

-
氮是蛋白质、酶、核酸、氨氧化细菌、硝化细菌和反硝化细菌所必须的[1]。然而过量的氨氮会损害水生环境,导致水体富营养化和溶解氧(dissolved oxygen,DO)下降,最终导致鱼类和其它水生生物死亡[2-3]。MFC现在被认为是一种很有前景的技术,与其它好氧技术和厌氧技术相比,MFC具有污泥产量较低以及去除污染物同时进行能源回收等优点[4]。近些年,通过将MFC与传统的脱氮工艺相结合,从而实现污水同步脱氮产电引起人们的广泛关注[5-7]。王佳琪等[8]探讨了碳氮比对单室MFC产电及污染物去除效果的影响,然而当碳氮比为7时,MFC的脱氮功能受到严重的抑制。与单室MFC相比,双室MFC中阴极室和阳极室发生的反应相对独立,相互干扰较小。因此当MFC与传统脱氮工艺相结合时更多采用的是双室MFC,可以依据功能的不同进行灵活的配置。
生物电营养反硝化是近年来一种新兴技术,其可以用电而不是有机物作为电子供体[9-10]。2007年CLAUWAERT等[11]以乙酸盐为阳极底物,不含有机物的NO3−污水为阴极底物构建双室MFC实现阴极完全脱氮。DING等[12]探究了阴极不同接种污泥对电营养反硝化的影响,优化了接种污泥选取策略。虽然电营养反硝化MFC中NO3−/NO2−可以从阴极中获得电子,然而这些电子来自于阳极上的碳氧化。从来源上看,碳仍然是反硝化过程的主要电子供体来源。因此,电营养反硝化MFC的阳极底物通常为含氨废水而阴极底物为单一的硝酸盐废水,而实际污水往往含有有机物、NH4+-N、NO3−-N、NO2−-N等多种污染物。VIRDIS等[13]在废水中应用完全脱氮的MFC,使用1个独立的硝化反应器与1个双室MFC结合。含乙酸钠和氨氮的合成废水被连续送入MFC的阳极室,然后阳极室出水流入外部硝化反应器进行硝化反应,最后进入MFC的阴极室利用阳极传递的电子进行反硝化。然而,采用外部硝化反应器的系统设计额外增加了建设成本。VIRDIS等[14]通过控制阴极室中的曝气强度,克服了这一弱点,将同步硝化反硝化与MFC进行结合。ZHANG等[15]通过构建阴极混合生物膜MFC,在碳源充足的盛宴阶段实现同步硝化反硝化,但在碳源缺乏的饥荒阶段则需要进行停曝利用电营养反硝化进行脱氮,然而低DO虽然有利于脱氮但对产电不利。同步硝化反硝化需要精确控制DO、pH等操作条件,操作复杂。为解决反硝化菌在好氧阴极难以富集和脱氮效果差的问题,ZHANG等[16]构建阳极反硝化MFC进行脱氮,使阴极硝化产生的NO3−/NO2−通过AEM迁移到厌氧阳极室中进行反硝化。黄丽巧等[17]通过构建阴极硝化耦合阳极反硝化的双室MFC,探究了以AEM作为分隔膜的MFC(AEM-MFC)和以CEM作为分隔膜的MFC(CEM-MFC)的脱氮性能,结果表明当阴极NH4+-N投加200 mg·L−1时,AEM-MFC只需要66 h即可完全去除总氮,而相同条件下,CEM-MFC需要26 d才能达到相同的脱氮效果。虽然阳极反硝化MFC具有良好的脱氮效果,但由于AEM的阻隔作用,阳极室中的NH4+无法得到有效去除,仍需要进一步处理。
目前双室MFC的阴阳极室通常以不同成分的污水作为底物。阴极脱氮型MFC为处理同一种污水,有的增设外部硝化反应器或蠕动泵等设施从而增加了建设成本,有的对DO进行调控,通过调节曝气强度/曝气时间实现硝化与反硝化,操作复杂。而阳极脱氮型MFC阳极室与阴极室的分隔膜通常采用AEM,由于AEM对阳离子的阻隔作用,阳极室中的NH4+无法得到有效去除。因此,本研究构建了一种CEM与AEM交替排列的FC-MFC,操作简单且无需外部硝化反应器等外部设施。阳极室与阴极室之间用CEM与AEM进行交替分隔,在浓度差作用下离子进行迁移,最终实现阳极室中有机物和氨氮的同步去除。通过改变阳极室COD(P1、P2、P3和P4分别为500、700、900和1 100 mg·L−1),设置不同进水碳氮比,探究不同阳极COD对FC-MFC污染物去除及产电性能的影响。并以P4工况为例,分析FC-MFC氮去除途径。
-
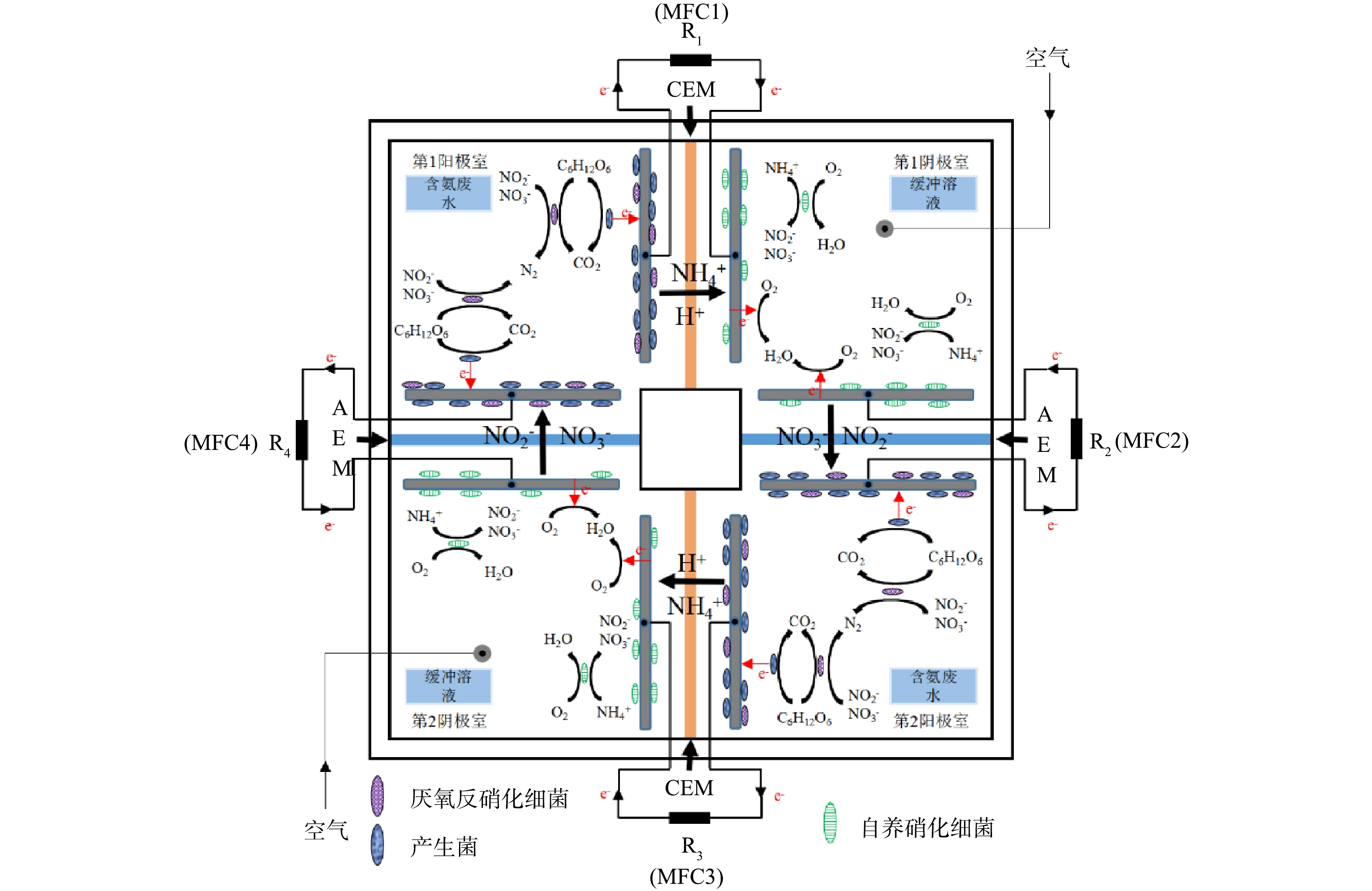
FC-MFC由2个阳极室与2个阴极室组成。序列的首位为阳极室,序列的末位为阴极室,且末位阴极室与首位阳极室相连形成循环序列。按照序列次序,位于前序位的阳极室与后序位阴极室之间安装有CEM,位于前序位的阴极室与后序位的阳极室之间安装有AEM。电极室序列排布顺序分别为第1阳极室、第1阴极室、第2阳极室和第2阴极室。按照上述排布顺序将4个电极室依次连接,形成如图1所示的外形呈方形的FC-MFC。
阳极室和阴极室的外观呈方形,材质为有机玻璃。反应器顶端设有电解液更换孔道、参比电极孔道与电极安置孔道,电解液更换孔道与参比电极孔道为8 mm,电极孔道尺寸为2 mm。阳极室和阴极室的有效工作体积均为500 mL,CEM和AEM横截面积为42 cm2(6 cm×7 cm)。阳极和阴极电极均为碳毡,碳毡尺寸为5 cm×6 cm×3 mm。电极相邻安置在每个CEM和AEM的两侧,相邻的电极通过连接1000 Ω的外部电阻形成闭合回路,形成4个MFC模块MFC1、MFC2、MFC3和MFC4。外部电路连接采用独立电路连接,每个MFC模块单独与1个外部电阻连接。阴极室采用鼓风曝气,阴极室DO通过转子流量计进行调节,控制DO在(5.5±0.5) mg·L−1。在阳极室和阴极室电解液施加磁力搅拌以避免短流。所有实验均在(25±1) ℃的室温下进行。
-
该实验采用间歇培养的模式,将实验室驯化的30 mL反硝化污泥和30 mL自养硝化污泥分别注入阳极室和阴极室中进行接种。阳极电解液为模拟含氨废水,阴极电解液为含无机盐的缓冲溶液。电解液主要成分为每升水中包含:0.1 g KCl、0.5 g NaCl、0.015 g CaCl2、0.02 g MgCl2·6H2O、14.15 g K2HPO4·3H2O、5.166 g KH2PO4、1 mL的微量元素和1 mL维生素[17]。除此之外,阳极电解液含有0.4684 g·L−1 C6H12O6(COD为500 mg·L−1)、0.3824 g·L−1 NH4Cl(氨氮为100 mg·L−1),阴极电解液中含有1 g·L−1 NaHCO3。当其中1个MFC模块低于50 mV时即认定产电周期结束,此时更换阳极和阴极电解液,在至少连续3个产电周期电池均能达到相似电压(相差不超过5%)时,认为FC-MFC启动成功。
FC-MFC成功启动后实验分为2个阶段:第1阶段,改变C6H12O6质量浓度(即改变C/N比),设置4个COD水平:500(P1)、700(P2)、900(P3)和1 100 mg·L−1(P4)。每改变1次工况,先运行1个周期使微生物适应该工况,再连续监测1个周期阳极室和阴极室中污染物和pH的变化,并考察系统产电性能。第2阶段,待第1阶段实验结束,以P4工况为例,探究FC-MFC氮去除途径和各部分的贡献度。
-
由电压采集系统(DAS,PISO-813,台湾泓格)每隔1 min自动记录一次MFCi(i=1、2、3、4)模块输出电压。功率密度曲线在电压稳定阶段采用变电阻法测定,输出电流和功率密度的计算按照已有研究进行[18],根据功率密度曲线可得MFCi模块最大功率密度。电流通过式(1)进行计算。功率密度根据式(2)计算。
式中:Ii分别为MFCi外部电路通过的电流,mA;Ui分别为MFCi模块输出电压,mV;Ri分别为MFCi模块外部电阻,Ω。
式中:Pi分别为MFCi模块输出功率密度,mW·m2;Ui分别为MFCi模块输出电压,V;Ii分别为MFCi外部电路通过的电流,mA;A为电极双侧表面积,m2。
库伦效率(coulombic efficiency,CE)为回收的电子与有机物质能提供的电子之比。根据图1所示,与传统双室MFC有所不同,以FC-MFC中的第1阳极室为例,第1阳极室中微生物氧化有机物的电子一部分通过R1,一部分通过R4。因此间歇运行时CE按式(3)计算。系统发电过程的能量输出通过式(4)计算。阴极室曝气消耗的能量通过式(5)计算。
式中:Q为库伦效率,%;MCOD为基于O2的COD摩尔质量,32 g·mol−1;t为产电周期,s;I1为MFC1外部电阻R1通过的电流,A;I4为MFC4外部电阻R4通过的电流,A;F为法拉第常数,96 485.34 C·mol−1;b为每摩尔O2还原转换的电子摩尔数,取4;Van为第1阳极室有效容积,m3;△COD为第1阳极室COD的变化量,g·m−3。
式中:W为系统发电过程的能量输出,KJ;U为电池输出电压,V;I为电池输出电流,A;t为产电周期,s。
式中:Wae为阴极室曝气消耗的能量,KJ;P1为标准大气压,101 325 Pa;P2为鼓风机入口压力,Pa;T为温度,298 K;λ为曝气常数,取1.4;ϛ为鼓风机效率,取0.8;ρ0为标准条件下的空气密度,1.29 kg·m−3;Qg为曝气流量,mol·s−1;tae为曝气时间,s。
所有常规水质指标均按照《水和废水监测分析方法》(第4版)检测,采用微机型酸度计监测pH。
-
以不同COD的含氨废水为阳极底物时FC-MFC的输出电压如图2所示。P1、P2、P3和P4时的产电周期和各MFC模块的峰值电压存在差异。在P1、P2、P3和P4工况下,FC-MFC的产电周期依次为103、128、162和189 h;MFC1的最高输出电压依次321、317、442和526 mV;MFC2的最高输出电压依次为325、318、436和525 mV;MFC3的最高输出电压依次为304、314、603和619 mV;MFC4的最高输出电压依次为313、316、603和619 mV。虽然MFC1、MFC2、MFC3和MFC4采用独立电路连接方式,但阳极室与阴极室中的电解液等效于导线对MFC1、MFC2、MFC3和MFC4进行连接。MFC1、MFC2、MFC3和MFC4的阳极电极电势基本没有差异,随后对阴极电极电势进行监测。共用第1阴极室的MFC1和MFC2的阴极电极电势基本一致,而共用第2阴极室的MFC3和MFC4的阴极电极电势基本一致,这也使得MFC1和MFC2的输出电压高度相似,而MFC3和MFC4的输出电压高度相似。但(MFC1、MFC2)与(MFC3、MFC4)之间又存在着差异,这可能因为2个阴极室中的生物膜丰度产生了一定的差异。
整体来说,随着阳极COD的升高,产电周期与各MFC模块的峰值电压呈上升趋势,其原因可能是COD的增加使电极微生物可利用的底物质量浓度增加,同时使阳极室的厌氧环境得到加强,从而促进其对底物的利用[8]。对于P3和P4工况,各MFC模块输出电压经历了3个阶段:延滞期、上升期和稳定期。分析原因主要归为以下2点:首先,有相关研究表明[19-20],当阳极存在硝酸盐时会对产电性能产生一定的影响。产电菌和反硝化细菌争夺电子供体,有机物被用于反硝化而未能传至阳极电极表面,随着系统运行,迁移至阳极室中硝酸盐质量浓度逐渐降低,阳极生物产生的电子大部分又被传递至阳极,因此系统电压呈上升趋势。其次,在反应初期,在浓度差作用下前序位阳极室中的大量氨氮迁移至后序位阴极室,硝化反应会争夺O2[21],阴极获取O2作为电子受体的能力会受到抑制,导致前期产电性能受到抑制。综合两者原因,P1、P2工况下阳极底物质量浓度较低,输出电压迅速上升到稳定电压就维持直到产电结束。而P3、P4工况下阳极底物质量浓度较高,前期与P1、P2时的输出电压基本持平,后期由于阳极充足的底物质量浓度和阴极O2更多用于产电,因此,输出电压经历了延滞期、上升期和稳定期3个阶段。
通过功率密度实验研究了产电性能,总结了开路电压和最大功率密度。如图3所示,P1和P2下的开路电压接近,在623~690 mV。P3和P4下的开路电压接近,在810~848 mV之间。在P1、P2、P3和P4工况下,MFC1模块最大功率密度分别为44.08、47.20、76.28和112.23 mW·m−2;MFC2模块最大功率密度分别为65.03、72.00、77.36和118.01 mW·m−2;MFC3模块最大功率密度分别为68.27、70.94、74.60和103.47 mW·m−2;MFC4模块最大功率密度分别为33.33、36.40、89.47和121.00 mW·m−2。
理论上,开路电压与MFC中氧化还原物质质量浓度密切相关[18]。在FC-MFC这个体系中,O2是在阴极处被还原的主要电子受体,阴极室中进行的硝化反应会与阴极争夺O2从而影响阴极电极电势。此外,COD作为主要的还原剂,在开路电压的变化趋势中起着关键作用[22]。
一般来说,功率输出由开路电压和过电压共同决定[18]。由于不同工况下溶液的电导率几乎相同,欧姆损失作为过电压的一部分在该系统中表现出微小的差异[18]。然而活化损失和浓度损失,在很大程度上导致了过电压[18]。事实上,一些非电力过程可能加剧了过电位[23]。例如,在硝化过程中,可能会消耗相当数量的O2[24]。有机物可能用于反硝化还原,而不是发电[5]。因此,氧化还原物质的“无效消耗”增加了活化损失和浓度损失,进一步导致严重的过电位。这可能是P1和P2功率输出较低的重要原因,而P3和P4在电压稳定阶段,硝化过程与反硝化过程的对电池的影响较小。整体来说,各MFC模块的最大功率密度随着阳极COD的增加而增加,表明阳极COD的增加有利于提升电池性能。同一工况下4个MFC模块最大功率密度有所差异,这表明在共用电解液的4个MFC模块之间,很难保证所有电极腔室间发生的氧化还原反应速率完全一致,某一电极产生的反应会影响到相邻的电极,因此4个MFC模块之间的差异是内在的和不可避免的。
在P1工况下COD出水介于65.80~76.83 mg·L−1,比P2、P3和P4的COD出水略高,可能原因是运行初期产电菌富集的较少,因此,在产电结束时出水COD略高于其它工况(51.61~63.99 mg·L−1)。由图4可知,随着COD的升高,阳极室COD去除率呈逐渐上升趋势,最高可达94%以上。而库伦效率呈现先下降后上升再下降的趋势,这个原因与图2的输出电压息息相关。在P1、P2阶段FC-MFC的输出电压相近,P3、P4阶段的输出电压相近,随着阳极COD的升高,相比因产电周期延长而产生的电荷量,更多的COD被非产电微生物所利用,因此,库伦效率反而有所下降。而根据图2,P3阶段的输出电压峰值远远高于P2,P3阶段产电周期内利用有机物产生的电荷量高于P2,因此,库伦效率反而有所上升。库伦效率是测量总净电输出量的关键指标,其变化趋势受到氧化还原物质“无效消耗”的影响,生化过程(如硝化、反硝化)对库伦效率的变化起着关键作用[25-26]。此外,库伦效率低于以往的研究[25],这是因为本实验的反应器体积比以往的装置体积大。
-
在MFC中,由于阳离子(Na+、NH4+、K+、Ca2+和Mg2+)的质量浓度通常是质子的105倍,因此除质子外,其它主要的阳离子负责正电荷通过膜的运输。此外,质子在阴极反应被消耗,除质子以外的阳离子运输导致了阴极室中pH的增加和MFC性能的下降[27]。KUNTKE等[28]开发了一种可以同时生产能量和回收氨氮的MFC,氨氮向阴极进行迁移,在高pH条件下阴极室中NH4+转化为挥发性氨。FENG等[29]和ZHANG等[16]构建的AEM-MFC,也证实了阴极室中的硝酸盐可以迁移至阳极室进行反硝化。这使得FC-MFC中前序位阳极室氨氮迁移至后序位阴极室进行硝化反应,硝化产物NO3−/NO2−迁移至后序位阳极室进行反硝化成为可能。
不同COD水平下,FC-MFC可以实现同步脱氮。由图5可知,同一时刻P1阶段的氨氮和硝酸盐氮略高于P2、P3和P4。原因可能是反应器运行前期阴极富集的自养硝化细菌丰度较低,阴极室氨氮有少量积累,影响了前序位阳极室中氨氮的迁移速率,进而影响了硝酸盐氮向后序位阳极室迁移的量。与先前的研究一致[6],当外部电阻为1000 Ω时,NH4+的迁移主要归因于浓度驱动的过程,尽管外部施加的电压可以略微增强这一过程。这可能原因是外部电阻阻值较大导致FC-MFC处于低电流密度,同时阴极室硝化反应的进行使阴阳极室的氨氮浓度差处于最大限值,因此浓度差占主导地位。产电周期结束后,在P1、P2、P3和P4工况下,第1阳极室的TN出水质量浓度分别为(20.03±3.59)、(9.60±0.27)、(5.60±0.16)和(3.71±0.42) mg·L−1;第1阴极室的TN出水质量浓度分别为(12.92±2.11)、(6.97±0.53)、(4.78±0.75)和(3.17±0.14) mg·L−1;第2阳极室的TN出水质量浓度分别为(14.31±2.84)、(9.62±0.30)、(5.70±0.86)和(2.64±0.29) mg·L−1;第2阴极室的TN出水质量浓度分别为(14.27±3.31)、(7.18±0.64)、(3.84±0.28)和(2.80±0.14) mg·L−1。
王佳琪等[8]探讨了碳氮比对单室MFC产电及污染物去除效果的影响,随碳氮比升高,NH4+和TN的去除率先上升后下降,当碳氮比为7时,NH4+和TN的去除率仅为(22.21±1.2)%和(22.18±1.3)%。而随着进水COD的增加(即进水碳氮比的升高),FC-MFC的产电周期延长,产电结束时,阳极室中的氨氮和阴极室中的硝酸盐氮出水质量浓度越低。阳极室TN去除率最高可达96%以上,同时阴极室中的TN残留量低于4 mg·L−1。这表明了FC-MFC对高C/N比污水具有较好的抵抗负荷,这是因为CEM与AEM的阻隔作用,阳极室中的COD对阴极室的干扰较小,有利于自养硝化细菌的生长,整个过程中阴极室中的氨氮基本低于检测限。高DO会抑制阴极反硝化反应的进行,导致NO3−的积累,而低DO影响硝化反应。而FC-MFC通过CEM与AEM的交替排列,硝化和反硝化反应在不同腔室中进行,通过离子之间迁移即可实现脱氮,维持高DO以保持产电输出,无需对DO进行调控(曝气强度/曝气时间),无需泵送设施和外部硝化反应器,简化了操作。同时阳极反硝化产生的碱度会中和厌氧降解氧化有机物产生的H+,阴极硝化反应会缓解O2作为阴极电子受体而产生的OH−,从而维持系统pH的稳定[17]。
-
以P4工况下为例,探究氮去除途径及各去除途径对氮去除总量的贡献度。由于阳极室电解液低DO环境不适合电极微生物进行硝化过程[30]。此外,高COD和低NO2−浓度的溶液条件下,厌氧氨氧化也不大可能发生[31]。因此,阳极室电解液中氨氮的减少最可能通过CEM迁移至阴极室所致[32]。当然,阳极室电解液中氨氮质量浓度的变化也可能与微生物吸附以及生长繁殖代谢作用有关,因此进行图6中的实验,将CEM与AEM都更换为有机玻璃板。
由图6可知,在前12 h内阳极室对氨氮的去除主要是吸附作用,由于阳极室两侧的离子交换膜被更换为有机玻璃板,阳极室中的氨氮无法进行膜扩散,之后很长一段时间基本保持稳定状态。第1阳极室和第2阳极室中微生物吸附代谢作用对氨氮的去除率分别为24.20%和24.76%。
将FC-MFC中的AEM替换为有机玻璃板,阻碍了阴极室中NOX−(NO3−和NO2−)的迁移,故在阴极室中的硝酸盐呈现累积状态。假设前序位阳极室中的氨氮通过CEM迁移至后序位阴极室进行硝化反应后完全转化为NOX−。由图7可知,第1阴极室中残留的NOX−占前序位阳极室迁移至第1阴极室中氨氮的比重为98.79%,第2阴极室中残留的NOX−占前序位阳极室迁移至第2阴极室中氨氮的比重为93.21%。
如图8所示,以闭路状态下分析各部分去除途径的贡献度,根据图6计算的百分数,第1阳极室和第2阳极室中微生物吸附代谢氨氮的量分别为25.76 mg·L−1和26.3 mg·L−1。理想情况下,阳极室中氨氮去除量扣除微生物吸附代谢作用后即为通过CEM迁移至阴极室的量。因此,第1阳极室和第2阳极室通过CEM迁移氨氮的量分别为76.81 mg·L−1和77.82 mg·L−1。根据图7计算结果所得的百分比,则第1阴极室和第2阴极室中NOX−的残留量分别为75.88 mg·L−1和72.54 mg·L−1。阴极室中NOX−的残留量减去闭路工况下阴极室中NOX−的残留量即为通过AEM迁移的量。故第1阴极室和第2阴极室中NOX−通过AEM迁移至后序位阳极室中的量分别为72.61 mg·L−1和69.91 mg·L−1。由第1阳极室中的氨氮通过CEM迁移至第1阴极室中的量减去第1阴极室中NOX−通过AEM迁移的量和阴极室中本身残留的氮污染物即为第1阴极室NOX−在阴极室自身去除的量。同理,第2阴极室也是如此。这部分比重仅占氮去除总量的0.91%~5.18%。
如图8所示,对FC-MFC进行开路,以评估生物电营养反硝化对阴极室脱氮作用的影响。其中TN质量浓度大约为氨氮、亚硝酸盐氮和硝酸盐氮的质量浓和。在考虑NH4+从阳极室到阴极室的迁移量后,闭路时阴极室电解液TN去除率可分别高达95.71%(第1阴极室)、96.59%(第2阴极室)。开路时阴极室电解液TN去除率可分别高达95.44%(第1阴极室)、98.47%(第2阴极室)。整体而言,开路时系统对TN的去除率与闭路时系统对TN的去除率基本一致。原因可能是在高DO水平下,O2的氧化还原电位高于NO3−,故O2更容易作为阴极电极受体[33],因此,生物电营养反硝化基本可以忽略不计。如图5通过对阴极室中的pH进行监测,阴极室中的pH在7~7.3,故氨氮在电解液中绝大部分以离子形态存在,且迁移至阴极室中的氨氮立刻进行了硝化反应,故氨气提的情况也很难发生。因此,阴极室更有可能进行的反应是利用微生物自身有机物进行内源反硝化。
综上所述,阳极室微生物吸附代谢作用、阴极室内源反硝化、阴极室通过AEM迁移至后序位阳极室进行反硝化过程分别贡献了25.96%~25.97%、0.91%~5.18%、68.87%~73.20%。值得注意的是,尽管定量分析存在一些偏差,但上述过程对于理解FC-MFC氮的去除机理是十分必要的。
-
成本计算方法主要以1 L废水处理能力为基准。以往研究的单室MFC有效反应器体积为250 mL[8],本研究的阳极室与阴极室有效体积均为500 mL,2个阳极室与2个阴极室构成4个MFC模块,大致相当于4个单室MFC。FC-MFC与单室MFC工艺成本构成见表1。
从建设成本角度来看,单室MFC总计为2 825.60元而FC-MFC总计为2 553.87元。单室MFC因省去阴极室,阴极直接暴露在空气中,从而减少了离子交换膜和曝气成本。单室MFC省去离子交换膜虽然有利于功率密度的提高,但由于O2的渗入会对阳极生物膜产生负面影响,同时也会消耗大量的COD,导致电池的库伦效率下降。此外单室MFC通常构建同步硝化反硝化用于含氨废水的处理,臧华生等[34]研究了碳氮比对单室MFC的影响,研究结果表明,除去C/N比为1,氨氮的去除率随C/N比的增加而降低,该结论与王佳琪等一致[8]。造成该现象的原因可能是在碳源充足的情况下,异养菌会迅速繁殖,与硝化细菌竞争时占据优势。离子交换膜虽然增加了FC-MFC的建设成本,但离子交换膜的存在使得阴阳极室之间的干扰较小,离子交换膜一方面减少阴极室中的O2扩散到阳极,一方面减少阳极室有机物对阴极室自养硝化细菌的影响。为了更深入地了解FC-MFC的经济性,进行了初步的能量平衡分析。FC-MFC最主要的能量输出来源是电力产生过程,不同进水碳氮下每去除1kg氨氮其能量产出为1301.85~4759.46 kJ·kg−1,阴极室曝气的能耗为498.31~744.32 kJ·kg−1。因此,在FC-MFC中实现了803.54~4015.14 kJ·kg−1的净能量输出。
-
1) FC-MFC中阴阳极室的分隔膜通过CEM与AEM进行交替排列,前序位阳极室中的氨氮通过CEM迁移至后序位阴极室中进行硝化反应,硝化后的产物通过AEM迁移至后序位阳极室中进行反硝化,最终实现系统同步脱碳除氮,浓度差是导致离子迁移的主要动力。
2) FC-MFC中的反硝化反应和硝化反应分别在阳极室和阴极室进行,由于CEM和AEM的阻隔作用,阳极室COD对阴极室干扰较小,给阴极室提供了适宜自养硝化细菌繁衍的环境,这使得FC-MFC对高C/N比污水具有良好的抵抗负荷。
3)随着阳极COD的增大(即进水碳氮比的增加),各MFC模块的产电周期、峰值输出电压和最大功率密度随之增大,阳极室COD和TN去除率也随着增大,阴极室TN残留量则越低。
4)当进水COD和NH4+分别为1 100 mg·L−1和100 mg·L−1时,FC-MFC的污染物去除效果和产电性能最佳。阳极室COD和TN去除率分别高达94%和96%以上。阳极室微生物吸附代谢作用、阴极室内源反硝化、阴极室通过AEM迁移至后序位阳极室进行反硝化过程分别贡献了25.96%~25.97%、0.91%~5.18%、68.87%~73.20%。
四室微生物燃料电池同步脱氮除碳及产电性能
Simultaneous nitrogen and carbon removal and electricity generation in four-chamber microbial fuel cell
-
摘要: 微生物燃料电池近年来被证实可以用来同步脱氮,然而微生物燃料电池中阴阳极室通常以不同成分的污水作为底物。为了实现废水脱氮,往往需要进行出水调配或停曝等复杂的操作。为解决上述问题,本研究构建了阴极硝化耦合阳极反硝化的四室微生物燃料电池(four chamber microbial fuel cell,FC-MFC),阳极室与阴极室之间用阳离子交换膜(cation exchange membrane,CEM)与阴离子交换膜(anion exchange membrane,AEM)进行交替分隔。在浓度差作用下离子进行定向迁移,最终实现阳极室有机物和氨氮的同步去除。探讨了阳极COD(即进水碳氮比)对FC-MFC产电及污染物去除效果的影响,并分析FC-MFC的氮去除途径。结果表明:随着阳极室COD的增加,各MFC模块的产电周期、峰值输出电压和最大功率密度随之增加,同时阳极室COD和TN的去除率也呈上升趋势,该系统对高碳氮比污水具有良好的抵抗负荷。当进水COD和NH4+-N质量浓度分别为1 100 mg·L−1和100 mg·L−1时,4个MFC模块的峰值输出电压介于526~619 mV,最大功率密度为103.47~121.00 mW·m−2,阳极室COD去除率和TN去除率分别高达94%和96%以上。氮去除途径分析结果表明,阳极室微生物吸附代谢作用、阴极室内源反硝化、阴极室通过AEM迁移至后序位阳极室进行反硝化过程分别贡献了25.96%~25.97%、0.91%~5.18%、68.87%~73.20%。Abstract: The microbial fuel cell(MFC) has been demonstrated to be a promising method for nitrogen removal. However, in traditional MFC, the anode and cathode chambers utilize distinct wastewater components as substrates, requiring intricate processes, such as effluent allocation or stopping aeration, to achieve nitrogen removal. Here, we show the concept of simultaneous nitrification and denitrification that occurs in separate anode and cathode chambers rather than in the same cathode chamber. Cathodic nitrification coupled to anode denitrification for nitrogen removal was achieved in a four-chamber microbial fuel cell(FC-MFC). This system employed cation exchange and anion exchange membranes to alternate between anode and cathode chambers. This promoted the directional migration of ions under concentration gradients, which facilitated the concurrent removal of organic matter and ammonia in the anode chamber. The impact of anode COD on MFC power generation and pollutant removal efficiency was investigated and the nitrogen removal pathway of this FC-MFC system was examined. The results showed that the power generation cycles, peak output voltages and maximum power densities of each MFC module increased with the increase of anode COD, along with the increased removal rates of COD and TN in the anode chamber. Notably, this system demonstrated an excellent resilience to high carbon-nitrogen ratio wastewater. When the influent COD and NH4+-N concentrations were 1100 mg·L−1 and 100 mg·L−1, respectively, the peak output voltages were 526~619 mV and maximum power densities were 103.47~121.00 mW·m−2 for four MFC modules, the COD and TN removal rates in the anode chamber were over 94% and 96%, respectively. Nitrogen removal pathway analysis revealed that microbial adsorption and metabolism in the anode chamber, endogenous denitrification in the cathode chamber, and the AEM-mediated denitrification process in the post-order anode chamber contributed 25.96%~25.97%, 0.91%~5.18%, and 68.87%~73.20% to nitrogen removal, respectively.
-
危险废物是指具有腐蚀性、毒性、易燃性、反应性、感染性等危险特性,会对环境或人群健康带来有害影响的固体废物(包括液态废物)[1]。目前,我国危险废物无害化处理或处置仍以焚烧或填埋为主,其中焚烧处理的占比超过45%[2]。在危废产量逐步上升、城市土地资源紧张的背景下,焚烧仍是我国危废无害化处理的主要方式[3]。
目前,危废焚烧产生的烟气主要采用“选择性非催化还原(selective non-catalytic reduction,SNCR)+急冷塔+干法脱酸+活性炭+袋式除尘+湿法脱酸+烟气加热”工艺处理[4]。该工艺可脱除约50%的NOx,排放值一般为200~300 mg·m−3,很难满足NOx最新排放标准《危险废物焚烧污染控制标准》(GB 18484-2020)。此外,对烟气中重金属和二恶英的处理主要采用“活性炭吸附+布袋”工艺。该工艺可将气态二恶英吸附至活性炭中,而产生的废活性炭(颗粒状)被布袋拦截并转至飞灰中[5]。然而,飞灰仍为危险废物,需经预处理后再送至安全填埋场处置。另外,该工艺还存在活性炭与烟气混合不均的问题,亦会带来超标排放的风险。
近年来,由于污染物排放标准日趋严格、投资运营成本的增大,烟气净化技术逐步从“单一污染物控制”发展至“多污染物协同控制”[6-9],一体化协同脱除技术成为热点[10-14]。已被成功开发的一体化净化技术有活性炭吸附法[15]、脉冲电晕等离子体法[16]、催化布袋法等。然而,单独采用上述方法脱除NOx仍无法满足超低排放要求,故应组合成本较低、技术成熟的多种工艺对污染物进行脱除。
本团队提出了一种基于催化陶纤维滤管的烟气一体化净化技术,并依托江苏省某危废焚烧厂搭建中试示范工程,考察其对SO2、NOx、颗粒物及二恶英等污染物的去除效果,以期为危废焚烧烟气的集约、高效处理提供参考。
1. 工程实施方案及关键技术
1.1 工程概况
江苏省南通市某危废焚烧厂2018年建成投产,主要处理南通市、盐城市的危险废物。该厂的危废处理规模为10 000 t·a−1,采用“回转窑+二燃室”焚烧处理技术。目前,该焚烧厂的烟气净化系统采用“SNCR+急冷+干法脱酸+活性炭喷射+布袋除尘器+湿法脱酸+烟气再热”处理工艺。该危废焚烧处理生产线的余热锅炉出口烟气温度540~560℃,烟气污染物排放按照《危险废物焚烧污染控制标准》(GB 18484—2020)标准执行,余热锅炉出口烟气常规污染物浓度检测值及排放限值(24 h)见表1。另外,余热锅炉出口烟气二恶英浓度检测值(毒性当量值)为1.3~3.4 ng·Nm−3,其规定的排放限值为0.5 ng·Nm−3。在自主研发的基础上,通过工艺技术的集成优化,在该危废厂搭建了每小时烟气处理量为1 500 Nm3的多污染物一体化净化中试示范工程。该示范工程采用“喷淋急冷降温+高效消石灰干法脱酸+喷氨+催化陶纤管一体化”工艺。
表 1 余热锅炉出口烟气污染物浓度检测值及排放限值(24 h)Table 1. Values of pollutant concentration in exhaust gas of waste heat boiler and mission concentration limits (24 h)mg·Nm−3 污染物项目 余热锅炉出口浓度质量 烟气污染物排放浓度限值 颗粒物 18 200~26 500 20 SO2 240~1 160 80 NOx 250~480 mg 250 HCl 3 600~13 800 50 注:烟气污染物排放浓度限值执行国标《危险废物焚烧污染控制标准》(GB 18484-2020)。 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 工程关键技术及反应机理
该示范工程可实现烟气中SO2、NOx、颗粒物及二恶英等污染物的去除。其中,最重要的处理过程为脱酸、脱硝、除尘。脱酸处理即对烟气中酸性气体的去除:先用干法脱酸去除烟气中的主要酸性气体(SO2、HCl、HF等);之后残余酸性气体经过预喷涂NaHCO3或Ca(OH)2(消石灰)的滤管预涂层,实现进一步的脱酸。脱酸工艺反应见式(1)~(4),其过程参数应考虑合适的钙硫比、钙氯比,以达到较高的处理效率。
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) 脱硝处理主要发生在喷氨系统和催化陶瓷纤维滤管(以下简称“陶纤管”)反应器2个工艺段。首先喷入氨水雾化成氨气,并设定一定的氨氮比,将其与NOx充分混合。然后,在管壁外附着的SCR催化剂作用下,进行选择性催化还原反应,使得系统可保持较高的脱硝效率。脱硝处理的反应见式(5)和(6)。
stringUtils.convertMath(!{formula.content}) (5) stringUtils.convertMath(!{formula.content}) (6) 陶瓷纤维滤管以陶瓷纤维为原材料,在高温高压下用模具压制而成。普通陶纤管为白管,具有高温除尘功能,并可定期反吹再生。催化陶纤管即黄管,是在白管内部附着SCR系列催化剂制得。这表明催化陶纤管可同时实现高效除尘和脱硝的功能,因此,催化陶纤管为本示范工程的关键技术部件。催化陶纤管附着的负载催化剂为V2O5-WO3/TiO2,使得该反应器具有与传统SCR工艺接近的脱硝效率。而催化剂颗粒又具有多孔性、体积小、比表面积大等优点,可高效地催化气态反应且无扩散限制。再加上结构的原因,烟气在催化陶纤管中的流速(0.8~1.2 m·min−1)远远低于烟气在蜂窝式催化剂里的流速(6 m·s−1)。另外,在该示范工程中,烟气的停留时间较长,亦相当于增加了活性表面积,从而使得催化剂的利用率可逼近100%[16]。
1.3 处理工艺流程及工况
该示范工程的流程图及现场照片见图1,工程设计参数见表2,工况见表3。工艺流程主要包含如下4部分。1)降温脱酸处理。经余热锅炉出来的高温烟气(550℃)引出先降温,然后进入干式除酸系统进行净化。在脱酸连接烟道处设有石灰粉喷射装置,大比表面积的高效消石灰药剂被输送至此。此处的物料温度已降至350~370℃。在干法脱酸系统中,烟气中的SO2、HCl、HF等酸性成分被去除。2)随后为烟气喷淋氨水阶段。此阶段物料降温至200℃,且使得NH3与其他气态污染物得以充分混合,以发生进一步的反应。3)之后烟气进入催化陶纤管反应器。烟气中滞留的细微粉尘、前段脱酸系统的脱硫副产物及未反应完的脱硫剂粉末在进入反应器后,被吸附在陶纤管上,而未反应完全的脱硫剂则继续反应并被吸收。除尘后烟气经过陶纤管时与管壁附着的催化充分接触,烟气中的二恶英被催化分解最终实现达标排放。4)催化陶纤管反应器出口烟气再通过引风机返回布袋除尘器入口,并进入主烟道。
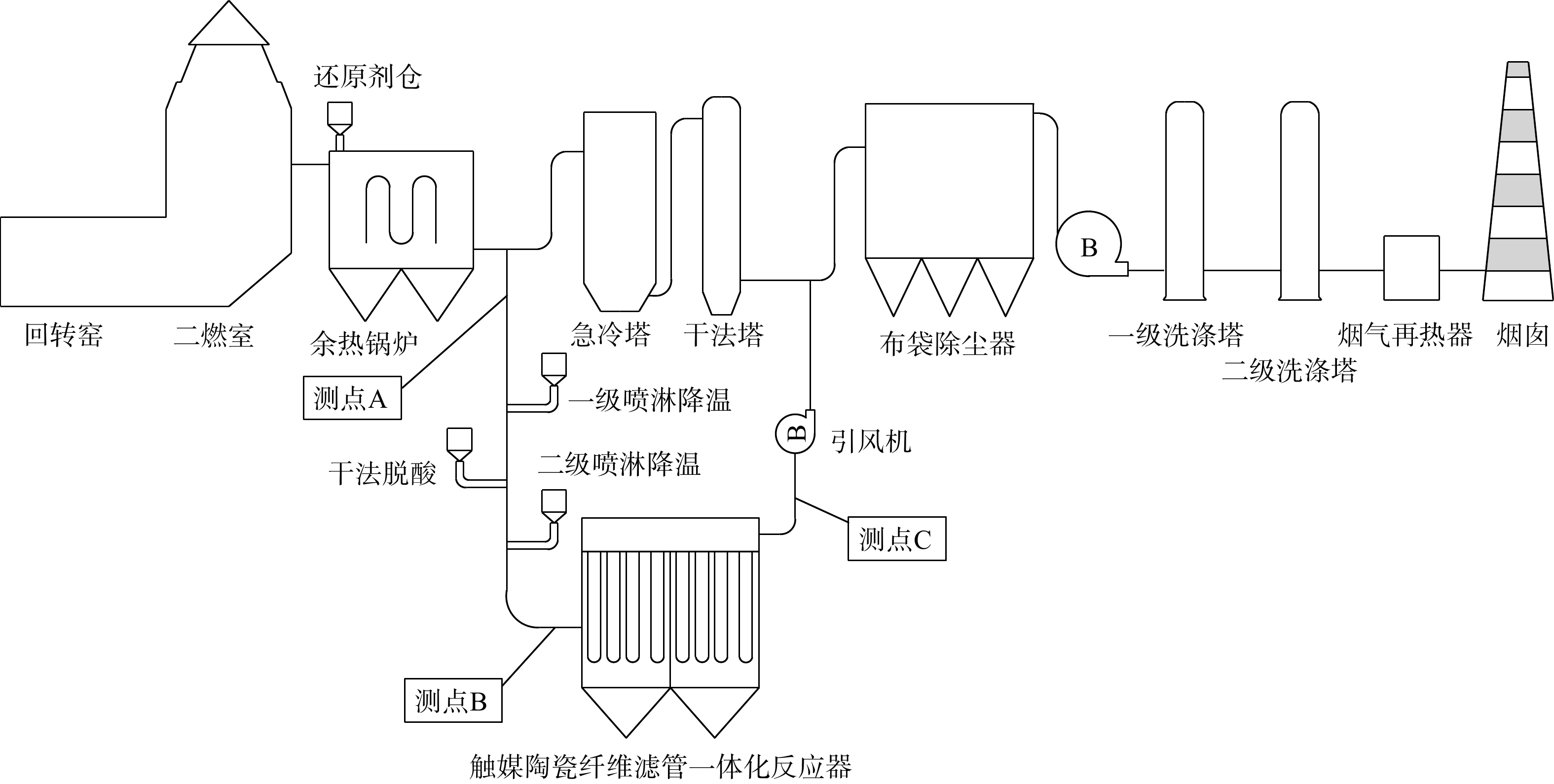 图 1 危废焚烧烟气一体化净化中试示范工程的工艺流程图Figure 1. Process flow chart of the pilot plant for co-processing of multi-pollutants of hazardous waste incineration gas表 2 示范工程的主要设计参数Table 2. Main design parameters of demonstration projects
图 1 危废焚烧烟气一体化净化中试示范工程的工艺流程图Figure 1. Process flow chart of the pilot plant for co-processing of multi-pollutants of hazardous waste incineration gas表 2 示范工程的主要设计参数Table 2. Main design parameters of demonstration projects流程单元 主要部件 工艺参数 喷淋急冷降温系统 水泵 流量300 L·h−1,压力1.0 MPa 喷枪 二流体喷枪,材质316 L 干法脱酸系统 干粉料仓 容积3 m3 螺旋喂料机 最大给料量50 kg·h−1 真空上料机 型号QVC-3,上料量1 500 kg·h−1 催化陶纤管反应器 催化陶纤管 规格L×D=3 000 mm×150 mm;数量36根;过滤风速0.8 m·min−1;压力损失1 300~1 800 Pa 氨水喷淋系统 仓室 1个 喷枪 二流体喷枪,材质316 L | Show TableDownLoad:
CSV
表 3 示范工程的工况Table 3. Working conditions of p ilot projects参数 取值 烟气量 1 000~2 000 Nm³·h−1 余热锅炉出口温度 540~560 ℃ 陶瓷纤维滤管入口温度 190~200 ℃ 钙硫比(Ca/S) 2 钙氯比(Ca/Cl) 1 氨氮比(NH3/NOx) 1.2 | Show TableDownLoad:
CSV
1.4 各工艺段内的运行及操作要点
1)喷淋急冷降温系统。本项目设置两段式喷淋降温,一用一备。一段设置在余热锅炉出口,将烟气温度由550℃降至中温(370℃),以满足干喷脱硫的最佳反应温度,提高脱硫效率,减少物料的消耗;二段喷淋降温将中温(370℃)烟气的温度降至200℃,以满足催化陶瓷纤维管分解二恶英的最佳反应温度,从而提高或保证二恶英的脱除效率。
2)干法脱酸系统。脱酸剂通过吨袋方式运至厂内吸收剂区域,再通过吨袋卸料站、真空上料机运送至粉仓内储存备用。粉仓中的脱酸剂再由粉体气力输送泵送至干喷脱酸段。吨包上料系统由配套导轨及2 t的电动葫芦组成,并配备振动器及除尘设备。料仓容积为3 m3,用于储存脱酸剂。螺旋喂料机的电机为变频电机系统。
3)氨水喷淋系统。由氨水喷射系统将氨水(体积分数25%)引至催化陶纤管一体化反应器前烟道中,完成对烟气的喷淋过程。该过程使得烟气与氨充分混合并进入催化陶纤管反应器。
4)催化陶纤管反应器。该段反应器由陶纤管、仓室、灰斗、钢结构支架、喷吹系统及卸灰系统等组成。本工程共配置36支催化陶纤管。单仓陶纤管采用6×6布置,共1个仓室。烟气在仓室中为下进上出。仓室和整个反应器均为直立式焊接钢结构容器。其中,内部设有高温复合陶瓷滤筒支撑结构,能承受内部压力、地震负荷、烟尘负荷、催化负荷及热应力等;外部设有加固肋及保温层。仓体入口设气流均布装置,即在仓体入口及出口段设导流板,在入口设整流装置。清灰采用压缩空气低压脉冲方式。仓体灰斗设有1个排灰口(尺寸为200 mm×200 mm),以避免灰尘搭桥,使反应器能承受长期的温度、湿度变化及振动等。同时,反应器中接触高温烟气部件均采用Q345B标准的钢材制作。
2. 运行效果分析
2.1 系统的除尘特性
在陶纤管入口烟气的温度为190~200 ℃、过滤速度为0.8 m·min−1的条件下,分别在余热锅炉出口A、陶纤管入口B、陶纤管出口C布置采样点(采样点布置下同),监测烟气颗粒物的含量,并取算术平均值进行分析。结果表明:A点颗粒物平均质量浓度为26 427.8 mg·Nm−3;喷射干粉后的B点颗粒物平均质量浓度为60 928.2 mg·Nm−3(实际干粉喷射量为38 000 mg·Nm−3,故此处数据比A点高);经陶纤管除尘器除尘后C点颗粒物平均质量浓度为8.99 mg·Nm−3。计算得到陶纤管对颗粒物去除效率达99.9%。因此,本项目采用的陶纤管具有极高的除尘效率,可保证烟气出口颗粒物质量浓度低于20 mg·Nm−3。
陶纤管具有高孔隙率结构,其孔隙直径为2~3 μm。烟气进入其中后通过表面过滤,粒径较大的粉尘在重力作用下沉降,粒径小的粉尘停留在滤料表面,形成尘饼层。当进行反向脉冲清灰时,附着在表面的尘饼层被剥离落入灰斗。因此,陶纤管除了具备较好的除尘效果,还可通过反向脉冲实现循环使用。
2.2 系统脱酸脱硝效果
干法脱酸采用高效消石灰进行。在反应温度350~370 ℃时,考察了Ca/S=2、Ca/Cl=1条件下工程的干法脱酸性能。另外,在喷淋阶段采用25%氨水做还原剂,同时将陶纤管进口烟气温度降至190~200℃,以考察其在NH3/NOx=1.2条件下的脱硝特性。分别在A、B、C点进行3次采样测试烟气中的SO2、HCl、NOx含量(取算术平均值进行分析),结果见表4。
表 4 SO2、HCl和NOx脱除效果Table 4. The removal efficiency of SO2, HCl and NOx污染物类型 不同采样点的污染物质量浓度/(mg·Nm−3) 脱除效率 余热锅炉出口A 陶纤管入口B 陶纤管出口C SO2 865.8 129.6 63.9 92.6% HCl 3 310.8 16.1 10.2 99.7% NOx 364.6 355.8 39.8 89.1% | Show TableDownLoad:
CSV
在干法脱酸阶段,温度为350~370 ℃的烟气被喷入脱酸药剂后,药剂中的Ca(OH)2与SO2、HCl等酸性污染物反应,发生一次脱酸过程。烟气中的大部分酸性污染物在此阶段被去除。在烟气进入陶纤管反应器后,携带脱酸药剂的小粒径粉尘沉积在滤管表面,形成尘饼。当烟气冲刷陶纤管外表面时,烟气中的酸性气体在陶瓷管表面的尘饼层上进行二次脱酸过程。两次脱酸过程后,烟气出口处的SO2质量浓度低于80 mg·Nm−3,HCl质量浓度低于50 mg·Nm−3。
本工程中主要脱硝工艺在“喷氨+催化陶纤管”两段,其脱硝效率可达到传统蜂窝状SCR体系的脱硝水平。在烟气出口处,NOx质量浓度远低于200 mg·Nm−3。对运行寿命进行对比,传统SCR催化剂寿命仅约为2 a,而催化陶纤管可正常运行超过8 a,故本工程采用的脱硝工艺比传统工艺更有优势。
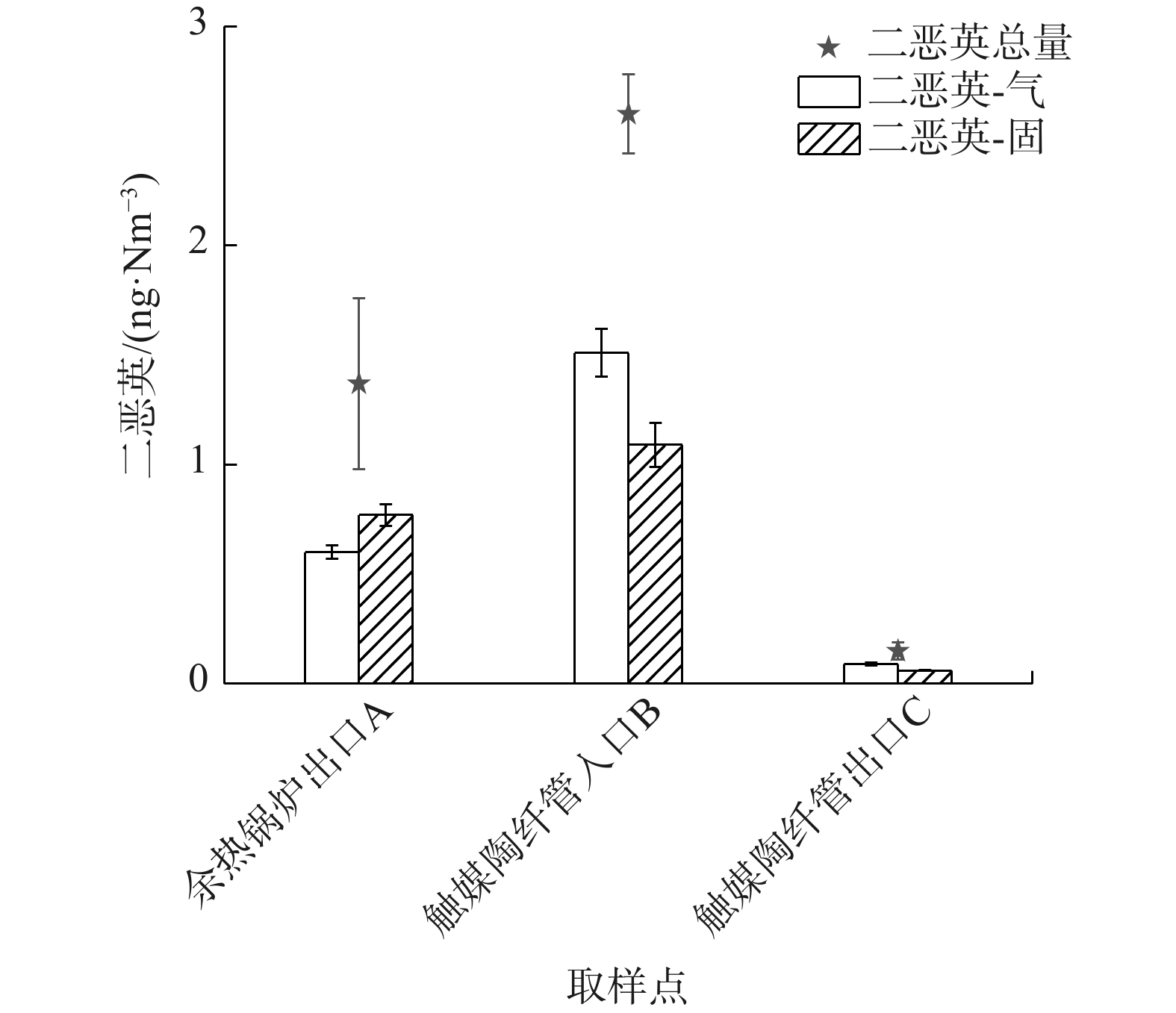
2.3 系统对二恶英的去除效果
图2为A、B、C 3个测点的二恶英浓度检测结果。其中,A点二恶英质量浓度为1.37 ng·Nm−3;在降温至200℃后于B点发生二恶英低温合成后,其质量浓度为2.6 ng·Nm−3;在C点二恶英质量浓度为0.15 ng·Nm−3。计算得到催化陶纤管对二恶英去除效率为94.2%,且其质量浓度低于目标值0.5 ng·Nm−3。
烟气中二恶英以气态和固态2种形态存在。A点数据表明,气态和固态二恶英质量浓度分别为0.60和 0.77 ng·Nm−3。由余热锅炉出口喷淋降温至200℃以下后,在B点测得气态和固体二恶英质量浓度分别为1.51和 1.09 ng·Nm−3。这表明降温后气态二恶英的合成占比更多。其中,气态二恶英合成2.52倍、固态二恶英合成1.42倍。烟气经过催化陶纤管反应器后,在C点测得气态和固态二恶英质量浓度分别为0.09和 0.06 ng·Nm−33。这表明陶纤管对气态二恶英的分解效率为94.0%,对固态二恶英的截留效率为94.5%。
气态二恶英去除机理见式(7)。气态二恶英在高温状态下穿过陶纤管,并与附着的催化剂接触,在钒基催化剂的作用下,与氧气发生反应,被氧化分解[16]。而固态二恶英的去除则为截留和阻断其二次生成的过程。固态二恶英存在于焚烧飞灰表面,经过陶纤管时被拦截,最终进入灰斗。因此,陶纤管也阻隔了飞灰表面金属氧化物与烟气中其他污染物,避免其进一步反应生成二恶英。
stringUtils.convertMath(!{formula.content}) (7) 3. 示范工程的运行成本
将本示范工程的“喷淋急冷降温+高效消石灰干法脱酸+喷氨+催化陶纤管一体化”烟气净化工艺,与同等处理规模的传统“急冷+干法脱酸+布袋除尘器+湿法脱酸+SGH升温”工艺进行对比,分析其运行经济性,结果见表5。
表 5 本工艺与传统工艺的运行费用对比Table 5. Operation cost comparison of this process and the traditional process项目 本工艺费用/万元 传统工艺费用/万元 备注 关键部件的维护 1.26 1.2 本工艺每8年更换陶纤管;传统工艺每3年更换布袋 消石灰的消耗 3.6 3.6 药剂种类和消耗均相同 压缩空气的使用 2.0 2.0 药剂种类和消耗均相同 SCR催化剂的成本 0 5.0 本工艺的催化剂附着于管壁,无需单独再购买催化剂,此处成本已含入关键部件维护中;传统工艺用到的催化剂每2年需更新 蒸汽的使用 0 2.4 传统工艺需额外使用蒸汽对SCR催化剂进行升温 氨水的使用 1.3 1.3 药剂种类和消耗均相同 NaOH的使用 0 4.0 传统工艺需额外使用NaOH为湿法药剂 电费 8.2 14.4 本工艺的电耗主要在风机、电动葫芦等,无需升温;传统工艺需加热使用蒸汽 人工成本 18 18 均设置2人即可 合计 34.36 51.9 — | Show TableDownLoad:
CSV
该示范工程的运行成本主要包括:陶纤管成本与维护费用、药剂费用、电耗和人工费等。与传统工艺相比,没有催化剂的更新、升温及NaOH药剂的使用等成本,在操作上也更简便。在考虑折旧等消耗后,按年运行8 000 h计算,得到该示范工程的年平均运行成本为34.36万元。这表明本示范工程与传统工艺相比,运行成本降低了至少30%。
另外,本示范工程为一体化净化工艺,具有处理效率更高、占地面积小、投资费用低等优点,可降低危废处置企业的投资运行成本,并确保危废焚烧烟气的达标排放。因此,本示范工程为危废烟气的净化提供了一种集约、高效的一体化解决方案,亦可为现役危废焚烧烟气净化工程的改造及新建项目实现深度净化提供参考。
-
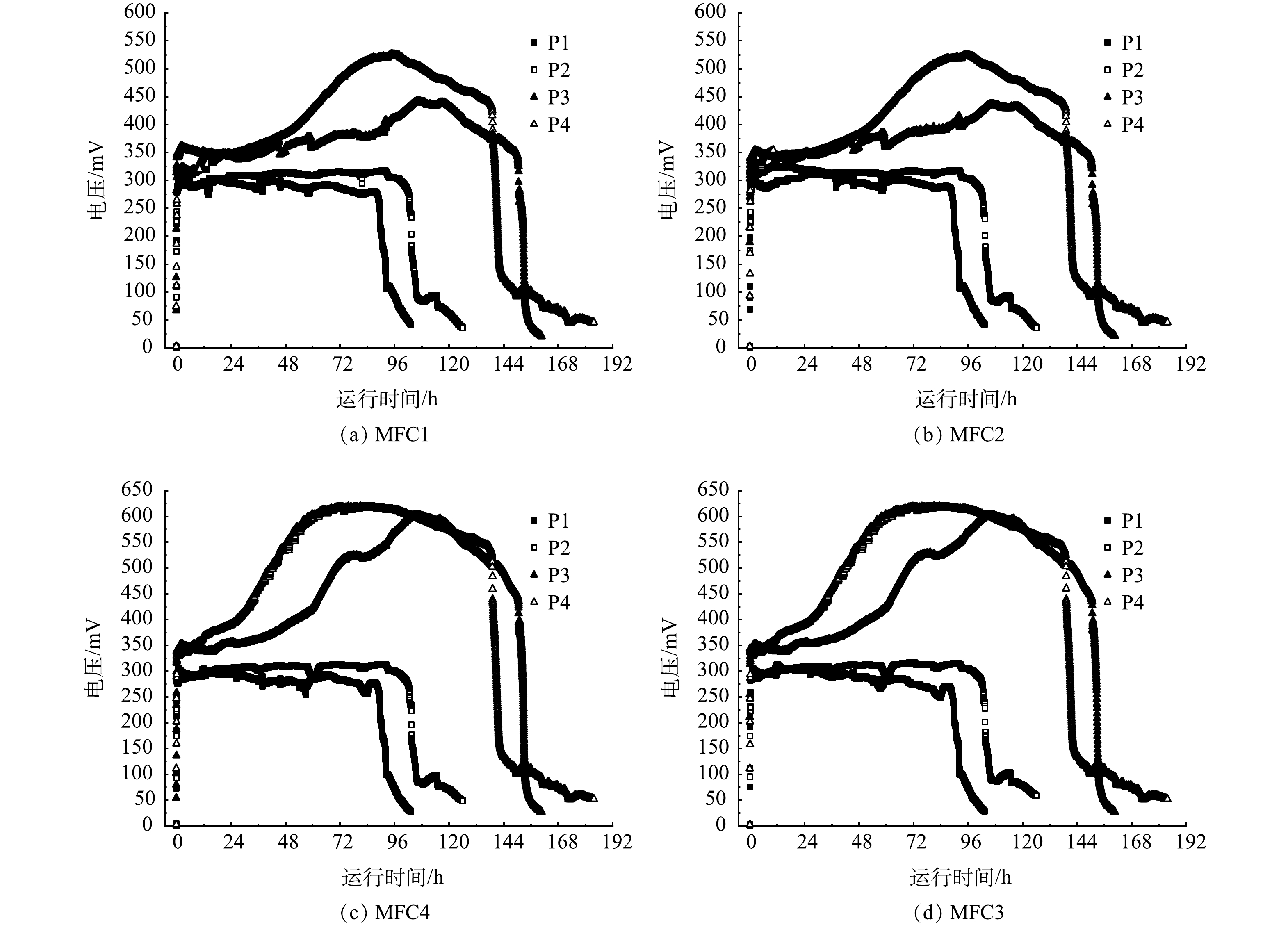
图 2 不同阳极COD下FC-MFC中各MFC模块的电压输出
Figure 2. Voltage outputs of each MFC module in FC-MFC at different anode COD
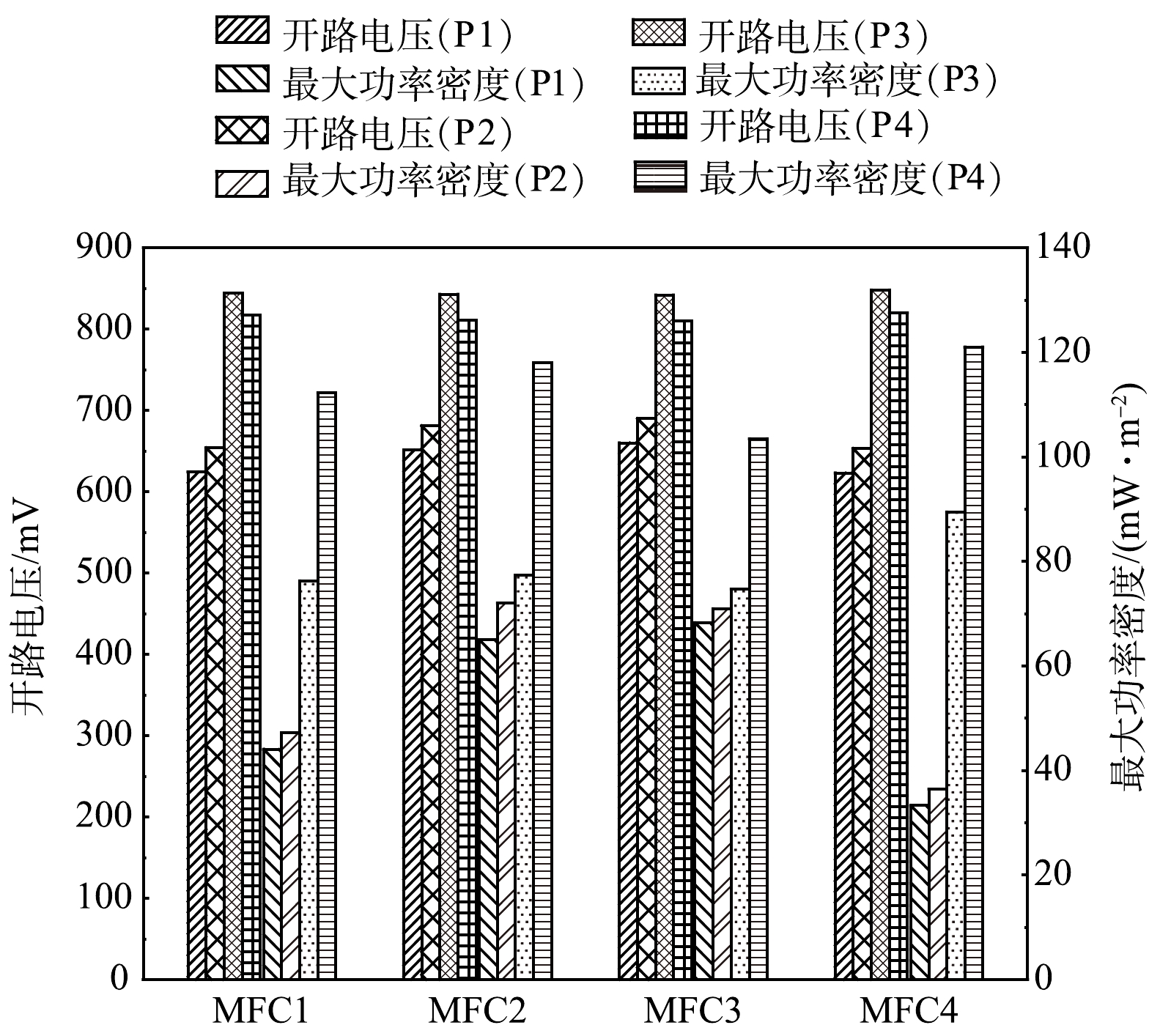
图 3 各个MFC模块在不同阳极COD下的开路电压和最大功率密度
Figure 3. Open circuit voltages and maximum power densities of each MFC module at different anode COD
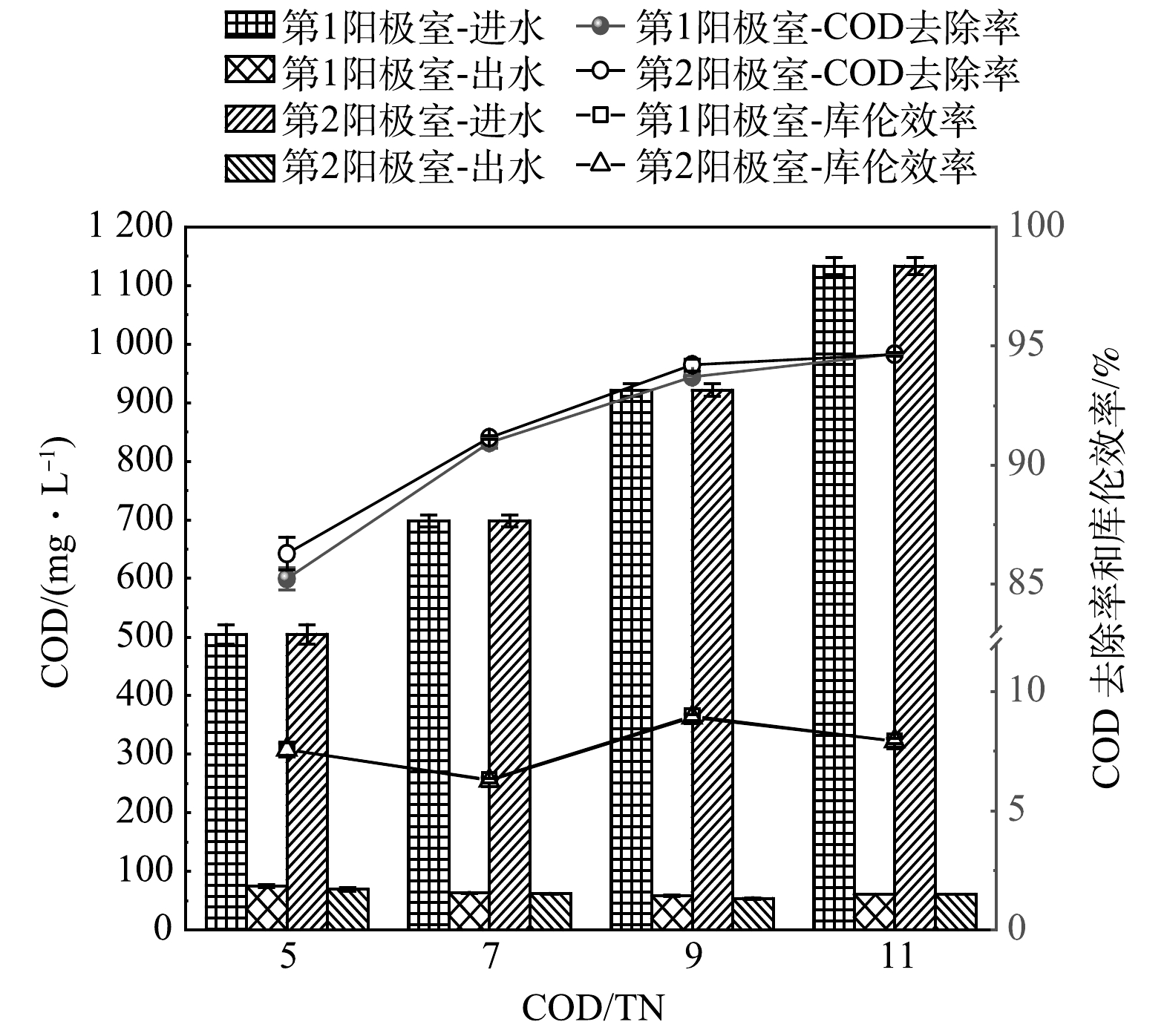
图 4 不同进水碳氮比下COD去除情况和库伦效率
Figure 4. COD removal and coulomb efficiencies at different carbon-nitrogen ratios
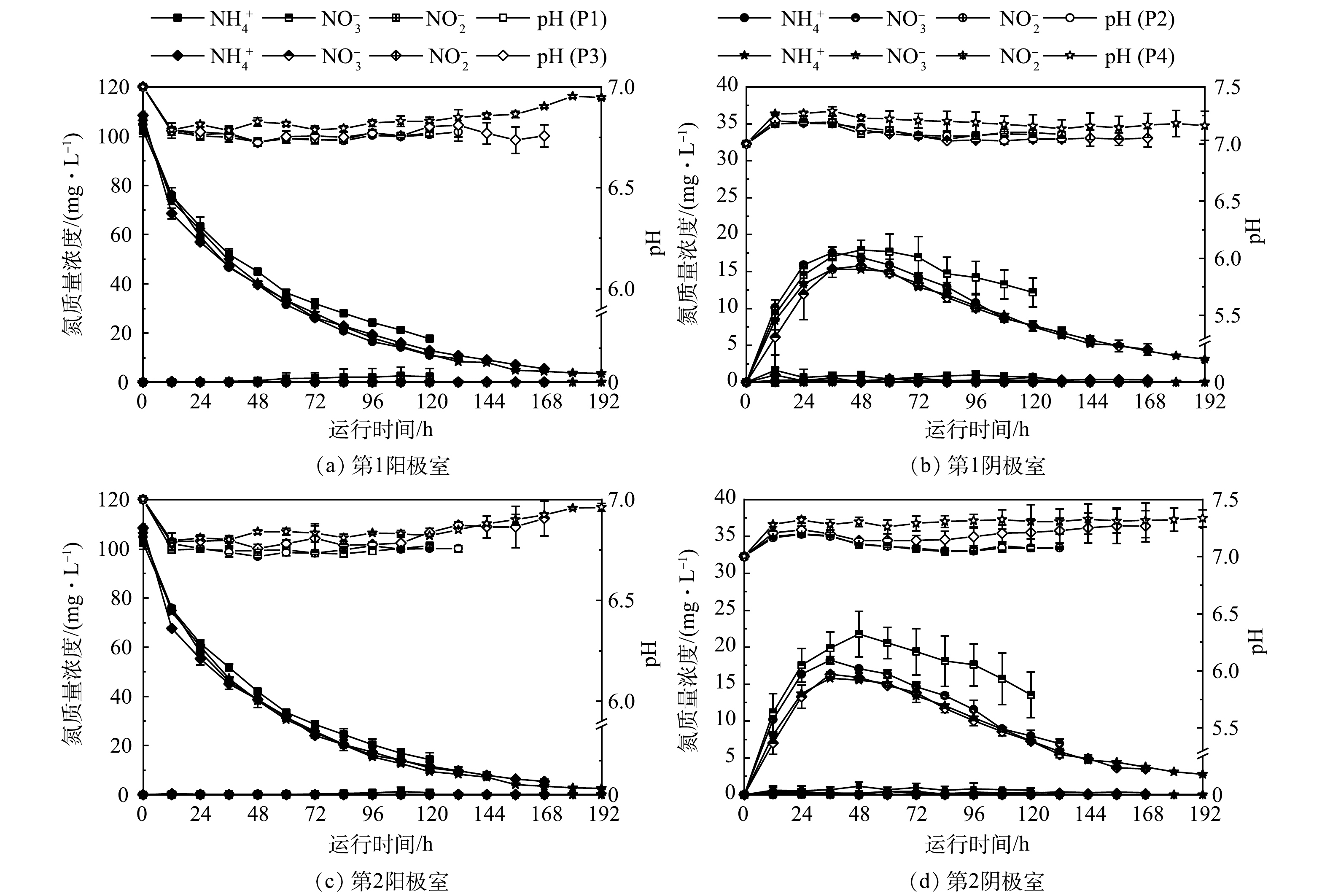
图 5 不同阳极COD下FC-MFC电极室电解液氮质量浓度和pH的变化
Figure 5. The Changes in electrolyte nitrogen concentration and pH in FC-MFC electrode chambers at different anode COD
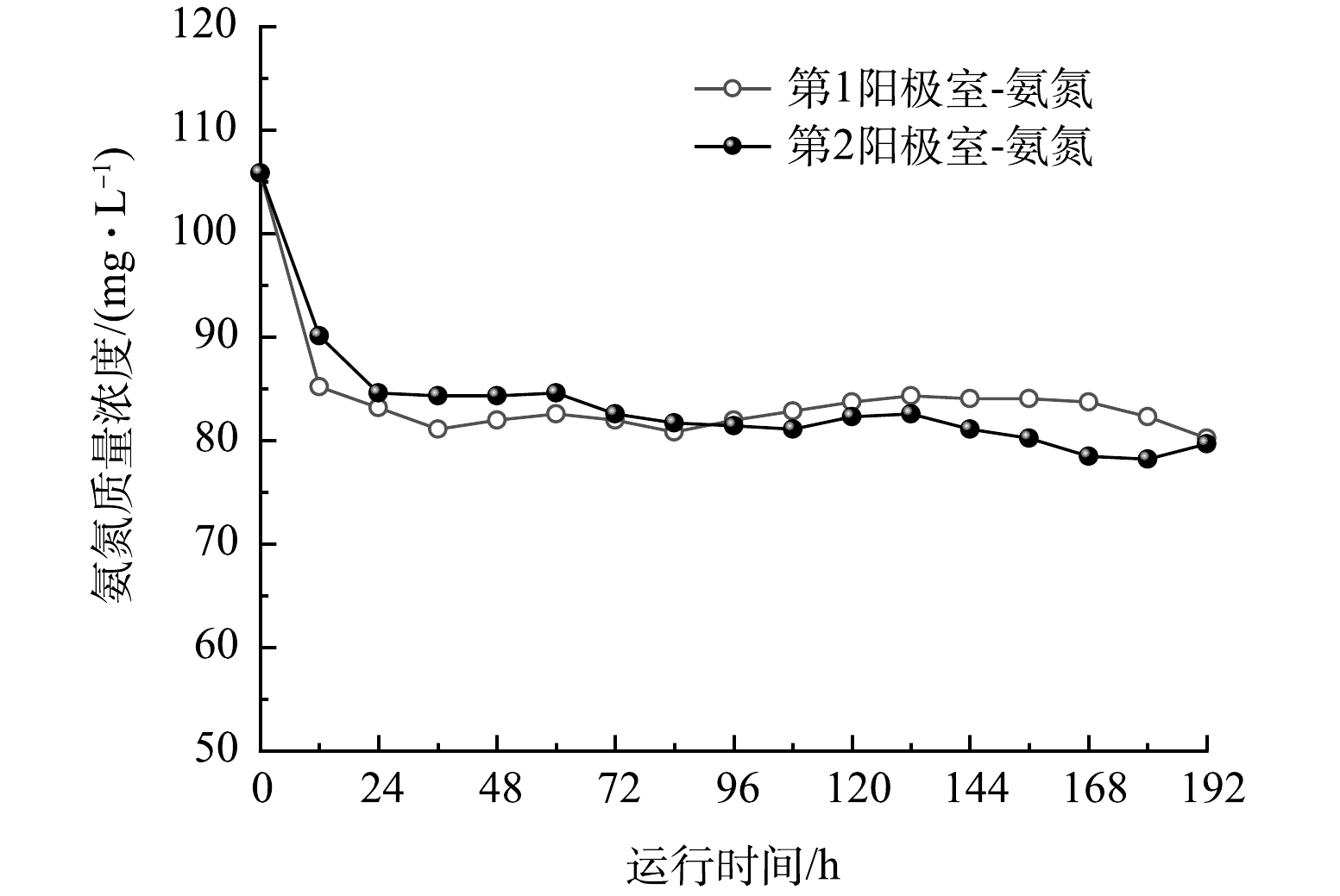
图 6 阳极微生物吸附代谢作用对氨氮质量浓度的影响
Figure 6. Effect of microbial adsorption metabolism on ammonia nitrogen concentration
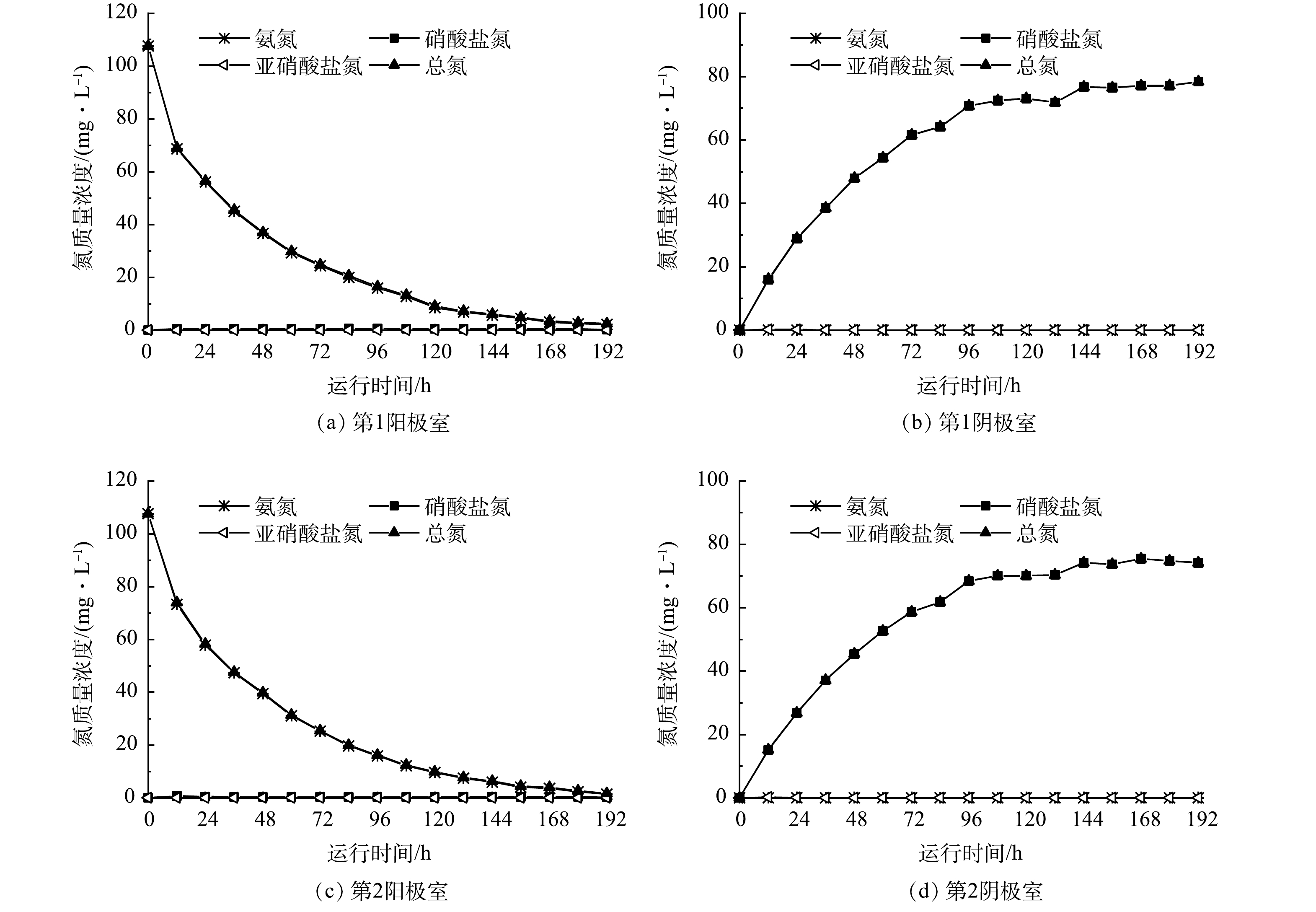
图 7 阴离子传输通道阻隔下FC-MFC电极室电解液氮质量浓度的变化
Figure 7. Changes in electrolyte nitrogen concentration in FC-MFC electrode chambers under the barrier of the anion transport channel
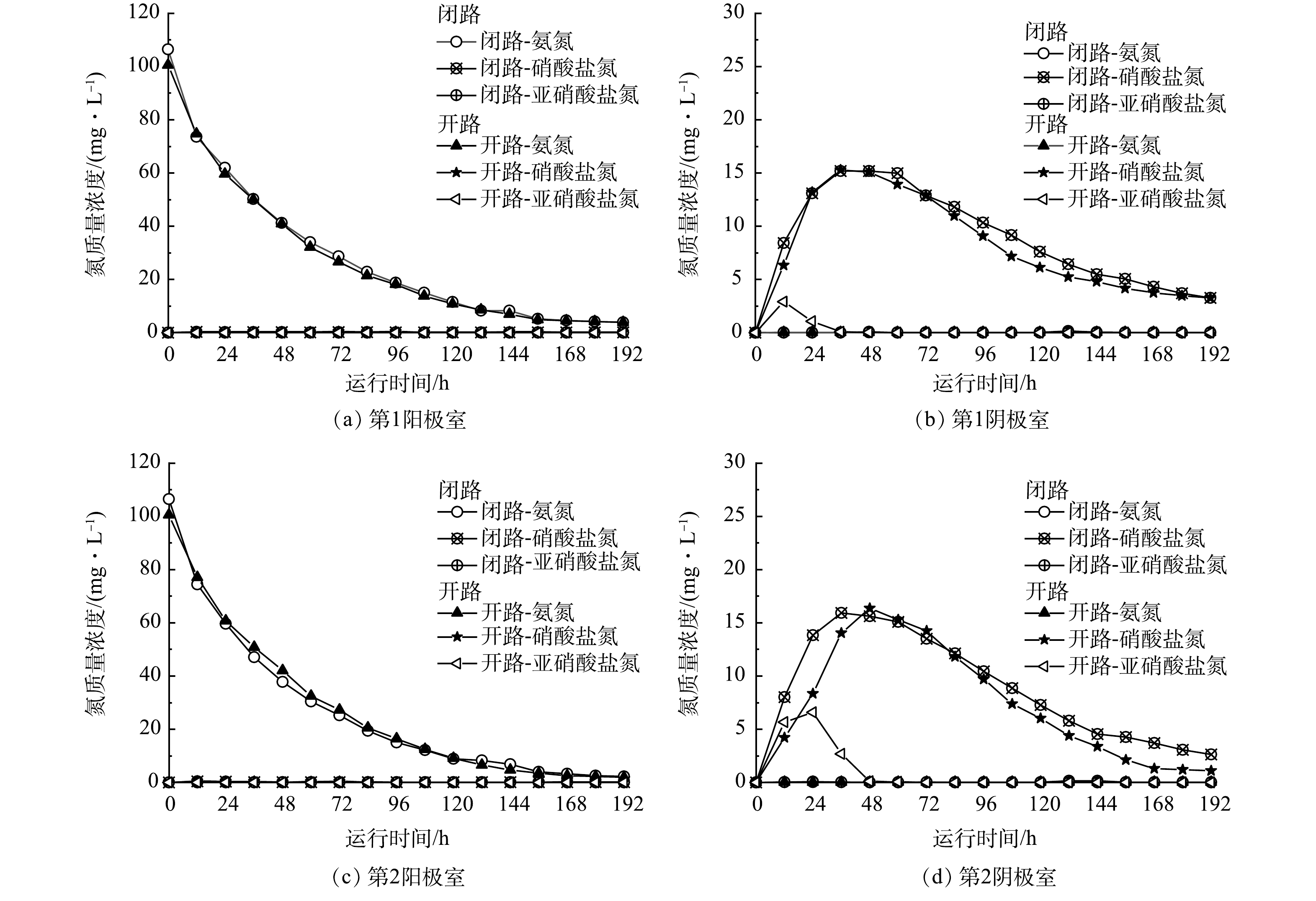
图 8 开路与闭路状态下FC-MFC电极室电解液氮质量浓度的变化
Figure 8. Changes in electrolyte nitrogen concentration in FC-MFC electrode chambers under open-circuit and closed-circuit conditions
表 1 FC-MFC和单室MFC工艺成本构成
Table 1. The cost structure of FC-MFC and single chamber MFC
部件 FC-MFC 单室MFC(4个) 材料 成本/元 材料 成本/元 框架 有机玻璃 2 400 有机玻璃 2 400 阳极 碳毡 5.94 碳毡 5.94 阴极 碳毡 5.94 碳布(20%的聚 四氟乙烯溶液和20%的Pt/C催化剂等) 147.93 曝气设施 空气泵/转子流量计 113 — — 离子交换膜 CEM/AEM 300.72 — —
下载: 导出CSV
-
[1] PARSONS C, STUEKEN E E, ROSEN C J, et al. Radiation of nitrogen-metabolizing enzymes across the tree of life tracks environmental transitions in Earth history[J]. Geobiology, 2021, 19(1): 18-34. doi: 10.1111/gbi.12419 [2] AHMED S F, KUMAR P S, KABIR M, et al. Threats, challenges and sustainable conservation strategies for freshwater biodiversity[J]. Environment Research, 2022, 214(1): 113808. [3] YE Y, NGO H H, GUO W, et al. Impacts of hydraulic retention time on a continuous flow mode dual-chamber microbial fuel cell for recovering nutrients from municipal wastewater[J]. Science of the Total Environment, 2020, 734: 139220. doi: 10.1016/j.scitotenv.2020.139220 [4] ASAI Y, MIYAHARA M, KOUZUMA A, et al. Comparative evaluation of wastewater-treatment microbial fuel cells in terms of organics removal, waste-sludge production, and electricity generation[J]. Bioresources and Bioprocessing, 2017, 4(1): 30. doi: 10.1186/s40643-017-0163-7 [5] YANG N, ZHAN G, LI D, et al. Complete nitrogen removal and electricity production in Thauera-dominated air-cathode single chambered microbial fuel cell[J]. Chemical Engineering Journal, 2019, 356: 506-515. doi: 10.1016/j.cej.2018.08.161 [6] JIN X, YANG N, LIU H, et al. Membrane penetration of nitrogen and its effects on nitrogen removal in dual-chambered microbial fuel cells[J]. Chemosphere, 2022, 297: 134038. doi: 10.1016/j.chemosphere.2022.134038 [7] NGUYEN H D, BABEL S. A novel coupled microbial fuel cell operation for organic and nitrogen removal with simultaneous energy recovery from wastewater[J]. Sustainable Energy Technologies and Assessments, 2023, 55:102981. [8] 王佳琪, 付国楷, 黄梓良, 等. 碳氮比对高盐废水单室MFCs产电、污染物去除及微生物群落结构的影响[J]. 环境工程学报, 2021, 15(4): 1354-1366. doi: 10.12030/j.cjee.202009094 [9] HUANG G, ZHANG Y, QU L, et al. Denitrification performance of ce-doped birnessite modified cathode in bioelectrochemical system[J]. Journal of Electroanalytical Chemistry, 2020, 871:114313. [10] PUIG S, SERRA M, VILAR-SANZ A, et al. Autotrophic nitrite removal in the cathode of microbial fuel cells[J]. Bioresource Technology, 2011, 102(6): 4462-4467. doi: 10.1016/j.biortech.2010.12.100 [11] CLAUWAERT P R K, AELTERMAN P. Biological denitrification in microbial fuel cells[J]. Environmental Science & Technology, 2007, 41(9): 3354-3360. [12] DING A, ZHAO D, DING F, et al. Effect of inocula on performance of bio-cathode denitrification and its microbial mechanism[J]. Chemical Engineering Journal, 2018, 343: 399-407. doi: 10.1016/j.cej.2018.02.119 [13] VIRDIS B, RABAEY K, YUAN Z, et al. Microbial fuel cells for simultaneous carbon and nitrogen removal[J]. Water Research, 2008, 42(12): 3013-3024. doi: 10.1016/j.watres.2008.03.017 [14] VIRDIS B, RABAEY K, ROZENDAL R A, et al. Simultaneous nitrification, denitrification and carbon removal in microbial fuel cells[J]. Water Research, 2010, 44(9): 2970-2980. doi: 10.1016/j.watres.2010.02.022 [15] ZHANG L, FU G, ZHANG Z. Long-term stable and energy-neutral mixed biofilm electrode for complete nitrogen removal from high-salinity wastewater: Mechanism and microbial community[J]. Bioresource Technology, 2020, 313: 123660. doi: 10.1016/j.biortech.2020.123660 [16] ZHANG Y, XU Q, HUANG G, et al. Effect of dissolved oxygen concentration on nitrogen removal and electricity generation in self pH-buffer microbial fuel cell[J]. International Journal of Hydrogen Energy, 2020, 45(58): 34099-34109. doi: 10.1016/j.ijhydene.2020.09.110 [17] 黄丽巧, 易筱筠, 韦朝海, 等. 阴极硝化耦合阳极反硝化实现微生物燃料电池技术脱氮[J]. 环境工程学报, 2015, 9(10): 5118-5124. doi: 10.12030/j.cjee.20151081 [18] R L B E H B R. Microbial fuel cells: Methodology and technology[J]. Environmental Science & Technology, 2006, 40: 5181-5192. [19] YI T, HARPER W F. The effect of nitrate and sulfate on mediator-less microbial fuel cells with high internal resistance[J]. Water Environment Research, 2009, 81(11): 2320-2328. doi: 10.2175/106143009X407267 [20] 张吉强, 郑平, 何崭飞. 废水中硝氮和 COD 浓度对 AD-MFC 脱氮产电性能的影响[J]. 环境工程学报, 2014, 8(10): 4508-4514. [21] VIRDIS B, READ S T, RABAEY K, et al. Biofilm stratification during simultaneous nitrification and denitrification (SND) at a biocathode[J]. Bioresource Technology, 2011, 102(1): 334-341. doi: 10.1016/j.biortech.2010.06.155 [22] DI LORENZO M, CURTIS T P, HEAD I M, et al. A single-chamber microbial fuel cell as a biosensor for wastewaters[J]. Water Research, 2009, 43(13): 3145-3154. doi: 10.1016/j.watres.2009.01.005 [23] JIN X, GUO F, MA W, et al. Heterotrophic anodic denitrification improves carbon removal and electricity recovery efficiency in microbial fuel cells[J]. Chemical Engineering Journal, 2019, 370: 527-535. doi: 10.1016/j.cej.2019.03.023 [24] ZHANG L, FU G, ZHANG Z. High-efficiency salt, sulfate and nitrogen removal and microbial community in biocathode microbial desalination cell for mustard tuber wastewater treatment[J]. Bioresource Technology, 2019, 289: 121630. doi: 10.1016/j.biortech.2019.121630 [25] HUANG H, CHENG S, YANG J, et al. Effect of nitrate on electricity generation in single-chamber air cathode microbial fuel cells[J]. Chemical Engineering Journal, 2018, 337: 661-670. doi: 10.1016/j.cej.2017.12.150 [26] ZHANG L, FU G, ZHANG Z. Simultaneous nutrient and carbon removal and electricity generation in self-buffered biocathode microbial fuel cell for high-salinity mustard tuber wastewater treatment[J]. Bioresource Technology, 2019, 272: 105-113. doi: 10.1016/j.biortech.2018.10.012 [27] ROZENDAL R A H H V M, BUISMAN C J N. Effects of membrane cation transport on pH and microbial fuel cell[J]. Environmental Science & Technology, 2006, 40(17): 5206-5211. [28] KUNTKE P, SMIECH K M, BRUNING H, et al. Ammonium recovery and energy production from urine by a microbial fuel cell[J]. Water Research, 2012, 46(8): 2627-2636. doi: 10.1016/j.watres.2012.02.025 [29] FENG C, HUANG L, YU H, et al. Simultaneous phenol removal, nitrification and denitrification using microbial fuel cell technology[J]. Water Research, 2015, 76: 160-170. doi: 10.1016/j.watres.2015.03.001 [30] TAO Q, LUO J, ZHOU J, et al. Effect of dissolved oxygen on nitrogen and phosphorus removal and electricity production in microbial fuel cell[J]. Bioresource Technology, 2014, 164: 402-407. doi: 10.1016/j.biortech.2014.05.002 [31] CHEN C, SUN F, ZHANG H, et al. Evaluation of COD effect on anammox process and microbial communities in the anaerobic baffled reactor (ABR)[J]. Bioresource Technology, 2016, 216: 571-578. doi: 10.1016/j.biortech.2016.05.115 [32] PARK W, NAM Y K, LEE M J, et al. Simultaneous nitrification and denitrification in a CEM (cation exchange membrane)-bounded two chamber system[J]. Water Research, 2009, 43(15): 3820-3826. doi: 10.1016/j.watres.2009.05.039 [33] ZHANG Y, ANGELIDAKI I. Bioelectrode-based approach for enhancing nitrate and nitrite removal and electricity generation from eutrophic lakes[J]. Water Research, 2012, 46(19): 6445-6453. doi: 10.1016/j.watres.2012.09.022 [34] 臧华生, 周新国, 李会贞, 等. pH值和碳氮比对微生物燃料电池脱氮除磷效果的影响[J]. 灌溉排水学报, 2019, 38(2): 49-55. doi: 10.13522/j.cnki.ggps.20180084 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2610
- HTML全文浏览数: 2610
- PDF下载数: 75
- 施引文献: 0
