸盐(包括过二硫酸盐PS和过一硫酸盐PMS)由于操作简单、氧化性高、应用广泛,被认为是处理OPEs的有前景的技术,其原理主要是活化产生硫酸根自由基(
⋅SO−4 )氧化降解OPEs。以⋅SO−4 为主要反应物种相比以·OH为主要反应物种有一些优势,比如氧化电位更高、对目标物污染物选择性强、产生更少的有害副产物等。以Co2+或Fe2+为典型的过渡金属、紫外光照射和加热都能激发PM/PMS从而产生⋅SO−4 ,但是外加重金属有二次污染的风险,加热涉及能耗问题,因此在实际水处理中,UV/PS和UV/PMS的可行性更高。从降解效率来看,UV/过硫酸盐在所有方法里是最高效的,在目标污染物浓度都为1 mg·L−1的情况下,不同研究显示出的降解效率结果较为一致。Ou等研究了TCEP在254 nm UV/PS体系下的降解效果,结果表明该方法对TCEP有较高的转化效率(0.1217 min−1),30 min降解率达99%,证明UV/PS是一种高效的方法[91]。类似地,Xu等研究了UV/PMS体系下TCEP的降解情况,30 min降解率达94.6%,同时发现Fe3+可以提高降解效果,而阴离子和HA则会抑制降解作用,降解效果在纯水、自来水、合成水(含NaHCO3、CaSO4·2H2O、MgSO4和KCl)、二沉池水、九乡河水中依次递减[92]。Yu等利用UV/PS降解TCPP,反应符合准一级动力学,25 min降解率为98.2%,表观速率常数kobs为0.1653 min−1,随着光催化反应的进行,TCPP通过活化的硫酸根诱导的选择性电子转移反应转化为12种降解中间体。阴离子的存在和高pH对降解效率有显著的抑制作用,说明实际水处理过程中TCPP很难完全矿化,与使用UV/H2O2方法时得出的结论一致[93]。 -
在光催化降解污染物的过程中,材料的选择与制备对于降解性能至关重要。尽管无毒、低成本的TiO2具有良好的化学稳定性,但其禁带宽度较大(−3.2 eV)只能吸收紫外光,造成太阳光的利用率严重受限(小于5%)。尽管利用可见光降解Cl-OPEs的研究非常有限,但其体现了光催化发展从紫外光向可见光转变的趋势。在光源选择发生转变的同时,学者也在积极探索光催化剂和光催化体系的优化,催化剂的优化表现为从以TiO2为典型的无机催化剂转向表面掺杂高聚合TiO2或有机高分子MOF材料,体系优化表现为从较为简单的UV/TiO2、UV/H2O2体系转向光/复合金属或半导体基光催化材料/过硫酸盐的多相光催化体系。例如,Antonopoulou等通过模拟太阳光,使用TiO2作为催化剂降解溶解在纯水中的TCPP,降解速率在0.0141—0.142 min−1(C0=25—500 μg·L−1)之间,处理后毒性降低[97]。通过掺杂改性等手段可以拓宽TiO2的光响应至可见波段,采用N、S掺杂改性TiO2对TCPP也进行了催化降解[94],在UV-Vis照射下,N、S共掺杂的TiO2是去除TCPP的最具光活性的催化剂,但6 h内,TCPP的去除率也不足65%。Lin等则在MIL-101(Fe)的基础上附载了氧化石墨烯,制备MOF材料GO@MIL-101(Fe),并在此基础上开发了可见光/MOF/H2O2的光催化体系增强可见光条件下对TCEP的降解能力,附载了氧化石墨烯后,MIL-101(Fe)材料电位从2.41eV降低到2.71 eV,从而对光的吸收范围从520 nm扩展到570 nm。GO的高电导率实现了快速活化和电子转移,30 min TCEP降解率达95%,且GO@MIL-101(Fe)在水中稳定[95]。
-
随着研究的深入,Cl-OPEs降解的安全风险越来越受到关注,这种风险主要源于降解过程中产生的高毒性含氯中间产物。由于pH依赖和矿物催化的水解方法研究尚不充分,微生物降解方法虽然选育菌种较受限但本身可以完全解毒代谢产物,对Cl-OPEs降解的安全风险探究主要聚焦在UV/H2O2、UV/TiO2、UV/PS等光降解方法研究方面。
UV/H2O2降解Cl-OPEs前后生物毒性的变化最早引发注意,但毒性结论并不统一,其升高或降低可能取决于受试生物。例如,用盐湖卤虫卵进行UV/H2O2降解TCEP前后的急性毒性测试,不论是单一TCEP还是混合OPEs降解,LC50都升高,表明急性毒性降低[82]。使用大肠杆菌评估TCPP在UV/H2O2降解前后的毒性,显示活性氧(ROS)和细胞凋亡减少,大肠杆菌的膜电位(MP)升高,生物分子功能得到修正,说明TCPP降解后毒性降低[84]。然而,根据发光菌实验数据,随着反应的进行,TCPP产物的毒性明显增加[75]。上述结果表明,需要更多降解前后的毒性变化案例佐证和完善现有的研究结果。
UV/TiO2降解Cl-OPEs的安全风险评估的研究热点是中间产物的蛋白质组学分析,因为蛋白质相互作用是揭示Cl-OPEs及其中间体在蛋白质组水平上生物分子影响的重要视角。Ye等研究了UV/TiO2非均相光催化降解水中TCEP及其对细菌蛋白质组的影响[86],自组装紫外辐射装置在投加70 mg·L−1 TiO2的情况下,对1 mg·L−1 TCEP 10 min降解率达到99%,·OH是主要的反应物种,降解过程符合准一级动力学模型,kobs=0.3167 min−1;对代谢反应、途径和网络的蛋白质组学分析发现初步降解产物能被大肠杆菌通过细胞代谢进行转运和利用;观察到抗逆性下降趋势,处理后产物毒性明显减弱,表明UV/TiO2体系下矿化不完全的TCEP的羟基化和脱氯作用对其解毒同样有效。随后Yu等将目标转向TCPP,发现Cl−和
PO3−4 的释放产生了6个稳定的中间产物,将蛋白质组学分析与KEGG代谢网络分析相结合,同样得出了毒性降低的结论,说明UV/TiO2也是一种安全高效的TCPP控制方法[90]。UV/PS降解Cl-OPEs不但高效,且毒理学评价证实其安全风险较小。例如,Ou等观察到TCEP在254 nm UV/PS体系下降解中间产物的毒性降低[91]。Xu等使用流式细胞术(FCM)分析对降解中间混合物进行的毒理学评价显示,细胞内活性氧(ROS)和细胞凋亡率明显下降,细胞膜电位(MP)较原TCPP升高,细胞周期分析表明这些降解产物对大肠杆菌DNA生物合成的负面影响也有所减弱,说明经过UV/PS处理后降解产物的毒性明显降低[92]。UV/过硫酸盐能在部分矿化Cl-OPEs的同时进行有效解毒,提示其能控制废/污水中Cl-OPEs的污染。
另外,对于MOFs材料或可见光催化降解Cl-OPEs,因为降解效果不够理想,降解机制尚不清楚,所以研究还未拓展到安全风险评估方面。
-
Cl-OPEs在环境中的高丰度、高毒性和难降解性使其去除技术成为研究热点。本文主要介绍了3种代表性的去除技术,包括pH依赖的水解、特异性磷酸水解酶促进的微生物降解和光驱动的高级氧化降解,另外还讨论了各种降解方法的安全风险。总的来说,碱性条件和矿物催化有利于Cl-OPEs水解,但这方面研究仅限于实验室领域;微生物降解具有彻底矿化和完全解毒Cl-OPEs的潜力,不足在于前期选育菌种和反应用时过长,且需要依靠两种菌株共同解毒,在实际水生或土壤环境中还可能面临低温制约;光驱动的高级氧化技术通过激发催化剂或氧化剂生成具有高度活性的·OH和
⋅SO−4 以降解Cl-OPEs,优点为成本低、二次污染小、有希望将阳光用作光源、可在现场进行,是未来主要的研究方向。在诸多光催化氧化方法中,UV/H2O2方法有效、可靠、成熟,相比O3氧化和UV/HOCl更高效和低耗,但用该方法去除TCPP矿化度较低;UV/M(光催化剂)方法的优势是不需外加氧化剂,然而,近年广受关注的MOFs材料作为降解Cl-OPEs的催化剂时性能不够理想,受pH影响大且稳定性不足;UV/PS和UV/PMS方法操作简单,对污染物选择性强,无二次污染,降解效率最高,且能在部分矿化条件下实现有效解毒,提示其能控制实际废/污水中Cl-OPEs的污染;可见光降解Cl-OPEs的优点在于对太阳光的利用,但由于其能量较低,往往需要耦合性能优良的光催化剂或外加高浓度的氧化剂才能保证降解效果,实际环境中杂质能级的引入和催化剂本身性能缺陷可能增加空穴-电子复合的几率,进而影响光催化性能,该方法同时面临技术和成本的挑战。中间产物毒性评价已经成为Cl-OPEs去除研究的重要一环,其中UV/TiO2、UV/PS已经被证实是高效且安全的降解方法,UV/H2O2降解前后的生物毒性变化仍有争议,常用的风险评估方法包括急性毒性测试、蛋白质组学分析和流式细胞术等。因此,未来Cl-OPEs的降解研究应集中开发和优化高效、绿色、无二次污染的技术。水解方面要补充基于实际水生环境和土壤基质的实验数据,尤其关注天然pH条件下降解稳态Cl-OPEs的新方法、新材料研发。微生物降解方面应注意选育能在降解目标物的同时降低其毒性的高效菌株。当然,研究着力点仍应放在光催化和高级氧化技术的联用方面,侧重点包括:(1)光催化剂性能的提升,通过改性和引入适当的有机高分子材料优化催化效果;(2)反应体系的设计优化;(3)含氯中间产物的毒性评价方法优化;(4)开发新型可见光驱动光催化剂以拓展光响应范围,并促进电荷有效利用。
氯代有机磷酸酯阻燃剂的去除技术研究进展
Advances in the removal technology of chlorinated organophosphate flame retardants
-
摘要: 有机磷酸酯类阻燃剂(organophosphate flame retardants, OPFRs)作为溴代阻燃剂的替代品得到广泛使用,其中氯代有机磷酸酯(chlorinated organophosphates, Cl-OPEs)因其在多种环境介质中检出浓度较高、生物毒性强且难以生化降解越来越受到关注。目前Cl-OPEs的降解技术研究集中在水解、微生物降解和光降解三个方面;同时,以复合金属/半导体基光催化剂、活化过硫酸盐和可持续光能体系为优化方向的高级氧化技术(AOPs)日渐成为主流。本文综述了Cl-OPEs不同降解途径的研究进展,其中重点介绍了4种光降解Cl-OPEs方法的原理、特点、效果和影响因素,通过分析比较不同降解途径的优势和不足,对未来Cl-OPEs降解的研究方向和前景提出了展望。
-
关键词:
- 氯代有机磷酸酯阻燃剂 /
- 降解途径 /
- 光催化 /
- 水解 /
- 生物降解
Abstract: Organophosphate flame retardants (OPFRs) have been widely used as an alternative to brominated flame retardants, of which chlorinated organophosphates (Cl-OPEs) have received increasing attention because of their high abundance in various environmental media, significant biotoxicity and strong resistance to conventional biochemical degradation. Efforts on Cl-OPEs degradation techniques are mainly focused on three aspects as follows: hydrolysis, microbial degradation and photodegradation. Meanwhile, advanced oxidation technologies (AOPs) with composite metal/semiconductor based photocatalysts, activated persulphates and sustainable light energy systems are becoming the mainstream. In this paper, advances in different degradation pathways of Cl-OPEs are reviewed. The principles, characteristics, effects and influencing factors of four photodegradation methods of Cl-OPEs were emphatically introduced. By analyzing and comparing the advantages and disadvantages of different degradation systems, the research direction and prospect of future degradation of Cl-OPEs are put forward. -
有机磷酸酯(organophosphorus esters, OPEs)是一类重要的有机磷阻燃剂(organophosphate flame retardants, OPFRs),主要以磷作为支架,用链烃、芳香烃或卤代烃取代磷酸上的氢原子组成。按性质的差异,OPEs可划分为卤代和非卤代有机磷阻燃剂[1],前者主要是氯代有机磷酸酯(Chlorinated organophosphates, Cl-OPEs),主要包括磷酸三(2-氯乙基)酯(TCEP),磷酸三(2-氯丙基)酯(TCPP)和磷酸三(1,3-二氯-2-丙基)酯(TDCPP)等。20世纪80年代之后,随着溴代阻燃剂在世界范围内的禁用,OPFRs因具有良好的阻燃效果及其低烟、低毒、低卤等特点,得到了广泛应用[2],2015年全球OPFRs消耗量达到68万t[3]。鉴于OPEs是以简单物理添加而非键合的方式添加到材料中,可以通过挥发和表面磨损进入环境,近年来,OPEs在室内灰尘[4-5]、大气[6-7]、水体[8-13] 、土壤[14]、沉积物[11,15]、生物体[12,16]、污泥[11,17]和垃圾填埋场[18]等多种环境介质中频繁检出。尽管确切的人体暴露数据还比较有限,大量利用斑马鱼、大型溞、小鼠等模式生物或借助体外细胞进行的试验表明,OPEs具有多种毒性效应或指向了明显的致毒趋势,如急性毒性[19-22]、生殖与发育毒性[22-30]、神经毒性[24,31-34]、脏器毒性[35]、基因毒性和致突变性[19,36-38]、内分泌干扰性[39-43]和致癌性[20,44]。表1列出了主要OPEs的基本信息。
表 1 主要OPEs名称和理化性质Table 1. Names and Physicochemical properties of the major OPEs化合物名称及简写Compounds and Abbreviation CAS 分子式Molecular formula 取代基Substituent lgKow Vp(Torr) 磷酸三乙酯(Triethyl phosphate, TEP) 78-40-0 C6H15O4P 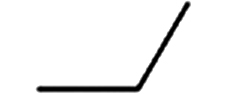
0.80 3.93×10−1 磷酸三正丁酯(Tri-n-butyl phosphate, TnBP) 126-73-8 C13H27O5P 
4.00 1.13×10−3 磷酸三异丁酯(Tri-iso-butyl phosphate, TiBP) 126-71-6 C12H27O4P 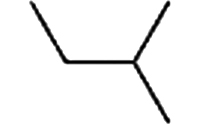
3.60 1.28×10−3 磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP) 78-51-3 C18H39O7P 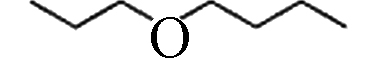
3.75 2.50×10−8 磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP) 115-96-8 C6H12Cl3O4P 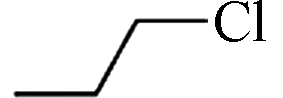
1.44 6.13×10−2 磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP) 13674-84-5 C9H18Cl3O4P 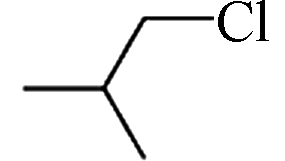
2.59 2.02×10−5 磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP) 13674-87-8 C9H15Cl6O4P 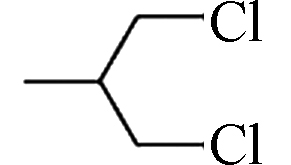
3.65 7.36×10−8 磷酸三苯酯(Triphenyl phosphate, TPhP) 115-86-6 C18H15O4P 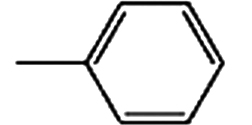
4.59 6.28×10−6 磷酸三甲苯酯(Tricresyl phosphate, TCrP) 1330-78-5 C21H21O4P 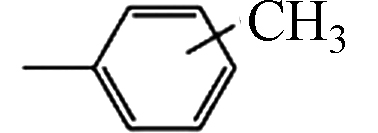
5.11 6.00×10−7 三苯基氧化膦(Triphenylphosphine oxide, TPPO) 791-28-6 C18H15OP 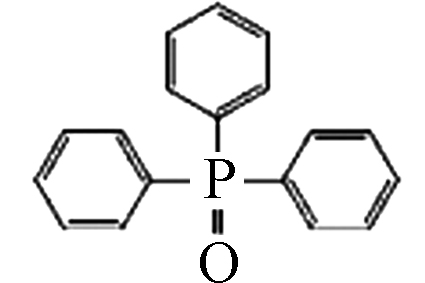
2.83 2.62×10−8 注:Kow为正辛醇-水分配系数,Vp为蒸汽压。Note: Kow is the n-octyl alcohol-water distribution coefficient, Vp is the vapor pressure. | Show Table DownLoad:
CSV
DownLoad:
CSV
随着研究的深入,Cl-OPEs作为特殊的一类OPEs越来越受到学界关注。首先,Cl-OPEs是环境赋存最广的一类OPEs,在各介质的检出组成中占据主导地位,其浓度水平为ng·L−1—μg·L−1(水体和垃圾渗滤液),pg·m−3(空气),ng·g−1(土壤、室内灰尘、生物体和沉积物)。例如,TCPP和TCEP被证实是水环境中丰度最高的两种OPEs[8-10],浓度分别可达921 ng·L−1 (TCPP)和268 ng·L−1 (TCEP)[10];以TCPP为主的Cl-OPEs在我国土壤OPEs中占74%以上[14];TCEP在我国城市家庭和大学宿舍室内灰尘中的含量高达208 μg·g−1[5];TCPP在德国北海大气OPEs中的占比高达60%±16%[7],TCPP和TDCPP是韩国石洼人工湖沉积物中的主导OPEs[45],其中TCPP浓度达到2500 ng·g−1。其次,Cl-OPEs具有显著、广泛的生物毒性效应。一方面,在所有OPEs表现出的生物毒性种类中均观察到Cl-OPEs,且只有Cl-OPEs被报道具有致癌性[44];另一方面,研究也证实Cl-OPEs中的TDCPP生物毒性极强,例如TDCPP对虹鳟鱼的96 h-LC50仅为1.1 mg·L−1[20],对斑马鱼仔鱼和胚胎的116 h-LC50仅为7 mg·L−1[19],室内灰尘中的TDCPP可能引起男性激素水平改变和精液质量显著下降[29],其代谢物双(1,3-二氯-2-丙基)磷酸盐(BDCPP)对斑马鱼胚胎是比本体强4个数量级的致畸剂[31]等。另外,Cl-OPEs是最难降解的一类OPEs,由于其对生物和化学降解的抵制倾向,以活性污泥法为基础的常规污水处理技术收效甚微[46],改性沸石[47]和碳纳米管[48]等吸附剂被报道可以有效地吸附水中的Cl-OPEs,但后续处理较为棘手,在此背景下,高级氧化技术(AOPs)在Cl-OPEs降解上得到应用。然而,Cristale等的研究表明TCEP、TDCPP和TCPP在AOPs的应用中也是最顽固的OPFRs[49],例如,UV/H2O2过程中,TCPP、TCEP和TDCPP烷基链上的Cl的存在显著降低了·OH对烷基磷酸酯的反应活性,有机物如腐殖质对UV的辐射吸收抑制了H2O2的消耗,增大了污水处理厂(WWTP)废水中Cl-OPEs的去除难度。
由于Cl-OPEs在环境中检出浓度较高、毒性效应显著且难以生化降解,亟需研究控制Cl-OPEs的替代处理技术。目前,在Cl-OPEs降解领域三种具有代表性的探索方向是水解、微生物降解和光降解。近来,以复合金属/半导体基光催化剂、强自由基发生基质(例如过硫酸盐的活化溶液)和可持续光能体系为优化方向的光催化氧化技术日渐流行。本文综述了Cl-OPEs的3种主要降解途径和机理,其中重点介绍了光降解Cl-OPEs的方法、特点和效果,通过分析比较不同降解途径的优势与不足,对未来Cl-OPEs降解的研究方向和前景提出展望。
1. 氯代有机磷酸酯的水解(Hydrolysis of chlorinated organophosphate)
1.1 Cl-OPEs的水解途径
有机磷物质的水解可以通过两条途径进行:(1) OH−和H2O攻击P原子,该亲电位点较“硬”,反应条件较为苛刻;(2) H2O攻击烷氧基上的α-碳,该亲电位点较“软”,反应较易进行[50]。这两条途径的断键机理如图1所示[51]。

1.2 Cl-OPEs水解的pH依赖性
鉴于OPEs的基本分子结构(即酯键),磷酸酯与磷酸的非生物水解与它们在水环境中的稳定性密切相关,已有研究证实OPEs的水解具有显著的pH依赖性,而对于含有特征官能团Cl原子的Cl-OPEs,这种依赖性表现为碱性pH有利于水解。早年间,曾有报道称TCPP在海洋固体废物处理场的渗滤液中不会发生非生物降解[52]。近来,Su等将16种OPEs划分为烷基侧链(通式CnH2(n + 1))、氯化烷烃侧链(通式CnHxCly,x+y = 2(n+1))、芳基侧链等类型,分别在pH=7、9、11、13时水解[53],降解动力学表明不同支链类型OPEs的稳定性排序:烷基>含氯烷烃>芳基,3种主要Cl-OPEs即TCEP、TCPP、TDCPP半衰期分别为0.083、11、0.044 d,降解速率常数分别为8.36、0.0621、15.6 d−1,三者均在35 d内被广泛降解。在pH=13的溶液中观察到Cl-OPEs降解速度比烷基OPEs快得多,例如,尽管每个取代基只有一个原子(Cl原子或H原子)的差异,TEP的t1/2,pH=13为16 d,而TCEP的t1/2,pH=13仅为0.083 d。水解产物筛选和质量平衡研究表明Cl-OPEs碱基催化水解的最终产物是二酯而非推测中的单酯,可能是二酯磷酸在碱性条件下被脱质子到相应的共轭碱后性质相当稳定导致的。近来,Wu等通过化合物特异性同位素分析方法(CSIA)表征了OPEs在水生环境中依赖于水解过程的同位素分馏模式[54],发现在较低的pH (pH=2−7)下有显著的碳分馏,但在较高的pH (pH=12)下无明显的碳分馏,在任何水解实验中都没有观察到氢的分馏,表明酸性或中性条件下造成C—O键的断裂,碱性水解则造成P—O键的断裂,进一步区分了两种不同的pH依赖的水解途径。
1.3 矿物催化水解Cl-OPEs
近两年的研究发现矿物催化水解是实际土壤或水生环境Cl-OPEs控制中一个重要的潜在途径。Fang等报道了TDCPP、TCPP、TCEP等8种OPEs在没有和存在金属氧化物矿物情况下的水解[55]。通常Cl-OPEs在中性pH条件的均相溶液中水解缓慢,半衰期t1/2>2年,但在矿物悬浮液中(pH=6,25℃,矿物表面积负载为15 m2·L−1),t1/2可缩短至不到10 d,其中三价铁系氢氧化物和氧化物(α-FeOOH、γ-FeOOH和α-Fe2O3)催化效果最好,铝和硅氧化物(Al2O3和SiO2)催化作用不明显。以代表性的铁系矿物针铁矿为例证实了表面相互作用是促进水解反应的关键,这种相互作用取决于矿物表面和OPFR的结构,进一步阐明了溶解金属离子和矿物表面催化有机磷酸酯水解反应的3种机理,包括表面金属原子与磷酸盐中心直接配位使SN2@P反应更易进行,表面金属原子与酯基直接配位削弱其与磷中心的键合从而促进裂解,以及形成表面Fe(III)羟基物种来提高矿物−水界面上的亲核剂浓度从而促进水解反应。另外,转化产物的高效液相色谱-四极杆飞行时间串联质谱分析(HPLC-Q-TOF-MS)证实酯水解为活性降解途径,单个OPE的kB值是高度可变的,并且与相应的醇离去基的酸离解常数(pKa)密切相关,矿物与OPEs的相互作用是速率控制的,可以通过调控反应速率促进Cl-OPEs的矿物水解。
2. 氯代有机磷酸酯的微生物降解(Microbial degradation of chlorinated organophosphates)
2.1 Cl-OPEs生物降解性能
微生物降解方法因能有效避免由化学降解方法产生的二次污染而受到关注。WWTPs中使用的活性污泥法对芳基和非卤代烷基OPFRs有一定的降解效果,但无法降解Cl-OPEs,因为活性污泥菌胶团中的微生物没有能使Cl-OPEs磷酯键断裂的特异性酶。微生物降解法的突出特点是选择性好、降解率高、成本低廉和环境友好,研究的重难点在于筛选出能降解目标化合物的菌种。
近十年来,被报道的可生物降解的OPEs主要是非卤代烷基磷酸酯中的TBP[56-62]和芳香族磷酸酯中的TPhP[63-66],针对Cl-OPEs的生物降解展开研究的主要是日本学者Takahashi领导的课题组,主要目标化合物为TCEP和TDCPP[67-72]。虽然研究规模较小,但该团队在筛选、培养、分离有效菌种,优化降解条件,细胞系统设计,解毒技术开发,基因工程,产物鉴定等方面做了一系列细致而深入的工作。具有降解Cl-OPEs能力的微生物及其降解性能总结见表2。由于TCEP和TDCPP各自的代谢产物2-氯乙醇(2-CE)和1,3-二氯-2-丙醇(1,3-DCP)也具有毒性,仅用目标化合物的去除率不足以完整评估其微生物降解效果,相关研究中特别引入“解毒率”这一概念来量化用微生物方法去除目标化合物以及各自的致毒代谢产物的程度。
表 2 具有降解Cl-OPEs性能的微生物及其降解性能总结Table 2. Summary of (Cl-OPEs)-degrading bacterial species and their performance菌种Bacterial species Cl-OPEs及代谢物Cl-OPEs and metabolites 降解/解毒率Degradation/detoxification rate 参考文献Reference (TCEP) Acidovorax spp.,Sphingomonas spp.(TDCPP) Acidovorax spp.,Aquabacterium spp.,Sphingomonas spp. TCEP TDCPP 100% (6 h, [TCEP]0=20 µmol·L−1)100% (3 h, [TDCPP]0=20 µmol·L−1) [67] (TCEP) Sphingobium sp.strain TCM1,(2-CE) Xanthobacter autotrophicus strain GJ10 TCEP2-CE 最佳条件测试:降解100% (30 ℃, pH=8.5, [Co2+]=50 μmol·L−1, OD660=0.8, [TCEP]0=10 µmol·L−1) 解毒100%(24 h, [2-CE]0=180 µmol·L−1)分步解毒:降解/解毒100%(①4 h,[TCEP]0=10 µmol·L−1; ②144 h,[2-CE]0=29 µmol·L−1) [68] (TDCPP)Sphingobium sp.strain TCM1,(1,3-DCP)Arthrobacter sp.strain PY1 TDCPP1,3-DCP 最佳条件测试:降解100% (30 ℃, pH=8.5, [MOPS]=50 mmol·L−1, [TDCPP]0=50 µmol·L−1)解毒100% (35 ℃, pH=9.5,[Tris-H2SO4]=50 mmol·L−1, [1,3-DCP]0=5 mmol·L−1)同时解毒:降解/解毒率100% (12 h, 30 ℃, pH=9, [Tris-H2SO4]=50 mmol·L−1,[TDCPP]0=53.2 µmol·L−1,) [69] Pseudomonas spp.Sphingobium spp. TCEP TCPP (48.37±9.52)%—(82.28±7.48)% [73] | Show TableDownLoad:
CSV
2008年,Takahashi等以TCEP或TDCPP为唯一磷源,尝试富集具有降解Cl-OPEs性能的菌种,首次报道了能降解TCEP和TDCPP的微生物[67],降解TCEP和TDCPP的优势菌种Acidovorax-和Sphingomonas-属可分别催化氯醇脱卤和OPEs的磷酸酯键裂解。接着,该团队就TCEP和TDCPP及各自代谢产生的氯醇2-CE和1,3-DCP分别设计了完全解毒的细胞系统[68-69]。一方面,利用TCM1菌株和2-CE降解菌(Xanthobacter autotrophicus菌株GJ10)开发了TCEP的两步反应解毒技术[68],包括TCEP在静息细胞TCM1的作用下24 h内水解成2-CE,然后在生长细胞GJ10的作用下经过144 h降解所有生成的2-CE,联合反应完全解毒9.6 μmol·L−1 TCEP。另一方面,开发了使用菌株TCM1和Arthrobacter sp.株PY1同时降解1,3-DCP而使TDCPP完全解毒的技术[69]。由于TCM1静息细胞对TDCPP脱磷和菌株PY1静息细胞对1,3-DCP脱卤的最佳条件相似,采用两种菌株同时对TDCPP及其代谢物进行降解以缩短反应时间,在30 ℃、pH=9.0、[Tris–H2SO4]=50 mmol·L−1菌株PY1细胞OD660=4.0的情况下,12 h内实现了TDCPP的完全解毒。这两个系统的设计为微生物解毒潜在的有毒持久性有机磷化合物开辟了一条道路。
此外,该团队在基因工程和降解路径推导方面也取得了一些研究成果,降解路径的推导依赖于产物鉴定,主要手段有离子色谱法[57]或衍生化气相色谱法[60]。Abe等从能够降解TCEP和TDCPP的两种菌株Sphingobium sp.TCM1和Sphingobium sp.TDK1中纯化克隆出两种卤代烷基磷水解酶,并分别命名为TDK-HAD和TCM-HAD,这两种磷酸三酯酶成功降解了Cl-OPEs,同时也能广泛降解溴化有机磷(如TDBPP)和三芳基磷酸盐(如TCP和TPhP)[70]。有机磷化合物对微生物细胞生长的利用被认为是由三种类型的磷酸酯酶介导的:磷酸三酯酶(PTE),磷酸二酯酶(PDE)和磷酸单酯酶(PME)。Takahashi等利用TCEP对Sphingobium sp.菌株TCM1进行碱性磷酸酶基因鉴定并推导了TCEP分步降解的路径[72],如图2所示,TCEP在菌株TCM1中的代谢包括全部3种磷酸酯酶,在每一步中,磷酸酯键中的一个被水解生成2-CE,最后一步生成Pi用于细胞生长。

2.2 Cl-OPEs生物修复案例
Cl-OPEs生物降解的最新进展源于针对混合OPFRs废水的实地生物修复案例。Liu等建立了综合垂直流人工湿地(IVCWs)以研究短期暴露于3种OPFRs(TCrP,TCEP,TCPP)后的环境行为和根际微生物反应[73],揭示了利用IVCWs处理以Cl-OPEs为主的OPFRs混合污染的潜力。研究发现,在处理低碳/氮比废水时,IVCWs对OPFRs废水既表现出良好的TN去除效果((628.13±110.63) mg·m−2·d−1),又具有相当的OPFRs去除效率((48.37%±9.52%)%—(82.28%±7.48%));对比高疏水化合物磷酸三甲苯(TCrP)在土壤、火成岩、沸石和植物组织中的吸附,Cl-OPEs则更容易被吸收并转移到植物体内;OPFRs与根际微生物的丰度、多样性和氮相关的微生物之间存在显著的相互作用;Pseudomonas属和Sphingobium属可能对混合OPFRs具有生物降解功能。
3. 氯代有机磷酸酯的光降解(Photodegradation of chlorinated organophosphate)
高级氧化技术(AOPs)是去除水溶液中难降解有机污染物的有效技术,包括臭氧化、光催化氧化、芬顿氧化、硫酸根自由基氧化等[74],其本质是通过各种途径生成羟基自由基(·OH)和硫酸盐自由基(
⋅SO−4 ⋅SO−4 按光源的不同,光氧化降解Cl-OPEs可分为紫外光(UV)降解和可见光(Vis)降解。鉴于Cl-OPEs的降解难度和高能量UV降解的稳定、高效性能,UV是学者开展Cl-OPEs降解研究的首选光源。然而,由于Cl-OPEs侧链烷基缺乏发色团,没有吸收波峰,很难直接光解,通常需要采用基于UV的复合光降解技术。按研究历程有三种复合思路:UV结合生成·OH的氧化剂如H2O2、O3[49,54,75,77-84],UV结合催化剂如TiO2和MOF材料[85-90],UV结合生成
⋅SO−4 ⋅SO−4 3.1 紫外光降解
3.1.1 UV/H2O2
在含有氧化剂H2O2的UV照射水中能形成大量的·OH,引发了UV/H2O2在全规模的水处理厂降解微污染有机物中的设计和实施[96]。Cl-OPEs在UV-C范围内是较差的光吸收剂,空白实验也表明H2O2不能单独促进化合物的降解,故UV/H2O2是UV和H2O2光解生成·OH氧化降解污染物的协同作用[80]。
多项研究表明,UV/H2O2是降解Cl-OPEs有效、可靠的技术,其光降解性能参数和效率见表3。目前,使用UV/H2O2降解OPEs的研究已经比较体系化,内容包括探究支链结构、光强、pH、底物浓度、氧化剂浓度、天然有机物的竞争性等因素对降解效率的影响,用C/C0、Cl−和
PO3−4 表 3 光降解Cl-OPEs的方法和效率总结Table 3. Summary of methods, efficiency and kinetics of photodegradation of Cl-OPEs方法Methods Cl-OPEs浓度Cl-OPEs concentration 关键试剂Key reagents 照射波长Irradiation wavelength 光降解效率Photodegradation efficiency 参考文献Reference UV/H2O2 5 mg·L−1 TCEP 50 mg·L−1,30% H2O2 254 nm >95% [77] UV/H2O2 143 mg·L−1 TCEP 5 mmol·L−1,30% H2O2 200—420 nm (1 h)>85% [80] UV/H2O2 50 μg·L−1 TCPP50 μg·L−1 TCEP50 μg·L−1 TDCPP 30% H2O2 254 nm (1 h)97%(1 h)91%(1 h)84% [81] UV/H2O2 500 μg·L−1 TCEP 1.5 mg·L−1,30% H2O2 200—400 nm (6 h)100% [82] UV/H2O2 500 mg·L−1 TCEP 30% H2O2,50 mg·L−1 185—400 nm (13 h)97% [54] UV/H2O2 5 mg·L−1 TCPP 30% H2O2,0.1mmol·L−1 200—400 nm (15 h)96% [75] UV/H2O2 4 mg·L−1 TCPP 30% H2O2 200—400 nm (25 min)96.1% [84] UV/TiO2(P25) 4 mg·L−1 TCPP 1000 mg·L−1 TiO2-P25 365 nm (12 h)80% [85] UV/TiO2-001 4 mg·L−1 TCPP 1000 mg·L−1 TiO2-001 365 nm (6 h)100% [85] UV/TiO2(P25) 1 mg·L−1 TCEP 70 mg·L−1 TiO2 254 nm (10 min)99% [86] UV/TiO2(P25) 1 mg·L−1 TCPP 100 mg·L−1 TiO2 254 nm (25 min)100% [90] UV/MIL-101(Fe)/PS 1 mg·L−1 TCEP 500 mg·L−1 MIL-101(Fe)500 mg·L−1 PS 420 nm (3 h)>80% [89] UV/PS 1 mg·L−1 TCEP 500 mg·L−1 PS 280 nm (3 h)>95% [89] UV/PS 1 mg·L−1 TCEP 175 μmol·L−1 PS 254 nm (30 min)99% [91] UV/PMS 1 mg·L−1 TCEP 20 mg·L−1 PMS 365 nm (30 min)94.6% [92] UV/PS 1 mg·L−1TCPP 75 mg·L−1 PS 254 nm (25 min)98.2% [93] 模拟太阳光/TiO2(P25) 250 μg·L−1 TCPP 50 mg·L−1 TiO2 290—800 nm (2 h)95% [97] Vis/N、-TiO2 100 μg·L−1 TCPP 250 mg·L−1 N、S-TiO2 400—800 nm (2 h)65%—70% [94] Vis/GO@MIL-101(Fe)/H2O2 1 mg·L−1 TCEP 500 mg·L−1 GO@MIL-101(Fe)165 mmol·L−1 30% H2O2 420 nm (30 min)95% [95] | Show TableDownLoad:
CSV
OPEs支链的分子结构显著影响UV/H2O2条件下·OH对目标物的降解速率。Watts等研究了TCEP、TCPP、TBP和TBEP等4种磷酸三酯的二阶反应速率,kOH, TCEP=5.6×108 mol·L−1·s−1,kOH, TCPP=1.98×108 mol·L−1·s−1,kOH, TBP=6.4×109 mol·L−1·s−1,kOH, TBEP=1.03×1010 mol·L−1·s−1,排序为TBEP>TBP>TCEP>TCPP[78],即烷基OPEs的降解速率高于Cl-OPEs,与水解时的规律一致。Cl-OPEs支链上的Cl原子显著降低了H原子的可用性,导致降解速率较慢。另外,随着烷基链的卤化,碳链数和加成物α链上的杂原子(O)也影响了·OH攻击的速率。理论上,TCPP在每个碳链上比TCEP多一个碳,降解速率应高于TCEP,然而实验结果正相反,很可能是受TCPP的α-碳处附加的—CH3的影响。此后,He等在超纯水中,用250 W紫外光照射50 mg·L−1 H2O2降解TCPP[75],动力学计算表明速率常数为0.0035 min−1(R2=0.9871),降解率达到96%,但TOC去除率仅有43.02%,证实了TCPP的矿化难度较大。
UV/H2O2体系中TCEP的去除效果与H2O2的投加量及其氧化特性密切相关。一方面,调控初始H2O2浓度对TCEP的去除非常关键。在5 mg·L−1 TCEP,pH=7碳酸盐缓冲溶液中外加50 mg·L−1 30% H2O2,用低压汞灯(λ=254 nm)照射,降解率达95%以上[77]。但31P核磁共振波谱法对UV/H2O2体系的监测表明,使用高浓度的氧化剂来改善混合OPEs中TCEP的去除可能会导致·OH氧化的抑制,使得TCEP无法完全降解[82]。以反应时间1 h为基本条件考察H2O2用量对UV/H2O2降解143 mg·L−1TCEP的影响[80],发现增加H2O2的用量提高了Cl−和
PO3−4 PO3−4 3.1.2 UV/M(光催化剂)
光催化技术及光催化材料的研究是目前最为活跃的研究方向之一。使用光催化技术降解Cl-OPEs的优势在于不需使用昂贵的氧化剂,也不易产生毒性更高的中间产物。
TiO2及其改性产品是最常用和最早实现商品化的光催化剂,具有催化活性高、应用范围广、清洁无二次污染、价格低廉等优点。TiO2的禁带宽度大(3.2 eV),产生的光生电子和空穴氧化还原电势高,且稳定性强,因而在光催化剂向有机半导体基/金属基高分子纵深发展的今天,TiO2仍然被广泛应用于实际工程领域中污染物的去除。UV/TiO2在降解Cl-OPEs上的有效性很早就得到了证实。刘青等采用商品化的TiO2-P25在UV下降解TCPP[85],UV和P25联合作用下12 h降解率达到80%,用高温醇热法进一步合成暴露(001)活性晶面TiO2,与其特征波长365 nm的紫外光联合作用,90 min TCPP降解率超过50%,360 min内完全降解,最优催化加投加量为1 g·L−1。中性和酸性时的降解率要略微大于碱性条件,但不同pH下TCPP在6 h内降解率都达到90%,表明改性催化剂TiO2-001的pH适应性更强,有潜力应用于不同酸碱度TCPP废液的处理。
天然有机物、无机离子和腐殖酸能够显著影响UV/TiO2对Cl-OPEs的降解。有报道称光催化降解TCEP的效率受pH、天然有机物和阴离子影响很大,在实际的水处理过程中,TCEP很难完全矿化[86]。Tang等在使用UV/TiO2光催化降解TCEP、TCPP和TDCPP时重点考察了无机离子和腐殖酸的作用[87],结果表明无机离子如
PO3−4 SO2−4 NO−3 PO3−4 SO2−4 NO−3 金属-有机骨架(MOFs)是一类具有超高的比表面积、较高且可调的孔隙率和开放的金属位点的新型多孔材料,作催化剂时具有多相催化后易回收分离、可循环使用的优点。光和MOFs的结合是降解Cl-OPEs的新方向。Hu等制备了禁带宽度为2.41 eV的高纯度规整晶体MIL-101(Fe),结合UV在过硫酸盐体系中降解TCEP[89],合成的MIL-101(Fe)可利用短波可见光(400—520 nm)和紫外光(200—400 nm),被光激活的MIL-101(Fe)使Fe(Ⅲ)转化为Fe(Ⅱ),进而使
S2O2−8 SO−4 S2O2−8 3.1.3 UV/PS和UV/PMS(过硫酸盐)
活化过硫酸盐(包括过二硫酸盐PS和过一硫酸盐PMS)由于操作简单、氧化性高、应用广泛,被认为是处理OPEs的有前景的技术,其原理主要是活化产生硫酸根自由基(
⋅SO−4 ⋅SO−4 ⋅SO−4 3.2 可见光降解
在光催化降解污染物的过程中,材料的选择与制备对于降解性能至关重要。尽管无毒、低成本的TiO2具有良好的化学稳定性,但其禁带宽度较大(−3.2 eV)只能吸收紫外光,造成太阳光的利用率严重受限(小于5%)。尽管利用可见光降解Cl-OPEs的研究非常有限,但其体现了光催化发展从紫外光向可见光转变的趋势。在光源选择发生转变的同时,学者也在积极探索光催化剂和光催化体系的优化,催化剂的优化表现为从以TiO2为典型的无机催化剂转向表面掺杂高聚合TiO2或有机高分子MOF材料,体系优化表现为从较为简单的UV/TiO2、UV/H2O2体系转向光/复合金属或半导体基光催化材料/过硫酸盐的多相光催化体系。例如,Antonopoulou等通过模拟太阳光,使用TiO2作为催化剂降解溶解在纯水中的TCPP,降解速率在0.0141—0.142 min−1(C0=25—500 μg·L−1)之间,处理后毒性降低[97]。通过掺杂改性等手段可以拓宽TiO2的光响应至可见波段,采用N、S掺杂改性TiO2对TCPP也进行了催化降解[94],在UV-Vis照射下,N、S共掺杂的TiO2是去除TCPP的最具光活性的催化剂,但6 h内,TCPP的去除率也不足65%。Lin等则在MIL-101(Fe)的基础上附载了氧化石墨烯,制备MOF材料GO@MIL-101(Fe),并在此基础上开发了可见光/MOF/H2O2的光催化体系增强可见光条件下对TCEP的降解能力,附载了氧化石墨烯后,MIL-101(Fe)材料电位从2.41eV降低到2.71 eV,从而对光的吸收范围从520 nm扩展到570 nm。GO的高电导率实现了快速活化和电子转移,30 min TCEP降解率达95%,且GO@MIL-101(Fe)在水中稳定[95]。
4. 氯代有机磷酸酯降解的安全风险评估(Safety risk assessment of chlorinated organophosphate degradation)
随着研究的深入,Cl-OPEs降解的安全风险越来越受到关注,这种风险主要源于降解过程中产生的高毒性含氯中间产物。由于pH依赖和矿物催化的水解方法研究尚不充分,微生物降解方法虽然选育菌种较受限但本身可以完全解毒代谢产物,对Cl-OPEs降解的安全风险探究主要聚焦在UV/H2O2、UV/TiO2、UV/PS等光降解方法研究方面。
UV/H2O2降解Cl-OPEs前后生物毒性的变化最早引发注意,但毒性结论并不统一,其升高或降低可能取决于受试生物。例如,用盐湖卤虫卵进行UV/H2O2降解TCEP前后的急性毒性测试,不论是单一TCEP还是混合OPEs降解,LC50都升高,表明急性毒性降低[82]。使用大肠杆菌评估TCPP在UV/H2O2降解前后的毒性,显示活性氧(ROS)和细胞凋亡减少,大肠杆菌的膜电位(MP)升高,生物分子功能得到修正,说明TCPP降解后毒性降低[84]。然而,根据发光菌实验数据,随着反应的进行,TCPP产物的毒性明显增加[75]。上述结果表明,需要更多降解前后的毒性变化案例佐证和完善现有的研究结果。
UV/TiO2降解Cl-OPEs的安全风险评估的研究热点是中间产物的蛋白质组学分析,因为蛋白质相互作用是揭示Cl-OPEs及其中间体在蛋白质组水平上生物分子影响的重要视角。Ye等研究了UV/TiO2非均相光催化降解水中TCEP及其对细菌蛋白质组的影响[86],自组装紫外辐射装置在投加70 mg·L−1 TiO2的情况下,对1 mg·L−1 TCEP 10 min降解率达到99%,·OH是主要的反应物种,降解过程符合准一级动力学模型,kobs=0.3167 min−1;对代谢反应、途径和网络的蛋白质组学分析发现初步降解产物能被大肠杆菌通过细胞代谢进行转运和利用;观察到抗逆性下降趋势,处理后产物毒性明显减弱,表明UV/TiO2体系下矿化不完全的TCEP的羟基化和脱氯作用对其解毒同样有效。随后Yu等将目标转向TCPP,发现Cl−和
PO3−4 UV/PS降解Cl-OPEs不但高效,且毒理学评价证实其安全风险较小。例如,Ou等观察到TCEP在254 nm UV/PS体系下降解中间产物的毒性降低[91]。Xu等使用流式细胞术(FCM)分析对降解中间混合物进行的毒理学评价显示,细胞内活性氧(ROS)和细胞凋亡率明显下降,细胞膜电位(MP)较原TCPP升高,细胞周期分析表明这些降解产物对大肠杆菌DNA生物合成的负面影响也有所减弱,说明经过UV/PS处理后降解产物的毒性明显降低[92]。UV/过硫酸盐能在部分矿化Cl-OPEs的同时进行有效解毒,提示其能控制废/污水中Cl-OPEs的污染。
另外,对于MOFs材料或可见光催化降解Cl-OPEs,因为降解效果不够理想,降解机制尚不清楚,所以研究还未拓展到安全风险评估方面。
5. 结论与展望(Conclusion and prospects)
Cl-OPEs在环境中的高丰度、高毒性和难降解性使其去除技术成为研究热点。本文主要介绍了3种代表性的去除技术,包括pH依赖的水解、特异性磷酸水解酶促进的微生物降解和光驱动的高级氧化降解,另外还讨论了各种降解方法的安全风险。总的来说,碱性条件和矿物催化有利于Cl-OPEs水解,但这方面研究仅限于实验室领域;微生物降解具有彻底矿化和完全解毒Cl-OPEs的潜力,不足在于前期选育菌种和反应用时过长,且需要依靠两种菌株共同解毒,在实际水生或土壤环境中还可能面临低温制约;光驱动的高级氧化技术通过激发催化剂或氧化剂生成具有高度活性的·OH和
⋅SO−4 因此,未来Cl-OPEs的降解研究应集中开发和优化高效、绿色、无二次污染的技术。水解方面要补充基于实际水生环境和土壤基质的实验数据,尤其关注天然pH条件下降解稳态Cl-OPEs的新方法、新材料研发。微生物降解方面应注意选育能在降解目标物的同时降低其毒性的高效菌株。当然,研究着力点仍应放在光催化和高级氧化技术的联用方面,侧重点包括:(1)光催化剂性能的提升,通过改性和引入适当的有机高分子材料优化催化效果;(2)反应体系的设计优化;(3)含氯中间产物的毒性评价方法优化;(4)开发新型可见光驱动光催化剂以拓展光响应范围,并促进电荷有效利用。
-
表 1 主要OPEs名称和理化性质
Table 1. Names and Physicochemical properties of the major OPEs
化合物名称及简写Compounds and Abbreviation CAS 分子式Molecular formula 取代基Substituent lgKow Vp(Torr) 磷酸三乙酯(Triethyl phosphate, TEP) 78-40-0 C6H15O4P </td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr> <td class="table_top_border" align="center" valign="middle">化合物名称及简写Compounds and Abbreviation</td><td class="table_top_border" align="center" valign="middle">CAS</td><td class="table_top_border" align="center" valign="middle">分子式Molecular formula</td><td class="table_top_border" align="center" valign="middle">取代基Substituent</td><td class="table_top_border" align="center" valign="middle">lg<i>K</i><sub>ow</sub></td><td class="table_top_border" align="center" valign="middle"><i>V</i><sub>p</sub>(Torr)</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">磷酸三乙酯(Triethyl phosphate, TEP)</td><td class="table_top_border2" align="center" valign="middle">78-40-0</td><td class="table_top_border2" align="center" valign="middle">C<sub>6</sub>H<sub>15</sub>O<sub>4</sub>P</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:1cm"><img class="graphic" src="2020111501-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">0.80</td><td class="table_top_border2" align="center" valign="middle">3.93×10<sup>−1</sup></td></tr><tr><td align="center" valign="middle">磷酸三正丁酯(Tri-n-butyl phosphate, TnBP)</td><td align="center" valign="middle">126-73-8</td><td align="center" valign="middle">C<sub>13</sub>H<sub>27</sub>O<sub>5</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.02cm"><img class="graphic" src="2020111501-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">4.00</td><td align="center" valign="middle">1.13×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三异丁酯(Tri-<i>iso</i>-butyl phosphate, TiBP)</td><td align="center" valign="middle">126-71-6</td><td align="center" valign="middle">C<sub>12</sub>H<sub>27</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:0.9cm"><img class="graphic" src="2020111501-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">3.60</td><td align="center" valign="middle">1.28×10<sup>−3</sup></td></tr><tr><td align="center" valign="middle">磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP)</td><td align="center" valign="middle">78-51-3</td><td align="center" valign="middle">C<sub>18</sub>H<sub>39</sub>O<sub>7</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.58cm"><img class="graphic" src="2020111501-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">3.75</td><td align="center" valign="middle">2.50×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP)</td><td align="center" valign="middle">115-96-8</td><td align="center" valign="middle">C<sub>6</sub>H<sub>12</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.2cm"><img class="graphic" src="2020111501-Tab1-T5.jpg"></styled-content></td><td align="center" valign="middle">1.44</td><td align="center" valign="middle">6.13×10<sup>−2</sup></td></tr><tr><td align="center" valign="middle">磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP)</td><td align="center" valign="middle">13674-84-5</td><td align="center" valign="middle">C<sub>9</sub>H<sub>18</sub>Cl<sub>3</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.21cm"><img class="graphic" src="2020111501-Tab1-T6.jpg"></styled-content></td><td align="center" valign="middle">2.59</td><td align="center" valign="middle">2.02×10<sup>−5</sup></td></tr><tr><td align="center" valign="middle">磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP)</td><td align="center" valign="middle">13674-87-8</td><td align="center" valign="middle">C<sub>9</sub>H<sub>15</sub>Cl<sub>6</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.23cm"><img class="graphic" src="2020111501-Tab1-T7.jpg"></styled-content></td><td align="center" valign="middle">3.65</td><td align="center" valign="middle">7.36×10<sup>−8</sup></td></tr><tr><td align="center" valign="middle">磷酸三苯酯(Triphenyl phosphate, TPhP)</td><td align="center" valign="middle">115-86-6</td><td align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.07cm"><img class="graphic" src="2020111501-Tab1-T8.jpg"></styled-content></td><td align="center" valign="middle">4.59</td><td align="center" valign="middle">6.28×10<sup>−6</sup></td></tr><tr><td align="center" valign="middle">磷酸三甲苯酯(Tricresyl phosphate, TCrP)</td><td align="center" valign="middle">1330-78-5</td><td align="center" valign="middle">C<sub>21</sub>H<sub>21</sub>O<sub>4</sub>P</td><td align="center" valign="middle"><styled-content style="width:1.6cm"><img class="graphic" src="2020111501-Tab1-T9.jpg"></styled-content></td><td align="center" valign="middle">5.11</td><td align="center" valign="middle">6.00×10<sup>−7</sup></td></tr><tr><td class="table_bottom_border" align="center" valign="middle">三苯基氧化膦(Triphenylphosphine oxide, TPPO)</td><td class="table_bottom_border" align="center" valign="middle">791-28-6</td><td class="table_bottom_border" align="center" valign="middle">C<sub>18</sub>H<sub>15</sub>OP</td><td class="table_bottom_border" style="padding-bottom:3pt;" align="center" valign="middle"><styled-content style="width:1.8cm"><img class="graphic" src="2020111501-Tab1-T10.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">2.83</td><td class="table_bottom_border" align="center" valign="middle">2.62×10<sup>−8</sup></td></tr></tbody>
<tfoot><tr><td colspan="6" align="left" valign="middle"> 注:<i>K</i>ow为正辛醇-水分配系数,<i>V</i><sub>p</sub>为蒸汽压<i>。</i>Note: <i>K</i>ow is the <i>n</i>-octyl alcohol-water distribution coefficient, <i>V</i><sub>p</sub> is the vapor pressure.</td></tr></tfoot>
</table></div></foreignObject></svg>)
0.80 3.93×10−1 磷酸三正丁酯(Tri-n-butyl phosphate, TnBP) 126-73-8 C13H27O5P 4.00 1.13×10−3 磷酸三异丁酯(Tri-iso-butyl phosphate, TiBP) 126-71-6 C12H27O4P 3.60 1.28×10−3 磷酸三丁氧基乙基酯(Tributoxyethyl phosphate, TBEP) 78-51-3 C18H39O7P 3.75 2.50×10−8 磷酸三(2-氯乙基)酯(Tri(2-chloroethyl)phosphate, TCEP) 115-96-8 C6H12Cl3O4P 1.44 6.13×10−2 磷酸三(2-氯丙基)酯(Tri(1-chloro-2-propy)phosphate, TCPP) 13674-84-5 C9H18Cl3O4P 2.59 2.02×10−5 磷酸三(1,3-二氯-2-丙基)酯(Tris(1,3-dichloro-2-propyl)phosphate, TDCPP) 13674-87-8 C9H15Cl6O4P 3.65 7.36×10−8 磷酸三苯酯(Triphenyl phosphate, TPhP) 115-86-6 C18H15O4P 4.59 6.28×10−6 磷酸三甲苯酯(Tricresyl phosphate, TCrP) 1330-78-5 C21H21O4P 5.11 6.00×10−7 三苯基氧化膦(Triphenylphosphine oxide, TPPO) 791-28-6 C18H15OP 2.83 2.62×10−8 注:Kow为正辛醇-水分配系数,Vp为蒸汽压。Note: Kow is the n-octyl alcohol-water distribution coefficient, Vp is the vapor pressure.
下载: 导出CSV
表 2 具有降解Cl-OPEs性能的微生物及其降解性能总结
Table 2. Summary of (Cl-OPEs)-degrading bacterial species and their performance
菌种Bacterial species Cl-OPEs及代谢物Cl-OPEs and metabolites 降解/解毒率Degradation/detoxification rate 参考文献Reference (TCEP) Acidovorax spp.,Sphingomonas spp.(TDCPP) Acidovorax spp.,Aquabacterium spp.,Sphingomonas spp. TCEP TDCPP 100% (6 h, [TCEP]0=20 µmol·L−1)100% (3 h, [TDCPP]0=20 µmol·L−1) [67] (TCEP) Sphingobium sp.strain TCM1,(2-CE) Xanthobacter autotrophicus strain GJ10 TCEP2-CE 最佳条件测试:降解100% (30 ℃, pH=8.5, [Co2+]=50 μmol·L−1, OD660=0.8, [TCEP]0=10 µmol·L−1) 解毒100%(24 h, [2-CE]0=180 µmol·L−1)分步解毒:降解/解毒100%(①4 h,[TCEP]0=10 µmol·L−1; ②144 h,[2-CE]0=29 µmol·L−1) [68] (TDCPP)Sphingobium sp.strain TCM1,(1,3-DCP)Arthrobacter sp.strain PY1 TDCPP1,3-DCP 最佳条件测试:降解100% (30 ℃, pH=8.5, [MOPS]=50 mmol·L−1, [TDCPP]0=50 µmol·L−1)解毒100% (35 ℃, pH=9.5,[Tris-H2SO4]=50 mmol·L−1, [1,3-DCP]0=5 mmol·L−1)同时解毒:降解/解毒率100% (12 h, 30 ℃, pH=9, [Tris-H2SO4]=50 mmol·L−1,[TDCPP]0=53.2 µmol·L−1,) [69] Pseudomonas spp.Sphingobium spp. TCEP TCPP (48.37±9.52)%—(82.28±7.48)% [73]
下载: 导出CSV
表 3 光降解Cl-OPEs的方法和效率总结
Table 3. Summary of methods, efficiency and kinetics of photodegradation of Cl-OPEs
方法Methods Cl-OPEs浓度Cl-OPEs concentration 关键试剂Key reagents 照射波长Irradiation wavelength 光降解效率Photodegradation efficiency 参考文献Reference UV/H2O2 5 mg·L−1 TCEP 50 mg·L−1,30% H2O2 254 nm >95% [77] UV/H2O2 143 mg·L−1 TCEP 5 mmol·L−1,30% H2O2 200—420 nm (1 h)>85% [80] UV/H2O2 50 μg·L−1 TCPP50 μg·L−1 TCEP50 μg·L−1 TDCPP 30% H2O2 254 nm (1 h)97%(1 h)91%(1 h)84% [81] UV/H2O2 500 μg·L−1 TCEP 1.5 mg·L−1,30% H2O2 200—400 nm (6 h)100% [82] UV/H2O2 500 mg·L−1 TCEP 30% H2O2,50 mg·L−1 185—400 nm (13 h)97% [54] UV/H2O2 5 mg·L−1 TCPP 30% H2O2,0.1mmol·L−1 200—400 nm (15 h)96% [75] UV/H2O2 4 mg·L−1 TCPP 30% H2O2 200—400 nm (25 min)96.1% [84] UV/TiO2(P25) 4 mg·L−1 TCPP 1000 mg·L−1 TiO2-P25 365 nm (12 h)80% [85] UV/TiO2-001 4 mg·L−1 TCPP 1000 mg·L−1 TiO2-001 365 nm (6 h)100% [85] UV/TiO2(P25) 1 mg·L−1 TCEP 70 mg·L−1 TiO2 254 nm (10 min)99% [86] UV/TiO2(P25) 1 mg·L−1 TCPP 100 mg·L−1 TiO2 254 nm (25 min)100% [90] UV/MIL-101(Fe)/PS 1 mg·L−1 TCEP 500 mg·L−1 MIL-101(Fe)500 mg·L−1 PS 420 nm (3 h)>80% [89] UV/PS 1 mg·L−1 TCEP 500 mg·L−1 PS 280 nm (3 h)>95% [89] UV/PS 1 mg·L−1 TCEP 175 μmol·L−1 PS 254 nm (30 min)99% [91] UV/PMS 1 mg·L−1 TCEP 20 mg·L−1 PMS 365 nm (30 min)94.6% [92] UV/PS 1 mg·L−1TCPP 75 mg·L−1 PS 254 nm (25 min)98.2% [93] 模拟太阳光/TiO2(P25) 250 μg·L−1 TCPP 50 mg·L−1 TiO2 290—800 nm (2 h)95% [97] Vis/N、-TiO2 100 μg·L−1 TCPP 250 mg·L−1 N、S-TiO2 400—800 nm (2 h)65%—70% [94] Vis/GO@MIL-101(Fe)/H2O2 1 mg·L−1 TCEP 500 mg·L−1 GO@MIL-101(Fe)165 mmol·L−1 30% H2O2 420 nm (30 min)95% [95]
下载: 导出CSV
-
[1] ANDRESEN J A, GRUNDMANN A, BESTER K. Organophosphorus flame retardants and plasticisers in surface waters [J]. Science of the Total Environment, 2004, 332(1-3): 155-166. doi: 10.1016/j.scitotenv.2004.04.021 [2] DODSON R E, PEROVICH L J, COVACI A, et al. After the PBDE phase-out: A broad suite of flame retardants in repeat house dust samples from california [J]. Environmental Science & Technology, 2012, 46(24): 13056-13066. [3] YAN S H, WU H M, QIN J H, et al. Halogen-free organophosphorus flame retardants caused oxidative stress and multixenobiotic resistance in Asian freshwater clams (Corbicula fluminea) [J]. Environmental Pollution, 2017, 225: 559-568. doi: 10.1016/j.envpol.2017.02.071 [4] ALI N, DIRTU A C, VAN DEN EEDE N, et al. Occurrence of alternative flame retardants in indoor dust from New Zealand: Indoor sources and human exposure assessment [J]. Chemosphere, 2012, 88(11): 1276-1282. doi: 10.1016/j.chemosphere.2012.03.100 [5] HE C T, ZHENG J, QIAO L, et al. Occurrence of organophosphorus flame retardants in indoor dust in multiple microenvironments of southern China and implications for human exposure [J]. Chemosphere, 2015, 133: 47-52. doi: 10.1016/j.chemosphere.2015.03.043 [6] SALAMOVA A, HERMANSON M H, HITES R A. Organophosphate and halogenated flame retardants in atmospheric particles from a european arctic site [J]. Environmental Science & Technology, 2014, 48(11): 6133-6140. [7] MOLLER A, XIE Z Y, CABA A, et al. Organophosphorus flame retardants and plasticizers in the atmosphere of the North Sea [J]. Environmental Pollution, 2011, 159(12): 3660-3665. doi: 10.1016/j.envpol.2011.07.022 [8] BOLLMANN U E, MOLER A, XIE Z Y, et al. Occurrence and fate of organophosphorus flame retardants and plasticizers in coastal and marine surface waters [J]. Water Research, 2012, 46(2): 531-538. doi: 10.1016/j.watres.2011.11.028 [9] REGNERY J, PUTTMANN W. Occurrence and fate of organophosphorus flame retardants and plasticizers in urban and remote surface waters in Germany [J]. Water Research, 2010, 44(14): 4097-4104. doi: 10.1016/j.watres.2010.05.024 [10] WANG R M, TANG J H, XIE Z Y, et al. Occurrence and spatial distribution of organophosphate ester flame retardants and plasticizers in 40 rivers draining into the Bohai Sea, north China [J]. Environmental Pollution, 2015, 198: 172-178. doi: 10.1016/j.envpol.2014.12.037 [11] PANTELAKI I, VOUTSA D. Organophosphate flame retardants (OPFRs): A review on analytical methods and occurrence in wastewater and aquatic environment [J]. Science of the Total Environment, 2019, 649: 247-263. doi: 10.1016/j.scitotenv.2018.08.286 [12] SUNDKVIST A M, OLOFSSON U, HAGLUND P. Organophosphorus flame retardants and plasticizers in marine and fresh water biota and in human milk [J]. Journal of Environmental Monitoring, 2010, 12(4): 943-951. doi: 10.1039/b921910b [13] REEMTSMA T, QUINTANA J B, RODIL R, et al. Organophosphorus flame retardants and plasticizers in water and air I. Occurrence and fate [J]. Trac-Trends in Analytical Chemistry, 2008, 27(9): 727-737. doi: 10.1016/j.trac.2008.07.002 [14] WANG Y, YAO Y, LI W, et al. A nationwide survey of 19 organophosphate esters in soils from China: Spatial distribution and hazard assessment [J]. Science of the Total Environment, 2019, 671: 528-535. doi: 10.1016/j.scitotenv.2019.03.335 [15] TAN X X, LUO X J, ZHENG X B, et al. Distribution of organophosphorus flame retardants in sediments from the Pearl River Delta in South China [J]. Science of the Total Environment, 2016, 544: 77-84. doi: 10.1016/j.scitotenv.2015.11.089 [16] KIM J W, ISOBE T, CHANG K H, et al. Levels and distribution of organophosphorus flame retardants and plasticizers in fishes from Manila Bay, the Philippines [J]. Environmental Pollution, 2011, 159(12): 3653-3659. doi: 10.1016/j.envpol.2011.07.020 [17] FU L, DU B, WANG F, et al. Organophosphate triesters and diester degradation products in municipal sludge from wastewater treatment plants in China: Spatial patterns and ecological implications [J]. Environmental Science & Technology, 2017, 51(23): 13614-13623. [18] QI C D, YU G, ZHONG M M, et al. Organophosphate flame retardants in leachates from six municipal landfills across China [J]. Chemosphere, 2019, 218: 836-844. doi: 10.1016/j.chemosphere.2018.11.150 [19] LIU C S, WANG Q W, LIANG K, et al. Effects of tris (1, 3-dichloro-2-propyl) phosphate and triphenyl phosphate on receptor-associated mRNA expression in zebrafish embryos/larvae [J]. Aquatic Toxicology, 2013, 128: 147-157. [20] ORGANIZATION W H. Environmental Health Criteria 209: Flame retardants: Tris-(chloropropyl) phosphate and tris-(2-chloroethyl) phosphate [J]. Geneva: WHO, 1998 [21] CRISTALE J, GARCIA VAZQUEZ A, BARATA C, et al. Priority and emerging flame retardants in rivers: Occurrence in water and sediment, Daphnia magna toxicity and risk assessment [J]. Environment International, 2013, 59: 232-243. doi: 10.1016/j.envint.2013.06.011 [22] 皮天星, 蔡磊明, 蒋金花, 等. 新型阻燃剂TCPP对斑马鱼的毒性研究 [J]. 生态毒理学报, 2016, 11(2): 247-256. PI T S, CAI L M, JIANG J H, et al. Toxicity of a new flame retardant TCPP to zebrafish [J]. Journal of Ecotoxicology, 2016, 11(2): 247-256(in Chinese).
[23] WANG Q, LAI N L S, WANG X, et al. Bioconcentration and transfer of the organophorous flame retardant 1, 3-dichloro-2-propyl phosphate causes thyroid endocrine disruption and developmental neurotoxicity in zebrafish larvae [J]. Environmental Science & Technology, 2015, 49(8): 5123-5132. [24] WANG Q, LAM J C W, MAN Y C, et al. Bioconcentration, metabolism and neurotoxicity of the organophorous flame retardant 1, 3-dichloro 2-propyl phosphate (TDCPP) to zebrafish [J]. Aquatic Toxicology, 2015, 158: 108-115. doi: 10.1016/j.aquatox.2014.11.001 [25] OLIVERI A N, BAILEY J M, LEVIN E D. Developmental exposure to organophosphate flame retardants causes behavioral effects in larval and adult zebrafish [J]. Neurotoxicology And Teratology, 2015, 52: 220-227. doi: 10.1016/j.ntt.2015.08.008 [26] WANG Q, LAM J C W, HAN J, et al. Developmental exposure to the organophosphorus flame retardant tris (1, 3-dichloro-2-propyl) phosphate: Estrogenic activity, endocrine disruption and reproductive effects on zebrafish [J]. Aquatic Toxicology, 2015, 160: 163-171. doi: 10.1016/j.aquatox.2015.01.014 [27] MCGEE S P, COOPER E M, STAPLETON H M, et al. Early zebrafish embryogenesis is susceptible to developmental TDCPP exposure [J]. Environmental Health Perspectives, 2012, 120(11): 1585-1591. doi: 10.1289/ehp.1205316 [28] LIJ H, SU G, ZOU M, et al. Effects of tris (1, 3-dichloro-2-propyl) phosphate on growth, reproduction, and gene transcription of daphnia magna at environmentally relevant concentrations [J]. Environmental Science & Technology, 2015, 49(21): 12975-12983. [29] MEEKER J D, STAPLETON H M. House dust concentrations of organophosphate flame retardants in relation to hormone levels and semen quality parameters [J]. Environmental Health Perspectives, 2010, 118(3): 318-323. doi: 10.1289/ehp.0901332 [30] FARHAT A, CRUMP D, CHIU S, et al. In ovo effects of two organophosphate flame retardants-TCPP and TDCPP-on pipping success, development, mrna expression, and thyroid hormone levels in chicken Embryos [J]. Toxicological Sciences, 2013, 134(1): 92-102. doi: 10.1093/toxsci/kft100 [31] NOYES P D, HAGGARD D E, GONNERMAN G D, et al. Advanced morphological-behavioral test platform reveals neurodevelopmental defects in embryonic zebrafish exposed to comprehensive suite of halogenated and organophosphate flame retardants [J]. Toxicological Sciences, 2015, 145(1): 177-195. doi: 10.1093/toxsci/kfv044 [32] DISHAW L V, POWERS C M, RYDE I T, et al. Is the PentaBDE replacement, tris (1, 3-dichloropropyl) phosphate (TDCPP), a developmental neurotoxicant? Studies in PC12 cells [J]. Toxicology and Applied Pharmacology, 2011, 256(3): 281-289. doi: 10.1016/j.taap.2011.01.005 [33] YUAN L, LI J, ZHA J, et al. Targeting neurotrophic factors and their receptors, but not cholinesterase or neurotransmitter, in the neurotoxicity of TDCPP in Chinese rare minnow adults (Gobiocypris rarus) [J]. Environmental Pollution, 2016, 208: 670-677. doi: 10.1016/j.envpol.2015.10.045 [34] 顾杰, 吴江, 王宏烨, 等. 有机磷酸酯对斑马鱼的早期神经毒性作用研究 [J]. 生态毒理学报, 2019, 14(5): 152-158. Gu J, WU J, WANG H Y, et al. Study on the early neurotoxicity of organophosphate on zebrafish [J]. Journal of Ecotoxicology, 2019, 14(5): 152-158(in Chinese).
[35] LIU C, SU G, GIESY J P, et al. Acute exposure to tris (1, 3-dichloro-2-propyl) phosphate (TDCIPP) causes hepatic inflammation and leads to hepatotoxicity in zebrafish [J]. Scientific Reports, 2016, 6: 19054. doi: 10.1038/srep19054 [36] LI F, CAO L, LI X, et al. Affinities of organophosphate flame retardants to tumor suppressor gene p53: An integrated in vitro and in silico study [J]. Toxicology Letters, 2015, 232(2): 533-541. doi: 10.1016/j.toxlet.2014.12.006 [37] LI J, GIESY J P, YU L, et al. Effects of Tris (1, 3-dichloro-2-propyl) Phosphate (TDCPP) in tetrahymena thermophila: Targeting the ribosome [J]. Scientific Reports, 2015, 5: 10562. doi: 10.1038/srep10562 [38] KRIVOSHIEV B V, DARDENNE F, BLUST R, et al. Elucidating toxicological mechanisms of current flame retardants using a bacterial gene profiling assay [J]. Toxicology In Vitro, 2015, 29(8): 2124-2132. doi: 10.1016/j.tiv.2015.09.001 [39] LIU X, CAI Y, WANG Y, et al. Effects of tris (1, 3-dichloro-2-propyl) phosphate (TDCPP) and triphenyl phosphate (TPP) on sex-dependent alterations of thyroid hormones in adult zebrafish [J]. Ecotoxicology and Environmental Safety, 2019, 170: 25-32. doi: 10.1016/j.ecoenv.2018.11.058 [40] LIU X, JI K, CHOI K. Endocrine disruption potentials of organophosphate flame retardants and related mechanisms in H295R and MVLN cell lines and in zebrafish [J]. Aquatic Toxicology, 2012, 114: 173-181. [41] CHEN G, JIN Y, WU Y, et al. Exposure of male mice to two kinds of organophosphate flame retardants (OPFRs) induced oxidative stress and endocrine disruption [J]. Environmental Toxicology and Pharmacology, 2015, 40(1): 310-318. doi: 10.1016/j.etap.2015.06.021 [42] KOJIMA H, TAKEUCHI S, ITOH T, et al. In vitro endocrine disruption potential of organophosphate flame retardants via human nuclear receptors [J]. Toxicology, 2013, 314(1): 76-83. doi: 10.1016/j.tox.2013.09.004 [43] FERNIE K J, PALACE V, PETERS L E, et al. Investigating endocrine and physiological parameters of captive american kestrels exposed by diet to selected organophosphate flame retardants [J]. Environmental Science & Technology, 2015, 49(12): 7448-7455. [44] VAN DER VEEN I, DE BOER J. Phosphorus flame retardants: Properties, production, environmental occurrence, toxicity and analysis [J]. Chemosphere, 2012, 88(10): 1119-1153. doi: 10.1016/j.chemosphere.2012.03.067 [45] LEE S, CHO H J, CHOI W, et al. Organophosphate flame retardants (OPFRs) in water and sediment: Occurrence, distribution, and hotspots of contamination of Lake Shihwa, Korea [J]. Marine Pollution Bulletin, 2018, 130: 105-112. doi: 10.1016/j.marpolbul.2018.03.009 [46] MEYER J, BESTER K. Organophosphate flame retardants and plasticisers in wastewater treatment plants [J]. Journal of Environmental Monitoring, 2004, 6(7): 599-605. doi: 10.1039/b403206c [47] GRIECO S A, RAMARAO B V. Removal of TCEP from aqueous solutions by adsorption with zeolites [J]. Colloids and Surfaces a-Physicochemical and Engineering Aspects, 2013, 434: 329-338. [48] 严炜, 景传勇. 有机磷阻燃剂在不同含氧碳纳米管上的吸附行为 [J]. 环境化学, 2014, 33(10): 1692-1699. doi: 10.7524/j.issn.0254-6108.2014.10.003 Yan W, JING C Y. Adsorption behavior of organophosphate flame retardants on different oxygen-containing carbon nanotubes [J]. Environmental Chemistry, 2014, 33(10): 1692-1699(in Chinese). doi: 10.7524/j.issn.0254-6108.2014.10.003
[49] CRISTALE J, RAMOS D D, DANTAS R F, et al. Can activated sludge treatments and advanced oxidation processes remove organophosphorus flame retardants? [J]. Environmental Research, 2016, 144: 11-18. doi: 10.1016/j.envres.2015.10.008 [50] WU T, GAN Q, JANS U. Nucleophilic substitution of phosphorothionate ester pesticides with bisulfide (HS-) and polysulfides (Sn2-) [J]. Environmental Science & Technology, 2006, 40(17): 5428-5434. [51] 刘佳. 有机磷酸酯阻燃剂污染现状及降解过程研究进展 [J]. 应用化工, 2018, 47(12): 2705-2710,2714. doi: 10.3969/j.issn.1671-3206.2018.12.036 LIU J. Pollution status and degradation process of organophosphate flame retardants [J]. Applied Chemical Engineering, 2018, 47(12): 2705-2710,2714(in Chinese). doi: 10.3969/j.issn.1671-3206.2018.12.036
[52] KAWAGOSHI Y, FUKUNAGA I, ITOH H. Distribution of organophosphoric acid triesters between water and sediment at a sea-based solid waste disposal site [J]. Journal of Material Cycles & Waste Management, 1999, 1(1): 53-61. [53] SU G Y, LETCHER R J, YU H X. Organophosphate flame retardants and plasticizers in aqueous solution: pH-dependent hydrolysis, kinetics, and pathways [J]. Environmental Science & Technology, 2016, 50(15): 8103-8111. [54] WU L, CHLADKOVA B, LECHTENFELD O J, et al. Characterizing chemical transformation of organophosphorus compounds by 13C and 2H stable isotope analysis [J]. Science of the Total Environment, 2018, 615: 20-28. doi: 10.1016/j.scitotenv.2017.09.233 [55] FANG Y, KIM E, STRATHMANN T J. Mineral and base-catalyzed hydrolysis of organophosphate flame retardants: potential major fate-controlling sink in soil and aquatic environments [J]. Environmental Science & Technology, 2018, 52(4): 1997-2006. [56] KULKARNI S V, MARKAD V L, MELO J S, et al. Biodegradation of tributyl phosphate using Klebsiella pneumoniae sp. S3 [J]. Applied Microbiology and Biotechnology, 2014, 98(2): 919-929. doi: 10.1007/s00253-013-4938-2 [57] NANCHARAIAH Y V, REDDY G K K, MOHAN T V K, et al. Biodegradation of tributyl phosphate, an organosphate triester, by aerobic granular biofilms [J]. Journal of Hazardous Materials, 2015, 283: 705-711. doi: 10.1016/j.jhazmat.2014.09.065 [58] RANGU S S, BASU B, MURALIDHARAN B, et al. Involvement of phosphoesterases in tributyl phosphate degradation in Sphingobium sp. strain RSMS [J]. Applied Microbiology and Biotechnology, 2016, 100(1): 461-468. doi: 10.1007/s00253-015-6979-1 [59] XIONG J, LI G, AN T. The microbial degradation of 2, 4, 6-tribromophenol (TBP) in water/sediments interface: Investigating bioaugmentation using Bacillus sp. GZT [J]. Science of the Total Environment, 2017, 575: 573-580. doi: 10.1016/j.scitotenv.2016.09.017 [60] RANGU S S, MURALIDHARAN B, TRIPATHI S C, et al. Tributyl phosphate biodegradation to butanol and phosphate and utilization by a novel bacterial isolate, Sphingobium sp strain RSMS [J]. Applied Microbiology and Biotechnology, 2014, 98(5): 2289-2296. doi: 10.1007/s00253-013-5158-5 [61] BERNE C, MONTJARRET B, GUOUNTTI Y, et al. Tributyl phosphate degradation by serratia odorifera [J]. Biotechnology Letters, 2004, 26(8): 681-686. doi: 10.1023/B:BILE.0000023030.69207.c0 [62] 刘佳. 有机磷酸酯类阻燃剂化学氧化及微生物降解过程的机理研究[D]. 北京: 北京科技大学, 2019. Liu J. Mechanism of chemical oxidation and microbial degradation of organophosphate flame retardants [D]. Beijing : University of Science and Technology Beijing, 2019 (in Chinese).
[63] WEI K, YIN H, PENG H, et al. Bioremediation of triphenyl phosphate in river water microcosms: Proteome alteration of Brevibacillus brevis and cytotoxicity assessments [J]. Science of the Total Environment, 2019, 649: 563-570. doi: 10.1016/j.scitotenv.2018.08.342 [64] HOU R, LUO X, LIU C, et al. Enhanced degradation of triphenyl phosphate (TPHP) in bioelectrochemical systems: Kinetics, pathway and degradation mechanisms [J]. Environmental Pollution, 2019, 254: 113040. doi: 10.1016/j.envpol.2019.113040 [65] JURGENS S S, HELMUS R, WAAIJERS S L, et al. Mineralisation and primary biodegradation of aromatic organophosphorus flame retardants in activated sludge [J]. Chemosphere, 2014, 111: 238-242. doi: 10.1016/j.chemosphere.2014.04.016 [66] 卫昆. 磷酸三苯酯的微生物降解机制及其降解产物毒性研究[D]. 广州: 华南理工大学, 2018. WEI K. Microbial degradation mechanism of triphenyl phosphate and toxicity of its degradation products [D]. Guangzhou: South China University of Technology, 2018 (in Chinese).
[67] TAKAHASHI S, KAWASHIMA K, KAWASAKI M, et al. Enrichment and characterization of chlorinated organophosphate ester-degrading mixed bacterial cultures [J]. Journal of Bioscience and Bioengineering, 2008, 106(1): 27-32. doi: 10.1263/jbb.106.27 [68] TAKAHASHI S, MIURA K, ABE K, et al. Complete detoxification of tris (2-chloroethyl) phosphate by two bacterial strains: Sphingobium sp. strain TCM1 and Xanthobacter autotrophicus strain GJ10 [J]. Journal of Bioscience and Bioengineering, 2012, 114(3): 306-311. doi: 10.1016/j.jbiosc.2012.04.010 [69] TAKAHASHI S, OBANA Y, OKADA S, et al. Complete detoxification of tris(1, 3-dichloro-2-propyl) phosphate by mixed two bacteria, Sphingobium sp. strain TCM1 and Arthrobacter sp. strain PY1 [J]. Journal of Bioscience and Bioengineering, 2012, 113(1): 79-83. doi: 10.1016/j.jbiosc.2011.08.020 [70] ABE K, YOSHIDA S, SUZUKI Y, et al. Haloalkylphosphorus hydrolases purified from Sphingomonas sp Strain TDK1 and Sphingobium sp strain TCM1 [J]. Applied and Environmental Microbiology, 2014, 80(18): 5866-5873. doi: 10.1128/AEM.01845-14 [71] ABE K, MUKAI N, MOROOKA Y, et al. An atypical phosphodiesterase capable of degrading haloalkyl phosphate diesters from Sphingobium sp. strain TCM1 [J]. Scientific Reports, 2017, 7(1): 2842. doi: 10.1038/s41598-017-03142-9 [72] TAKAHASHI S, KATANUMA H, ABE K, et al. Identification of alkaline phosphatase genes for utilizing a flame retardant, tris (2-chloroethyl) phosphate, in Sphingobium sp strain TCM1 [J]. Applied Microbiology and Biotechnology, 2017, 101(5): 2153-2162. doi: 10.1007/s00253-016-7991-9 [73] LIU T, LU S Y, WANG R W, et al. Behavior of selected organophosphate flame retardants (OPFRs) and their influence on rhizospheric microorganisms after short-term exposure in integrated vertical-flow constructed wetlands (IVCWs) [J]. Science of the Total Environment, 2020, 710: 136403. doi: 10.1016/j.scitotenv.2019.136403 [74] DIAO Z H, LIU J J, HU Y X, et al. Comparative study of Rhodamine B degradation by the systems pyrite/H2O2 and pyrite/persulfate: Reactivity, stability, products and mechanism [J]. Separation and Purification Technology, 2017, 184: 374-383. doi: 10.1016/j.seppur.2017.05.016 [75] HE H, JI Q, GAO Z, et al. Degradation of tri (2-chloroisopropyl) phosphate by the UV/H2O2 system: Kinetics, mechanisms and toxicity evaluation [J]. Chemosphere, 2019, 236: 124388. doi: 10.1016/j.chemosphere.2019.124388 [76] LOHSE S, ROSENTRETER J J. Photooxidation of aqueous trichloroethylene using a buoyant photocatalyst with reaction progress monitored via micro-headspace GC/MS [J]. Microchemical Journal, 2006, 82(1): 66-72. doi: 10.1016/j.microc.2005.09.002 [77] WATTS M J, LINDEN K G. Photooxidation and subsequent biodegradability of recalcitrant tri-alkyl phosphates TCEP and TBP in water [J]. Water Research, 2008, 42(20): 4949-4954. doi: 10.1016/j.watres.2008.09.020 [78] WATTS M J, LINDEN K G. Advanced oxidation kinetics of aqueous trialkyl phosphate flame retardants and plasticizers [J]. Environmental Science & Technology, 2009, 43(8): 2937-2942. [79] SANTORO D, RAISEE M, MOGHADDAMI M, et al. Modeling hydroxyl radical distribution and trialkyl phosphates oxidation in UV-H2O2 photoreactors using computational fluid dynamics [J]. Environmental Science & Technology, 2010, 44(16): 6233-6241. [80] RUAN X C, AI R, JIN X, et al. Photodegradation of Tri (2-chloroethyl) phosphate in aqueous solution by UV/H2O2 [J]. Water Air and Soil Pollution, 2013, 224(1): 1406. doi: 10.1007/s11270-012-1406-z [81] YUAN X J, LACORTE S, CRISTALE J, et al. Removal of organophosphate esters from municipal secondary effluent by ozone and UV/H2O2 treatments [J]. Separation and Purification Technology, 2015, 156: 1028-1034. doi: 10.1016/j.seppur.2015.09.052 [82] ROCHA O R S D, DANTAS R F, NASCIMENTO JúNIOR W J, et al. Organophosphate esters removal by UV/H2O2 process monitored by 31p nuclear magnetic resonance spectroscopy [J]. Brazilian Journal of Chemical Engineering, 2018, 35(2): 521-530. doi: 10.1590/0104-6632.20180352s20160568 [83] TANG T, LU G N, WANG R, et al. Rate constants for the reaction of hydroxyl and sulfate radicals with organophosphorus esters (OPEs) determined by competition method [J]. Ecotoxicology and Environmental Safety, 2019, 170: 300-305. doi: 10.1016/j.ecoenv.2018.11.142 [84] YU X, YIN H, PENG H, et al. Degradation mechanism, intermediates and toxicology assessment of tris-(2-chloroisopropyl) phosphate using ultraviolet activated hydrogen peroxide [J]. Chemosphere, 2020, 241: 124991. doi: 10.1016/j.chemosphere.2019.124991 [85] 刘青. TiO2光催化降解水体中氯代有机磷酸酯类阻燃剂研究[D]. 南京: 南京大学, 2013. Liu Q. Photocatalytic degradation of chlorinated organophosphate flame retardants in water by TiO2 [D]. Nanjing : Nanjing University, 2013 (in Chinese).
[86] YE J, LIU J, LI C, et al. Heterogeneous photocatalysis of tris (2-chloroethyl) phosphate by UV/TiO2: Degradation products and impacts on bacterial proteome [J]. Water Research, 2017, 124: 29-38. doi: 10.1016/j.watres.2017.07.034 [87] TANG T, LU G, WANG W, et al. Photocatalytic removal of organic phosphate esters by TiO2: Effect of inorganic ions and humic acid [J]. Chemosphere, 2018, 206: 26-32. doi: 10.1016/j.chemosphere.2018.04.161 [88] ABDULLAH A M, O’SHEA K E. TiO2 photocatalytic degradation of the flame retardant tris (2-chloroethyl) phosphate (TCEP) in aqueous solution: A detailed kinetic and mechanistic study [J]. Journal of Photochemistry and Photobiology A:Chemistry, 2019, 377: 130-137. doi: 10.1016/j.jphotochem.2019.03.026 [89] HU H, ZHANG H X, CHEN Y, et al. Enhanced photocatalysis degradation of organophosphorus flame retardant using MIL-101(Fe)/persulfate: Effect of irradiation wavelength and real water matrixes [J]. Chemical Engineering Journal, 2019, 368: 273-284. doi: 10.1016/j.cej.2019.02.190 [90] YU X, YIN H, YE J S, et al. Degradation of tris-(2-chloroisopropyl) phosphate via UV/TiO2 photocatalysis: Kinetic, pathway, and security risk assessment of degradation intermediates using proteomic analyses [J]. Chemical Engineering Journal, 2019, 374: 263-273. doi: 10.1016/j.cej.2019.05.193 [91] OU H S, LIU J, YE J S, et al. Degradation of tris (2-chloroethyl) phosphate by ultraviolet-persulfate: Kinetics, pathway and intermediate impact on proteome of Escherichia coli [J]. Chemical Engineering Journal, 2017, 308: 386-395. doi: 10.1016/j.cej.2016.09.076 [92] XU X, CHEN J, QU R, et al. Oxidation of Tris (2-chloroethyl) phosphate in aqueous solution by UV-activated peroxymonosulfate: Kinetics, water matrix effects, degradation products and reaction pathways [J]. Chemosphere, 2017, 185: 833-843. doi: 10.1016/j.chemosphere.2017.07.090 [93] YU X, YIN H, PENG H, et al. Oxidation degradation of tris-(2-chloroisopropyl) phosphate by ultraviolet driven sulfate radical: Mechanisms and toxicology assessment of degradation intermediates using flow cytometry analyses [J]. Science of the Total Environment, 2019, 687: 732-740. doi: 10.1016/j.scitotenv.2019.06.163 [94] ANTONOPOULOU M, GIANNAKAS A, BAIRAMIS F, et al. Degradation of organophosphorus flame retardant tris (1-chloro-2-propyl) phosphate (TCPP) by visible light N, S-codoped TiO2 photocatalysts [J]. Chemical Engineering Journal, 2017, 318: 231-239. doi: 10.1016/j.cej.2016.06.124 [95] LIN J, HU H, GAO N, et al. Fabrication of GO@MIL-101(Fe) for enhanced visible-light photocatalysis degradation of organophosphorus contaminant [J]. Journal of Water Process Engineering, 2020, 33: 101010. doi: 10.1016/j.jwpe.2019.101010 [96] KRUITHOF J C, KAMP P C, MARTIJN B J. UV/H2O2 treatment: A practical solution for organic contaminant control and primary disinfection [J]. Ozone-Science & Engineering, 2007, 29(4): 273-280. [97] ANTONOPOULOU M, KARAGIANNI P, KONSTANTINOU I K. Kinetic and mechanistic study of photocatalytic degradation of flame retardant Tris (1-chloro-2-propyl) phosphate (TCPP) [J]. Applied Catalysis B:Environmental, 2016, 192: 152-160. doi: 10.1016/j.apcatb.2016.03.039 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5229
- HTML全文浏览数: 5229
- PDF下载数: 69
- 施引文献: 0





















