

-
河流是淡水生态系统的重要组成部分,河道生态系统中的浮游微生物是河道生物多样性的重要组成部分,且与河流中溶解氧、pH、硝态氮、总磷和重金属等众多环境因子息息相关[1-3],做好河流微生物多样性研究是河流健康评价、水系连通与生态修复、供水安全保障等工作的重要基础。
近 30 年来,各国形成了侧重点不同、各具特色的评价方法,如,美国环境保护局(US EPA)提出的快速生物监测协议(Rapid Bio-assessment Protocols,RBP)[4]、欧盟的《水框架法令》(WFD) [5]等都已在河流水生态系统健康管理中得到应用。2020年世界自然基金会联合我国8家科研院所共同发布《长江生命力报告2020》,提出长江生命力指数。这些评价方法均将水生态作为重要维度。
微生物作为有机化合物的主要分解和矿化者[6-7],在食物链和食物网中所处的营养级比大型水生动植物更低[8],水体微生物网络的复杂性一定程度上能反应水体中大型生物的网络复杂性与社区对扰动的抵抗力和恢复力[9]。微生物群落结构、丰度等也可以作为有机物分解、反硝化功能的指标[10]。
近一个世纪以来,地表水微生物安全主要是通过检测粪便污染的细菌指标来确定[11] ,但除致病菌外,在地表水微生物监测中污染指示微生物更能反应水体受污染的现状[12]。有研究者[13]提出可以创建可靠、标准化的参考数据库,作为指示健康微生物群落的基线指标。通过相关性计算可以分析环境因子与物种分布间的相关关系[14]。已有研究发现微生物多样性指数与总磷和磷酸盐含量呈极显著负相关关系[15]。SOUFFREAU et al[16]也发现总磷和氨氮对湖泊的微生物群落组成有显著影响。
微生物多样性研究的传统方法是微生物分离纯化培养,存在如培养周期长、分类学鉴定不准确等弊端[17]。分子生物学技术可以高效、准确地从遗传物质水平上研究环境中微生物的群落结构[18]。高通量测序技术是近年来微生物群落分析的常用技术[19-21],具备测序快速、通量高等优势,且可对多样品的多个可变区同时测序[22]。通过高通量测序技术研究三江干流水体微生物群落结构及其多样性,可以为构建三江流域生态系统管理方案提供科学依据。
宁波市的余姚江、奉化江和甬江合称“三江”。三江口由于其独特的自然地理条件,成为宁波市的重要标志[23-24]。三江河道的治理方案主要为水利工程建设[20],需要对河道水生态进行长期综合监测以提供数据支撑,极有必要对其中微生物群落结构与多样性展开全面研究。当前对于宁波三江流域微生物群落结构与多样性的研究较为缺乏,本文开展的研究说明通过高通量测序的方法开展河流的生态指标监测具有可行性。
-
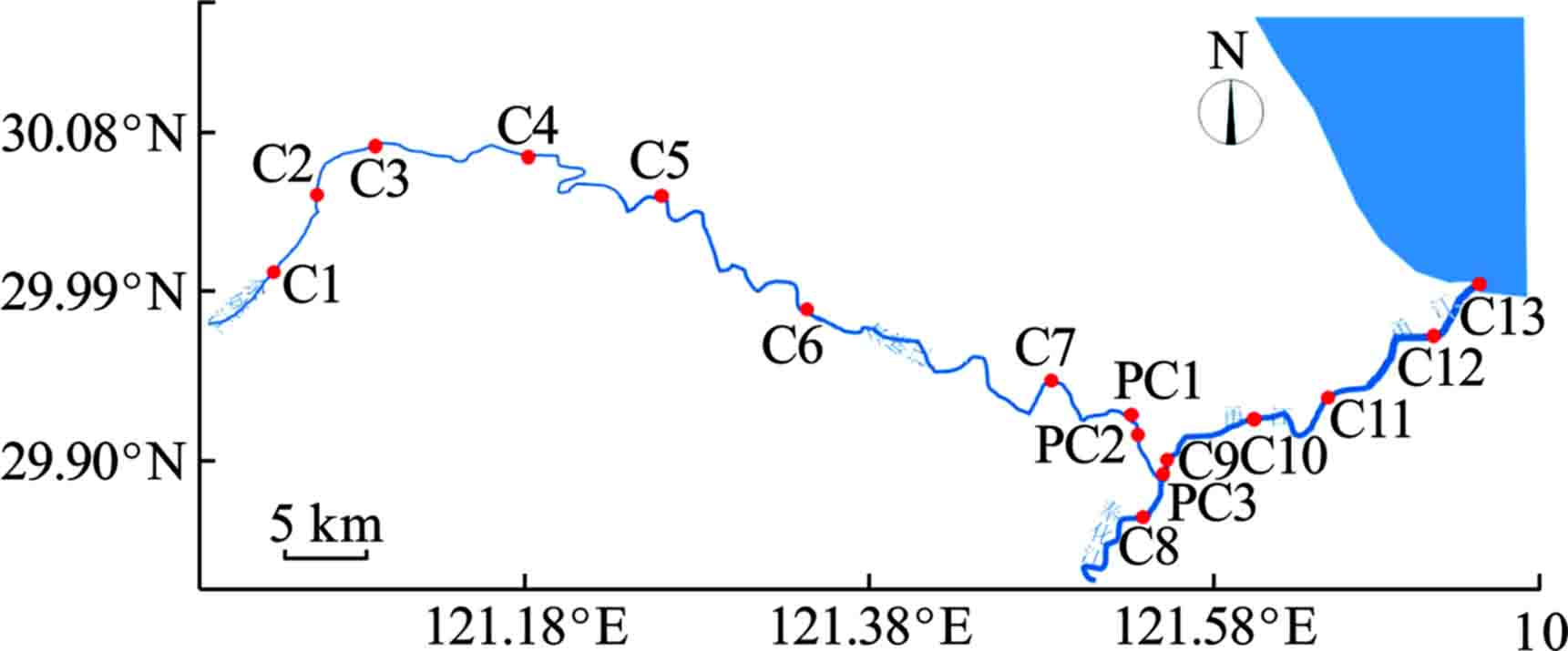
此次三江干流巡航测量范围为余姚江(牟山闸/永思桥~蜀山大闸~姚江大闸~三江口)、奉化江(三江口~澄浪堰)、甬江(三江口~入海口)。航测总长度约105 km。以下称三江干流均为上述范围。巡航测量时间为2021年5月11~14日。测量方式为使用航测船只沿余姚江、奉化江和甬江深泓航道线单向巡航一次。途中在三江干流12处点位停船进行水体取样测量。测量断面及取样点位置分布,见表1和图1。
取样过滤装置含真空泵或蠕动泵、六联过滤器和直径为47 mm、孔径0.45 µm的混合纤维素酯膜。
-
DNA提取和测序在Personal Biotechnology Co. Ltd.(中国上海)进行。按照制造商的说明,通过 DNeasy PowerSoil Kit(QIAGEN,Hilden,Germany)提取水样中总微生物基因组 DNA。通过琼脂糖凝胶电泳和 NanoDrop ND-1000 分光光度计 (Thermo Fisher Scientific, Waltham, MA, USA) 评估提取的 DNA 的质量和数量。
使用引物对515F_926R和18SV4F_18SV4R来分别对 16S rRNA基因、18S rRNA基因的可变区进行PCR扩增。
采用Illumina HiSeq X-10(Illumina,San Diego,CA,USA)平台对群落DNA片段进行双端(Paired-end)测序。使用DADA2方法进行去引物,质量过滤,去噪(denoise),拼接和去嵌合体等步骤。用QIIME2 (2019.4)进行物种分类学注释。将有效数据在97%的相似水平上划分为不同的运算分类单位(operational taxonomic unit, OTU), 将OTU代表序列与相应的微生物数据库比对, 利用Greengene数据等数据库对物种进行注释, 从而得到每个样本所含的物种信息。通过对OTUs进行Alpha多样性指数、丰度等分析, 并对物种注释在各个分类水平上进行群落结构的统计分析, 得到细菌群落的组成信息, 再通过Beta多样性分析, 系统分析三江干流不同流域样本间的微生物群落结构差异。
使用Alpha多样性来衡量局域均匀生境下的物种数目。α多样性使用Hill’s number family多样性计算[25],主要包括Chao指数;Faith’s pd指数;Simpson指数;Shannon 指数;Good’s coverage 指数;Pielou’s evenness指数;Observed species指数。
Chao1 是基于丰度的估计量,计算见式(1)[26]:
式中,
$ {S}_{\mathrm{c}\mathrm{h}\mathrm{a}\mathrm{o}1} $ 为预测丰富度,$ {S}_{\mathrm{o}\mathrm{b}\mathrm{s}} $ 为观测到的物种数量,$ {n}_{1} $ 为单序列OTUs数目,$ {n}_{2} $ 为双序列OTUs数目。Faith’s pd指数反映了谱系多样性,等于生命发育树(The Tree of Life Web)的分支数量[27]。
Simpson指数是衡量个体在分类时的集中程度,见式(2)[25] :
式中,
$ {S}_{\mathrm{o}\mathrm{b}\mathrm{s}} $ 为观测到的OTUs数量,$ {n}_{i} $ 为第i个OUT对应的个体数目,N为群落中所含的总个体数目(所有 OTU 的丰度之和)。Shannon 指数量化了预测从数据集中随机抽取的个体物种身份的不确定性,见式(3)[28] :
式中,
$ {S}_{\mathrm{o}\mathrm{b}\mathrm{s}} $ 为观测到的OTUs数量,$ {n}_{i} $ 为第i个OUT对应的个体数目,N为群落中所含的总个体数目。Good’s coverage 指数用于评估测序结果代表的community物种的总数[29] ,见式(4):
式中,F是单序列 OTU 的数量,N是群落中所含的总个体数目。
Pielou’s evenness指数用于描述物种中的个体的相对丰富度或所占比例[30],见式(5):
式中
$ ,H\mathrm{’} $ 为Shannon 指数的值,$ H{\mathrm{’}}_{\mathrm{m}\mathrm{a}\mathrm{x}} $ 为$ H\mathrm{’} $ 的最大值。Observed species指数为群落中丰度﹥0的物种数之和,可以反映物种种类信息。
Beta多样性是指沿着环境梯度变化的不同群落之间,物种组成的相异性或物种沿环境梯度的更替速率,因此也被称为生境间多样性(between-habitat diversity)。β多样性分析所使用的Bray-curtis距离基于OTUs的计数统计,比较2个群落微生物的组成差异,见式(6)[31]:
式(6)中
$ {,S}_{A,i} $ 和$ {S}_{B,i} $ 表示第i个OTU分别在A群落和B群落中的计数。min表示取两者最小值。 -
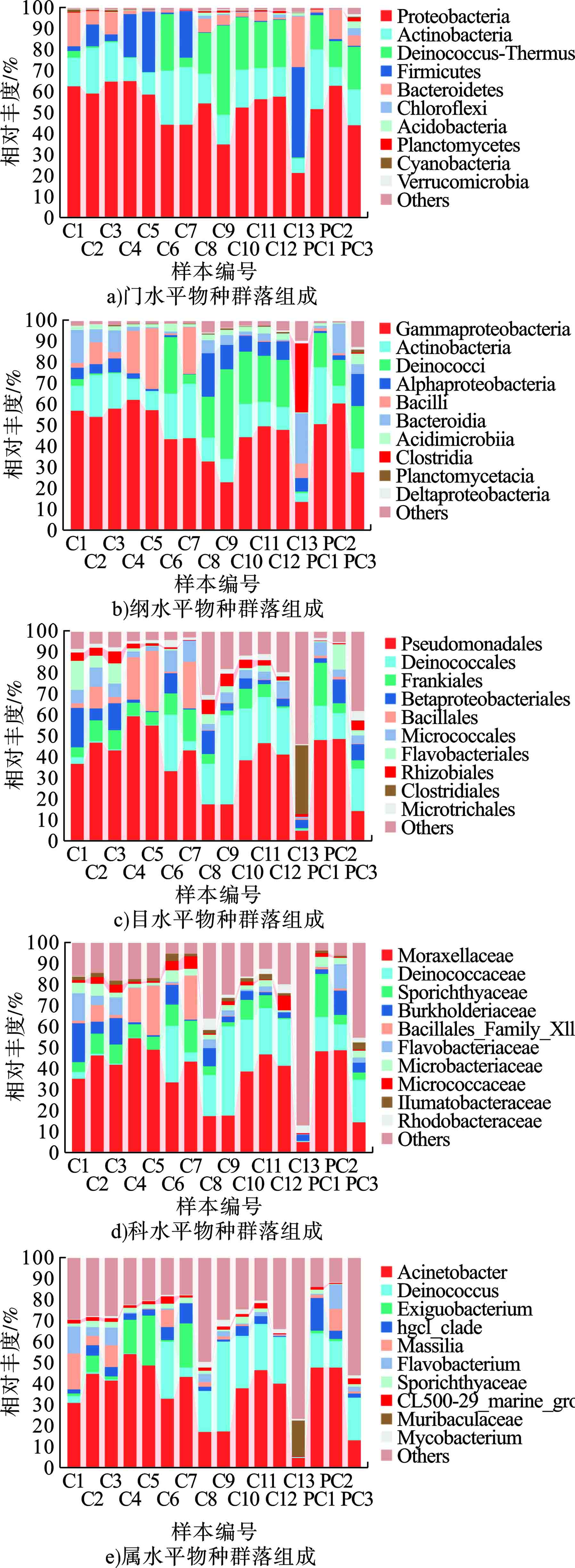
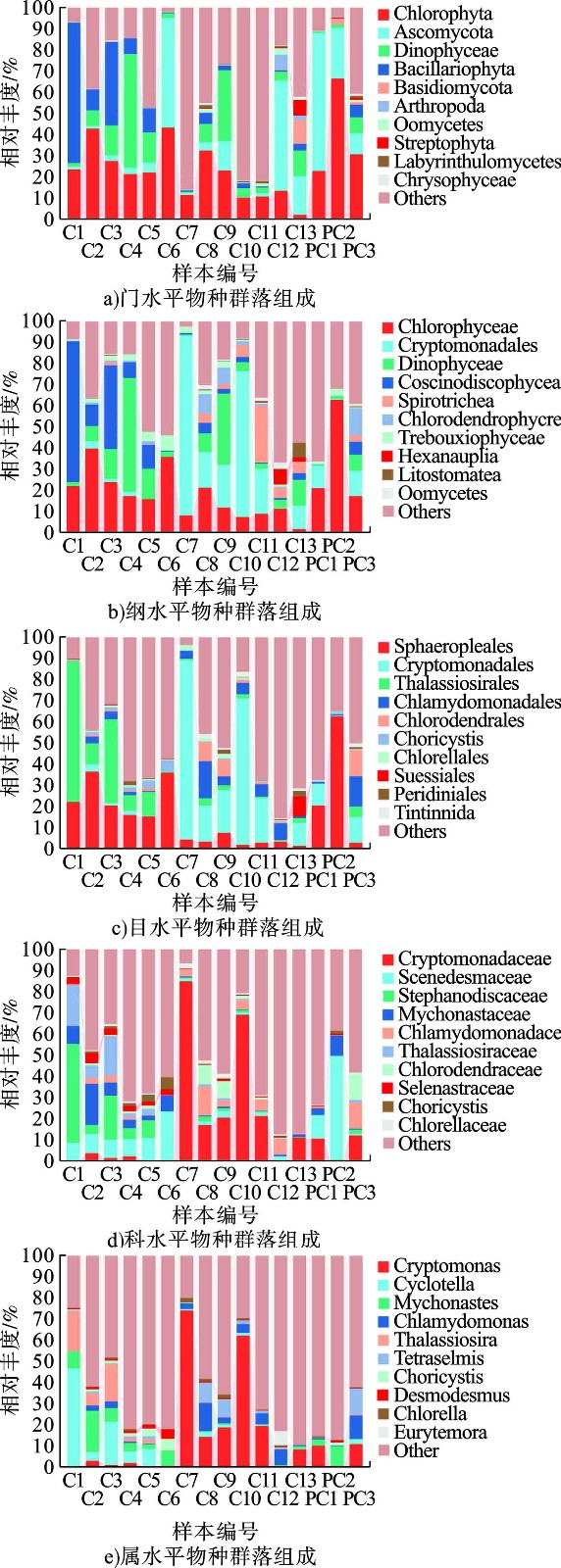
通过Silva 数据库对所得数据进行比对,根据序列物种分类学注释的结果对物种组成进行分类,展示相对丰度最高的10个分类,其余合并显示为others。统计样本的物种注释结果中门(Phylum) 、纲(Class) 、目(Order) 、科(Family) 和属(Genus)水平上含有的分类单元的数量。
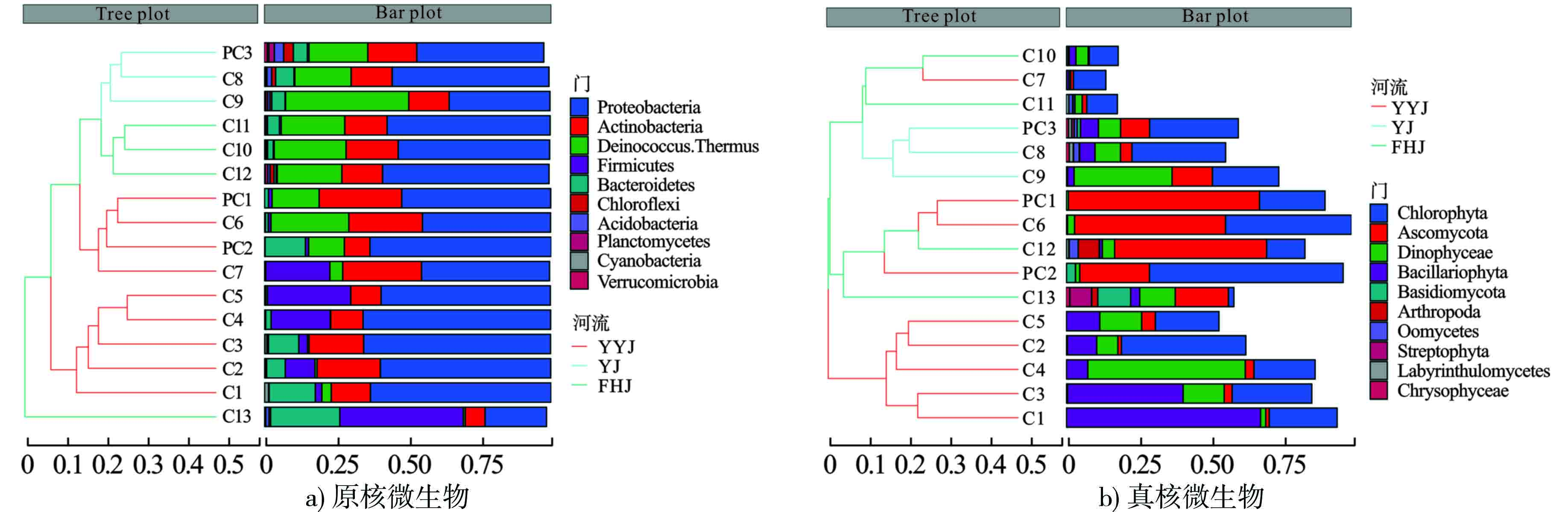
对于原核微生物,在门水平上,变形菌门(Proteobacteria)的平均丰度最高(图2a),在C1、C3、C4和PC2点位处相对丰度均超过60%,但在靠近入海口的C13点位处拟杆菌门(Firmicutes)的相对丰度最高,达到42.8%。需要注意在C4、C5和C7等采样水域厚壁菌门(Firmicutes) 表现出较高丰度,说明已经存在污水排放导致的DO含量下降,存在厌氧环境[32] 。在纲水平上(图2b),γ-变形菌纲(Gammaproteobacteria) 的平均丰度最高,其中在C1-C5、PC1、PC2的7个点位处相对丰度均超过50%,在临近入海口的C13处γ-变形菌纲的丰度明显下降,拟杆菌纲(Bacteroidia)、梭菌纲(Clostridia)的相对丰度呈现上升趋势。在门水平与纲水平上,优势菌种与其他研究中的结果一致[33-34]。在目水平上(图2c ),在余姚江河段,假单胞菌目(Pseudomonadales)平均相对丰度较高,达到46.4%,在奉化江和甬江河段异常球菌目(Deinococcales)相对丰度可达到20%以上,在C9处达到最高值42.7%。在科水平上(图2d),莫拉菌科(Moraxellaceae)在余姚江河段各采样点处都表现出较高的丰度,在C2-C5点位处丰度均高于40%,尤其在C4处达到54.5%,异常球菌科(Deinococcaceae)在甬江流域也表现出较高丰度,在C9处达到最高值42.7%。在属水平上,不动杆菌属(Acinetobacter)的平均丰度最高,异常球菌属(Deinococcus)在甬江流域也表现出了较高的丰度,最高在C9支流交汇处,达到42.7%。最丰富的细菌种类包括奇异球菌(Deinococcus grandis)(9.44%)、鲁氏不动杆菌(Acinetobacter lwoffii) (1.18%)、波西米亚不动杆菌(Acinetobacter bohemicus) (1.09%)等。
对于真核微生物,在门水平上,绿藻门(Chlorophyta)的平均丰度最高(图3a),在C2、C6和PC2点位处相对丰度均超过40%,在C6、PC1、PC2和C12等采样点处子囊菌门(Ascomycota) 也表现出较高的相对丰度,在PC1点位处子囊菌门(Ascomycota)的相对丰度最高,达到65.5%。在纲水平上(图3b),绿藻纲 (Chlorophyceae)的平均丰度最高,为25.6%,其次为引藻纲(Cryptomonadales)(12.7%)、甲藻纲(Dinophyceae)(7.8%)等分类。在临近入海口的C12、C13处绿藻纲的丰度明显下降,拟杆菌纲(Bacteroidia)、梭菌纲(Clostridia)的相对丰度呈现上升趋势。在目水平上(图3c),在C1~C6,环藻目(Sphaeropleales)、海链藻目(Thalassiosirales)相对丰度较高,达到46.4%,在C7、C10处,隐藻目(Cryptomonadales)相对丰度分别达到85.0%、69.1%,表现出显著优势。在科水平上(图3d),隐鞭藻科(Cryptomonadaceae) 平均丰度达到13.2%,在C7、C10处表现出显著的高丰度,尤其在C7处达到85.0%,但在C1~C5各点位处其丰度并不显著。在属水平上,隐藻属(Cryptomonas)的平均丰度最高,主要在C7、C10处表现出显著的高丰度,小环藻属(Cyclotella)在C1处相对丰度达到46.6%。最丰富的真核微生物种类包括弯隐藻(Cryptomonas curvata) (6.85%)、寄生索囊藻(Choricystis parasitica)(0.79%)和马索隐藻(Cryptomonas marssonii) (0.62%)等。
-
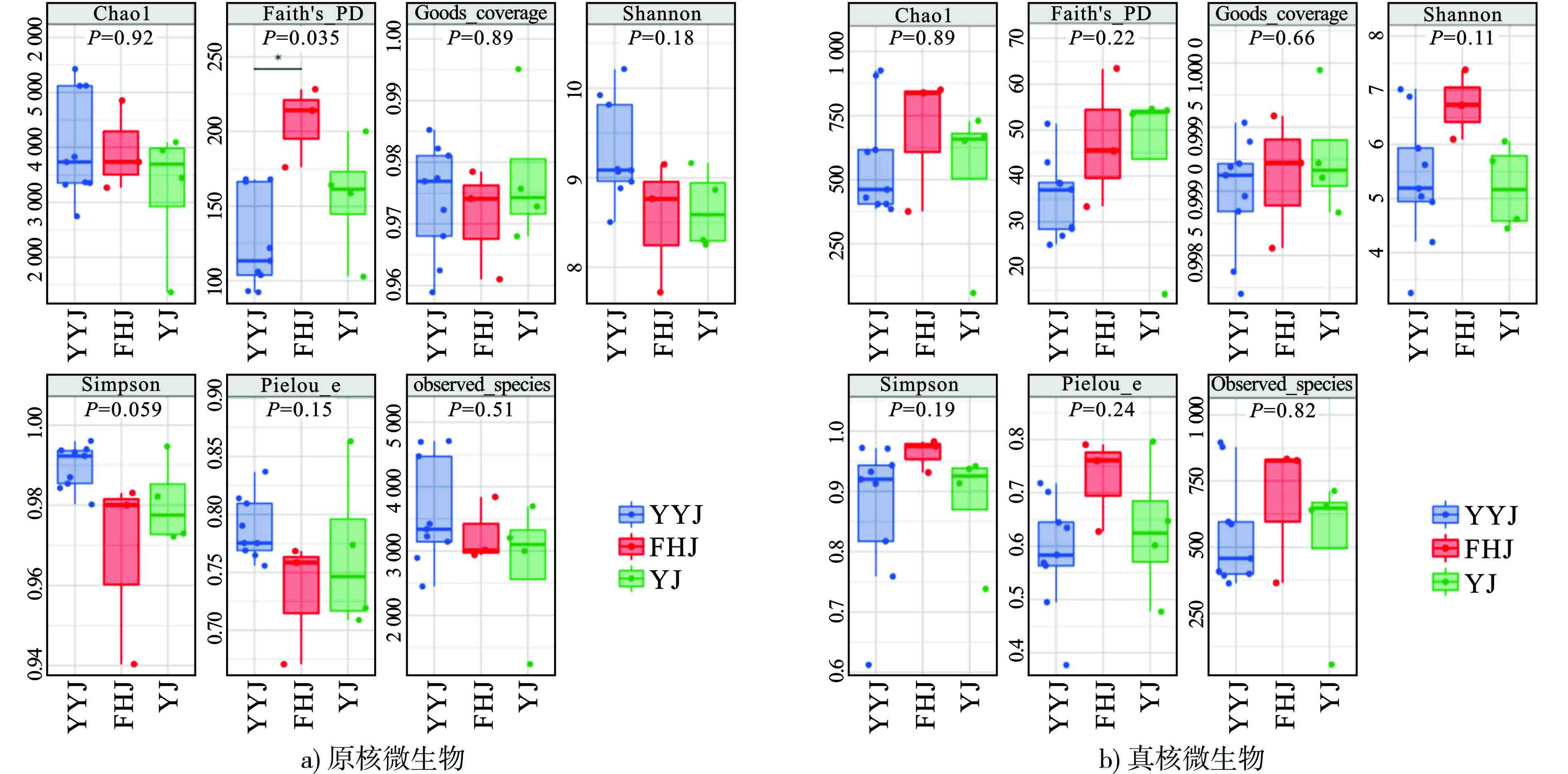
以Chao1和Observed species指数表征丰富度,以Shannon和Simpson指数表征多样性,以Faith’s PD指数表征基于进化的多样性,以Pielou’s evenness指数表征均匀度,以Good’s coverage指数表征覆盖度,微生物群落Alpha多样性分析结果,见图4。
图4a可知,对于原核微生物,YYJ样品的Chao1和Observed species指数最高, FHJ和YJ样品的Chao1和Observed species指数值差异不大,说明余姚江流域的群落丰富度最高,Chao1值在3 000以上。YYJ样品的Shannon指数值相对较高,但3组样品整体差异不显著,Simpson 指数基本稳定在0.98以上,说明三江干流流域各采样点原核微生物群落多样性较丰富,稳定性较好,其中余姚江流域的原核微生物多样性相对更高。YYJ流域样品的Faith’s PD指数较小,而YJ流域样品Faith’s PD指数较大,FHJ居中,说明三江干流自上游至下游原核微生物的亲缘关系逐渐复杂化,基于进化的多样性较高。YYJ流域样品的Pielou’s evenness指数较高,FHJ其次,YJ最小,说明3个流域的物种均匀度自上游向下游逐渐减低。Good’s coverage指数值均在0.97以上,说明测序对群落中物种的覆盖度较高,未被检测出的物种所占的比例很少。与澜沧江[35]、科希河[36] 和博斯腾湖流域[37]等流域相比,宁波三江流域原核微生物群落丰富度与多样性更高。
图4b可知,对于真核微生物,FHJ样品的Chao1和Observed species指数最高, YJ其次,YYJ样品最低,说明奉化江采样监测流域的群落丰富度最高。FHJ样品的Shannon指数值相对较高,67%的采样点的Simpson 指数在0.9以上,3组样品Simpson 指数值无显著差异,说明三江干流流域各采样点真核微生物群落多样性较丰富,稳定性较好,其中FHJ流域的真核微生物多样性相对更高。YYJ流域样品的Faith’s PD指数较小,而YJ流域样品Faith’s PD指数较大,FHJ居中,说明三江干流自上游至下游真核微生物的亲缘关系与原核微生物的发展一致,也呈现逐渐复杂化的趋势,基于进化的多样性较高。FHJ流域样品的Pielou’s evenness指数较高,YJ其次,YYJ最小,说明3个流域的物种均匀度自上游向下游逐渐升高。Good’s coverage指数值均在0.999以上,说明测序对群落中物种的覆盖度较高,未被检测出的物种所占的比例极少。与九龙江[38]、西安渭河、灞河和浐河[39]等流域相比,宁波三江流域真核微生物丰富度更低,多样性相近。
-
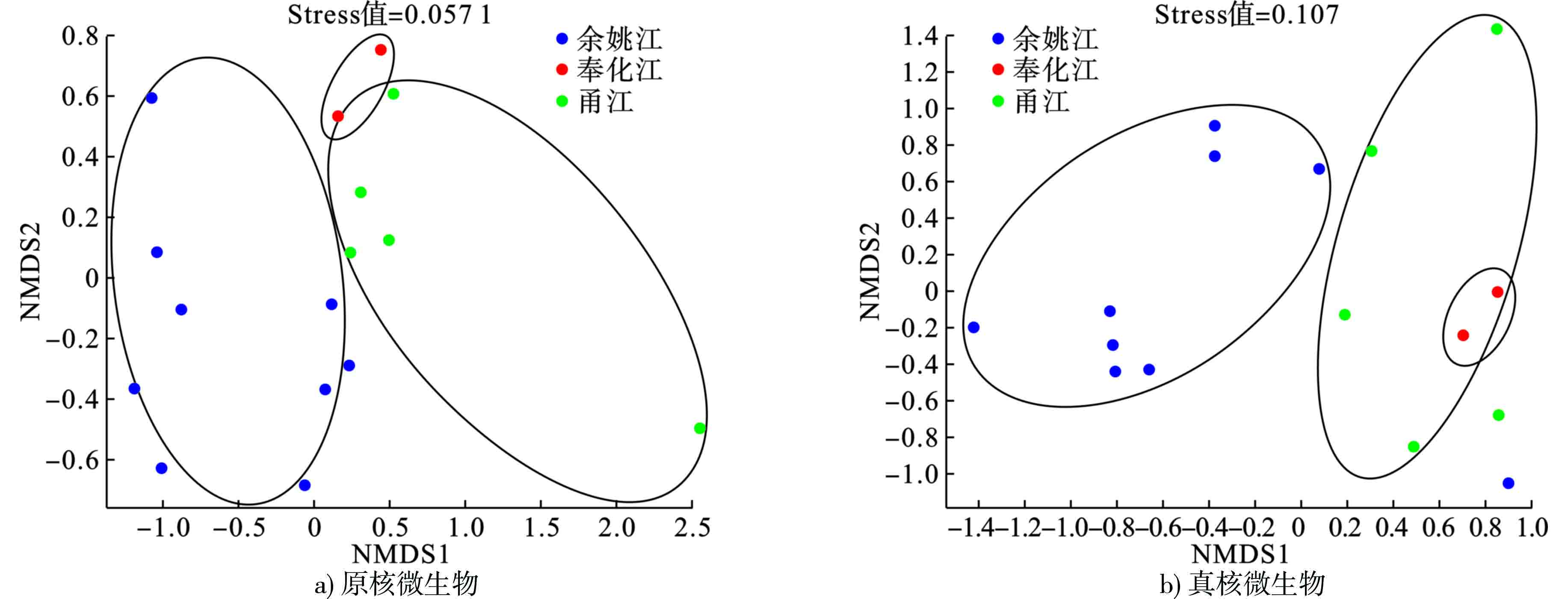
根据Bray-curtis 距离算法,对物种进行非度量多维尺度(non-metric multidi-mensional scaling,NMDS)分析(图5),发现YYJ的样品在维度NMDS1 和维度NMDS2 能够很好地与FHJ、YJ区分开,说明余姚江流域样品的原核、真核微生物种类与FHJ、YJ有较为明显的区分性。其中Stress 值分别为0.0571、0.107,均﹤0.2,很好地证明了数据的可靠性。
采用层次聚类(hierarchical clustering)的分析方法,以等级树的形式展示样本间的相似度,通过聚类树的分枝长度衡量聚类效果的好坏。对Bray-curtis距离矩阵采用UPGMA算法(即聚类方法为average)在门水平上进行聚类分析,见图6。可以看出,三江干流的YYJ、FHJ和YJ的3部分采集的样品中的原核微生物在不同河段呈现出鲜明的聚类效果,说明三江干流自上游向下游原核微生物在门水平上有比较明显的变化,不同流域的原核微生物相似度较高,入海口C13处则展示出完全不同的群落组成。位置相近的采样点的真核微生物的聚类也比较相似,如干流上游方位的C1~C5点位处样本之间的相似度很高,但与相对处于下游方向的点位之间的相似度较低。在支流汇入后的样品的群落构成会出现比较明显的变化,如C9采样点与其他点位的相似度就相对较低。
-
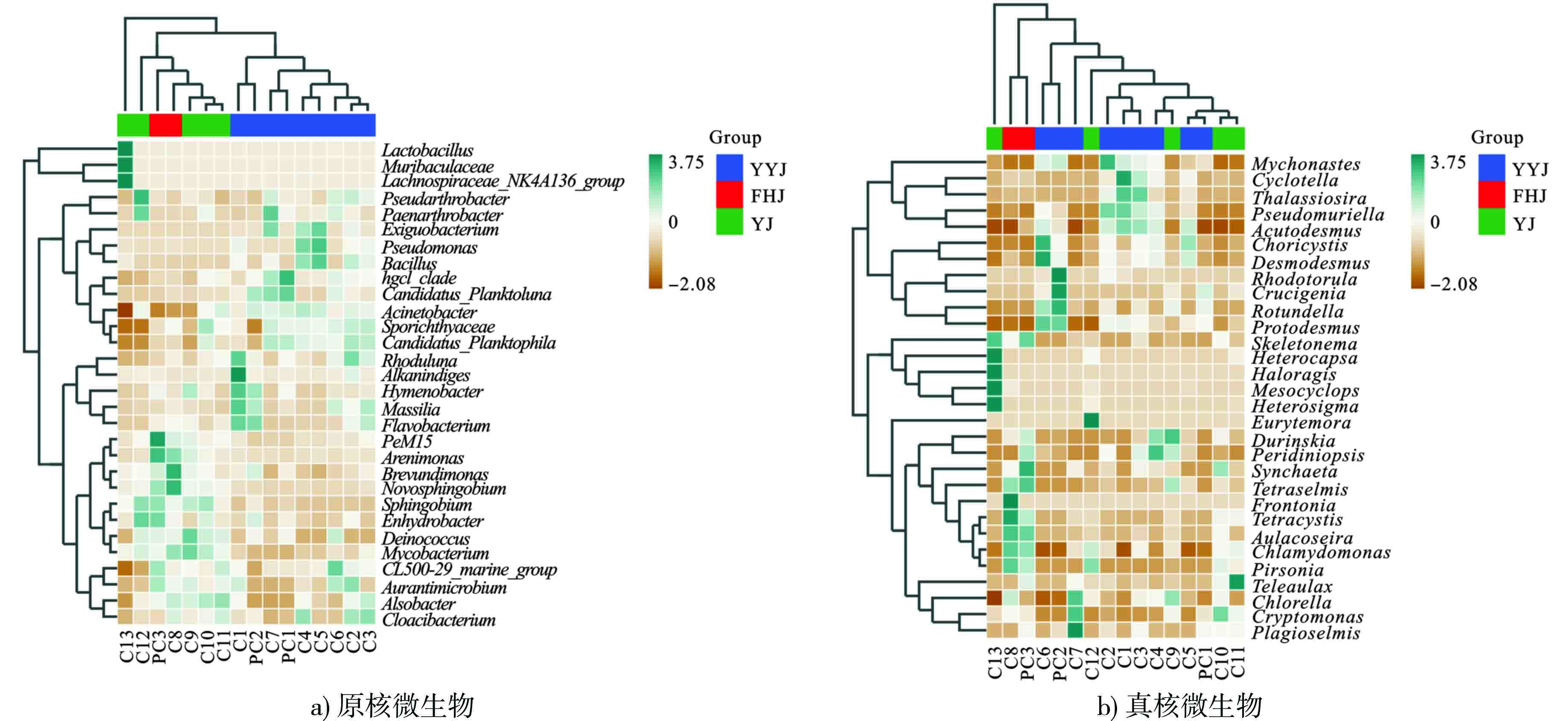
根据测序结果和注释OTU 数据进行样本间丰度和相似性聚类,构建宁波三江干流微生物属层次物种分类Heatmap 图谱,以颜色梯度变化表现丰度值的高低,见图7。使用平均丰度前30位的属的丰度数据绘制热图。样本按照物种组成数据的欧式距离(euclidean distance)进行UPGMA聚类。
原核微生物在属上的Heatmap 图谱显示,YYJ流域样品中微小杆菌属(Exiguobacterium) 的丰度最高,次为马赛菌属(Massilia)、黄杆菌属(Flavobacterium)、芽孢杆菌属(Bacillus)和不动杆菌属(Acinetobacter)等;FHJ流域样品中丰度最高的属为新鞘脂菌属(Novosphingobium)、砂单胞菌属(Arenimonas)和短波单胞菌属 (Brevundimonas)等;YJ流域样品中分支杆菌属(Mycobacterium)、鞘脂菌属(Sphingobium)和异常球菌属(Deinococcus)等丰度更高。原核乳杆菌属(Lactobacillus)、鼠杆菌属(Muribaculaceae)等只在入海口C13处测得,该处组成与其余点位有明显的区别。
真核微生物在属水平上的Heatmap图谱显示,YYJ流域样品中绿藻属(Pseudomuriella)、麦可属(Mychonastes) 和海链藻属(Thalassiosira)的丰度最高,FHJ样品中囊藻属(Tetracystis)、扁藻属(Tetraselmis)和直链藻属(Aulacoseira)表现出较高的丰度,YJ样品中尖尾藻属(Teleaulax)、隐藻属(Cryptomonas)相对丰度较高。入海口C13采样点处测得铜绿微囊藻属(Hererocapsa)、小二仙草属(Haloragis)、剑水蚤属(Mesocyclops)和赤潮异弯藻属(Heterosigma)等仅在此点位显示出较高丰度的真核生物属。
-
通过高通量测序,发现三江干流优势菌门主要有变形菌门、放线菌门、异常球菌-栖热菌门和拟杆菌门,真核微生物优势门包括子囊菌门、甲藻门、绿藻门和硅藻门等,这些均为典型淡水微生物门类,与其他水域微生物群落组成检测结构相似[40-42]。如刘峰等[43]发现汾河入黄口水体中变形菌门、放线菌门和厚壁菌门处于优势地位,在湖泊水体中变形菌门、蓝细菌门、拟杆菌门、放线菌门和疣微菌门也是最主要的5个原核生物门类[44],这些典型的淡水菌门在淡水生态系统中的广泛分布已经得到研究证实[45-46]。在三江干流下游异常球菌-栖热菌门丰度的增加可能意味着该河段受到较多的热污染影响[45]。在西安渭河、沣河、灞河和浐河的研究中,绿藻和真菌界子囊菌门也是在各类群中的优势物种[39],与宁波三江流域真核微生物优势门一致,个别点位处(如C4、C9等处)甲藻门丰度较高可能与温度的升高或pH的降低有关[46],与原核微生物的优势菌门中异常球菌-栖热菌门的出现共同说明宁波三江干流受到热污染的可能性很高。
同时通过Beta多样性分析也注意到,不同流域微生物群落组成表现出明显的区分性,如异常球菌-栖热菌门、子囊菌门在三江干流的中下游水域丰度更高,而在上游的采样点C1~C5处的样品中丰度较低。厌氧性厚壁菌的出现说明存在工业或生活废水的排入,已经造成水生态环境的破坏[47]。入海口处水域的微生物组成也与其他采样点处明显不同。由物种组成热图发现,微生物组成在相邻采样点间相似性较高,YYJ、FHJ和YJ 3组样品中也有不同的优势物种,可以作为流域水环境状况的指示生物。
Alpha 多样性分析反映了宁波三江干流流域微生物多样性的综合指标,自三江干流上游向下游进行采样检测,各采样点水域处的原核微生物丰富度指数和多样性指数均较高,YYJ样品与FHJ样品、YJ样品相比较,原核微生物丰富度差异不大,YYJ流域样品多样性最高。真核微生物方面,三江干流流域整体原核微生物丰富度与其他流域研究结果相比偏低,多样性维持在较高的水平,其中FHJ样品的物种丰富度和多样性相对更高。自上游至下游微生物的亲缘关系呈现逐渐复杂化的趋势。
-
本实验结果仅就宁波三江干流流域微生物多样性进行表征,水体微生物多样性受到多种环境因子的影响,一方面水体微生物多样性与环境因子之间的关系,不同水域所处的地理位置对于水体微生物群落的影响都值得进一步探索。
同时,水体的微生物作为重要的环境指示生物也可以作为水生态环境现状的指示,通过研究流域微生物群落结构多样性与群落结构,可以为构建完善的监测与管理体系提供理论支持。在常态化生态监测工作中微生物多样性的监测与分析工作也具备可行性。通过对微生物群落结构多样性与其驱动因子的研究,可以开发宁波三江干流流域特殊功能菌群,为区域水环境治理提供针对性支持。
基于高通量测序的宁波三江干流微生物多样性分析
Analysis of microbial diversity of main stream of Yuyao-Fenghua-Yongjiang rivers watershed in Ningbo based on high throughput sequencing
-
摘要: 为了解宁波三江干流水域微生物多样性,文章通过 16S rDNA、18S rDNA 基因扩增和高通量测序分析了自上游向下游方向的16 个采样点水域微生物群落结构组成及多样性差异。结果显示,不同采样点原核、真核微生物 Chao1和Observed species指数值较大,覆盖率均高于0.97。奉化江样品的真核微生物物种丰富度和多样性相对更高。门水平上,优势菌门包括变形菌门(Proteobacteria)、放线菌门(Actinobacteria)等,真核生物绿藻门(Chlorophyta)的平均丰度最高。Beta多样性分析显示,不同流域微生物群落组成表现出明显的区分性,余姚江、奉化江和甬江3组样品的微生物种群结构以及优势门有显著差异。结果表明,三江干流水域整体微生物丰富度与多样性较高,但是少数采样水域厚壁菌门(Firmicutes)的高丰度说明存在工业或生活污水排放导致的水体污染。Abstract: To have a full understanding of the microbial diversity in the main stream of Yuyao River, Fenghua River and Yongjiang River in Ningbo, the microbial community structure composition and diversity of 16 sampling points from upstream to downstream were analyzed by 16S rDNA, 18S rDNA gene amplification and high-throughput sequencing. The results showed that the Chao1 and Observed species index values of prokaryotic and eukaryotic microorganisms at different sampling points were large, and the coverage rate for both prokaryotic and eukaryotic microorganisms were higher than 0.97. The species richness and diversity of eukaryotic microorganisms in Fenghua River samples were relatively higher. As for the phylum level, the dominant bacteria included Proteobacteria, Actinobacteria, etc. For eukaryotic, the average abundance of Chlorophyta was the highest. Beta diversity analysis showed that the microbial community composition in different watersheds would show an obvious differentiation, and there were differences in the microbial population structure and dominant gate among Yuyao River, Fenghua River and Yongjiang River. The conclusion was the overall microbial richness and diversity in the main stream of the three rivers were high. However, the high abundance of Firmicutes in a few sampling watersheds indicated that there was water pollution caused by the industrial or domestic sewage discharge.
-

-
表 1 三江干流航测断面及取样点位置分布
编号 位置描述 河流 C1 永思桥 YYJ C2 支流交汇处 YYJ C3 河流分叉前 YYJ C4 河流分叉汇聚后 YYJ C5 支流交汇处和弯曲处 YYJ C6 河流弯曲处,临近河姆渡遗址 YYJ C7 河流弯曲处 YYJ C8 澄浪堰 FHJ C9 支流后,甬江起点 YJ C10 支流交汇处,临近泛迪码头 YJ C11 支流交汇处 YJ C12 河流弯曲处,临近镇电新村 YJ C13 入海口 YJ PC1 国控断面补测点1 YYJ PC2 国控断面补测点2 YYJ PC3 水文站补测点 FHJ 注:余姚江(YYJ),奉化江(FHJ),甬江(YJ)。  下载: 导出CSV
下载: 导出CSV
-
[1] 刘亚军, 刘欣, 牟晓真, 等. 大型浅水湖泊鄱阳湖湿地微生物的研究现状[J]. 微生物学通报, 2019, 46(12): 279 − 286. [2] 秦宇, 郑望, 张曦, 等. 三峡库区中段水体微生物群落结构与环境因子相关性研究[J]. 长江流域资源与环境, 2021, 30(05): 160 − 169. [3] YUAN T M, MCCARTHY A J, ZHANG Y X, et al. Impact of temperature, nutrients and heavy metals on bacterial diversity and ecosystem functioning studied by freshwater microcosms and high-throughput DNA sequencing[J]. Current Microbiology, 2020, 77(11): 3512 − 3525. doi: 10.1007/s00284-020-02138-5 [4] USEPA. Rapid bioassessment protocols for use in wadeable streams and rivers, 2nd Edition. EPA 841-B-99-002[R]. U. S. Environmental Protection Agency; Office of Water; Washington, D. C, 1999. [5] Common Implementation Strategy for the Water Framework Directive (2000/60/EC)[S/OL]. (2022-11-01). https://ec.europa.eu/environment/water/water-framework/objectives/pdf/strategy3.pdf. [6] COLWELL R R. Microbial diversity: the importance of exploration and conservation[J]. Journal of Industrial Microbiology and Biotechnology, 1997, 18(5): 302 − 307. doi: 10.1038/sj.jim.2900390 [7] NOLD S C, ZWART G. Patterns and governing forces in aquatic microbial communities[J]. Aquatic Ecology, 1998, 32(1): 17 − 35. doi: 10.1023/A:1009991918036 [8] SHERR E B, SHERR B F. Heterotrophic dinoflagellates a significant component of microzooplankton biomass and major grazers of diatoms in the sea[J]. Marine Ecology Progress Series, 2007, 352: 187 − 197. doi: 10.3354/meps07161 [9] ISIBOR P O, IMOOBE T O T, DEDEKE G A, et al. Health risk indices and zooplankton-based assessment of a tropical rainforest river contaminated with iron, lead, cadmium, and chromium[J]. Scientific Reports, 2020, 10(1): 16896. doi: 10.1038/s41598-020-72526-1 [10] KARIMI B, MARON P A, CHEMIDLIN-PREVOST B N, et al. Microbial diversity and ecological networks as indicators of environmental quality[J]. Environmental Chemistry Letters, 2017, 15(2): 265 − 281. doi: 10.1007/s10311-017-0614-6 [11] 杨蓉, 李垒, 霍晓芹, 等. 水质标准中指示微生物的发展及现状[J]. 中国环境监测, 2020, 36(4): 1 − 10. [12] 满江红. 中国华北西北地区地表水微生物指标调查及研究[D]. 合肥: 安徽医科大学, 2013. [13] ASTUDILLO-GARCÍA C, HERMANS S M, STEVENSON B, et al. Microbial assemblages and bioindicators as proxies for ecosystem health status: potential and limitations[J]. Applied Microbiology and Biotechnology, 2019, 103(16): 6407 − 6421. doi: 10.1007/s00253-019-09963-0 [14] 谢煜, 邢雅丽, 沈钧亮, 等. 淡水水体中异养细菌丰度研究进展[J]. 浙江万里学院学报, 2017, 30(2): 89 − 95. [15] 朱金山, 秦海兰, 孙启耀, 等. 冬季小流域水体微生物多样性及影响因素[J]. 环境科学, 2020, 41(11): 255 − 265. [16] SOUFFREAU C, VAN DER GUCHT K, VAN GREMBERGHE I, et al. Environmental rather than spatial factors structure bacterioplankton communities in shallow lakes along a > 6000 km latitudinal gradient in South America[J]. Environmental Microbiology, 2015, 17(7): 2336 − 2351. doi: 10.1111/1462-2920.12692 [17] AMANN R I, LUDWIG W, SCHLEIFER K H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation[J]. Microbiological Reviews, 1995, 59(1): 143 − 169. doi: 10.1128/mr.59.1.143-169.1995 [18] 刘国华, 叶正芳, 吴为中. 土壤微生物群落多样性解析法: 从培养到非培养[J]. 生态学报, 2012, 32(14): 133 − 146. [19] LIU Q, ZHAO Q N, MCMINN A, et al. Planktonic microbial eukaryotes in polar surface waters: recent advances in high-throughput sequencing[J]. Marine Life Science & Technology, 2021, 3(1): 94 − 102. [20] GAO W L, CHEN Z J, LI Y Y, et al. Bioassessment of a Drinking Water Reservoir Using Plankton: High Throughput Sequencing vs. Traditional Morphological Method[J]. Water, 2018, 10(1): 82 [21] SONG P X, TANABE S, YI R, et al. Fungal community structure at pelagic and littoral sites in Lake Biwa determined with high-throughput sequencing[J]. Limnology, 2018, 19(2): 241 − 251. doi: 10.1007/s10201-017-0537-8 [22] ZHAN A B, MACISAAC H J. Rare biosphere exploration using high-throughput sequencing: research progress and perspectives[J]. Conservation Genetics, 2015, 16(3): 513 − 522. doi: 10.1007/s10592-014-0678-9 [23] 胡安谊, 李姜维, 杨晓永, 等. 宁波三江口水域原核生物群落结构分析[J]. 环境科学, 2015(7): 163 − 171. [24] 郑建根, 张松达, 谢敏, 等. 宁波市三江河道水沙变化规律及成因分析[J]. 长江科学院院报, 2018, 35(5): 6 − 11. doi: 10.11988/ckyyb.20171259 [25] ARMSTRONG J S. Colonisation of New Zealand by hemicordulia australiae, with notes on its displacement of the indigenous procordulia grayi (Odonata: Corduliidae)[J]. New Zealand Entomologist, 1978, 6(4): 381 − 384. doi: 10.1080/00779962.1978.9722297 [26] CHAO A, CHIU C. H. Species Richness: Estimation and Comparison[R]. Wiley StatsRef: Statistics Reference Online, 2016: 1-26. [27] FAITH D P, RICHARDS Z T. Climate change impacts on the tree of life: Changes in phylogenetic diversity illustrated for acropora corals[J]. Biology, 2012, 1(3): 906 − 932. doi: 10.3390/biology1030906 [28] ALLEN B, KON M, BAR‐YAM Y. A new phylogenetic diversity measure generalizing the shannon index and its application to phyllostomid bats[J]. The American Naturalist, 2009, 174(2): 236 − 243. doi: 10.1086/600101 [29] STEELE W A. Good coverage[J]. Nature, 1992, 355(6358): 310 − 310. [30] TUCKER C, AZE T, CADOTTE M, et al. Assessing the utility of conserving evolutionary history[J]. Biological Reviews, 2019, 94(5): 1740 − 1760. doi: 10.1111/brv.12526 [31] BRAY J R, CURTIS J T. An ordination of the upland forest communities of Southern Wisconsin[J]. Ecological Monographs, 1957, 27(4): 325 − 349. doi: 10.2307/1942268 [32] NELSON M C, MORRISON M, YU Z T. A meta-analysis of the microbial diversity observed in anaerobic digesters[J]. Bioresource Technology, 2011, 102(4): 3730 − 3739. doi: 10.1016/j.biortech.2010.11.119 [33] 韩雪梅, 龚子乐, 杨晓明, 等. 汛期前后老鹳河干流人类干扰下浮游细菌多样性及功能预测[J]. 环境科学, 2021, 42(2): 831 − 841. [34] 于小彦, 张平究, 张经纬, 等. 城市河流沉积物微生物量分布和群落结构特征[J]. 环境科学学报, 2020, 40(2): 227 − 238. [35] 程豹, 望雪, 徐雅倩, 等. 澜沧江流域浮游细菌群落结构特征及驱动因子分析[J]. 环境科学, 2018, 39(8): 3649 − 3659. [36] NAMITA, YONGQIN L, KESHAO L, et al. Bacterial community composition and diversity in Koshi River, the largest river of Nepal[J]. Ecological Indicators, 2019, 104: 501 − 511. doi: 10.1016/j.ecolind.2019.05.009 [37] TANG X, XIE G, SHAO K, et al. Contrast diversity patterns and processes of microbial community assembly in a river-lake continuum across a catchment scale in northwestern China[J]. Environmental Microbiome, 2020, 15(1): 10. doi: 10.1186/s40793-020-00356-9 [38] 黄亚玲. 河流氮输出与真核微生物群落结构对流域土地利用模式和水文状况的响应研究[D]. 厦门: 厦门大学, 2019. [39] 何晓乐. 西安四条河流急流—深潭—河滩系统底泥微生物多样性及退化河流恢复策略[D]. 西安: 长安大学, 2020. [40] YANG J, JIANG H C, DONG H L, et al. A comprehensive census of lake microbial diversity on a global scale[J]. Science China-Life Sciences, 2019, 62(10): 1320 − 1331. doi: 10.1007/s11427-018-9525-9 [41] SALMASO N, BOSCAINI A, PINDO M. Unraveling the diversity of eukaryotic microplankton in a large and deep perialpine lake using a high throughput sequencing approach[J]. Frontiers in Microbiology, 2020: 11. [42] 彭柯, 董志, 邸琰茗, 等. 基于16S rRNA高通量测序的北运河水体及沉积物微生物群落组成对比分析[J]. 环境科学, 2021, 42(11): 5424 − 5432. [43] 刘峰, 冯民权, 王毅博. 汾河入黄口夏季微生物群落结构分析[J]. 微生物学通报, 2019, 46(1): 60 − 70. [44] 任丽娟, 何聃, 邢鹏, 等. 湖泊水体细菌多样性及其生态功能研究进展[J]. 生物多样性, 2013, 21(4): 36 − 47. [45] THEODORAKOPOULOS N, BACHAR D, CHRISTEN R, et al. Exploration of Deinococcus-Thermus molecular diversity by novel group-specific PCR primers[J]. MicrobiologyOpen, 2013, 2(5): 862 − 872. [46] SÆTRE R, AURE J, DANIELSSEN D S. Long-term hydrographic variability patterns off the Norwegian coast and in the Skagerrak[J]. ICES Journal of Marine Science, 2003, 219: 150 − 159. [47] JOSHI A A, KANEKAR P P, KELKAR A S, et al. Cultivable bacterial diversity of alkaline Lonar Lake, India[J]. Microbial Ecology, 2008, 55(2): 163 − 172. doi: 10.1007/s00248-007-9264-8 -
 点击查看大图
点击查看大图
图( 7) 表( 1)
计量
- 文章访问数: 1960
- HTML全文浏览数: 1960
- PDF下载数: 15
- 施引文献: 0













